
Submitted:
18 March 2024
Posted:
18 March 2024
You are already at the latest version
Abstract
Hemangioblasts give rise to endothelial progenitor cells (EPCs), which also express the cell surface markers CD133 and C-kit. They may differentiate into the outgrowth endothelial cells (OECs) that control neovascularization in the developing embryo. Reduced levels of EPC in circulation have been linked to human cardiovascular disorders, according to numerous studies. Furthermore, preeclampsia and senescence have been linked to levels of EPCs produced from cord blood. Uncertainties surround how preeclampsia affects the way EPCs function. It is reasonable to speculate that preeclampsia may have an impact on the function of fetal EPCs during the in utero period; however, given the present literature's suggestion that maternal vasculopathies, including preeclampsia, damage fetal circulation.
Additionally, the differentiation potential and general activity of EPCs may serve as an indicator of the health of the fetal vascular system since they promote neovascularization and repair during pregnancy. Thus, the purpose of this review is to compare, through assessment of their quantity, differentiation potency, angiogenic activity, and senescence, the angiogenic function of fetal EPCs obtained from cord blood for normal and pregnancy problems. This will shed light on the relationship between the angiogenic function of fetal EPCs and pregnancy complications, which could have an effect on the management of long-term health issues like metabolic and cardiovascular disorders in offspring with abnormal vasculature development.
Keywords:
Endothelial progenitor cell
; pre-eclampsia
; Gestational diabetes mellitus
; Fetal growth restriction
; angiogenesis
1. Introduction
A new paradigm for endothelial regeneration involving angiogenesis and vasculogenesis was established in 1997 with the identification of endothelial progenitor cells (EPCs) by Asahara et al. [1]. EPCs have been found in cord blood and circulating mononuclear cells (MNCs) [1,2] , and have been identified as being crucial to vascular homeostasis. A crucial finding is that the quantity and function of endothelial proliferators (EPCs) are reduced as a result of senescence in conditions involving endothelial dysfunction, such as diabetes mellitus and cardiovascular disease [3,4], where the EPC count is a reliable indicator of future cardiovascular events [5]. Accelerated aging of these cells has also been linked to decreased EPC activity and number [6,7]. These data led to the hypothesis that diseases associated with endothelial dysfunction might be caused by the reduction of circulating EPC.
Pre-eclampsia (PE) and intrauterine growth restriction in particular are regarded to be pathogenesis-related conditions that are influenced by aberrant vasculature and impaired endothelial function on both the maternal and fetal sides. The etiology of abnormal vessel development could perhaps result from decreased EPC function or bioavailability. Consequently, these pregnancy problems may be a result of impaired EPC function.
Pregnancies affected by preeclampsia or fetal growth restriction have been found in multiple studies to have a decreased number and abnormal function of cord blood-derived EPCs [8,9].
When the fetus is growth constrained, PE is linked to poor chorionic villous development and fetoplacental angiogenesis [10]. Significant decreases in cord blood EPCs and cord plasma free VEGF, which is known to be involved in EPC mobilization, were observed in severe preeclampsia [11]. Furthermore, in comparison to the normal-derived EPCs, the preeclampsia-derived CD133+/C-kit+/Lin− EPCs showed reduced OEC migration, adhesion, and tube formation activities, as well as delayed differentiation times and a significant decrease in the number of generated OEC colonies [8].
Maternal hyperglycemia caused by glucose intolerance that initially manifests during pregnancy is known as gestational diabetes mellitus (GDM). Hyperglycemia inhibits the clonogenic growth of cord blood endothelial colony-forming cells (ECFCs) by causing premature senescence. Additionally, ECFCs exposed to a diabetic environment during pregnancy display lower proliferation and premature senescence. [12,13].
One pregnancy condition that is clinically relevant is fetal growth restriction (FGR). A birth weight that is at or below the 10th percentile for gestational age is commonly used to determine FGR. When compared to the normal control group, the fetal EPC numbers in the FGR group were considerably lower. The FGR group had a longer EPC differentiation period and significantly fewer OEC colonies [14].
These pregnancy disease are linked to both maternal and perinatal morbidity as well as bad long-term results for the offspring, making them one of the major concerns for fetal development and the health of the offspring. Numerous research studies have demonstrated that children exposed to prenatal disorders are more vulnerable to long-term conditions such obesity, metabolic syndrome, type 2 diabetes, and hypertension, all of which are subtypes of cardiovascular disease (CVD) [8,11,15,16,17,18,19].
In this review, we highlighted the role that fetal EPCs may play in the increased life-long cardiovascular risk that is associated with pre-eclampsia (PE), gestational diabetes (GDM), and fetal growth restriction (FGR) as well as their number, senescence, differentiation, and angiogenic function in the pathophysiology of these conditions.
2. Preeclampsia
2.1. Number of EPC in Preeclampsia
About 5–8% of pregnancies are complicated by preeclampsia (PE), a pregnancy disease with unclear etiology that greatly increases maternal and neonatal morbidity and mortality [20]. When the fetus is growth constrained, PE is linked to poor chorionic villous development and fetoplacental angiogenesis [10]. Normal fetal growth is associated with increased branching angiogenesis and hyperramified capillary loops with uneven and narrow lumina [21,22]. It is appropriate to speculate that disturbances in EPCs may be implicated in this vasculopathy given their involvement in vascular homeostasis.
Recent research by Fadini et al. [23] revealed a decreased number of circulating EPC in patients with chronic hypoxia with severe lung disease, and the reduction was related to impaired alveolo-arterial diffusion. Studies by Vasa et al. [3] and Hill et al. [24] also demonstrated a negative correlation between circulating EPCs and coronary artery disease risk factors. These data led to the hypothesis that diseases associated with endothelial dysfunction might be caused by the reduction of circulating EPC. Additionally, a number of animal studies and modest clinical trials have shown the therapeutic efficacy of EPC transplantation in tissue ischemia, indicating a potential role for EPCs in the management of vascular disease [25,26,27,28].
We examined the circulating EPC number of umbilical cord blood in the control and preeclamptic groups in earlier research. Table 1 displays the features of the patient. There were no discernible variations between the two groups' gestational ages at delivery or patient ages. On the other hand, the severe preeclampsia group had a much greater rate of SGA, lower birth weight, and a significantly higher maternal systolic blood pressure. The preeclampsia group's circulating EPC number in the umbilical cord blood of the control group showed a significant decrease [11,29]. Concurrently, the same group's umbilical cord plasma showed a decline in the free VEGF level. Nevertheless, the rise of sVEGFR-1 did not attain statistical significance [11]. Additionally, Xia et al. demonstrated that in the pre-eclampsia group as opposed to the control, there is a significant decrease in the placental/fetal circulating EPC numbers (median, 200; range, 100-440 cells/mL vs 390; 270-440 cells/mL, p < 0.001) [30]. Following in vitro culture, the pre-eclampsia group's EPC counts likewise declined (19.5; 5.0-32.0 vs 39.5; 31.2-52.0 EPC/ x 200 field; p < 0.001). There was an inverse correlation between the level of soluble fms-like tyrosine kinase 1 (sFlt-1) in cord blood and both circulating and cultured EPC [30].
