
Submitted:
18 March 2024
Posted:
22 March 2024
You are already at the latest version
Abstract
Background: Amyloid beta peptides (Aβ) have been identified as the main pathogenic agents in Alzheimer’s disease (AD). Soluble Aβ oligomers, rather than monomer or insoluble amyloid fibrils, show membrane-binding capacity to red blood cells (RBCs) and trigger several morphological and functional alterations in RBCs that can result in impaired oxygen transport and delivery. Since bioactive lipids have been recently proposed as potent protective agents against Aβ toxicity, we investigated the role of sphingosine-1-phosphate (S1P) in signaling pathways involved in the mechanism underlying ATP release in Aβ-treated RBCs.
Methods: In RBCs following different treatments, ATP, 2,3 DPG, cAMP levels, and caspase 3 activity were determined by spectrophotometric and immunoassay.
Results: S1P rescued the inhibition of ATP release from RBCs triggered by Aβ through a mechanism involving caspase-3 and restoring 2,3 DPG and cAMP levels within the cell.
Conclusions: These findings reveal the molecular basis of S1P protection against Aβ in RBCs and suggest new therapeutic avenues in AD.
Keywords:
Sphingosine-1-phosphate
; Band 3
; erythrocyte
; caspase-3
; beta-amyloid
; Alzheimer's disease
1. Introduction
Sphingosine-1-phosphate (S1P) is a potent lipid mediator that performs several roles [1]. Sphingosine kinase 1 (Sphk1) or sphingosine kinase 2 produce S1P from its precursor sphingosine; meanwhile, S1P phosphatase and S1P lyase (Sgpl) revert into sphingosine and 2-hexadecenal and phospho-ethanolamine, respectively [2]. Red blood cells (RBCs) uptake S1P [3,4,5], while S1P may also be produced within the cells through Sphk1 [2]. Since RBCs contain Sphk1 but no S1P-degrading enzymes [6], S1P is abundantly stored in RBCs [7], as well as in platelets [8] and the endothelium [9,10]. S1P performs several functions and regulates many cellular processes, including cell growth, proliferation, migration, and apoptosis [11,12,13,14]. In recent papers, S1P has been discussed concerning the RBC adaptation mechanism to SARS-COVID-19 infection [15]. In RBCs, S1P promotes deoxygenated haemoglobin (deoxyHb), which anchors to band 3, the most abundant membrane protein in RBCs, thereby increasing glycolysis flux, 2,3-diphosphoglycerate (2,3 DPG) levels [16], and ATP release [17]. RBCs release ATP under reduced oxygen tensions and following deformation, to modulate vasodilation [18]. The pathway underlying ATP release from RBCs involves several proteins, such as G proteins, adenylyl cyclase (AC) and cyclic AMP-dependent protein kinase A [18], which are a cystic fibrosis transmembrane conductance regulator and protein pannexin, respectively [19,20]. Alzheimer's disease (AD) is a pathology characterised by senile plaques in several regions of the central nervous system (CNS), which are frequently correlated with areas of neurodegeneration [21]. Amyloid beta (Aβ) peptides, which are major protein components in the plaques, consist of 39–43 amino acid peptides that originate from a more significant transmembrane protein, amyloid precursor protein (APP). Aβ neurotoxicity has been associated with peptide self-aggregation, which leads to the formation of amyloid-like fibrils [22] and eventually to neuronal cell death through apoptosis. However, recent studies have shown that soluble forms of Aβ exhibit stronger neurotoxicity, and in the monomeric form, Aβ may be responsible for the neurodegeneration observed in AD [23,24]. Aβ has been found in blood at nanomolar concentrations and is abundantly produced by platelets [25]. RBCs encounter Aβ peptides at the luminal surface level of brain capillaries [26] and seem to only interact with monomeric Aβ peptides [27]. Aβ alters RBC metabolism and induces RBC death [28,29,30,31,32,33,34] through a signaling pathway involving protein kinase C [35,36]. Evidence from epidemiological data indicates a close association between vascular and AD pathology [37]. However, experimental studies suggest that Aβ can reduce cerebral blood flow (CBF), inducing neurovascular dysfunction and increasing the brain's susceptibility to ischemia [38]. Therefore, we are interested in determining whether RBCs contribute to the AD pathogenesis. Previous studies have reported decreased S1P levels in AD tissues and plasma [39,40]. S1P protects neuronal cells from apoptosis [41], notably in response to Aβ [42]. Moreover, a recent paper demonstrated that S1P abrogates neuronal Ca2+ dyshomeostasis induced by toxic Aβ cells [43].
Based on the importance of vascular dysfunction in AD pathology, in this study, we investigated the protective role of S1P against Aβ peptides on ATP release in RBCs.
2. Results
2.1. Protective Role of Sphingosine-1-Phosphate on ATP Release
It is known that RBCs can readily uptake exogenous S1P, up to 5 μmol, l−1 in an in vitro system [3]. Firstly, we assessed whether S1P affected the mechanism responsible for ATP release from RBCs. Here, RBCs were treated at high and low oxygen tensions with S1P at concentrations of 0.1 and 0.5 μM for 24 h. The ATP values were significantly higher for control cells with S1P at 0.5 μM compared to 0.1 μM (Figure 1A). When 0.1 μM Aβ was added to RBCs at low and high oxygen tensions for 24 hours, it inhibited the release of ATP from RBCs at the low oxygen tension (Figure 1B), as previously reported [31]. Next, to verify the protective role of S1P against Aβ, S1P was pre-incubated with RBCs for 30 min before Aβ exposure at a low oxygen tension. As shown in Figure 1B, ATP values were fully restored in the presence of 0.5 μM S1P, with a slight protective effect at 0.1 μM. It is known that caspase-3 is involved in the mechanism responsible for the inhibition of ATP release from RBCs by Aβ [31]. Next, we examined whether the protective effect of S1P against Aβ was mediated by caspase 3. Pre-treatment of RBCs exposed to Aβ with a caspase-3 inhibitor, i.e., Z-DEVD-FMK, was able to rescue ATP levels back to control levels (Figure 1B), evidencing the involvement of caspase-3 in the protective mechanism elicited by S1P. In RBCs, it has been shown that ATP release is linked to a pathway including Gi and adenylyl cyclase (AC) [18]. Mastoparan 7, an activator of Gi, was used to clarify the involvement of the Gi-related pathway in the protective role of S1P against Aβ. As reported in Figure 2, in the experiments with mastoparan 7, ATP release values remained similar between RBCs in the presence and absence of S1P, demonstrating that Gi proteins do not mediate S1P action.
