
Submitted:
15 June 2024
Posted:
17 June 2024
You are already at the latest version
Abstract
According to the ECCB, cancer represents the transition of a genomically damaged multicellular cell to a lower level of cell organization, controlled by the ancestral gene regulatory network (GRN) from the ancestral compartment of human and metazoan genomes. Cancer adopts ancient mechanisms and rules of the common ancestor, similar to those observed in parasitic amoebae. In cancer, the productive cell line generating CSCs is a non-gametogenic cancer germline. This germline proliferates through asymmetric cell division (ACD phenotype). CSCs lack proliferative capacity; they persist in hypoxic niches in a quiescent cell state or transition to an amplifying cell state that performs cyst-like polyploidization cycles, giving rise to precursors and CSCs capable of differentiation. Malignization arises from genomically dysfunctional tetraploid germline cells (DSCD phenotype) that proliferate in a precancerous stage through symmetric cell cycles. DSCD progeny undergo homotypic fusion, forming multinucleated genome repair syncytia (MGRS pathway) driving carcinogenesis. During tumorigenesis, metastasis, and recurrence, subsequent germline clones initiate a cascade of increasingly pathogenic CSCs through mechanisms such as heterotypic cell fusion, soma-to-germ transition, and polyploid giant cell structures (PGCCs). Irradiation and chemotherapies drive some of the very few surviving but dysfunctional CSCs through a PGCC repair pathway homologous to the MGRS pathway of carcinogenesis. The large number of PGCCs during metastasis and recurrence results from the perpetual germline cell damage and CSC poverty induced by oxygen levels above 6.0% O2 (germline hyperoxia).
Keywords:
Cancer
; Entamoeba
; germline
; asymmetric cell division
; CSCs
; loss of function
; MGRS/PGCC
1.0. Introduction
1.1. Requirements for a Modern CSC State Concept
According to the most common definitions to day, stem cells and cancer stem cells are either (i) undifferentiated or partially differentiated cells that can differentiate into various types of cells and proliferate indefinitely to produce more stem cells, or (ii) undifferentiated cells that continuously divide to produce some offspring that remain as stem cells and others that are destined to differentiate. However, the findings of recent years have shown that these definitions are no longer adequate.
Very recently, Loh and Ma (1) discussed the need to re-evaluate the origin, hallmarks, and characteristics of cancer stem cells. In their opinion, there is substantial evidence that cancer cells possess a plastic state that can be influenced by the interplay of stressors, the environment, and CSC niches. The researchers consider that the features acquired through de-differentiation require a re-evaluation of the basic attributes of the CSC state. Cellular plasticity allows several cancer cells to adopt a stem-like state, enabling the tumor to expand its malignant properties, including resistance to therapy and metastasis.
The author of the present work also advocates for a reevaluation of the cancer stem cell (CSC) concept, specifically in relation to the germline and its plasticity, as proposed by evolutionary cancer cell biology (ECCB) (2). In the view of the ECCB, current CSC definitions above are vague, biologically and evolutionarily imprecise, and require readjustment.
1.2. The Evolutionary Origin of Stemness
According to the evolutionary cancer cell biology (ECCB), the evolutionary origin of cancer stem and progenitor cells dates back approximately 2000 million years ago (Mya), i.e., to the time before the metazoans. During this period, the common unicellular ancestor of amoebozoans, metazoans and fungi (AMF) evolved a dual life cycle consisting of a non-gametogenic oxygen-sensitive germline (Urgermline) and a somatic oxygen-resistant cell line. The non-gametogenic Urgermline, capable of generating unipotent germline stem cells (GSCs), serves as the ancestral blueprint for all modern stem cell lineages and also plays a central role in cancer, which largely mirrors the mechanisms of the AMF ancestor also observed in protists parasiting humans (Entamoeba) (3).
In contrast, oxygen resistant somatic cancer cells are not damaged by excess oxygen and participate to the reconstruction of functional germline clones through soma-to-germ transition (SGT),
1.3. Dysfunctional Multicellularity and Reversal to a Stable Unicellular Cell System
Around 1750 Mya, with the onset of the multicellularity, the number of ancestral genes underwent a significant increase. Evolutionary constraints led to the suppression of the unicellular AMF life cycle. However, when early multicellular progress became unstable and dysfunctional, there was a reversal to the stable AMF life cycle. This evolutionarily conserved strategy persisted during the evolution of metazoans and led to cancer. According to the ECCB, cancer is the transition from a higher level of organization of the cell system (multicellularity) to a lower, unicellular level (4, 5).
Suppressor and antisuppressor genes from the transition period between unicellularity and multicellularity, along with the AMF life cycle, were retained in the genomes of metazoans. Over time, these genes evolved into tumor suppressor genes (TSGs) and oncogenes. Genes from early multicellular dead-end experiments were also retained as additional genes and could be repurposed during metazoan evolution, such as for the transition to multi- and pluripotent stem cells.
