
Submitted:
17 April 2024
Posted:
18 April 2024
Read the latest preprint version here
Abstract
The evolutionary cancer cell biology ECCB explains the deep homology existent between the life cycle of cancer and protists. Due to their origin and sensitivity to oxygen, all non-gametogenic germ lines and germ stem cells (GSCs), including human adult stem cells (ASCs) and cancer stem cells (CSCs), suffer severe DNADSB damage when exposed to stress oxygen content above 6.0% O2 (Urgermline hyperoxia) as occurring in tissue and bloodstream. As a result, GSCs/ASCs/ CSCs lose their stemness potential and convert from a functional asymmetric cyclic phenotype (ACD phenotype) to a dysfunctional symmetric phenotype capable of defective symmetric proliferation (DSCD) that is, however, unable to repair genomic degradation. Genome reconstruction occurs through evolutionary mechanisms evolved by the common ancestor of amoebozoans, metazoans, and fungi. They complete the cycle by removing the damage, reconstructing the functional genome, and reinstating the ASC phenotype. The stemness life cycle acts not only during carcinogenesis, tumorigenesis, and recurrence but also in the life cycle of protists.
Keywords:
stem cell function
; dysfunctionality
; stress
; DNA DSB
; repair
; genomic integrity
The Unicellular Life Cycle of Cancer as Derived from the Urgermline
According to the evolutionary cancer cell biology (ECCB), the evolutionary origin of stem and progenitor cells dates back approximately 2000 million years ago (Mya), i.e. to the time before the metazoans. During this period, the common unicellular ancestor of amoebozoans, metazoans and fungi (AMF) evolved a dual life cycle consisting of a non-gametogenic oxygen-sensitive germline (Urgermline) and a somatic oxygen-resistant cell line. The non-gametogenic (NG) Urgermline, capable of generating progenitor and unipotent germline stem cells (GSCs), serves as the ancestral blueprint for all modern stem cell lineages and also plays a central role in cancer [1,2,3].
Around 1750 Mya, with the onset of the multicellularity, the number of ancestral genes underwent a significant increase. Evolutionary constraints led to the suppression of the unicellular AMF life cycle. However, when early multicellular progress became unstable and dysfunctional, there was a reversal to the stable AMF life cycle. Suppressor and antisupressor genes, along with the AMF life cycle, were retained in the genomes of metazoans. Over time, these genes evolved into tumor suppressor genes (TSG) and oncogenes. Genes from the early multicellular dead-end experiments are also retained as additional genes and could be repurposed during metazoan evolution, such as for the transition to multi- and pluripotent stem cells.
All modern progenitor and stem cell lineages have inherited the Urgermline's sensitivity to oxygen levels exceeding AMF normoxia. Consequently, oxygen levels above 6.0% O2 (AMF hyperoxia), as found in tissues and the bloodstream, far from the stem cell niche, cause damage to progenitors and stem cells in all animal cell systems, regardless of whether they are humans or mammals, vertebrates or invertebrates, or protists such as parasitic amoebae. The damage is consistent: oxygen-stressed NG germlines and stem cells respond with a decrease in the gene activity of homologous recombination (HR) repair, leading to DNA double-strand breaks (DNA DSBs) and loss of function.
DNA DSB damaged cells of protists and cancers continue proliferation through defective symmetric cell division (3), which involves endomitosis and binucleation, mature and immature nuclei, cytokinetic failure, and polyploidization-depolyploidization cycles with diploidy reversal and repolyploidization to tetraploidy (4n>2n>4n) [4], whereas DNA DSB damaged stem cells and cells related to the NG_germline related cells manage to evade apoptosis and persist in a state of quiescence, as low-grade polyploids ( e.g., ≤ 8n hepatocytes) or middle-grade polyploids (e.g., ≤ 64n cardiomyocytes).
Not all DNA DSB cells have a carcinogenic outcome, and not all cells that undergo polyploidization progress to transition from the higher level of organization (such as humans and mammals) to a lower level of unicellular imprinting (cancer). Among those that do progress, the majority are low-grade polyploids, such as cyclic hepatic tetraploids (4n). Middle grade tetraploids such as the <64n cardiomyocytes, do not follow this trajectory. Instead, they tend to enter a state of terminal-like quiescence.