These results imply that in cases of severe preeclampsia, ineffective EPC mobilization from the bone marrow due to a reduction in free VEGF as a consequence of increased binding to excess sVEGFR-1 is the cause of the decreased EPCs in the umbilical cord blood. We were unable to show a correlation between decreased cord plasma free VEGF and increased cord plasma sVEGFR-1, despite the fact that the EPC count was lower in the cord blood from preeclamptic pregnancy.
2.2. Senescence of EPC in Preeclampsia
Senescent cell burden in adipose, skeletal muscle, kidney, and skin tissues is low in young people but rises with age [31,32,33]. Specifically, among the many diseases linked to elevated senescent cell load are parts of the metabolic syndrome, such as diabetes, atherosclerosis, hypertension, and abdominal obesity [34,35,36].
Maternal vascular/endothelial malfunction and placental deficient angiogenesis are the hallmarks of pre-eclampsia [37,38]. Numerous investigations revealed that vascular endothelial dysfunction is significantly influenced by cellular senescence [39,40,41,42].
Senescence of EPC in maternal circulation associated with pre-eclampsia has been observed recently. Patients with preeclampsia had a considerably higher rate of cellular senescence (33.9%) than controls (22.9%; P < 0.05) [43]. Furthermore, patients diagnosed with preeclampsia were separated into two subgroups: those with CRP levels less than or equal to 0.1 mg/dl (n = 4) and those with CRP levels greater than or equal to 0.1 mg/dl (n = 4). It is interesting to note that the CRP-positive group had higher median values for cellular senescence than the CRP-negative group did, however this difference did not reach statistical significance (43.5% and 33.3%, respectively; p = 0.12) [43].
Additionally, the in vitro features of EPC cellular aging were evaluated using the Senescence-associated β-galactosidase (SA-β-gal) activity assay for fetal EPCs obtained from women with preeclampsia and normal pregnancies.
Compared to normal pregnant women (32.4%; range, 25.2-39.6%; p < 0.001), patients with PE had a considerably higher number of SA-β-gal-positive cells (55.8%; range, 49.5-62.1%) [29]. These results show that endothelial dysfunction may be linked to the cellular aging of fetal EPCs in preeclamptic individuals.
2.3. Differentiation Activity and Angiogenic Function of EPC in Preeclampsia
In addition to the immediate implications on the fetus, such as preterm or fetal growth restriction, maternal preeclampsia may also result in potential problems with the fetus's vascular health due to effects sustained in utero. The findings of several additional studies that have previously documented a higher prevalence of adult-onset metabolic and cardiovascular disorders, including hypertension, cardiovascular disorders, cerebrovascular disorders, impaired neural development, type 2 diabetes, metabolic syndrome, and hypercortisolism, in people born to preeclamptic women, provide support to this. [44,45,46,47,48].
Since EPCs are endothelial cell progenitors and can therefore regulate angiogenesis, neovascularization, and vasculogenesis postnatally [26,49], it is likely that a variety of vascular disorders are caused by their malfunction. Indeed, previous research has demonstrated a decrease in the quantity of circulating EPCs in vascular and metabolic conditions, including metabolic syndrome, coronary artery disease, diabetic vasculopathy, atherosclerosis, and systemic lupus erythematosus [50,51,52,53,54]. It has been demonstrated that preeclampsia, which can be defined as a hypertensive disease, is linked to a lower level of circulating EPCs in peripheral blood vessels [55].
Since cord blood is a part of the fetal circulation, the angiogenic property of cord blood EPCs most likely resembles the fetus's total angiogenic potential. The findings of an earlier investigation, which demonstrated that OECs differentiated from cord blood EPCs and had angiogenic potential [56], are consistent with this. It is known that there are fewer cord blood-derived EPCs in preeclamptic pregnancies and that the few EPCs that are found are primarily senescent, despite the fact that little research has been done to date to examine the function of fetal EPCs exposed to preeclampsia. [11,29,57,58].
Proliferation, migration, and vasculogenesis were all markedly compromised in the AC133+/KDR+ EPC from cord blood of pre-eclamptic individuals [30]. Furthermore, compared to normal-derived CKL− EPCs, Preeclampsia-derived cord-blood CD133+/C-kit+/Lin−(CKL−) EPCs and the resulting differentiated OECs showed decreased differentiation capacity and angiogenic function, respectively [8]. Immature differentiation of OEC for angiogenesis is shown by diminished migration, adhesion, and tube formation function of preeclampsia generated OECs in comparison to normal OECs. Moreover, exposure to normal serum conditions did not restore the preeclampsia-derived CKL− EPCs' reduced differentiation potency, indicating that their impaired function might be irreversible [8]. Conversely, CKL− EPCs produced from normal donors were able to differentiate even in the presence of preeclamptic serum in culture media [8]. Therefore, via influencing fetal vasculogenic/angiogenic function, the preeclamptic intrauterine environment appears to permanently alter pathways that significantly regulate and maintain fetal vascular health.
Environmental factors can induce permanent alterations in gene expression and/or protein function through a common process known as epigenetic modifications. A high frequency of transcriptionally competent yet changed genes, characterized by both active and repressive histone modifications linked to significant processes of differentiation and development, is a defining feature of stem and progenitor cells [59]. H3K4me3 and H3K9me3 levels were observed to be lower in the CKL− EPCs obtained from preeclampsia in the previous investigation [8]. Histone alterations have the potential to trigger pro-angiogenic (H3K4me3) and/or repressive (H3K9me3) signaling pathways, perhaps signifying a change in the proangiogenic/antiangiogenic balance in favor of anti-angiogenesis.
Collectively, these results demonstrated that the differentiation potency of cord blood EPCs produced from preeclampsia was irreversibly lower than that of EPCs derived from normal. Also, there was a notable reduction in the angiogenic potential of OECs that differentiated from EPCs produced from preeclampsia. To determine if the reduced function of preeclampsia-derived cord blood EPCs lasts after delivery and to clarify the mechanisms by which preeclampsia-induced effects on EPCs affect the exposed fetus's long-term vascular health, more research is necessary.
3. Gestational Diabetes Mellitus (GDM)
3.1. Number of EPC in GDM
Maternal hyperglycemia caused by glucose intolerance that initially manifests during pregnancy is known as gestational diabetes mellitus, or GDM. It has been steadily rising globally along with the increase in obesity among women of reproductive age and older mothers, complicating 5-31.5% of pregnancies. [60,61]. Offspring of women with gestational diabetes mellitus (GDM) are more likely to develop obesity, metabolic syndrome, and cardiovascular disease. A growing amount of research indicates that intrauterine hyperglycemia affects endothelial function in several fetal tissues and permanently changes genes' epigenetic alterations.