2.2. Effect of Sphingosine-1-Phosphate on the Accumulation of cAMP
Then, we investigated whether cAMP was involved in the protective effect of S1P against Aβ in deoxygenated RBCs. Here, in deoxygenated RBCs treated for 24 h with S1P alone at 0.1 and 0.5 μM, cAMP values were significantly higher for control cells treated with S1P at 0.5 μM. As previously reported [18], cAMP levels in RBCs are significantly higher when the cells are deoxygenated compared to oxygenated conditions (Figure 3A). Next, further, to verify the protective role of S1P against Aβ, S1P was pre-incubated with RBCs for 30 min before Aβ exposure at low oxygen tension. As shown in Figure 3B, cAMP values were fully restored in the presence of 0.5 μM S1P, with no effects observed at 0.1 μM. Pre-treatment of Aβ-exposed RBCs with a caspase-3 inhibitor, i.e., Z-DEVD-FMK, rescued cAMP levels to those shown by control cells, thereby demonstrating the involvement of caspase-3 in the protective mechanisms elicited by S1P.
2.3. Effect of Sphingosine-1-Phosphate on 2,3 DPG Levels
When RBCs were treated with Aβ for 24 h, the 2,3 DPG levels observed in the deoxygenated RBCs were significantly reduced compared to the control cells (Figure 4). S1P alone at 0.5 μM increased 2,3 DPG levels compared to the control, demonstrating that S1P could increase metabolic fluxes through glycolysis to generate 2,3-BPG, as previously reported [16]. In RBCs pre-incubated for 30 min with S1P at 0.5 μM before Aβ, the 2,3 DPG levels were significantly higher than those shown by the Aβ-treated cells. Pre-treatment of Aβ-A-exposed RBCs with Z-DEVD-FMK fully restored the 2,3 DPG levels, thereby demonstrating that, among other mechanisms, caspase is involved in the protective mechanism elicited by S1P.
2.4. Effect of Sphingosine-1-Phosphate on Caspase-3 Activity
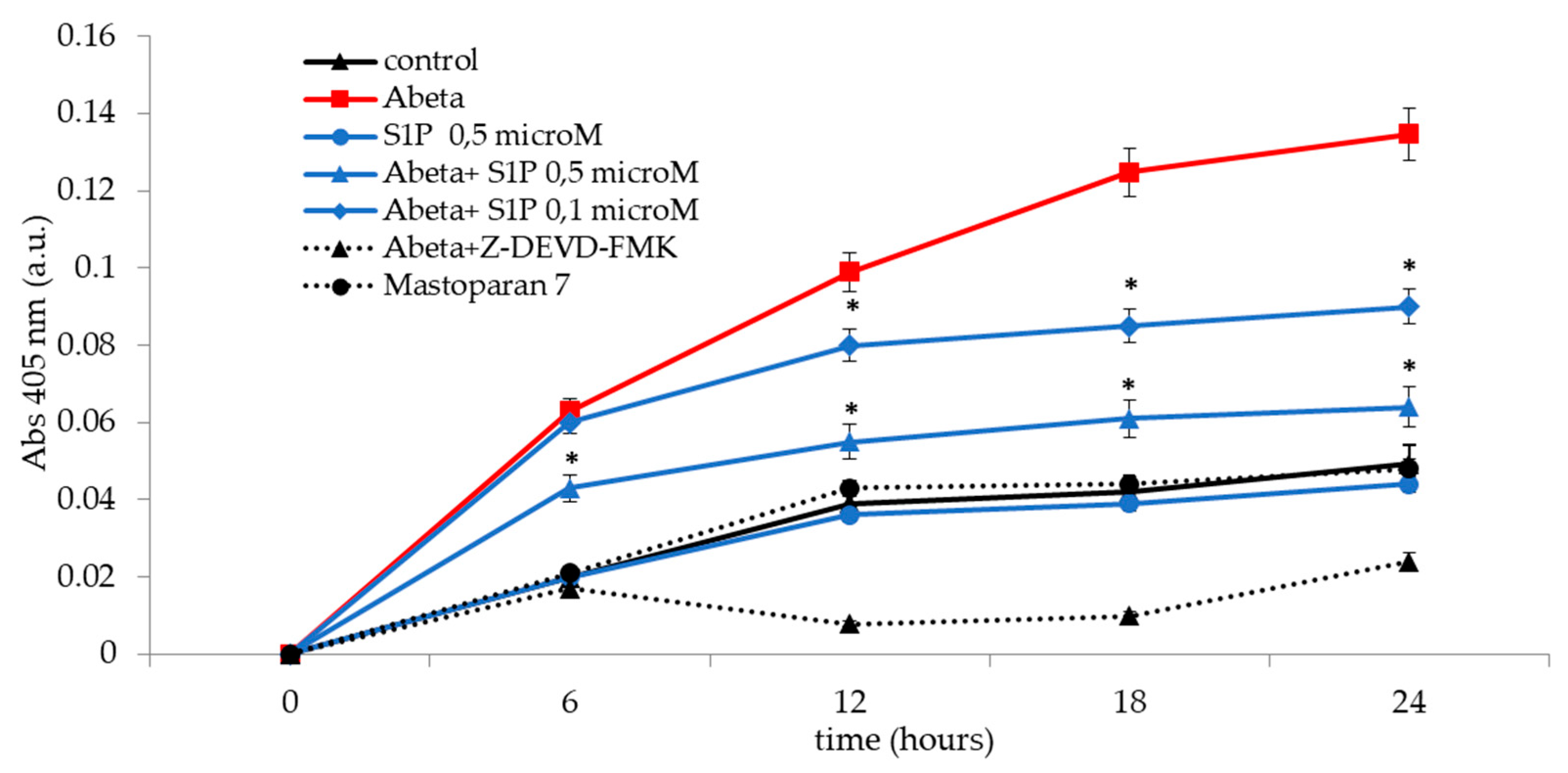
Band 3 degradation by caspase-3 has been suggested to induce cdb3/deoxyHb binding site disruption in RBCs [29,30]. Cdb3/deoxyHb binding activated the pathway responsible for ATP release from deoxygenated RBCs [18]. Aβ inhibits ATP release from RBCs through a pathway involving the activation of caspase-3 [31]. As shown in Figure 5, Aβ treatment dramatically increased caspase-3 activity in a time-dependent manner. Aβ-mediated caspase-3 activation was significantly rescued by pre-incubation with S1P at 0.5 μM for 30 min, with only a minor protective effect observed with 0.1 μM. Pre-incubation of Aβ-treated RBCs with Z-DEVD-FMK inhibited the Aβ-mediated caspase-3 activation. However, this observation excluded the presence of unspecified proteolytic activities. Moreover, S1P alone at 0.5 μM did not affect caspase-3 activity. Then, we examined the effects of mastoparan 7, an activator of Gi, to determine whether Gi mediated the observed protective effect of S1P against the activation of caspase-3 by Aβ. As reported in Figure 5, caspase-3 was unaffected in the presence of mastoparan 7, demonstrating that Gi proteins do not mediate S1P action.
2.5. Hemolysis Degree
The spontaneous lysis of RBCs is another potential source of extracellular ATP. Thus, the RBC suspensions were analyzed to evaluate hemoglobin concentrations in the supernatants and determine hemolysis after experiments [40]. In all experiments, hemolysis was less than ~3%.
