
Submitted:
19 March 2024
Posted:
21 March 2024
You are already at the latest version
Abstract
Cervical cancer is caused by a persistent and high-grade infection, caused by the Human Papilloma Virus (HPV), which, when entering cervical cells, alters their physiology and generates serious lesions. HPV 18 - is among those most involved in carcinogenesis in this region, but there are still no drug treatments that cause cure or total remission of lesions caused by HPV. Knowing that L-Asparaginase is an amidohydrolase, which plays a significant role in the pharmaceutical industry, particularly in the treatment of specific cancers, due to its antitumor properties, some studies have demonstrated its cytotoxic effect against cervical cancer cells. However, the commercial version of this enzyme has side effects, such as hypersensitivity, allergic reactions and silent inactivation due to the formation of antibodies. To mitigate these adverse effects, several alternatives have been explored, including the use of L-asparaginase from other microbiological sources, which is the case with the use of the fungus Aspergillus niger, a high producer of L-asparaginase. The study investigated the influence of the type of fermentation, precipitant, purification, characterization and in vitro cytotoxicity of L-asparaginase. The results revealed that semisolid fermentation produced higher enzymatic activity and protein concentration of A. niger. The characterized enzyme showed excellent stability at pH 9.0, temperature of 50ºC, resistance to surfactants and metallic ions, and an increase in enzymatic activity was also noted with the organic solvent ethanol. Furthermore, it exhibited low cytotoxicity in GM and RAW cells, and significant cytotoxicity in HeLa cells. These findings indicate that L-asparaginase derived from Aspergillus niger may be a promising alternative for pharmaceutical production. Its attributes, including stability, activity and low toxicity in healthy cells, suggest that this modified enzyme could overcome challenges associated with antitumor therapy.
Keywords:
Câncer cervical
; L-asparaginase
; Aspergillus niger
1. Introduction
Cervical cancer is caused by a persistent and high-grade infection, which can be caused by the Human Papilloma Virus (HPV), which, when entering cervical cells, alters their physiology and generates serious lesions [1]. HPV 18 is among those most involved in carcinogenesis in this region [2], but there are still no drug treatments that cause cure or total remission of lesions caused by HPV [3].
Knowing that L-Asparaginase is an amidohydrolase [4], which plays a significant role in the pharmaceutical industry, particularly in the treatment of specific cancers, due to its antitumor properties, some studies have demonstrated its cytotoxic effect against HeLa cells [5]. This enzyme is industrially produced and commercially available, constituting 40% of global enzyme demand, representing US$380 million in sales in 2017. This number is predicted to increase to US$420 million by 2025 [6]. L-Asparaginase, originating from bacteria, is available in three formulations, two from Escherichia coli and one from Erwinia chrysanthemi [7].
Despite its therapeutic applications, commercially available L-asparaginase has side effects such as hypersensitivity, allergic reactions, silent inactivation due to the formation of anti-asparaginase antibodies, and a short serum half-life [8]. These problems arise in part from their microbiological origin and strong immunological response [9]. To minimize adverse effects, several alternatives have been explored, including L-asparaginase from different sources, protein engineering and enzyme immobilization [10].
Aspergillus niger, a vital industrial fermentation strain, is crucial in biotechnology. Its metabolic compounds are widely used in the fermentation of animal feed, food additives, industrial enzyme preparations and biotransformation [11,12,13,14,15]. Due to its ability to produce enzymes in high concentrations and beneficial pharmaceutical supplies, A. niger is a focus of biotechnological research [16,17].
Given these considerations, the search for a new, highly efficient and stable source of L-asparaginase for pharmaceutical and biotechnological applications continues. This study employed strategies to optimize the production of L-asparaginase through fermentation using A. niger, aiming to explore its catalytic properties, to achieve a stable enzyme with high activity and efficiency, testing its antitumor activity in HeLa cells.
2. Materials and Methods
2.1. Materials
Agar Potato Dextrose (PDA) - Merck KGaA, Brazil, Tris/Borate/EDTA (TBE) - Ludwig Biotecnologia, Methanol, and Acetone purchased from Vetec Brazil, Isopropanol, and Sodium Hydroxide acquired from Quimex Brazil, Aluminum Sulfate (AlSO4), Barium Chloride (BaCl2), Copper Sulfate (CuSO4), Calcium Chloride (CaCl2), Magnesium Chloride (MgCl2), Iron Sulfate (FeSO4), Potassium Chloride (KCl), Zinc Sulfate (ZnSO4), Iron Chloride (FeCl), Manganese Chloride (MnCl2), Peptone, Citric Acid, Potassium Phosphate, Tween 20 and 80 were procured from ISOFAR Brazil, Dipotassium Phosphate, L-asparagine, Hydrochloric Acid (HCl), Triton-X100, Tris(hydroxymethyl)aminomethane (TRIS), Sodium Dodecyl Sulfate (SDS), Ammonium Sulfate ((NH₄)₂SO₄), Ethylenediaminetetraacetic Acid (EDTA), Sodium Chloride (NaCl), Human Fibroblast (GM), and Macrophage (RAW 264.7) cell lines were purchased from Sigma-Aldrich Brazil. Human cancer cells HeLa (cervix adenocarcinoma; ATCC CCL-2).
2.2. Comparative Study of Different Fungi in the Production of L-Asparaginase
With the aim of identifying potential sources of production and high enzymatic activity of L-asparaginase, the enzymatic activities of crude extracts produced by Aspergillus niger, Aspergillus flavus, Curvularia, Fusarium solani, Fusarium oxysporum, Penicillium decumbens and Rhizopus sp. All microorganisms in the study were obtained from the mycotheque at the Federal University of Maranhão, São Luís, Brazil. The production of L-asparaginanse in these microorganisms was investigated through two types of fermentation: solid (SsF) and submerged (SmF).
2.2.1. Solid-State Fermentation (SsF) of L-Asparaginase
For the production of L-asparaginase using SsF technique, the methodology described by Dias et al. [21] was employed with some adaptations. For each microorganism, a mixture of 50 g of wheat bran with 20 mL of 100 mMol/L-1 L-asparagine solution diluted in 50 mM Tris-HCl buffer at pH 8.0 was prepared. The mixture was sterilized, and approximately 5 fragments of the microorganism culture medium, each about 5 mm2, were added. Fermentation was maintained at a temperature of 30°C for 120 hours.
After the fermentation period, the cultures were treated with 50 mL of 50 mMol/L-1 Tris-HCl buffer at pH 8.0. Agitation was carried out at 190 rpm at a temperature of 25°C for 60 minutes. Subsequently, the cultures were filtered and centrifuged at 14,000 rpm at 4°C, and the supernatant was considered as the crude extract of SsF.
2.2.2. Submerged Fermentation (SmF) of L-Asparaginase
For SmF, the methodology described by Mahajan et al. [22] was used with adaptations. Flasks containing 50 mMol/L-1 Tris-HCl buffer at pH 8.0 were supplemented with NH4NO3 (2 g/L), KH2PO4 (1.52 g/L), KCl (0.52 g/L), MgSO4 (0.52 g/L), CuSO4 (0.001 g/L), ZnSO4 (0.001 g/L), FeSO4 (0.001 g/L), and L-asparagine (10 g/L). Approximately 5 fragments of the culture medium, each about 5 mm2, were added. The liquid media were kept under orbital agitation at 190 rpm and a temperature of 30°C for 7 days. Samples were collected every 24 hours to determine the time of maximum enzymatic activity. The collected samples were filtered, centrifuged at 10,000 rpm at 4°C for 25 minutes, and the supernatant was considered as the crude extract of SmF.
2.3. Fungal Culture and Isolation
The environmental sample of the filamentous fungus used for these analyses, previously identified as belonging to the species Aspergillus niger, was obtained from the mycoteca ( at the Federal University of Maranhão, São Luís, Maranhão, Brazil. Fungal cultivation was carried out in Petri dishes of 4% Sabouraud Dextrose culture medium (MERCK) and incubated in a bacteriological oven (SOLAB SL-101) at 37 ºC for 5 days. All cultivation and subculture procedures took place in a laminar flow hood close to the Bunsen burner.
2.3.1. DNA Extraction
The biochemical DNA extraction protocol was used following the methodology of Valenzuela-Lopez et al. [18] with the addition of glass spherules and adaptations to perform cell lysis of the chitin wall. Subsequently, the quality of the DNA was checked, followed by DNA quantification and purity using a Nanodrop One C spectrophotometer (Thermo Scientific). The samples contained between 100-200 ƞg/µl and were sent for amplification through polymerase chain reaction.
2.3.2. Primers and Polymerase Chain Reaction (PCR)
For molecular analysis, species-specific primers (Thermofisher-Scientific) of the internal transcribed spacer region (ITS) and ITS1-5.8S-ITS2 fragments were used [19]. The nucleotide sequences used were ITS 1 (5'- GCTCATTAAATCAGTTATCG-3') and ITS 2 (5'- GTTATTATGATTCACCAAGG-3' -3') according to Alabdalall et al. [20].
Polymerase chain reaction (PCR) of the regions had a final volume of 25ul, using a MasterMix PCR Set (Ludwig Biotechnology) containing Tris-KCl, pH 8.4; 2.0 mM MgCl2; 0.2 mM DNTP mix and 2.5 U of Taq DNA polymerase; 12.5 µl of Pre-Mix; 6.5 µl of ultra pure water; 2.5 µl of ITS1-5.8S-ITS2 primers (Termofisher Invitrogen 10 pmol mL-1) 1.0 µl DNA (5 ng mL-1) and following the following thermocycling conditions in the Biocycler MJ96G Thermocycling equipment.
