
Submitted:
20 March 2024
Posted:
21 March 2024
You are already at the latest version
Abstract
Survivin was initially identified as a member of the inhibitor apoptosis (IAP) protein family and has been shown to play a critical role in regulation of both apoptosis and mitosis. Survivin has emerged as an attractive target for cancer therapy because its overexpression has been found in most human cancers and is frequently associated with chemotherapy resistance, recurrence, and poor survival rate in cancer patients. In this review, we discuss our current understanding of how survivin mediates various aspects of malignant transformation and drug resistance, as well as efforts that have been made to develop therapeutics targeting survivin for the treatment of cancer.
Keywords:
survivin
; cancer
; mitosis
; apoptosis
; Cancer therapy
Survivin and Cancer
Survivin overexpression has been found in most human cancers and is associated with poor prognosis [1,2,3,4,5,6,7,8]. In particular, high levels of survivin expression are linked with metastasis of various forms of human cancers, including presence in circulating tumor cells [9,10,11,12,13]. Overexpression of survivin can facilitate bypassing of cell cycle checkpoints and promote survival of aneuploid cells [14,15]. Survivin renders cancer cells resistance to radiation [16,17].
While survivin plays an essential role in early embryogenesis [18,19], its expression levels are very low or undetectable in adult tissues, and usually restricted to stems cells and progenitor cells [15,20,21,22,23]. As shown in conditional knockout mice, survivin is required for T cell development and homeostasis, and triggers p53-dependent cell cycle arrest [24,25]. Similarly, suvivin also plays an essential role in B cell expansion [26]. Moreover, survivin is essential for pancreatic beta cell expansion [27,28,29], early brain development [30], and intestinal epithelial progenitor cells [31].
Survivin, encoded by the BIRC5 gene, is a polypeptide of 142 amino acid residues. The transcription of the BIRC5 gene is mediated through a TATA-less promoter that contains multiple Sp1 sites, a CpG island subjected to potential epigenetic modifications, and canonical CDE/CHR boxes that mediate cell cycle-dependent gene expression [32,33]. Survivin is up regulated by multiple pathways that are commonly activated in human cancers, such as EGFR, p185Her2/neu, PI-3 kinase, MAPK, NF-κB and mTOR [34,35,36,37,38]. The transcriptional events from the survivin promoter can also be modulated by Wnt/β-catenin [39], notch [40], YAP [41] and hedgehog signaling pathways [42,43]. In addition, survivin is regulated by Forkhead box m1 (Foxm1), a transcriptional factor critical for G1/S transition and mitotic progression [44]. Conversely, survivin expression can be downregulated by several tumor suppressors, such as TP53, PTEN, Rb, and BRCA1 [45,46,47,48], which are frequently silenced in human cancers.
At least five splicing variants of survivin have been described, which include survivin-ΔEx3 [49,50], survivin-2α [51], survivin-3α [52], survivin-2B [53], and survivin-3B [54]. The differential splicing events lead to generation of proteins with shortened BIR domain, or truncated polypeptides that do not have the intact NES or the coiled coil region in the c-terminus, which can exhibit distinct localization patterns. For example, survivin-Ex3 lacks the NES but contains a distinct bipartite nuclear localization signal (NLS) that mediates its localization to the nucleus [49]. In contrast, in survivin-2B, the BIR motif is interrupted by an in-frame insertion of a cryptic exon, generating a protein predominantly localized to the cytoplasm. Survivin-Ex3 and survivin-2B showed reduced affinity to CPC and cannot compensate for loss of survivin functions [54]. The survivin splicing variants have been reported to associate with certain transformed phenotypes and clinical outcome [55].
Role of Survivin in Cell Division
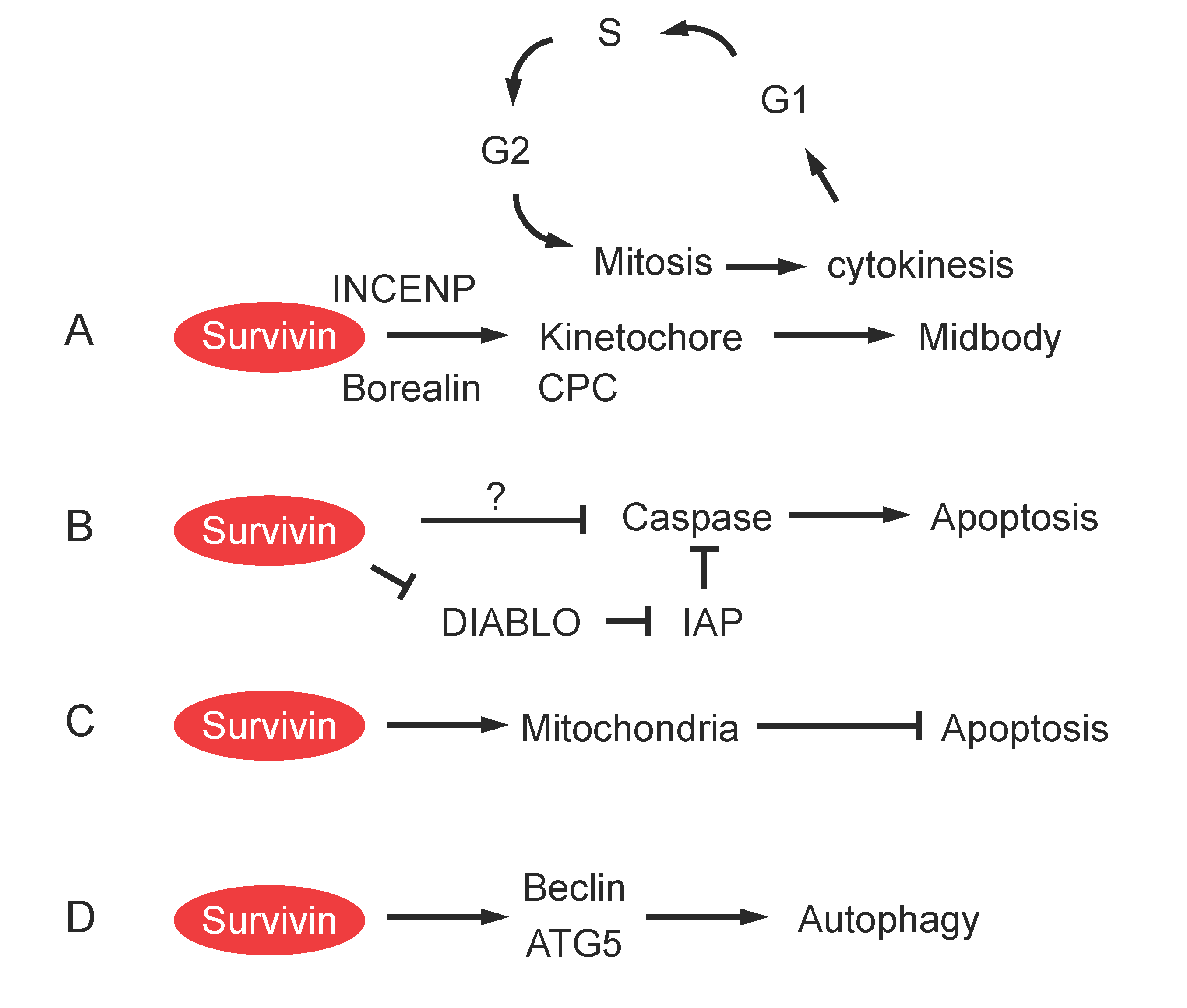
Survivin plays an essential in cell division, and loss of survivin leads to mitotic failure and cell death [32,56,57]. Survivin participates in mitotic checkpoint regulation, as a component of the chromosomal passenger protein complex that also includes Aurora kinase B, INCENP, TD-60, and Borealin [58]. The BIR domain of survivin can interact with a mitotically phosphorylated form of histone H3, which leads to recruitment of the other CPC proteins to the inner centromere [59,60,61]. Interference with the survivin-histone H3 interaction leads to mis localized aurora kinase B and mitotic defects [59,60,61]. Upon entry into mitosis, survivin is localized to the kinetochore in a manner that is dependent on inner centromere protein (INCENP) and cooperates with Aurora kinase B other CPC proteins to mediate spindle formation and proper chromosome alignment [62,63].
When chromosome segregation occurs at the initiation of anaphase, survivin is separated from the kinetochore but remains in the spindle midzone, subsequently becoming associated with the midbody. The molecular mechanism involved in the relocation of survivin to the midbody is not well understood. Nonetheless, survivin has been shown to interact with non-muscle myosin II and the midbody-localized survivin is implicated in playing a role in the formation of the contractile ring during cytokinesis [64]. Because of its essential role in mitotic checkpoint and cytokinesis, survivin abnormality is commonly accompanied with aneuploidy. Notably, loss of p53 function is required for re-entry to the cell cycle following depletion of survivin [65,66].
