
Submitted:
21 March 2024
Posted:
22 March 2024
You are already at the latest version
Abstract
Metformin, a widely used anti-diabetic drug, has garnered attention for its potential in cancer management, particularly in breast and colorectal cancer. It is established that metformin reduces mitochondrial respiration, but its specific molecular targets within mitochondria vary. Proposed mechanisms include inhibiting mitochondrial respiratory chain Complex I and/or Complex IV, and mitochondrial glycerophosphate dehydrogenase, among others. These actions lead to cellular energy deficits, redox state changes, and several molecular changes that reduce hyperglycemia in type 2 diabetic patients.
Clinical evidence supports metformin’s role in cancer prevention in type 2 diabetes mellitus patients. Moreover, in these patients with breast and colorectal cancer, metformin consumption leads to an improvement of survival outcomes and prognosis. The synergistic effects of metformin with chemotherapy and immunotherapy highlights its potential as an adjunctive therapy for breast and colorectal cancer. However, nuanced findings underscore the need for further research and stratification by molecular subtype, particularly for breast cancer.
This comprehensive review integrates metformin-related findings from epidemiological, clinical, and preclinical studies in breast and colorectal cancer. Here, we discuss current research addressed to define metformin's bioavailability and efficacy, exploring novel metformin-based compounds and drug delivery systems, including derivatives targeting mitochondria, combination therapies, and novel nanoformulations, showing enhanced anticancer effects.
Keywords:
Metformin
; Diabetes
; Mitochondria
; Breast Cancer
; Colorectal Cancer
1. Introduction
Metformin is widely recognized as a primary medication in the global management of type 2 diabetes mellitus (T2DM), earning its inclusion on the World Health Organization’s list of essential medicines since 2019 [1]. Its low cost, excellent tolerability, and safety profile, with low risk of hypoglycemia, make it a preferred choice either alone or in combination with other drugs for managing millions of patients with T2DM [2]. Metformin belongs to the biguanide family (1,1-dimethylbiguanida hydrochloride) originating from the plant known as French lilac (Galega officinalis) [3]. Historically, this plant has been utilized in Europe since the Middle Ages to alleviate symptoms of diabetes mellitus, owing to its abundance of galegine, an isoprenyl guanidine compound. While monoguanidines and diguanidines exhibit toxicity, biguanides, composed of two N-linked guanidine molecules, have been employed for diabetes treatment since the late 1950s [4]. Metformin gained approval for use in Europe and Canada in 1957, not being introduced in the United States until 1995 [3]. The more potent biguanides, phenformin and buformin, were widely used in the United States and Europe in the 1960s, however, they were withdrawn from the market in the late 1970s due to a higher risk of lactic acidosis compared to metformin (approximately 3-9 cases per 100,000 person-years). Notably, the risk of lactic acidosis is elevated in patients with chronically impaired renal function or acute kidney disease, populations for whom metformin is contraindicated [5]. Consequently, the most prevalent adverse effect associated with metformin is gastrointestinal intolerance [6].
Although metformin has been administered to millions of patients with T2DM for over 60 years, the exact mechanism (or mechanisms) of action remains a subject of debate. Metformin is described as an anti-hyperglycemic agent that does not induce clinical hypoglycemia in patients with T2DM or disturb glucose homeostasis in non-diabetic individuals [2,7]. It is known that metformin primarily acts by suppressing hepatic glucose production, which is increased in individuals with T2DM, through a decrease of 25-40% in the hepatic gluconeogenesis rate [8]. Additionally, some euglycemic-hyperinsulinemic clamp studies suggest it may also have a beneficial effect on insulin sensitivity at the skeletal muscle level, although this effect is not consistently observed across all studies [9,10,11,12]. In recent years, a growing body of evidence points to the gut as a key target of metformin action, promoting glucose utilization, GDF15 secretion, which reduces appetite, and regulating intestinal microbiota, all of which collectively contribute to its potential benefits (reviewed in (Barroso et al., 2023) [13]).
Controversy surrounds the primary targets of metformin to promote the reduction in hepatic gluconeogenesis. For over 50 years, it has been known that biguanides decrease mitochondrial respiration, thus placing mitochondria at the core of their action [14]. However, the precise molecular targets within this organelle and their subsequent effects are diverse, leading to a plethora of proposed mechanisms, some of which may overlap, in explaining metformin’s antigluconeogenic effects in the liver. Notably, inhibition of mitochondrial respiratory chain Complex I and mitochondrial glycerophosphate dehydrogenase are prominent among these targets, although other respiratory chain complexes have also been suggested as potential targets [15,16,17]. Metformin’s impact on these targets manifests in a complex array of interconnected effects, including cellular energy deficits, changes in the redox state, activation of AMP-activated protein kinase (AMPK), inhibition of cAMP-mediated glucagon signaling, allosteric modulation of gluconeogenic enzymes, as well as epigenetic alterations [18,19]. Untangling which of these events truly drives the reduction in hepatic gluconeogenesis induced by metformin remains a subject of intense debate. Moreover, the fact that many of metformin’s effects seen in in vitro and preclinical studies occur at suprapharmacological concentrations, coupled with discrepancies in effects between acute and chronic administration in in vivo models, further complicates the interpretation of these findings and, consequently, the elucidation of the underlying molecular mechanisms of this drug. Investigation into the underlying molecular mechanisms of action of metformin has garnered significant interest beyond its applications in diabetes treatment. Metformin’s capacity to alter cellular bioenergetics and modulate crucial aspects of mitochondrial function, such as oxidative stress and apoptosis, has sparked curiosity about its potential repurposing in treating various diseases [18]. Notably, there is growing enthusiasm for exploring metformin’s potential as a therapeutic agent against certain types of cancer, such as breast and colon cancer [20,21,22]. One of the hallmarks of cancer is the reprogramming of cellular energy metabolism, allowing tumor cells to sustain continuous growth and proliferation by substituting the metabolic program typically found in normal tissues. Drugs like metformin, with the ability to exploit specific metabolic vulnerabilities in tumor cells, present a promising avenue for cancer treatment [23]. Indeed, lower incidences of certain types of cancer and/or improved overall survival has been reported in T2DM patients treated with metformin [22,24,25]. Repurposing existing drugs for other diseases offers significant time and cost-saving advantages, as their pharmacokinetics, pharmacodynamics, and safety profiles are already established, thus allowing preclinical studies to be streamlined. Given the considerable interest in metformin, this review aims to delve into the current understanding of its mechanisms of action and its potential applications in breast and colon cancer treatment. Furthermore, we will examine the primary challenges associated with repurposing metformin for cancer therapy and discuss the strategies being contemplated to address these challenges.
1.1. Pharmacokinetics
Metformin is characterized chemically as a highly hydrophilic compound with an acid dissociation constant (pKa) of 11.5, meaning that at physiological pH the drug exists as a monoprotonated cation. The presence of charge at physiological pH results in two main consequences: drug transport across biological membranes involves uptake via specific transporters, and organelles such as energized mitochondria, can slowly accumulate the drug driven by their transmembrane electrochemical potential (Δψ) [26]. Thus, it is known that the absorption, distribution, and excretion of metformin primarily rely on organic cation transporters (OCTs), multidrug and toxin extruders (MATEs), and plasma membrane monoamine transporter (PMAT) [27,28].