Reduced number and impaired function of EPCs was shown in patients with endothelial dysfunction, and impaired EPC-mediated vascular repair allows further progression of vascular disease. This is because the number of EPCs and their functional capacity determine maintenance of endothelial homeostasis and de novo vessel formation [62,63,64,65]. Cord blood-derived EPCs, which are also referred to as fetal EPCs, can play a role in the development of the fetal vasculature and the preservation of vascular integrity. As a result, dysfunction of these fetal EPCs may be a reflection of stem cell damage, which may lead to vascular dysfunction in the offspring later in life. It is still unclear, nevertheless, if fetal EPC dysfunction is a result of intrauterine hyperglycemia driven on by gestational diabetes mellitus (GDM) and what the underlying mechanisms involve.
EPC have been demonstrated to be reduced in diabetes [4] and play an essential role in the neovascularization process that occurs following damage. When comparing the amount of maternal EPCs isolated from GDM to a normal pregnancy, Penno G et al. found that there was a substantial decrease in circulating EPCs in GDM [62]. Additionally, compared to non-diabetic patients, GDM patients had a statistically significant decrease in the percentage of circulating EPC from maternal blood (0.26% vs. 0.41%, respectively; p < 0.05) [66]. According to Gui J et al., there was a decrease in the number of progenitor cells called endothelial colony-forming cells (ECFCs) generated from umbilical cord blood in an intrauterine environment that was diabetic as compared to healthy pregnancies [67].
Although there was a difference in umbilical cord EPC between GDM patients and control patients, it was not statistically significant (1.76% vs. 1.46%, respectively) [66]. Furthermore, in our earlier research, the quantity of fetal EPCs produced from cord blood did not alter in GDM pregnancies (data not shown). These results suggested that prenatal exposure to GDM does not affect the quantity of fetal EPCs, and more research is required.
3.2. Senescence of EPC in GDM
The function of EPCs may be reduced by premature senescence associated with gestational diabetes mellitus (GDM) [68]. In tissue stem cells, aging is a universal, progressive, and irreversible loss of function [69]. Furthermore, diabetes has been frequently linked to lower levels of circulating EPCs [70,71,72,73]. Endothelial progenitor cells (EPCs) in circulation are crucial for vascular regeneration. Nevertheless, it is still unclear how high hyperglycemia (HG) causes cord blood EPC aging. According to Wu et al. and Ingram et al., HG markedly raised the proportion of cord blood CD34+ EPC senescent cells [74] and cord blood endothelial colony-forming cells (ECFCs) that underwent premature senescence [12], respectively. Additionally, we examined the impact of GDM on fetal EPC senescence in our earlier research, and the results indicated a considerable rise in SA-β-gal positive CKL− EPCs (Data not shown). These results show that endothelial dysfunction may be linked to the cellular aging of fetal EPCs in GDM patients.
3.3. Differentiation and Angiogenic Function of EPC in GDM
Because GDM is linked to both maternal and perinatal morbidity as well as unfavorable long-term effects in offspring, it is one of the major issues for fetal development and offspring health. Numerous studies have revealed that children exposed to gestational diabetes mellitus (GDM) are more vulnerable to long-term conditions such obesity, metabolic syndrome, type 2 diabetes, and hypertension, all of which are subtypes of cardiovascular disease (CVD) [15,16,17,75,76].
Several investigations have indicated that pregnancies complicated by preeclampsia or fetal growth restriction were associated with a lower number and abnormal function of cord blood-derived EPCs [8,9]. It is yet unclear, nevertheless, if intrauterine hyperglycemia brought on by GDM influences fetal EPC development and angiogenic activity.
According to Gui J et al., compared to those from healthy pregnancies, the number and function of endothelial colony-forming cells (ECFCs) produced from umbilical cord blood were decreased in a diabetic intrauterine environment, including proliferation, migration, and tube formation [67]. They further showed that long-term cardiovascular disorders in the offspring of GDM pregnancies may be linked to lower SIRT expression and activity in fetal ECFCs and human umbilical vein endothelial cells (HUVECs) from GDM pregnancies [77].
Based on prior research, we postulate that intrauterine exposure to gestational diabetes mellitus (GDM) induces endothelial dysfunction by modifying the function of fetal endothelial progenitor cells (EPCs) via epigenetic modifications in gene expression, eventually affecting the offspring's subsequent developmental processes [13,66,78,79]. The objective of our study was to explore the programming effects of GDM on fetal EPCs and clarify the underlying mechanisms that may be associated with unfavorable outcomes in progeny. We used the cell migration, adhesion, tube formation, and proliferation assay to measure the angiogenic activity of OECs that were differentiated from cord blood-derived EPCs in order to investigate at how GDM affected the fetal EPCs. When compared to normal EPCs, GDM-EPCs showed comparable differentiation activity in terms of both number and time of differentiation to OECs [80]. However, GDM-OECs demonstrated significantly lower migration capacity, fibronectin adherence, tube formation, and proliferative activity [80].
Fetal EPCs from normal pregnancies were grown in high glucose (30 mM) during differentiation into OECs in order to mimic an intrauterine hyperglycemic circumstance. These cultures were then compared with those cultured in normal glucose (5 mM). Similar to GDM-EPCs and GDM-OECs, OECs differentiated by exposure to HG had reduced capacity in cell migration, adhesion, tube formation, and proliferation compared with those under NG conditions, even though there was no discernible difference in the differentiation from EPCs to OECs between NG and HG conditions [80].
In addition, we identified a number of genes linked to decreased fetal EPC function in GDM and discovered epigenetic modifications affecting gene expression to further comprehend the pathology of endothelial dysfunction in GDM-exposed offspring.
These results showed that, whereas fetal EPCs exposed to GDM can differentiate into OECs under normal circumstances, in utero exposure to GDM reduces the angiogenic potential of OECs, including migration, adhesion, tube formation, and proliferation. It makes sense to assume that an intrauterine hyperglycemic environment at crucial stages of fetal vascular development causes persistent damage to the offspring's vascular system.
4. Number and Senescence of EPC in FGR
One pregnancy complication that is clinically relevant is fetal growth restriction (FGR). A birth weight that is at or below the 10th percentile for gestational age is commonly used to determine FGR. The fact that 13% of instances do not exhibit postnatal catch-up growth is the reason for the growing interest in FGR [81]. Up to 5% of pregnancies may be affected by the common condition known as FGR, which has a significant morbidity and mortality rate. Low birth weight for gestation babies are more likely to have long-term developmental disorders as well as neonatal morbidity and mortality [18]. A significant risk of perinatal problems is often linked to FGR [82]. A increasing amount of research on animals and epidemiology has shown that the effects of FGR persist throughout adulthood [19]. The majority of FGR cases are idiopathic, meaning their cause is unclear, and treatment approaches are ineffective [83].
Adult disorders like diabetes mellitus and cardiovascular disease have been the focus of EPC-related research up to now. In actuality, fetal blood contains a higher concentration of EPCs than adult blood does, and pathologic diseases related to endothelial cells, as FGR and preeclampsia, provide a serious clinical problem during pregnancy.