The description of the PCR cycles performed for A. niger samples were 95ºC for 8 min, 34 cycles 94ºC for 1 min, 57ºC for 1 min and 72ºC for 1 min and then 72ºC for 7 min for extension and finishing at 4 ºC. The amplicons were subjected to electrophoresis in 1.5% agarose gel at 90 V for 120 minutes in TBE 1X (Ludwig Biotechnology) and molecular marker (Ladder 100 bp – 0.1 µg/µl-Ludwig Biotechnology). The products were evaluated for quality using a transluminator (Loccus L-PIX TOUCH) (Vilber Lourmat ECX-F20.M).
2.4. Purification of Crude L-Asparaginase Extracts
The crude extracts from SmF and SsF were precipitated according to the methodology of Vala et al. [23], adapted for different fractions of L-asparaginase: (0 - 40%) and (40 - 80%) ethanol, isopropanol, and ammonium sulfate, aiming to minimize significant protein losses and determine the most suitable precipitation method to optimize L-asparaginase extraction. The precipitants were added to the crude L-asparaginase extracts, which were kept at 4°C for 24 hours with gentle agitation, and then centrifuged at 14,000 rpm for 25 minutes at 4°C.
The precipitates were resuspended in a volume corresponding to 1/4 of the total volume of the centrifuged extract, using 50 mMol/L-1 Tris-HCl buffer at pH 7.0. Subsequently, dialysis was performed; the suspension was dialyzed against the same buffer used for crude extract preparation at 4°C for 6 hours, with two buffer changes every 3 hours, with the last change kept overnight. After dialysis, the precipitates were centrifuged at 10,000 rpm for 25 minutes at 4°C and resuspended in the same buffer used for extract preparation. They were then stored at -34°C for subsequent studies. These fractions were classified as L-asparaginase F1 (fraction 0 – 40%) and L-asparaginase F2 (fraction 40 – 80%). Fraction purifications were performed using ion exchange chromatography, utilizing a diethylaminoethyl cellulose (DEAE cellulose) column.
2.5. Protein Concentration
Protein concentration was estimated using the method described by Warburg & Christian [24] with a spectrophotometer at absorbances of 260 nm and 280 nm, using the following formula:
2.6. Determination of L-Asparaginase Activity
L-Asparaginase activity was assessed by the formation of β-hydroxamate aspartic acid from asparagine and hydroxylamine, following the method described by Drainas et al. [25]. The reaction mixture consisted of 300 μL of 20 mmol L-1 Tris-HCl buffer at pH 8.0, 100 μL of 100 mmol L-1 L-asparagine, 100 μL of 1M hydroxylamine, and 500 μL of the enzyme sample. The mixture was incubated in a water bath at 37°C for 15 minutes, and the reaction was stopped by adding 250 μL of a solution containing HCl (2.4%), ferric chloride (10%), and TCA (5%).
The enzymatic reaction between β-hydroxamate aspartic acid and ferric chloride produced a reddish color, and its absorbance was measured at 500 nm using a spectrophotometer. An analytical curve was constructed using a solution of β-hydroxamate aspartic acid (0.1 μmol/mL to 3 μmol/mL). One unit of L-asparaginase activity (U) was defined as the amount of enzyme required to form one μmol of β-hydroxamate aspartic acid per minute under the assay conditions. The specific activity of L-asparaginase was expressed as μmol of β-hydroxamate aspartic acid formed per minute per milligram of protein.
2.7. Biochemical Characterization of Purified L-Asparaginase from Fungus Extract
2.7.1. Fungo Optimal pH and Stability
Different pH ranges from 3.0 to 9.0 were tested using 0.1M acetate buffer, 0.1M Tris-HCl, and 0.1M citrate-phosphate solutions. For stability, the reaction at each pH was maintained for 1 hour at 37°C, and the tests were performed in triplicate.
2.7.2. Optimal Temperature and Stability
To determine the optimal temperature, L-asparaginase solution was incubated for 30 minutes at temperatures ranging from 10 to 90 ºC. For stability, the enzyme was kept for 1 hour at the same temperature ranges. The tests were performed in triplicate.
2.7.3. Effect of Surfactants and Metal Ions in Salts
To evaluate stability against different surfactants and ions, the method adapted from Vala et al. [23] was used. L-asparaginase was incubated for 1 hour with different proportions of non-ionic surfactants: Triton X-100, Tween-20, and Tween-80 (0.01%, 0.10%, and 0.50% w/v) and ionic surfactant sodium dodecyl sulfate (SDS). The effect of metal ions on activity was determined by pre-incubating L-asparaginase in 50 mM Tris-HCl buffer (pH 8.0) with 0.01 mM CoSO4, FeCl3, CaCO3, NaCl, MgCl2, MnSO4, and ZnSO4 at 35 °C for 1 hour. The tests were performed in triplicate, and residual activity was measured.
2.7.4. Stability in Organic Solvents
To determine the stability of A. niger L-asparaginase in acetone, ethanol, methanol, and isopropanol, the method adapted from Oliveira et al. [26] was used. The enzyme was incubated for 1 hour at 37°C in different solvent concentrations (25%, 50%, 80%, and 100% v/v). After incubation, residual activity was measured.
2.8. Cytotoxic Activity of L-Asparaginase
The cytotoxicity of the enzyme was evaluated using the methods described by El-Gendy et al. [27] with adaptations. L-Asparaginase was tested at concentrations ranging from 100 to 6.25 μg/mL-1 against cells derived from human fibroblasts (GM cells), macrophage cell line (RAW 264.7), and HeLa (human cervical cancer). Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as follows:
2.9. Morphological Viability in HeLa Cells by Inverted Light Microscopy
Cellular morphology was analyzed by inverted light microscopy (Optika Microscope, Eco, United States of America), after treatment with L-asparaginase at a concentration of 100 ug/mL. Image analysis was performed using Axiovision Release 4.8.1 software (Carl Zeiss Inc., Jena, Germany). Cells were cultured in 12-well plates in the presence and absence of L-asparaginase for 24, 48 and 72 hours and then observed under a microscope.
2.10. Statistical Analysis
The results of this study were analyzed using ANOVA and Tukey's post hoc test (p < 0.05) with GraphPad Prism 8.0.1 software. The results were reported as mean ± standard deviation (SD) of triplicate determinations.
3. Results
3.1. Tracking and Optimization of L-Asparaginase production by Fungi
3.1.1. Screening for L-Asparaginase Producers
The results indicated that, through the SsF process, the studied fungi exhibited higher L-asparaginase production, suggesting that this method was the most suitable for enzyme production. Among the tested species, Aspergillus niger demonstrated the highest activity, with 46.4 U/mL, compared to other fungal species: Aspergillus flavus (30.23 U/mL), Curvularia (23.41 U/mL), Fusarium solani (37.64 U/mL), Fusarium oxysporum (21.69 U/mL), Rhizopus sp. (11.75 U/mL), and Penicillium decumbens (24.09 U/mL) (Figure 1). Due to its higher enzymatic activity, Aspergillus niger's L-asparaginase was further studied in subsequent steps.
3.1.2. Fermentation Type for L-Asparaginase Production
In order to achieve high enzymatic activity and concentration of L-asparaginase produced by Aspergillus niger, the production of this enzyme was analyzed in two types of fermentation. SsF exhibited higher activity in all stages of purification. Consequently, L-asparaginase showed superior activity compared to SmF in the Fraction (0 – 40%) and Fraction (40 – 80%) stages, displaying specific activity (187.19 U/mg) with yields of 52.25% and purification of 17.08 in the Fraction (40 – 80%), respectively, compared to the SmF fractions (Table 1)
3.2. Molecular Identification of A. niger
After molecular analysis, amplification of the sample resulted in a band of approximately 295 bp (base pairs), which corroborates what is described in the literature for the species (Figure 2).
3.3. Precipitant Type for L-Asparaginase Purification
Since Solid-State Fermentation (SsF) exhibited higher specific activity of L-asparaginase and a higher protein concentration per milliliter of extract produced by A. niger, purification and optimization of the precipitation method were performed using different types of precipitants to determine which one enhances the concentration of L-asparaginase. The results showed that ethanol, when used as a protein precipitant to obtain L-asparaginase produced by A. niger, led to a higher concentration of L-asparaginase in the 40-80% fraction, with a higher specific activity of 34.35 U/mg, compared to isopropanol (12.33 U/mg) and ammonium sulfate (6.39 U/mg) used as precipitants (Table 2).
3.4. Biochemical Characterization of L-Asparaginase from A.niger
The biochemical characterization of L-asparaginase precipitated by ethanol was conducted to assess the stability of the enzyme's catalytic activity under various conditions, aiming to apply the enzyme in biochemical processes effectively.
3.4.1. Optimal pH and Stability
The enzymatic activity of L-asparaginase produced by A. niger was tested across the pH range of 3.0 to 9.0. The results indicated that the optimal pH for L-asparaginase activity was pH 9.0, with an enzymatic activity of 47.25 U/mL (Figure 3A). The enzyme exhibited activity above 80% in the pH range of 5.5 to 8.5 compared to its optimal pH. Additionally, the enzyme did not exhibit significant resistance to different pH ranges in this study, differing from the findings in previous research (Figure 3 A). In terms of pH stability (Figure 3 B), the L-asparaginase produced by A. niger demonstrated maximum activity at pH 9.0, consistent with its optimal pH.