Metformin is typically administered orally and exhibits a low bioavailability ranging from 40% to 60% [29]. The drug is not metabolized and is excreted unchanged through urine. In the treatment of T2DM, patients are typically prescribed a dosage of 25-30 mg/kg per day, typically divided into two or three oral doses of 500-850 mg each. However, it’s important to note that the absorption of metformin is reliant on specific transporters and, therefore, administering higher doses can slow the absorption rate and decrease overall bioavailability [30]. In mouse models, higher doses (200-250 mg/kg) are often necessary due to their more efficient renal clearance, resulting in a shorter half-life of 1-2 hours compared to the 4-9 hours observed in humans [31]. Biodistribution studies in humans, employing positron emission tomography (PET) with 11C-labeled metformin, demonstrate its primary distribution in the small intestine, liver, and kidneys. This distribution pattern aligns with both the expression profile of previously discussed specific transporters and the major target organs (liver and gut), as well as its high rate of renal elimination [27,32]. Following intestinal absorption, metformin attains high concentrations in the portal vein (40-70 µM), leading to the accumulation of higher levels of metformin in the liver than in surrounding organs, as confirmed by PET in in both humans and mice [17,33,34]. It is noteworthy that studies in mice indicate that the liver can reach concentrations even higher than those in portal vein plasma [35].
In this sense, it is important to emphasize that antineoplastic effects of metformin depend on drug concentration within neoplastic tissue. This concentration is influenced not only by plasma level, but also by cellular uptake in cancer cells, which depends on the expression of relevant transporters including OCT1 [36]. It is worth noting that serum levels of metformin achieved in diabetic patients and in vivo models are in the micromolar range, while in vitro antitumoral activity is observed at millimolar concentrations [37]. Hence, a fundamental research inquiry is to determine the metformin concentrations achieved in tumors of patients receiving conventional antidiabetic metformin dose.
2. Mechanisms of Action
2.1. Complex I Inhibition
In the early 2000s, the inhibition of the mitochondrial respiratory chain Complex I emerged as a prominent mechanism explaining metformin’s anti-gluconeogenic effect in the liver of T2DM patients. Metformin binds to Complex I, inducing a mild and reversible non-competitive inhibition [26,38], contrary to typical inhibitors of Complex I, such as rotenone and piericidin A, which are uncharged, hydrophobic molecules that exhibit high efficiency (IC50 around 2μM), being able to halve Complex I activity at very low concentrations [39].
The precise mechanism by which metformin targets Complex I and exerts its function remains under investigation. In 2014, Bridges et al. [40] demonstrated that metformin interacts with the Cys39-containing matrix loop of the respiratory chain subunit ND3. They observed that metformin binds to Complex I in a deactive-like open-loop conformation, stabilizing the enzyme in an inactive state. Consequently, this inhibition leads to a lower proton gradient and reduced ATP synthesis due to diminished NADH oxidation, proton pumping across the inner mitochondrial membrane, and oxygen consumption rates. In 2023, the same group identified up to three potential binding sites for biguanides within various protein subunits of Complex I [41]. The primary inhibitory site is situated within the amphipathic region of the quinone binding channel (Q-channel), adjacent to a mobile structural element within the NDUFS7 subunit [18]. Mechanistically, these studies revealed that synthetic biguanide binding at this site prevents Complex I reactivation. Consequently, this inhibition leads to a moderate reduction in ATP synthesis in the liver, as NADH cannot transfer its electrons to Complex I. This limitation thereby reduces mitochondrial respiratory chain activity, leading to a decrease in the ATP-dependent gluconeogenic process (Figure 1).
Several mechanisms have been proposed to explain how Complex I inhibition decreases hepatic gluconeogenesis (Figure 1). These include alterations in hepatic energy state characterized by a moderate decrease in ATP/AMP and ATP/ADP ratios. These hepatic energy alterations underlie the inhibition of gluconeogenesis through AMP-activated protein kinase (AMPK)-dependent and AMPK-independent pathways. Indeed, given that gluconeogenesis is a highly energy-consuming process, the hepatic energy charge alteration could be sufficient to reduce gluconeogenic pathway [42,43,44].
2.2. AMPK-Dependent Mechanisms
AMPK activation may be one of the mechanisms explaining metformin’s effect on gluconeogenesis. It is suggested that alterations in hepatic energy status and increased AMP levels induce allosteric activation of AMPK [45,46], which leads to the phosphorylation and nuclear exclusion of the cAMP response element-binding protein (CREB)-regulated transcription co-activator 2 (CRTC2) with the subsequent gluconeogenic gene inhibition [47]. Of note, other mechanisms to inactivate CRTC2 by metformin have been described [34,48]. AMPK has also been shown to enhance the upregulation of the orphan nuclear receptor small heterodimer partner (SHP), which through direct or indirect interaction with CREB, suppresses CREB-dependent gene expression involved in hepatic gluconeogenesis [48].
AMPK activation by metformin remains a subject of controversy, with some studies suggesting its independence from energy status, as indicated by findings that activation relies on liver kinase B1 (LKB1) or calcium/calmodulin protein kinase kinase 2 (CAMKKbeta) [45]. Nonetheless, Foretz et al. (2019) [4], demonstrated that LKB1 knockout hepatocytes still respond to metformin’s effects. Furthermore, they demonstrate that specific deletion of hepatic AMPK does not suffice to impede metformin’s action, suggesting the existence of AMPK-independent mechanisms facilitating metformin’s efficacy. Controversy surrounds these findings due to variations in methodology, using different concentrations and experimental models, particularly in in vitro studies where metformin concentrations often exceed those used in clinical practice (millimolar vs. micromolar range). Noteworthy is the fact that only suprapharmacological concentrations of metformin led to Complex I inhibition in isolated mitochondria [49]. Consequently, extrapolating these results to clinical scenarios becomes challenging. In addition, discrepancies extend to the localization of metformin within hepatic cells, with some studies proposing mitochondrial accumulation and others emphasizing cytosolic predominance [50]. The prevailing accepted hypothesis regarding metformin accumulation in the mitochondrial matrix suggests that, due to its positive charge, it slowly enters energized mitochondria driven by the electrochemical gradient. Consequently, despite existing at micromolar concentrations in the cytosol, metformin can reach millimolar concentrations within the matrix. This gradual accumulation is believed to contribute to the mild inhibitory effect observed on Complex I and modulate the drug’s impact. Finally, its own concentration into the mitochondrial matrix drives a reduction in the membrane potential, thereby self-limiting further accumulation [15,26,40].
