
Submitted:
20 March 2024
Posted:
20 March 2024
You are already at the latest version
Abstract
Visual exploration of complex data helps interpretation, especially in the case of omics data analysis in life sciences and clinical research. Generating and analysing omics data requires bioinformatic skills, while they are interpreted by domain experts, for whom parsing large and complex data structures may be challenging. However, outcomes of visual analytics are often difficult to quantify, which emphasises precision and reproducibility, especially for research on human diseases. Here, we propose a workflow combining a reproducible computational environment with a dedicated visualisation functionality for systems biology diagrams. By linking the Galaxy with the MINERVA Platform, we visualise and explore COVID-19 transcriptomic data to demonstrate the utility of our workflow. Visualised data recapitulate findings of the original publication and offer new insights about the COVID-19 pathology. Our results offer a blueprint for quick prototyping of computational workflows that facilitate communication and exploration of complex data in biomedical research.
Keywords:
Galaxy workflows
; disease maps
; visual exploration
; systems biomedicine
1. Introduction
Visual exploration is a useful approach to gain insight into complex molecular data. ‘Omics’ readouts usually produce large sets of data with several variables that complicate their interpretation by domain experts. However, such an interpretation is necessary to understand what the results mean. Which again is needed to generate new hypotheses, design further experiments, or, for instance in the case of clinical research, develop diagnostic or treatment strategies.
There are many approaches to visualise large-scale omics data. Clustering and heatmap-based solutions are data-oriented, repartitioning similar values to discover meaningful patterns [1]. Data interpretation can be further supported by their integration into graph-structured representations of evidence-based molecular interactions [PMID:35880747]. These networks, created based on prior knowledge from interaction databases [2], enable the inference of causal relationships between differentially regulated molecules. Pathway databases [2] or disease maps [3] are particularly useful in this task, as they provide molecular networks with diagrammatic representations and functional or spatial modularization. Data from ‘omics’ analyses can be directly displayed on such diagrams [4] or combined with network analysis to infer regulations across multiple biological levels [5].
Standardised data processing pipelines are essential to ensure the reproducibility of results and coherence of their visual exploration. Standardisation is particularly important when processing omics data, whose complex pipelines consist of different tools with specific parameters [PMID: 26738481]. Moreover, such ‘omics’ tools constantly evolve as a response to technological advances that enable greater quantities and resolution of data. Consequently, it is necessary to properly document workflows used in ‘omics’ data processing and to ensure that their repeated execution yields stable and reliable results. To address this challenge, workflow environments and web-based analytical platforms are developed, allowing to encode and reproduce computational pipelines [6]. Galaxy is an example of such a platform, supported by a large bioinformatics community and featuring a versatile set of tools that can be combined into a functional workflow [7]. Galaxy allows the integration of generic software tools developed in common languages like R and Python, this also allows easy deployment of tools available in Galaxy on other platforms or uptake from there.
In this work, we combine a reproducible Galaxy workflow for transcriptomic data analysis with a streamlined visualisation of the generated molecular profiles. Results of the workflow are projected on a disease map, hosted on the MINERVA Platform [8]. This allows the user to create a reproducible ‘omics’ pipeline coupled with visual exploration and analytics in diagrams illustrating concrete molecular mechanisms. To demonstrate our work, we present a Galaxy workflow analysing SARS-CoV-2 RNASeq dataset [9], which is then visualised and interpreted using the COVID-19 Disease Map resource [10]. The workflows and visualisation use and interpretation are discussed in an associated tutorial created in the Galaxy Training Network which will help a researcher unfamiliar with Galaxy run the workflow [11].
2. Results
2.1. Reproducible Visualisation Workflow
We developed a workflow enabling reproducible visual analysis of ‘omics’ data by coupling the environment of the Galaxy server, specialising in encoding bioinformatic workflows, with the MINERVA Platform that interactively visualises large diagrams of molecular pathways. A key workflow component is a MINERVA plugin that is launched from within the Galaxy environment and dynamically visualises data on a MINERVA project. The workflow is illustrated in Figure 1, and described in detail below. To demonstrate the utility of the workflow, we used a publicly available bulk RNASeq dataset of blood samples of severe cases of COVID-19 [9] and projected the processed data on the COVID-19 Disease Map [10].