3.4.2. Optimal Temperature and Stability
The L-asparaginase produced by A. niger exhibited maximum activity at 50°C and demonstrated resistance to other temperatures within the range of 30 to 45°C (Figure 4 A). Regarding temperature stability, the enzyme maintained its maximum activity up to 50°C, similar to its optimal temperature. Subsequently, there was a decline in activity, leading to complete loss of enzymatic activity, as depicted in Figure 4 B. These results highlight the enzyme's preference for higher temperatures but also indicate a limited stability range beyond its optimal temperature.
3.4.3. Effect of Surfactants and Metal Ions in Salts
The objective of this study was to understand the L-asparaginase produced by A. niger and identify compounds that might cause a decrease in enzymatic activity. Various surfactants and metal ions were tested to assess their impact on the enzyme's activity, crucial for its application in pharmaceutical compounds and biotechnological processes. The residual activity of A. niger produced L-asparaginase was examined concerning different surfactants. There was a significant increase in enzymatic activity with Tween 20, showing a 16.41% increase at a concentration of 0.10% and an 8.15% increase at a concentration of 0.50%. However, in the case of EDTA, enzymatic activity decreased at the higher concentration of 0.50%, with a residual activity of 40.29% (Table 3). These findings provide valuable insights into the enzyme's response to various surfactants and metal ions, aiding in its tailored application in specific contexts.
In the presence of ions, the enzymatic activity of L-asparaginase exhibited a 53.94% increase when in contact with Co2+ and a 48.20% increase with Mg+2. Conversely, the enzyme's activity decreased by 52.67% upon contact with Ca2+ (Table 4).
3.4.4. Stability in Organic Solvents
L-asparaginase exhibited increased enzymatic activity when in contact with organic solvents such as ethanol, showing a 46.24% increase in activity at a concentration of 50% (v/v). Similar trends were observed with isopropanol, where a 41.41% increase was observed at a concentration of 25% (v/v), and with methanol, which displayed the highest activity, showing a 77.20% increase at a concentration of 100% (Table 5).
3.5. Cytotoxic Activity of L-Asparaginase in GM, RAW and HeLa Cells
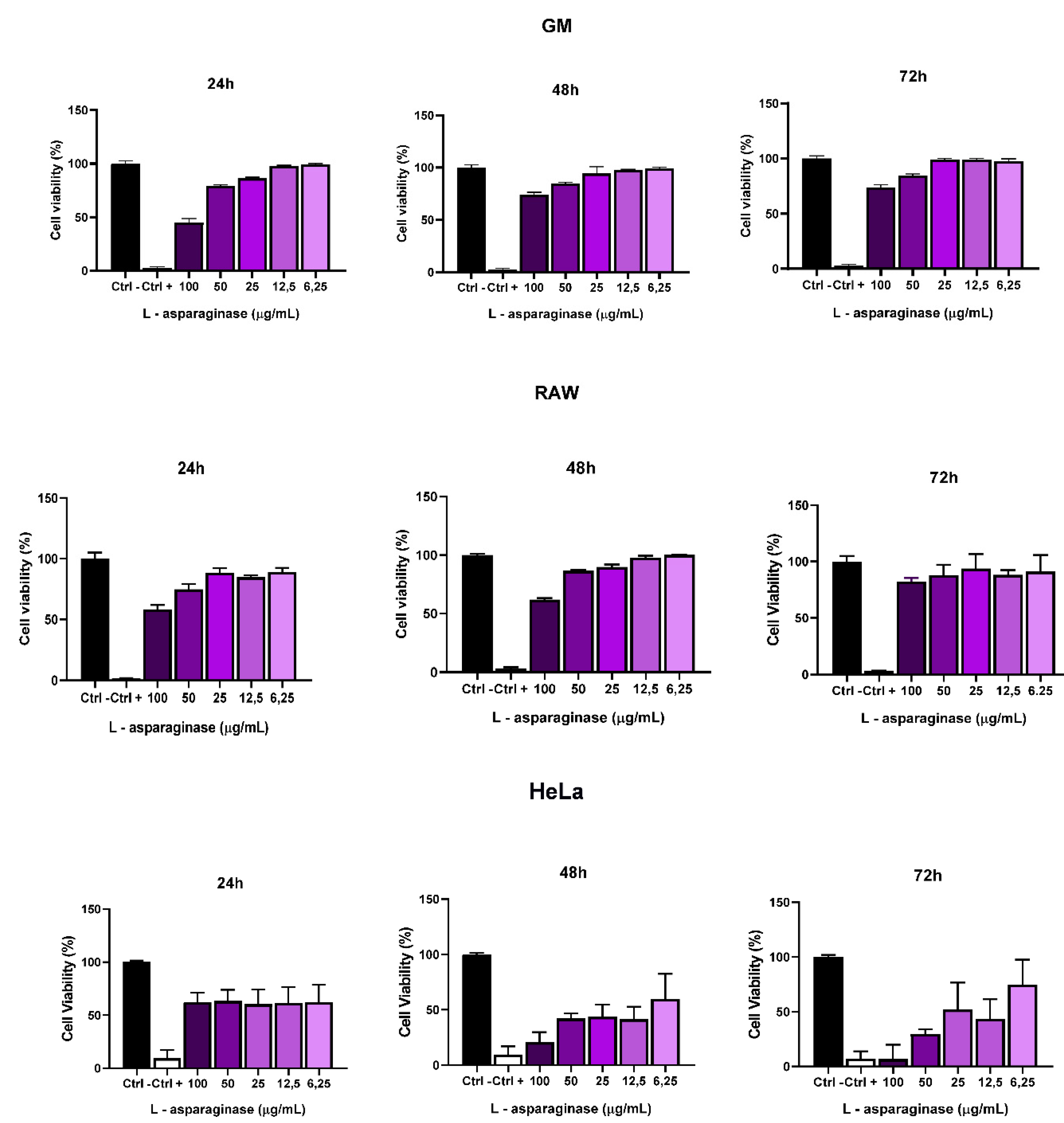
The cytotoxic activity of L-asparaginase from A.niger was evaluated in GM, RAW and HeLa cells (Figure 5). These cells were treated with 6.25 up 100 ug/mL of the enzyme, at 24, 48 and 72h. L-asparaginase in GM and RAW cells showed a slight cytotoxic activity at 24 and 48 hours only at maximum concentrations of 100 and 50 ug/mL, however, within 72 hours the tested GM and RAW cells recovered and began to proliferate again. In HeLa cells, in the first 24h L-asparaginase did not show significant cytotoxic activity, however, after 48h, L-asparaginase cytotoxicity was observed at concentrations up to 12.5 ug/mL, and within 72h cytotoxicity remained at concentrations of 100 and 50 ug/mL.
Figure 7.
Cell Viability after Treatment with A. niger L-asparaginase A. niger. RAW Cells, GM Cells and HeLa Cells. MTT test analyzing the viability of RAW, GM and HeLa cells after treatment with A. niger L-asparaginase. Significant differences (p < 0.05) made by One-way analysis of variance (ANOVA). Triplicate tests. C- (Negative control) without treatment with L-asparaginase and C+ (Positive control) cell death caused by 10% DMSO.
Figure 7.
Cell Viability after Treatment with A. niger L-asparaginase A. niger. RAW Cells, GM Cells and HeLa Cells. MTT test analyzing the viability of RAW, GM and HeLa cells after treatment with A. niger L-asparaginase. Significant differences (p < 0.05) made by One-way analysis of variance (ANOVA). Triplicate tests. C- (Negative control) without treatment with L-asparaginase and C+ (Positive control) cell death caused by 10% DMSO.
3.6. Morphological Viability of the HeLa Cell Line under L-Asparaginase Treatment
The morphology of the HeLa cell line treated with the enzyme L-asparaginase at a concentration of 100 µg/mL after 72h of treatment was analyzed using an inverted microscope and analyzed using ImageJ software (NIH, Bethesda, MD) for surface area and perimeter. Control HeLa cells (Figure 6 – A) present their typical morphology with an elongated structure, with growth forming a monolayer and adhesion to each other.
The cells treated with the enzyme (Figure 6 – B) showed morphological changes compared to the control cells, showing circular formations and reduction in size, an increase in the area without the presence of cells, a rough appearance on their surface, which may indicate the loss of its cytoplasmic material, indicating that the enzyme affects its morphological structure by the exposure time and the characteristics of the enzyme with the cells.