Role of Survivin in Apoptosis
Survivin was first identified as a member of the Inhibitor-of-Apoptosis (IAP) protein family, also known as the Baculoviral IAP repeat containing (BIRC) proteins, based on the presence of a Baculovirus IAP Repeat (BIR) in the N-terminus [32,56]. The IAP family proteins share the common feature of having at least one BIR domain, which consist of ~70 amino acid residues and are involved in mediating protein-protein interactions [67]. While ablation of survivin can lead to apoptosis, overexpression of survivin can protect cells from apoptosis under various experimental conditions [32,56,68,69].
Some IAP family proteins can inhibit apoptosis by directly bind to the activated form of caspase and abolish its activities [70,71]. For example, the second BIR domain and a linker region of XIAP can directly bind to caspase and block substrate access [72,73,74,75]. In addition, some IAPs also contain either a RING domain - which functions as an E3 ubiquitin ligase – or a ubiquitin-associated domain, which mediate ubiquitin-mediated proteolytic degradation of caspase [76]. In comparison, survivin contains only a BIR domain and, to date, no compelling evidence is available to show that survivin can directly bind and inhibit caspase activities.
Survivin has been reported to bind to DIABLO/SMAC [77,78], a mitochondrial protein that can potentiate certain forms of apoptosis, by blocking the action of IAPs and thereby activate caspases [79,80]. Thus, survivin may inhibit apoptosis by neutralizing an IAP antagonist, namely, DIABLO/SMAC. However, it remains to demonstrate the survivin-DIABLO interaction indeed participate in protection from apoptosis [81].
Role of Survivin in Mitochondrial Function and Autophagy
Survivin has been shown to inhibit mitochondrial-dependent apoptotic events [69]. It has been reported that the N-terminus of survivin contain a mitochondria-targeting sequence that can direct protein localization to the mitochondria when fused with a reporter protein [82]. Interestingly, survivin can be detected in the mitochondrial fraction in cancer cells but not in non-transformed cells [83,84]. Overexpression of mitochondria-targeted survivin can protect cells from apoptosis and enhance transformation, which may involve its binding to another IAP family member XIAP [83,85]. Localization of survivin to the mitochondria can also promote cancer cell invasion and metastasis [86]. Overexpression of survivin appears to alter the dynamic of mitochondrial fission and fusion [87], or inhibit mitophagy [84].
Paradoxically, it has been reported that both knockdown [86] and overexpression of survivin [84,87] can disable mitochondrial functions and reduce oxidative phosphorylation in cancer cells. It should be noted that the studies of the mitochondrial survivin were conducted using a fusion protein of survivin with the mitochondrial targeting sequence of cytochrome c and the knockdown approach used in the study does not specifically target the mitochondrial pool of the protein [83,84,86].
Survivin has been reported to physically interact with several proteins that are involved in autophagy. For example, beclin was found to bind to survivin, which may be involved in regulating survivin protein levels [88]. Intriguingly, ATG5 can form a complex with survivin in the nucleus upon exposure to DNA damage, leading to mitotic catastrophe in an autophagy-independent manner [89]. Conversely, the interaction between survivin and ATG5 may also impact on autophagy-mediated events responsive to DNA damage [90]. Ectopic expression of survivin enables autophagy, which renders the cells more sensitive to inhibition of glycolysis [91]. However, these observations were made only in cell lines with ectopic expression of survivin.
Survivin Localization
Survivin contains a nuclear export signal (NES) that binds to the nuclear export receptor Crm1, which is required for survivin cytoplasmic localization during the interphase [49,92,93,94]. Alteration of the NES, which is located between BIR and the c-terminal coiled coil structures, can disrupt survivin’s nuclear export and localization to the kinetochore and midbody, but not homodimerization or binding to several CPC proteins [93,94,95]. The shift from a cytoplasmic localization pattern to a nuclear one caused by the NES mutations is associated with loss of survivin function to protect cells from apoptosis induced by genotoxic damage or external stimulus [92,94]. In addition, nucleus-directed survivin protein appears to enhance cancer cell sensitivity to apoptosis [96,97].
These observations led to the notion that cytoplasmic survivin is primarily involved in protection from apoptosis. However, it should be noted that the survivin localization patterns in the cytoplasm or nucleus may not a reliable biomarker for clinical outcomes, as it has been linked to both favorable [98,99,100], pancreatic cancer [8], and unfavorable prognosis of cancer patients [101,102,103,104,105,106].
Figure 1.
Schematic representation of survivin structure features involved in dimerization (red), chromosome passenger complex (CPC) binding (green), histone H3 threonine 3 phosphorylated peptide (H3T3ph) and DIABLO binding (black), nuclear export (NES, blue), and mitochondria targeting (MTS, arrowhead). The amino acid residues that have been reported to be modified by acetylation or phosphorylation, as well as the kinases involved, are also indicated.
Figure 1.
Schematic representation of survivin structure features involved in dimerization (red), chromosome passenger complex (CPC) binding (green), histone H3 threonine 3 phosphorylated peptide (H3T3ph) and DIABLO binding (black), nuclear export (NES, blue), and mitochondria targeting (MTS, arrowhead). The amino acid residues that have been reported to be modified by acetylation or phosphorylation, as well as the kinases involved, are also indicated.
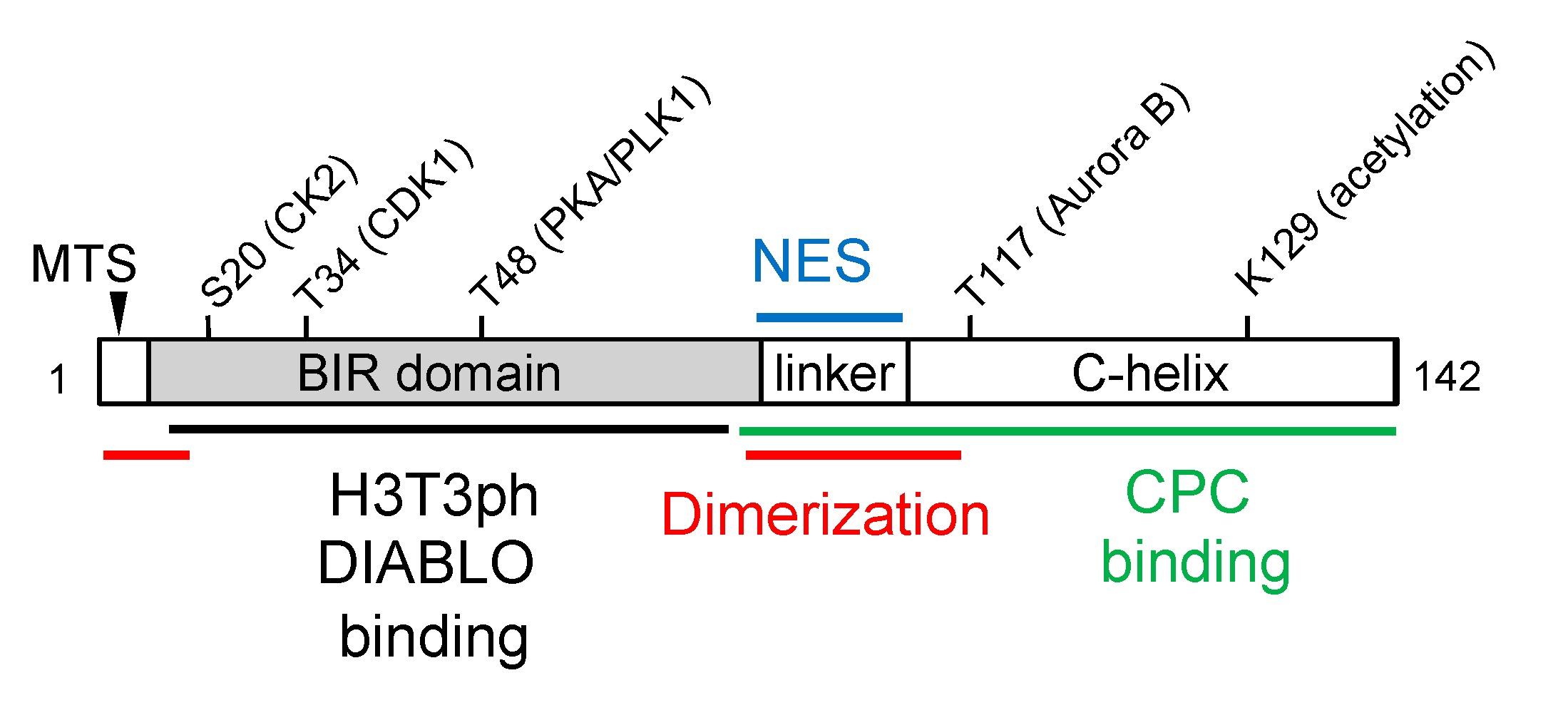
Survivin Protein Structure and Post-Translational Modification
The survivin protein structure in the form of a homodimer has been determined by both crystallography [107,108,109] and by nuclear magnetic resonance (NMR) [110]. The N-terminal BIR domain consists of a three-stranded β-sheet and four α-helices, with a zinc-binding fold, and the survivin protein forms a bow-tie shaped dimer via part of the N-terminal region and the linker region between the BIR domain and the C-terminal helix [107,108]. Notably, ubiquitination of survivin on several lysine residues within the BIR domain are implicated in playing a role in modulating localization to the centromere [111].