Metformin-induced AMPK activation may reduce gluconeogenesis by enhancing hepatic insulin sensitivity. This activation leads to the phosphorylation and inhibition of acetyl-CoA carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2), resulting in decreased hepatic lipogenesis. Additionally, it promotes hepatic fatty acid oxidation by activating carnitine palmitoyltransferase 1 (CPT1), facilitating the transport of acyl-CoA into mitochondria [19]. As a result, metformin treatment would alleviate hepatic steatosis, thereby enhancing insulin sensitivity and consequently, insulin’s ability to inhibit gluconeogenesis.
2.2.1. AMPK-Dependent Lysosomal Pathway
Low doses of metformin have been reported to activate AMPK through an AMP-independent mechanism involving lysosomal protein complex v-ATPase-Ragulator in hepatocytes (Figure 2). This complex was described as an endosomal docking site for AXIN/LKB1 to mediate AMPK activation [51,52]. This mechanism relies on the interaction of presenilin enhancer 2 (PEN2) with the biguanide moiety of metformin, PEN2 is then recruited to the accessory protein v-ATPase ATP6P1 and v-ATPase is inhibited. As a result of v-ATPase inhibition, AXIN-LKB1 translocation to the lysosome surface is promoted, which leads to AMPK activation in the lysosome [53]. This novel mechanism may be particularly relevant in the intestine and liver, since the deletion of PEN2 abolishes the beneficial effects of glucagon-like peptide 1 (GLP1) secretion in mice enterocytes and blunts the reduction in hepatic lipid content in mice fed a high fat diet.
This AMP-independent mechanism is of key importance, as it explains how low doses of metformin can activate AMPK, while AMP-dependent mechanisms require higher doses. As previously emphasized by Foretz et al. in a prior review, numerous unanswered questions remain to be addressed. This is particularly relevant, given that lysosomal activation of AMPK could potentially elucidate the effects of low doses of metformin in the intestine but not in the liver [18].
2.3. AMPK-Independent Mechanisms
Metformin also diminishes hepatic gluconeogenesis by directly inhibiting mitochondrial glycerol-3-phosphate dehydrogenase (mGPDH) in an energy- and AMPK-independent manner [17,54]. This enzyme, a component of the glycerol-phosphate shuttle system, works in conjunction with its cytosolic isoform (cGPDH) to channel electrons from cytosolic NADH to the mitochondrial respiratory chain (Figure 3). Thus, inhibition of mGPDH leads to the accumulation of NADH in the cytosol, which hinders gluconeogenesis from redox-dependent substrates such as glycerol and lactate, but not from redox-independent substrates like pyruvate, alanine, and DHAP. It is noteworthy that this selective inhibition of certain substrates’ utilization for gluconeogenesis may explain why metformin has a low risk of inducing hypoglycemia or does not alter gluconeogenesis in non-diabetic patients, where amino acid contribution to gluconeogenesis is higher. Nonetheless, some studies suggest that inhibiting the glycerol-phosphate shuttle alone in the liver may not be sufficient to lower blood glucose levels. This discrepancy may result from the liver’s reliance on the malate-aspartate shuttle as the primary NADH transporter, which effectively reduces blood glucose levels when inhibited by metformin [55,56,57].
The mechanism by which metformin increases the NADH/NAD+ ratio remains debated. Conflicting findings suggest that this effect may stem from Complex I or mGPDH inhibition. Alternative proposals have emerged, including one positing that metformin accumulation in the mitochondrial matrix depolarizes the membrane, inhibiting the aspartate transporter of the aspartate-malate shuttle. Consequently, this inhibition may stimulate glycerol-phosphate shuttle activity, reducing glycerol-3-phosphate (G3P) concentration, a potent allosteric inhibitor of phosphofructokinase 1 (PFK1), thus alleviating its inhibition and favoring glycolytic activity over gluconeogenesis [33].
Another AMPK-independent mechanism is based on the fact that the mild increase in intracellular AMP levels caused by metformin is sufficient to decrease hepatic gluconeogenesis stimulated by glucagon. AMP inhibits the glucagon-induced activation of adenylate cyclase, thereby reducing intracellular cyclic AMP levels. This action diminishes protein kinase A (PKA) activity and lowers the phosphorylation levels of key gluconeogenesis-related enzymes such as fructose-2,6-biphosphatase 1, inositol trisphosphate receptor, and CREB1 [43,44]. Consequently, these events result in a reduction in glucagon-stimulated glucose production. Additionally, Miller et al. proposed that AMP allosterically inhibits fructose-1,6-biphosphatase, a key gluconeogenic enzyme [58]. Indeed, a recent study revealed that the expression of a mutant fructose 1,6-bisphosphatase enzyme, unresponsive to AMP regulation, nullified the glucose-lowering impact of metformin in vivo [43].
2.4. Complex IV Inhibition
Another proposed mechanism for the interaction of metformin with mitochondria suggests that metformin inhibits Complex IV, leading to the disruption of the OXPHOS system. This disruption indirectly reduces the ubiquinone pool, which serves as the electron acceptor for mGPDH, thus inhibiting mGPDH activity (Figure 3). The interaction between metformin and Complex IV may be facilitated by the ability of biguanides to bind metal ions, such as iron and copper, both of which are present in Complex IV [59]. However, it’s worth noting that all complexes of the ETC contain iron and/or copper ions, essential for electron transfer. Consequently, further research is needed to confirm whether metformin is capable of inhibiting the mitochondrial respiratory chain at different complexes beyond Complex I.
2.5. Epigenetic Effects of Metformin
Metformin has been shown to influence epigenetic modifications, particularly through its effects on DNA methylation and histone modifications. These metformin-induced epigenetic alterations underscore its potential as a promising therapeutic intervention with wide-ranging implications for disease management and prevention.
2.5.1. Histone Acetylases
Metformin exerts regulatory effects on histones via AMPK activation. In particular, histone acetylation is a critical epigenetic modification that alters chromatin architecture and regulates gene expression by either opening or closing the chromatin structure. Histone acetyltransferases (HATs) depend on acetyl-CoA as a substrate for histone acetylation, therefore, intracellular levels of acetyl-CoA can modulate histone modification and subsequently, gene expression. Metformin can regulate histone function by activating AMPK, which inhibits ACC. This inhibition prevents the conversion of acetyl-CoA to malonyl-CoA, resulting in elevated acetyl-CoA levels available for histone acetylation [60].
Metformin can induce changes in the epigenetic landscape through the HAT CREB-binding protein (CBP), a critical transcriptional co-activator of gluconeogenesis genes. AMPK-mediated phosphorylation of CBP at Ser436 has been shown to decrease its binding to the promoter regions of target genes, such as peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and phosphoenolpyruvate carboxykinase 1 (Pck1), potentially contributing to the antigluconeogenic effects of metformin [61]. More recently, the glycogen-targeting regulatory subunit of protein phosphatase 1 (PPP1R3C), has emerged as a potential target of metformin in suppressing cAMP-stimulated hepatic gluconeogenesis. Xueying Ji et al. propose that PPP1R3C overexpression could enhance PPI1 activity and modulate the phosphorylation of the CREB coactivator Target of Rapamicyn complex-2 (TORC2) to upregulate the transcription of gluconeogenic genes [62].