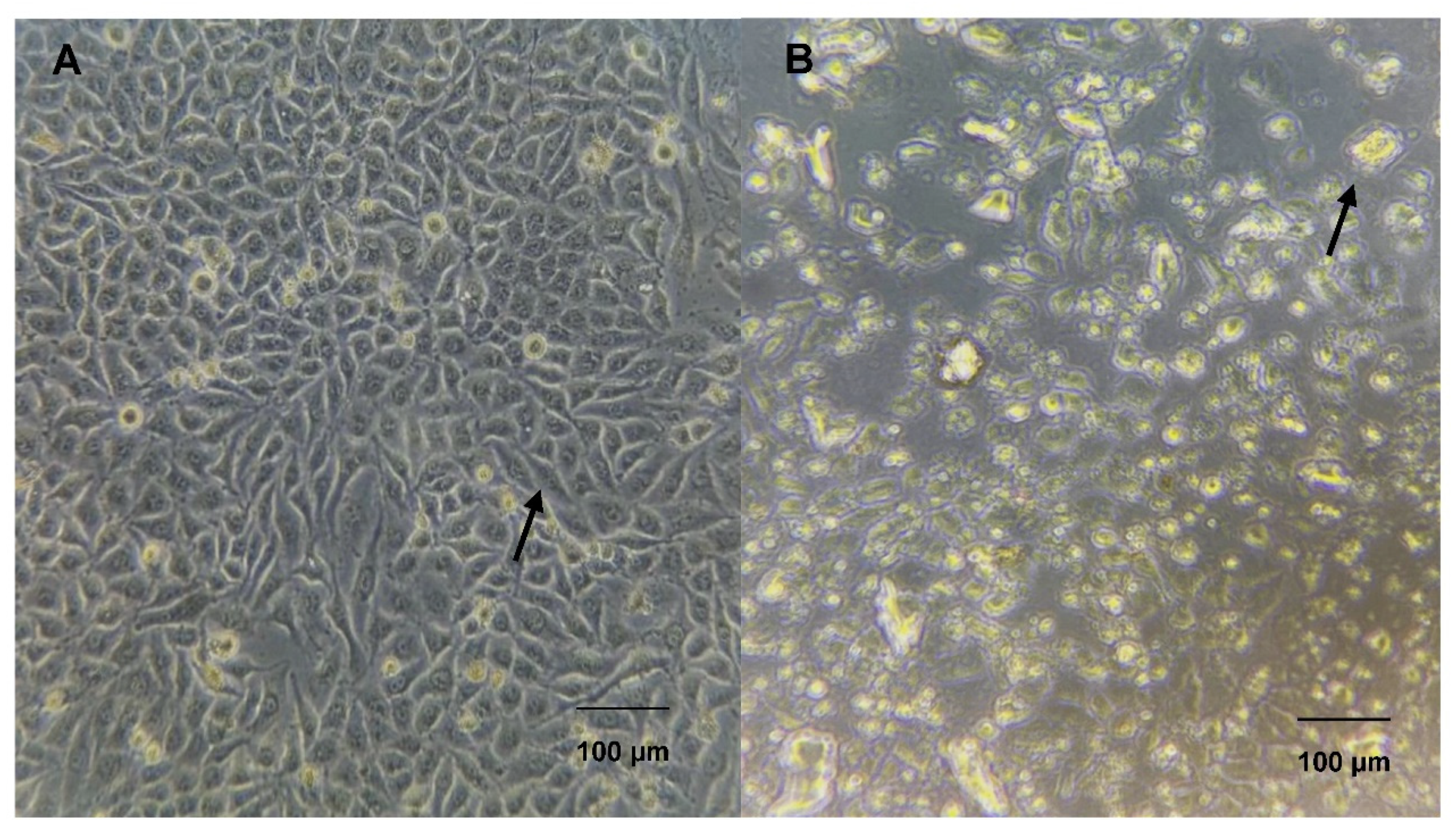
Control HeLa cell line (A) and after treatment with A. niger L-asparaginase for 72h (B). HeLa cell line treated with 100µg/mL in DMEM low glucose medium with 10% fetal bovine serum and 1% antibiotic.
4. Discussion
With an enzyme activity of 46.4 U/mL using the semi-solid fermentation method, A. niger in this study exhibited higher enzyme activity compared to other fungal genera. Profitability is a crucial aspect to consider when contemplating large-scale enzyme production. Using industrial waste as substrates for enzyme production not only leads to profitable production but also promotes cleaner production methods. For the production of L-asparaginase, agro-industrial residues have been reported as suitable substrates in several studies [23,30,31], such as wheat bran in this study. Simulating the natural habitat of A. niger facilitates the production of enzymes and minimizes the risk of contamination. It is easy to maintain and allows a high concentration of proteins. This demonstrates that A. niger, through semisolid fermentation, can be a new alternative to produce high-activity and high-concentration L-asparaginase [32].
The Aspergillus niger in this study previously demonstrated potential in the production of L-asparaginase compared to other genera chosen in this work. Studies carried out by Vala et al. [23] and Babu et al. [28]. They showed that Aspergillus niger has a high capacity for L-asparaginase production, significant enzymatic activity, resistance to various pH levels and temperatures, obtaining results similar to those of this work, justifying its selection for this study. Showing its ability to be an alternative for producing this enzyme on a large scale.
In enzyme purification, the precipitation technique is used to remove the protein of interest from the sample and reduce interactions with other biomolecules that could affect enzyme activity. Therefore, the enzyme can be used in biotechnological processes without interference. Sample preparation becomes a challenge to develop faster methods that require smaller volumes and more efficient protein concentration. Among the commonly used steps, protein precipitation using solvents such as ethanol is often employed due to its high ability to concentrate the target molecule and its profitability, making it part of almost all purification processes [33].
The advantage of ethanol precipitation lies in the fact that the solvent can be rapidly evaporated from the sediment at room temperature, eliminating the need for subsequent processes to remove the precipitant [34]. Thus, ethanol proves to be the most suitable precipitant for the precipitation step of L-asparaginase produced by A. niger, as demonstrated in the results of this study. Vala et al. [23] also achieved superior results using ethanol to precipitate L-asparaginase for purification purposes. According to El-Naggar [35], any protein, once produced by a biological entity, must be purified to characterize its physical and biological properties. A protein must be free of contaminants before being used in any application. Implementing an enzyme in pharmacological studies or other industrial processes depends on understanding its biochemical conditions and how the surrounding environment affects the enzyme.
The process of biochemical characterization of the enzyme, including optimum temperature, optimum pH, kinetics and influence of compounds on its enzymatic activity, is crucial. This characterization ensures that the enzyme can be properly applied in pharmacological compounds without significant loss of activity [36].
The pH of the solution affects the generation of hydroxyl radicals and influences the surface charge and potential interface properties of the catalyst, making it one of the essential factors. Each enzyme has an optimum pH at which its activity is at its maximum [37]. The results obtained in this study were similar to the findings of Vala et al. [23], where purified L-asparaginase from A. niger AKV-MKBU obtained from seawater samples had an optimum pH of 9.0. Similar optimum pH values of 8.0 and 9.0 were found for Escherichia coli [38]. Regarding temperature and stability, unlike other L-asparaginases produced by A. niger AKV-MKBU [23], Brevibacillus borstelensis ML12 [39] showed an optimum temperature of 30°C. Bacillus sp. was reported to have an optimum temperature of 40°C [40], and 37°C for Rhizobium etli [41] but did not exhibit resistance to other temperature ranges. L-asparaginase produced by Brevibacillus borstelensis ML12 [39] demonstrated stability at a temperature similar to that of A. niger studied in this work. These results highlight the distinct characteristic of L-asparaginase produced by A. niger, where it showed greater resistance to different temperatures compared to L-asparaginases produced by other microorganisms. This demonstrates that L-asparaginase produced by A. niger has the potential to serve as a new alternative source of this enzyme.
In the presence of surfactants, SDS exhibits a retarding effect on enzyme activity progressively as the concentration is increased to 20 mM, as supported by Krishnapura and Belur [42]. Studies such as L-asparaginase produced by Brevibacillus borstelensis ML12 reported a significant increase in enzymatic activity with Tween 20, probably due to greater exposure of the enzyme's active site due to the reduction in surface tension. Another study reported an increase in L-asparaginase activity from Fusarium culmorum by Meghavarnam & Janakiraman [43].
The cytotoxic activity of A. niger L-asparaginase was tested in RAW, GM and HeLa cells in this work. L-asparaginase produced by microorganisms and plants demonstrated cytotoxic potential in vitro against several types of cells, as shown in the study by Asthana et al. [44]. In the research by Rani et al. [45], L-asparaginase produced by Aspergillus flavus at a concentration of 131.25 µg/ml, inhibited about 50% of HeLa cell growth, and in another later study, the dose-dependent oncogenic activity of L-asparaginase produced by Aspergillus oryzae occurred up to a concentration of 2 µg/mL in HeLa cells [46].
L-asparaginase produced from microbial cells is capable of reaching HeLa cells, according to Fátima et al. [47], who presented in his research that L-asparaginase isolated from P. aeruginosa (P31, P32 and P34) at concentrations 86.7, 40.3 and 57.6 µg/mL respectively, had IC50 in HeLa cells, with increased cytotoxicity at enzyme concentrations of 5, 10, 25, 50, 75 and 100 µg/mL, with maximum viability of 46.08% and minimum viability of 28.33%.
In the current study, RAW, GM, HeLa and SiHa cells were evaluated with lyophilized purified L-asparaginase, at concentrations: 100, 50, 25, 12.5 and 6.25 ug/mL, therefore L-asparaginase in GM and RAW cells showed a slight cytotoxic activity in 24 and 48 hours only at maximum concentrations of 100 and 50 ug/mL, however, in 72 hours GM and RAW cells recovered. In HeLa cells, in the first 24h L-asparaginase did not show significant cytotoxic activity, however, after 48h, L-asparaginase cytotoxicity was observed at concentrations up to 12.5 ug/mL, and within 72h cytotoxicity remained at concentrations of 100 and 50 ug/mL. L-asparaginase from A. niger was able to effectively inhibit the growth of human cervical cancer cells in vitro of HeLa origin, which in the future could form a therapeutic agent capable of treating cervical cancer caused by HPV 18.