3. Discussion
RBCs release ATP in response to low oxygen tension [18]. The starting event in the release of ATP from RBCs involves an interaction between deoxyHb and the cytoplasmic domain of the anion exchange protein band 3, i.e., the cdb3–deoxyHb/band 3 complex induces stress in the membrane components, triggering the downstream pathway responsible for ATP release. It has been shown that ATP release and cAMP accumulation are strongly reduced in RBCs in the presence of Aβ and associated with caspase-3 activation [31], thus decreasing tissue oxygenation, particularly in cerebral microvascular circulation, and aggravating AD pathology. Here, we report that Aβ-mediated inhibition of ATP release from deoxygenated RBCs was abolished when cells were pre-incubated with sphingosine-1-phosphate (S1P) before treatment with Aβ. The signalling pathway underlying ATP release from RBCs includes the heterotrimeric G proteins Gs and Gi, adenylyl cyclase (AC), and cyclic AMP-dependent protein kinase A [18]. In the presence of S1P, comparable amounts of intracellular cAMP were measured following incubation with mastoparan 7 (i.e., stimulatory agent of Gi), both in the presence and absence of Aβ peptides; this finding suggests that the activity of the Gi subunit in heterotrimeric G proteins could not explain the protective effect induced by S1P in Aβ-treated RBCs. The possible role of S1P In AD is controversial, with some studies suggesting a causative role in AD while others propose a protective role [44]. We observed that the pre-treatment with a caspase-3 inhibitor, i.e., Z-DEVD-FMK, before Aβ, could rescue ATP and cAMP levels to those observed in control cells. We suggest that S1P inhibited the Aβ-mediated activation of caspase-3 activity, protecting the cytoplasmic domain of the anion exchange protein band 3, i.e., cdb3, through caspase-3 cleavage.
Since the release of ATP from RBCs occurs in response to low oxygen tension and consists of an interaction between deoxyHb and cdb3 [18], our findings indicate that the mechanism underlying the protective role of S1P on the inhibition of ATP release, triggered by Aβ, partially involves the S1P-mediated abrogation of caspase-3 activation. These findings align with a previous paper, which showed that the S1P agonist SEW2871 decreased Aβ-induced caspase-3 activation, neuronal death, and cognitive damage in rats with AD [45]. Furthermore, we showed that S1P increased 2,3-DPG levels within the cell. In a previous study [16], it has been suggested that S1P induces 2,3 DPG production by binding directly to deoxy-Hb, thereby stabilizing Hb in the deoxygenated state. DeoxyHb binds to cdb3, triggering the release of some glycolytic enzymes to the cytosol, thereby increasing glycolysis flux to produce more 2,3-DPG. Thus, the increase in 2,3 DPG can bind more oxyHb molecules, meaning S1P promotes deoxyHb anchoring to cdb3 and triggers the mechanism responsible for ATP release from RBCs in response to low oxygen tension.
4. Materials and Methods
4.1. Chemicals
Aβ peptide (1-42) purity > 98%. was purchased by Peptide Speciality Laboratories GmbH (Heidelberg, Germany). Peptides were solubilized in 100% 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; Sigma, St. Louis, MO, USA). The HFIP was then removed by vacuum evaporation, and the remaining disaggregated peptide was dissolved in dimethylsulphoxide (DMSO). Sphingosine-1-phosphate (S1P) and other chemicals were purchased from Sigma Aldrich (St. Louis, MO, USA).
4.2. Preparation of Red Blood Cells and Incubation Conditions
Blood samples were collected in citrate and washed three times with an iso-osmotic NaCl solution. Low-speed centrifugation (800x g,5 min) was performed to separate plasma, avoiding mechanical stress that could determine RBC morphological alterations. Ficoll was used to isolate mature RBC for a density gradient centrifugation. RBCs were incubated at 37 °C for 24h with or without 0.1 μM Aβ peptide, pre-incubated in the presence and absence of S1P at 0.1 and 0.5 μM. In experiments performed under low oxygen conditions, the measured percentage of deoxyHb was 60% ± 0.32%. RBCs were sedimented by centrifugation at 500g for 10 min to exclude the possibility that RBC lysis affects our determinations. Oxygenated hemoglobin in the supernatant was determined by light absorption at 405 nm (Cary 3E, Varian, Palo Alto, CA) [46]. Although this method does not measure methemoglobin and oxidized forms of hemoglobin (about 1-3% of the total hemoglobin), it is commonly used when measuring experimentally induced RBC lysis [47].
4.3. ATP Assay
The luciferin-luciferase technique was used to measure ATP, as reported [48], which uses the ATP concentration dependence of light generated by the reaction of ATP with firefly tail extract.
4.4. Measurement of cAMP
After RBCs exposure to different experimental conditions, cAMP's concentration was then determined, as previously described [49], with a cAMP Biotrak enzyme immunoassay system (Amersham Biosciences).
4.5. Determination of 2,3 DPG
2,3-DPG in 20 μl RBC pellet was isolated with 100 μl, 0.6 M cold perchloric acid on ice, vortexed, and centrifuged. A volume of 80 μl supernatant was transferred to a new tube, neutralized, and centrifuged. An aliquot of supernatant was used to measure 2,3-DPG levels using a commercially available kit (Sigma Aldrich, St. Louise, USA).
4.6. Caspase-3 Activity Determination
After RBCs exposure to different experimental conditions, caspase activity has been carried out as previously described [50].
4.7. Statistical Analysis
All data are expressed as means ± SD. Statistical analyses (Student’s-test and ANOVA) were performed with SYSTAT 10.2 software (Statcom, Inc., Richmond, CA, USA). The level of significance was set at 0.05.
5. Conclusions
We provide evidence that S1P rescued the inhibition of ATP release from RBCs triggered by Aβ. Among several signalling pathways mediated by S1P, our results suggest that the protective path is mediated by caspase-3 and deoxyHb. The protective role of S1P could be relevant to supporting energy demands in tissues, particularly in cerebral microvascular regions after ischemia or where a deposition may cause the cerebral vessel lumen to narrow. While this is a promising finding, this study is limited because it did not use AD models; therefore, future studies that use blood cells from AD patients are warranted. Lately, fingolimod (FTY720), a synthetic analog of S1P, was observed to revert memory deficits in a rat model of AD, suggesting a crucial role of S1P in neuroprotection against Aβ toxicity [51]. Taken together, data from this study indicate that restoring S1P plasma levels may be an attractive therapy to treat or prevent AD.
Author Contributions
FM was responsible for designing, collecting data, analyzing, and writing the manuscript; PD and GEL were responsible for analyzing data; ET was responsible for analyzing data and writing the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Human blood was collected, treated, and used according to the ethical and safety guidelines and regulations approved by the “Institutional Review Board” of the University of Messina. (Prot 71-23 del 05.04.2023).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All data points generated or analyzed during this study are included in this article, and no further underlying data is necessary to reproduce the results.
