
Submitted:
18 March 2024
Posted:
21 March 2024
You are already at the latest version
Abstract
Colorectal cancer (CRC) with KRAS mutation is prone to develop metachronous metastasis. However, effective measures for prediction and prevention of such clinical issue are currently unavailable. In this study, we confirmed that KRAS mutation is a risk factor for CRC metachronous metastasis through meta-analysis and public database analysis. Based on the ICGC-AGRO database, we have constructed a risk scoring model for predicting metachronous metastasis in KRAS-mutant CRC using machine learning, which was validated in public datasets. Furthermore, we discovered that KRAS inhibitors can effectively suppress the migration and invasion capabilities of high risk CRC cells by wound healing assay and transwell experiment, which was validated ex vivo and in vivo using organoids and splenic liver metastatic mouse model. Finally, RNA sequencing, RT-qPCR, and western blots have proven that KRAS inhibitors can suppress the epithelial-mesenchymal transition (EMT) and transforming growth factor beta (TGF-beta) signaling pathway in CRC. Collectively, our study suggests that KRAS inhibitors may inhibit the metastatic ability of CRC by suppressing EMT and the TGF-beta pathway, suggesting their potential for the prevention of metachronous metastasis in CRC with KRAS mutation.
Keywords:
KRAS inhibitor
; metastasis
; colorectal cancer
; machine learning
; meta analysis
1. Introduction
Colorectal cancer (CRC) is one of the most prevalent malignancies worldwide, with nearly 2 million new cases reported annually[1]. Despite comprehensive treatments including surgery, neoadjuvant and adjuvant chemoradiotherapy were applied to patients according to guideline, up to 23% of patients still develop distant recurrence after curative treatment, known as metachronous metastasis[2]. Patients with metachronous metastasis often have a poor prognosis, with 5-year survival rate less than 30%, which is a severe challenge in the clinical practice of CRC[3,4]. Since the effectiveness of first-line chemotherapy in preventing metachronous metastasis of CRC is limited[5,6], there is an urgent need to identify a biomarker that can predict the risk of distant recurrence of CRC after curative surgery, and to provide interventions other than chemotherapy for high-risk patients.
KRAS is a well-known oncogene involved in the development and progression of CRC, with about 40% of CRC cases harboring KRAS mutation[7]. Abundant evidence has indicated a strong correlation between KRAS mutation and unfavorable prognosis in CRC[8,9,10,11]. Also, research has suggested that it may have an impact on the effectiveness of chemotherapy and targeted therapy[12,13]. Furthermore, certain studies have observed that KRAS mutation might serve as a risk factor for metachronous metastasis in CRC[14,15,16]. Hence, the development of novel assessment and adjuvant treatment strategies specifically tailored to KRAS mutations may offer new perspectives in preventing metachronous metastasis in CRC.
KRAS has long been considered an undruggable target. The emergence of KRAS inhibitors in recent years has broken this impasse. Small molecule inhibitors such as sotorasib and adagrasib can directly bind to KRASG12C with specificity, whose efficacy have been demonstrated by clinical trials in CRC[17]. Currently, the aforementioned KRAS inhibitors have obtained FDA approval. however, such novel therapy is only limited to heavily pre-treated stage IV CRC[18]. A lack of research concerning whether KRAS inhibitors can prevent CRC metastasis still exists.
This study validated the correlation between KRAS mutations and metachronous metastasis of CRC through meta-analysis and public database investigation. Furthermore, we developed a tool to identify high-risk CRC patients with KRAS mutation who are likely to suffer from metachronous metastasis. Through in vitro, in vivo and ex vivo experiments, we proved that KRAS inhibitors could effectively suppress the metastatic ability of high-risk CRC by inhibiting epithelial to mesenchymal transition and the TGF-beta pathway.
2. Materials and Methods
2.1. Meta Analysis
A systemic literature review was performed using PubMed database, Embase and Cochrane library. The following syntax was used for the search: ([KRAS] or [K-RAS]) and ([colorectal cancer] or [colon cancer] or [rectal cancer]) and ([metachronous metastasis] or [distant recurrence] or [distant relapse]). The review adhered to the guidelines outlined in the PRISMA statement[19]. Literature that meets the following criteria will be included in the meta analysis:study includes patients who were initially diagnosed with stage I-III colorectal cancer and underwent curative surgery; KRAS mutational status was provided; follow-up information including distant recurrence after surgery was provided. Duplicates, non-English literature, reviews, editorials, case reports, abstracts, as well as studies unrelated to colorectal cancer, will be excluded. A systematic full-text review was conducted on eligible literature, data including the first author's name, publication date, sample size, KRAS mutation rate, and metachronous survival rate were extracted for meta-analysis. The odds ratio (OR) and its 95% confidence interval (CI) were used as the outcome measures for the study. OR greater than 1 indicates that KRAS mutation is a risk factor for metachronous metastasis. Heterogeneity was assessed using the I2 and Cochran’s Q statistics. I2 greater than 50% was considered as substantial heterogeneity. Statistical significance of the summary estimates was based on Wald test P value. Result of the meta-analysis were presented using a forest plot. Publication bias in the studies was assessed using a funnel plot. Figures were plotted using R(Version:4.0.3) (http://www.R-project.org).
2.2. Data Acquisition
Clinical information, KRAS mutation status, and transcriptome sequencing of 1001 colorectal cancer patients were downloaded from the COCC (Omics Study of Colorectal Cancer in China, https://www.icgc-argo.org/page/114/cgcc cohort of the ICGC-ARGO project (https://www.icgc-argo.org) of Sixth Affiliated Hospital, Sun Yat-sen University. Additionally, clinical and KRAS mutational data from MSKCC cohort reported by Chatila et al. and the Sidra-LUMC AC-ICAM cohort reported by Roelands et al were retrieved from cbioportal (https://www.cbioportal.org)[20,21]. Transcriptome sequencing were obtained from the GEO database (GSE209746) and the dbP database (phs002978.v1.p1), respectively. The transcriptome data of cell lines were acquired from the Cancer Cell Line Encyclopedia (CCLE) database (https://sites.broadinstitute.org/ccle/datasets).
2.3. Model Construction
To develop the metachronous metastatic risk prediction model for the patients with KRAS-mutant CRC, 286 cases with KRAS mutation from ICGC-ARGO cohort were selected to serve as training cohort. This study also collected 77 KRAS-mutant cases from the Sidra-LUMC AC-ICAM cohort and 32 KRAS-mutant cases from the MSKCC cohort with eligible gene-expression data as two independent validation cohorts, respectively. In the training cohort, univariate Cox regression was performed to identify metastasis-associated genes, and then these genes were further utilized in the Lasso regression to establish the metastatic risk prediction model. Generating metastasis risk score for each patient, patients were stratified into high-risk and low-risk groups based on the risk score threshold determined by the Youden index. Kaplan-Meier analysis, differential gene expression analysis, and gene set enrichment analysis (GSEA) were conducted to compare the differences in metastasis, gene expression and pathway activation pattern between high and low risk groups.
2.4. Cell Culture
All cell lines were purchased from Meisen CTCC (Zhejiang Meisen Cell Technology, Ltd.) and authenticated using short tandem repeat (STR) analysis. Cells were cultured in DMEM (SW480, HCT116 and SW837) or RPMI1640 (DLD1 and HCT15) supplemented with 10% fetal bovine serum (HyClone, Cytiva) and 1% penicillin-streptomycin (Gibco) in a 5% CO2 environment maintained at 37°C. Monthly mycoplasma testing was performed on the cells. BI-2865, adagrasib, oxaliplatin, and 5-fluorouracil were purchased from TargetMol.
For colony formation assay, cells were dissociated using trypsin and resuspended in complete culture medium as single cells, then seeded into six-well plates (20,000 cells per well). The medium was changed every 3 days, and the cells were cultured for 12 days. Colonies were fixed with methanol and stained with crystal violet (0.5% crystal violet, 20% methanol), followed by photography.
2.5. Wound-Healing Assay
Seed 5x10^5 cells into a six-well plate and incubate overnight to form a confluent monolayer. Replace complete medium with serum-free medium. Scratches were made using a 200uL pipette tip. After 48 hours, the scratches were photographed. Images were processed using ImageJ.
2.6. Transwell
Resuspend 1x10^5 cells in 200uL serum-free medium and then seed them into the upper chamber (8μm pore size, Corning). For cell invasion assay, the upper chamber was pre-coated with Matrigel (1:20, cat. no. 356234; Corning, NY, USA). 500uL of medium containing 10% serum was added to the lower chamber. After 48 hours, cells underneath the upper chamber were fixed with 4% paraformaldehyde and then stained using crystal violet solution. Photographs of 5 random fields of each chamber were taken under Olympus IX73 inverted microscope. Images were processed using ImageJ.