A second structure of survivin, which features heteromeric complex with borealin and INCENP has also been resolved [112]. In this structure, the C-terminus of survivin, which contains an extended α-helical coiled-coil domain, forms a three-helical bundle with elements of borealin and INCENP in 1:1:1 stoichiometry [112]. These interactions are essential for central spindle and midbody localization of the complex.
Figure 2.
Functions of survivin. (A) Survivin is required for mitosis and cytokinesis. Survivin is associated with INCENP and borealin as components of the chromosome passenger complex (CPC) localized to the kinetochore during mitosis. Survivin remains associated with the spindle midbody from the anaphase of mitosis until the end of cytokinesis. (B) Survivin can protect cells from apoptosis. The binding of survivin to DIABLO may prevent the latter to inactivate other inhibitor of apoptosis (IAP) family proteins. Alternatively, survivin may directly inhibit caspase activity. (C) Survivin can be localized to the mitochondria and protect cells from mitochondria-mediated apoptosis (D) Survivin can associate with beclin or ATG5 and is postulated to be involved in aspects of autophagy.
Figure 2.
Functions of survivin. (A) Survivin is required for mitosis and cytokinesis. Survivin is associated with INCENP and borealin as components of the chromosome passenger complex (CPC) localized to the kinetochore during mitosis. Survivin remains associated with the spindle midbody from the anaphase of mitosis until the end of cytokinesis. (B) Survivin can protect cells from apoptosis. The binding of survivin to DIABLO may prevent the latter to inactivate other inhibitor of apoptosis (IAP) family proteins. Alternatively, survivin may directly inhibit caspase activity. (C) Survivin can be localized to the mitochondria and protect cells from mitochondria-mediated apoptosis (D) Survivin can associate with beclin or ATG5 and is postulated to be involved in aspects of autophagy.
An accumulating body of evidence show that survivin can be regulated by phosphorylation. Phosphorylation of survivin at T34 by CDK1 has been shown to be important for its anti-apoptotic role [113,114]. In addition, phosphorylation by PKA at Ser20 is also involved in protection against apoptosis by mediating the interaction with XIAP [85]. Moreover, survivin can be phosphorylated by aurora B at T117 and negatively regulates its localization to the kinetochore and function in mitosis [115]. CK2 can phosphorylate survivin at T48 in the BIR domain, which is critical for its mitotic and antiapoptotic functions [116]. Furthermore, PLK1 phosphorylates survivin at T20, which seems to be involved in chromosome orientation during mitosis [117].
Survivin is subject to other forms of post-translational modification. For example, survivin can be acetylated at K129, which affects its homodimerization, binding to Crm1 and nuclear export [118]. Survivin can also be modified via K48- and K63-linked ubiquitination during mitosis, which mediates survivin localization to the kinetochore and mitotic progression [111].
Therapeutic Strategies to Target Survivin
Efforts have been made in recent years to develop therapeutic strategies to disable survivin functionally. However, suvivin is an unconventional drug target, due to its unique structure and lack of enzymatic activity. Currently only a limited number of survivin inhibitors have been developed with success.
YM155, a small-molecule imidazolium-based compound identified by high-throughput screen, is one of the first selectively antagonists for inhibition of survivin expression. YM155 distinctively targets a 269-bp survivin promoter to inhibit gene transcription [119]. Preclinical research showed that YM155 can effectively decrease survivin expression in various cancer cell lines and xenografts mouse models, including prostate, non-Hodgkin lymphoma, and lung [119,120]. However, recent studies indicate that YM155 can elicit DNA damage in cell [121,122], which indicate that the compound may target other proteins and signaling events. Several phase I and phase II studies showed that YM155 generally show low toxicity but has limited antitumor efficacy when used as either a single agent or in combination with other therapeutic agents [123,124,125,126,127,128].
Several other molecules that suppress surivivn expression levels have been described. For example, FL-118 has been identified by HTS screen of a library of compound as a molecule that can reduce expression of survivin, as well as other IAP family members [129]. By using a similar approach to screen for drugs that can inhibit surviving promoter activities, a cytotoxic molecule, termed WM127, was also found to be capable of reducing survivin expression levels and causing cell cycle delay in G2/M the stage, which is accompanied with apoptosis [130]. In addition, EM-1421 (also known as terameprocol), a small-molecule that targets Sp1-dependent promoters, can reduce survivin expression levels and induce apoptosis [131]. Moreover, GDP366 is another a small molecule that can reduce survivin expression at both mRNA and protein levels [132], although the mechanism involved remains to be elucidated.
Strategies designed to reduce survivin protein stability have been reported. Based Heat shock protein 90 can bind to survivin and protect it from proteasomal degradation, and disruption of this interaction can lead to apoptosis [133]. Sheperdin, a peptidomimetic derived from the survivin region that is sufficient to bind to Hsp90, showed the ability to bind the ATP pocket of HSP90 and disrupt the interaction with several of its client proteins, including survivin [134]. This causes the degradation of survivin, among other proteins, leading to apoptosis in tumor cells [134].
In another example, by virtual screen of compounds that mimic the DIABLO/SMAC-IAP interaction, a series of small molecule IAP inhibitors have been developed [135,136]. These molecules can inhibit survivin and, to a lesser extent, XIAP, by downregulate their protein levels and showed efficacy to inhibit tumor growth [135,136]. Other small molecules that block the interaction between survivin and DIABLO/SMAC have been described and showed anti-cancer activities [137,138].
A high-throughput, affinity-based NMR screen has led to identification of several survivin-binding molecules that bind to the dimer interface [139,140,141,142]. Several of the compounds identified in the screen displayed activities to inhibit growth of tumor cells and appeared to cause cell cycle delay in the G0/G1 stage, rather in the mitotic stage [140]. One was of molecules was shown to sensitize colon cancer cells to topoisomerase inhibitor irinotecan [142].
We employed a unique structure-based approach to identify survivin inhibitors that bind directly to the protein and modulate its functions [143,144]. The method, termed as Cavity-Induced Allosteric Modulation (CIAM), was previously used successfully to develop inhibitors for other targets such as TNFR1 [145]. With the CIAM method, we have identified a cavity close to the survivin dimeric interface. The compounds that fit into this cavity in silico were further tested for the ability to bind surviving protein and effect mitotic arrest, as one would expect from loss of survivn function. Several compounds identified by this approach, including S12, exhibits efficacy to inhibit growth of human cancer cell lines both in vitro and in vivo [26,43,143,144]. Notably, knockdown of YAP can increase sensitivity of cancer cell to S12 [144]. Finally, S12 can be modified and potentially used for imaging survivin in tumors by single-photon emission computerized tomography [146].
More recently, a separate group performed a virtual screen of molecules that target the survivin dimeric interface and identified a series of molecules (e.g. LQZ-7F and LQZ-7I) that can induce proteasomal degradation of survivin [147,148]. These molecules were also shown cause apoptosis and inhibit tumor growth in xenograft tumor models [147,148]. It is not clear how disruption of the survivin homodimer by these small molecule leads to reduction of protein stability.
Survivin based immunotherapy has been developed. Cytotoxic T lymphocyte (CTL) response to survivin can be detected in patients [149]. Survivin-derived, MHC-restricted T cell epitope has been identified and can be harnessed to trigger CTL response against survivin expressing cancer cells [150]. DNA vaccine encoding survivin and CCL12 can trigger strong immune response against lung cancer cells in an animal model [151]. Recent studies showed that long synthetic peptides derived from the survivin protein can generate both cytotoxic CD8+ and CD4+ T-cell responses, leading to tumor regression and prevention of relapse in animal models [152]. In particular, the survivin peptide mimic SurVaxM showed efficacy to stimulate anti-tumor immune responses against brain tumors in animal models and early promise in clinical trials [153,154,155]. A whole protein survivin dendritic cell vaccine has also been developed and tested for the treatment of myeloma patients [156,157].
