
Submitted:
18 March 2024
Posted:
19 March 2024
You are already at the latest version
Abstract
INTRODUCTION : In this scoping review, we delve into the transformative potential of artificial intelligence (AI) in addressing challenges inherent in whole genome sequencing (WGS) analysis, with a specific focus on its implications in surgical oncology. METHODS: Scoping review of whole genomic sequencing and artificial intelligence.DISCUSSION : Unveiling the limitations of existing sequencing technologies, the review illuminates how AI-powered methods emerge as innovative solutions to surmount these obstacles. The evolution of DNA sequencing technologies, progressing from Sanger sequencing to next-generation sequencing, sets the backdrop for AI's emergence as a potent ally in processing and analyzing the voluminous genomic data generated by these technologies. Particularly, deep learning methods play a pivotal role in extracting knowledge and discerning patterns from the vast landscape of genomic information. In the context of oncology, AI-powered methods exhibit considerable potential across diverse facets of WGS analysis, including variant calling, structural variation identification, and pharmacogenomic analysis. CONCLUSIONS : This review underscores the significance of multimodal approaches in diagnoses and therapies, highlighting the imperative for ongoing research and development in AI-powered WGS techniques. Integrating AI into the analytical framework empowers scientists and clinicians to unravel the intricate interplay of genomics within the realm of multi-omics research, paving the way for more personalized and targeted treatments in surgical oncology and perhaps beyond.
Keywords:
whole genomic sequencing
; proteomics
; transcriptomics
; machine learning
; deep learning
; modalities
1. Introduction
Currently, most advanced genomic studies for patients with cancers involves panels that analyze approximately 500 genes; however, the human genome contains approximately 20,000 genes. The reality is that cost is no longer the main obstacle to whole genomic sequencing (WGS) tumors with prices ranging as low as $1,000. The limitation is how to analyze the massive amounts of data created (Figure 1). It could be argued that if there are no chemotherapeutic targets outside of the known genes in tumor panels that are being used, then more genomic data wouldn’t be able to provide any useful data to help guide therapeutic treatment decisions. Luckily, over the last 10-15 years computer scientists have developed machine learning (ML) algorithms with deep learning (DL) architectures that have paved the way for viable artificial intelligence (AI). This review will discuss the reasons why WGS is not routinely done in the management of the oncologic patient and discuss how AI can potentially overcome many of the limitations in WGS analysis of cancers.
2. The Evolution of DNA Sequencing
Sanger and Maxam Gilbert sequencing technologies were first-generation sequencing that revolutionized DNA sequencing with their 1977 publication. Sanger sequencing, also known as the chain termination, dideoxynucleotide or sequencing by synthesis method, involves utilizing one strand of double-stranded DNA as a template. This method employs chemically modified dideoxy-nucleotides (ddNTPs), marked as ddG, ddA, ddT, and ddC for each DNA base. Incorporating these ddNTPs prevents further elongation, resulting in DNA fragments of varied sizes. Gel electrophoresis separates these fragments, visible through imaging systems (X-ray or UV light) [1,2,3,4]. Applied Biosystems automated Sanger sequencing in the late 1980s with the ABI Prism 370, utilizing capillary electrophoresis for fast and accurate sequencing. This method played a crucial role in sequencing projects for Bacteriophage [5] and various plant species,[6,7] with its most notable accomplishment being the decoding of the first human genome [8].
While Sanger sequencing remained prominent for single or low-throughput DNA sequencing over three decades, challenges arose in speeding up the analysis for complex genomes, such as those of plant species, while still being expensive and time-consuming [9]. In contrast, Maxam-Gilbert sequencing, another first-generation method known as the chemical degradation method, relies on the chemical cleaving of nucleotides, which is particularly effective for small nucleotide polymers [10,11]. This method, performed without DNA cloning, generates marked fragments separated by electrophoresis. Despite its initial use, Maxim-Gilbert sequencing faced challenges, and development and refinements of Sanger sequencing favored the latter method. Moreover, Maxim-Gilbert sequencing was deemed unsafe due to its use of toxic and radioactive chemicals [9].
For three decades Sanger sequencing, faced significant challenges in terms of cost and time. However, a transformative shift occurred after 2005 with the advent of a new generation of sequencers, overcoming the limitations of the earlier generations. Second-generation sequencing (SGS) technologies rapidly produce vast amounts of sequence data at a relatively low cost, enabling the completion of a human genome in just a few weeks. This approach involves generating millions of short reads from amplified individual DNA fragments through iterative cycles of nucleotide extensions. However, the extensive data generated pose challenges in terms of interpretation, analysis, and management.
The widespread adoption of SGS technologies has significantly influenced biomedical research with applications ranging from WGS and target resequencing to the characterization of structural and copy number variations, profiling epigenetic modifications, transcriptome sequencing and identification of infectious agents. Ongoing developments involve the creation of new methodologies and instruments aimed at sequencing the entire human genome in less than a day [12]. Short read sequencing methods are broadly categorized into sequencing by ligation (SBL) and sequencing by synthesis (SBS), with major platforms such as Roche/454 (launched in 2005), Illumina/Solexa (in 2006), and ABI/SOLiD (in 2007).
These platforms marked significant advancements in sequencing technology [9]. Roche/454 sequencing, introduced in 2005, utilizes pyrosequencing based on the detection of pyrophosphate released after each nucleotide incorporation. This method involves random fragmentation of DNA samples, bead attachment with primers, emulsion PCR amplification, and pyrosequencing on a picotiter plate, enabling parallel reactions. The latest instrument, GS FLX+, generates reads of up to 1000 bp [13,14,15]. Ion Torrent semiconductor sequencing, acquired by Life Technologyies in 2010, employed a chip with micro wells and detected hydrogen ion release during sequencing instead of fluorescent labeled nucleotides. Ion Torrent sequencers produce reads of lengths 200 bp, 400 bp, and 600 bp, offering advantages in read length and fast sequencing times [16,17].
Solexa, later acquired by Illumina, commercialized the Genome Analyzer (GA) in 2006. Illumina's sequencing by synthesis approach, currently the most widely used technology, involves random DNA fragmentation, adapter ligation, cluster amplification, and sequencing using reversible terminators. Illumina sequencers yield high data outputs exceeding 600 Gbp, with short read lengths initially around 35 bp but now reaching about 125 bp [18]. Supported Oligonucleotide Ligation and Detection SOLiD, developed by Applied Biosystems (ABI) after acquiring Solexa, utilizes sequencing by ligation. The process involves multiple sequencing rounds, adapter attachment, emulsion PCR, ligation of 8-mers with fluorescent labels, and recording emitted colors [19]. ABI/SOLiD produces short reads with lengths initially at 35 bp, improving to 75 bp with high accuracy due to each base being read twice. However, drawbacks include relatively short reads and long run times, with errors attributed to noise during the ligation cycle, mainly causing substitution errors [9,20,21].