2.7. Organoid Culture
The origin and culture method of organoids can be referred to our previously published literature[22].
2.8. Establishment of Cell Lines with Acquired Resistance to Adagrasib
SW837 cells were grown with increasing concentrations of adagrasib. Subconfluent cells were treated with trametinib at IC50 dose (60 nM based on 96-hour cytotoxicity assays) for two 3-day periods, and surviving cells were maintained for another 4 days. The process was repeated once, and the remaining cells were cultured in the presence of drug for 3 additional passages. Resistant cells were maintained in the presence of adagrasib, and the same passaged parental cells were used in various cellular assays.
2.9. Western Blotting
Protein extracts were prepared with RIPA cell lysis buffer (150 mm NaCl, 50 mm Tris-HCl, 0.5% deoxychlorate sodium, 200mmNaF, 200mmPMSF, 1.0%NP40, and 1mmEDTA) with a protease inhibitor cocktail (Roche, Basel, Switzerland). Lysates and prestained protein marker (M221, GenStar, Beijing, China) were subjected to SDS-PAGE and transferred to PVDF membranes for immunoblotting analysis. The antibodies used in this study are provided in supplementary table s2.
2.10. Cell Viability Assay
For the cell viability assay, cells were seeded in 96-well plates at the optimal seeding density in triplicate. After 24 h, cells were treated with different concentrations of drugs and cultured at 37 °C for 96 h, and the number of viable cells was measured by using the CellTiter-Glo Luminescent Cell Viability Assay (#G7573, Promega, Madison, WI) according to the manufacturer’s instructions.
2.11. Real-Time Quantitative PCR
Total RNA was extracted from cells using an RNeasy Mini Kit (QIAGEN, Duesseldorf, Germany) according to the manufacturer’s protocol. One microgram of RNA was used for RT-qPCR using a reverse transcription kit (#AT341-02, Transgen Biotech, Beijing, China) and quantitative kit (#N30920, Transgen Biotech, Beijing, China) on a Biorad CFX Real-time PCR machine. All qPCR experiments were performed in triplicate, and mean values were used to determine mRNA levels. Relative quantification was performed using the comparative CT method with 18S as the reference gene and with the formula 2-ΔΔCT. Error bars showed standard deviation. The primer sequences are listed in supplementary table S2.
2.12. RNA Sequencing
Total RNA was extracted using the RNAeasy Mini Kit according to the manufacturer’s protocol (QIAGEN, Duesseldorf, Germany). The mRNA libraries were prepared using a TruSeq Stranded Total RNA Sample Preparation kit with Ribo Zero Gold (Illumina), followed by sequencing on a NovaSeq sequencer (Illumina).
2.13. Mice Experiment
Male BALB/c nude mice (6 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd and housed in the Biological Resource Centre. SW480 cells were treated with either DMSO or BI2865(1μM) for 96. After drug withdrawn and cultured in complete medium for 48h, equal number of cells (2x10^6) cells in 50uL sterile saline were injected into the spleen of nude mice (6 mice per group). After 12 weeks, the mice were euthanized, and the livers were collected for photography and immunohistochemistry hematoxylin–eosin staining.
2.14. Statistical Analysis
For survival analysis, clinical data obtained from the ICGC-ARGO, MSKCC, and Sidra-LUMC AC-ICAM databases were screened and excluded the following cases: cases without KRAS mutation status; initially diagnosed with stage IV CRC; distant recurrence information missing; local recurrence. The remaining patients were included in the subsequent analysis of metachronous-metastasis free survival (MFS) (Supplementary figure S2A-C). Metachronous metastasis is defined as distant metastasis occurring after initial diagnosis or primary surgery[23]. To evaluate the effect of chemotherapy on metachronous metastasis in colorectal cancer, propensity score matching was performed considering the influence of postoperative pathological staging on clinical decision-making of chemotherapy. Matched cases were included in subsequent survival analysis. Kaplan-Meier curves were plotted using R (Version:4.0.3). Propensity score matching was performed using SPSS 26.0 (SPSS Inc., Chicago, IL, USA).
Experimental data were presented as the mean ± SD, unless otherwise stated. Statistical significance between two groups was evaluated by two-tailed Student’s t-test, while statistical significance among multiple groups was analyzed by two-way ANOVA with Dunnett’s multiple comparisons test using GraphPad Prism software. Statistical significance was considered at p < 0.05.
3. Results
3.1. KRAS Mutation is a Risk Factor for CRC Metachronous Metastasis
Whether KRAS mutation is a predictive factor for metachronous metastasis of CRC remains controversial[24,25,26]. Here, we conducted a meta-analysis focused on this issue. The initial literature search returned 210 records (Supplementary figure S1A); after exclusions, 117 articles remained for assessment of eligibility. After application of the other exclusion criteria and review of reference lists for missed articles, 11 unique articles remained and were pooled in the meta-analysis (Figure 1A). These articles reported on 1296 patients, of whom 387 had mutant KRAS[16,25,27,28,29,30,31,32,33,34,35]. The results from these 11 studies were generally consistent, with KRAS positively correlate with CRC metachronous metastasis (OR 2.57, 95% CI: 1.96-3.37). The funnel plot did not suggest notable publication bias (Supplementary figure S1B). Further investigation using ICGC-ARGO database validated that KRAS mutation is negatively correlated with postoperative MFS of CRC patients (HR 2.73, 95%CI 1.47-5.07, p<0.01) (Figure 1B)(Supplementary figure S2A)(Supplementary table S1A). Similar trends can still be observed in two other online databases, although the significance is not less than 0.05 (Sidra-LUMC AC-ICAM: HR 1.72, 95%CI 0.88-3.37, p=0.11; MSKCC: 1.55, 95%CI 0.92-2.61, p=0.10)(Figure 1C-D)(Supplementary figure S2B-C)(Supplementary table S1B,C)[20,21]. By comparing MFS between patients with or without chemotherapy after propensity score matching (Supplementary table S1D-I), we found that adjuvant chemotherapy can significantly prevent distant recurrence in KRASwt CRC, while the efficacy was quite limited in KRASmut (Figure 1E-J) patients, which was consistent with previous report[12]. These results suggested that KRAS mutation is a risk factor for CRC metachronous metastasis and that first-line chemotherapy cannot profoundly improve metastasis-free survival in KRASmut CRC patients after curative surgery. However, according to the above data, KRAS mutational status is far from an accurate predictive factor for CRC metachronous metastasis, hence the need of improvement of such biomarker.
3.2. Machine-Learning Method Stratifies KRASmut CRC according to the Risk of Metachron Ous Metastasis
Utilizing complete RNA sequencing data and follow-up information from 403 KRASmut CRC cases across three cohorts, we constructed and validated a metastatic risk prediction model consisting of 36 genes (Supplementary Figure 3). With the metastatic risk score, which will be referred to as KRAS metachronous metastatic (KMM) score, for each patient in the training cohort (COCC, n = 286) and two validation cohorts (Sidra-LUMC AC-ICAM, n = 82; MSKCC, n = 35), we stratified patients into high-risk and low-risk groups. Kaplan-Meier curve analysis showed that patients in the high-risk group had significantly higher risk for metachronous metastasis (Figure 2A-C). Furthermore, through differential gene expression analysis, we discovered a significant upregulation of genes related to the EMT (epithelial-mesenchymal transition) pathway and TGF-beta pathway in the high-risk group compared to the low-risk group (Figure 2D). Additionally, subsequent gene set enrichment analysis (GSEA) revealed that the EMT and TGF-beta pathways were also highly activated in the high-risk group (Figure 2E). These findings suggest that the highly activated EMT pathway and TGF-beta pathway may be closely associated with tumor metastasis in patients with KRASmut CRC.
3.3. KRAS Inhibitors Suppress Migration and Invasion Ability of KRASmut CRC Cells
Using the above-mentioned KMM scoring model, we stratified KRAS-mutant colorectal cancer cell lines based on the gene expression profile in CCLE database (Figure 3A). To begin with, we treated the high-risk cell lines (SW480 and HCT116) and the low-risk cell lines (DLD1 and HCT15) with 5-fluorouracil plus oxaliplatin to assess the effect of first-line chemotherapy on the migration and invasion ability of CRC. Wound healing assay and Boyden-Chamber transwell experiments both indicate that chemotherapy can significantly reduce the migration and invasion ability of KMM low-risk CRC cell lines(Supplementary figure 4A-D), while having no significant effect in KMM high-risk CRC cell lines (Supplementary figure 4E-H). Therefore, alternative adjuvant treatment options need to be explored for KMM high-risk CRC.