The enzyme object of this study also demonstrated significant results with regard to cell viability in HeLa cells, which presented, after the use of 100ug/mL of L-asparaginase for 72h, important morphological changes compared to control cells. After the experiment, due to the time of exposure to the enzyme, HeLa cells exhibited circular formations with a significant reduction in size and a rough appearance on their surface, indicating loss of cytoplasmic material and relevant degeneration of cell bodies, making it possible to see empty spaces in the analyzed fields. Similar results were found in the study by Fátima et al. [47], with L-asparaginase isolated from P. aeruginosa, demonstrating that the enzyme could be used as an effective therapeutic agent in the treatment of cervical cancer, requiring additional in vivo studies.
In the near future, a significant increase in the demand for L-asparaginase is expected, due to its increasing application in both clinical and industrial contexts, covering several sectors of the biotechnology industry. The A. niger L-asparaginases purified and characterized in this study proved to be highly efficient and low-cost in this research. Therefore, it is important to explore new sources for L-asparaginase production.
5. Conclusion
These results demonstrate the ability of A. niger to produce the L-asparaginase enzyme, highlighting its low cost, easy obtainment from the environment and resistance to different pH levels, temperatures, different ions and surfactants. These findings suggest that this L-asparaginase could serve as an alternative for applications in pharmaceutical and biotechnological processes. Using simple enzymatic purification techniques, this L-asparaginase was obtained with satisfactory purity, high protein concentration, activity and low cytotoxicity in healthy cells. It was observed that L-asparaginase has the ability to significantly inhibit the proliferation of HeLa cells, even in a short period of enzymatic reaction, indicating that L-asparaginase from A. niger may represent a new alternative source for future treatments. oncology, requiring additional in vivo studies.
Acknowledgments
The authors express their gratitude to the Federal University of Maranhão (UFMA), the Foundation for Research and Technological Development of Maranhão (FAPEMA), and the Coordination for the Improvement of Higher Education Personnel - Brazil (CAPES: Financing code 001: Procad Amazonia number 23038015702/2018 – M.D.S.B.N) for their support.
References
- Okunade, K.S. (2020). Human papillomavirus and cervical cancer. Journal of Obstetrics and Gynaecology, 40(5), 602-608. [CrossRef]
- Oliveira, M A A, Mascarenhas, G M S, de Sousa Lima, A A, Carneiro, V M S, Silva, C D C M, Silva, M V C M (2022). Correlation of genetic factors of hpv 16/18 virus and cervical cancer. Advanced Studies on Health and Nature, 3. [CrossRef]
- El-Salem, F, Mansour, M, Gitman, M, Miles, B A, Posner, M R, Bakst,. L, Genden E M, Westra, W H. Real-time PCR HPV genotyping in fine needle aspirations of metastatic head and neck squamous cell carcinoma: Exposing the limitations of conventional p16 immunostaining. Oral Oncology, 2019; 90, 74–79.
- Pedreschi F, Mariotti S, Granby K, Risum, J. (2011). Acrylamide reduction in potato chips using commercial asparaginase in combination with conventional bleaching. LWT - Food Sci Technol 44(6):1473–1476. [CrossRef]
- Shakambari, G, Birendranarayan, A K, Lincy, M J A, Rai, S K, Ahamed, Q T, Ashokkumar, B, Varalakshmi, P (2016). Hemocompatible glutaminase free L-asparaginase from marine Bacillus tequilensis PV9W with anticancer potential modulating p53 expression. RSC advances, 6(31), 25943-25951. [CrossRef]
- Nunes, J.C.; Cristóvão, R.O. Santos-Ebinuma, V.C. Faria, J.L. Silva, C.G. Neves, M.C. Freire, M.G. Tavares, A.P. (2021). L-Asparaginase-based biosensors. Encyclopedia 2021, 1, 848–858. [Google Scholar] [CrossRef]
- Alam, S. Pranaw, K. Tiwari, R. Khare, S.K. (2019). Recent development in the uses of asparaginase as food enzyme. Green Bio-processes: Enzymes in Industrial Food Processing 2019, 55–81. [Google Scholar]
- Hijiya, N.Van Der Sluis, I.M. (2016). Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leukemia & lymphoma, 57(4), 748-757. [CrossRef]
- De Melo, D.W. Fernandez-Lafuente, R. Rodrigues, R.C. (2023). Enhancing biotechnological applications of l-asparaginase: Immobilization on amino-epoxy-agarose for improved catalytic efficiency and stability. Biocatalysis and Agricultural Biotechnology, 52, 102821. [CrossRef]
- Feenstra, L. R. Gehring, R van Geijlswijk, I. M., König, T. Prinsen, H. C. M. T. Vandemeulebroecke, et al. (2022). Evaluation of PEG-L-asparaginase in asparagine suppression and anti-drug antibody development in healthy Beagle dogs: A multi-phase preclinical study. The Veterinary Journal, 286, 105854. [CrossRef]
- Fonseca, M.H.G. da Silva Fiúza, T. de Morais, S.B. Trevizani, R. (2021). Circumventing the side effects of L-asparaginase. Biomedicine & Pharmacotherapy, 139 111616. [CrossRef]
- Khokhar, I. Haider, M.S. Mukhtar, I. Mushtaq, S. (2012). Biological control of Aspergillus niger, the cause of Black-rot disease of Allium cepa L.(onion), by Penicillium species. Journal of Agrobiology 2012, 29, 23. [Google Scholar] [CrossRef]
- Kim, T. Mullaney, E.J. Porres, J. M, Roneker, K.R. Crowe, S., Rice, S. Lei, X.G. (2006). Shifting the pH profile of Aspergillus niger PhyA phytase to match the stomach pH enhances its effectiveness as an animal feed additive. Applied and environmental microbiology 2006, 72, 4397–4403. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, G. Arif, R. Bukhari, S.A. Ali, M., Sharif, S. Atta, A. (2018). Structural and functional annotation of citrate synthase from Aspergillus niger ANJ-120. Pakistan journal of pharmaceutical sciences, 31.
- Qin, S. Zhou, M. Wang, Z. Li, P. Huang, S. Meng, J. (2023). Effect of pulsed electric field on spore germination rate and enzyme activity of Aspergillus niger. Innovative Food Science & Emerging Technologies, 89, 103473. [CrossRef]
- Cairns, T.C. Nai, C. Meyer, V. (2018). How a fungus shapes biotechnology: 100 years of Aspergillus niger research. Fungal biology and biotechnology, 5, 1-14. [CrossRef]
- Bellaouchi, R. Abouloifa, H. Rokni, Y. Hasnaoui, A. Ghabbour, N. Hakkou, A. Asehraou, A. (2021). Characterization and optimization of extracellular enzymes production by Aspergillus niger strains isolated from date by-products. Journal of Genetic Engineering and Biotechnology, 19(1), 1-8. [CrossRef]
- Valenzuela-Lopez, N. Sutton, D.A. Cano-Lira, J.F. Paredes, K., Wiederhold, N. Guarro, J. Stchigel, A.M. (2017). Coelomycetous fungi in the clinical setting: morphological convergence and cryptic diversity. Journal of clinical microbiology, 55 (2), 552-567. [CrossRef]
- White, T.J. Bruns, T. Lee, S.J.W.T. Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR protocols: a guide to methods and applications 1990, 18, 315–322. [Google Scholar]
- labdalall, A H, ALanazi, N A, Aldakeel, S A, AbdulAzeez, S, Borgio, J F. 2020. Molecular, physiological, and biochemical characterization of extracellular lipase production by Aspergillus niger using submerged fermentation. PeerJ, 8, e9425. [CrossRef]
- Dias, F.F.G. Ruiz, A.L.T.G. Della Torre, A. Sato, H.H. (2016). Purification, characterization and antiproliferative activity of L-asparaginase from Aspergillus oryzae CCT 3940 with no glutaminase activity. Asian Pacific Journal of Tropical Biomedicine, 6(9), 785-794. [CrossRef]
- Mahajan, R.V. Saran, S. Saxena, R.K. Srivastava, A.K. (2013). A rapid, efficient and sensitive plate assay for detection and screening of l-asparaginase-producing microorganisms. FEMS microbiology letters, 341(2), 122-126. [CrossRef]
- Vala, A.K. Sachaniya, B. Dudhagara, D. Panseriya, H.Z. Gosai, H., Rawal, R., & Dave, B.P. (2018). Characterization of L-asparaginase from marine-derived Aspergillus niger AKV-MKBU, its antiproliferative activity and bench scale production using industrial waste. International journal of biological macromolecules, 108, 41-46. [CrossRef]
- Warburg, O and Christian, W. Isolation and Crystallization of Enolase. Biochemische Zeitschrift 1942, 310, 384–421.