In adulthood, an increasing amount of research indicates that endothelial dysfunction-related disorders such cardiovascular disease [3], diabetes mellitus [4] and preeclamptic pregnancy [43] are associated with impairments in the number and function of EPCs. Similar to this, numerous studies have proposed a link between children born with FGR and the development of the metabolic syndrome in adulthood, which includes diabetes, cardiovascular disease, asthma, intellectual disabilities such as depression and schizophrenia, and a lower intelligence quotient. This link has been considered "fetal programming" [84,85,86]. Therefore, if there was any impairment of fetal EPCs in FGR, it may have affected future adult life as well as intrauterine fetal growth, even if it is unclear whether the depletion and enhanced aging of EPC in these diseases are causes or outcomes.
The cord blood from the FGR group had a mean EPC count per 50 mL that was considerably lower than that of the normal group, as demonstrated by Hwang et al. [14]. Furthermore, the FGR group's 8.8 ± 1.5-day differentiation period from EPC to OEC was longer than the normal group's 6.0 ± 1.2-day differentiation period (p < 0.001). The staining intensity of SA-β-gal-positive cells was relatively higher in the FGR group compared to normal (141.3 ± 13.6 % vs. 100.0 ± 14.82 %; p < 0.001), and the OEC colony number from 1 × 105 fetal EPCs in each group was significantly decreased in the FGR group (7.9 ± 0.9 × 105 EPC) compared with normal (11.8 ± 1.6 × 105 EPC) (p < 0.001). Additionally, the FGR group's telomerase activity was noticeably lower than that of the normal group (70.3 ± 6.5 % vs. 100.0 ± 9.4 %; p < 0.001).
In summary, circulating fetal EPCs in FGR are more senescent, have a lower differentiation capacity, and are fewer in number. These findings imply that the functional and numerical impairment of fetal EPC in idiopathic FGR offers a likely explanation and could point to a practical intrauterine growth restriction treatment approach. Additionally, a number of studies found that during a FGR pregnancy, the concentrations of several known inflammatory, hypoxic, and antiangiogenic markers were higher in the cord blood [87,88]. These findings could help to explain why fetal EPCs in FGR pregnancies have lower in number, decreased functional ability, and higher senescence.
5. Conclusion
In this review, we address the importance of fetal EPCs in the pathophysiology of fetal growth restriction (FGR), gestational diabetes (GDM), and pre-eclampsia (PE) (Figure 1). At first, we proposed that preeclamptic patients had a considerably higher rate of fetal EPC senescence than did controls. Furthermore, in comparison to the normal-derived EPCs, the preeclampsia-derived EPCs showed lower angiogenic activity, delayed differentiation periods, and a significant decrease in the number of produced OECs colonies. At second, we concluded that prenatal exposure to GDM reduces the angiogenic potential of EPCs and causes a marked increase in fetal EPC senescence in the GDM group. Nonetheless, there was no difference in the number of fetal EPCs obtained from cord blood or in the differentiation activity of EPCs during GDM pregnancy. At third, we postulated that there are less circulating fetal EPCs in FGR, they have lower angiogenic activity and differentiation potential, and they are more senescent. As a result, this study offers a thorough explanation for the role that cord blood fetal EPC plays in pregnancy problems and suggests that EPC may play a role in the elevated lifetime cardiovascular risk that offspring of pregnancy disorders may face.
Author Contributions
J.-Y.K. and Y.-S.M. have contributed to the writing of the manuscript. Y.-S.M. conceptualized the review. Y.-S.M. drawn the images. J.-Y.K. edited language. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2019R1I1A1A01059738, NRF-2021R1A2C2014591, and NRF-2022R1I1A1A01064011).
Data Availability Statement
The data that support the finding of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J. M. , Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, (5302), 964–7. [Google Scholar] [CrossRef]
- Murohara, T.; Ikeda, H.; Duan, J.; Shintani, S.; Sasaki, K.; Eguchi, H.; Onitsuka, I.; Matsui, K.; Imaizumi, T. , Transplanted cord blood-derived endothelial precursor cells augment postnatal neovascularization. J Clin Invest 2000, (11), 1527–36. [Google Scholar] [CrossRef] [PubMed]
- Vasa, M.; Fichtlscherer, S.; Aicher, A.; Adler, K.; Urbich, C.; Martin, H.; Zeiher, A. M.; Dimmeler, S. , Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res 2001, (1), E1–7. [Google Scholar] [CrossRef] [PubMed]
- Tepper, O. M.; Galiano, R. D.; Capla, J. M.; Kalka, C.; Gagne, P. J.; Jacobowitz, G. R.; Levine, J. P.; Gurtner, G. C. , Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002, (22), 2781–6. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Lucke, C.; Rossig, L.; Fichtlscherer, S.; Vasa, M.; Britten, M.; Kamper, U.; Dimmeler, S.; Zeiher, A. M. , Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation 2005, (22), 2981–7. [Google Scholar] [CrossRef] [PubMed]
- Assmus, B.; Urbich, C.; Aicher, A.; Hofmann, W. K.; Haendeler, J.; Rossig, L.; Spyridopoulos, I.; Zeiher, A. M.; Dimmeler, S. , HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ Res 2003, (9), 1049–55. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, T.; Hano, T.; Sawamura, T.; Nishio, I. , Oxidized low-density lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin Exp Pharmacol Physiol 2004, (7), 407–13. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Lee, H. J.; Jung, Y. J.; Kwon, H. Y.; Kim, H.; Lee, J.; Kim, Y. H.; Kim, H. O.; Maeng, Y. S.; Kwon, J. Y. , CD133+/C-kit+Lin(-) endothelial progenitor cells in fetal circulation demonstrate impaired differentiation potency in severe preeclampsia. Pregnancy Hypertens 2019, 15, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Monga, R.; Buck, S.; Sharma, P.; Thomas, R.; Chouthai, N. S. , Effect of preeclampsia and intrauterine growth restriction on endothelial progenitor cells in human umbilical cord blood. J Matern Fetal Neonatal Med 2012, (11), 2385–9. [Google Scholar] [CrossRef]