BI-2865 is recently reported pan-KRAS inhibitor that has demonstrated effective inhibition against SW480 (KRASG12V) and H116 (KRASG13D)[36]. Both wound healing assay and transwell experiment revealed that BI-2865 effectively inhibits the migration and invasion ability of SW480 cells (Figure 3B-C). Similar results can be observed in HCT116 (Figure 3D-E). Meanwhile, we noticed the mesenchymal morphology of the cells gradually transformed into an epithelial-like morphology under the treatment of BI2865 in SW480 and HCT116, suggesting the potential of KRAS inhibitor to induce mesenchymal-epithelial transition (Supplementary figure 5). As BI-2865 has not been used clinically, we conducted further research on the impact of the FDA-approved KRAS inhibitor, adagrasib, on the invasion and migration ability of CRC[18]. We conducted long-term in vitro drug treatment on SW837, a KMM high-risk CRC cell line harboring KRASG12C mutation (Figure 3A), to simulate the extended application of adagrasib in clinical scenario (Supplementary figure S6A). Acquired resistance to KRAS inhibitors is a common issue in colorectal cancer[37]. As expected, resistance of SW837-R occurred after prolonged drug treatment, characterized by a significant increase in IC50 and the maintenance of MAPK pathway under relatively high dosage of adagrasib compared with SW837-P (Supplementary figure S6B-D). Despite the development of acquired resistance to adagrasib, SW837-R showed significantly reduced ability of migration and invasion compared with SW837-P cells (Figure 3F-G), indicating the potential of adagrasib in stabilizing tumor cells and preventing their metastasis. Furthermore, we applied BI-2865 on a previously established organoid, PDO-CC0117, harboring KRASG12D[22]. While treatment-naive PDO-CC0117 organoid showed a solid structure with spike-formation which indicates migration activity, BI-2865 treatment maintained a relatively normal cystic morphology, suggesting the stabilizing ability of KRAS inhibitor (Figure 3H). We further investigated the effect of KRAS inhibitors on the metastatic ability of CRC in vivo. Formation of liver metastases in BI2865 treated group was significantly reduced compared with control group (Figure 3I-J). Collectively, the above results indicate that KRAS inhibitors can suppress the metastatic ability of high-risk CRC and may have potential applications in preventing metachronous metastasis of KRAS-mutant colorectal cancer.
3.4. KRAS Inhibitors Repress EMT and TGF-Beta Pathway in CRC Cell Lines
It has been reported that mutant KRAS can enhance the migration and invasion capabilities of CRC via EMT and TGF-beta signaling pathway[38,39,40,41]. GSEA enrichment analysis using GSE228010 dataset showed that SW480 cells[36],after BI2865 treatment, exhibited down-regulation in the EMT and TGF-beta pathways, which was verified by RT-qPCR (Figure 4A-B). Similar results can be observed in HCT116 (Figure 4C-D). Further enrichment analysis using GSE116823 dataset indicates that KRAS knockdown also leads to EMT and TGF-beta down-regulation in HCT116 (Supplementary figure S4A-B). Consistent with RT-qPCR results, western-blot showed that BI-2865 can result in a decrease of EMT markers such as N-cadherin, Vimentin and MMP1 while up-regulate E-cadherin (Figure 4E). We further investigated the transcriptomic alterations between SW837-P and SW837-R. RNA-seq identified 2,531 differentially expressed genes, and pathway enrichment indicated a significant down-regulation of the EMT and TGF-beta pathways in SW837-R compared with SW837-P (Figure 4F-J), which was verified by RT-qPCR (Figure 4K-L). Consistently, western blot showed down-regulation of N-cadherin, Vimentin and MMP1 and an upregulation of E-cadherin in SW837-R(Figure 4M). This indicates that even acquired resistance occurred, prolonged treatment with adagrasib led to the down-regulation of the EMT and TGF-beta pathways in SW837 and thus, decreasing its migration and invasion abilities. Collectively, the above results indicate that KRAS inhibitors might inhibit CRC metastatic capability through suppressing EMT and TGF-beta pathways.
4. Discussion
Colorectal cancer is one of the most prevalent malignancy worldwide[1]. Despite comprehensive treatments were applied to non stage IV patients as a standard procedure, up to 23% of patients still develop metachronous metastasis after curative treatments[2]. Overall, we proved that KRAS mutation is a risk factor for CRC metachronous metastasis and further developed a model for assessing metachronous metastasis risk in KRAS-mutant CRC patients. In vitro and in vivo experiments showed that KRAS inhibitors can effectively suppress metastatic capabilities of high-risk CRC, providing a new approach for adjuvant therapy in colorectal cancer(Figure 5).
The first tool predicting CRC metachronous metastasis is reported by Hao et al, who developed a nomogram based on clinical data[16]. However, this tool is limited to metachronous liver metastasis and cannot assess metachronous lung metastasis, which has a relatively worse outcome[6]. Besides, specific intervention for high risk patients was not proposed, therefore, more efforts are still required to address the clinical challenge of metachronous metastasis in CRC. Currently, multi-omics molecular diagnostics have gradually been integrated into clinical practice, boosting personalized diagnoses and treatment[42]. Utilizing high-throughput data to create a predictive model for CRC metachronous metastasis is a promising approach to enhance prediction efficiency and on this basis, identify novel postoperative intervention strategies.
KRAS is the oncogene with the highest mutation frequency in CRC, with a cumulative 49% mutational rate reported by a recent study[43]. There is ample evidence suggesting the correlation between KRAS mutation and CRC metastasis; compared with wildtype, KRAS mutant patients have a higher incidence of stage IV CRC[44,45,46]. However, whether KRAS mutation contributes to CRC metachronous metastasis remains controversial. Studies have indicated that KRAS mutations are only associated with metachronous lung metastasis rather than liver metastasis in CRC[47]. Some research also suggest that codon 13 mutation are prognostically significant, whereas mutations on other hotspots of KRAS do not exhibit a clear association with CRC metachronous metastasis[14,15]. In light of this, we conducted a meta-analysis without taking into account the location of metastasis and the type of mutation, and proved that KRAS mutation is indeed a risk factor for CRC metachronous metastasis, which was confirmed by subsequent survival analysis, indicating the role of KRAS in promoting metastasis. As a single factor, KRAS mutation itself is not a robust predictive factor, suggesting the association between KRAS and metastasis exhibits individual variability[48]. Therefore, we utilized public omic databases to develop a tool to predict metachronous metastasis risk in KRAS-mutant CRC. Anyway, the prediction efficacy of such model is not that satisfactory according to its performance in validation cohorts. Considering the KMM tool was based solely on gene expression omics, it would be necessary to incorporate clinical data, radiomics, genomics and proteomics information to establish multiomics models in our future work, thereby optimizing the KMM scoring system.
KRAS-mutant CRC has long been consider as a clinical conundrum for its lack of specific therapeutic approach. Research indicates that KRAS mutations may affect the efficacy of chemotherapy[12,13], and causes EGFR-targeted therapy resistance[49,50,51]. Furthermore, studies found that KRAS mutation can reduce tumor infiltrated T lymphocytes, resulting in a cold tumor immune microenvironment in CRC, which suggest that immunotherapy may not be a preferred option for KRAS-mutant CRC[52,53]. In recent years, a variety of KRAS inhibitors have emerged and shown promising results in cancer treatment, providing specific and effective options for KRAS-mutant CRC patients[54]. However, current KRAS inhibitors are only approved for the use of metastatic CRC that has failed first-line treatment[18], and there are no studies to date reporting on their ability to prevent tumor metastasis[55]. Recently, Miao et al. discovered that erianin specifically inhibits the metastasis of colorectal cancer harboring KRASG13D mutation, providing an alternative for the prevention of metachronous metastasis in KRAS-mutant CRC[56]. However, KRASG13D only accounts for 8.1% of all CRC patients, suggesting the use of erianin might be limited. In this study, we proved that adagrasib, an FDA-approved KRASG12C inhibitor, as well as the recently reported pan-KRAS inhibitor BI-2865, which targets multiple mutational subtypes, both exhibit inhibitory effects on the migration and invasion capabilities of KRAS-mutant CRC. This marks the first instance of such work in preclinical studies involving KRAS inhibitors and offers a prophylactic strategy for metachronous metastasis in KRAS-mutant CRC.
The mechanisms by which KRAS promotes tumor metastasis have been documented in several studies. It has been reported that KRAS rewires TGF-beta pathway and acts as the upstream of EMT, driving the migration and invasion behavior of CRC[38,39,40,41]. In this study, we found that KRAS inhibitors can effectively suppress EMT and the TGF-beta pathway, suggesting KRAS inhibitors may suppress the metastatic capabilities of CRC through this manner. In-depth investigation is required to shed light on how KRAS inhibitors exert inhibitory effect on these pathways. Perhaps the 36 genes in the KMM signature may provide clues to this end (Figure 4Supplementary C).