1.4. The Ancestral Genome Compartment and the Hybryd Genome of Cancer
During malignization and carcinogenesis a large number of silenced unicellular genes (UGs) from the ancestral genome compartment are reactivated and many of the interfering multicellular genes are downloaded or inactivated (MGs) (04-07). This results in a hybrid genome that changes to an primitive germ and soma genome of unicellular imprinting with a malignant non-gametogenic germline (NG germline). The cancer germline proliferates in an environment-dependent manner through asymmetric and symmetric cell division (ACD and SCD). The ACD-phenotype of the germline has stemness and differentiation potential, generating malignant cancer stem cells (4) and considered the progenitor the progenitor of CSCs.
1.5. The Risks of Well-Oxygenated Tissue and Blood Flow for the Cancer Germline Genome
All modern germlines, including progenitor and stem cell lineages are predominantly hypoxic, with normoxic ranges below 6.0 % O2 (germline physoxia). They are highly sensitive to excess oxygen. Oxygen levels above 6.0% O2, as found in tissues and the bloodstream, far from the hypoxic stem cell niche, cause damage to progenitors and stem cells in all animal cell systems and cancer, regardless of whether they are humans or mammals, vertebrates or invertebrates, or protists such as parasitic amoebae. The damage is consistent: oxygen-stressed NG germlines producing stem cells respond with a decrease in the gene activity of homologous recombination (HR) repair, leading to DNA double-strand breaks (DNA DSBs) and loss of function. (4, 5)
2.0. CSC Phenotypes as Non-Proliferating Germline Cells:
According to the ECCB, germline cell exiting ACD proliferation cycles are termed CSCs. They can be committed for differentiation (C-CSCs) or not (NC-CSCs). NC-CSCs can remain in a state of G0 quiescence for extended periods and can reenter the G1 phase and cell cycle as self renewing germline cells. C-CSCs are also non proliferative; however, they can progress into a phase of cyst-like amplification through polyploidization-depoliploidization cycles and multiple genome duplications, resulting in an increased niumber of CSCs by 32 fold or more. The amplified CSCs pool follow the same dichotomous cell fate: differentiation processes or amplification by cyst-like polyploidization CSC amplification is more effectively than multiplication through repeated ACD cell cycles. (Figure 1 and Figure 2).
In tumors, there are a large number of CSCs of different origins. On the one hand, there are primary CSCs (pCSCs), which are formed by the primary cancer germline through homotypic cell fusion and malignization (carcinogenesis), and primary germline clones. During tumorigenesis, the diversity of CSCs also increases through the formation of secondary stem cells (sCSCs) by heterotypic cell fusion with somatic, non-cancerous cells, soma-to-germ transition, and the formation of secondary germline clones with stemness and differentoiation potential.
CSC – as a product of the germline - and the germline itself build a strong inseparable environmental dependant unit that can be disrupted under unfavorable environmental conditions. Harmful environmental conditions, such as oxygen stress and germline hyperoxia with more as 6.0% O2 damage irreversibly the germline (4, 5) and the dysfunctional germline loses its ACD phenotype and stemness capacity.
This is the major misunderstanding of the current CSC biology, which assumes that “stem cells proliferate indefinitely to produce more of the same stem cells.” The current CSC biology does not understand that CSC biology is the primitive stem cell biology of a primitive, unicellular germ and soma systems different from the more evolved stem cell biology of humans and higher metazoans. Moreover, it confuses proliferating ACD germline cells with non-proliferating quiescent CSCs and amplifying CSC. Primitive, unicellular stem cells CSCs are never proliferative; instead, they are capable of conversion into proliferative germline cells that keep producing new CSCs as long as environmental conditions favor germline proliferation and asymmetric cell division.
3.0. Functional and Dysfunctional Germline States, and the Cessation of CSC Production
As previously mentioned, all germlines and germline clones are oxygen-sensitive and can be damaged by germline hyperoxia, which occurs at oxygen levels above 6.0% O2 (oxygen stress). They irreversibly lose their ACD potential due to severe DNA damage caused by altered HR repair genes and resulting in the cessation of CSC production. This damage occurs when oxygen sensitive germline cells and CSCs leave the strongly hypoxic ranges and translocate via the bloodstream to well-oxygenated tissue. Under excess oxygen pressure, germline cells transform into an irreparably dysfunctional germline cell state.