Acknowledgments
not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Yatomi, Y. Sphingosine 1-phosphate in vascular biology: possible therapeutic strategies to control vascular diseases. Curr Pharm Des 2006, 12, 575–587. [Google Scholar] [CrossRef]
- Le Stunff, H.; Milstien, S.; Spiegel, S. Generation and metabolism of bioactive sphingosine-1-phosphate. J Cell Biochem 2004, 92, 882–899. [Google Scholar] [CrossRef]
- Hanel, P.; Andreani, P.; Graler M., H. Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J 2007, 21, 1202–1209. [Google Scholar] [CrossRef]
- Bode, C.; Sensken, S.C.; Peest, U. Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J Cell Biochem 2010, 109, 1232–1243. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Vu, T.M.; Tukijan, F. Erythrocytes efficiently utilize exogenous sphingosines for S1P synthesis and export via Mfsd2b. J Biol Chem 2022, 96, 100201. [Google Scholar] [CrossRef]
- Ito, K.; Anada, Y.; Tani, M.; Ikeda, M.; Sano, T.; Kihara, A. Lack of sphingosine 1-phosphate-degrading enzymes in erythrocytes. Biochem Biophys Res Commun 2007, 357, 212–217. [Google Scholar] [CrossRef]
- Pappu, R.; Schwab, SR.; Cornelissen, I.; Pereira, JP.; Regard, JB.; Xu, Y.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Yatomi, Y.; Ruan, F.; Hakomori, S.; Igarashi, Y. Sphingosine-1-phosphate: a platelet-activating sphingolipid released from agonist-stimulated human platelets. Blood 1995, 86, 193–202. [Google Scholar] [CrossRef]
- Venkataraman, K.; Le, YM.; Michaud, J.; Thangada, S.; Ai Y Bonkovsky, HL.; et al. Vascular endothelium is a contributor to plasma sphingosine 1-phosphate. Circ Res 2008, 102, 669–676. [Google Scholar] [CrossRef]
- Fukuhara, S.; Simmons, S.; Kawamura, S.; Inoue, A.; Orba, Y.; Tokudome, T. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Investig, 2012; 122, 1416–1426. [Google Scholar]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; et al. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as a second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef]
- Kupperman, E.; An, S.; Osborne, N.; Waldron, S.; Stainier, D.Y. A sphingosine-1-phosphate receptor regulates cell migration during vertebrate heart development. Nature 2000, 406, 192–195. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol 2003, 4, 397–407. [Google Scholar] [CrossRef]
- Winkler, M.S.; Claus, R.A.; Schilder, M.; Pöhlmann, S.; Coldewey, S.M.; Grundmann, J.; Fricke, T.; Moerer, O.; Meissner, K.; Bauer, M.; Hofmann-Winkler, H.; Gräler, MH. Erythrocytes increase endogenous sphingosine 1-phosphate levels as an adaptive response to SARS-CoV-2 infection. Clin Sci (Lond) 2021, 135, 2781–2791. [Google Scholar] [CrossRef]
- Sun, K.; Zhang, Y.; D'Alessandro, A.; Nemkov, T.; Song, A.; Wu, H.; Liu, H.; Adebiyi, M.; Huang, A.; Wen, Y.E.; Bogdanov, M.V.; Vila, A.; O'Brien, J.; Kellems, R.E.; Dowhan, W.; Subudhi, A.W.; Jameson-Van Houten, S.; Julian, C.G.; Lovering, A.T.; Safo, M.; Hansen, K.C.; Roach, R.C.; Xia, Y. Sphingosine-1-phosphate promotes erythrocyte glycolysis and oxygen release for adaptation to high-altitude hypoxia. Nat Commun 2016, 7, 12086. [Google Scholar] [CrossRef]
- Misiti, F. Sphingosine Increases ATP Release from Red Blood Cells. The Open Biochem J 2022, 16, 1–5. [Google Scholar] [CrossRef]
- Ellsworth, M.L.; Ellis, C.G.; Sprague, R.S. Role of erythrocyte-released ATP in the regulation of microvascular oxygen supply in skeletal muscle. Acta Physiol (Oxf) 2016, 216, 265–276. [Google Scholar] [CrossRef]
- Liang, G.; Stephenson, A.H.; Lonigro, A.J.; Sprague, R.S. Erythrocytes of humans with cystic fibrosis fail to stimulate nitric oxide synthesis in isolated rabbit lungs. Am J Physiol 2004, 288, H1580–H1585. [Google Scholar] [CrossRef]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett 2004, 572, 65–68. [Google Scholar] [CrossRef]
- Selkoe, D. A central role for amyloid. Journal of neuropathology and experimental neurology. J. Alzheimer’s Disease 1994, 53, 438–447. [Google Scholar]
- Pike C., J.; Walencewicz-Wasserman A., J.; Kosmosi, J.; Cribbs, D.H.; Glabe C., G.; Cotman, C.W. Structure–activity analyses of beta-amyloid peptides: Contributions of the b35-35 region to aggregation and neurotoxicity. Journal of Neurochemistry 1995, 64, 253–265. [Google Scholar] [CrossRef]
- Pillot, T.; Drouet, B.; Queille, S.; Labeur, C.; Vandekerchkhove, J.; Rosseneu, M.; et al. The nonfibrillar amyloid betapeptide induces apoptotic neuronal cell death: Involvement of its C-terminal fusogenic domain. Journal of Neurochemistry 1999, 73, 1626–1634. [Google Scholar] [CrossRef]
- Kim H., J.; Chae S., C.; Lee, D. K:; Chromy B.; Lee S. C.; Park Y. C. et al. Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. The FASEB Journal 2003, 17, 118–120. [Google Scholar] [CrossRef]
- Kiko, T.; Nakagava, K.; Satoh, A.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. Amyloid b levels in human red blood cells. PLoS One 2012, e49620. [Google Scholar] [CrossRef]
- Grammas, P.; Yamada, M.; Zlokovic, B. The cerebromicrovasculature: a key player in the pathogenesis of Alzheimer's disease. J. Alzheimers Dis 2002, 4, 217–223. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Kokjohn, T.A.; Beach, T.G.; Sue, L.I.; Brune, D.; Lopez, J.C. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer's disease brains. J. Biol. Chem 2001, 276, 12991–12998. [Google Scholar] [CrossRef]
- Carelli-Alinovi, C.; Dinarelli, S.; Sampaolese, B.; Misiti, F.; Girasole, M. Morphological changes induced in erythrocyte by amyloid beta peptide and glucose depletion: A combined atomic force microscopy and biochemical study. Biochim Biophys Acta Biomembr 2019, 1861, 236–244. [Google Scholar] [CrossRef]
- Carelli-Alinovi, C.; Pirolli, D.; Giardina, B.; Misiti, F. Protein kinase C mediates caspase 3 activation: a role for erythrocyte morphology changes. Clin. Hemorheol. Microcirc 2015, 59, 345–354. [Google Scholar] [CrossRef]