The SGS technologies discussed earlier have significantly transformed DNA analysis and have been widely adopted compared to the first-generation sequencing technologies. However, SGS technologies often necessitate a time-consuming and expensive PCR amplification step. Moreover, the intricate nature of genomes, featuring numerous repetitive regions, poses challenges for SGS technologies, especially with their relatively short reads, complicating genome assembly. In response to these challenges, scientists have introduced a new era of sequencing known as "third-generation sequencing" (TGS). TGA technologies address the limitations of SGS by offering lower sequencing costs, streamlined sample preparation without the need for PCR amplification, and significantly faster execution times. TGS also excels in generating long reads, surpassing several kilobases, which proves instrumental in resolving assembly issues and dealing with repetitive regions in complex genomes [9].
Two primary approaches characterize TGS [22]: the single-molecule real-time sequencing approach (SMRT) [23] and the synthetic approach. The SMRT approach, developed by the Quake laboratory [24,25,26], is widely used and implemented by Pacific Biosciences and Oxford Nanopore sequencing, particularly the MinION sequencer. Pacific Biosciences, a leading developer in TGS introduced the first genomic sequencer using the SMRT approach. Unlike other technologies, Pacific Biosciences detects signals in real-time during nucleotide incorporations instead of executing amplification cycles. The system employs SMRT cells, each containing zero-mode waveguides (ZMWs), nanostructures with diameters in the tens of nanometers [27,28]. These ZMWs utilize light properties, preventing its propagation through openings with diameters less than the wavelength, and the resulting decrease in light intensity along the wells illuminates the bottom. Each ZMW houses a DNA polymerase and the target DNA fragment for sequencing. As nucleotides are incorporated, they emit a luminous signal recorded by sensors, enabling the determination of the DNA sequence. Pacific Bioscience technology offers several advantages over SGS. Sample preparation is remarkably swift, taking 4 to 6 hours instead of days. Additionally, the technology produces long-read lengths, averaging around 10 kbp, with individual reads extending up to 60 kbp—surpassing the capabilities of any SGS technology. Despite its high error rate of approximately 13%, dominated by insertions and deletions, these errors are randomly distributed along the long reads [29,30,31,32].
Oxford Nanopore sequencing (ONT) was devised as a method for determining the sequence of nucleotides in DNA. In 2014, Oxford Nanopore Technologies introduced the MinION, a compact single-molecule Nanopore sequencing device measuring four inches in length and connecting to a laptop computer via a USB 3.0 port. Released for testing through the MinION Access Program (MAP), the MinION sequencer garnered attention for its potential to generate longer reads, facilitating improved resolution of structural genomic variants and repeat content. [33,34,35] The MinION sequencer presents several advantages, including its cost-effectiveness, compact size, and real-time data display on the device screen without the need to wait for the run's completion. Notably, the MinION can yield very long reads, surpassing 150 kbp, which enhances the contiguity of de novo assembly. However, the MinION does exhibit a relatively high error rate of approximately 12%, distributed across ~3% mismatches, ~4% insertions, and ~5% deletions [36].
3. What Is Whole Genomic Sequencing?
WGS provides the most comprehensive data about a given organism. Next Generation Sequencing (NGS) can deliver large amounts of data in a short amount of time. Profiling the entire genome facilitates the discovery of novel genes and variants associated with disease, particularly those in non-coding areas of the genome. The initial phase of NGS crucially involves the extraction and isolation of nucleic acids, whether it be total RNA, genomic DNA, or various RNA types. The DNA (or cDNA) sample undergoes a process that results in relatively short double-stranded fragments, typically ranging from 100 to 800 base pairs. Depending on the specific application, DNA fragmentation can be achieved through various methods such as physical shearing, enzyme digestion, or PCR-based amplification of specific genetic regions. These resulting DNA fragments are then linked to technology-specific adaptor sequences, creating a fragment library. These adaptors may also carry a distinctive molecular "barcode" to uniquely tag each sample with a specific DNA sequence.
Library preparation is the subsequent step, involving the preparation of DNA or RNA samples for processing and reading by sequencers. This is accomplished by fragmenting the samples to produce a pool of appropriately sized targets and adding specialized adapters at both ends, which will later interact with the NGS platform. The resulting prepared samples, referred to as “libraries,” represent a collection of molecules ready for sequencing. The specific library preparation procedure may vary based on the reagents and methods used, but the ultimate NGS libraries must consist of DNA fragments of desired lengths with adapters at both ends. Before sequencing, the DNA library must be affixed to a solid surface and clonally amplified to enhance the detectable signal from each target during sequencing. Throughout this process, each unique DNA molecule in the library is attached to the surface of a bead or a flow cell and subjected to PCR amplification, generating a set of identical clones. These libraries are then subjected to further quality control steps before sequencing to ensure accuracy. Subsequently, all the DNA in the library is sequenced simultaneously using a sequencing instrument.
Each NGS experiment results in substantial quantities of intricate data comprising short DNA reads. While different technology platforms have their distinct algorithms and data analysis tools, they generally follow a similar analysis 'pipeline' and employ common metrics to assess the quality of NGS datasets. The analysis can be categorized into three stages: primary, secondary, and tertiary analysis. Primary analysis involves the conversion of raw signals from instrument detectors into digitized data or base calls. Raw data, collected during each sequencing cycle, are processed into files containing base calls assembled into sequencing reads (FASTQ files) along with their associated quality scores (Phred quality score). Secondary analysis encompasses read filtering and trimming based on quality, followed by the alignment of reads to a reference genome or the assembly of reads for novel genomes, concluding with variant calling. The primary output is a BAM file containing aligned reads. Tertiary analysis is the most intricate phase, requiring the interpretation of results and extraction of meaningful information from the data [37,38,39,40,41].
4. AI-Powered Whole Genomic Sequencing
Genomics is progressing into an era of data-driven science. With the emergence of high-throughput technologies in human genomics, we find ourselves inundated with a vast amount of genomic data. AI particularly DL methods, plays a crucial role in extracting knowledge and patterns from this wealth of genomic information [70]. The proper execution of the variant calling step is pivotal for the success of numerous studies in clinical, association, or population genetics. The array of contemporary genomics protocols, techniques, and platforms complicates the selection of methods and algorithms, as there is no universal solution applicable to all scenarios. The accurate identification of gene variants in a person's genome from tens of millions of small, error-prone reading sequences is still an ongoing challenge despite the fast progress made by sequencing technologies. Poplin et al. have shown that the deep convolutional neuronal network, called DeepVariant, was able to effectively identify gene variations within a concurrent NGS reading. This is achieved through the model learning statistical relationships from images of read pileups around potential variants and true genotype calls.