- Mayhew, T. M.; Wijesekara, J.; Baker, P. N.; Ong, S. S. , Morphometric evidence that villous development and fetoplacental angiogenesis are compromised by intrauterine growth restriction but not by pre-eclampsia. Placenta 2004, (10), 829–33. [Google Scholar] [CrossRef]
- Kwon, J. Y.; Maeng, Y. S.; Kwon, Y. G.; Kim, Y. H.; Kang, M. H.; Park, Y. W. , Decreased endothelial progenitor cells in umbilical cord blood in severe preeclampsia. Gynecol Obstet Invest 2007, (2), 103–8. [Google Scholar] [CrossRef] [PubMed]
- Ingram, D. A.; Lien, I. Z.; Mead, L. E.; Estes, M.; Prater, D. N.; Derr-Yellin, E.; DiMeglio, L. A.; Haneline, L. S. , In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes 2008, (3), 724–31. [Google Scholar] [CrossRef] [PubMed]
- Buemi, M.; Allegra, A.; D'Anna, R.; Coppolino, G.; Crasci, E.; Giordano, D.; Loddo, S.; Cucinotta, M.; Musolino, C.; Teti, D. Concentration of circulating endothelial progenitor cells (EPC) in normal pregnancy and in pregnant women with diabetes and hypertension. Am J Obstet Gynecol 2007, 196, (1),, 68 e1-6. [Google Scholar] [CrossRef]
- Hwang, H. S.; Kwon, Y. G.; Kwon, J. Y.; Won Park, Y.; Maeng, Y. S.; Kim, Y. H. , Senescence of fetal endothelial progenitor cell in pregnancy with idiopathic fetal growth restriction. J Matern Fetal Neonatal Med 2012, (9), 1769–73. [Google Scholar] [CrossRef] [PubMed]
- Wright, C. S.; Rifas-Shiman, S. L.; Rich-Edwards, J. W.; Taveras, E. M.; Gillman, M. W.; Oken, E. , Intrauterine exposure to gestational diabetes, child adiposity, and blood pressure. Am J Hypertens 2009, (2), 215–20. [Google Scholar] [CrossRef]
- Boney, C. M.; Verma, A.; Tucker, R.; Vohr, B. R. , Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 2005, (3), e290–6. [Google Scholar] [CrossRef]
- Bianco, M. E.; Josefson, J. L. , Hyperglycemia During Pregnancy and Long-Term Offspring Outcomes. Curr Diab Rep 2019, (12), 143. [Google Scholar] [CrossRef]
- Low, J. A.; Handley-Derry, M. H.; Burke, S. O.; Peters, R. D.; Pater, E. A.; Killen, H. L.; Derrick, E. J. , Association of intrauterine fetal growth retardation and learning deficits at age 9 to 11 years. Am J Obstet Gynecol 1992, (6), 1499–505. [Google Scholar] [CrossRef]
- Godfrey, K. M.; Barker, D. J. , Fetal nutrition and adult disease. Am J Clin Nutr 2000, (5 Suppl), 1344s–52s. [Google Scholar] [CrossRef]
- National High Blood Pressure Education Program Working Group Report on High Blood Pressure in Pregnancy. Am J Obstet Gynecol 1990, (5 Pt 1) Pt 1, 1691–712.
- Mayhew, T. M.; Charnock-Jones, D. S.; Kaufmann, P. Aspects of human fetoplacental vasculogenesis and angiogenesis. III. Changes in complicated pregnancies. Placenta 2004, 25, (2-3), 127-39. [Google Scholar] [CrossRef]
- Resta, L.; Capobianco, C.; Marzullo, A.; Piscitelli, D.; Sanguedolce, F.; Schena, F. P.; Gesualdo, L. Confocal laser scanning microscope study of terminal villi vessels in normal term and pre-eclamptic placentas. Placenta 2006, 27, (6-7), 735-9. [Google Scholar] [CrossRef]
- Fadini, G. P.; Schiavon, M.; Cantini, M.; Baesso, I.; Facco, M.; Miorin, M.; Tassinato, M.; de Kreutzenberg, S. V.; Avogaro, A.; Agostini, C. , Circulating progenitor cells are reduced in patients with severe lung disease. Stem Cells 2006, (7), 1806–13. [Google Scholar] [CrossRef]
- Hill, J. M.; Zalos, G.; Halcox, J. P.; Schenke, W. H.; Waclawiw, M. A.; Quyyumi, A. A.; Finkel, T. , Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 2003, (7), 593–600. [Google Scholar] [CrossRef]
- Kalka, C.; Masuda, H.; Takahashi, T.; Kalka-Moll, W. M.; Silver, M.; Kearney, M.; Li, T.; Isner, J. M.; Asahara, T. , Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci U S A 2000, (7), 3422–7. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kalka, C.; Masuda, H.; Chen, D.; Silver, M.; Kearney, M.; Magner, M.; Isner, J. M.; Asahara, T. , Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med 1999, (4), 434–8. [Google Scholar] [CrossRef] [PubMed]
- Stamm, C.; Westphal, B.; Kleine, H. D.; Petzsch, M.; Kittner, C.; Klinge, H.; Schümichen, C.; Nienaber, C. A.; Freund, M.; Steinhoff, G. , Autologous bone-marrow stem-cell transplantation for myocardial regeneration. Lancet 2003, (9351), 45–6. [Google Scholar] [CrossRef]
- Tateishi-Yuyama, E.; Matsubara, H.; Murohara, T.; Ikeda, U.; Shintani, S.; Masaki, H.; Amano, K.; Kishimoto, Y.; Yoshimoto, K.; Akashi, H.; Shimada, K.; Iwasaka, T.; Imaizumi, T. , Therapeutic angiogenesis for patients with limb ischaemia by autologous transplantation of bone-marrow cells: a pilot study and a randomised controlled trial. Lancet 2002, (9331), 427–35. [Google Scholar] [CrossRef]
- Hwang, H. S.; Maeng, Y. S.; Park, Y. W.; Koos, B. J.; Kwon, Y. G.; Kim, Y. H. Increased senescence and reduced functional ability of fetal endothelial progenitor cells in pregnancies complicated by preeclampsia without intrauterine growth restriction. Am J Obstet Gynecol 2008, 199, (3), 259.e1-7. [Google Scholar] [CrossRef]
- Xia, L.; Zhou, X. P.; Zhu, J. H.; Xie, X. D.; Zhang, H.; Wang, X. X.; Chen, J. Z.; Jian, S. , Decrease and dysfunction of endothelial progenitor cells in umbilical cord blood with maternal pre-eclampsia. J Obstet Gynaecol Res 2007, (4), 465–74. [Google Scholar] [CrossRef]
- Tchkonia, T.; Morbeck, D. E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M. D.; Kirkland, J. L. , Fat tissue, aging, and cellular senescence. Aging Cell 2010, (5), 667–84. [Google Scholar] [CrossRef]
- Melk, A.; Schmidt, B. M.; Vongwiwatana, A.; Rayner, D. C.; Halloran, P. F. , Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am J Transplant 2005, (6), 1375–82. [Google Scholar] [CrossRef]
- Waaijer, M. E.; Parish, W. E.; Strongitharm, B. H.; van Heemst, D.; Slagboom, P. E.; de Craen, A. J.; Sedivy, J. M.; Westendorp, R. G.; Gunn, D. A.; Maier, A. B. , The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, (4), 722–5. [Google Scholar] [CrossRef]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; Ishikawa, F.; Komuro, I. , A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med 2009, (9), 1082–7. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I. , Vascular cell senescence: contribution to atherosclerosis. Circ Res 2007, (1), 15–26. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, J. H.; Hilgers, K. F.; Steinbach, M. P.; Hartner, A.; Klanke, B.; Amann, K.; Melk, A. , Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension 2008, (1), 123–9. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J. M.; Lain, K. Y. , Recent Insights into the pathogenesis of pre-eclampsia. Placenta 2002, (5), 359–72. [Google Scholar] [CrossRef] [PubMed]