However, dysfunctional cancer germ cells do not initiate apoptosis programs or undergo cell death; instead, they continue to proliferate through aberrant symmetric cell cycles and defective symmetric cell division (DSCD) in a unicellular-like pattern. The DSCD- phenotype is usually tetraploid and includes endomitosis and binucleation, mature and immature nuclei, cytokinetic failure, and polyploidization/depolyploidization cycles with diploidy reversal and repolyploidization to tetraploidy (4n>2n>4n) (4,5)
Loss of function and irreparable germline dysfunctionality occur repeatedly in cancer and tumorigenesis, as well as in protists. In amoebiasis, the infectious disease caused by parasitic amoebae, there is a constant alternation between functional and dysfunctional germlines and clones. This includes alternation of ACD phenotypes, stem cells, and cyst differentiation processes (cyst-positive phases), as well as negative phases of dysfunctional germlines without cyst differentiation, when amoebiasis cannot be detected by means of coprologic analysis.
4.0. The Life Cycle of Stemness
Comparing data on the fate of NG germlines and stem cell lineages from lower organization systems (cancers and protists) with data from higher organization systems (humans and metazoans) reveals an evolutionarily homology across all life cycles of germline and their stemness potential (4,5). Stemness cycles (Figure 3) have three distinct phases dependant of the germline cell state:
(i) The phase of germline functionality: Functional ACD phenotypes with stemness and aCLS differentiation potential accelerate the production of cancer stem cells (CSCs) via cyst-like polyploidization.
(ii) The phase of germline dysfunctionality: Triggered by stress factors and DNA DSB damage, resulting in the loss of stemness and differentiation potential and the cessation of CSC production. The damaged cancer germline cells bypass the senescence/apoptosis program and proliferate as DSCD phenotypes.
(iii) The phase of genome repair and stem cell restoration: In this phase, the ACD phenotype is restored. This phase, mediated by hyperpolyploid MGRS or PGCC programs with or without cell fusion, is absent in healthy humans and metazoans. MGRS’ and PGCCs are homologous germline cell restoration programs. The MGRS/PGCC pathway has two polyploidization phases: a cyst-like phase for genome multiplication and the formation of several identical daughter nuclei, followed by a phase of nuclear fusion and the formation of hyperpolyploid giant nuclei. These processes reconstruct the genome architecture of damaged germline cells, recover the ACD phenotype, restore its functionality, and enable subsequent CSC generation.
Der MGRS/PGCC repair pathway of cancer erinnert an die human amoebiasis verursacht durch Entamoeba histolytica. Auch in der amoebiasis alternieren phasen of cyst production and cyst excretion, with negative phases, as cyst can not be found und umgekehrt. Es ist der gleich homolge germline life cycle, with alternating phases of productive ACD phenotypes and negative DSCD phases with repair through the MGRS pathway (4).
5.0. CSC Heterogeneity in Tumors
5.1. Primary CSCs (pCSCs)
According to evolutionary cancer cell biology (ECCB), the carcinogenic ACD phenotype initiating malignization generates naive, primary cancer stem cells (pCSCs), as well as primary somatic cancer cells through cell line conversion and epigenomic changes induced by varying environmental conditions. These somatic cancer cells preserve the germline genome from excess oxygen damage and can regenerate through soma-to-germ transition (SGT) clones with the same genomic architecture, forming further pCSCs. SGTs are induced when the ACD germline phenotype loses functionality, transforms into the dysfunctional DSCD phenotype, and ceases the production of pCSCs. However, the cancer cell system requires continuous CSC production. In this situation, SGT processes are initiated to generate new germline clones and reverse the loss of pCSCs. These processes are known in current cancer research as somatic dedifferentiation, epithelial-mesenchymal transition (EMT), and cancer cell plasticity.
5.2. Secondary CSCs (sCSCs)
As cancer evolves, primary somatic cells can fuse with non-cancerous host cells (e.g., macrophages), thereby enriching their genome with multicellular MG genes acquired through heterotypic cell fusion. Parallel to the existing primary ACD germline, which continues to produce pCSCs, secondary germline clones producing secondary cancer stem cells (sCSCs) with increasingly higher and more complex anti-host potential arise. This complex plasticity of cancer cells through SGT processes extends and clarifies the concept of CSC emergence through somatic cancer cell dedifferentiation (1, 8, 9). Each new heterotypic cell fusion process with non-cancerous host cells may contribute to the expansion of the germline genome and the production of more highly developed and potent sCSCs.
6.0. Function Recovery; the MGRS/PGCC Pathway
Dysfunctional tetraploid DSCD germlines do not express stemness and differentiation potential; these are exclusively expressed by the “healthy” diploid ACD phenotype. Genes ensuring the functional ACD phenotype are inactivated, while genes of the tetraploid DSCD phenotype are upregulated. Proliferating DSCD cell lines continue an aberrant symmetric cell proliferation, leading to the accumulation of DSCD cells, which replaces the previously termed “CSC accumulation” for symmetric cell cycles observed in tumors. This DSCD cell accumulation increases the number of defective DSCDs, in the hope of encountering environmental stimuli that may trigger homotypic DSCD cell fusion and repair by the MGRS/PGCC pathway. This process leads to the formation of multinucleated genome repair structures (MGRS) during carcinogenesis, and polyploid giant cancer cells (PGCCs) during tumorigenesis, metastasis, and recurrence.