Notably, DeepVariant outperforms existing state-of-the-art tools. The acquired model demonstrates generalization across genome builds and mammalian species, enabling nonhuman sequencing projects to leverage the extensive human ground-truth data. The study further illustrates DeepVariant's ability to adapt and call variants in various sequencing technologies and experimental designs, including deep whole genomes from 10X Genomics and Ion Ampliseq exomes. This underscores the advantages of employing automated and versatile techniques for variant calling [42].
Identifying genetic variants from NGS data presents a formidable challenge due to the inherent errors in NGS reads, which exhibit error rates ranging from approximately 0.1% to 10%. Moreover, these errors stem from a multifaceted process influenced by factors such as instrument characteristics, preceding data processing tools, and the genomic sequence itself. A central challenge in genomics involves identifying nucleotide variations within an individual's genome compared to a reference sequence, a process known as “variant calling”. Accurate and efficient variant calling is crucial for detecting genomic variations responsible for phenotypic disparities and diseases. Clairvoyante addresses this challenge by predicting variant type, zygosity, alternative allele, and indel length. Remarkably, Clairvoyante is agnostic to sample specifics and can identify variants in less than 2 hours on a standard server. Introduced to predict variant characteristics such as SNP or Indel type, zygosity, alternative allele, and indel length, the Clairvoyante model overcomes a limitation in the DeepVariant model, which lacks comprehensive variant details, including precise alternative allele and variant type. Notably, Clairvoyante is tailored for utilizing long-read sequencing data from technologies like Pacific Biosciences (PacBio) and Oxford Nanopore Technology (ONT), though it is versatile enough to be commonly applied to short-read datasets as well [43].
Lei Cai et al. expands this advanced approach to address the challenge of calling structural variations [44]. They introduce DeepSV, a DL-based method designed for calling long deletions from sequence reads. DeepSV utilizes a unique visualization method for sequence reads, strategically capturing multiple sources of information in the sequence data relevant to long deletions. Additionally, DeepSV incorporates techniques to handle noisy training data effectively. The model in DeepSV is trained using the visualized sequence reads, and deletion calls are made based on this trained model. The authors demonstrate that DeepSV surpasses existing methods in terms of the accuracy and efficiency of deletion calling, particularly on data from the 1000 Genomes Project. This study highlights the potential of DL in effectively calling various types of genetic variations that are more complex than single nucleotide polymorphisms (SNPs) [44].
Intelli-NGS, on the other hand, excels in discerning reliable variant calls from Ion Torrent sequencer data. IonTorrent is a second-generation sequencing platform with smaller capital costs than Illumina but is also prone to higher machine error than later. Given its lower costs, the platform is generally preferred in developing countries where NGS is still a very exclusive technique. There are many software tools available for other platforms but IonTorrent. This makes the already tricky analysis part more error prone. Intelli-NGS excels in discerning reliable variant calls from Ion Torrent sequencer data [45]. Additionally, models like DeepGestalt are designed to identify facial phenotypes associated with genetic disorders [46], DeepMiRGene predicts miRNA precursors [47], and DeepMILO (DL for Modeling Insulator Loops) forecasts the impact of non-coding sequence variants on 3D chromatin structure [48].
DeepPVP (PhenomeNet Variant Predictor) is proficient in identifying variants in both whole exome and whole genome sequence data [49], while ExPecto excels in accurately predicting tissue-specific transcriptional effects of mutations and functional Single Nucleotide Polymorphisms (SNPs) [50]. PEDIA (Prioritisation of exome data by image analysis) is instrumental in prioritizing variants and genes for diagnosing patients with rare genetic disorders [51].
Exome sequencing approach is extensively used in research and diagnostic laboratories to discover pathological variants and study the genetic architecture of human diseases. However, a significant proportion of identified genetic variants are actually false positive calls, and these pose a serious challenge for interpretation of variants. A new tool named Genomic vARiants FIltering by dEep Learning moDels in NGS (GARFIELD-NGS), which relies on DL models to dissect false and true variants in exome sequencing experiments. GARFIELD-NGS significantly reduces the proportion of false candidates, thus improving the identification of diagnostic relevant variants. These results define GARFIELD-NGS as a robust tool for all types of Illumina and ION exome data. GARFIELD-NGS script performs automated variant scoring on VCF files, and it can be easily integrated in existing analysis pipelines [52].
Other notable models, including DeepWAS [53], Basset [54], DanQ [55], and SPEID [56], focus on identifying disease-associated Single Nucleotide Polymorphisms (SNPs), predicting causative SNPs, extracting DNA function directly from sequence data, and enhancing promoter interaction (EPI) prediction, respectively [53,54,55,56]. In the realm of gene expression and regulation, a range of tools such as DeepExpression, DeepGSR, SpliceAI, Gene2vec, and MPRA-DragoNN serve distinct purposes, such as predicting gene expression, recognizing genomic signals and regions, identifying splice function, generating a representation of gene distribution, and predicting as well as analyzing regulatory DNA sequences and non-coding genetic variants [57,58,59,60,61]
Epigenetics is a field dedicated to exploring heritable alterations in gene expression that occur without modifying the DNA sequence. In past decades researchers have increasingly recognized the pivotal role of epigenetic regulation in processes such as cell growth, differentiation, autoimmune disease, and cancer. Key epigenetic mechanisms encompass well established phenomena like DNA methylation, histone modifications and the influence of non-coding RNAs. Non-coding RNAs, a relatively new focus of intensive investigation, were initially perceived as regulators of gene expression at the post-transcriptional level, without encoding functional proteins. Studies have unveiled that non-coding RNAs, including miRNAs, piRNAs, endogenous siRNAs and long non-coding RNAs, are prevalent regulators. Importantly, a growing body of evidence underscores the significant contribution of regulatory non-coding RNAs to the realm of epigenetic control, emphasizing the noteworthy role of RNA in governing gene expression [62].
Detecting the functional impacts of noncoding variants poses a significant hurdle in human genetics. To predict the effects of noncoding variants directly from sequencing data a DL-based algorithmic framework called DeepSEA was created. DeepSEA learns a regulatory sequence code directly from extensive chromatin-profiling data, allowing the precise prediction of chromatin effects resulting from sequence alterations at the single-nucleotide level. Other DL models in epigenomics, such as FactorNet, DeepCpG, and Basenji, are adept at predicting cell-type-specific transcriptional binding factors, methylation states from single-cell data, and cell-type-specific epigenetic and transcriptional profiles in large mammalian genomes [63].