Conclusions and Perspective
Because of its high expression levels in most tumors and absence in most normal tissues, survivin has been considered as a promising therapeutic target. Studies in the past 25 years showed that survivin plays a vital role in regulation of cell division as a component of the CPC on the mitotic apparatus. However, despite its structural similarities to the other IAP family proteins, how survivin acts as an inhibit of apoptosis remains elusive. Studies of the survivin protein ensembles in various subcellular pools, such as the mitochondria and the interphase or mitotic cytoplasm, may unravel a mechanism by which survivin links mitotic checkpoint regulation with apoptotic pathways. Progress has been made to develop survivin-targeted therapy, including small molecules that directly bind and disable survivin. Understand the signaling pathways that mediates cancer cell sensitivity to survivin targeted therapy may help to develop more effective therapeutic strategies. Conceivably, the combination of survivin inhibition with chemotherapy or other targeted therapeutics may achieve the maximal benefit clinically.
Author Contributions
Conceptualization, Q.W. and M.I.G.; writing—original draft preparation, Q.W.; writing—review and editing, Q.W. and M.I.G.; funding acquisition, M.I.G. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tanaka, K.; Iwamoto, S.; Gon, G.; Nohara, T.; Iwamoto, M.; Tanigawa, N. Expression of survivin and its relationship to loss of apoptosis in breast. Clin Cancer Res 2000, 6, 127–134. [Google Scholar]
- Cohen, C.; Lohmann, C.M.; Cotsonis, G.; Lawson, D.; Santoianni, R. Survivin expression in ovarian carcinoma: correlation with apoptotic markers and. Mod Pathol 2003, 16, 574–583. [Google Scholar] [CrossRef]
- Sarela, A.I.; Verbeke, C.S.; Ramsdale, J.; Davies, C.L.; Markham, A.F.; Guillou, P.J. Expression of survivin, a novel inhibitor of apoptosis and cell cycle regulatory. Br J Cancer 2002, 86, 886–892. [Google Scholar] [CrossRef]
- Grabowski, P.; Kuhnel, T.; Muhr-Wilkenshoff, F.; Heine, B.; Stein, H.; Hopfner, M.; Germer, C.T.; Scherubl, H. Prognostic value of nuclear survivin expression in oesophageal squamous cell. Br J Cancer 2003, 88, 115–119. [Google Scholar] [CrossRef]
- Sui, L.; Dong, Y.; Ohno, M.; Watanabe, Y.; Sugimoto, K.; Tokuda, M. Survivin expression and its correlation with cell proliferation and prognosis in epithelial ovarian tumors. Int J Oncol 2002, 21, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Kuwabara, Y.; Mitani, M.; Shinoda, N.; Sato, A.; Toyama, T.; Mitsui, A.; Nishiwaki, T.; Moriyama, S.; Kudo, J.; et al. Expression of survivin in esophageal cancer: correlation with the prognosis and response to chemotherapy. Int J Cancer 2001, 95, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Oparina, N.; Erlandsson, M.C.; Faldt Beding, A.; Parris, T.; Helou, K.; Karlsson, P.; Einbeigi, Z.; Bokarewa, M.I. Prognostic Significance of BIRC5/Survivin in Breast Cancer: Results from Three Independent Cohorts. Cancers 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Tonini, G.; Vincenzi, B.; Santini, D.; Scarpa, S.; Vasaturo, T.; Malacrino, C.; Coppola, R.; Magistrelli, P.; Borzomati, D.; Baldi, A.; et al. Nuclear and cytoplasmic expression of survivin in 67 surgically resected pancreatic cancer patients. Br J Cancer 2005, 92, 2225–2232. [Google Scholar] [CrossRef]
- Cao, M.; Yie, S.M.; Wu, S.M.; Chen, S.; Lou, B.; He, X.; Ye, S.R.; Xie, K.; Rao, L.; Gao, E.; et al. Detection of survivin-expressing circulating cancer cells in the peripheral blood of patients with esophageal squamous cell carcinoma and its clinical significance. Clin Exp Metastasis 2009, 26, 751–758. [Google Scholar] [CrossRef]
- Yie, S.M.; Lou, B.; Ye, S.R.; He, X.; Cao, M.; Xie, K.; Ye, N.Y.; Lin, R.; Wu, S.M.; Xiao, H.B.; et al. Clinical significance of detecting survivin-expressing circulating cancer cells in patients with non-small cell lung cancer. Lung Cancer 2009, 63, 284–290. [Google Scholar] [CrossRef]
- Goossens-Beumer, I.J.; Zeestraten, E.C.; Benard, A.; Christen, T.; Reimers, M.S.; Keijzer, R.; Sier, C.F.; Liefers, G.J.; Morreau, H.; Putter, H.; et al. Clinical prognostic value of combined analysis of Aldh1, Survivin, and EpCAM expression in colorectal cancer. Br J Cancer 2014, 110, 2935–2944. [Google Scholar] [CrossRef]
- Chen, J.; Li, T.; Liu, Q.; Jiao, H.; Yang, W.; Liu, X.; Huo, Z. Clinical and prognostic significance of HIF-1alpha, PTEN, CD44v6, and survivin for gastric cancer: a meta-analysis. PLoS One 2014, 9, e91842. [Google Scholar] [CrossRef]
- Ning, Y.; Hanna, D.L.; Zhang, W.; Mendez, A.; Yang, D.; El-Khoueiry, R.; Matsusaka, S.; Sunakawa, Y.; Stremitzer, S.; Parekh, A.; et al. Cytokeratin-20 and Survivin-Expressing Circulating Tumor Cells Predict Survival in Metastatic Colorectal Cancer Patients by a Combined Immunomagnetic qRT-PCR Approach. Mol Cancer Ther 2015, 14, 2401–2408. [Google Scholar] [CrossRef]
- Rosa, J.; Canovas, P.; Islam, A.; Altieri, D.C.; Doxsey, S.J. Survivin modulates microtubule dynamics and nucleation throughout the cell cycle. Mol Biol Cell 2006, 17, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Gianani, R.; Jarboe, E.; Orlicky, D.; Frost, M.; Bobak, J.; Lehner, R.; Shroyer, K.R. Expression of survivin in normal, hyperplastic, and neoplastic colonic mucosa. Hum Pathol 2001, 32, 119–125. [Google Scholar] [CrossRef]
- Chakravarti, A.; Zhai, G.G.; Zhang, M.; Malhotra, R.; Latham, D.E.; Delaney, M.A.; Robe, P.; Nestler, U.; Song, Q.; Loeffler, J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 2004, 23, 7494–7506. [Google Scholar] [CrossRef] [PubMed]
- Rodel, F.; Hoffmann, J.; Distel, L.; Herrmann, M.; Noisternig, T.; Papadopoulos, T.; Sauer, R.; Rodel, C. Survivin as a radioresistance factor, and prognostic and therapeutic target for radiotherapy in rectal cancer. Cancer Res 2005, 65, 4881–4887. [Google Scholar] [CrossRef]
- Uren, A.G.; Wong, L.; Pakusch, M.; Fowler, K.J.; Burrows, F.J.; Vaux, D.L.; Choo, K.H. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol 2000, 10, 1319–1328. [Google Scholar] [CrossRef]
- Conway, E.M.; Pollefeyt, S.; Steiner-Mosonyi, M.; Luo, W.; Devriese, A.; Lupu, F.; Bono, F.; Leducq, N.; Dol, F.; Schaeffer, P.; et al. Deficiency of survivin in transgenic mice exacerbates Fas-induced apoptosis via mitochondrial pathways. Gastroenterology 2002, 123, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997, 3, 917–921. [Google Scholar] [CrossRef]
- Zhang, T.; Otevrel, T.; Gao, Z.; Ehrlich, S.M.; Fields, J.Z.; Boman, B.M. Evidence that APC regulates survivin expression: a possible mechanism. Cancer Res 2001, 61, 8664–8667. [Google Scholar] [PubMed]
- Fukuda, S.; Pelus, L.M. Regulation of the inhibitor-of-apoptosis family member survivin in normal cord. Blood 2001, 98, 2091–2100. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Pelus, L.M. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther 2006, 5, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Conway, E.M.; Kang, C.; Winoto, A. Essential role of survivin, an inhibitor of apoptosis protein, in T cell development, maturation, and homeostasis. J Exp Med 2004, 199, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Bakal, C.; Shahinian, A.; Elia, A.; Wakeham, A.; Suh, W.K.; Duncan, G.S.; Ciofani, M.; Rottapel, R.; Zuniga-Pflucker, J.C.; et al. Survivin loss in thymocytes triggers p53-mediated growth arrest and p53-independent cell death. J Exp Med 2004, 199, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Miletic, A.V.; Jellusova, J.; Cato, M.H.; Lee, C.R.; Baracho, G.V.; Conway, E.M.; Rickert, R.C. Essential Role for Survivin in the Proliferative Expansion of Progenitor and Mature B Cells. J Immunol 2016, 196, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Nishimura, W.; Devor-Henneman, D.; Kusewitt, D.; Wang, H.; Holloway, M.P.; Dohi, T.; Sabo, E.; Robinson, M.L.; Altieri, D.C.; et al. Postnatal expansion of the pancreatic beta-cell mass is dependent on survivin. Diabetes 2008, 57, 2718–2727. [Google Scholar] [CrossRef]