Hyperpolyploid genome repair structures (MGRS) and polyploid giant cancer cells (PGCCs) are based on a process of “cyst-like polyploidization” (5, 10), which occurs in the reproductive cysts of parasitic amoebae and is developed by healthy germline cells. Cystic polyploidy in protists results in the formation of 4 or 8 diploid nuclei and 8 or 16 haploid cyst daughter cells (basal stem cells) that generate a functional germline capable of stem cell formation and differentiation. However, in contrast to multicellular organisms, protist differentiation is rudimentary. The haploid daughter cells of the cyst are the progenitors of germline stem cells (GSC).
Both in cancer and protists, MGRSs and PGCCs, resulting from the fusion of defective DSCDs, are two-phase processes.The first is a cyst-like phase of polyploidization that ends by the formation of defective daughter nuclei. During genome amplification and polyploidization, the intracystic HU genome repair genes cannot repair the dysfunctional germline genome, the resulting daughter nuclei remain dysfunctional, and fail to undergo cellularization. The DNA DSB damage of the DSCD nuclei persist (5, 10).
This is why the MGRS/PGCC pathway proceeds to a second phase of genomic repair. This phase involves the fusion of defective daughter nuclei and the formation of high-grade hyperpolyploid nuclei with an extensive DNA mass. These hyperpolyploid nuclei are capable of removing DNA fragments containing DSBs and reconstituting the functional germline genome architecture with functional HR repair genes.
7.0. Genotoxic Insults by Irradiation and Chemotherapy
There is evidence that CSCs surviving irradiation and chemotherapy exhibit significant genomic alterations. Genotoxic damage has far more severe consequences than the common degradation of CSCs and germline cells caused by hyperoxia. Irradiation and chemotherapeutic agents not only kill somatic cancer cells and proliferating germline cells, but also cause extensive damage to all types of committed and non-committed CSCs, including those in a quiescent state. NC-CSCs cannot revert to self-renewing ACD germline cells nor adopt the defective DSCD phenotype. Similarly, C-CSCs and quiescent CSCs can neither differentiate nor amplify through cyst-like polyploidization. Due to the insufficient number of damaged CSCs and their progeny, the MGRS/PGCC repair pathway cannot be induced via homotypic cell fusion.
Only C-CSCs with residual potential for CSC amplification have a chance to restructure the dysfunctional genome via the MGRS/PGCC pathway. Engaged C-CSCs can initiate the first phase of the MGRS/PGCC genome repair pathway via some still functional cyst-like polyploidization (amplification) genes. However, as expected, the daughter nuclei that emerge from the cyst-like polyploidization and depolyploidization are dysfunctional and must undergo the second MGRS/PGCC phase and fuse with each other to form a hyperpolyploid giant nucleus. This nucleus now has the opportunity to regenerate the architecture of the functional germline genome. This begins with the fusion of the defective daughter nuclei and the formation of a giant hyperpolyploid nucleus with a hyperpolyploidy level of up to 380. Hyperpolyploid nuclei can cut out the defective DNADSB fragments and restore the architecture and functionality of the genome. PGCC processes that occur during tumorigenesis, metastasis, and recurrence due to harmful environmental conditions such as germline hyperoxia are more “physiological” than post-genotoxic repair and take far less time. Otherwise, the MGRS/PGCC processes proceed as expected, except that they take much more time (delayed MGRS/PGCC pathway).
Recently, Wu et al. in 2023 confirmed that genotoxic cancer therapy inactivates and kills cancer cells through extensive DNA damage (11). Energy delivery triggers extensive DNA damage, typically in the form of double-strand breaks, single-strand breaks, base damage, and interstrand crosslinks, with DSBs being the most deleterious ((12, 13). In the same time, DNA damage checkpoints are activated. delaying the onset of mitosis and providing more time for DNA repair (14,15).
On the other hand, molecular studies on irradiated cancer stem cell (CSC) cultures indicate an enhanced DNA damage response (DDR) process and overexpression of genes involved in specific DSB-sensing proteins. Several authors have noted this difference between CSCs and the bulk of somatic tumor cells, suggesting that CSCs may have an efficient DDR capable of rapidly recognizing DNA damage (4-6).