5. Pharmacogenomic Deep Learning Models
In pharmacogenomics, models like DeepD, DNN-DTI, DeepBL, DeepDrug3D, DrugCell, and DeepSynergy are applied for translating pharmacogenomic features, predicting drug-target interactions, forecasting beta-lactamase, characterizing, and classifying protein 3D binding pockets, as well as predicting drug response and synergy in anticancer drugs. Collectively, these models form a comprehensive suite of DL tools crucial for understanding and analyzing genomics and related fields [63,64,65,66,67,68]. DL methods have proven to be highly effective in predicting treatment responses based on "-omics” datasets of cell lines. An illustrative example is Drugcell, a visible neural network (VNN) interpretation model designed to elucidate the structure and function of human cancer cells in response to therapy. This model aligns its central mechanisms with the organizational structure of human biology, enabling the prediction of drug responses across various cancers and intelligently planning successful treatment combinations.
DrugCell was specifically engineered to capture both aspects of therapy response within an interpretable model, comprising two divisions: the VNN, which integrates call genotype and artificial neural networks (ANN), which integrates drug design. The first VNN model takes as input text files detailing the hierarchical associations between molecular subsystems in human cells, incorporating 2086 biological process standards from the Gene Intology (GO) database. The second ANN model takes traditional ANN inputs, integrating text files representing the Morgan fingerprint of drugs- the chemical structure represented by canonical vector symbol. The outputs from these two divisions are amalgamated into single layer of neurons, generating the response of a given genotype to a specific therapy. The prediction accuracy of each drug individually demonstrated significant precision, revealing distinct drug sub-populations. This level of accuracy competes with state-of-the-art regression methods utilized in previous models for predicting drug responses. Notably, when compared to a parallel neural network model trained solely on drug design and labeled tissue. DrugCell significantly outperformed the tissue-based model. This underscores that DrugCell has effectively assimilated data from somatic mutations beyond the capabilities of a tissue-only approach [68].
6. Exploring AI-Powered Genomics in Multi-Omics Research
6.1. Radiomics, Pathomics and Surgomics
In addition to WGS, AI has also enabled the development of multiple new “-omics” fields such as Radiomics Pathomics and more recently Surgomics. A fusion of WGS, radiomics and pathomics has been proposed by the Artificial intelligence Radiomics Oncopathomics and Surgomics (AiRGOS) project [71]. It has been shown that radiomic analysis of single arterial phase cross-sectional images of hepatocellular cancers can be used to accurately predict which tumors will recur early [72]. By combing AI-powered analysis of WGS patterns of tumors with pre-operative 3-dimensional reconstructions of tumors and pathomic analysis of histopathology slides, it is believed that tumor boards enhanced with AI can be created once they are trained with retrospective chemotherapy, immunotherapy, and radiation treatment regimens. Hopefully an algorithm will be able to be created that can reduce the therapeutic non-responsive rate that many cancer patients suffer. By minimizing lost time to ineffective treatments and reducing exposure to side-effects of these treatments, it is believed that overall survival can be significantly improved (Figure 2).
Once this concept is validated post-operatively so that better decisions on adjuvant treatments can be determined, the project will be expanded to neoadjuvant treatment regimens and then real-time analysis in the actual operating room. Known as Video Surgomics, video data from the operating room can theoretically be analyzed in real time with enhanced imaging techniques such as multispectral imaging, narrow band imaging and biophotonics so that information that the human eye cannot comprehend during surgery can be captured, analyzed and interpreted so that the operating surgeon can make better decisions regarding decision for surgery and extent of resection. Hopefully video Surgomics will be able to reduce the number of patients operated on who have occult carcinomatosis, sarcomatosis or metastases. Conceivably, it could help guide surgeons to make real-time decisions for more advanced therapies such as choice for intra-peritoneal chemotherapy.
6.2. Proteomics, Transcriptomics and Genomics
The generation and processing of huge biological data sets (omics data) is made possible by technological and informatics advancements, which are driving a fundamental change in the field of biomedical sciences research. Even while the fields of proteomics, transcriptomics, genomics, bioinformatics, and biostatistics are gaining ground, they are still primarily evaluated separately using different methodologies, producing monothematic rather than integrated information. combining and applying (multi)omics data to improve knowledge of the molecular pathways, mechanisms, and processes that distinguish between health and disease[73]. Within the field of proteomics, transcriptomics and genomics dynamic partners that provide distinct insights into the complex regulation of biological functions.
Proteomics is the scientific study of the proteome, or the whole set of proteins expressed and altered by a biological system. Proteomes are extremely dynamic and constantly changing both within and between biological systems. The word "proteomics" was coined by Marc Wilkins in 1996 to emphasise how much more complex and dynamic researching proteins is than studying genomes. Using techniques like mass spectrometry (MS), protein microarrays, X-ray crystallography, chromatography-based methods, and Western blotting, proteomics analyses a range of factors related to protein content, function, regulation, post-translational modifications, expression levels, mobility within cells, and interactions. Mass spectrometry has become an essential high-throughput proteomics technique these days, especially when paired with liquid chromatography (LC-MS/MS). The way that protein structure is predicted has fundamentally changed as a result of DL advancements like the AlphaFold algorithm [74].
The use of AI technology has resulted in notable advancements in the field of proteomics. The exponential increase of biomedical data, particularly multiomics and genome sequencing datasets, has ushered in a new era of data processing and interpretation. AI-driven mass spectrometry-based proteomics research has progressed because of data sharing and open-access laws. At initially, AI was restricted to data analysis and interpretation, but recent advances in DL have transformed the sector and improved the accuracy and calibre of data. DL may be able to surpass the best-in-class biomarker identification processes that are currently available in predicting experimental peptide values from amino acid sequences. Proteomics and AI convergence presents a transformative paradigm for biomedical research, offering fresh perspectives on biological systems and ground-breaking approaches to illness diagnosis and treatment [75].
Though proteomics is not expressly discussed, the story alludes to its consequences. Understanding protein-level manifestations becomes crucial at the intersection of genetics and proteomics, as highlighted by the focus on PPAR proteins and their therapeutic potential in colonic disorders. Additionally, the integration of flow cytometry and genomics in haematological malignancies suggests a proteomic component, highlighting the importance of assessing protein expression for accurate diagnosis [76].
The transcriptome, or collection of all RNA transcripts, of an organism is studied by transcriptomics technology. An organism's DNA encodes its information, which is then expressed by transcription. Since its first attempt in the early 1990s, transcriptomics has seen substantial change. RNA sequencing (RNA-Seq) and microarrays are two important methods in the field. Measurements of gene expression in various tissues, environments, or periods of time shed light on the biology and regulation of genes. Understanding human disease and identifying wide-ranging coordinated trends in gene expression have both benefited greatly from the analysis [77].