Polyploidy and Stemness
Dysfunctional stem cell derivatives (DSCDs), resulting from persistent DNA DSB damage and proliferate as tetraploid/octoploid cells, are non-functional. They do not contribute to stemness recovery and differentiation processes, which are exclusively carried out by "healthy" diploid cells and asymmetric cell cycling (ACD phenotype) [4]. Similarly, the tetraploid DSCD cancer phenotypes are dysfunctional and unable to regain stemness. In cancer, tetraploidy is rather associated with subsequent aneuploidy.
Proliferating DSCDs continue aberrant proliferation with endomitosis, binucleation, mature and immature nuclei and cytokinesis failure in the hope of encountering environmental stimuli that may trigger unicellular repair programs, such as MGRS and PGCC [4]. Until then, germlines and germline clones can only regenerate through soma-to-germ transition (SGT) known as epithelial-mesenchymal transition (EMT) in cancer.
Multinucleated genome repair structures (MGRS') and polyploid giant cancer cells (PGCCs) initiate both with a phase of dysfunctional "cystic polyploidy" [4,5]. After homotypic cell fusion, the DSCD nuclei within MGRS begin with polyploidization-depolyploidization cycles similar to those occurring in the reproductive cysts of parasitic amoebae. However, due to the absence of effective homologous recombination (HR) repair, the resulting daughter nuclei remain dysfunctional and fail to undergo cellularization to form daughter cells.
As a result, the MGRS proceeds to a second phase of genomic repair. This phase entails the fusion of daughter nuclei and the formation of high-grade hyperpolyploid nuclei with an extensive DNA mass. These nuclei are capable of removing DNA fragments containing DSB and reconstituting the functional germline genome with functional HR genes.
Loss of Stemness and Stemness Recovery
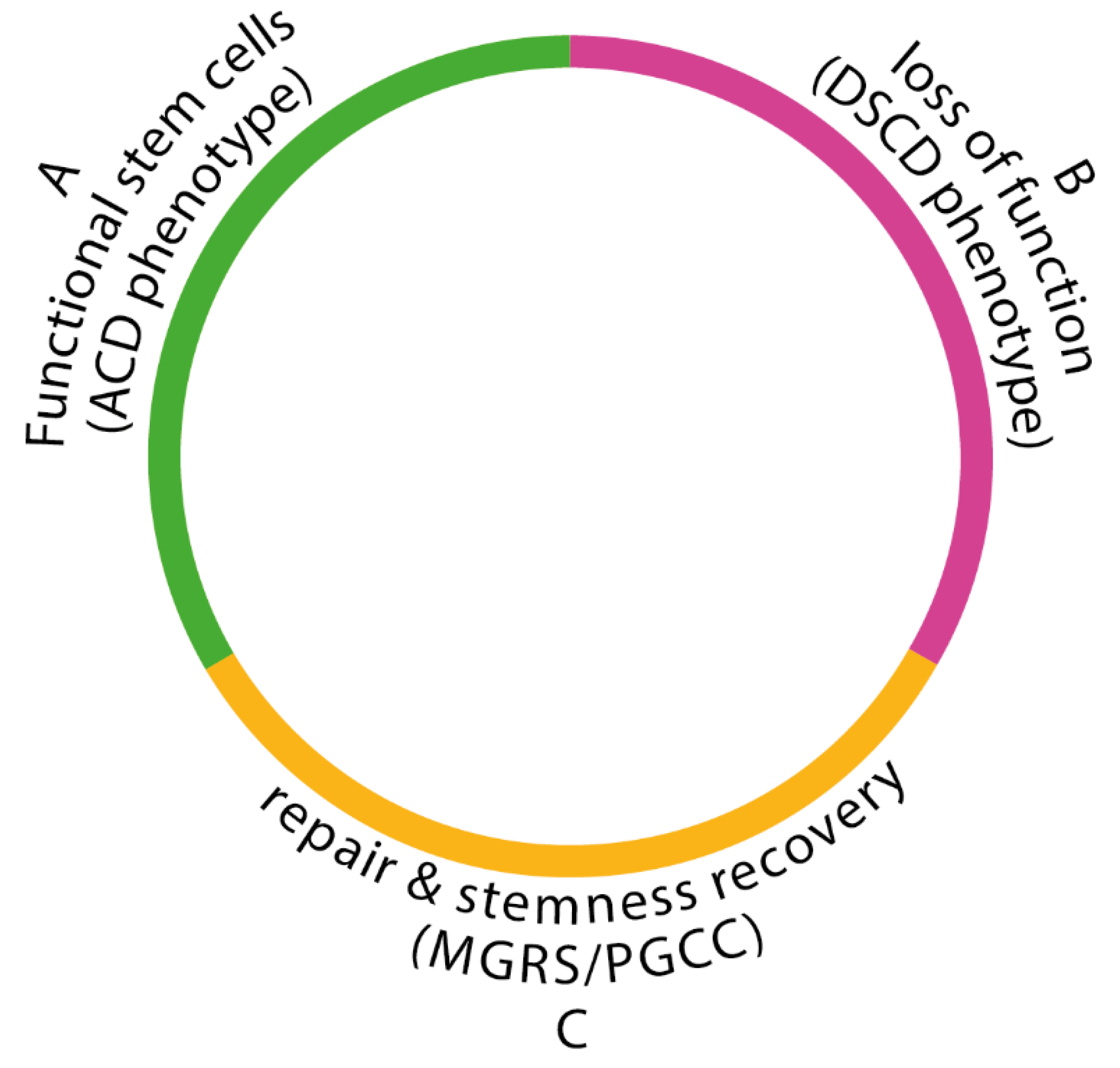
Comparing data on the fate of the NG germlines and stem cell lineages in lower organization systems (cancers and protists) with data from higher multicellular levels (i.e humans and metazoans) reveals an evolutionarily homology across all stemness life cycles [3,4]. All stemness cycles have three distinct phases (Figure 1):
(i) a phase of stem cell functionality, characterized by ACD phenotypes with stemness and differentiation potential, leading to the production of new stem cells (SCs) or cancer stem cells (CSCs);