The Galaxy workflow was divided into two parts as indicated in Figure 1, with the first part handling the processing of RNASeq data, and the second part focused on identifying differentially expressed genes between the conditions (see Methods for details). In the second step, read count data are aggregated, and genes are annotated with NCBI gene and HGNC identifiers. Next, the Voom method of the Limma package calculates the log2-fold change of gene expression and the associated statistical significance.
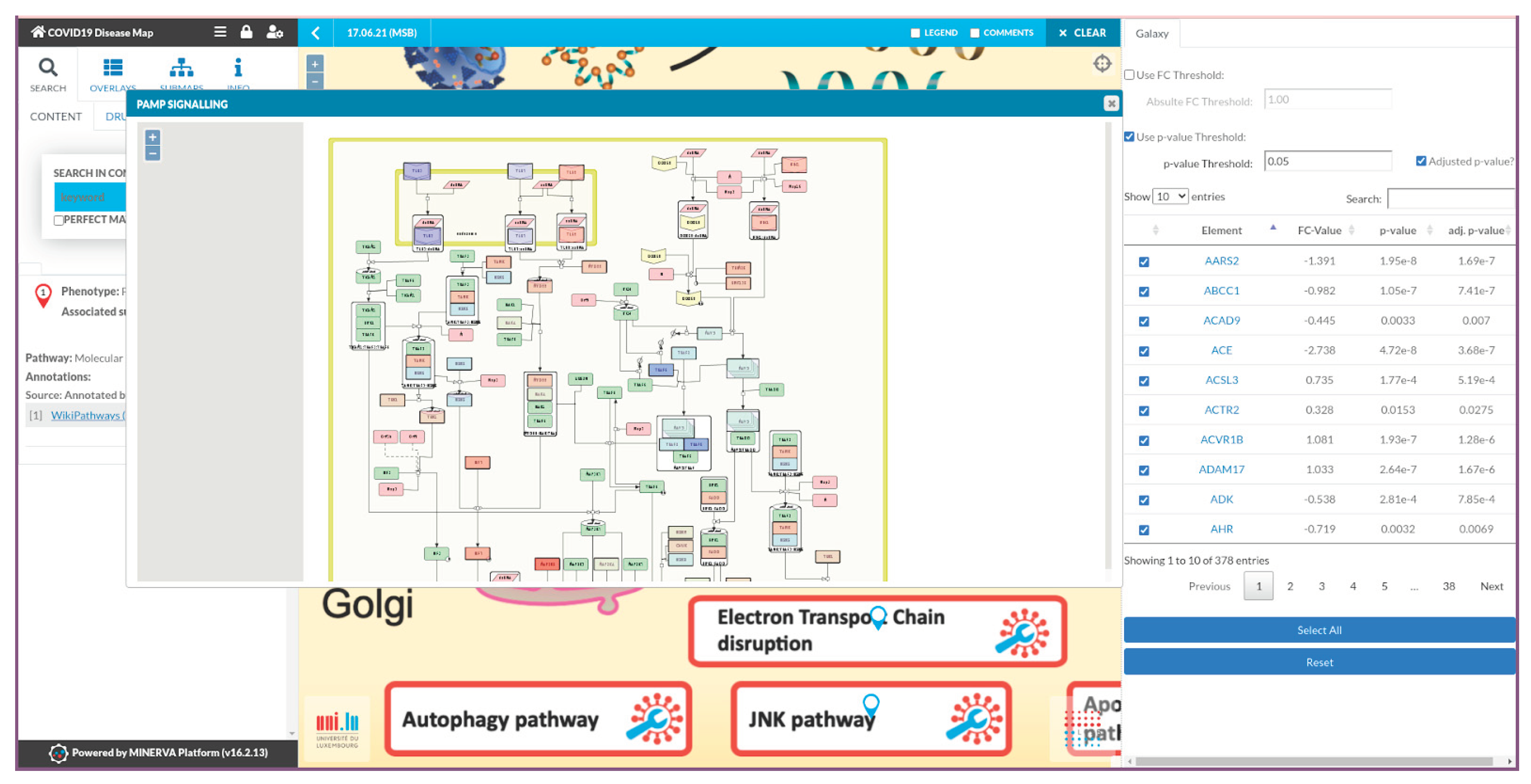
The final step of the workflow is the visualisation of RNASeq differentially expressed genes in the COVID-19 Disease Map for functional interpretation. The Galaxy workflow compiles a table with the HGNC symbol, log-fold change, and raw and adjusted p-values and parses the data to the MINERVA plugin. In the COVID-19 Disease Map, HGNC symbols are mapped to human genes for their visual exploration. Users can filter the data in the plugin interface to focus on the most significant or differentially expressed genes (Figure 2). The significantly differentially expressed genes are also used for enrichment analysis with the Gene Ontology terms from the Biological Process branch using GOSeq, aiming for an unbiased interpretation of the expression data.