Since the late 1990s, technological advancements have transformed the sector. Transcriptomics has come a long way since its inception because to techniques like SAGE, the emergence of microarrays, and NGS technologies in the 2000s. Because the transcriptome is dynamic, it is challenging to define and analyse, which calls for the application of AI methods such ML techniques like Random Forests and Support Vector Machines. Neural networks and other DL technologies have shown to be crucial in enhancing transcript categorization by unveiling intricate biological principles. Understanding the molecular mechanisms underlying differential gene expression requires a thorough analysis of gene expression data. AI tools such as Random Forests and Deep Neural Networks (DNNs) analyse massive datasets to distinguish between clinical groups and detect diseases. Gene expression becomes more complex due to polyadenylation and alternative splicing; AI aids in predicting AS patterns and comprehending splicing code [78].
Specific challenges are introduced by single-cell RNA sequencing (scRNA-seq), including a high proportion of zero-valued observations, or "dropouts." The visualisation and interpretation of high-dimensional scRNA-seq data has benefited from enhanced dimensionality reduction through the use of ML and DL techniques like Uniform Manifold Approximation and Projection (UMAP) and t-distributed Stochastic Neighbour Embedding (t-SNE) [78].
Transcriptomics research has been greatly impacted by AI, especially in the field of cancer research. Significant transcriptome data is produced by RNA-seq technology and can be obtained by AI-based algorithms [79].
The transcriptome is largely composed of non-coding RNAs (ncRNAs), which have become important actors with a variety of roles ranging from complex mRNA regulatory mechanisms to catalytic functions. The field of transcriptome analysis has continued to advance, as seen by the evolution of techniques from conventional Northern Blotting to sophisticated RNA sequencing (RNA-Seq) [76]. By enhancing the accuracy of identifying cancer states and stages, AI in transcriptome analysis contributes to the development of precision medicine. AI techniques like denoising and dropout imputation tackle problems in scRNA-seq research such as high noise levels and missing data. AI algorithms are essential for separating biological signals from noise and integrating multi-omics data as the profiles get more complicated.
Immunotherapy issues are resolved by AI-assisted transcriptome analysis, which analyses tumour heterogeneity, predicts responses, and identifies different cell types. Technologies will be continuously created and used in immunotherapy research as the era of precision medicine dawns. Combining it could boost the effectiveness of immunotherapies and alter the course of cancer research [79]. The integration of AI into transcriptomics has significantly enhanced our comprehension of the transcriptome, particularly considering the growing technologies and new challenges in single-cell research [78].
The study of genomics reveals the complexities encoded within an organism's entire set of genes. It emphasizes the importance of single-nucleotide polymorphisms (SNPs) in defining genetic loci that contribute to complicated disorders by delving into them. It does, however, address the difficulties, such as false-positive connections, highlighting the importance of precision in experimental design. The combined global efforts in genomics, particularly in the quick identification of the SARS-associated coronavirus, demonstrate the discipline's real-world effect in tackling new health concerns [76].
ML has made immense progress in genomics since the 1980s, particularly with the integration of DL techniques in the 2000s. In fact, ML has been instrumental in predicting DNA regulatory areas, annotating sequence elements, and discovering chromatin effects in genomics. To effectively handle the vast amount of sequences and diverse data sources, DL methods such as Convolutional Neural Networks (CNNs) and Generative Adversarial Networks (GANs) have been utilized. The success of unsupervised learning, specifically GANs and Autoencoders (AEs), in identifying intricate patterns in biological sequences through the extraction of representative features from genomics data further demonstrates the potential of these powerful techniques in genomics research.
The integration of ML and CRISPR-Cas9 technology is a pivotal coming together of experimental and computational biology, expediting research on large-scale genetic interactions. Approaches utilizing ML and DL have displayed potential in identifying connections between diseases and genes, as well as forecasting the genetic susceptibility of intricate disorders. The scope of methods is evident, ranging from SVM-based classification to comprehensive frameworks employing CNNs, as demonstrated by CADD, DANN, and ExPecto [74].
Studies have been greatly impacted by the emergence of AI, especially big data fields like functional genomics. Large amounts of data have been organised and abstracted using deep architectures, which has improved interpretability. Conversely, the lack of explainability in deep AI architectures casts doubt on the findings' applicability and transparency. The wider use of functional genomics depends on the free and open exchange of AI tools, resources, and knowledge. AI technologies are selected in the fields of biology and functional genomics to provide mechanistic understanding of biological processes. This enables systems biology to assess biological systems or develop theoretical models that forecast their behaviour. The use of AI in systems biology will see competition or cooperation between data-driven and model-driven methods. DeepMind's AlphaFold approach highlights the power of AI, particularly transformer-based models. Complex considerations of individual and communal rights are involved in functional genomics, and the application of AI necessitates navigating through different data, interpreting pertinent questions, and addressing legal, ethical, and moral issues. In the developing landscape of AI development, a cautious approach is required to ensure that the advantages outweigh the potential negative repercussions [74].
Diverse Interaction provides insight on the vast synergy present in molecular biology. Proteomics reveals expressions at the protein level, transcriptomics reveals the functional RNA environment, and genomics lays the groundwork by interpreting genetic data. The integration of various disciplines offers a thorough understanding of diseases, emphasising the value of a multimodal approach in diagnosis and therapy.
Ultimately, the dynamic interplay of transcriptomics, proteomics, and genomes holds the key to understanding the complexity of illnesses. This discussion highlights the dynamic nature of molecular biology, where each specialty contributes in a different way to the overarching narrative of health and disease [76].
While fields like proteomics, transcriptomics and genomics are making individual strides, they are often evaluated in isolation, leading to monothematic information. To overcome this limitation, efforts are being made to integrate (multi)omics data, aiming to enhance our understanding of molecular pathways, mechanisms, and processes related to health and disease. Within proteomics, transcriptomics, and genomics, synergistic partnerships provide unique insights into the intricate regulation of biological functions. the integration of proteomics, transcriptomics, and genomics offers a comprehensive understanding of diseases, emphasizing the significance of a multimodal approach in diagnosis and therapy. The dynamic interplay among these disciplines holds the key to unraveling the complexity of illnesses, showcasing the nuanced contributions of each specialty in the broader narrative of health and disease.