- Wu, X.; Wang, L.; Schroer, S.; Choi, D.; Chen, P.; Okada, H.; Woo, M. Perinatal survivin is essential for the establishment of pancreatic beta cell mass in mice. Diabetologia 2009, 52, 2130–2141. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Q.; Wang, X.; Zhu, J.; Xu, K.; Okada, H.; Wang, R.; Woo, M. Survivin is required for beta-cell mass expansion in the pancreatic duct-ligated mouse model. PLoS One 2012, 7, e41976. [Google Scholar] [CrossRef]
- Jiang, Y.; de Bruin, A.; Caldas, H.; Fangusaro, J.; Hayes, J.; Conway, E.M.; Robinson, M.L.; Altura, R.A. Essential role for survivin in early brain development. J Neurosci 2005, 25, 6962–6970. [Google Scholar] [CrossRef]
- Martini, E.; Wittkopf, N.; Gunther, C.; Leppkes, M.; Okada, H.; Watson, A.J.; Podstawa, E.; Backert, I.; Amann, K.; Neurath, M.F.; et al. Loss of Survivin in Intestinal Epithelial Progenitor Cells Leads to Mitotic Catastrophe and Breakdown of Gut Immune Homeostasis. Cell Rep 2016, 14, 1062–1073. [Google Scholar] [CrossRef]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef]
- Li, F.; Altieri, D.C. Transcriptional analysis of human survivin gene expression. Biochem J 1999, 344 Pt 2, 305–311. [Google Scholar] [CrossRef]
- Vaira, V.; Lee, C.W.; Goel, H.L.; Bosari, S.; Languino, L.R.; Altieri, D.C. Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene 2007, 26, 2678–2684. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, H.; Torigoe, T.; Kamiguchi, K.; Hirohashi, Y.; Ohmura, T.; Hirata, K.; Sato, M.; Sato, N. Survivin expression is regulated by coexpression of human epidermal growth factor. Cancer Res 2005, 65, 11018–11025. [Google Scholar] [CrossRef]
- Tracey, L.; Perez-Rosado, A.; Artiga, M.J.; Camacho, F.I.; Rodriguez, A.; Martinez, N.; Ruiz-Ballesteros, E.; Mollejo, M.; Martinez, B.; Cuadros, M.; et al. Expression of the NF-kappaB targets BCL2 and BIRC5/Survivin characterizes small. J Pathol 2005, 206, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Greene, M.I. EGFR enhances Survivin expression through the phosphoinositide 3 (PI-3) kinase. Exp Mol Pathol 2005, 79, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Bisi, J.; Strum, J.; Liu, L.; Carrick, K.; Graham, K.M.; Treece, A.L.; Hardwicke, M.A.; Dush, M.; Liao, Q.; et al. Regulation of survivin by ErbB2 signaling: therapeutic implications for. Cancer Res 2006, 66, 1640–1647. [Google Scholar] [CrossRef]
- Kim, P.J.; Plescia, J.; Clevers, H.; Fearon, E.R.; Altieri, D.C. Survivin and molecular pathogenesis of colorectal cancer. Lancet 2003, 362, 205–209. [Google Scholar] [CrossRef]
- Lee, C.W.; Raskett, C.M.; Prudovsky, I.; Altieri, D.C. Molecular dependence of estrogen receptor-negative breast cancer on a notch-survivin signaling axis. Cancer Res 2008, 68, 5273–5281. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Vlckova, K.; Ondrusova, L.; Vachtenheim, J.; Reda, J.; Dundr, P.; Zadinova, M.; Zakova, P.; Pouckova, P. Survivin, a novel target of the Hedgehog/GLI signaling pathway in human tumor cells. Cell Death Dis 2016, 7, e2048. [Google Scholar] [CrossRef] [PubMed]
- Brun, S.N.; Markant, S.L.; Esparza, L.A.; Garcia, G.; Terry, D.; Huang, J.M.; Pavlyukov, M.S.; Li, X.N.; Grant, G.A.; Crawford, J.R.; et al. Survivin as a therapeutic target in Sonic hedgehog-driven medulloblastoma. Oncogene 2015, 34, 3770–3779. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.C.; Chen, Y.J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, T.; Costa, R.H. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol 2005, 25, 10875–10894. [Google Scholar] [CrossRef]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human survivin is negatively regulated by wild-type p53 and participates in. Oncogene 2002, 21, 2613–2622. [Google Scholar] [CrossRef]
- Raj, D.; Liu, T.; Samadashwily, G.; Li, F.; Grossman, D. Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis 2008, 29, 194–201. [Google Scholar] [CrossRef]
- Guha, M.; Plescia, J.; Leav, I.; Li, J.; Languino, L.R.; Altieri, D.C. Endogenous tumor suppression mediated by PTEN involves survivin gene silencing. Cancer Res 2009, 69, 4954–4958. [Google Scholar] [CrossRef]
- Wang, R.H.; Zheng, Y.; Kim, H.S.; Xu, X.; Cao, L.; Luhasen, T.; Lee, M.H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell 2008, 32, 11–20. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Span, S.W.; Ferreira, C.G.; Kruyt, F.A.; Giaccone, G. CRM1-mediated nuclear export determines the cytoplasmic localization of the antiapoptotic protein Survivin. Exp Cell Res 2002, 275, 44–53. [Google Scholar] [CrossRef]
- Mahotka, C.; Wenzel, M.; Springer, E.; Gabbert, H.E.; Gerharz, C.D. Survivin-deltaEx3 and survivin-2B: two novel splice variants of the apoptosis inhibitor survivin with different antiapoptotic properties. Cancer Res 1999, 59, 6097–6102. [Google Scholar]
- Caldas, H.; Honsey, L.E.; Altura, R.A. Survivin 2alpha: a novel Survivin splice variant expressed in human malignancies. Mol Cancer 2005, 4, 11. [Google Scholar] [CrossRef]
- Mola, G.; Vela, E.; Fernandez-Figueras, M.T.; Isamat, M.; Munoz-Marmol, A.M. Exonization of Alu-generated splice variants in the survivin gene of human and non-human primates. J Mol Biol 2007, 366, 1055–1063. [Google Scholar] [CrossRef]
- Caldas, H.; Jiang, Y.; Holloway, M.P.; Fangusaro, J.; Mahotka, C.; Conway, E.M.; Altura, R.A. Survivin splice variants regulate the balance between proliferation and cell death. Oncogene 2005, 24, 1994–2007. [Google Scholar] [CrossRef]
- Noton, E.A.; Colnaghi, R.; Tate, S.; Starck, C.; Carvalho, A.; Ko Ferrigno, P.; Wheatley, S.P. Molecular analysis of survivin isoforms: evidence that alternatively spliced variants do not play a role in mitosis. J Biol Chem 2006, 281, 1286–1295. [Google Scholar] [CrossRef]
- Pavlidou, A.; Kroupis, C.; Dimas, K. Association of survivin splice variants with prognosis and treatment of breast cancer. World J Clin Oncol 2014, 5, 883–894. [Google Scholar] [CrossRef]
- Li, F.; Ackermann, E.J.; Bennett, C.F.; Rothermel, A.L.; Plescia, J.; Tognin, S.; Villa, A.; Marchisio, P.C.; Altieri, D.C. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol 1999, 1, 461–466. [Google Scholar] [CrossRef]
- Lens, S.M.; Wolthuis, R.M.; Klompmaker, R.; Kauw, J.; Agami, R.; Brummelkamp, T.; Kops, G.; Medema, R.H. Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension. Embo J 2003, 22, 2934–2947. [Google Scholar] [CrossRef]
- Vader, G.; Kauw, J.J.; Medema, R.H.; Lens, S.M. Survivin mediates targeting of the chromosomal passenger complex to the centromere and midbody. EMBO Rep 2006, 7, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.E.; Ghenoiu, C.; Xue, J.Z.; Zierhut, C.; Kimura, H.; Funabiki, H. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science 2010, 330, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Dai, J.; Daum, J.R.; Niedzialkowska, E.; Banerjee, B.; Stukenberg, P.T.; Gorbsky, G.J.; Higgins, J.M. Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science 2010, 330, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, Y.; Honda, T.; Tanno, Y.; Watanabe, Y. Two histone marks establish the inner centromere and chromosome bi-orientation. Science 2010, 330, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, S.P.; Carvalho, A.; Vagnarelli, P.; Earnshaw, W.C. INCENP is required for proper targeting of Survivin to the centromeres and the anaphase spindle during mitosis. Curr Biol 2001, 11, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Bolton, M.A.; Lan, W.; Powers, S.E.; McCleland, M.L.; Kuang, J.; Stukenberg, P.T. Aurora B kinase exists in a complex with survivin and INCENP and its kinase activity is stimulated by survivin binding and phosphorylation. Mol Biol Cell 2002, 13, 3064–3077. [Google Scholar] [CrossRef] [PubMed]