5. Conclusions
In summary, we developed a risk model for KRAS-mutant colorectal cancer that can predict the likelihood of metachronous metastasis. We further demonstrated that KRAS inhibitors are effective in suppressing the migration and invasion properties of colorectal cancer, offering novel post-surgical assessment and intervention strategies for colorectal cancer.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, G.-Y.Y., Y.-Z.L. and W.-X.J.; methodology, G.-Y.Y., L.-G.J., H.-X.X. and H.-P.S.; software, H.-C.L. and C.-D.; validation, G.-Y.Y., L.-G.J., H.-X.X. and H.-P.S.; formal analysis, G.-Y.Y.; investigation, Y.-Z.L.; resources, C.-Z.R.; data curation, C.-Y.F.; writing—original draft preparation, G.-Y.Y.; writing—review and editing, G.-Y.Y. and Y.-Z.L.; visualization, G.-Y.Y.; supervision, W.-X.J.; project administration, W.-X.J. and G.-F.; funding acquisition, Y.-Z.L., C.-Y.F. and W.-X.J.. All authors have read and agreed to the published version of the manuscript.
Funding
The project was supported by National Natural Science Foundation of China (Nos. 82203072).
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Sixth Affiliated Hospital, Sun Yat-sen University No. 2020[020] on 9 January 2020. (Guangzhou, China). The animal study protocol was approved by the Animal Care Committee of Sixth Affiliated Hospital, Sun Yat-sen University (SYSU-IACUC-2022-080601).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data analyzed during the present study are available from the corresponding author on reasonable request.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
CRC: colorectal cancer; KRAS, Kirsten Rat Sarcoma Viral Oncogene Homolog; RT-qPCR, real-time quantitative polymerase chain reaction; SDS-PAGE, sodium lauryl sulfate polyacrylamide gel electrophoresis; PVDF, polyvinylidene fluoride; MAPK, Mitogen-activated protein kinase; EMT, epithelial-mesenchymal transition; TGF-beta, transform growth factor beta; PRISMA, The Preferred Reporting Items for Systematic Reviews and Meta-Analyses; OR, odds ratio; CI, confidence interval; MFS, metachronous metastasis free survival; COCC, Omics Study of Colorectal Cancer in China; CCLE, Cancer Cell Line Encyclopedia; DMEM, Dulbecco's modified eagle medium; EDTA, Ethylenediaminetetraacetic acid; PMSF, Phenylmethanesulfonylfluoride.
References
- Siegel, R.L.; et al. Cancer statistics, 2023. CA Cancer J Clin 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Platell, C.F. , Metachronous colorectal cancer metastasis: Who, what, when and what to do about it. J Surg Oncol 2024, 129, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hansdotter, P.; et al. Treatment and survival of patients with metachronous colorectal lung metastases. J Surg Oncol 2023, 127, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Reboux, N.; et al. Incidence and Survival in Synchronous and Metachronous Liver Metastases From Colorectal Cancer. JAMA Netw Open 2022, 5, e2236666. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; et al. Colorectal liver metastases: Current management and future perspectives. World J Clin Oncol 2020, 11, 761–808. [Google Scholar] [CrossRef]
- Pfannschmidt, J.; Dienemann, H.; Hoffmann, H. , Surgical resection of pulmonary metastases from colorectal cancer: a systematic review of published series. Ann Thorac Surg 2007, 84, 324–38. [Google Scholar] [CrossRef] [PubMed]
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Dinu, D.; et al. Prognostic significance of KRAS gene mutations in colorectal cancer--preliminary study. J Med Life 2014, 7, 581–7. [Google Scholar]
- Abubaker, J.; et al. Prognostic significance of alterations in KRAS isoforms KRAS-4A/4B and KRAS mutations in colorectal carcinoma. J Pathol 2009, 219, 435–45. [Google Scholar] [CrossRef]
- Taieb, J.; et al. Prognostic Value of BRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. J Natl Cancer Inst 2017, 109. [Google Scholar] [CrossRef]
- Formica, V.; et al. KRAS and BRAF Mutations in Stage II and III Colon Cancer: A Systematic Review and Meta-Analysis. J Natl Cancer Inst 2022, 114, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Iseas, S.; et al. Prognostic Impact of An Integrative Landscape of Clinical, Immune, and Molecular Features in Non-Metastatic Rectal Cancer. Front Oncol 2021, 11, 801880. [Google Scholar] [CrossRef] [PubMed]
- De Dosso, S.; et al. Influence of KRAS mutations on clinical outcome in patients with curatively resected stage III colon cancer treated with adjuvant chemotherapy. Eur Rev Med Pharmacol Sci 2020, 24, 2994–3003. [Google Scholar] [PubMed]
- Feng, Q.; et al. A specific KRAS codon 13 mutation is an independent predictor for colorectal cancer metachronous distant metastases. Am J Cancer Res 2015, 5, 674–88. [Google Scholar] [PubMed]
- Ilm, K.; et al. High MACC1 expression in combination with mutated KRAS G13 indicates poor survival of colorectal cancer patients. Mol Cancer 2015, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; et al. Predicting metachronous liver metastasis in patients with colorectal cancer: development and assessment of a new nomogram. World J Surg Oncol 2022, 20, 80. [Google Scholar] [CrossRef] [PubMed]