7. Conclusion
In conclusion, in this scoping review we highlighted the potential of AI-powered WGS in overcoming the limitations of existing sequencing technologies, particularly in the context of oncology. We have provided an overview of the evolution of DNA sequencing technologies, the process of WGS, and the application of AI, specifically DL, in variant calling and pharmacogenomics. In light of this, we emphasize the importance of multimodal approaches in diagnoses and therapies, and the need for further research and development in AI-powered WGS techniques. We suggest that AI-powered WGS has the potential to revolutionize the field of genomics and improve patient outcomes when associated within the real of multi-omics data, but also acknowledge the challenges associated with the interpretation and management of the vast amount of genomic data generated by high-throughput technologies. Overall, as we have provided a comprehensive overview of the potential of AI in addressing the challenges associated with WGS analysis, we underscore the need for further research and development in this field to further improve patient outcomes through personalized and targeted treatments.
References
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. Journal of molecular biology 1975, 94, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Masoudi-Nejad, A.; Narimani, Z.; Hosseinkhan, N. (2013). Next generation sequencing and sequence assembly: methodologies and algorithms (Vol. 4). Springer Science & Business Media.
- El-Metwally, S.; Ouda, O.M.; Helmy, M. (2014). Next generation sequencing technologies and challenges in sequence assembly (Vol. 7). Springer Science & Business.
- Sanger, F.; Coulson, A.; Barrell, B.G.; Smith AJ, H.; Roe, B.A. Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing. Journal of molecular biology 1980, 143, 161–178. [Google Scholar] [CrossRef]
- Arabidopsis Genome Initiative genomeanalysis@ tgr. org genomeanalysis@ gsf. de. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. nature 2000, 408, 796–815. [CrossRef] [PubMed]
- Goff, S.A.; Ricke, D.; Lan, T.H.; Presting, G.; Wang, R.; Dunn, M.; Briggs, S.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Rm, D. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar]
- Kchouk, M.; Gibrat, J.F.; Elloumi, M. Generations of sequencing technologies: from first to next generation. Biology and Medicine 2017, 9. [Google Scholar]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proceedings of the National Academy of Sciences 1977, 74, 560–564. [Google Scholar] [CrossRef]
- Masoudi-Nejad, A.; Narimani, Z.; Hosseinkhan, N. (2013). Next generation sequencing and sequence assembly: methodologies and algorithms (Vol. 4). Springer Science & Business Media.
- Bayés, M.; Heath, S.; Gut, I.G. Applications of second generation sequencing technologies in complex disorders. Behavioral Neurogenetics 2012, 321–343. [Google Scholar]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Law, M.; et al. Comparison of next-generation sequencing systems. Journal of Biomedicine and Biotechnology 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- El-Metwally, S.; Ouda, O.M.; Helmy, M. (2014). Next generation sequencing technologies and challenges in sequence assembly (Vol. 7). Springer Science & Business.
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Molecular cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nature biotechnology 2012, 30, 434–439. [Google Scholar]
- Kulski, J.K. Next-generation sequencing—an overview of the history, tools, and “Omic” applications. Next generation sequencing-advances, applications and challenges 2016, 10, 61964. [Google Scholar]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 2008, 9, 387–402. [Google Scholar] [PubMed]
- Alic, A.S.; Ruzafa, D.; Dopazo, J.; Blanquer, I. Objective review of de novo stand-alone error correction methods for NGS data. Wiley Interdisciplinary Reviews: Computational Molecular Science 2016, 6, 111–146. [Google Scholar] [CrossRef]
- Masoudi-Nejad, A.; Narimani, Z.; Hosseinkhan, N. (2013) Next generation sequencing and sequence assembly. Methodologies and algorithms. Springer.
- Masoudi-Nejad, A.; Narimani, Z.; Hosseinkhan, N. (2013). Next generation sequencing and sequence assembly: methodologies and algorithms (Vol. 4). Springer Science & Business Media.
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Roe, P.M.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. nature 2008, 456, 53–59. [Google Scholar]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Turner, S.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Braslavsky, I.; Hebert, B.; Kartalov, E.; Quake, S.R. Sequence information can be obtained from single DNA molecules. Proceedings of the National Academy of Sciences 2003, 100, 3960–3964. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.D.; Buzby, P.R.; Babcock, H.; Beer, E.; Bowers, J.; Braslavsky, I.; Xie, Z.; et al. Single-molecule DNA sequencing of a viral genome. Science 2008, 320, 106–109. [Google Scholar] [CrossRef] [PubMed]
- McCoy, R.C.; Taylor, R.W.; Blauwkamp, T.A.; Kelley, J.L.; Kertesz, M.; Pushkarev, D.; Fiston-Lavier, A.S.; et al. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PloS one 2014, 9, e106689. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genomics, proteomics & bioinformatics 2015, 13, 278–289. [Google Scholar]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Law, M.; et al. Comparison of next-generation sequencing systems. Journal of Biomedicine and Biotechnology 2012, 2012. [Google Scholar] [CrossRef]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Schatz, M.C.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nature methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Kulski, J.K. Next-generation sequencing—an overview of the history, tools, and “Omic” applications. Next generation sequencing-advances, applications and challenges 2016, 10, 61964. [Google Scholar]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Phillippy, A.M.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nature biotechnology 2012, 30, 693–700. [Google Scholar] [CrossRef]
- Mikheyev, A.S.; Tin, M.M. A first look at the Oxford Nanopore MinION sequencer. Molecular ecology resources 2014, 14, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Laehnemann, D.; Borkhardt, A.; McHardy, A.C. Denoising DNA deep sequencing data—high-throughput sequencing errors and their correction. Briefings in bioinformatics 2016, 17, 154–179. [Google Scholar] [CrossRef] [PubMed]
- Laver, T.; Harrison, J.; O’neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the oxford nanopore technologies minion. Biomolecular detection and quantification 2015, 3, 1–8. [Google Scholar] [CrossRef]
- Ip, C.L.; Loose, M.; Tyson, J.R.; de Cesare, M.; Brown, B.L.; Jain, M.; Reference, Consortium; et al. MinION Analysis and Reference Consortium: Phase 1 data release and analysis. F1000Research 2015, 4. [Google Scholar]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Archives of Disease in Childhood-Education and Practice 2013, 98, 236–238. [Google Scholar] [CrossRef]
- Grada, A.; Weinbrecht, K. Next-generation sequencing: methodology and application. Journal of Investigative Dermatology 2013, 133, 1–4. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of next-generation sequencing technologies. Current protocols in molecular biology 2018, 122, e59. [Google Scholar] [CrossRef]