(ii) a phase of dysfunctionality, triggered by stress factors and DNA DSB damage, resulting in the loss of stemness, and differentiation potential. The damaged cells either become quiescent or undergo a senescence/ apoptosis program, or bypass these programs and proliferate as DSCDs; and
(iii) a phase of DSCD repair and stem cell restoration in which ACD phenotypes are restored. This phase, mediated by hyperpolyploid MGRS or PGCC programs with or without cell fusion, is absent in humans and metazoans, where DSCD cells persist. MGRS and PGCC have homologous stem cell restoration programs. They have two polyploidization phases, a phase of "cystic" polyploidization- depolyploidization that form multiple nuclear progeny, and a phase of nuclear progeny fusion to form hyperpolyploid giant nuclei capable of DNA repair and genome reconstruction.
Excess Oxygen Damages All Types of CSCs in Tumors
Excessive oxygen damages all types of cancer stem cells (CSCs) within tumors. Oxygen stress, induced by ancestral hyperoxia with a content exceeding 6.0% O2, leads to HR gene defects and compromises the HR repair mechanisms of CSCs, including primary, secondary, and tertiary stem cells. As elucidated in previous studies [1,2,3], primary CSCs (pCSCs) originate as naïve cancer stem cells from the ACD phenotype via "cystic polyploidization". Secondary CSCs (sCSCs) emerge from somatic cancer cells through soma-to-gene transition (SGT) called epithelial-mesenchymal transition (EMT).
Throughout tumorigenesis, metastasis, and recurrence, oxygen-damaged DSCD phenotypes originating from pCSCs or sCSCs contribute to the restoration of the normal stem cells through homotypic cell fusion and the formation of polyploid giant cancer cells (PGCCs). Tertiary CSCs (tCSCs), derived from previous PGCC repair processes, are also susceptible to HR gene damage caused by tissue and bloodstream oxygen. DSCDs originating from tCSCs can similarly undergo homotypic fusion and generate new PGCCs. Particularly in metastatic and recurrent settings, the abundance of PGCCs substantially increases due to the persistent oxygen stress within tumors and the continuous regeneration of the CSC population via the PGCC pathway.
Repair by Hyperpolyploidization
Oxygenic stress When dysfunctional tumor DSCDs caused by hyperoxia and their progeny become fusionable, they merge into MGRS/PGCC structures via homotypic cell fusion, similar to the oxygenic stressed DSCDs of protists. This fusion process leads to the accumulation of damaged CSC/DNA copies and giant hyperpolyploid nuclei capable of excising damaged DNA DSB segments reconstructing the functional genome of cancer stem cells [4].