Although there is lack of data, a study addressing the impact of histone acetylation in cancer research reported the ability of metformin to rectify specific histone H3 acetylation patterns in cancer-prone cells [63]. Nevertheless, further efforts need to be made to delve into the specific underlying mechanism and implications in clinical practice.
2.5.2. Histone Deacetylases
Histone deacetylases (HDACs) play a crucial role in gene expression by removing acetyl groups from histone proteins, impacting chromatin accessibility. HDACs also modulate cellular processes via interaction with repressor complexes and transcription factors as well as deacetylation of non-histone proteins [64]. Commonly known as sirtuins, class III HDACs are NAD+-dependent HDACs that emulate the metabolic effects of calorie restriction [65]. Importantly, emerging evidence suggests that metformin exerts its metabolic effects, at least partially, through sirtuin-mediated enhancement of mitochondrial function [66,67,68].
Sirtuin 1 (SIRT1), the most conserved mammalian class III HDAC [69], serves as an intracellular energy sensor regulated by the NAD+/NADH ratio. Metformin, through AMPK activation, can enhance the phosphorylation of nicotinamide phosphoribosyltransferase (NAMPT), thus promoting NAD+ synthesis and subsequently, stimulating SIRT1 activity [70]. Furthermore, in silico and in vivo assays suggest that metformin could directly interact with the activation domain of SIRT1 thereby increasing SIRT1 catalytic efficiency under low NAD+ conditions [65]. In turn, SIRT1 mediates AMPK activation through LKB1 Lys-48 deacetylation, potentially reinforcing a SIRT1-AMPK activation loop [70]. Previous studies indicate that metformin can also exert its anti-hyperglycemic effects by modulating SIRT1. Increased SIRT1 activity is linked to diminished gluconeogenesis through target of rapamycin kinase 2 (TORC2) inhibition and enhanced gluconeogenesis via PGC-1α deacetylation [71]. Moreover, SIRT1 activity confers benefits on hepatic metabolism, mitochondrial biogenesis [66], inflammation [72], and diabetic nephropathy [73]. Conversely, in cancer cells, it has been implicated in potential contributions to mitochondrial dysfunction and pyroptosis [67].
HDAC Sirtuin 3 (SIRT3) is also a target of metformin. Primarily localized in the mitochondrial matrix, SIRT3 modulates mitochondrial dynamics, the tricarboxylic acid cycle, respiratory chain, and fatty acid oxidation [68]. Additionally, antioxidant effects of SIRT3 have been reported [74]. Metformin has demonstrated the ability to enhance mitochondrial function [75], beta-oxidation [74,76], and antioxidant defenses [75,77] by modulating SIRT3 activity. Additionally, metformin-mediated induction of SIRT3 may confer protection against chemotoxicity and exacerbate metformin-induced apoptosis and mitochondrial dysfunction in cancer cells [78].
2.5.3. DNA Methylation
In addition to modulating histone acetylation/deacetylation, current data supports a role for metformin as an epigenetic regulator by linking cellular metabolism to the DNA methylation machinery. Metformin increases the ratio of SAM:SAH by indirectly activating S-adenosylhomocysteine (SAH) hydrolase, thereby relieving the inhibition of S-adenosylmethionine (SAM)-dependent methyltransferases [79,80]. Genome-wide alterations in the DNA methylome of non-cancerous [79] and cancerous cells [80] in response to metformin treatment have been reported. In T2DM patients, three methylation sites (PKNOX2, WDTC1 and MICB) have been linked to the effects of metformin on HbA1c levels [81]. Additionally, higher DNA methylation levels in OCT1 (SLC22A1), an epigenetic change associated with hyperglycemia and obesity, are modulated by metformin [82].
2.6. Effects of Metformin on microRNAs
Several microRNA (miRNA) circulating levels have been associated with diabetes and insulin resistance. Interestingly, an increase in DICER1 levels, an enzyme involved in miRNA maturation, has been observed in diabetic humans treated with metformin as well as in mice. This observation suggests that metformin may exert its effects through the modulation of miRNAs [83]. Recent studies have identified circulating miRNAs altered by metformin treatment in T2DM patients, with miR-194-5p, miR-148-3a being of special interest due to their impact on Wnt and NF-kB signaling pathways, both involved in T2DM development [84]. Additionally, in a cross-sectional study, metformin treatment was associated with changes in the circulating profiles of miR-192, miR-222, and mir-140-5p, paralleled by decreases in fasting glucose and HbA1c levels [85]. Circulating levels of miR-192 have previously been suggested as potential biomarkers of T2DM and miR-222 has been associated with the progression of obesity and insulin resistance [86]. Metformin also enhances hepatic redox balance by elevating the ratio of reduced glutathione to oxidized glutathione. Consequently, this alteration inhibits genes associated with gluconeogenesis through the let-7–TET3–HNF-4α pathway [87]. Moreover, in HepG2, miR-140-5p overexpression induced impairment of glucose consumption and glucose uptake [88].
Importantly, the potential anti-cancer properties of metformin have been linked to the regulation of numerous miRNAs. A great effort has been made in the discovery of metformin-modulated miRNAs and the associated downstream effectors in various malignancies. Of note, miR-192 is a known prognostic biomarker in gastric cancer and miR-194 direct target Bmi-1 overexpression has been reported in different malignancies [89]. Thus, the anti-tumoral mechanisms of metformin could be mediated through miRNAs simultaneously involved in the anti-hyperglycemic actions of metformin.
2.7. Effects of Metformin on the Microbiota
Preclinical research has shown that metformin induces changes in the composition and function of the gut microbiota [90]. Evidence suggests potential benefits for metabolic and immune health resulting from these changes. However, the impact of metformin on the human gut microbiota remains ambiguous and additional research is required to clarify the mechanisms by which the drug operates and to comprehend its role in fostering the growth of particular bacteria that offer advantages to the host.
In a 2023 systematic review encompassing 13 studies, the use of metformin was linked to changes in bacterial genera abundance. However, data across different phyla were inconsistent, possibly due to differences in metformin dosage and treatment duration. Nonetheless, prior investigations found an enrichment in bacterial genera linked to beneficial effects on glucose metabolism and insulin sensitivity, such as Lactobacillus and Akkermansia muciniphila [91], following metformin treatment. An enrichment in Akkermansia muciniphila has been documented in both mouse [92] and human treated with metformin [93]. Similarly, a study on T2DM mouse model revealed that metformin alone increases the abundance of Lactobacillus [94]. Moreover, in a randomized trial in overweight/obese adults, metformin treatment resulted in significant changes in the microbiota composition, increasing Escherichia coli and Ruminococcus torques, while decreasing Intestinibacter bartlettii and Ruminococcus. Remarkably, these changes in microbiota composition were associated to an increase in short chain fatty acids (SCFAs). The increased abundance of SCFA-producing bacteria has been postulated to facilitate SCFA-induced GLP1 secretion [95].