- Drainas, C. Kinghorn, J.R. Pateman, J.A. Aspartic hydroxamate resistance and asparaginase regulation in the fungus Aspergillus nidulans. Microbiology 1977, 98, 493–501. [Google Scholar]
- Oliveira, B.H. Coradi, G.V. de Oliva-Neto, P. do Nascimento, V.M.G. (2020). Biocatalytic benefits of immobilized Fusarium sp. (GFC) lipase from solid state fermentation on free lipase from submerged fermentation. Industrial Crops and Products, 147, 112235. [CrossRef]
- El-Gendy, M.M.A.A. Yahya, S.M., Hamed, A.R. Soltan, M.M. El-Bondkly, A.M.A. (2018). Phylogenetic analysis and biological evaluation of marine endophytic fungi derived from Red Sea sponge Hyrtios erectus. Applied biochemistry and biotechnology 185, 755–777. [CrossRef]
- Babu, U.K., Ramagopal, N. Reddy, D.R. (2010). Optimization of L-Asparaginase production from Isolated Aspergillus niger by using Solid State Fermentation on sesame cake via application of Genetic Algorithm, and Artificial Neural Network-based design model. Journal of Biotechnology, (150), 538-539. [CrossRef]
- Karanam, S.K. Medicherla, N.R. Application of Doehlert experimental design for the optimization of medium constituents for the production of L-asparaginase from Palm Kernal cake (Elaeis guineensis). J Microbial Biochem Technol 2010, 2, 1–7. [Google Scholar] [CrossRef]
- Elshafei, A.M. El-Ghonemy, D.H. (2015). Screening and media optimization for enhancing L-asparaginase production, an anticancer agent, from different filamentous fungi in solid state fermentation. British Biotechnology Journal, 9(3), 1-15. [CrossRef]
- Cardoso, S.L. de Freitas, M.M. de Souza, P.M. Homem-de-Mello, M. Silveira, D Fonseca-Bazzo, Y. M Magalhães, P.O. (2020). Optimization of aqueous two-phase micellar system for partial purification of L-asparaginase from Penicillium sp. grown in wheat bran as agro-industrial residue. Brazilian Journal of Microbiology, 51, 979-988. [CrossRef]
- Castro, A.M.D. Pereira Jr, N. (2010). Production, properties and application of cellulases in the hydrolysis of agro-industrial waste. Química Nova, 33, 181-188. [CrossRef]
- Tormo, M. A Gil-Exojo, I. de Tejada, A.R. Campillo, J.E. (2006). White bean amylase inhibitor administered orally reduces glycaemia in type 2 diabetic rats. British journal of nutrition, 96(3), 539-544. [CrossRef]
- Pereira, L.L.S. Santos, C.D.D. Pereira, C.A., Marques, T.R. Sátiro, L.C. Precipitation of α-amylase inhibitor from white beans: evaluation of methods. Food and Nutrition Araraquara 2010, 21, 15–20. [Google Scholar]
- El-Naggar, N.E.A. Deraz, S.F. Soliman, H.M. El-Deeb, N.M. El-Ewasy, S.M. (2016). Purification, characterization, cytotoxicity and anticancer activities of L-asparaginase, anti-colon cancer protein, from the newly isolated alkaliphilic Streptomyces fradiae NEAE-82. Scientific reports, 6(1), 32926. [CrossRef]
- Dunn, J.B., Mueller, S., Wang, M. Han, J. (2012). Energy consumption and greenhouse gas emissions from enzyme and yeast manufacture for corn and cellulosic ethanol production. Biotechnology letters, 34, 2259-2263. [CrossRef]
- Pourkhanali, K. Khayati, G. Mizani, F. Raouf, F. (2022). Characterization of free and immobilized lipase from Penicillium sp. onto three modified bentonites: A comparative study. Journal of Biotechnology, 344, 57-69. [CrossRef]
- El-Bessoumy, A.A. Sarhan, M. Mansour, J. Production, isolation, and purification of L-asparaginase from Pseudomonas aeruginosa 50071 using solid-state fermentation. BMB Reports 2004, 37, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R. Bera, D. (2022). Biochemical characterization and thermodynamic principles of purified L-asparaginase from novel Brevibacillus borstelensis ML12. Biocatalysis and Agricultural Biotechnology, 39, 102260. [CrossRef]
- Singh, Y. Gundampati, R.K. Jagannadham, M.V. Srivastava, S.K. (2013). Extracellular L-asparaginase from a protease-deficient Bacillus aryabhattai ITBHU02: purification, biochemical characterization, and evaluation of antineoplastic activity in vitro. Applied biochemistry and biotechnology, 171, 1759-1774. [CrossRef]
- Moreno-Enríquez, A. Evangelista-Martínez, Z. González-Mondragón, E.G. Calderón-Flores, A. Arreguín, R. Pérez-Rueda, E. Huerta-Saquero, A. (2012). Biochemical characterization of recombinant L-asparaginase (AnsA) from Rhizobium etli, a member of an increasing rhizobial-type family of L-asparaginases. [CrossRef]
- Krishnapura, P.R. Belur, P.D. (2016). Partial purification and characterization of L-asparaginase from an endophytic Talaromyces pinophilus isolated from the rhizomes of Curcuma amada. Journal of Molecular Catalysis B: Enzymatic, 124, 83-91. [CrossRef]
- Meghavarnam, A.K. Janakiraman, S. (2018). Evaluation of acrylamide reduction potential of l-asparaginase from Fusarium culmorum (ASP-87) in starchy products. Lwt, 89, 32-37. [CrossRef]
- Asthana, N, Azmi, W (2003). Microbial L-asparaginase: A potent antitumour enzyme.
- Rani, S A, Sundaram, L, Vasantha, P B. (). A study on in vitro antioxidant and anticancer activity of l-asparaginase. J Pharm Res 2012, 5, 1463–1466.
- Sudarkodi, C.; Sundar, S. Anticancer activity of L-asparaginase from Aspergillus oryzae against HEP-G2 and Hela cell lines. Int J Recent Sci Res 2018, 9, 25328–25330. [Google Scholar]
- Fatima, N, Khan, M M, Khan, I A. (2019). L-asparaginase produced from soil isolates of Pseudomonas aeruginosa shows potent anti-cancer activity on HeLa cells. Saudi Journal of Biological Sciences, 26(6), 1146-1153. [CrossRef]
Figure 1.
L-Asparaginase Activity Produced by Different Fungi. Results are presented as relative activity ± standard deviation, each measurement was performed in triplicate using asparagine as a substrate at 37°C for 15 minutes.
Figure 1.
L-Asparaginase Activity Produced by Different Fungi. Results are presented as relative activity ± standard deviation, each measurement was performed in triplicate using asparagine as a substrate at 37°C for 15 minutes.
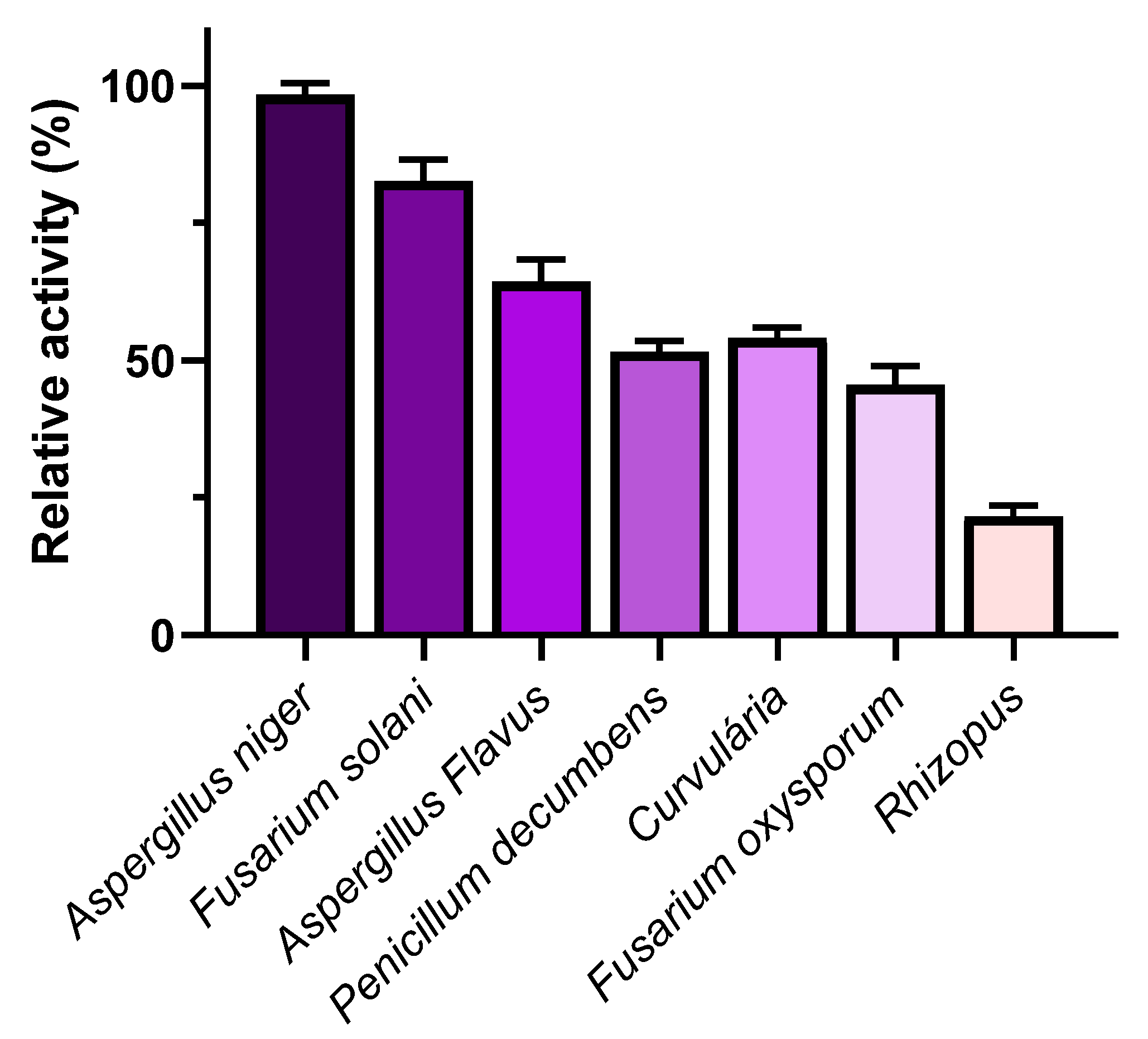
Figure 2.