Gentoxic insultsWhen exposed to irradiation or chemotherapeutics, tumor CSC experience more extensive DNA DSB damage and the repair process through polyploidization is prolonged. The few surviving CSCs, which are unable to replicate, initiate the PGCC repair process without cell fusion.
Nevertheless, genotoxic PGCCs also begins with a phase of "cystic polyploidization", followed by a second phase of nuclear fusion resulting in 128n/396n giant repair nuclei [4,5]. This type of induced self-repair is specific for severe damage caused by radiation and chemotherapy, as the few surviving CSCs are not capable of proliferation and cannot form fusionable progeny.
Alternative Programs
Molecular studies on irradiated cancer stem cell (CSC) cultures suggest enhanced DNA damage response (DDR) process and overexpression of genes involved in specific DNA DSB sensing proteins. This difference between CSCs and the bulk of somatic tumor cells has been noted by several authors, who suggest that CSCs may have an efficient DDR capable of quickly detecting DNA damage [6,7,8].
Whether really two different mechanisms are involved in irradiated cancer stem cell repair - namely, (i) a slow repair mechanism through hyperpolyploidization and (ii) a fast mechanism for DNA damage detection and repair - remains unclear. It is also uncertain whether these different processes would operate in a dose-dependent manner or in conjunction with each other. Further research is needed to elucidate these questions.
Humans and metazoans lose MGRS/PGCC repair mechanisms; normally, they cannot complete and restart the evolutionary stemness life cycle. Stem cells in aging, as well as cells of germline origin transitioning from the ACD phenotype into a dysfunctional phenotype incapable of proliferation, such as dysfunctional aging stem cells, and hepatic or heart polyploids, lack the ability to repair DNA damage and regain restaurating diploidy. Instead, they persist in a state of terminal dysfunctional quiescence.
However, Benjamin et al. in 2023 [9] suggested that in old muscle stem cells, a perturbed glutathione (GSH) metabolism is the cause of stem cell dysfunctionality that could be compensated through metabolic manipulation. They believe that manipulation of glutathione metabolism could become an accessible target for reversing stem cell aging, thereby preventing the deterioration or initiating a replacement repair program.
Conclusions
A better understanding of the functionality and dysfunctionality of stem cells and their repair mechanisms to restore genomic integrity could provide new targets for the fight against cancer and ageing.
References
- Niculescu, VF. Cancer genes and cancer stem cells in tumorigenesis: Evolutionary deep homology and controversies. [CrossRef]
- Niculescu, V.F. The evolutionary cancer genome theory and its reasoning. Genetics in Medicine Open 2023, 1, 100809. [Google Scholar] [CrossRef]
- Niculescu, V.F. Understanding cancer from an evolutionary perspective: high-risk reprogramming of genome-damaged stem cells. Academia Medicine 2024, 2. [Google Scholar] [CrossRef]
- Niculescu, V.; Niculescu, E. The Enigma of Cancer Polyploidy as Deciphered by Evolutionary Cancer Cell Biology (ECCB). Preprints 2024, 2024021693. [Google Scholar] [CrossRef]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; et al. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, M.H.; Kim, S.A.; Jae Suk Chang, J.S. Age-related Deterioration of Hematopoietic Stem Cells. International Journal of Stem Cells 2008, 1, 55–63. [Google Scholar] [CrossRef]
- Wang, Q.E. DNA damage responses in cancer stem cells: Implications for cancer therapeutic strategies. World J Biol Chem. 2015, 6, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Valencia-González, H.A.; Ruíz, G.; Ortiz-Sánchez, E.; García-Carrancá, A. Cancer Stem Cells from Tumor Cell Lines Activate the DNA Damage Response Pathway after Ionizing Radiation More Efficiently Than Noncancer Stem Cells. Stem Cells International (Hindawi), 2019; ID 7038953.
- Benjamin, D.I.; Brett, J.O.; Both, P.; et al. Multiomics reveals glutathione metabolism as a driver of bimodality during stem cell aging. Cell Metabolism 2023, 35, 472–486. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The evolutionary life cycle of stemness as develops in carcinogenesis and tumorigenesis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