- Frusca, T.; Morassi, L.; Pecorelli, S.; Grigolato, P.; Gastaldi, A. , Histological features of uteroplacental vessels in normal and hypertensive patients in relation to birthweight. Br J Obstet Gynaecol 1989, (7), 835–9. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. , Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 2002, (13), 1541–4. [Google Scholar] [CrossRef] [PubMed]
- Donato, A. J.; Morgan, R. G.; Walker, A. E.; Lesniewski, L. A. , Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol 2015, (Pt B), 122–35. [Google Scholar] [CrossRef]
- Tian, X. L.; Li, Y. , Endothelial cell senescence and age-related vascular diseases. J Genet Genomics 2014, (9), 485–95. [Google Scholar] [CrossRef] [PubMed]
- Regina, C.; Panatta, E.; Candi, E.; Melino, G.; Amelio, I.; Balistreri, C. R.; Annicchiarico-Petruzzelli, M.; Di Daniele, N.; Ruvolo, G. , Vascular ageing and endothelial cell senescence: Molecular mechanisms of physiology and diseases. Mech Ageing Dev 2016, 159, 14–21. [Google Scholar] [CrossRef]
- Sugawara, J.; Mitsui-Saito, M.; Hayashi, C.; Hoshiai, T.; Senoo, M.; Chisaka, H.; Yaegashi, N.; Okamura, K. , Decrease and senescence of endothelial progenitor cells in patients with preeclampsia. J Clin Endocrinol Metab 2005, (9), 5329–32. [Google Scholar] [CrossRef] [PubMed]
- Gumina, D. L.; Su, E. J. , Endothelial Progenitor Cells of the Human Placenta and Fetoplacental Circulation: A Potential Link to Fetal, Neonatal, and Long-term Health. Front Pediatr 2017, 5, 41. [Google Scholar] [CrossRef]
- Kajantie, E.; Osmond, C.; Eriksson, J. G. , Gestational hypertension is associated with increased risk of type 2 diabetes in adult offspring: the Helsinki Birth Cohort Study. Am J Obstet Gynecol.
- Kajantie, E.; Eriksson, J. G.; Osmond, C.; Thornburg, K.; Barker, D. J. , Pre-eclampsia is associated with increased risk of stroke in the adult offspring: the Helsinki birth cohort study. Stroke 2009, (4), 1176–80. [Google Scholar] [CrossRef] [PubMed]
- Warshafsky, C.; Pudwell, J.; Walker, M.; Wen, S. W.; Smith, G. N. , Prospective assessment of neurodevelopment in children following a pregnancy complicated by severe pre-eclampsia. BMJ Open 2016, (7), e010884. [Google Scholar] [CrossRef]
- Henley, D.; Brown, S.; Pennell, C.; Lye, S.; Torpy, D. J. , Evidence for central hypercortisolism and elevated blood pressure in adolescent offspring of mothers with pre-eclampsia. Clin Endocrinol (Oxf) 2016, (4), 583–9. [Google Scholar] [CrossRef]
- Eguchi, M.; Masuda, H.; Asahara, T. , Endothelial progenitor cells for postnatal vasculogenesis. Clin Exp Nephrol 2007, (1), 18–25. [Google Scholar] [CrossRef]
- Kunz, G. A.; Liang, G.; Cuculi, F.; Gregg, D.; Vata, K. C.; Shaw, L. K.; Goldschmidt-Clermont, P. J.; Dong, C.; Taylor, D. A.; Peterson, E. D. , Circulating endothelial progenitor cells predict coronary artery disease severity. Am Heart J 2006, (1), 190–5. [Google Scholar] [CrossRef]
- Fadini, G. P.; Sartore, S.; Albiero, M.; Baesso, I.; Murphy, E.; Menegolo, M.; Grego, F.; Vigili de Kreutzenberg, S.; Tiengo, A.; Agostini, C.; Avogaro, A. , Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler Thromb Vasc Biol 2006, (9), 2140–6. [Google Scholar] [CrossRef]
- Fadini, G. P.; Coracina, A.; Baesso, I.; Agostini, C.; Tiengo, A.; Avogaro, A.; de Kreutzenberg, S. V. , Peripheral blood CD34+KDR+ endothelial progenitor cells are determinants of subclinical atherosclerosis in a middle-aged general population. Stroke 2006, (9), 2277–82. [Google Scholar] [CrossRef] [PubMed]
- Ebner, P.; Picard, F.; Richter, J.; Darrelmann, E.; Schneider, M.; Strauer, B. E.; Brehm, M. , Accumulation of VEGFR-2+/CD133+ cells and decreased number and impaired functionality of CD34+/VEGFR-2+ cells in patients with SLE. Rheumatology (Oxford) 2010, (1), 63–72. [Google Scholar] [CrossRef]
- Jialal, I.; Devaraj, S.; Singh, U.; Huet, B. A. , Decreased number and impaired functionality of endothelial progenitor cells in subjects with metabolic syndrome: implications for increased cardiovascular risk. Atherosclerosis 2010, (1), 297–302. [Google Scholar] [CrossRef]
- Beasley, K. M.; Lovering, A. T.; Gilbert, J. S. , Decreased endothelial progenitor cells in preeclampsia and consequences for developmental programming. Hypertension 2014, (1), 23–5. [Google Scholar] [CrossRef]
- Cardenas, C.; Kwon, J. Y.; Maeng, Y. S. , Human Cord Blood-Derived CD133(+)/C-Kit(+)/Lin(-) Cells Have Bipotential Ability to Differentiate into Mesenchymal Stem Cells and Outgrowth Endothelial Cells. Stem Cells Int 2016, 2016, 7162160. [Google Scholar] [CrossRef]
- Muñoz-Hernandez, R.; Miranda, M. L.; Stiefel, P.; Lin, R. Z.; Praena-Fernández, J. M.; Dominguez-Simeon, M. J.; Villar, J.; Moreno-Luna, R.; Melero-Martin, J. M. , Decreased level of cord blood circulating endothelial colony-forming cells in preeclampsia. Hypertension 2014, (1), 165–71. [Google Scholar] [CrossRef] [PubMed]
- Gumina, D. L.; Black, C. P.; Balasubramaniam, V.; Winn, V. D.; Baker, C. D. , Umbilical Cord Blood Circulating Progenitor Cells and Endothelial Colony-Forming Cells Are Decreased in Preeclampsia. Reprod Sci 2017, (7), 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Tee, W. W.; Reinberg, D. , A double take on bivalent promoters. Genes Dev 2013, (12), 1318–38. [Google Scholar] [CrossRef]
- Metzger, B. E.; Buchanan, T. A.; Coustan, D. R.; de Leiva, A.; Dunger, D. B.; Hadden, D. R.; Hod, M.; Kitzmiller, J. L.; Kjos, S. L.; Oats, J. N.; Pettitt, D. J.; Sacks, D. A.; Zoupas, C. , Summary and recommendations of the Fifth International Workshop-Conference on Gestational Diabetes Mellitus. Diabetes Care 2007, 30 Suppl 2, S251–60. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, C. , Prevalence of Gestational Diabetes and Risk of Progression to Type 2 Diabetes: a Global Perspective. Curr Diab Rep 2016, (1), 7. [Google Scholar] [CrossRef] [PubMed]