1.5% agarose gel with Aspergillus niger samples (fragments highlighted in red), demonstrating approximately 295bp.
Figure 2.
1.5% agarose gel with Aspergillus niger samples (fragments highlighted in red), demonstrating approximately 295bp.

Figure 3.
Optimal pH (A) and Stability (B) of A. niger L-Asparaginase. Results are presented as relative activity ± standard deviation. Enzymatic activity was measured using asparagine as a substrate at 37°C for 15 minutes to determine the optimal pH and for 1 hour to assess stability. All tests were conducted in triplicate.
Figure 3.
Optimal pH (A) and Stability (B) of A. niger L-Asparaginase. Results are presented as relative activity ± standard deviation. Enzymatic activity was measured using asparagine as a substrate at 37°C for 15 minutes to determine the optimal pH and for 1 hour to assess stability. All tests were conducted in triplicate.
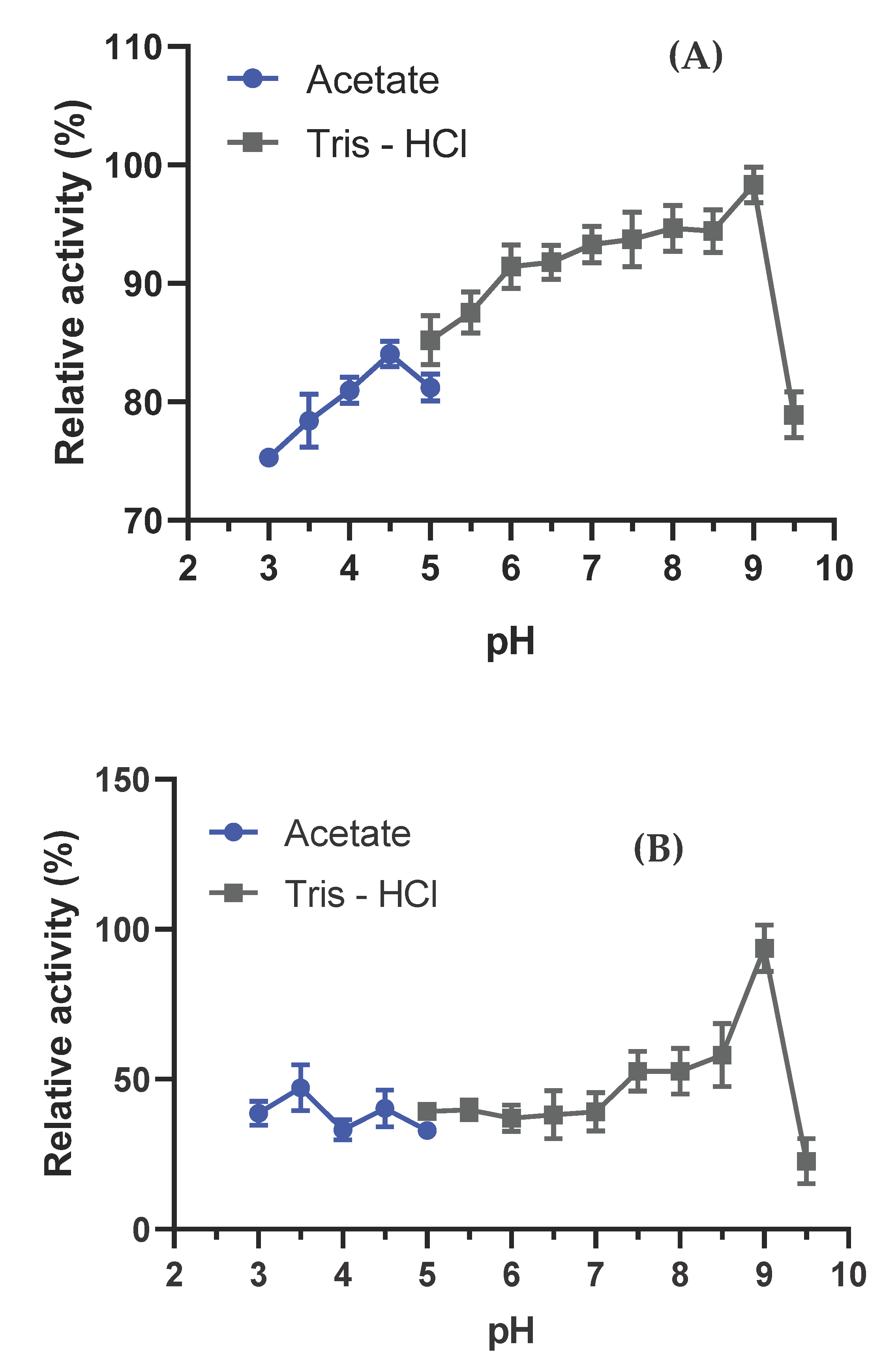
Figure 4.
Optimal Temperature (A) and Stability (B) of A. niger L-Asparaginase. Results are presented as relative activity ± standard deviation. Enzymatic activity was measured using asparagine as a substrate for 15 minutes to determine the optimal temperature and for 1 hour to assess stability. All tests were conducted in triplicate.
Figure 4.
Optimal Temperature (A) and Stability (B) of A. niger L-Asparaginase. Results are presented as relative activity ± standard deviation. Enzymatic activity was measured using asparagine as a substrate for 15 minutes to determine the optimal temperature and for 1 hour to assess stability. All tests were conducted in triplicate.
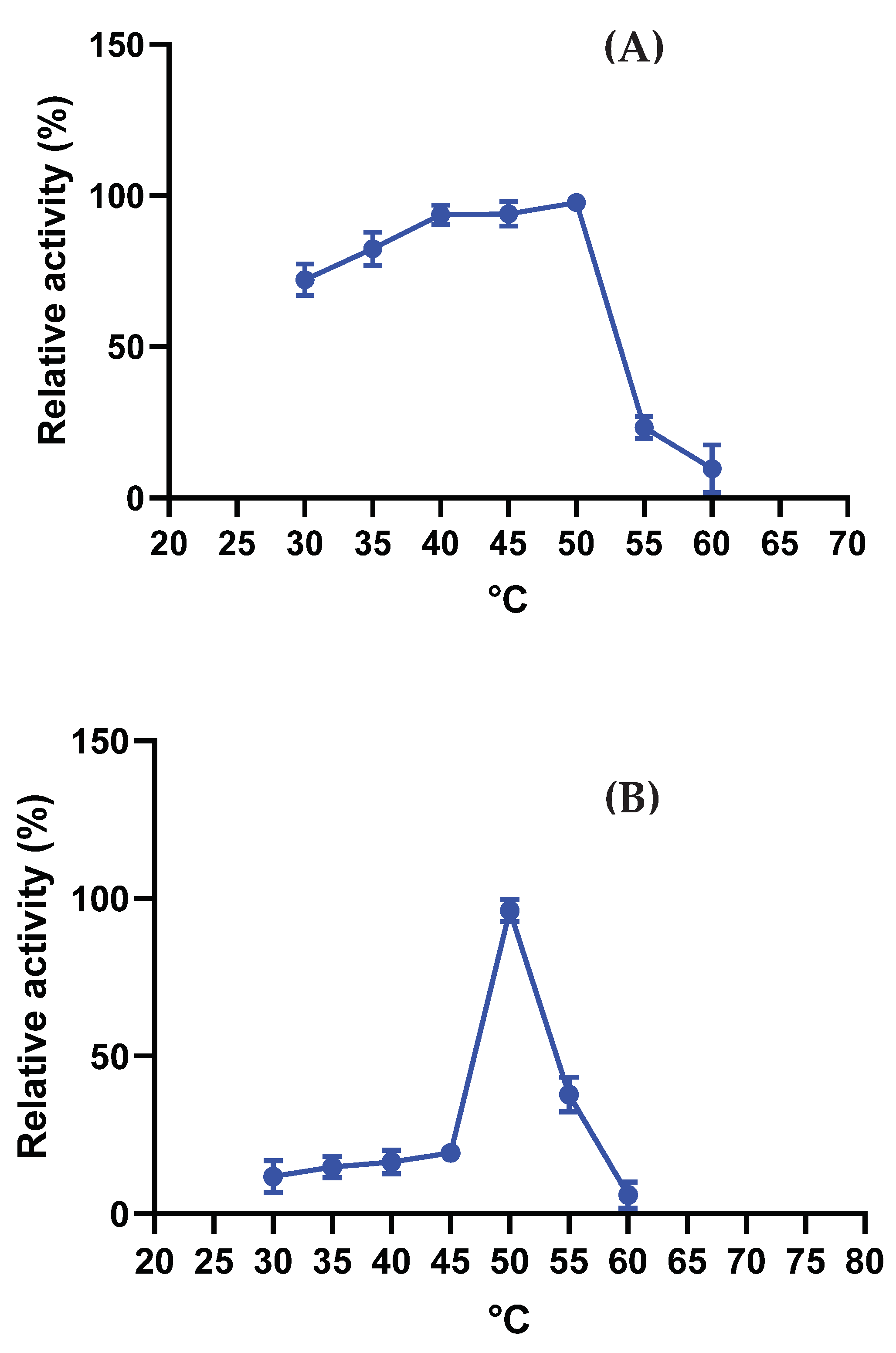

Table 1.