8.0. Contradictory Opinions on a Delayed Cell Cycle Progression of Damaged CSCs
Current CSC research presents some questionable narratives regarding the supposed “proliferation of CSCs“, which is, in fact, the proliferation of the germline. For example, the statements on the delayed cell cycle progression of CSCs. It has been reported that CSCs respond to ionizing radiation exposure in tumor tissue by activating checkpoint signaling pathways, upregulating the expression of genes mediating DNA repair, and delaying cell cycle progression to gain time for repair, thereby escaping the harmful effects of radiotherapy (16). However, according to the ECCB, CSCs are not proliferative either before or after irradiation. What can be proliferative is only the NG germline or clones that arise after irradiation through the MGRS/PGCC-repair pathway.
Mechanisms such as activation of DNA damage checkpoints to control the cell cycle, increased DNA repair capacity, and reduced radiosensitivity by upregulating the expression of homologous recombination (HR) genes all contribute to the development of radioresistance in gliomas (17,18). However, we agree with the idea that DNA repair capacity must indeed be considered a measure of radioresistance (19,20).
9. Conclusions
A better understanding of the origin and relation between CSCs as quiescent, non- proliferative germline stem cells with dichotomous cell fate potential, as well as their ability to repair and restore genomic integrity and functionality, could provide new targets in the fight against cancer.. The major statements of the present work are:
1. CSC are formed by the non-gametogenic (NG) cancer germline. The ECCB differentiates between (i) non-committed, quiescent CSCs capable of transforming back into self-renewing germline cells (ACD phenotype) and committed CSCs capable of differentiation and amplification. According to the ECCB, CSCs are never proliferative; only the germline clones generated by quiescent CSC that transform back into proliferative germline clones are proliferative.
2. The ECCB differentiate between primary and secondary CSCs. Primary pCSC are generated by the primary NG cancer germline through homotypic cell fusion from dysfunctional tetraploid DSCD cells. Secondary sCSCs are initiated by heterotypic cell fusion between somatic cancer cells and non-cancerous somatic cells, which enriches the cancer cell genome with functional multicellular MG genes; somat-to-germ transition processes (SGT) lead to the formation of new proliferating germline clones capable of generating sCSCs with higher genomic potential.
3. Stemness is a hallmark of the NG germline, which can change from dysfunctionality to functionality (stemness life cycle) via the MGRS/PGCC pathway. Dysfunctional DSCD do not express stemness, but MGRSs and PGCCs do. MGRS are formed during carcinogenesis from dysfunctional, oxygen-damaged DSCD cells, and PGCCs are formed during tumorigenesis from dysfunctional germline cells, clones and CSCs.
4. MGRSs and PGCCs are two- phase genome repair processes. The fisrt phase is a cyst-like polyploidization and depolyploidization phase that forms multiple CSC precursor cells by cystic polyploidization, as observed in protists. During this phase, genomically damaged nuclei of the dysfunctional DSCDs undergo the cystic polyploidization process, forming daughter cell nuclei that are not capable of cellularization into CSC precursor cells. In the second repair phase, these dysfunctional daughter nuclei fuse together, forming hyperpolyploid nuclei (≤ 380n) capable to reconstructing the functional genomic architecture.
5. Genotoxic damage from irradiation or chemotherapeutics destroys most cancer cells. The few CSCs that survive genotoxicity are committed CSCs with a residual potential for proliferation through cyclic polyploidization and the capacity for autonomous genome restoration via PGCCs, without cell fusion.
Abbreviations
ACD, asymmetric cell division; aCLS,amplifying cyst like structure; AMF, amoebozoans, metazoans and fungi; C-CSCs, commited cancer stem cells; DSCD, dysfunctional symmetric cell division; HR, homologous recombination; GSC, germline stem cells; MG, multicelularian genes; MGRS, multinucleated genome repair syncytia; NC-CSCs, non-committed cancer stem cells; NG, non gametogenic; SGT, soma-to-germ transition; UG, unicellularity genes.