- Misiti, F.; Carelli-Alinovi, C.; Sampaolese, B.; Giardina, B. b-amyloid decreases detectable endothelial nitric oxide synthase in human erythrocytes: a role for membrane acetylcholinesterase. Cell Biochem. Funct 2012, 30, 474–479. [Google Scholar] [CrossRef]
- Misiti, F.; Orsini, F.; Clementi, M.E.; Masala, D.; Tellone, E.; Galtieri, A.; Giardina, B. Amyloid peptide inhibits ATP release from human erythrocytes. Biochem. Cell Biol 2008, 86, 501–508. [Google Scholar] [CrossRef]
- Kosenko, E.A.; Solomadin, I.N.; Tikhonova, L.A.; Ready, V.P.; Aliev, G.; Kaminsky, Y.G. Pathogenesis of Alzheimer's disease: role of oxidative stress,amyloid-b peptides, systemic ammonia, and RBC energy metabolism. CNS Neurol. Disord. Drug Targets 2014, 13, 112–119. [Google Scholar] [CrossRef]
- Jayakumar, R. : Kusiak J.W.; Chrest F.J.;Femehin A.A.; Murali J. West R.P. Red cell perturbations by amyloid beta-protein. Biochim. Biophys. Acta 2003, 1622, 20–28. [Google Scholar] [CrossRef]
- Clementi, M.E.; Giardina, B.; Colucci, D.; Galtieri, A.; Misiti, F. Amyloid-beta peptide affects the oxygen dependence of RBC metabolism: a role for caspase 3. Int. J. Biochem. Cell Biol 2007, 39, 727–735. [Google Scholar] [CrossRef]
- Mandal, D.; Baudin-Creuza, V.; Bhattacharyya, A.; Pathak, S.; Delaunay, J.; Kundu, M.; Basu, J. Caspase 3-mediated proteolysis of the N-terminal cytoplasmic domain of the human erythroid anion exchanger 1 (band 3). J Biol Chem 2003, 278, 52551–52558. [Google Scholar] [CrossRef]
- Carelli-Alinovi, C.; Giardina, B.; Misiti, F. Amyloid beta peptide (1-42)-mediated antioxidant imbalance is associated with activation of protein kinase C in red blood cells. Cell Biochem. Funct 2015, 33, 196–201. [Google Scholar] [CrossRef]
- Gottesman, RF.; Albert, MS.; Alonso, A.; Coker, LH.; Coresh, J.; Davis, SM.; Deal, JA.; McKhann, GM.; Mosley, TH.; Sharrett, AR.; Schneider, ALC.; Windham, BG.; Wruck, LM.; Knopman, DS. Associations between midlife vascular risk factors and 25-year incident dementia in the Atherosclerosis Risk in Communities (ARIC) cohort. JAMA Neurol 2017, 74, 1246–1254. [Google Scholar] [CrossRef]
- Iadecola, C.; Gottesman, RF. Cerebrovascular Alterations in Alzheimer Disease. Circ Res 2018, 123, 406–408. [Google Scholar] [CrossRef]
- He, X.; Huang Y Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging 2010, 2, 398–408. [Google Scholar] [CrossRef]
- Oizumi, H.; Sugimura, Y.; Totsune, T.; Kawasaki, I.; Ohshiro, S.; Baba, T.; Kimpara, T.; Sakuma, H.; Hasegawa, T.; Kawahata, I.; Fukunaga, K.; Takeda, A. Plasma sphingolipid abnormalities in neurodegenerative diseases. PLoS One 2022, 16, e0279315. [Google Scholar] [CrossRef]
- Edsall, L.C.; Cuvillier, O.; Twitty, S.; Spiegel, S.; Milstien, S. Sphingosine kinase expression regulates apoptosis and caspase activation in PC12 cells. J Neurochem 2001, 2, 1573–1584. [Google Scholar] [CrossRef]
- Gomez-Brouchet, A.; Pchejetski, D.; Let, B. Critical role for sphingosine kinase-1 in regulating survival of neuroblastoma cells exposed to amyloid-beta peptide. Mol Pharmacol 2007, 2, 341–349. [Google Scholar] [CrossRef]
- Bigi, A.; Cascella, R.; Fani, G.; Bernacchioni, C.; Cencetti, F.; Bruni, P.; Chiti, F.; Donati, C.; Cecchi, C. Sphingosine 1-phosphate attenuates neuronal dysfunction induced by amyloid-β oligomers through endocytic internalisation of NMDA receptors. FEBS J 2023, 290, 112–133. [Google Scholar] [CrossRef]
- Grassi, S.; Mauri, L.; Prioni, S.; Cabitta, L.; Sonnino, S.; Prinetti, A.; Giussani, P. Sphingosine 1-Phosphate Receptors and Metabolic Enzymes as Druggable Targets for Brain Diseases. Front Pharmacol 2019, 23, 807. [Google Scholar] [CrossRef]
- Asle-Rousta, M.; Oryan, S.; Ahmadiani, A.; Rahnema, M. Activation of sphingosine 1-phosphate receptor-1 by SEW2871 improves cognitive function in Alzheimer's disease model rats. Excli J 2013, 3, 449–461. [Google Scholar]
- Zijlstra, W.G.; Buursma, A.; Meeuwsen-van der Roest, W.P. Absorption spectra of human fetal and adult oxyhemoglobin, de-oxyhemoglobin, carboxyhemoglobin, and methemoglobin. Clin Chem 1991, 37, 37,1633–1638. [Google Scholar] [CrossRef]
- Blasi, B.; D'Alessandro, A.; Ramundo, N.; Zolla, L. Red blood cell storage and cell morphology. Transfus Med 2012, 22, 90–96. [Google Scholar] [CrossRef]
- Bergfeld, G. R, Forrester T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc. Res 1992, 26, 40–47. [Google Scholar] [CrossRef]
- Sprague, R.S.; Stephenson, A.H.; Bowles, E.A.; Stumpf, M.S.; Lonigro, A.J. Reduced expression of G(i) in erythrocytes of humans with type 2 diabetes is associated with impairment of both cAMP generation and ATP release. Diabetes 2006, 55, 3588–3593. [Google Scholar] [CrossRef]
- Ficarra, S.; Misiti, F.; Russo, A.; Carelli-Alinovi, C.; Bellocco, E.; Barreca, D.; Laganà, G.; Leuzzi, U.; Toscano, G.; Giardina, B.; Galtieri, A.; Tellone, E. Antiepileptic carbamazepine drug treatment induces alteration of membrane in red blood cells: possible positive effects on metabolism and oxidative stress. Biochimie 2013, 95, 833–841. [Google Scholar] [CrossRef]
- Hemmati, F.; Dargahi, L.; Nasoohi, S.; Omidbakhsh, R.; Mohamed, Z.; Chik, Z. Neurorestorative effect of FTY720 in a rat model of Alzheimer’s disease: comparison with memantine. Behav Brain Res 2013, 252, 415–421. [Google Scholar] [CrossRef]
Figure 1.
(A) Effect of sphingosine-1-phosphate (S1P) on ATP release in oxygenated (white) and deoxygenated (black) red blood cells (RBCs). Values are presented as the mean ± SD (N = 5). *p < 0.05 compared with control. (B) Protective role of S1P against amyloid beta (Aβ) peptides .Values are presented as the mean ± SD (N = 5). #p < 0.05 compared with deoxygenated cells, *p < 0.05 compared with Aβ.
Figure 1.