Partial Purification Results of L-Asparaginase from A. niger from Submerged Fermentation (SmF) and Solid-State Fermentation (SsF) .
Table 1.
Partial Purification Results of L-Asparaginase from A. niger from Submerged Fermentation (SmF) and Solid-State Fermentation (SsF) .
| Type of Fermentation | Purification Stage | Total Activity (U/mL) | Total Protein (mg) | Specific Activity (U/mg) | Yield (%) | purification |
| SmF | Crude Extract | 3711,50 | 480,00 | 7,73 | 100 | 1 |
| Fraction 1 (0 - 40%) | 363,96 | 18,95 | 19,21 | 9,81 | 2,48 | |
| Fraction 2 (40 - 80%) | 767,20 | 44,93 | 17,07 | 20,67 | 2,21 | |
| SsF | Crude Extract | 5261 | 480 | 10,96 | 100,00 | 1,00 |
| Fraction 1 (0 - 40%) | 888,58 | 84,63 | 10,50 | 16,89 | 0,96 | |
| Fraction 2 (40 - 80%) | 2748,9 | 14,685 | 187,19 | 52,25 | 17,08 |
Total protein and enzymatic activities were determined according to Warburg & Christian [24] and Drainas et al. [25], respectively, using L-asparagine as substrate. Samples included crude extract, Fraction 1 (F1 (0-40%)), and Fraction 2 (F2 (0-80%)) from Submerged Fermentation (SmF) and Solid-State Fermentation (SsF).
Table 2.
Influence of Precipitation Agents in A. niger Solid-State Fermentation (SsF). Total protein and enzymatic activities were determined, respectively, according to Warburg and Christian [24] and Drainas et al., [25], using L-asparagine as a substrate. Samples included crude extract, Fraction 1 (0-40%), Fraction 2 (0-80%), and DEAE cellulose column fractions from Solid-State Fermentation (SsF).
Table 2.
Influence of Precipitation Agents in A. niger Solid-State Fermentation (SsF). Total protein and enzymatic activities were determined, respectively, according to Warburg and Christian [24] and Drainas et al., [25], using L-asparagine as a substrate. Samples included crude extract, Fraction 1 (0-40%), Fraction 2 (0-80%), and DEAE cellulose column fractions from Solid-State Fermentation (SsF).
| Type of precipitation | Purification Stage | Total Activity (U/mL) | Total Protein (mg) | Specific Activity (U/mg) | Yield | purification (%) |
| Crude Extract | 5520,50 | 3367,5 | 1,64 | 100,00 | 1,00 | |
| Ethanol | Fraction 1 (0 - 40%) | 1205,68 | 84,63 | 14,25 | 21,84 | 8,69 |
| Fraction 2 (40 - 80%) | 2671,90 | 114,18 | 23,40 | 48,40 | 14,27 | |
| DEAE Cellulose Column | 1092,30 | 31,8 | 34,35 | 19,79 | 20,95 | |
| Ammonium Sulfate | Fraction 1 (0 - 40%) | 182,16 | 57,09 | 3,19 | 3,30 | 1,95 |
| Fraction 2 (40 - 80%) | 826,20 | 152,01 | 5,44 | 14,97 | 3,32 | |
| DEAE Cellulose Column | 265,00 | 21,5 | 12,33 | 4,80 | 7,52 | |
| Isopropanol | Fraction 1 (0 - 40%) | 113,40 | 64,35 | 1,76 | 2,05 | 1,07 |
| Fraction 2 (40 - 80%) | 306,60 | 143,955 | 2,13 | 5,55 | 1,30 | |
| DEAE Cellulose Column | 104,10 | 16,3 | 6,39 | 1,89 | 3,90 |
Table 3.
Effect of Surfactants on the Activity of A. niger L-Asparaginase. The percentage values correspond to the amount of surfactant in 50 mMol phosphate buffer at pH 7.0. Tests were conducted in triplicate. L-asparaginase samples were mixed with surfactants (0.01%, 0.10%, and 0.50% w/v) and incubated for 1 hour at 37°C before determining residual activity. Results are presented as enzymatic activity ± standard deviation.
Table 3.
Effect of Surfactants on the Activity of A. niger L-Asparaginase. The percentage values correspond to the amount of surfactant in 50 mMol phosphate buffer at pH 7.0. Tests were conducted in triplicate. L-asparaginase samples were mixed with surfactants (0.01%, 0.10%, and 0.50% w/v) and incubated for 1 hour at 37°C before determining residual activity. Results are presented as enzymatic activity ± standard deviation.
| Type | Concentrations | U/mL | Relative Activity (%) ± SD |
| Control | 47,48 | 100 ± 0,014 | |
| Tween 20 | |||
| 0,01% | 44,98 | 94,73 ± 0,035 | |
| 0,10% | 55,27 | 116,41± 0,084 | |
| 0,50% | 51,35 | 108,15 ± 0,028 | |
| Triton X – 100 | |||
| 0,01% | 45,91 | 96,69 ± 0,013 | |
| 0,10% | 36,56 | 77,00 ± 0,018 | |
| 0,50% | 33,92 | 71,44 ± 0,020 | |
| Tween 80 | |||
| 0,01% | 41,61 | 87,64 ± 0,011 | |
| 0,10% | 42,45 | 89,41± 0,006 | |
| 0,50% | 38,01 | 80,05 ± 0,027 | |
| SDS | |||
| 0,01% | 35,02 | 73,76 ± 0,012 | |
| 0,10% | 24,13 | 50,82 ± 0,009 | |
| 0,50% | 24,43 | 51,45 ± 0,025 | |
| E.D.T. A | |||
| 0,01% | 31,1 | 65,50 ± 0,005 | |
| 0,10% | 22,12 | 46,59 ± 0,004 | |
| 0,50% | 19,13 | 40,29 ± 0,003 | |
Table 4.
Influence of Ions on the Activity of A. niger L-Asparaginase. The L-asparaginases were incubated with ions (1mM) in 50mM Tris-HCl buffer at pH 7.0 and then incubated for 1 hour at 37°C before determining residual activity. Tests were conducted in triplicate. Results are presented as residual activity ± standard deviation.
Table 4.
Influence of Ions on the Activity of A. niger L-Asparaginase. The L-asparaginases were incubated with ions (1mM) in 50mM Tris-HCl buffer at pH 7.0 and then incubated for 1 hour at 37°C before determining residual activity. Tests were conducted in triplicate. Results are presented as residual activity ± standard deviation.
| Type | U/mL | Relative Activity (%) ± SD |
| Control | 31,31 | 100 ± 0,04 |
| Mg+2 | 46,4 | 148,20 ± 0,02 |
| Co2+ | 16,89 | 153,94 ± 0,01 |
| SO42- | 15,05 | 48,07 ± 0,03 |
| Zn+2 | 16,21 | 51,77 ± 0,01 |
| Ca2+ | 46,13 | 47,33 ± 0,01 |
| Fe3+ | 27,63 | 88,25 ± 0,01 |
| Na+ | 28,47 | 90,93 ± 0,02 |
Table 5.
Effect of Organic Solvents on A. niger L-Asparaginase. The percentage values associated with polar organic solvents correspond to the amount of solvent in 50 mMol/L phosphate buffer, pH 7.0. Tests were conducted in triplicate. L-asparaginase samples were mixed with organic solvents (25%, 50%, 80%, and 100% v/v) and incubated for 1 hour at 37°C before determining residual activity. Results are presented as enzymatic activity ± standard deviation.
Table 5.
Effect of Organic Solvents on A. niger L-Asparaginase. The percentage values associated with polar organic solvents correspond to the amount of solvent in 50 mMol/L phosphate buffer, pH 7.0. Tests were conducted in triplicate. L-asparaginase samples were mixed with organic solvents (25%, 50%, 80%, and 100% v/v) and incubated for 1 hour at 37°C before determining residual activity. Results are presented as enzymatic activity ± standard deviation.
| Type | Concentrations | U/mL | Relative Activity (%) ± SD |
| Control | 32,48 | 100 ± 0,014 | |
| Acetone | |||
| 25 | 16,79 | 51,72 ± 0,05 | |
| 50 | 14,53 | 44,72 ± 0,02 | |
| 80 | 5,76 | 17,73 ± 0,03 | |
| 100 | 2,49 | 7,67 ± 0,04 | |
| Ethanol | |||
| 25 | 24,15 | 74,38 ± 0,04 | |
| 50 | 15,02 | 46,18 ± 0,01 | |
| 80 | 9,54 | 29,34 ± 0,02 | |
| 100 | 6,28 | 19,33 ± 0,02 | |
| Isopropanol | |||
| 25 | 13,45 | 41,37 ± 0,03 | |
| 50 | 10,26 | 31,59 ± 0,01 | |
| 80 | 6,75 | 20,78 ± 0,03 | |
| 100 | 4,14 | 12,75 ± 0,01 | |
| Methanol | |||
| 25 | 8,55 | 26,32 ± 0,09 | |
| 50 | 7,82 | 24,05 ± 0,03 | |
| 80 | 5,19 | 16,00 ± 0,02 | |
| 100 | 2,34 | 7,20 ± 0,07 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