- Nusrat, M.; Yaeger, R. KRAS inhibition in metastatic colorectal cancer: An update. Curr Opin Pharmacol 2023, 68, 102343. [Google Scholar] [CrossRef]
- Dhillon, S. , Adagrasib: First Approval. Drugs 2023, 83, 275–285. [Google Scholar] [CrossRef]
- Moher, D.; et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Chatila, W.K.; et al. Genomic and transcriptomic determinants of response to neoadjuvant therapy in rectal cancer. Nat Med 2022, 28, 1646–1655. [Google Scholar] [CrossRef]
- Roelands, J.; et al. An integrated tumor, immune and microbiome atlas of colon cancer. Nat Med 2023, 29, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; et al. Inhibition of the PLK1-Coupled Cell Cycle Machinery Overcomes Resistance to Oxaliplatin in Colorectal Cancer. Adv Sci (Weinh) 2021, 8, e2100759. [Google Scholar] [CrossRef] [PubMed]
- Engstrand, J.; et al. Synchronous and metachronous liver metastases in patients with colorectal cancer-towards a clinically relevant definition. World J Surg Oncol 2019, 17, 228. [Google Scholar] [CrossRef] [PubMed]
- Kuan, T.C.; et al. Prognosticators of Long-Term Outcomes of TNM Stage II Colorectal Cancer: Molecular Patterns or Clinicopathological Features. World J Surg 2019, 43, 3207–3215. [Google Scholar] [CrossRef] [PubMed]
- Jo, P.; et al. KRAS mutation status concordance between the primary tumor and the corresponding metastasis in patients with rectal cancer. PLoS One 2020, 15, e0239806. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; et al. The prognostic role of microsatellite instability, codon-specific KRAS, and BRAF mutations in colon cancer. J Surg Oncol 2014, 110, 451–7. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, A.; et al. Association of K-ras mutations with liver metastases from colorectal carcinoma. Anticancer Research 2004, 24, 2471–2476. [Google Scholar]
- Vakiani, E.; et al. Local recurrences at the anastomotic area are clonally related to the primary tumor in sporadic colorectal carcinoma. Oncotarget 2017, 8, 42487–42494. [Google Scholar] [CrossRef]
- Sperlich, A.; et al. Genetic and immunological biomarkers predict metastatic disease recurrence in stage III colon cancer. BMC Cancer 2018, 18, 998. [Google Scholar] [CrossRef]
- Nitsche, U.; et al. Integrative marker analysis allows risk assessment for metastasis in stage II colon cancer. Annals of surgery 2012, 256, 763–771, discussion 771. [Google Scholar] [CrossRef]
- Nakamura, Y.; et al. Preoperative detection of KRAS mutated circulating tumor DNA is an independent risk factor for recurrence in colorectal cancer. Sci Rep 2021, 11, 441. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; et al. Overexpression of MutL homolog 1 and MutS homolog 2 proteins have reversed prognostic implications for stage I-II colon cancer patients. Biomedical Journal 2017, 40, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; et al. A specific KRAS codon 13 mutation is an independent predictor for colorectal cancer metachronous distant metastases. American Journal of Cancer Research 2015, 5, 674–688. [Google Scholar] [PubMed]
- El Otmani, I.; et al. Analysis of Molecular Pretreated Tumor Profiles as Predictive Biomarkers of Therapeutic Response and Survival Outcomes after Neoadjuvant Therapy for Rectal Cancer in Moroccan Population. Disease Markers 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Birgisson, H.; et al. Microsatellite instability and mutations in BRAF and KRAS are significant predictors of disseminated disease in colon cancer. BMC Cancer 2015, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature 2023, 619, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Flum, M.; et al. Canonical TGFbeta signaling induces collective invasion in colorectal carcinogenesis through a Snail1- and Zeb1-independent partial EMT. Oncogene 2022, 41, 1492–1506. [Google Scholar] [CrossRef]
- Boutin, A.T.; et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev 2017, 31, 370–382. [Google Scholar] [CrossRef]
- Sanchez-Tillo, E.; et al. The EMT factor ZEB1 paradoxically inhibits EMT in BRAF-mutant carcinomas. JCI Insight 2023, 8. [Google Scholar] [CrossRef]
- Lemieux, E.; et al. Oncogenic KRAS signalling promotes the Wnt/beta-catenin pathway through LRP6 in colorectal cancer. Oncogene 2015, 34, 4914–27. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; et al. Multi-Omics Approaches in Colorectal Cancer Screening and Diagnosis, Recent Updates and Future Perspectives. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; et al. Comprehensive pan-cancer genomic landscape of KRAS altered cancers and real-world outcomes in solid tumors. NPJ Precis Oncol 2022, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, L.; et al. Recent Updates on the Significance of KRAS Mutations in Colorectal Cancer Biology. Cells 2021, 10. [Google Scholar] [CrossRef]
- Yaeger, R.; et al. RAS mutations affect pattern of metastatic spread and increase propensity for brain metastasis in colorectal cancer. Cancer 2015, 121, 1195–203. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev 2018, 37, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; et al. KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clin Cancer Res 2011, 17, 1122–30. [Google Scholar] [CrossRef]