References
- Loh JJ, Ma S. Hallmarks of cancer stemness. Cell stem cell 2024, 31, 617–639. [Google Scholar] [CrossRef] [PubMed]
- Niculescu VF. Introduction to Evolutionary Cancer Cell Biology(ECCB) and Ancestral Cancer Genomics.2023. Preprints.org. 2023. [Google Scholar] [CrossRef]
- Niculescu VF. The stem cell biology of the protist pathogen <italic>Entamoebainvadens </italic>in the context of eukaryotic stem cell evolution. Stem Cell Biology and Research 2015. [Google Scholar] [CrossRef]
- Niculescu VF. Understanding cancer from an evolutionary perspective: high-risk reprogramming of genome-damaged stem cells. Academia Medicine 2024, 2. [Google Scholar]
- Niculescu VF, Niculescu ER. The Enigma of Cancer Polyploidy as Deciphered by Evolutionary Cancer Cell Biology (ECCB). Academia Medicine in press. 2024. [Google Scholar]
- Trigos AS, Pearson RB, Papenfuss AT, Goode DL. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc Natl Acad SciU S A. 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [PubMed]
- Trigos A, Pearson R, Papenfuss A, Goode DL. How the evolution of multicellularity set the stage for cancer. Br J Cancer 2018, 118, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gonzalez A, Bevant K, Blanpain. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer 2023, 4, 1063–1082. [Google Scholar] [CrossRef] [PubMed]
- Salmina K, Huna A, Kalejs M, Pjanova D, Scherthan H, et al. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef]
- Wu Y, Song, Y, Wang R et al. Molecular mechanisms of tumor resistance to radiotherapy. Mol Cancer. 2023, 22, 96. [Google Scholar] [CrossRef] [PubMed]
- Li J, Sun H, Huang Y, Wang Y, Liu Y, Chen X. Pathways and assays for DNA double-strand break repair by homologous recombination. Acta Biochim Biophys Sin (Shanghai). 2019, 51, 879–89. [Google Scholar] [CrossRef] [PubMed]
- Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Gavande NS, VanderVere-Carozza PS, Hinshaw HD, et al. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol Ther 2016, 160, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Schulz A, Meyer F, Dubrovska A, Borgmann K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers (Basel). 2019, 11, 862. [Google Scholar] [CrossRef] [PubMed]
- Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–60. [Google Scholar] [CrossRef] [PubMed]
- Balbous A, Cortes U, Guilloteau K, et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer 2016, 16, 604. [Google Scholar] [CrossRef] [PubMed]
- Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–3. [Google Scholar] [CrossRef] [PubMed]
- Szumiel, I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int J Radiat Biol 2015, 91, 1–12. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The ancestral germ and soma life cycle of cancer and protist, including the oxygen- sensitive ACD phenotype of the non-gametogenic germline (green) as observed under normoxic conditions of life with less as 6.0%O2, and the oxygen resistant somatic cell line (blue) that proliferates under conditions of ancestral hyperoxic values (>6.0%O2) through the symmetric SCD phenotype. In protists, functional ACD germline cells proliferate by asymmetric cell cycles, producing self renewing cells and cells exiting the cell cycle, which (i) can be committed to cyst differentiation (C-SCs) and genome amplification through polyploidization –depolyploidization cycles, forming 16-32 daughter cells as precursors of unipotent stem cells, and (ii) uncommitted cells (NC-SCs) that, after a state of G0 quiescence, can transform back into a self-renewing cell state capable of continuing ACD proliferation. In cancer, germline cells exiting ACD cell cycles are cancer stem cells (CSCs). CSCs are non-proliferative and cannot multiply, neither by symmetric nor by asymmetric cell cycles. Homologous to the non commited NC-SCs of protists, cancer NC-CSCs can enter a state of variable G0 quiescence, with the perspective to convert back into the proliferating ACD germline phenotype, becomming a self-renewing germline cells. Commited C-CSCs have a dichotomous cell fate: they can differentiate or enter a state of genome and CSC amplification (cyst-like amplification) resulting in an amplified CSC pool that generates new germline clones. In contrast, differentiation and stem cell amplification are linked in protists.
Figure 1.
The ancestral germ and soma life cycle of cancer and protist, including the oxygen- sensitive ACD phenotype of the non-gametogenic germline (green) as observed under normoxic conditions of life with less as 6.0%O2, and the oxygen resistant somatic cell line (blue) that proliferates under conditions of ancestral hyperoxic values (>6.0%O2) through the symmetric SCD phenotype. In protists, functional ACD germline cells proliferate by asymmetric cell cycles, producing self renewing cells and cells exiting the cell cycle, which (i) can be committed to cyst differentiation (C-SCs) and genome amplification through polyploidization –depolyploidization cycles, forming 16-32 daughter cells as precursors of unipotent stem cells, and (ii) uncommitted cells (NC-SCs) that, after a state of G0 quiescence, can transform back into a self-renewing cell state capable of continuing ACD proliferation. In cancer, germline cells exiting ACD cell cycles are cancer stem cells (CSCs). CSCs are non-proliferative and cannot multiply, neither by symmetric nor by asymmetric cell cycles. Homologous to the non commited NC-SCs of protists, cancer NC-CSCs can enter a state of variable G0 quiescence, with the perspective to convert back into the proliferating ACD germline phenotype, becomming a self-renewing germline cells. Commited C-CSCs have a dichotomous cell fate: they can differentiate or enter a state of genome and CSC amplification (cyst-like amplification) resulting in an amplified CSC pool that generates new germline clones. In contrast, differentiation and stem cell amplification are linked in protists.