- Babkoff, A.; Cohen-Kfir, E.; Aharon, H.; Ronen, D.; Rosenberg, M.; Wiener, R.; Ravid, S. A direct interaction between survivin and myosin II is required for cytokinesis. J Cell Sci 2019, 132. [Google Scholar] [CrossRef]
- Yang, D.; Welm, A.; Bishop, J.M. Cell division and cell survival in the absence of survivin. Proc Natl Acad Sci U S A 2004, 101, 15100–15105. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, E.; Plescia, J.; Wilkinson, J.C.; Duckett, C.S.; Altieri, D.C. Acute ablation of survivin uncovers p53-dependent mitotic checkpoint functions and control of mitochondrial apoptosis. J Biol Chem 2004, 279, 2077–2084. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Ashwell, J.D. IAPs: what’s in a name? Mol Cell 2008, 30, 123–135. [Google Scholar] [CrossRef]
- Grossman, D.; Kim, P.J.; Blanc-Brude, O.P.; Brash, D.E.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J Clin Invest 2001, 108, 991–999. [Google Scholar] [CrossRef]
- Blanc-Brude, O.P.; Mesri, M.; Wall, N.R.; Plescia, J.; Dohi, T.; Altieri, D.C. Therapeutic targeting of the survivin pathway in cancer: initiation of mitochondrial apoptosis and suppression of tumor-associated angiogenesis. Clin Cancer Res 2003, 9, 2683–2692. [Google Scholar]
- Cetraro, P.; Plaza-Diaz, J.; MacKenzie, A.; Abadia-Molina, F. A Review of the Current Impact of Inhibitors of Apoptosis Proteins and Their Repression in Cancer. Cancers 2022, 14. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Reed, J.C. IAP family proteins--suppressors of apoptosis. Genes Dev 1999, 13, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Renatus, M.; Schwarzenbacher, R.; Zhou, Q.; Sun, C.; Fesik, S.W.; Liddington, R.C.; Salvesen, G.S. Structural basis for the inhibition of caspase-3 by XIAP. Cell 2001, 104, 791–800. [Google Scholar] [CrossRef]
- Huang, Y.; Park, Y.C.; Rich, R.L.; Segal, D.; Myszka, D.G.; Wu, H. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 2001, 104, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Shiozaki, E.; Srinivasula, S.M.; Wu, Q.; Datta, P.; Alnemri, E.S.; Shi, Y. Structural basis of caspase-7 inhibition by XIAP. Cell 2001, 104, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Deveraux, Q.; Tamm, I.; Welsh, K.; Assa-Munt, N.; Salvesen, G.S.; Reed, J.C. A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem 1998, 273, 7787–7790. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Nakabayashi, Y.; Takahashi, R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci U S A 2001, 98, 8662–8667. [Google Scholar] [CrossRef]
- Song, Z.; Yao, X.; Wu, M. Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J Biol Chem 2003, 278, 23130–23140. [Google Scholar] [CrossRef]
- Du, J.; Kelly, A.E.; Funabiki, H.; Patel, D.J. Structural basis for recognition of H3T3ph and Smac/DIABLO N-terminal peptides by human Survivin. Structure 2012, 20, 185–195. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- McNeish, I.A.; Lopes, R.; Bell, S.J.; McKay, T.R.; Fernandez, M.; Lockley, M.; Wheatley, S.P.; Lemoine, N.R. Survivin interacts with Smac/DIABLO in ovarian carcinoma cells but is redundant in Smac-mediated apoptosis. Exp Cell Res 2005, 302, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Dunajova, L.; Cash, E.; Markus, R.; Rochette, S.; Townley, A.R.; Wheatley, S.P. The N-terminus of survivin is a mitochondrial-targeting sequence and Src regulator. J Cell Sci 2016, 129, 2707–2712. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Beltrami, E.; Wall, N.R.; Plescia, J.; Altieri, D.C. Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J Clin Invest 2004, 114, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Townley, A.R.; Wheatley, S.P. Mitochondrial survivin reduces oxidative phosphorylation in cancer cells by inhibiting mitophagy. J Cell Sci 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Xia, F.; Altieri, D.C. Compartmentalized phosphorylation of IAP by protein kinase A regulates cytoprotection. Mol Cell 2007, 27, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Rivadeneira, D.B.; Caino, M.C.; Seo, J.H.; Angelin, A.; Wallace, D.C.; Languino, L.R.; Altieri, D.C. Survivin promotes oxidative phosphorylation, subcellular mitochondrial repositioning, and tumor cell invasion. Sci Signal 2015, 8, ra80. [Google Scholar] [CrossRef]
- Hagenbuchner, J.; Kuznetsov, A.V.; Obexer, P.; Ausserlechner, M.J. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene 2013, 32, 4748–4757. [Google Scholar] [CrossRef]
- Niu, T.K.; Cheng, Y.; Ren, X.; Yang, J.M. Interaction of Beclin 1 with survivin regulates sensitivity of human glioma cells to TRAIL-induced apoptosis. FEBS Lett 2010, 584, 3519–3524. [Google Scholar] [CrossRef]
- Maskey, D.; Yousefi, S.; Schmid, I.; Zlobec, I.; Perren, A.; Friis, R.; Simon, H.U. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun 2013, 4, 2130. [Google Scholar] [CrossRef]
- Lin, T.Y.; Chan, H.H.; Chen, S.H.; Sarvagalla, S.; Chen, P.S.; Coumar, M.S.; Cheng, S.M.; Chang, Y.C.; Lin, C.H.; Leung, E.; et al. BIRC5/Survivin is a novel ATG12-ATG5 conjugate interactor and an autophagy-induced DNA damage suppressor in human cancer and mouse embryonic fibroblast cells. Autophagy 2020, 16, 1296–1313. [Google Scholar] [CrossRef]
- Hagenbuchner, J.; Kiechl-Kohlendorfer, U.; Obexer, P.; Ausserlechner, M.J. BIRC5/Survivin as a target for glycolysis inhibition in high-stage neuroblastoma. Oncogene 2016, 35, 2052–2061. [Google Scholar] [CrossRef]
- Colnaghi, R.; Connell, C.M.; Barrett, R.M.; Wheatley, S.P. Separating the anti-apoptotic and mitotic roles of survivin. J Biol Chem 2006, 281, 33450–33456. [Google Scholar] [CrossRef] [PubMed]
- Knauer, S.K.; Bier, C.; Habtemichael, N.; Stauber, R.H. The Survivin-Crm1 interaction is essential for chromosomal passenger complex localization and function. EMBO Rep 2006, 7, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Stauber, R.H.; Rabenhorst, U.; Rekik, A.; Engels, K.; Bier, C.; Knauer, S.K. Nucleocytoplasmic shuttling and the biological activity of mouse survivin are regulated by an active nuclear export signal. Traffic 2006, 7, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Knauer, S.K.; Bier, C.; Schlag, P.; Fritzmann, J.; Dietmaier, W.; Rodel, F.; Klein-Hitpass, L.; Kovacs, A.F.; Doring, C.; Hansmann, M.L.; et al. The survivin isoform survivin-3B is cytoprotective and can function as a chromosomal passenger complex protein. Cell Cycle 2007, 6, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Temme, A.; Rodriguez, J.A.; Hendruschk, S.; Gunes, S.; Weigle, B.; Schakel, K.; Schmitz, M.; Bachmann, M.; Schackert, G.; Rieber, E.P. Nuclear localization of Survivin renders HeLa tumor cells more sensitive to apoptosis by induction of p53 and Bax. Cancer Lett 2007, 250, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Connell, C.M.; Colnaghi, R.; Wheatley, S.P. Nuclear survivin has reduced stability and is not cytoprotective. J Biol Chem 2008, 283, 3289–3296. [Google Scholar] [CrossRef] [PubMed]
- Okada, E.; Murai, Y.; Matsui, K.; Isizawa, S.; Cheng, C.; Masuda, M.; Takano, Y. Survivin expression in tumor cell nuclei is predictive of a favorable prognosis in gastric cancer patients. Cancer Lett 2001, 163, 109–116. [Google Scholar] [CrossRef]
- Kennedy, S.M.; O’Driscoll, L.; Purcell, R.; Fitz-Simons, N.; McDermott, E.W.; Hill, A.D.; O’Higgins, N.J.; Parkinson, M.; Linehan, R.; Clynes, M. Prognostic importance of survivin in breast cancer. Br J Cancer 2003, 88, 1077–1083. [Google Scholar] [CrossRef]
- Vischioni, B.; van der Valk, P.; Span, S.W.; Kruyt, F.A.; Rodriguez, J.A.; Giaccone, G. Nuclear localization of survivin is a positive prognostic factor for survival in advanced non-small-cell lung cancer. Ann Oncol 2004, 15, 1654–1660. [Google Scholar] [CrossRef]
- Mohamed, S.; Yasufuku, K.; Nakajima, T.; Hiroshima, K.; Chiyo, M.; Yoshida, S.; Suzuki, M.; Sekine, Y.; Shibuya, K.; Agamy, G.; et al. Nuclear Survivin in pN2 Non-small Cell Lung Cancer: Prognostic and Clinical Implications. Eur Respir J 2008.