- Podnar, J.; Deiderick, H.; Huerta, G.; Hunicke-Smith, S. Next-Generation sequencing RNA-Seq library construction. Current protocols in molecular biology 2014, 106, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer science 2018, 109, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; DePristo, M.A.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nature biotechnology 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Sedlazeck, F.J.; Lam, T.W.; Schatz, M.C. A multi-task convolutional deep neural network for variant calling in single molecule sequencing. Nature communications 2019, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Wu, Y.; Gao, J. DeepSV: accurate calling of genomic deletions from high-throughput sequencing data using deep convolutional neural network. BMC bioinformatics 2019, 20, 1–17. [Google Scholar] [CrossRef]
- Singh, A.; Bhatia, P. Intelli-NGS: Intelligent NGS, a deep neural network-based artificial intelligence to delineate good and bad variant calls from IonTorrent sequencer data. bioRxiv 2019, 2019–12. [Google Scholar]
- Gurovich, Y.; Hanani, Y.; Bar, O.; Nadav, G.; Fleischer, N.; Gelbman, D.; Gripp, K.W.; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nature medicine 2019, 25, 60–64. [Google Scholar] [CrossRef]
- Park, S., Min, S., Choi, H., & Yoon, S. (2016). deepMiRGene: Deep neural network based precursor microrna prediction. arXiv preprint arXiv:1605.00017.Boudellioua I, Kulmanov M, Schofield PN, Gkoutos GV, Hoehndorf R. DeepPVP: phenotype-based prioritization of causative variants using deep learning. BMC Bioinform. 2019;20(1):65. [CrossRef]
- Trieu, T.; Martinez-Fundichely, A.; Khurana, E. DeepMILO: a deep learning approach to predict the impact of non-coding sequence variants on 3D chromatin structure. Genome biology 2020, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boudellioua, I.; Kulmanov, M.; Schofield, P.N.; Gkoutos, G.V.; Hoehndorf, R. DeepPVP: phenotype-based prioritization of causative variants using deep learning. BMC bioinformatics 2019, 20, 1–8. [Google Scholar] [CrossRef]
- Zhou, J.; Theesfeld, C.L.; Yao, K.; Chen, K.M.; Wong, A.K.; Troyanskaya, O.G. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nature genetics 2018, 50, 1171–1179. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Mensah, M.A.; Pantel, J.T.; Aguilar, D.; Bar, O.; Bayat, A.; Krawitz, P.M.; et al. PEDIA: prioritization of exome data by image analysis. Genetics in Medicine 2019, 21, 2807–2814. [Google Scholar] [CrossRef]
- Ravasio, V.; Ritelli, M.; Legati, A.; Giacopuzzi, E. Garfield-ngs: Genomic variants filtering by deep learning models in NGS. Bioinformatics 2018, 34, 3038–3040. [Google Scholar] [CrossRef] [PubMed]
- Arloth, J.; Eraslan, G.; Andlauer, T.F.; Martins, J.; Iurato, S.; Kühnel, B.; Mueller, N.S.; et al. DeepWAS: Multivariate genotype-phenotype associations by directly integrating regulatory information using deep learning. PLoS computational biology 2020, 16, e1007616. [Google Scholar] [CrossRef]
- Kelley, D.R.; Snoek, J.; Rinn, J.L. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome research 2016, 26, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Xie, X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic acids research 2016, 44, e107–e107. [Google Scholar] [CrossRef]
- Singh, S.; Yang, Y.; Póczos, B.; Ma, J. Predicting enhancer-promoter interaction from genomic sequence with deep neural networks. Quantitative Biology 2019, 7, 122–137. [Google Scholar] [CrossRef]
- Zeng, W.; Wang, Y.; Jiang, R. Integrating distal and proximal information to predict gene expression via a densely connected convolutional neural network. Bioinformatics 2020, 36, 496–503. [Google Scholar] [CrossRef]
- Kalkatawi, M.; Magana-Mora, A.; Jankovic, B.; Bajic, V.B. DeepGSR: an optimized deep-learning structure for the recognition of genomic signals and regions. Bioinformatics 2019, 35, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Farh KK, H.; et al. Predicting splicing from primary sequence with deep learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef]
- Du, J.; Jia, P.; Dai, Y.; Tao, C.; Zhao, Z.; Zhi, D. Gene2vec: distributed representation of genes based on co-expression. BMC genomics 2019, 20, 7–15. [Google Scholar] [CrossRef]
- Movva, R.; Greenside, P.; Marinov, G.K.; Nair, S.; Shrikumar, A.; Kundaje, A. Deciphering regulatory DNA sequences and noncoding genetic variants using neural network models of massively parallel reporter assays. PLoS One 2019, 14, e0218073. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovascular research 2011, 90, 430–440. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nature methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Chiu, Y.C.; Chen HI, H.; Zhang, T.; Zhang, S.; Gorthi, A.; Wang, L.J.; Chen, Y.; et al. Predicting drug response of tumors from integrated genomic profiles by deep neural networks. BMC medical genomics 2019, 12, 143–155. [Google Scholar]
- Xie, L.; He, S.; Song, X.; Bo, X.; Zhang, Z. Deep learning-based transcriptome data classification for drug-target interaction prediction. BMC genomics 2018, 19, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, F.; Bharathwaj, M.; Rosas, N.C.; Leier, A.; Akutsu, T.; Song, J.; et al. DeepBL: a deep learning-based approach for in silico discovery of beta-lactamases. Briefings in Bioinformatics 2021, 22, bbaa301. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Govindaraj, R.G.; Lemoine, J.M.; Wu, H.C.; Brylinski, M. DeepDrug3D: classification of ligand-binding pockets in proteins with a convolutional neural network. PLoS computational biology 2019, 15, e1006718. [Google Scholar] [CrossRef] [PubMed]
- Kuenzi, B.M.; Park, J.; Fong, S.H.; Sanchez, K.S.; Lee, J.; Kreisberg, J.F.; Ideker, T.; et al. Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer cell 2020, 38, 672–684. [Google Scholar] [CrossRef]
- Preuer, K.; Lewis, R.P.; Hochreiter, S.; Bender, A.; Bulusu, K.C.; Klambauer, G. DeepSynergy: predicting anti-cancer drug synergy with Deep Learning. Bioinformatics 2018, 34, 1538–1546. [Google Scholar] [CrossRef]
- Alharbi, W.S.; Rashid, M. A review of deep learning applications in human genomics using next-generation sequencing data. Human Genomics 2022, 16, 1–20. [Google Scholar] [CrossRef]
- Gumbs, A.A.; Croner, R.; Abu-Hilal, M.; Bannone, E.; Ishizawa, T.; Spolverato, G.; Frigerio, I.; Siriwardena, A.; Messaoudi, N. Surgomics and the Artificial intelligence, Radiomics, Genomics, Oncopathomics and Surgomics (AiRGOS) Project. Art Int Surg 2023, 3, 180–185. [Google Scholar] [CrossRef]
- Kinoshita, M.; Ueda, D.; Matsumoto, T.; Shinkawa, H.; Yamamoto, A.; Shiba, M.; Okada, T.; Tani, N.; Tanaka, S.; Kimura, K.; Ohira, G.; Nishio, K.; Tauchi, J.; Kubo, S.; Ishizawa, T. Deep Learning Model Based on Contrast-Enhanced Computed Tomography Imaging to Predict Postoperative Early Recurrence after the Curative Resection of a Solitary Hepatocellular Carcinoma. Cancers (Basel). 2023, 15, 2140. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Chen, Z.; Hood, L. Systems medicine: the future of medical genomics and healthcare. Genome Med [Internet]. 2009, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Caudai, C.; Galizia, A.; Geraci, F.; Le, L.; Morea, V.; Salerno, E.; et al. AI applications in functional genomics. Comput Struct Biotechnol J. 2021, 19, 5762–5790. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kumar, C.; Zeng, W.; Strauss, M.T. Perspective Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Kiechle, F.L.; Holland-Staley, C.A. Genomics, transcriptomics, proteomics, and numbers. Arch Pathol Lab Med. 2003, 127, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. 2017;(Fig 1):1–23.