- Guo, L.; et al. Molecular Profiling Provides Clinical Insights Into Targeted and Immunotherapies as Well as Colorectal Cancer Prognosis. Gastroenterology 2023, 165, 414–428. [Google Scholar] [CrossRef]
- Misale, S.; et al. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov 2014, 4, 1269–80. [Google Scholar] [CrossRef]
- De Roock, W.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010, 11, 753–62. [Google Scholar] [CrossRef]
- Misale, S.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–6. [Google Scholar] [CrossRef] [PubMed]
- Edin, S.; et al. Opposing roles by KRAS and BRAF mutation on immune cell infiltration in colorectal cancer - possible implications for immunotherapy. Br J Cancer 2024, 130, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Lal, N.; et al. KRAS Mutation and Consensus Molecular Subtypes 2 and 3 Are Independently Associated with Reduced Immune Infiltration and Reactivity in Colorectal Cancer. Clin Cancer Res 2018, 24, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.A.; Strickler, J.H. Targeting KRAS-Mutated Gastrointestinal Malignancies with Small-Molecule Inhibitors: A New Generation of Breakthrough Therapies. Drugs 2024, 84, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Webster, K.R. Targeting mutant KRAS. Curr Opin Chem Biol 2021, 62, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; et al. Erianin inhibits the growth and metastasis through autophagy-dependent ferroptosis in KRAS(G13D) colorectal cancer. Free Radic Biol Med 2023, 204, 301–312. [Google Scholar] [CrossRef]
Figure 1.
KRAS mutation is associated with CRC metachronous metastasis. (A) Forest plot of association between KRAS mutation status and CRC metachronous metastasis in eleven studies. A random-effects model with inverse-variance method was used for meta-analysis. Odds ratios are shown with 95% CI. Results of comparison of MFS between KRAS-MUT and KRAS-WT CRC patients in the ARGO cohort (B), Sidra-LUMC AC-ICAM cohort (C) and MSKCC cohort (D). Results of comparison of MFS between KRAS-MUT patients with or without chemotherapy in ARGO (E), Sidra-LUMC AC-ICAM (F) and MSKCC cohort (G). Results of comparison of MFS between KRAS-WT patients with or without chemotherapy in ARGO (E), Sidra-LUMC AC-ICAM (F) and MSKCC cohort (G). CRC, colorectal cancer; MFS, metachronous-metastasis free survival; KRAS-MUT, KRAS-mutant; KRAS-WT, KRAS-wildtype.
Figure 1.
KRAS mutation is associated with CRC metachronous metastasis. (A) Forest plot of association between KRAS mutation status and CRC metachronous metastasis in eleven studies. A random-effects model with inverse-variance method was used for meta-analysis. Odds ratios are shown with 95% CI. Results of comparison of MFS between KRAS-MUT and KRAS-WT CRC patients in the ARGO cohort (B), Sidra-LUMC AC-ICAM cohort (C) and MSKCC cohort (D). Results of comparison of MFS between KRAS-MUT patients with or without chemotherapy in ARGO (E), Sidra-LUMC AC-ICAM (F) and MSKCC cohort (G). Results of comparison of MFS between KRAS-WT patients with or without chemotherapy in ARGO (E), Sidra-LUMC AC-ICAM (F) and MSKCC cohort (G). CRC, colorectal cancer; MFS, metachronous-metastasis free survival; KRAS-MUT, KRAS-mutant; KRAS-WT, KRAS-wildtype.
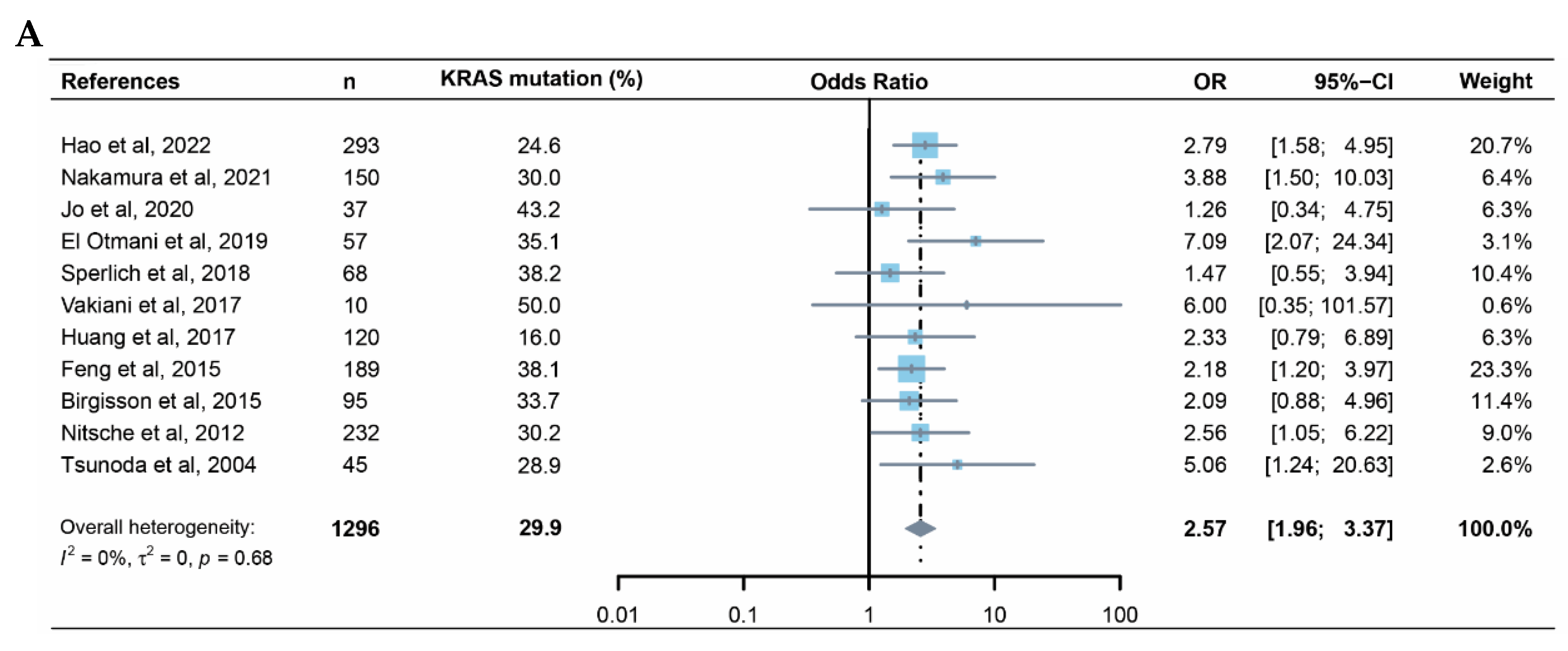
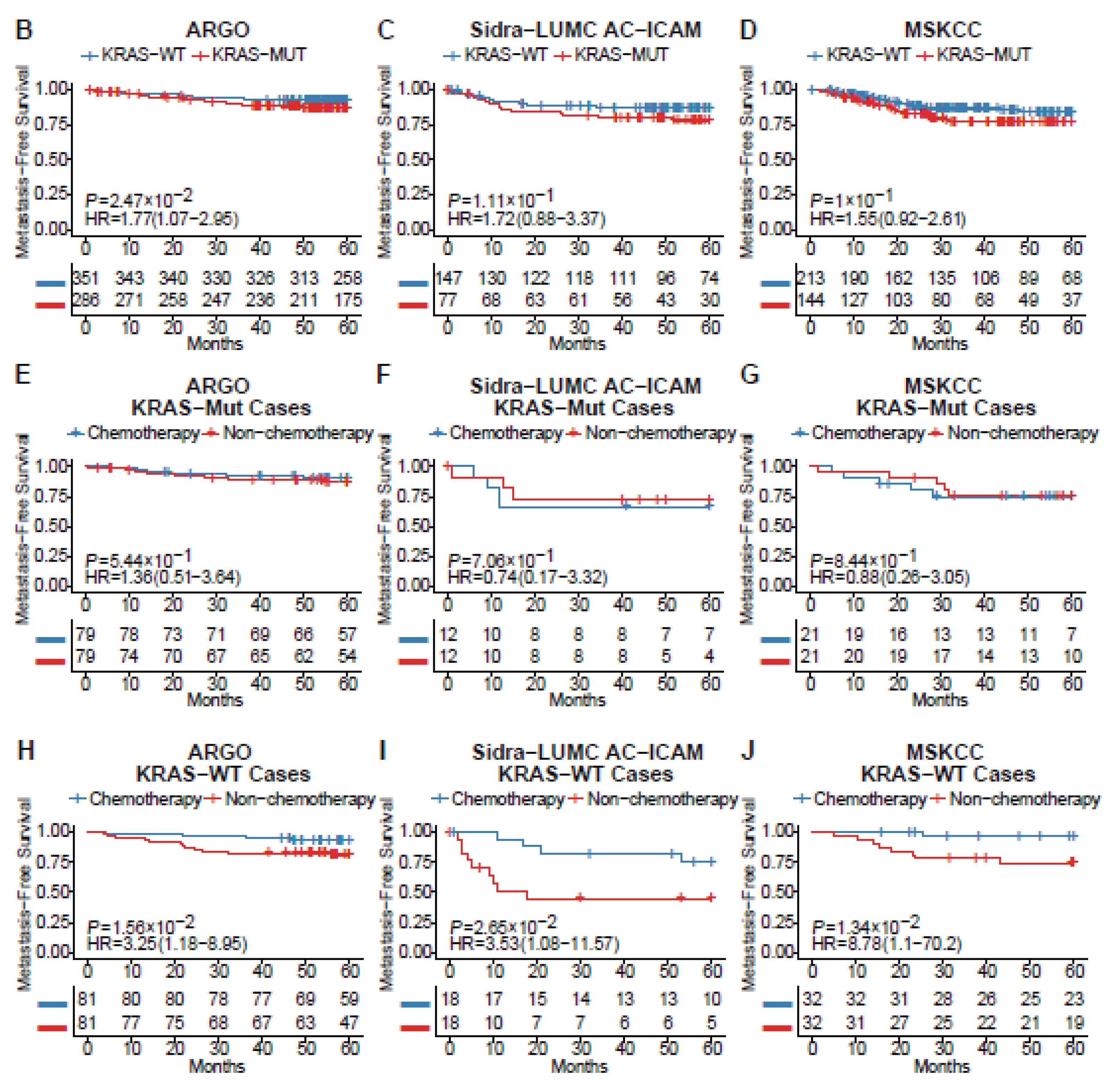
Figure 2.
Machine-learning method stratifies KRASmut CRC according to the risk of metachronous metastasis. MFS analysis comparing KMM high and low risk patients in (A) training cohort (ICGC-ARGO) and (B-C) validation cohorts (Sidra-LUMC AC-ICAM cohort and MSKCC cohort). (D) Genes of EMT and TGF-beta pathways differentially expressed between high and low risk groups in ICGC-ARGO KRASmut CRC patients. (E) Hallmark pathway analysis enriched by upregulated genes in ICGC-ARGO high risk group showed that the EMT pathway was significantly enriched. (F) KEGG pathway analysis enriched by upregulated genes in ICGC-ARGO high risk group showed that the TGF-beta pathway was significantly enriched. KRASmut, KRAS mutant; CRC, colorectal cancer; MFS, metachronous-metastasis free survival; KMM, KRAS metachronous-metastasis score; EMT, epithelial-mesenchymal transition; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Figure 2.
Machine-learning method stratifies KRASmut CRC according to the risk of metachronous metastasis. MFS analysis comparing KMM high and low risk patients in (A) training cohort (ICGC-ARGO) and (B-C) validation cohorts (Sidra-LUMC AC-ICAM cohort and MSKCC cohort). (D) Genes of EMT and TGF-beta pathways differentially expressed between high and low risk groups in ICGC-ARGO KRASmut CRC patients. (E) Hallmark pathway analysis enriched by upregulated genes in ICGC-ARGO high risk group showed that the EMT pathway was significantly enriched. (F) KEGG pathway analysis enriched by upregulated genes in ICGC-ARGO high risk group showed that the TGF-beta pathway was significantly enriched. KRASmut, KRAS mutant; CRC, colorectal cancer; MFS, metachronous-metastasis free survival; KMM, KRAS metachronous-metastasis score; EMT, epithelial-mesenchymal transition; KEGG, Kyoto Encyclopedia of Genes and Genomes.
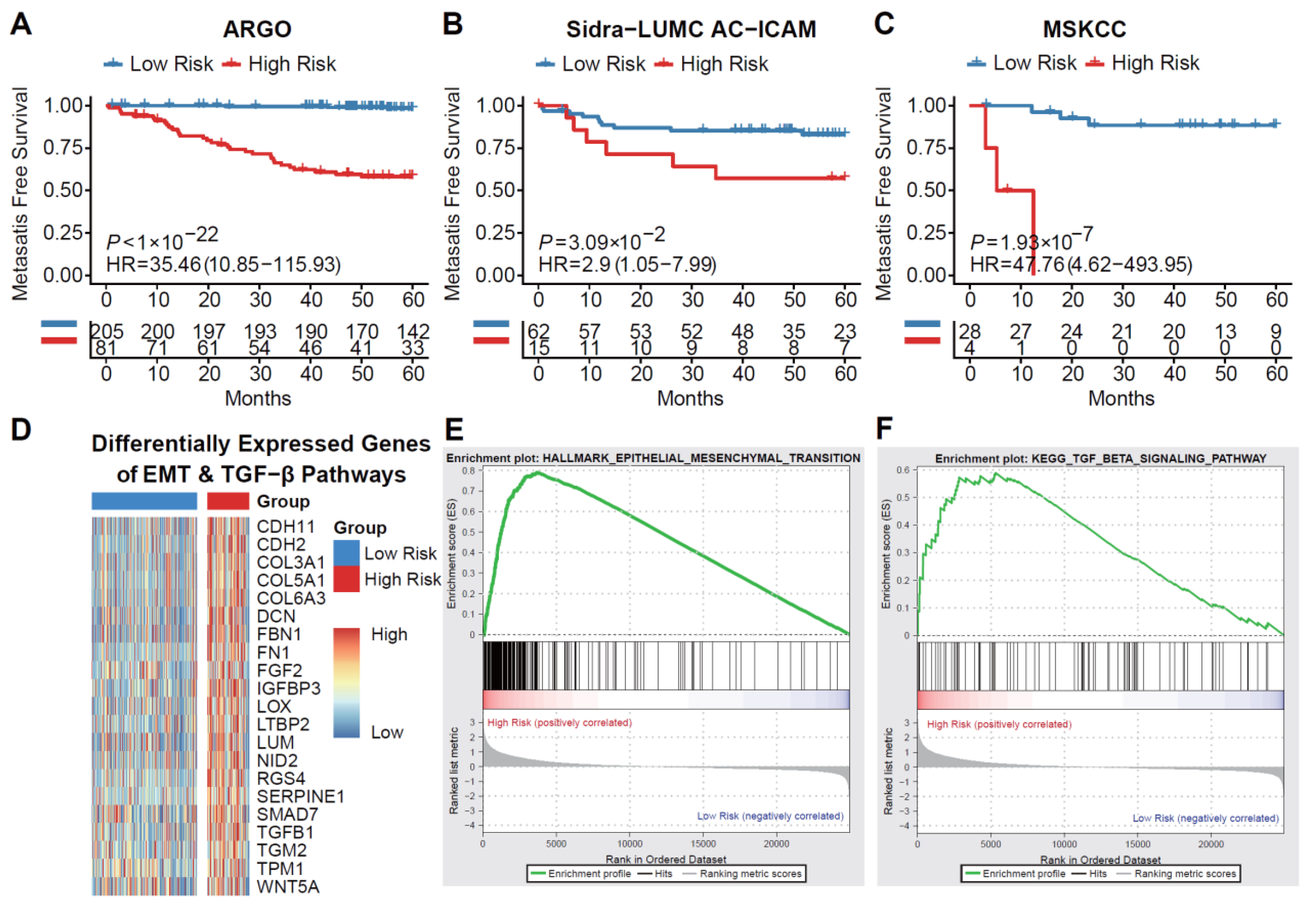
Figure 3.