Several studies show a decrease in the abundance of Bacteroides fragilis [94,96,97]. Bacteroides fragilis produces the bile acid glycoursodeoxycholic acid (GUDCA), an endogenous antagonist of FXR and it is hypothesized to be able to modulate GLP1 [97]. Notably, available data suggests that metformin can directly inhibit the growth of Bacteroides fragilis through the inhibition of the bacterial NADH–menaquinone oxidoreductase (NDH1) complex, a mechanism closely related to the eukaryotic mitochondrial respiratory chain Complex I, thereby interfering with ATP production [4,98].
3. Metformin and Cancer
An increasing number of epidemiological and clinical investigations have indicated that metformin decreases the risk of cancer in individuals with T2DM and enhances the survival outcomes of cancer patients diagnosed with breast, ovarian, liver, and colorectal tumors [22,24,25,99]. One proposed mechanism for the antineoplastic action of metformin involves inhibition of mitochondrial respiratory Complex I. This inhibition compromises ATP production, consequently activating AMPK in an LKB1-dependent manner. This AMPK activation leads to mTOR inhibition, leading to anticancer effects, including reduced protein and lipid synthesis, slow proliferation rates, activation of autophagy, and inhibition of inflammatory responses.
According to public statistics from 2020, breast and colorectal cancers rank among the most prevalent types of cancer. Data from the Global Cancer Observatory (GLOBOCAN) of the International Agency for Research on Cancer (IARC) predicts a significant increase in the global burden of breast and colorectal cancers by 2040, with over 3 million new cases per year and 1 million and 1.6 million deaths per year, respectively [100,101,102]. These findings emphasize the urgent need to advance new therapies and strategies to enhance cancer patient management. Repurposing metformin for cancer treatment holds promise for rapid progress in this regard, given its approved status, well-established pharmacokinetics, and tolerability profile.
3.1. Breast Cancer
The estimated global incidence in 2020 of female breast cancer was around 2.3 million new cases, accounting for 11.6% of the total new cases, and emerged as the leading cause of cancer-related deaths, surpassing lung cancer [103].
The complex relationship between diabetes and breast cancer, both significant global health concerns, has stimulated extensive research to reduce risk and improve outcomes. Diabetes, a prevalent chronic disease, significantly increases the risk of breast cancer, which remains the leading malignancy among women worldwide [104]. Within this complex interplay, metformin, a widely used anti-diabetic drug, has become a focal point due to its potential anti-cancer properties.
3.1.1. Clinical Studies
Epidemiological studies form the basis for understanding the relationship between diabetes, breast cancer and metformin. Notable, women with T2DM are more likely to be diagnosed with breast cancer. This association persists even after adjustment for body mass index (BMI) and menopausal status [105].
Significantly, several observational studies have consistently shown a decreased risk of breast cancer and lower recurrence rates among individuals who are overweight and/or have T2DM and use metformin [106,107]. Metformin emerges as a significant player associated with improved disease-free survival (DFS) and overall survival (OS), suggesting its potential positive impact on survival outcomes among invasive breast cancer patients with T2D [108]. In addition, a meta-analysis of 31,031 breast cancer patients suggests a potential benefit of metformin therapy, showing reductions in all-cause mortality and progression-free survival compared with non-metformin users [109].
However, while certain studies show beneficial effects of metformin on breast cancer risk, others report no association or nuanced effects compared with other cancers [110].
Lu et al. observed a higher likelihood of breast cancer diagnosis among women with T2D but found no statistically significant reduction in breast cancer risk among metformin users [105].
Despite these nuanced findings, a consistent association between metformin exposure and reduced risk of HR+/HER2- breast cancer is evident [107]. Another study exposes the significant limitations of real-world observational studies in accurately assessing metformin’s effects on cancer incidence and outcomes due to avoidable biases that, surprisingly, persist today [111]. These controversial studies underscore the importance of stratifying patients by molecular subtype to gain a clearer understanding of the effects of metformin within more homogeneous cohorts. Moreover, these conflicting results are similarly observed in studies investigating the potential of metformin as a chemopreventive or adjuvant agent [111,112].
Clinical trials are the main platform for evaluating the efficacy of metformin in breast cancer. These trials shed light on its impact on DFS and OS, particularly among invasive breast cancer patients with T2D. These studies consistently show significant associations, suggesting that metformin is a potential game-changer in adjuvant therapy [107]. A systematic meta-analysis study of nine clinical trials and over 1,000 patients, provides nuanced insights, highlighting metformin’s capacity to decrease insulin levels, fasting blood sugar, BMI, and Ki-67 in patients with breast and endometrial cancer [113]. Thus, clinical analyses go beyond traditional survival metrics to examine plasma biomarkers, shedding light on metformin’s role in modulating hormonal and metabolic factors that affect breast cancer. Leptin, HOMA-IR, SHBG, and estradiol levels show positive responses to metformin, especially when combined with lifestyle interventions [114].
Clinical trials, exploring metformin’s potential in combination therapies, present a mixed picture. While some trials demonstrate safety and tolerability [115], others, like a phase I clinical trial evaluating erlotinib and metformin, reveal no significant clinical benefits in metastatic triple-negative breast cancer (TNBC) patients [116]. Research also uncovers metformin’s therapeutic opportunity in controlling toxicities caused by neoadjuvant chemotherapy in non-diabetic breast cancer patients [117]. However, the intricacies of its impact on cholesterol levels after neoadjuvant treatment hint at broader metabolic effects [118].
3.1.2. Animal Studies
Recent in vivo experiments conducted by Schmidt et al. provided dynamic insights into metformin’s effects on breast cancer progression. Combined treatment with a ketogenic diet and metformin in mouse models of TNBC shows significantly reduced tumor burden, increased tumor latency and slower tumor growth. This synergistic approach is emerging as a promising avenue for potentially prolonging survival [119].
In addition, TNBC xenografts show discernible changes in tumor imaging metrics following metformin treatment, highlighting its impact on tumor biology [120]. Furthermore, metformin has been shown to be synergistic with immunotherapy against TNBC in mouse models [121]. Studies conducted in metformin-treated mice underscore its immunomodulatory effects. Specifically, a reduction in M2-like macrophages, monocytic myeloid-derived suppressor cells (M-MDSCs), and regulatory T cells (Tregs) indicates the ability of metformin to enhance local antitumor activity [122]. Taken together, these studies suggest avenues for metformin’s potential in combination treatments. Finally, metformin’s cardioprotective effects are underscored in experiments aimed at attenuating doxorubicin-induced cardiotoxicity [123].
3.1.3. In Vitro Studies
In vitro experiments are exploring the intricate cellular and molecular mechanisms underlying the effects of metformin on breast cancer cells. It is well known that altering mitochondrial function by inhibiting Complex I is an outstanding anticancer mechanism [124]. A remarkable consideration is that metformin antiproliferative effects may depend on glucose concentration in culture media [125]. It acts through mTOR-dependent and mTOR-independent pathways, demonstrating versatility in different cellular environments [125].