(A) Effect of sphingosine-1-phosphate (S1P) on ATP release in oxygenated (white) and deoxygenated (black) red blood cells (RBCs). Values are presented as the mean ± SD (N = 5). *p < 0.05 compared with control. (B) Protective role of S1P against amyloid beta (Aβ) peptides .Values are presented as the mean ± SD (N = 5). #p < 0.05 compared with deoxygenated cells, *p < 0.05 compared with Aβ.
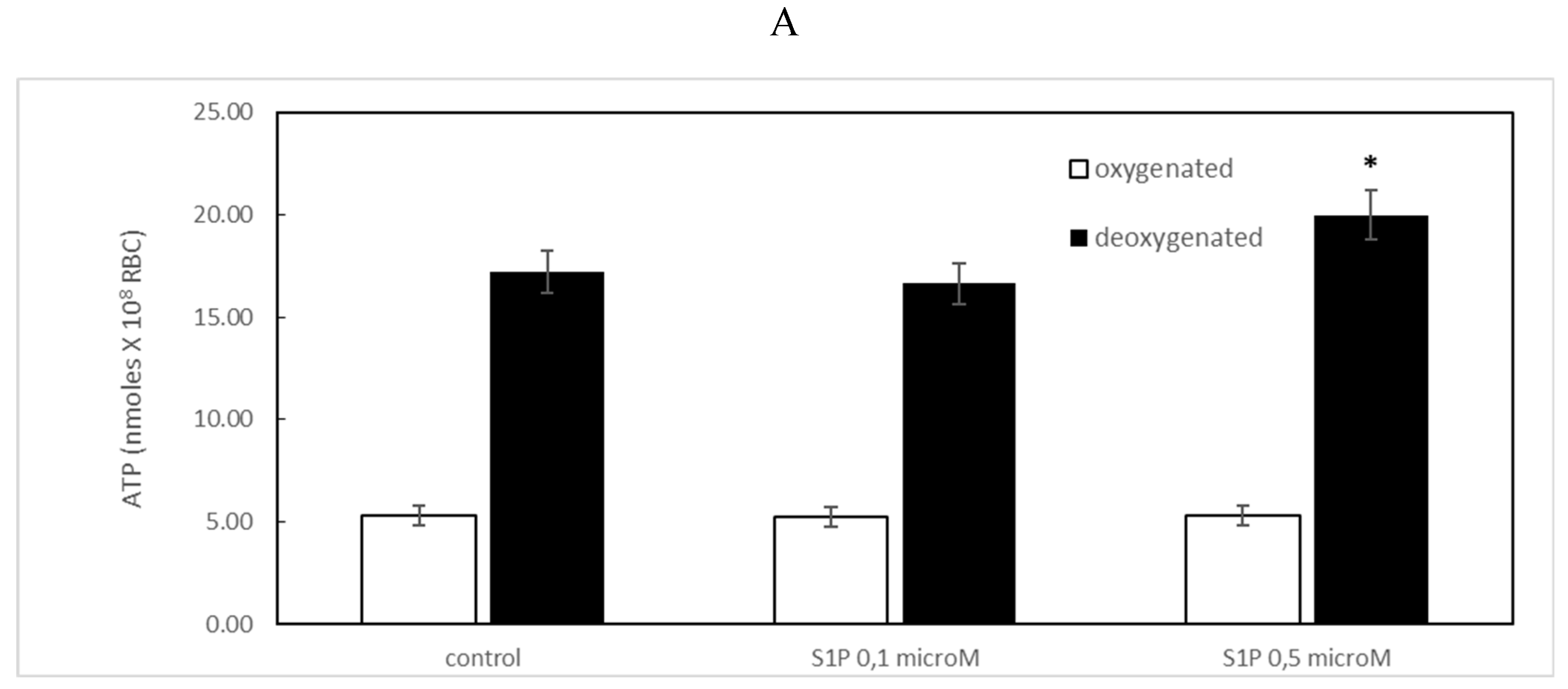
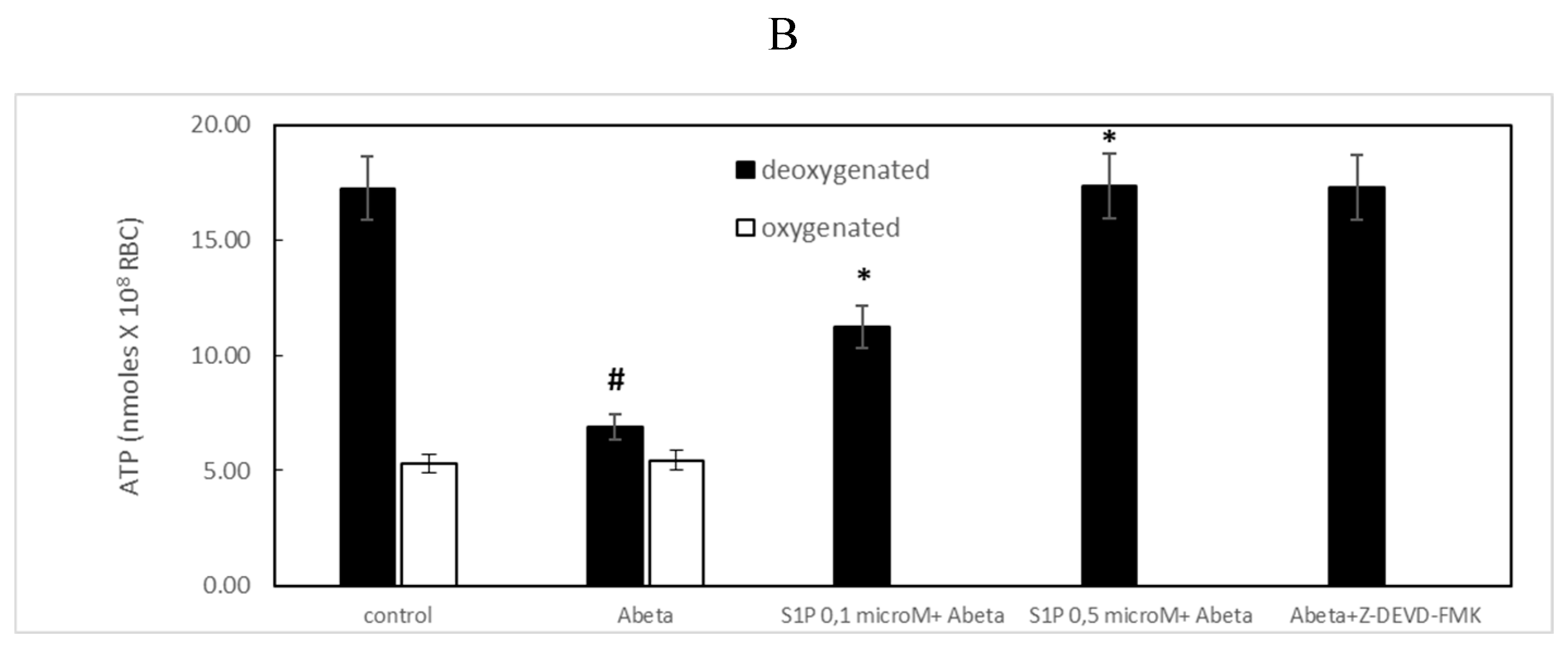
Figure 2.