- Shinohara, E.T.; Gonzalez, A.; Massion, P.P.; Chen, H.; Li, M.; Freyer, A.S.; Olson, S.J.; Andersen, J.J.; Shyr, Y.; Carbone, D.P.; et al. Nuclear survivin predicts recurrence and poor survival in patients with resected nonsmall cell lung carcinoma. Cancer 2005, 103, 1685–1692. [Google Scholar] [CrossRef]
- Shirai, K.; Suzuki, Y.; Oka, K.; Noda, S.E.; Katoh, H.; Suzuki, Y.; Itoh, J.; Itoh, H.; Ishiuchi, S.; Sakurai, H.; et al. Nuclear survivin expression predicts poorer prognosis in glioblastoma. J Neurooncol 2008. [CrossRef] [PubMed]
- Preuss, S.F.; Weinell, A.; Molitor, M.; Stenner, M.; Semrau, R.; Drebber, U.; Weissenborn, S.J.; Speel, E.J.; Wittekindt, C.; Guntinas-Lichius, O.; et al. Nuclear survivin expression is associated with HPV-independent carcinogenesis and is an indicator of poor prognosis in oropharyngeal cancer. Br J Cancer 2008, 98, 627–632. [Google Scholar] [CrossRef]
- Kim, J.; McNiff, J.M. Nuclear expression of survivin portends a poor prognosis in Merkel cell carcinoma. Mod Pathol 2008, 21, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, P.; Griss, S.; Arnold, C.N.; Horsch, D.; Goke, R.; Arnold, R.; Heine, B.; Stein, H.; Zeitz, M.; Scherubl, H. Nuclear survivin is a powerful novel prognostic marker in gastroenteropancreatic neuroendocrine tumor disease. Neuroendocrinology 2005, 81, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chantalat, L.; Skoufias, D.A.; Kleman, J.P.; Jung, B.; Dideberg, O.; Margolis, R.L. Crystal structure of human survivin reveals a bow tie-shaped dimer with two unusual alpha-helical extensions. Mol Cell 2000, 6, 183–189. [Google Scholar] [CrossRef]
- Verdecia, M.A.; Huang, H.; Dutil, E.; Kaiser, D.A.; Hunter, T.; Noel, J.P. Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat Struct Biol 2000, 7, 602–608. [Google Scholar] [CrossRef]
- Muchmore, S.W.; Chen, J.; Jakob, C.; Zakula, D.; Matayoshi, E.D.; Wu, W.; Zhang, H.; Li, F.; Ng, S.C.; Altieri, D.C. Crystal structure and mutagenic analysis of the inhibitor-of-apoptosis protein survivin. Mol Cell 2000, 6, 173–182. [Google Scholar] [CrossRef]
- Sun, C.; Nettesheim, D.; Liu, Z.; Olejniczak, E.T. Solution structure of human survivin and its binding interface with Smac/Diablo. Biochemistry 2005, 44, 11–17. [Google Scholar] [CrossRef]
- Vong, Q.P.; Cao, K.; Li, H.Y.; Iglesias, P.A.; Zheng, Y. Chromosome alignment and segregation regulated by ubiquitination of survivin. Science 2005, 310, 1499–1504. [Google Scholar] [CrossRef]
- Jeyaprakash, A.A.; Klein, U.R.; Lindner, D.; Ebert, J.; Nigg, E.A.; Conti, E. Structure of a Survivin-Borealin-INCENP core complex reveals how chromosomal passengers travel together. Cell 2007, 131, 271–285. [Google Scholar] [CrossRef]
- Barrett, R.M.; Osborne, T.P.; Wheatley, S.P. Phosphorylation of survivin at threonine 34 inhibits its mitotic function and enhances its cytoprotective activity. Cell Cycle 2009, 8, 278–283. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.S.; Grossman, D.; Plescia, J.; Li, F.; Zhang, H.; Villa, A.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A 2000, 97, 13103–13107. [Google Scholar] [CrossRef]
- Wheatley, S.P.; Barrett, R.M.; Andrews, P.D.; Medema, R.H.; Morley, S.J.; Swedlow, J.R.; Lens, S.M. Phosphorylation by aurora-B negatively regulates survivin function during mitosis. Cell Cycle 2007, 6, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.M.; Colnaghi, R.; Wheatley, S.P. Threonine 48 in the BIR domain of survivin is critical to its mitotic and anti-apoptotic activities and can be phosphorylated by CK2 in vitro. Cell Cycle 2011, 10, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Colnaghi, R.; Wheatley, S.P. Liaisons between survivin and Plk1 during cell division and cell death. J Biol Chem 2010, 285, 22592–22604. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Holloway, M.P.; Ma, L.; Cooper, Z.A.; Riolo, M.; Samkari, A.; Elenitoba-Johnson, K.S.; Chin, Y.E.; Altura, R.A. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J Biol Chem 2010, 285, 36129–36137. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Kita, A.; Yamanaka, K.; Mori, M.; Amino, N.; Takeuchi, M.; Tominaga, F.; Hatakeyama, S.; Kinoyama, I.; Matsuhisa, A.; et al. YM155, a novel small-molecule survivin suppressant, induces regression of. Cancer Res 2007, 67, 8014–8021. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, T.; Okamoto, I.; Suzuki, M.; Nakahara, T.; Yamanaka, K.; Hatashita, E.; Yamada, Y.; Fukuoka, M.; Ono, K.; Nakagawa, K. Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in. Clin Cancer Res 2008, 14, 6496–6504. [Google Scholar] [CrossRef]
- Arora, R.; Shuda, M.; Guastafierro, A.; Feng, H.; Toptan, T.; Tolstov, Y.; Normolle, D.; Vollmer, L.L.; Vogt, A.; Domling, A.; et al. Survivin is a therapeutic target in merkel cell carcinoma. Sci Transl Med 2012, 4, 133ra156. [Google Scholar] [CrossRef] [PubMed]
- Glaros, T.G.; Stockwin, L.H.; Mullendore, M.E.; Smith, B.; Morrison, B.L.; Newton, D.L. The "survivin suppressants" NSC 80467 and YM155 induce a DNA damage response. Cancer Chemother Pharmacol 2012. [CrossRef]
- Giaccone, G.; Zatloukal, P.; Roubec, J.; Floor, K.; Musil, J.; Kuta, M.; van Klaveren, R.J.; Chaudhary, S.; Gunther, A.; Shamsili, S. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol 2009, 27, 4481–4486. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Thomas, A.; Rajan, A.; Chun, G.; Lopez-Chavez, A.; Szabo, E.; Spencer, S.; Carter, C.A.; Guha, U.; Khozin, S.; et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann Oncol 2013, 24, 2601–2606. [Google Scholar] [CrossRef]
- Satoh, T.; Okamoto, I.; Miyazaki, M.; Morinaga, R.; Tsuya, A.; Hasegawa, Y.; Terashima, M.; Ueda, S.; Fukuoka, M.; Ariyoshi, Y.; et al. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin Cancer Res 2009, 15, 3872–3880. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Quinn, D.I.; Ferrari, A.; Ahmann, F.; Giaccone, G.; Drake, T.; Keating, A.; de Bono, J.S. A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Ann Oncol 2012, 23, 968–973. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Mita, A.; Lewis, L.D.; Garrett, C.R.; Till, E.; Daud, A.I.; Patnaik, A.; Papadopoulos, K.; Takimoto, C.; Bartels, P.; et al. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J Clin Oncol 2008, 26, 5198–5203. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Zhang, L.; Boufraqech, M.; Liu-Chittenden, Y.; Zhang, Y.; Patel, D.; Davis, S.; Rosenberg, A.; Ylaya, K.; Aufforth, R.; et al. Inhibition of Survivin with YM155 Induces Durable Tumor Response in Anaplastic Thyroid Cancer. Clin Cancer Res 2015, 21, 4123–4132. [Google Scholar] [CrossRef]
- Ling, X.; Cao, S.; Cheng, Q.; Keefe, J.T.; Rustum, Y.M.; Li, F. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS One 2012, 7, e45571. [Google Scholar] [CrossRef]
- Yin, H.; Que, R.; Liu, C.; Ji, W.; Sun, B.; Lin, X.; Zhang, Q.; Zhao, X.; Peng, Z.; Zhang, X.; et al. Survivin-targeted drug screening platform identifies a matrine derivative WM-127 as a potential therapeutics against hepatocellular carcinoma. Cancer Lett 2018, 425, 54–64. [Google Scholar] [CrossRef]