The metabolic reprogramming observed in cancer cells, particularly their ability to adapt metabolism to support rapid proliferation, is discussed in the light of metformin’s inhibition of mitochondrial Complex I. Mitochondrial pathways are identified as potential targets for therapeutic intervention, in line with the premise that understanding the metabolic context is essential [126,127].
The concentration- and time-dependent induction of apoptosis by metformin in breast cancer cells was demonstrated in a study by Gao et al., implicating the intrinsic mitochondria-mediated apoptosis pathway [128]. This highlights the potential of metformin as a cytotoxic agent against breast cancer. Furthermore, metformin’s influence on the antioxidant system and its modulation of various molecular factors, including pro- and anti-apoptotic proteins, MMP-2, MMP-9, miR-21, and miR-155, are highlighted by Sharma & Kumar [129]. These findings hold potential implications for targeted treatment and clinical management of breast cancer.
Metformin’s suppression of ROS/EGF-induced breast cancer cell migration, invasion, and epithelial–mesenchymal transition (EMT) through inhibition of the PI3K/Akt/NF-κB pathway highlights its potential as an anti-metastatic agent [130]. Studies are further elucidating its effects on breast cancer stem cells, highlighting its potential in targeting these initiating cells. Shi, et al. highlighted the ability of metformin to suppress triple-negative breast cancer stem cells by targeting KLF5 for degradation, offering a promising avenue for treating aggressive forms of breast cancer [131].
In addition, the adjuvant potential of metformin in radiotherapy is emerging as a promising facet, as evidenced by its ability to increase radiosensitivity by modulating specific genes such as miR-21-5p and SESN1 [132].
Finally, cellular NAD+ depletion is emerging as a critical aspect of metformin sensitivity in breast cancer cells, as demonstrated in the study [133]. NAMPT, a key player in maintaining NAD+ levels, influences metformin resistance and provides insights into potential targets for overcoming resistance in breast cancer treatment.
3.2. Colorectal Cancer
Colorectal cancer (CRC) ranks as the third most common cancer worldwide, with almost 2 million new cases diagnosed each year, accounting 9.6% of all cancer cases, and is the second leading cause of cancer-related deaths worldwide [120,121].
Obesity and T2D are among the risk factors associated with CRC. Recent studies have proposed incorporating T2D patients into CRC screening programs to mitigate their elevated risk [134,135]. Hyperinsulinemia and high levels of insulin-like growth factor 1 (IGF-1) are associated with increased proliferation of colon cells, resulting in malignancy [136]. The risk is even higher in patients treated with sulfonylureas and insulin [136].
3.2.1. Clinical Studies
As previously mentioned, metformin is the first-line treatment for T2D and emerges as a potential candidate for chemoprevention in cancer, including CRC. This is supported by epidemiological studies demonstrating that patients with T2D not only have reduced risk incidence of tumor development but also show a lowered risk of mortality when undergoing metformin treatment after CRC diagnosis [137,138,139]. More concretely, long-term consumption of metformin has been associated with a decreased risk of CRC in diabetic patients [140]. However, further studies are needed to demonstrate whether the observed chemopreventive effects in T2DM patients is due to the direct influence of metformin on cancer cells or is due to the metabolic status provoked by the disease. In this sense, some studies have explored the effects of metformin in nondiabetic CRC patients with promising results [141]. Nonetheless, in other cancer type such as prostate and breast cancer, the impact of metformin on nondiabetic patients appears to be non-significant [142,143,144].
Another interesting aspect to consider is the potential synergistic effect of metformin when combined with conventional chemotherapeutic agents or immunotherapy. In a recent study focusing on patients with stage IV microsatellite-stable CRC who had experienced progression on prior therapies (NCT03800602), Akce et al. demonstrated that the combination of nivolumab, a medication that binds to the PD-1 protein to enhance immune cell activity against cancer cells, along with metformin, did not exhibit significant immune modulation compared to metformin alone. However, the study revealed promising trends in tumorous T-cell infiltration following the dual treatment of metformin and PD-1 blockade, despite disease progression observed in the majority of patients. patients [145].
Another study carried out in China by Zhang et al. analyzed a total of 187 T2DM patients who underwent resection of CRC [146]. The results unveiled inverse correlations between metformin treatment and the incidence of distant metastasis as well as poorly differentiated adenocarcinoma grade. Furthermore, metformin inversely correlated with positive staining for CD133 and β-catenin protein expression, a marker for cancer stem cells and an upregulator of epithelial-to-mesenchymal transition, in patients with CRC and T2DM [146]. In addition to this, the combination of metformin with irinotecan resulted in disease control in 41% of patients within 12 weeks [147]. Moreover, synergistic effects of metformin plus fluorouracil (5-FU) have been reported in a phase 2 trial in patients with refractory CRC [148].
From another point of view, Kim et al. demonstrated that the glutamine metabolism could play an important role in the sensitivity of CRC patients-derived tumor-organoid cells to metformin [149]. It is known that glutamine metabolization by mitochondrial enzymes into alpha-ketoglutarate, promotes cell growth by the induction of mTORC1 activation, in contrast to metformin effects [150]. Thus, a combination of glutamine metabolism inhibitors with metformin could be an effective adjunctive treatment against CRC. In another study, metformin modulated the mitochondrial metabolism in CRC patients-derived organoids, with effects in mitochondria morphology, with deformation and partial loss of mitochondrial membranes and degradation of cristae [151]. Consequently, the authors also observed an accumulation of free radicals and a loss of membrane potential, suggesting that metformin is a good candidate to act synergistically with other drugs to induce cell death [151].
It is imperative to consider a recent review that underscores the controversy surrounding the impact of metformin treatment on tumor cells in human patients. This review posits that the concentrations of metformin required for substantial inhibition of Complex I are roughly 1000 times higher than those typically utilized for treating T2D, making its application as a neoadjuvant anticancer therapy impractical [37]. Despite the theoretical possibility of metformin accumulation in mitochondria owing to its cationic charge, there exists no definitive supportive evidence for this assertion. Conversely, metformin may exert effects through mechanisms distinct from Complex I inhibition. Studies have indeed suggested that while metformin diminishes the ADP/ATP ratio and activates AMPK, such effects alone might not be sufficient to provoke significant energy depletion and cellular demise [49,152].
Taking together these considerations in human studies on metformin effects on CRC chemoprevention and as a possible combined therapy with classical chemotherapeutic agents or in immunotherapy, it must be considered the basic in vitro and in vivo studies to elucidate the molecular mechanisms of metformin, with a special focus on mitochondria.
3.2.2. Animal Studies
Several animal studies support the potential anticancer effect of metformin against various types of cancer, including CRC. In murine models of CRC induced by chemical carcinogens or mutations in the APC gene, administration of metformin activated AMPK and inhibited the mTOR/S6K/S6 pathway. This led to a decrease in colonic mucosal epithelial cell proliferation and a reduction in the development of intestinal polyps [153,154]. These results suggest that metformin holds promise as a potential candidate for CRC chemoprevention.