KRAS inhibitors suppress the ability of migration and invasion of CRC. (A ) KRAS metachronous metastasis (KMM) score of Cancer Cell Line Encyclopedia (CCLE) colorectal cancer cell lines. (B-E) The effect of BI-2865 on the migration and invasion phenotype of SW480 and HCT116 was detected by wound healing assay and transwell experiment. Scale bar: 200μm. (F-G) Migratory and invasion ability was compared between SW837-P and SW837-R using wound healing assays and transwell experiments. Scale bar: 200μm. (H) Retention of the cystic structure with less spike-like formation by BI-2865 treatment in KRASG12D CRC organoid PDO-CC0117. Scale bar: 200μm. (I) Images showing the liver metastasis of SW480 cells treated with or without BI-2865 before trans-splenic injection into nude mice. (Arrows indicate metastatic nodules). Right: Plot showing the liver metastatic nodules per mouse in two groups. (J) H&E staining of liver tissue samples from the two groups. Scale bar: 2000μM. **P < 0.01; ***P < 0.001. KRASi, KRAS inhibitor (BI-2865, 1μM).
Figure 3.
KRAS inhibitors suppress the ability of migration and invasion of CRC. (A ) KRAS metachronous metastasis (KMM) score of Cancer Cell Line Encyclopedia (CCLE) colorectal cancer cell lines. (B-E) The effect of BI-2865 on the migration and invasion phenotype of SW480 and HCT116 was detected by wound healing assay and transwell experiment. Scale bar: 200μm. (F-G) Migratory and invasion ability was compared between SW837-P and SW837-R using wound healing assays and transwell experiments. Scale bar: 200μm. (H) Retention of the cystic structure with less spike-like formation by BI-2865 treatment in KRASG12D CRC organoid PDO-CC0117. Scale bar: 200μm. (I) Images showing the liver metastasis of SW480 cells treated with or without BI-2865 before trans-splenic injection into nude mice. (Arrows indicate metastatic nodules). Right: Plot showing the liver metastatic nodules per mouse in two groups. (J) H&E staining of liver tissue samples from the two groups. Scale bar: 2000μM. **P < 0.01; ***P < 0.001. KRASi, KRAS inhibitor (BI-2865, 1μM).
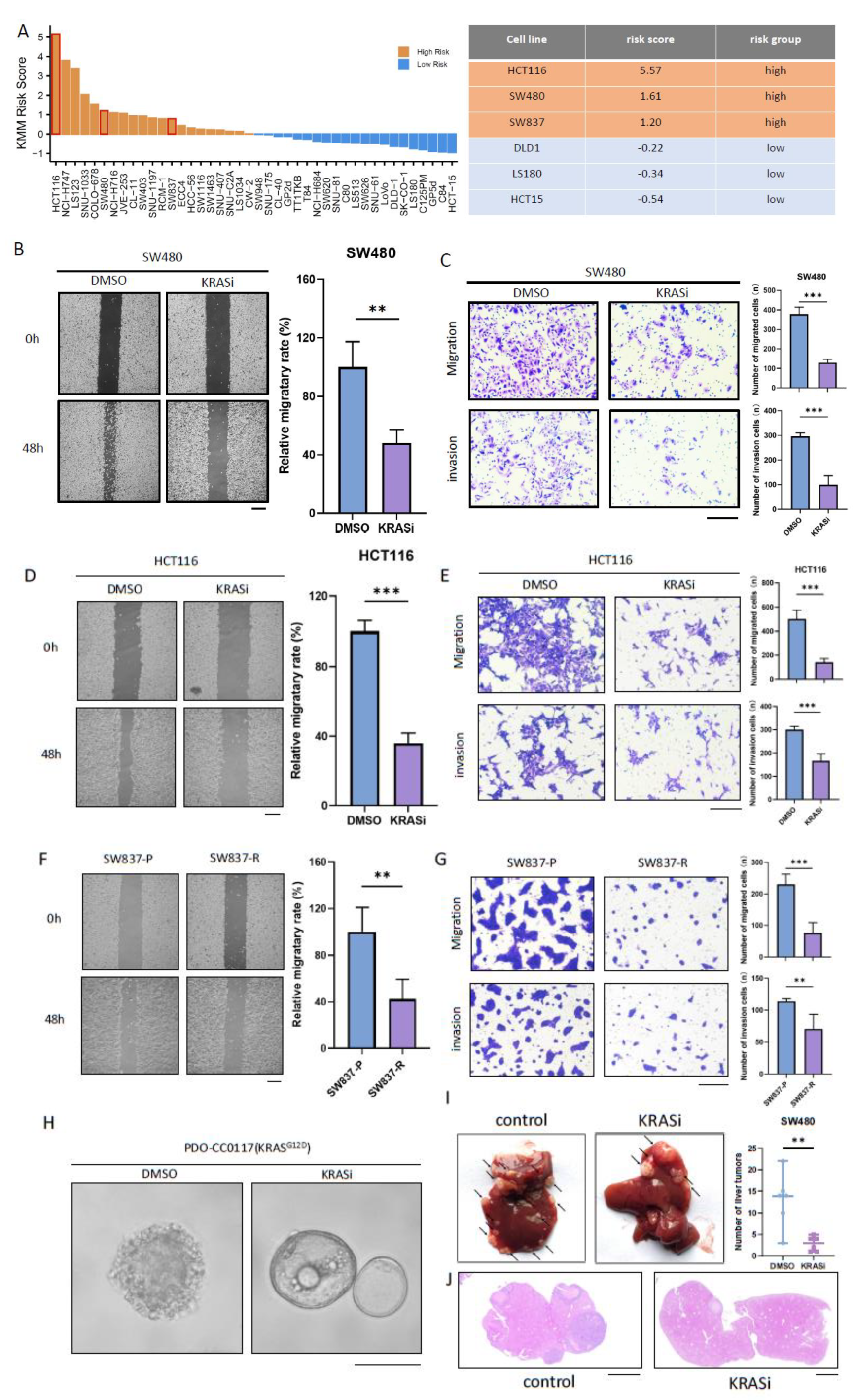
Figure 4.
KRAS inhibitor abrogates TGF-β and EMT pathway in CRC cell lines. (A-B) GSEA analysis of Hallmark in GSE228010 showing the epithelial-mesenchymal transition (EMT) and TGF-beta signaling pathways were enriched after BI-2865 treatment in SW480 and HCT116. NES, normalized enrichment score. (C-D) EMT and TGF-beta associated gene expression was validated by RT-qPCR in SW480 and HCT116. (E) Protein expression of EMT markers in SW480 and HCT116 upon treatment with DMSO or BI-2865. (F) The differentially expressed genes in SW837-P versus SW837-R are shown in the volcano plot. (G) The top five down enrichment pathways in SW837-R versus SW837-P. (H-I) Hallmark pathway analysis enriched by down-regulated genes in SW837-R (relative to SW837-P) showed that the EMT and TGF-beta pathways were significantly enriched. (J) Heatmap for significantly down-regulated EMT genes (P < 0.05) in SW837-R versus SW837-P. (K-L) Relative mRNA levels of EMT and TGF-beta pathways in SW837-P/R cells. (M) Western-blot of EMT markers in SW837-P/R cells. *P < 0.05 ; **P < 0.01; ***P < 0.001. KRASi, KRAS inhibitor (BI-2865, 1μM).
Figure 4.
KRAS inhibitor abrogates TGF-β and EMT pathway in CRC cell lines. (A-B) GSEA analysis of Hallmark in GSE228010 showing the epithelial-mesenchymal transition (EMT) and TGF-beta signaling pathways were enriched after BI-2865 treatment in SW480 and HCT116. NES, normalized enrichment score. (C-D) EMT and TGF-beta associated gene expression was validated by RT-qPCR in SW480 and HCT116. (E) Protein expression of EMT markers in SW480 and HCT116 upon treatment with DMSO or BI-2865. (F) The differentially expressed genes in SW837-P versus SW837-R are shown in the volcano plot. (G) The top five down enrichment pathways in SW837-R versus SW837-P. (H-I) Hallmark pathway analysis enriched by down-regulated genes in SW837-R (relative to SW837-P) showed that the EMT and TGF-beta pathways were significantly enriched. (J) Heatmap for significantly down-regulated EMT genes (P < 0.05) in SW837-R versus SW837-P. (K-L) Relative mRNA levels of EMT and TGF-beta pathways in SW837-P/R cells. (M) Western-blot of EMT markers in SW837-P/R cells. *P < 0.05 ; **P < 0.01; ***P < 0.001. KRASi, KRAS inhibitor (BI-2865, 1μM).