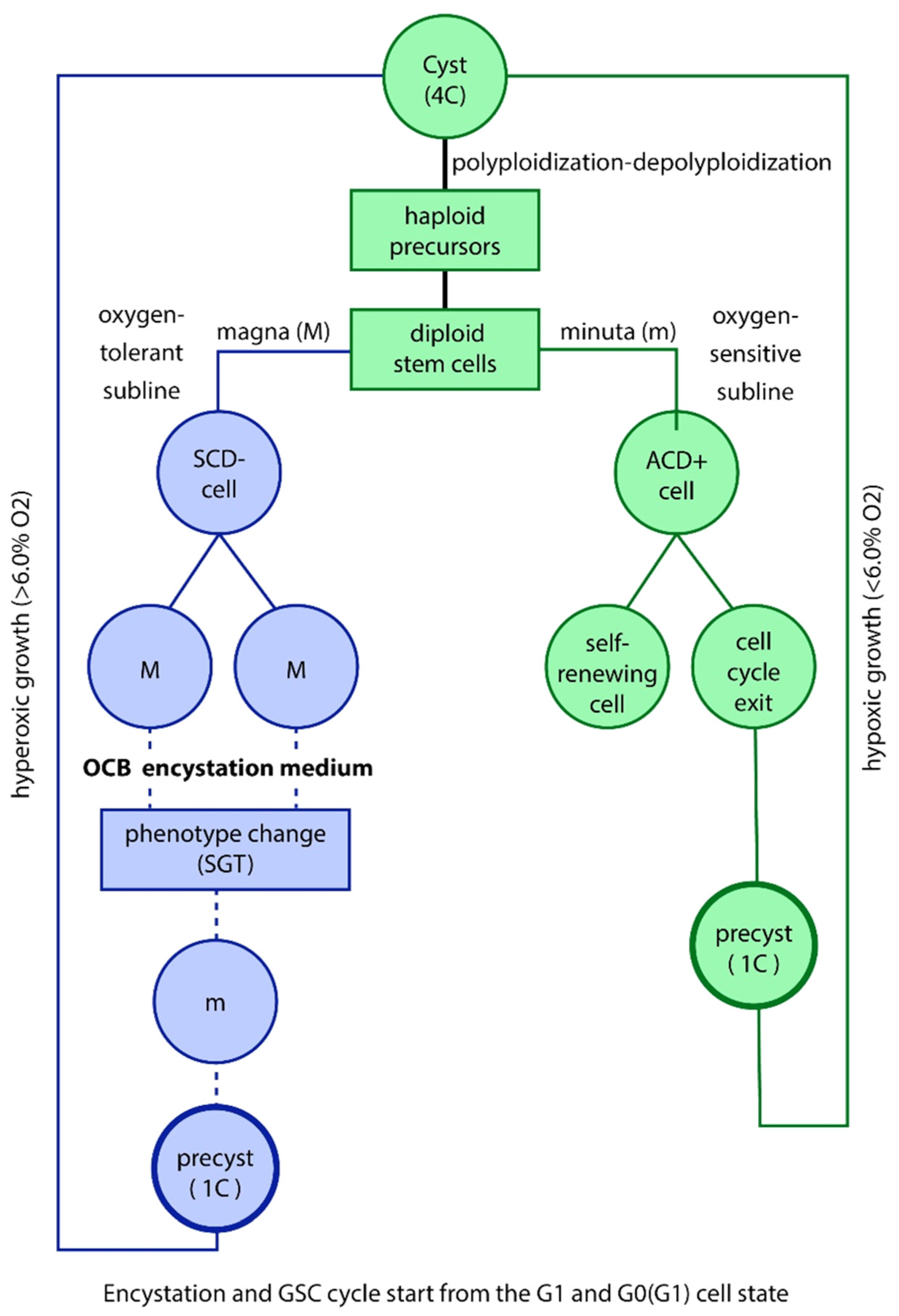
Figure 2.
Functional “cystic polyploidy“ through polyploidization- depolyploidization cycles forming in Entamoeba histolytica / E. invadens 4 polyploid nuclei, which give rise during ex-cystation to 16 haploid stem cell progenitors, and in E.coli to 8 polyploid nuclei, 16 diploid daughter cells and 32 stem cell progenitors. It is an ancestral program of genome multiplication and formation of multiple stem cell progenitor cells through the functional ACD phenotype. In cancer, this ancestral amplification program serve for the generation of A-CSCs and their precursor cells. (From V.F. Niculescu, Cancer genes and cancer stem cells in tumorigenesis: Evolutionary deep homology and controversies, Genes & Diseases, https://doi.org/10.1016/j.gendis.2022.03.010 (CC BY-NC-ND license) (11).
Figure 2.