In addition to the chemopreventive properties, other in vivo findings suggest that metformin combined with adjuvant chemotherapy might be associated with a better prognosis. In a chemical-induced CRC model using 1,2-dimethylhydrazine (DMH), the combination of metformin and oxaliplatin decreased DMH-induced CRC in diabetic and non-diabetic mice by downregulating tumor angiogenesis [155]. Similarly, in patient-derived xenograft models, the administration of metformin in combination with 5-FU or 5-FU plus oxaliplatin exhibited a tumor-suppressive effect by activating AMPK-mediated pathways, which was accompanied by a reduction in stem-like cells [156,157] A recent preclinical study has revealed that metformin suppressed liver metastasis of a CRC cancer xenograft mouse model, mediated by inhibition of mTOR phosphorylation through activation of AMPK [158].
It is noteworthy that the endocrine and metabolic alterations accompanying obesity are a factor to consider in promoting CRC, and the ability of metformin to correct some of these alterations may contribute to its antineoplastic effect. For instance, obese individuals exhibit reduced levels of adiponectin, which may contribute to the negative impact of obesity on neoplastic diseases. This adipokine activates AMPK, thereby inhibiting cell growth. [159]. Therefore, metformin could be particularly beneficial in mitigating the adverse effects of obesity on neoplasia [160]. In fact, Algire et al. demonstrated in vivo that metformin inhibits the stimulatory effect of a high-energy diet on colon carcinoma growth [161]. The authors observed that metformin treatment reduced insulin levels, attenuated diet-induced phosphorylation of AKT, decreased the expression of FASN, and activated AMPK. These findings suggest that metformin may have a potential in addressing colon cancer in the context of obesity.
One interesting study conducted by Scharping et al. has unveiled the role of metformin in altering the metabolism of the tumor microenvironment, thereby enhancing the response to PD-1 blockade immunotherapy in mouse tumor models [162]. Additionally, some studies have indicated that metformin can modulate the metabolic profile of gut microbiota [97,163]. Broadfield et al. (2022) showed that metformin reduces tumor growth in mice fed a high-fat diet through changes in the gut microbiome using a murine model of CRC and fecal transfer approaches [163]. In fact, the authors demonstrated the transfer of the gut microbiome from metformin-treated mice to drug-naïve, conventionalized fed mice on a high-fat diet results in suppressed growth of murine colon cancer cells. Increased levels of SCFAs, such as butyrate and propionate, were found, which may be associated with down-regulation of highly activated T cell clusters (CD8+, NK1.1+, Ki67+). Recent evidence also suggests the impact of butyrate in promoting CD8+ T cell long-term survival as memory cells [164].
3.2.3. In Vitro Studies
Numerous in vitro studies have explored the efficacy of metformin and its underlying molecular mechanism in colon cancer cells. Initial evidence was provided by Zakikhani et al., showing that metformin reduces the proliferation of HT-29 cells in dose- and time-dependent manner by activating AMPK a major regulator of cellular energy metabolism [160]. Another study found that metformin transiently inhibits CRC cell proliferation by inducing G0/G1 phase arrest. The anti-proliferative effect may involve AMPK activation or increased reactive oxygen species (ROS) production [165]. Metformin inhibits Complex I activity of the mitochondrial electron transport chain, resulting in the mitochondrial depolarization and release of ROS, which contribute to metformin antiproliferative effect.
Moreover, evidence suggests that metformin improves tumor sensitivity to chemotherapy drugs like 5-FU, oxaliplatin, or irinotecan, underscoring the involvement of mitochondria in the sensitization induced by metformin. Boyle and collaborators suggested that the activation of antitumor mitophagy by metformin could modulate the chemotherapy response in CRC cells [166]. This is also consistent with another study performed by Denise et al., showing that colon cancer cells reprogrammed their metabolism to OXPHOS in response to 5-FU and, only under these conditions (with 5-FU treatment), colon cancer cells are sensitive to metformin treatment [167]. These authors also determined that OXPHOS inhibition is the primary mechanism of action of metformin resulting in anti-tumor effects, while targeting AMPK plays a marginal role. Zhang and collaborators found that metformin enhanced the sensitivity of CRC cells to cisplatin by modulating mitochondrial function, resulting in decreased membrane potential and induction of ROS production. This ultimately promoted apoptosis through the PI3K/Akt pathway [168], providing an alternative mechanism to AMPK activation. Another study demonstrated that metformin-induced tumor suppression was prevented in cancer cells expressing NADH-ubiquinone reductase (H(+)-translocating), also known as NDI1, which is metformin-resistant yeast analogue of complex I. This suggests the crucial role of inhibiting this mitochondrial target in the drug’s anticancer effect [169]. This is also consistent with another work showing that metformin in CRC cells altered mitochondrial activity by increased ROS levels and SIRT3 activity, suggesting that these changes may be crucial for its cytotoxic effects. [170]. However, other studies suggest that metformin may mitigate DNA damage and mutagenesis by reducing ROS production [161,171,172].
Metformin in combination with 5-FU, a first-line drug in CRC treatment, significantly enhanced the antiproliferative effect, apoptosis, and cell-cycle arrestment in SW620 cells, likely due to the activation of LKB1/AMPK pathway, leading to mTOR activity inhibition [146]. This combination may selectively target cancer stem cells (CSC) through the inhibition of the β-catenin pathway [173] or by AMPK activation and the inhibition of DNA replication, and the NF-κB pathway [174]. CSC contribute to therapy resistance, thus, strategies targeting the depletion of this subpopulation of cancer cells are likely to be more effective in eradicating the tumor. In fact, synergistic effects of metformin plus 5-FU have been reported in a phase 2 trial [148]. Similarly, metformin combined with 5-FU and oxaliplatin restored sensitivity in chemoresistant CRC cells by reducing stemness and EMT through inhibition of the Wnt/β-catenin pathway [175].
4. Future Perspectives and Improvements in Cancer Therapy
Remarkable, metformin displays promising antitumor potential in clinical studies. However, its efficacy is primarily observed at concentrations higher than therapeutic, and its biodistribution to cancer cells remains uncertain. To address this challenge, researchers are exploring various metformin-based compounds to enhance its antitumor properties across different types of cancer.
Ongoing research is investigating novel, more lipophilic derivatives of metformin to target mitochondria. Gang Cheng et al. have developed metformin derivates called Mito-MET by attaching positively charged lipophilic substituents. These mitochondria-targeted analogues of metformin enhance antiproliferative and radiosensitizing effects in cancer cells [176].