2.2. Interpretation of Visualised Data
We processed and visualised a publicly available bulk RNASeq dataset of blood samples of severely ill COVID-19 patients [9]. We focused primarily on the interpretation of mRNA expression data. Differentially expressed genes were filtered using the plugin to only those with adjusted p-value < 0.05, and with the absolute log2-fold change of 1.0 and above. The resulting 154 entries were mapped to the COVID-19 Disease Map, as shown in Figure 1.
The visualisation confirmed previous results, with interferon-related pathways being prominently populated with differentially expressed genes [9]. An advantage was the visualisation of detailed expression patterns, leading to hypotheses about relationships between differentially expressed genes. For instance, TLR3 and TLR7 receptors are downregulated, while MYD88 gene downstream of TLR9 is upregulated, suggesting the recognition of viral particles as a potential signalling route. Moreover, STAT1, an interactor of a number of viral proteins, is prominently upregulated together with IRF9. These findings allow hypothesis building about activation of a particular pathway, and potential nuances of its signalling cascades.
Moreover, visual analysis revealed that other hallmark pathways are activated for these selected differentially expressed genes, including viral replication cycle, coagulation cascade, renin-angiotensin pathway, and pyrimidine deprivation. These observations can be made in a dedicated set of COVID-19 pathways, while they could be overlooked in a usual overexpression analysis, where such findings may be hidden in a long list of potentially significant pathways as expected during general infectious disease progression.
3. Methods
Galaxy
UseGalaxy.eu running Galaxy 23.1.5 was used for the development of the workflows and Galaxy features. As part of this work, we developed an external display application plugin [12] within Galaxy to enable an analyst to load a dataset directly into the MINERVA platform. This external display application plugin is now available for everyone by default with the latest release of the Galaxy platform (23.2) (github.com/galaxyproject/galaxy/pull/11880). This plugin provides one-click visualisation with MINERVA for any tabular dataset annotated as being from a human genome within Galaxy.
Workflows
The workflows are split into two portions, the first providing Quality Control and generating Counts, the second processing those Counts files for differential expression. Data: the primary input is a dataset collection of FASTQ files from the RNASeq experimental data; this can be most easily obtained via fasterq-dump (SRA Toolkit v3.0.8) and the two-column sample table with SRA identifiers and study conditions. Counts: The counts workflow processes the collection with FastQC (v0.74) before trimming with Cutadapt (v4.4). These reads are then checked for quality again with FastQC, before alignment with HISAT2 (v2.2.1) using default parameters, and counting via featureCounts (v2.0.3). Simultaneously the read_distribution.py and geneBody_coverage.py (RSeQC v5.0.1) process the outputs of HISAT2 to generate additional reports. All of these are aggregated with MultiQC (v1.11) to generate a full report for the analyst. Enrichment Analysis: in the second workflow, these count files are aggregated via join (GNU Coreutils v8.25) before their annotation with annotateMyIds using org.hs.eg.db (bioconductor release 3.16) to add NCBI Gene ID (Entrez), symbol, and gene name columns. After reformatting the count tables to merge them into a count matrix this dataset and the original factor table are analysed with Limma (v3.58.1) using the Voom method. The differential expression results are then processed for their use in goseq (v1.50.0) via the GNU Coreutils v8.25 suite and assorted Perl and Python scripts to process columnar data. Both of these workflows are made available on WorkflowHub (https://workflowhub.eu/workflows/688 and https://workflowhub.eu/workflows/689) to enable precise reproduction of our methods.