Functional “cystic polyploidy“ through polyploidization- depolyploidization cycles forming in Entamoeba histolytica / E. invadens 4 polyploid nuclei, which give rise during ex-cystation to 16 haploid stem cell progenitors, and in E.coli to 8 polyploid nuclei, 16 diploid daughter cells and 32 stem cell progenitors. It is an ancestral program of genome multiplication and formation of multiple stem cell progenitor cells through the functional ACD phenotype. In cancer, this ancestral amplification program serve for the generation of A-CSCs and their precursor cells. (From V.F. Niculescu, Cancer genes and cancer stem cells in tumorigenesis: Evolutionary deep homology and controversies, Genes & Diseases, https://doi.org/10.1016/j.gendis.2022.03.010 (CC BY-NC-ND license) (11).
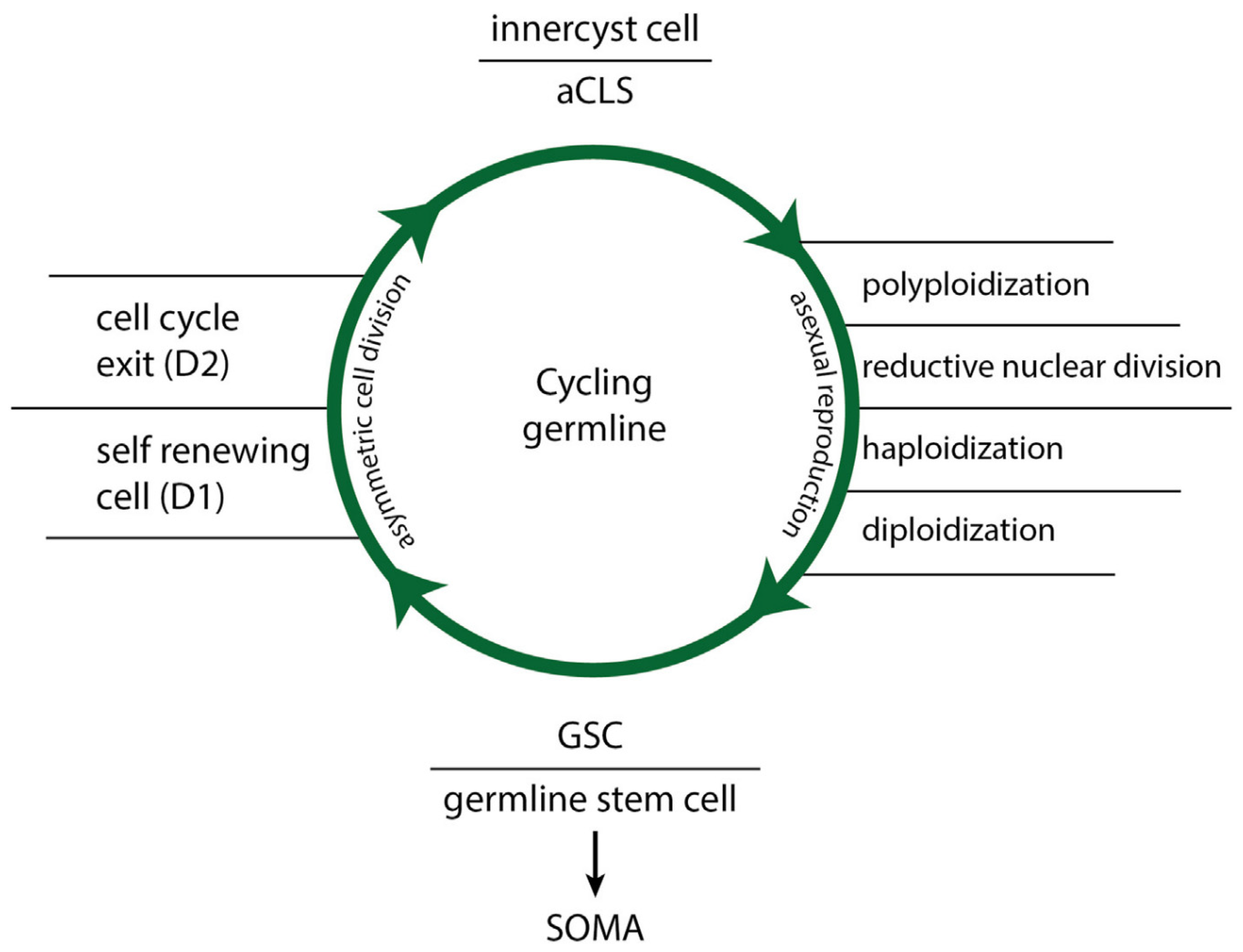
Figure 3.
The evolutionary life cycle of stemness as develops in carcinogenesis and tumorigenesis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