- Penno, G.; Pucci, L.; Lucchesi, D.; Lencioni, C.; Iorio, M. C.; Vanacore, R.; Storti, E.; Resi, V.; Di Cianni, G.; Del Prato, S. , Circulating endothelial progenitor cells in women with gestational alterations of glucose tolerance. Diab Vasc Dis Res 2011, (3), 202–10. [Google Scholar] [CrossRef]
- Altabas, V. , Diabetes, Endothelial Dysfunction, and Vascular Repair: What Should a Diabetologist Keep His Eye on? Int J Endocrinol 2015, 2015, 848272. [Google Scholar] [CrossRef]
- Rigato, M.; Avogaro, A.; Fadini, G. P. , Levels of Circulating Progenitor Cells, Cardiovascular Outcomes and Death: A Meta-Analysis of Prospective Observational Studies. Circ Res 2016, (12), 1930–9. [Google Scholar] [CrossRef]
- Rigato, M.; Bittante, C.; Albiero, M.; Avogaro, A.; Fadini, G. P. , Circulating Progenitor Cell Count Predicts Microvascular Outcomes in Type 2 Diabetic Patients. J Clin Endocrinol Metab 2015, (7), 2666–72. [Google Scholar] [CrossRef]
- Mordwinkin, N. M.; Ouzounian, J. G.; Yedigarova, L.; Montoro, M. N.; Louie, S. G.; Rodgers, K. E. , Alteration of endothelial function markers in women with gestational diabetes and their fetuses. J Matern Fetal Neonatal Med 2013, (5), 507–12. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Rohrbach, A.; Borns, K.; Hillemanns, P.; Feng, L.; Hubel, C. A.; von Versen-Hoynck, F. , Vitamin D rescues dysfunction of fetal endothelial colony forming cells from individuals with gestational diabetes. Placenta 2015, (4), 410–8. [Google Scholar] [CrossRef]
- Blue, E. K.; DiGiuseppe, R.; Derr-Yellin, E.; Acosta, J. C.; Pay, S. L.; Hanenberg, H.; Schellinger, M. M.; Quinney, S. K.; Mund, J. A.; Case, J.; Haneline, L. S. , Gestational diabetes induces alterations in the function of neonatal endothelial colony-forming cells. Pediatr Res 2014, (2), 266–72. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Depinho, R. A. , Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 2010, (7288), 520–8. [Google Scholar] [CrossRef]
- Egan, C. G.; Lavery, R.; Caporali, F.; Fondelli, C.; Laghi-Pasini, F.; Dotta, F.; Sorrentino, V. , Generalised reduction of putative endothelial progenitors and CXCR4-positive peripheral blood cells in type 2 diabetes. Diabetologia 2008, (7), 1296–305. [Google Scholar] [CrossRef] [PubMed]
- Loomans, C. J.; de Koning, E. J.; Staal, F. J.; Rookmaaker, M. B.; Verseyden, C.; de Boer, H. C.; Verhaar, M. C.; Braam, B.; Rabelink, T. J.; van Zonneveld, A. J. , Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 2004, (1), 195–9. [Google Scholar] [CrossRef]
- Fadini, G. P.; Pucci, L.; Vanacore, R.; Baesso, I.; Penno, G.; Balbarini, A.; Di Stefano, R.; Miccoli, R.; de Kreutzenberg, S.; Coracina, A.; Tiengo, A.; Agostini, C.; Del Prato, S.; Avogaro, A. , Glucose tolerance is negatively associated with circulating progenitor cell levels. Diabetologia 2007, (10), 2156–63. [Google Scholar] [CrossRef]
- Fadini, G. P.; Miorin, M.; Facco, M.; Bonamico, S.; Baesso, I.; Grego, F.; Menegolo, M.; de Kreutzenberg, S. V.; Tiengo, A.; Agostini, C.; Avogaro, A. , Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol 2005, (9), 1449–57. [Google Scholar] [CrossRef]
- Wu, Y.; Fu, C.; Li, B.; Liu, C.; He, Z.; Li, X. E.; Wang, A.; Ma, G.; Yao, Y. , Bradykinin Protects Human Endothelial Progenitor Cells from High-Glucose-Induced Senescence through B2 Receptor-Mediated Activation of the Akt/eNOS Signalling Pathway. J Diabetes Res 2021, 2021, 6626627. [Google Scholar] [CrossRef]
- Bunt, J. C.; Tataranni, P. A.; Salbe, A. D. , Intrauterine exposure to diabetes is a determinant of hemoglobin A(1)c and systolic blood pressure in pima Indian children. J Clin Endocrinol Metab 2005, (6), 3225–9. [Google Scholar] [CrossRef]
- Cho, N. H.; Silverman, B. L.; Rizzo, T. A.; Metzger, B. E. , Correlations between the intrauterine metabolic environment and blood pressure in adolescent offspring of diabetic mothers. J Pediatr 2000, (5), 587–92. [Google Scholar] [CrossRef]
- Gui, J.; Potthast, A.; Rohrbach, A.; Borns, K.; Das, A. M.; von Versen-Hoynck, F. , Gestational diabetes induces alterations of sirtuins in fetal endothelial cells. Pediatr Res 2016, (5), 788–98. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Dimmeler, S. , Endothelial progenitor cells functional characterization. Trends Cardiovasc Med 2004, (8), 318–22. [Google Scholar] [CrossRef] [PubMed]
- Robb, A. O.; Mills, N. L.; Newby, D. E.; Denison, F. C. , Endothelial progenitor cells in pregnancy. Reproduction 2007, (1), 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Jung, Y. J.; Lee, Y.; Son, G. H.; Kim, H. O.; Maeng, Y. S.; Kwon, J. Y. Impaired Angiogenic Function of Fetal Endothelial Progenitor Cells via PCDH10 in Gestational Diabetes Mellitus. Int J Mol Sci 2023, 24, (22). [Google Scholar] [CrossRef]
- Karlberg, J.; Albertsson-Wikland, K. , Growth in full-term small-for-gestational-age infants: from birth to final height. Pediatr Res 1995, (5), 733–9. [Google Scholar] [CrossRef] [PubMed]
- Illanes, S.; Soothill, P. , Management of fetal growth restriction. Semin Fetal Neonatal Med 2004, (5), 395–401. [Google Scholar] [CrossRef]
- Resnik, R. , Intrauterine growth restriction. Obstet Gynecol 2002, (3), 490–6. [Google Scholar]
- Rosso, I. M.; Cannon, T. D.; Huttunen, T.; Huttunen, M. O.; Lönnqvist, J.; Gasperoni, T. L. , Obstetric risk factors for early-onset schizophrenia in a Finnish birth cohort. Am J Psychiatry 2000, (5), 801–7. [Google Scholar] [CrossRef] [PubMed]
- Gale, C. R.; Martyn, C. N. , Birth weight and later risk of depression in a national birth cohort. Br J Psychiatry 2004, 184, 28–33. [Google Scholar] [CrossRef]
- Frisk, V.; Amsel, R.; Whyte, H. E. , The importance of head growth patterns in predicting the cognitive abilities and literacy skills of small-for-gestational-age children. Dev Neuropsychol 2002, (3), 565–93. [Google Scholar] [CrossRef] [PubMed]
- Amarilyo, G.; Oren, A.; Mimouni, F. B.; Ochshorn, Y.; Deutsch, V.; Mandel, D. , Increased cord serum inflammatory markers in small-for-gestational-age neonates. J Perinatol 2011, (1), 30–2. [Google Scholar] [CrossRef]
- Wallner, W.; Sengenberger, R.; Strick, R.; Strissel, P. L.; Meurer, B.; Beckmann, M. W.; Schlembach, D. , Angiogenic growth factors in maternal and fetal serum in pregnancies complicated by intrauterine growth restriction. Clin Sci (Lond) 2007, (1), 51–7. [Google Scholar] [CrossRef]
Figure 1.