The visualisation plugin for the MINERVA Platform
We developed a plugin for the MINERVA Platform that automatically fetches data from Galaxy and provides an intuitive visualisation of disease-specific pathways. The MINERVA Platform provides an extensive JavaScript API, enabling plugins developed in JavaScript to communicate with the disease map by fetching map contents and highlighting map components with colour-coded overlays. On plugin startup, the content of the disease map is fetched through the MINERVA API, collecting HGNC symbols, NCBI Gene IDs (Entrez), and UniProt IDs of MINERVA objects representing map elements. The plugin is loaded automatically on any disease map by providing a hash representation of the plugin in the disease map URL. This way, the output from Galaxy was fully automated, without requiring users to manually browse the disease map and find out how to load overlays. The data source, i.e. the data repository on Galaxy, is additionally provided as an URL parameter “datasource” containing the respective data’s URL string. The file is loaded and read by the plugin, mapping MINERVA objects based on the mapping type defined by the first column’s header “identifier_hgnc_symbol”, “identifier_entrez”, or “identifier_uniprot”. The entries are then displayed in a table, showing the HGNC gene symbol (independent of the mapping), log2 fold change (FC) values, p-values, and adjusted p-values. The gene icons are interactive and link to the position of the associated gene or gene product on the map. UI elements allow users to filter genes by setting the significance threshold (0.05 by default) for either the unadjusted or adjusted p-value and setting an absolute log2 FC threshold. Any changes automatically update the data table. Each row in the table is associated with a checkbox, which, upon selection, highlights the element on the map with a colour fitting the log2 FC value on a scale from blue (negative values) to red (positive values). Additional buttons allow users to automatically select all filtered elements in the table or reset the visualisation. The coloured overlays are generated through the MINERVA API by first normalising all log2 FC values by the highest absolute values, and then mapping the normalised values to respective string hex colour codes on the scale from white (0) to red (1) or blue (-1).
Tutorial
Both the Galaxy workflow, and MINERVA are best utilised with a thorough understanding of the materials and theory behind it, and with guidance and explanation. As such a tutorial was produced to introduce researchers to both the workflows’ analyses, and use of the MINERVA interface, and published in the Galaxy Training Network (GTN) under the topic “Transcriptomics”. This tutorial enables researchers to launch the workflow directly in their preferred Galaxy instance, and guides them through the correct configuration of the workflow, as well as discussion of its contents and analysis methods. The tutorial is available online: http://training.galaxyproject.org/training-material/topics/transcriptomics/tutorials/minerva-pathways/tutorial.html
4. Discussion
Here, we introduced a workflow for reproducible analysis and visualisation of RNASeq data, combining the Galaxy and MINERVA platforms. In Galaxy, we carried out data analysis and basic data interpretation tasks. The results - differentially expressed genes - were supplied to the MINERVA Platform using a dedicated connecting plugin.
Differentially expressed genes calculated by the Galaxy workflow and visualised in the COVID-19 Disease Map recapitulated the findings of the study by Togami et al [9], which we considered as our use-case. Marked de-regulation of the interferon pathways, both IFN-1 and IFN-lambda, was indicated in the Map. In some cases, our results were different from those reported by Togami and colleagues, including a pathway related to cancer immunotherapy. Such a result or other disease-related artefacts are expected from an unbiased gene expression analysis, which in turn offers a broader range of results. In the case of the COVID-19 Disease Map, only pathways related to SARS-CoV-2 infection were visible. This focused exploration identified a number of affected relevant pathways, together with a detailed pattern of their activation. Importantly, this improved focus offered by the Map does not give insight into potential novel pathways suggested by the data. To address this shortcoming, our workflow implements enrichment analysis of a larger pathway set, complementing this shortcoming.
Complex analytical workflows are inherently difficult to reproduce, given different tools that have to be combined together, and the challenges of the computational environment. On top, visual exploration of resulting complex datasets is challenging to reproduce, as it involves manual and individual parsing of the content to visualise. Our work addresses these two aspects by i) proposing the Galaxy platform as the environment to encode and execute the workflow, and ii) developing a seamless connector between the results produced by Galaxy and an online repository of molecular interaction diagrams. This reduces the effort required by setup of a computational pipeline, and guarantees the same starting point for visual exploration of the results, with clearly defined controls and a possibility of further automation [13].