- Chang, C.C.; Heller, J.D.; Kuo, J.; Huang, R.C. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. Proc Natl Acad Sci U S A 2004, 101, 13239–13244. [Google Scholar] [CrossRef]
- Shi, X.; Wang, D.; Ding, K.; Lu, Z.; Jin, Y.; Zhang, J.; Pan, J. GDP366, a novel small molecule dual inhibitor of survivin and Op18, induces cell growth inhibition, cellular senescence and mitotic catastrophe in human cancer cells. Cancer Biol Ther 2010, 9, 640–650. [Google Scholar] [CrossRef]
- Fortugno, P.; Beltrami, E.; Plescia, J.; Fontana, J.; Pradhan, D.; Marchisio, P.C.; Sessa, W.C.; Altieri, D.C. Regulation of survivin function by Hsp90. Proc Natl Acad Sci U S A 2003, 100, 13791–13796. [Google Scholar] [CrossRef]
- Plescia, J.; Salz, W.; Xia, F.; Pennati, M.; Zaffaroni, N.; Daidone, M.G.; Meli, M.; Dohi, T.; Fortugno, P.; Nefedova, Y.; et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005, 7, 457–468. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, Q.; Wu, Z.; Tian, X.; Yan, H.; Wang, B.; Dong, P.; Watari, H.; Pfeffer, L.M.; Guo, Y.; et al. Ovarian Primary and Metastatic Tumors Suppressed by Survivin Knockout or a Novel Survivin Inhibitor. Mol Cancer Ther 2019, 18, 2233–2245. [Google Scholar] [CrossRef]
- Wang, J.; Li, W. Discovery of novel second mitochondria-derived activator of caspase mimetics as selective inhibitor of apoptosis protein inhibitors. J Pharmacol Exp Ther 2014, 349, 319–329. [Google Scholar] [CrossRef]
- Oikawa, T.; Unno, Y.; Matsuno, K.; Sawada, J.; Ogo, N.; Tanaka, K.; Asai, A. Identification of a small-molecule inhibitor of the interaction between Survivin and Smac/DIABLO. Biochem Biophys Res Commun 2010, 393, 253–258. [Google Scholar] [CrossRef]
- Park, S.H.; Shin, I.; Park, S.H.; Kim, N.D.; Shin, I. An Inhibitor of the Interaction of Survivin with Smac in Mitochondria Promotes Apoptosis. Chem Asian J 2019, 14, 4035–4041. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.D.; Sun, C.; Kunzer, A.; Sauer, D.; Sarris, K.; Hoff, E.; Yu, L.; Nettesheim, D.G.; Chen, J.; Jin, S.; et al. Discovery of a novel small molecule binding site of human survivin. Bioorg Med Chem Lett 2007, 17, 3122–3129. [Google Scholar] [CrossRef] [PubMed]
- Guvenc, H.; Pavlyukov, M.S.; Joshi, K.; Kurt, H.; Banasavadi-Siddegowda, Y.K.; Mao, P.; Hong, C.; Yamada, R.; Kwon, C.H.; Bhasin, D.; et al. Impairment of glioma stem cell survival and growth by a novel inhibitor for Survivin-Ran protein complex. Clin Cancer Res 2013, 19, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Chettiar, S.N.; Cooley, J.V.; Park, I.H.; Bhasin, D.; Chakravarti, A.; Li, P.K.; Li, C.; Jacob, N.K. Design, synthesis and biological studies of survivin dimerization modulators that prolong mitotic cycle. Bioorg Med Chem Lett 2013, 23, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Steigerwald, C.; Rasenberger, B.; Christmann, M.; Tomicic, M.T. Sensitization of colorectal cancer cells to irinotecan by the Survivin inhibitor LLP3 depends on XAF1 proficiency in the context of mutated p53. Arch Toxicol 2018, 92, 2645–2648. [Google Scholar] [CrossRef] [PubMed]
- Berezov, A.; Cai, Z.; Freudenberg, J.A.; Zhang, H.; Cheng, X.; Thompson, T.; Murali, R.; Greene, M.I.; Wang, Q. Disabling the mitotic spindle and tumor growth by targeting a cavity-induced allosteric site of survivin. Oncogene 2012, 31, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Nagatomo, I.; Suzuki, E.; Mizuno, T.; Kumagai, T.; Berezov, A.; Zhang, H.; Karlan, B.; Greene, M.I.; Wang, Q. YAP modifies cancer cell sensitivity to EGFR and survivin inhibitors and is negatively regulated by the non-receptor type protein tyrosine phosphatase 14. Oncogene 2013, 32, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Murali, R.; Cheng, X.; Berezov, A.; Du, X.; Schon, A.; Freire, E.; Xu, X.; Chen, Y.H.; Greene, M.I. Disabling TNF receptor signaling by induced conformational perturbation of tryptophan-107. Proc Natl Acad Sci U S A 2005, 102, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Fuchigami, T.; Mizoguchi, T.; Yoshida, S.; Haratake, M.; Nakayama, M. Synthesis and characterization of radioiodinated 3-phenethyl-2-indolinone derivatives for SPECT imaging of survivin in tumors. Bioorg Med Chem 2018, 26, 3111–3116. [Google Scholar] [CrossRef]
- Qi, J.; Dong, Z.; Liu, J.; Peery, R.C.; Zhang, S.; Liu, J.Y.; Zhang, J.T. Effective Targeting of the Survivin Dimerization Interface with Small-Molecule Inhibitors. Cancer Res 2016, 76, 453–462. [Google Scholar] [CrossRef]
- Peery, R.; Kyei-Baffour, K.; Dong, Z.; Liu, J.; de Andrade Horn, P.; Dai, M.; Liu, J.Y.; Zhang, J.T. Synthesis and Identification of a Novel Lead Targeting Survivin Dimerization for Proteasome-Dependent Degradation. J Med Chem 2020, 63, 7243–7251. [Google Scholar] [CrossRef]
- Andersen, M.H.; Pedersen, L.O.; Becker, J.C.; Straten, P.T. Identification of a cytotoxic T lymphocyte response to the apoptosis inhibitor protein survivin in cancer patients. Cancer Res 2001, 61, 869–872. [Google Scholar]
- Hirohashi, Y.; Torigoe, T.; Maeda, A.; Nabeta, Y.; Kamiguchi, K.; Sato, T.; Yoda, J.; Ikeda, H.; Hirata, K.; Yamanaka, N.; et al. An HLA-A24-restricted cytotoxic T lymphocyte epitope of a tumor-associated protein, survivin. Clin Cancer Res 2002, 8, 1731–1739. [Google Scholar]
- Xiang, R.; Mizutani, N.; Luo, Y.; Chiodoni, C.; Zhou, H.; Mizutani, M.; Ba, Y.; Becker, J.C.; Reisfeld, R.A. A DNA vaccine targeting survivin combines apoptosis with suppression of angiogenesis in lung tumor eradication. Cancer Res 2005, 65, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Onodi, F.; Maherzi-Mechalikh, C.; Mougel, A.; Ben Hamouda, N.; Taboas, C.; Gueugnon, F.; Tran, T.; Nozach, H.; Marcon, E.; Gey, A.; et al. High Therapeutic Efficacy of a New Survivin LSP-Cancer Vaccine Containing CD4(+) and CD8(+) T-Cell Epitopes. Front Oncol 2018, 8, 517. [Google Scholar] [CrossRef]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother 2016, 65, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J Clin Oncol 2023, 41, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, M.J.; Ahluwalia, M.S.; Munich, S.A.; Orton, M.; Barone, T.; Chanan-Khan, A.; Fenstermaker, R.A. Antitumor cytotoxic T-cell response induced by a survivin peptide mimic. Cancer Immunol Immunother 2010, 59, 1211–1221. [Google Scholar] [CrossRef]
- Locke, F.L.; Menges, M.; Veerapathran, A.; Coppola, D.; Gabrilovich, D.; Anasetti, C. Survivin-specific CD4+ T cells are decreased in patients with survivin-positive myeloma. J Immunother Cancer 2015, 3, 20. [Google Scholar] [CrossRef]
- Freeman, C.L.; Atkins, R.; Varadarajan, I.; Menges, M.; Edelman, J.; Baz, R.; Brayer, J.; Castaneda Puglianini, O.; Ochoa-Bayona, J.L.; Nishihori, T.; et al. Survivin Dendritic Cell Vaccine Safely Induces Immune Responses and Is Associated with Durable Disease Control after Autologous Transplant in Patients with Myeloma. Clin Cancer Res 2023, OF1–OF11. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