Effect of mastoparan 7 (10 µM) on ATP release from RBCs. Values are presented as the mean ± SD (N = 6). *p < 0.05 compared with Aβ cells.
Figure 2.
Effect of mastoparan 7 (10 µM) on ATP release from RBCs. Values are presented as the mean ± SD (N = 6). *p < 0.05 compared with Aβ cells.
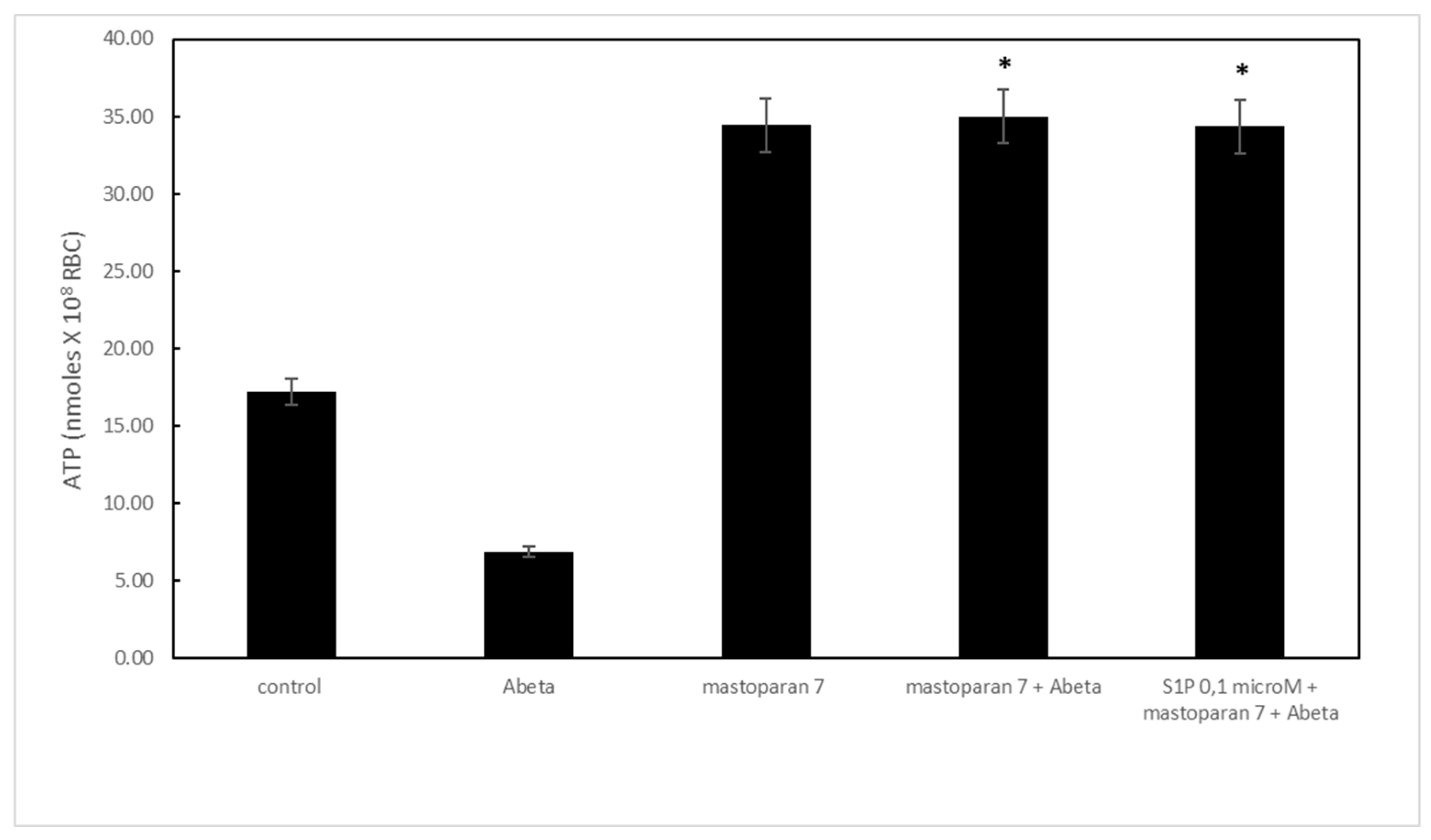
Figure 3.
(A) Effect of S1P on cyclic adenosine monophosphate (cAMP) levels in deoxygenated RBCs (black). Values are presented as the mean ± SD (N = 5). *p < 0.05 compared with control. (B) Protective role of S1P against Aβ in deoxygenated (black) RBCs. . Values are presented as the mean ± SD (N = 5). #p < 0.05 compared with deoxygenated cells, *p < 0.05 compared with Aβ.
Figure 3.
(A) Effect of S1P on cyclic adenosine monophosphate (cAMP) levels in deoxygenated RBCs (black). Values are presented as the mean ± SD (N = 5). *p < 0.05 compared with control. (B) Protective role of S1P against Aβ in deoxygenated (black) RBCs. . Values are presented as the mean ± SD (N = 5). #p < 0.05 compared with deoxygenated cells, *p < 0.05 compared with Aβ.