Reproducibility and automation in exploration of complex data is needed especially when preparing for future pandemics. The research on COVID-19 delivered a massive amount of molecular data, which are to this day challenging to mobilise, harmonise and interpret. Our capability to generate data was much higher than the ability to fully benefit from them. In this light, it is important to develop reproducible workflows that allow domain experts to explore data that would otherwise require advanced bioinformatic skills. An interdisciplinary environment is a necessary prerequisite for such a goal, which was made possible during the virtual BioHackathon Europe organised and funded by the ELIXIR Hub in November 2020 (https://2020.biohackathon-europe.org). We designed and prototyped our workflow during that productive event, focused on challenges of the COVID-19 pandemic.
Our work faces certain limitations. First, we focus on a single dataset in the COVID-19. This limitation can be addressed by re-using the workflow and by adapting its parameters to new inputs, using detailed documentation and deposition in the WorkflowHub repository. Second, the interpretation of the biological findings related to the dataset is rudimentary. Detailed comparison will require in-depth interpretation supported by relevant articles, which we considered to be out of scope of this article.
Acknowledgments
This work was conceptualised and prototyped during the virtual BioHackathon Europe, organised and funded by the ELIXIR Hub in November 2020. We thank the organisers for an opportunity to participate in such a productive and collaborative event. This work was funded as a part of the BY-COVID project (Grant agreement ID: 101046203). MK and CE received funding from the ZonMw COVID-19 programme (Grant No. 10430012010015).
References
- Engle S, Whalen S, Joshi A, Pollard KS. Unboxing cluster heatmaps. BMC Bioinformatics. 2017;18:63.
- Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022;50:D687–92.
- Mazein A, Ostaszewski M, Kuperstein I, Watterson S, Le Novère N, Lefaudeux D, et al. Systems medicine disease maps: community-driven comprehensive representation of disease mechanisms. NPJ Syst Biol Appl. 2018;4:21.
- Sidiropoulos K, Viteri G, Sevilla C, Jupe S, Webber M, Orlic-Milacic M, et al. Reactome enhanced pathway visualization. Bioinforma Oxf Engl. 2017;33:3461–7.
- Hoch M, Smita S, Cesnulevicius K, Lescheid D, Schultz M, Wolkenhauer O, et al. Network- and enrichment-based inference of phenotypes and targets from large-scale disease maps. NPJ Syst Biol Appl. 2022;8:13.
- Wratten L, Wilm A, Göke J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat Methods. 2021;18:1161–8.
- Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022;50:W345–51.
- Hoksza D, Gawron P, Ostaszewski M, Smula E, Schneider R. MINERVA API and plugins: opening molecular network analysis and visualization to the community. Bioinforma Oxf Engl. 2019;35:4496–8.
- Togami Y, Matsumoto H, Yoshimura J, Matsubara T, Ebihara T, Matsuura H, et al. Significance of interferon signaling based on mRNA-microRNA integration and plasma protein analyses in critically ill COVID-19 patients. Mol Ther Nucleic Acids. 2022;29:343–53.
- Ostaszewski M, Niarakis A, Mazein A, Kuperstein I, Phair R, Orta-Resendiz A, et al. COVID19 Disease Map, a computational knowledge repository of virus-host interaction mechanisms. Mol Syst Biol. 2021;17:e10387.
- Hiltemann S, Rasche H, Gladman S, Hotz H-R, Larivière D, Blankenberg D, et al. Galaxy Training: A powerful framework for teaching! Ouellette F, editor. PLOS Comput Biol. 2023;19:e1010752.
- Blankenberg D, Chilton J, Coraor N. Galaxy External Display Applications: closing a dataflow interoperability loop. Nat Methods. 2020;17:123–4.
- Gawron P, Smula E, Schneider R, Ostaszewski M. Exploration and comparison of molecular mechanisms across diseases using MINERVA Net. Protein Sci [Internet]. 2023 [cited 2023 Feb 8];32. Available from: https://onlinelibrary.wiley.com/doi/10.1002/pro. 4565.