- Supplitt, S.; Karpinski, P.; Sasiadek, M.; Laczmanska, I. Current Achievements and Applications of Transcriptomics in Personalized Cancer Medicine. 2021. [Google Scholar]
- Gui, Y.; He, X.; Yu, J.; Jing, J. Artificial Intelligence-Assisted Transcriptomic Analysis to Advance Cancer Immunotherapy. 2023. [Google Scholar]
Figure 1.
Whole genomic sequencing can lead to predictive chemotherapy response: Certain genetic markers or patterns may be associated with sensitivity or resistance to specific chemotherapeutic agents. By analyzing the entire genetic profile of the tumor, clinicians can predict the likelihood of response to different chemotherapy drugs and select the most effective treatment regimen for an individual patient.
Figure 1.
Whole genomic sequencing can lead to predictive chemotherapy response: Certain genetic markers or patterns may be associated with sensitivity or resistance to specific chemotherapeutic agents. By analyzing the entire genetic profile of the tumor, clinicians can predict the likelihood of response to different chemotherapy drugs and select the most effective treatment regimen for an individual patient.

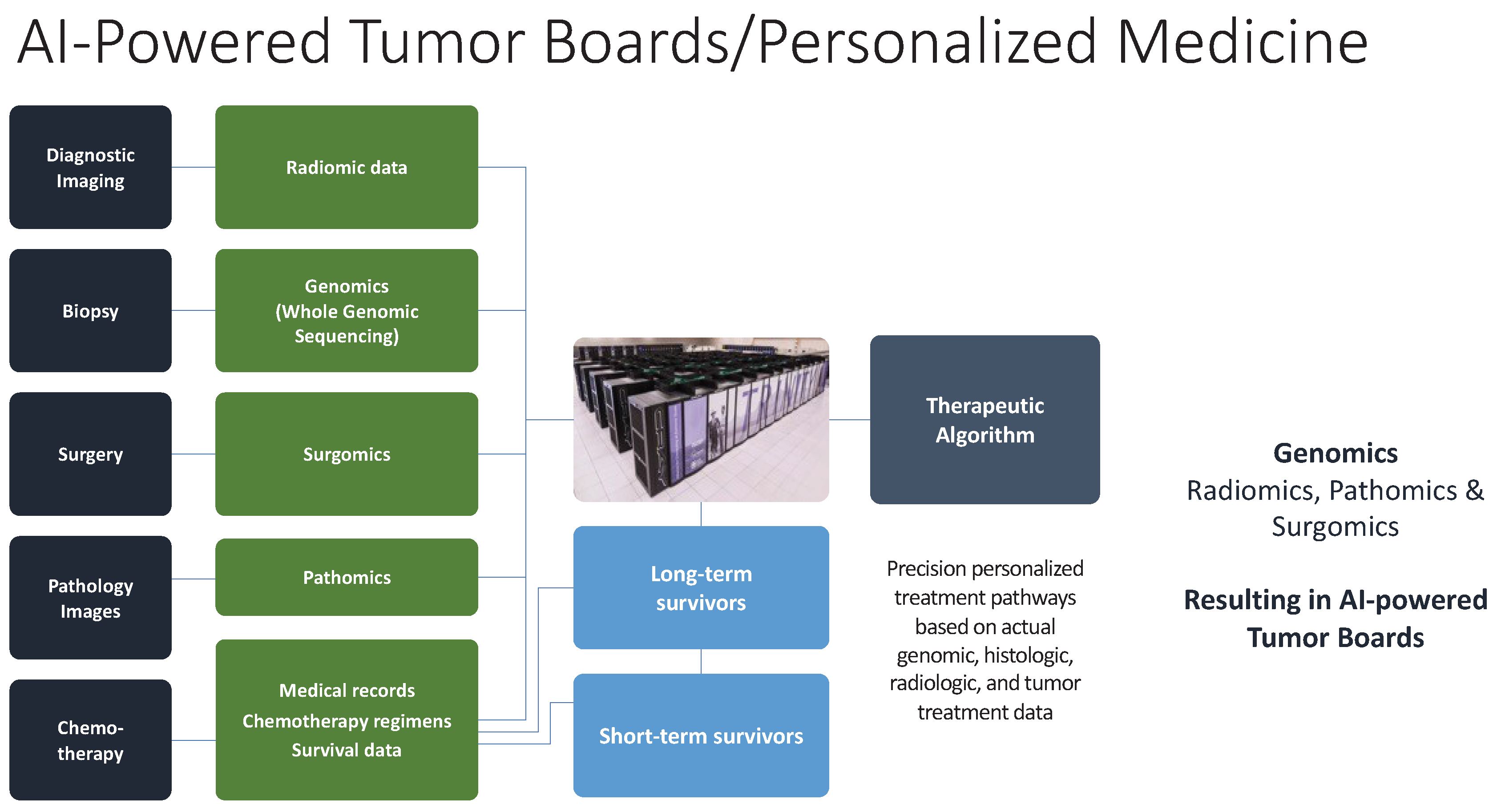
Figure 2.
Artificial Intelligence (AI) powered analysis of whole genomic sequences can create more powerful cancer treatment paradigms that can require integration and use of AI/deep learning to optimize surgical resection and neoadjuvant and adjuvant treatment options. Use of AI/deep learning algorithm based on comparative survivor outcomes to identify best chance chemotherapy/immunotherapy regimens and predictive long-term survival.
Figure 2.
Artificial Intelligence (AI) powered analysis of whole genomic sequences can create more powerful cancer treatment paradigms that can require integration and use of AI/deep learning to optimize surgical resection and neoadjuvant and adjuvant treatment options. Use of AI/deep learning algorithm based on comparative survivor outcomes to identify best chance chemotherapy/immunotherapy regimens and predictive long-term survival.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