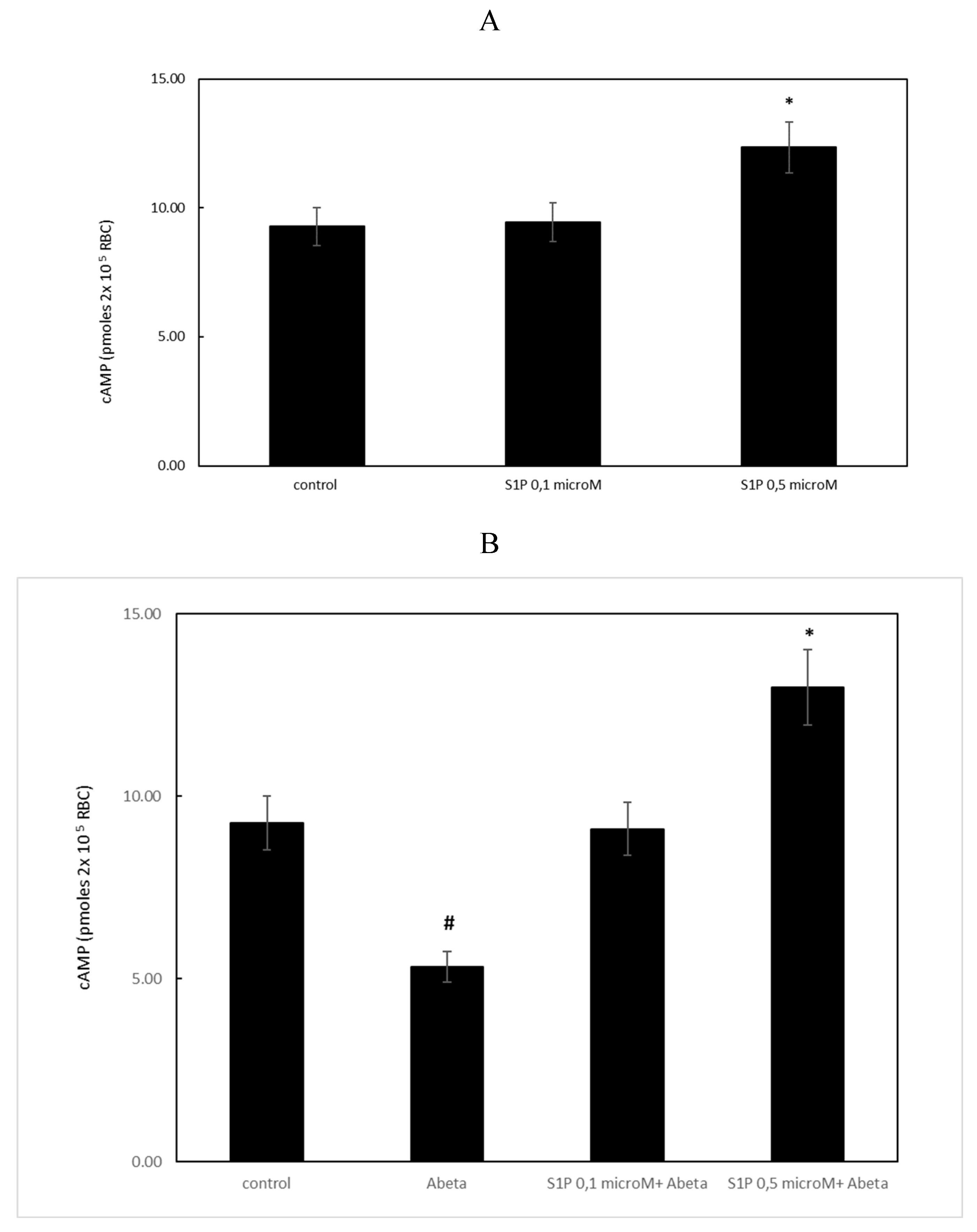
Figure 4.
Effect of S1P treatment on 2,3-diphosphoglycerate (2,3 DPG) levels in deoxygenated RBCs. Values are presented as the mean ± SD (N = 5). *p < 0.01 compared with control deoxygenated cells. #p < 0.01 compared with Aβ-treated cells.
Figure 4.
Effect of S1P treatment on 2,3-diphosphoglycerate (2,3 DPG) levels in deoxygenated RBCs. Values are presented as the mean ± SD (N = 5). *p < 0.01 compared with control deoxygenated cells. #p < 0.01 compared with Aβ-treated cells.
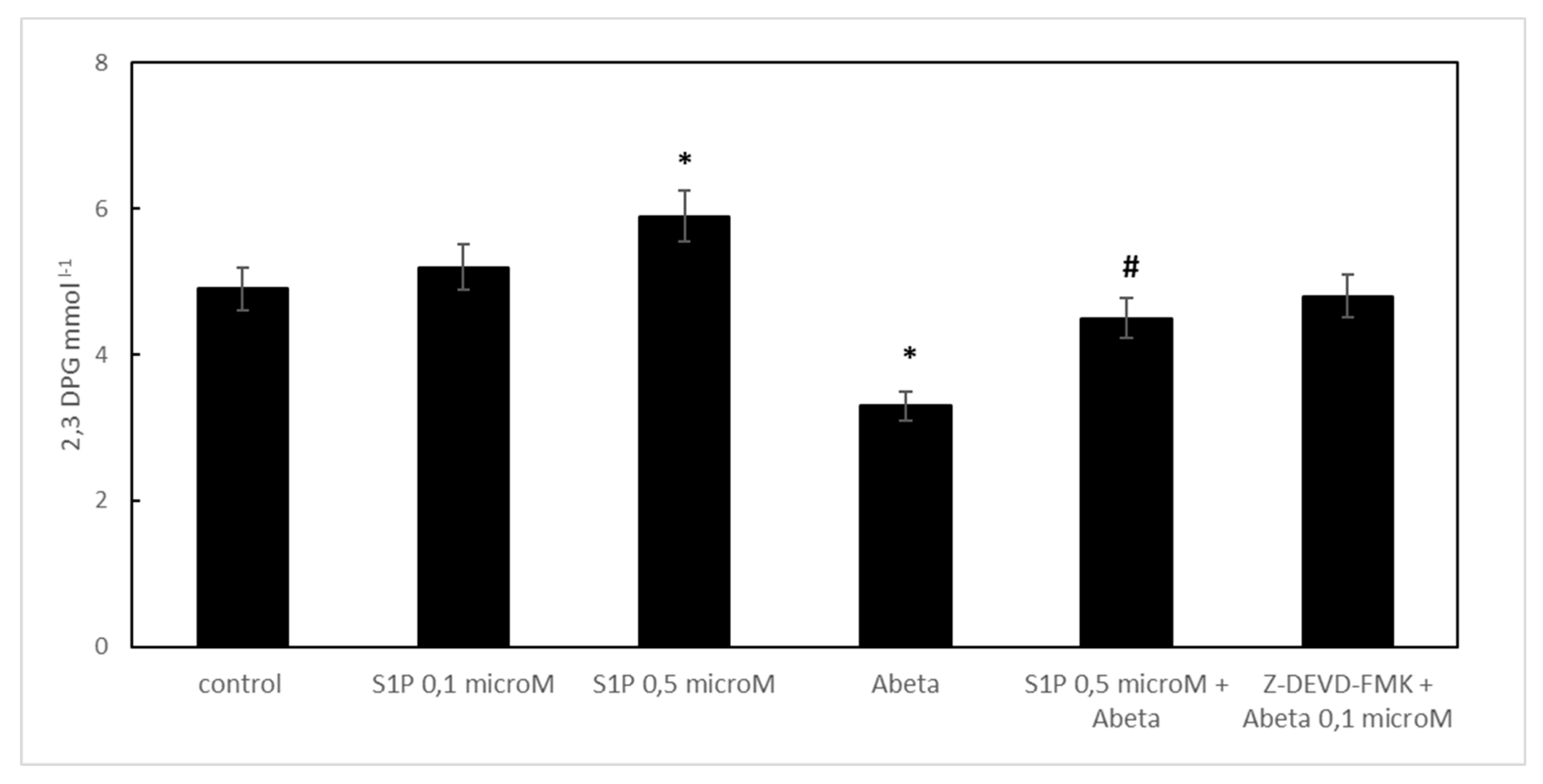
Figure 5.
Caspase-3 activity in deoxygenated RBCs following treatment under different conditions. Values are presented as the mean ± SD (N = 5). *p < 0.05 compared with Aβ-treated cells. a.u.
Figure 5.
Caspase-3 activity in deoxygenated RBCs following treatment under different conditions. Values are presented as the mean ± SD (N = 5). *p < 0.05 compared with Aβ-treated cells. a.u.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