Figure 1.
Reproducible visualisation workflow. The figure illustrates the steps of the workflow, and its outputs used in data interpretation. The Galaxy portion involves i) data processing steps such as quality control, alignment and counting, ii) calculation of differentially expressed genes (Limma), iii) interpretation using Gene Ontology overrepresentation analysis (GOSeq) and iv) making the data available via the MINERVA platform plugin. The MINERVA Platform part involves visualisation of the expressed genes and their interactive filtering by available criteria, including statistical significance.
Figure 1.
Reproducible visualisation workflow. The figure illustrates the steps of the workflow, and its outputs used in data interpretation. The Galaxy portion involves i) data processing steps such as quality control, alignment and counting, ii) calculation of differentially expressed genes (Limma), iii) interpretation using Gene Ontology overrepresentation analysis (GOSeq) and iv) making the data available via the MINERVA platform plugin. The MINERVA Platform part involves visualisation of the expressed genes and their interactive filtering by available criteria, including statistical significance.
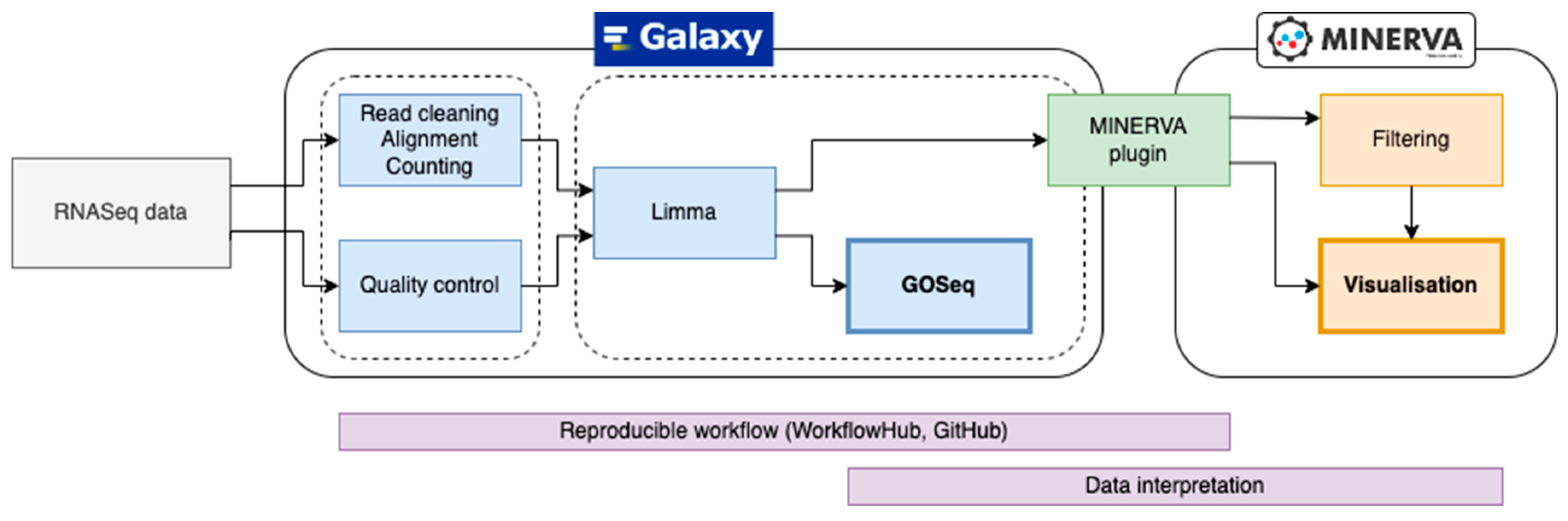
Figure 2.
Visualisation of the workflow results. The results of the RNASeq differential expression analysis, projected on the COVID-19 Disease Map. The MINERVA platform plugin allows filtering of the list of differentially expressed genes by significance and log-fold change values.
Figure 2.
Visualisation of the workflow results. The results of the RNASeq differential expression analysis, projected on the COVID-19 Disease Map. The MINERVA platform plugin allows filtering of the list of differentially expressed genes by significance and log-fold change values.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