Proposed model describing the vessel formation of EPCs in the pathophysiology of pre-eclampsia (PE), gestational diabetes (GDM), fetal growth restriction (FGR) and normal pregnancy conditions. The fetal EPCs of pregnancy complications displayed decreased blood vessel formation activity.
Figure 1.
Proposed model describing the vessel formation of EPCs in the pathophysiology of pre-eclampsia (PE), gestational diabetes (GDM), fetal growth restriction (FGR) and normal pregnancy conditions. The fetal EPCs of pregnancy complications displayed decreased blood vessel formation activity.
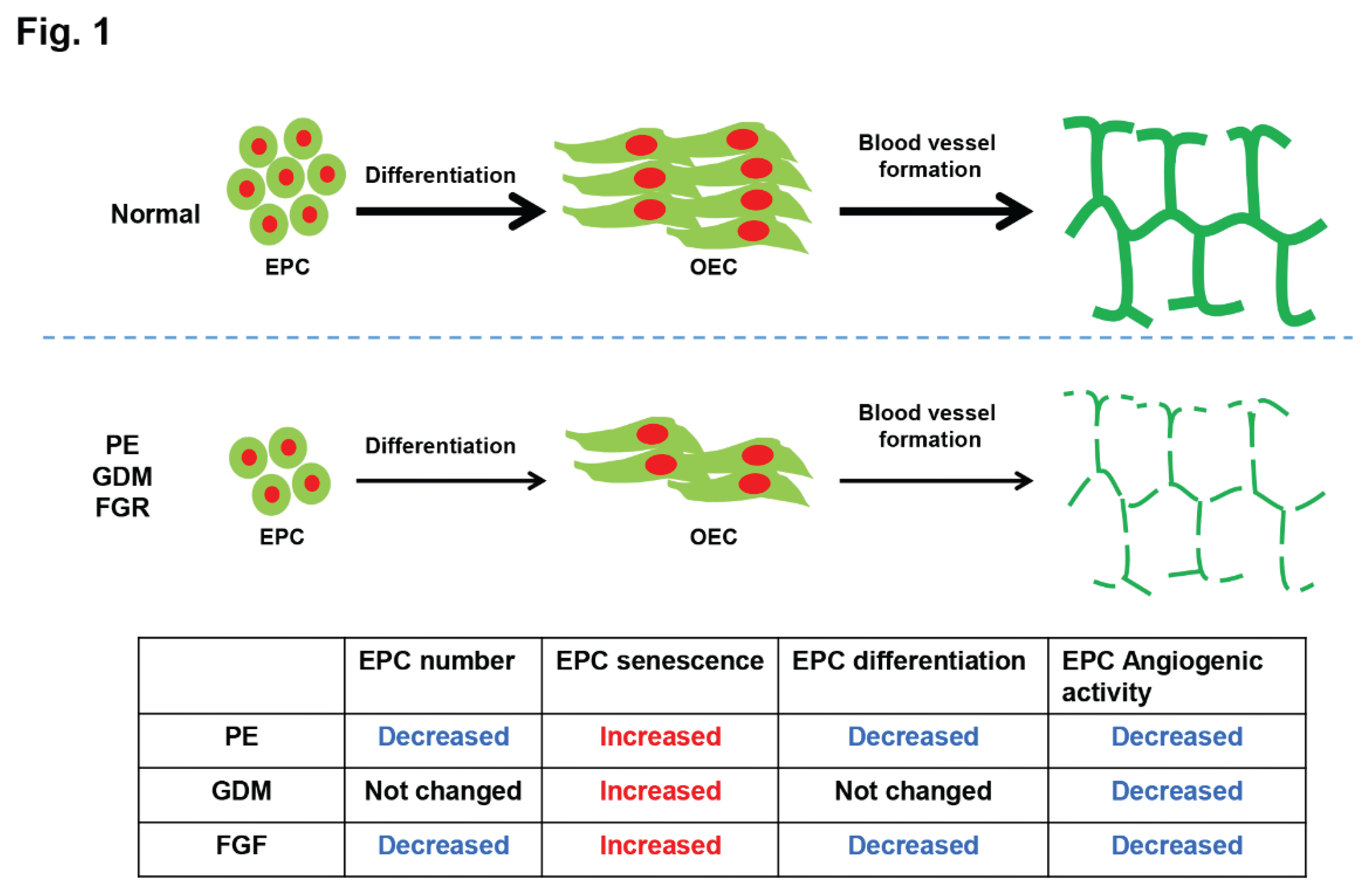

Table 1.
Patient characteristics.
| Normal pregnancy (n=30) |
Preeclampsia (n=30) |
P-value | |
|---|---|---|---|
| Maternal age (years) | 33.7 ± 5.3 | 32.75 ± 3.1 | 0.549 |
| Gestational age at delivery (weeks) | 38.65 ± 0.8 | 36.35 ± 2.1 | 0.386 |
| Gravida | 2.5 ± 1.4 | 1.55 ± 1.0 | 0.05 |
| Pre-pregnancy BMI (kg/m2) | 21.75 ± 4.7 | 22.1 ± 3.8 | 0.387 |
| Blood pressure (mmHg) | |||
| Systolic blood pressure | 115.1 ± | 151.05 ± | <0.015 |
| Diastolic blood pressure | 74.25 ± | 95.7 ± | <0.015 |
| Birth weight (g) | 3301.0 ± 365 | 2303.1 ± 536 | <0.001 |
| Small gestational age (n) | 0 | 21 | 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