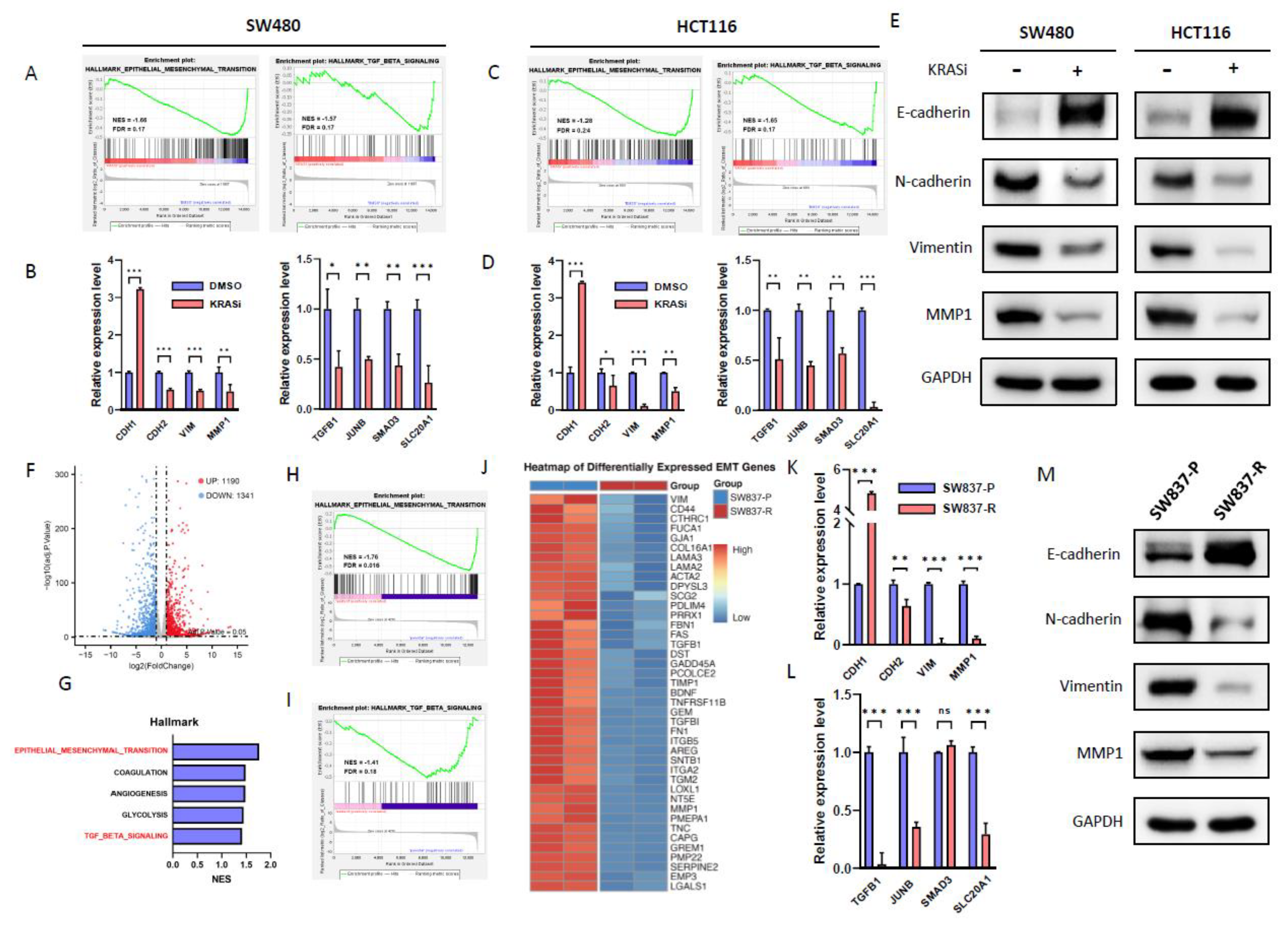
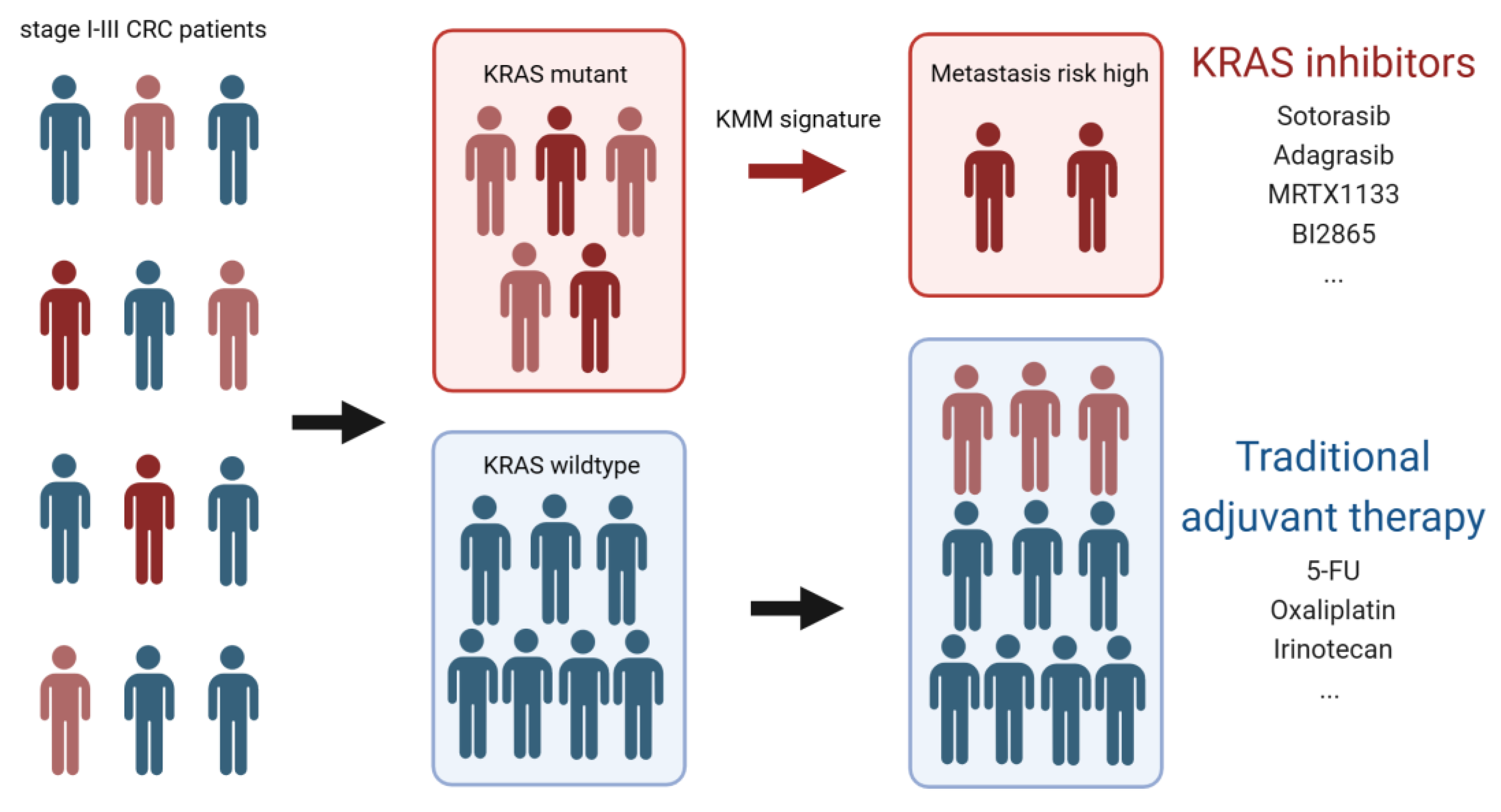
Figure 5.
Schematic of the potential use of KRAS inhibitors in preventing metachronous metastasis CRC. Colorectal cancer harboring KRAS mutation is resistant to traditional chemotherapy and is likely to develop metachronous metastasis after curative surgery. By using KMM signature, patients who are highly risky to suffer from distant recurrence can be identified and KRAS inhibitors can be used prophylactically.
Figure 5.
Schematic of the potential use of KRAS inhibitors in preventing metachronous metastasis CRC. Colorectal cancer harboring KRAS mutation is resistant to traditional chemotherapy and is likely to develop metachronous metastasis after curative surgery. By using KMM signature, patients who are highly risky to suffer from distant recurrence can be identified and KRAS inhibitors can be used prophylactically.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