Similarly, other researchers have developed metformin derivates obtained by combination with other antitumor compounds studied, obtaining promising results. A combination strategy involving WZB117 (a GLUT1 inhibitor), OCMC (O-carboxymethyl-chitosan), and metformin was proposed for breast cancer treatment. WZB117-OCMC-metformin exhibited improved efficacy by simultaneously targeting GLUT1 and mTOR, overcoming limitations of metformin monotherapy [177]. Moreover, polyethylene glycol niosomal nanoparticles co-loaded with metformin and the widely used phytochemical artemisinin have shown enhanced anticancer effects on A549 lung cancer cells [178]. Similar results were observed by Kumar et al. by combining metformin and gallic acid, a phenolic acid found in tea leaves and some fruits with anticancer properties [179]. A novel compound, Met-ITC, was developed by incorporating an isothiocyanate moiety to metformin. The isothiocyanate (ITC), as a hydrogen sulfide (H2S) donor, acts as an anticancer agent by affecting the cell cycle, inducing apoptosis, and inhibiting histone deacetylases. This hybrid molecule demonstrated enhanced efficacy and potency against various cancer cells (AsPC-1, MIA PaCa-2, MCF-7) while being less effective on non-tumorigenic cells (MCF 10-A) [180].
Modifications to the metformin molecule could also provide novel drugs for the treatment of cancer as well as other diseases. For instance, supformin (LCC-12), a rationally designed dimer of metformin, exhibited anti-inflammatory effects by targeting the cell surface glycoprotein CD44 and regulating copper (II) levels in mitochondria. This intervention led to reduced NAD(H) redox cycling, metabolic and epigenetic reprogramming, and decreased inflammation in macrophages. LCC-12 demonstrated potential therapeutic effects in mouse models of bacterial and viral infections, highlighting its role in modulating cell plasticity and controlling inflammatory responses deregulated in cancer [181].
Due to its poor oral absorption, therapeutic doses of metformin are relatively high, often causing unpleasant gastrointestinal adverse effects. Consequently, novel derivatives of metformin have been synthesized over the past decades to overcome this limitation or achieve a more targeted release. Specifically, metformin-loaded liposomes have been designed for targeted delivery to inflamed endothelia. Using a three-step pretargeting system based on biotin-avidin interaction, these liposomes effectively delivered metformin hydrochloride to inflamed endothelial cells, offering potential anti-inflammatory therapy for conditions such as neurodegenerative diseases, atherosclerosis, and cancer [182].
Moreover, Zhao et al. are studying Met-3BP-Lip@M1, a drug delivery system derived from M1 macrophage membranes, which targets breast cancer cells to kill them by simultaneously inhibiting glycolysis and oxygen consumption. This biomimetic nanomedicine induced cancer cell apoptosis through effective cellular uptake and demonstrated synergistic improvements in therapeutic efficiency against breast cancer, both in vitro and in vivo [183]. In the same spirit, sulfenamides and sulfonamides metformin derivatives have displayed their potential as prodrugs and inhibitors of various diseases, including cancer [184].
Additionally, to improve the antitumor effects of metformin while reducing dosage requirements, a novel polymer dot, MA-dots, has been developed. These dots, synthesized using metformin and L-arginine, exhibit dual targeting capabilities for tumor cell membranes and mitochondria, demonstrating a 12-fold increase in antitumor activity compared to raw metformin, with effective tumor growth suppression in vivo [185]. Similarly, combining Mito-MET and iron chelators, such as DFX and DXR, Cheng et al. showed synergistic inhibition of pancreatic and triple-negative breast cancer cell proliferation [186].
Addressing challenges in tumor immunotherapy, FCM@4RM, a tumor-specific nanovaccine, was developed to deliver tumor antigens and adjuvants while modulating the immune microenvironment. FCM@4RM demonstrates effective antigen presentation, stimulation of effector T cells, and modulation of the immune microenvironment through the synergistic effects of CpG, metformin, and a bioreconstituted cytomembrane, offering a novel approach for antitumor immunotherapy [187].
5. Concluding Remarks
In summary (Figure 4), the accuracy of drug levels achieved in cancer cells in laboratory data represents a challenge for applying findings to the clinical setting. Therefore, further clarification about mechanisms of action of metformin and its biodistribution to cancer cells is essential for designing novel metformin-based compounds, ultimately optimizing clinical applications and outcomes. Finally, combined therapies and targeted delivery strategies hold promise for improving the efficacy and minimizing the adverse effects of metformin in cancer treatment.
Author Contributions
Conceptualization, A.C.-E., P.M.M.-B., A.M.-C., A.V., D.G.P., J.S.-S., P.R., M.N.-S.; writing—original draft preparation, A.C.-E., P.M.M.-B., A.M.-C., A.V., D.G.P., J.S.-S., P.R., M.N.-S.; writing—review and editing, A.C.-E., A.V., D.G.P., J.S.-S., M.N.-S; supervision, A.C.-E., P.R., M.N.-S. All authors have read and agreed to the published version of the manuscript.
Funding
Andrea Morán-Costoya was funded by an FPI_043_2021 de la Conselleria de Fons Europeus, Universitat i Cultura del Govern de les Illes.
References
- World Health Organization Model List of Essential Medicines: 21st List 2019 Available online:. Available online: https://iris.who.int/handle/10665/325771 (accessed on 5 March 2024).
- Ahmad, E.; Sargeant, J.A.; Zaccardi, F.; Khunti, K.; Webb, D.R.; Davies, M.J. Where Does Metformin Stand in Modern Day Management of Type 2 Diabetes? Pharmaceuticals 2020, 13, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J. Metformin: Historical Overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the Glucoregulatory Mechanisms of Metformin in Type 2 Diabetes Mellitus. Nat Rev Endocrinol 2019, 15, 569–589. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Lipska, K.J.; Mayo, H.; Bailey, C.J.; McGuire, D.K. Metformin in Patients With Type 2 Diabetes and Kidney Disease: A Systematic Review. JAMA 2014, 312, 2668–2675. [Google Scholar] [CrossRef] [PubMed]
- Raičević, B.; Janković, S. Predictors of Gastrointestinal Complaints in Patients on Metformin Therapy. Open Med (Wars) 2023, 18, 20230871. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From Mechanisms of Action to Therapies. Cell Metab 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by Which Metformin Reduces Glucose Production in Type 2 Diabetes. Diabetes 2000, 49, 2063. [Google Scholar] [CrossRef]
- Cree-Green, M.; Bergman, B.C.; Cengiz, E.; Fox, L.A.; Hannon, T.S.; Miller, K.; Nathan, B.; Pyle, L.; Kahn, D.; Tansey, M.; et al. Metformin Improves Peripheral Insulin Sensitivity in Youth With Type 1 Diabetes. J Clin Endocrinol Metab 2019, 104, 3265–3278. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Barzilai, N.; Simonson, D.C. Mechanism of Metformin Action in Obese and Lean Noninsulin-Dependent Diabetic Subjects. J Clin Endocrinol Metab 1991, 73, 1294–1301. [Google Scholar] [CrossRef]
- Hother-Nielsen, O.; Schmitz, O.; Andersen, P.H.; Beck-Nielsen, H.; Pedersen, O. Metformin Improves Peripheral but Not Hepatic Insulin Action in Obese Patients with Type II Diabetes. Acta Endocrinol (Copenh) 1989, 120, 257–265. [Google Scholar] [CrossRef]
- Jahn, L.A.; Hartline, L.; Liu, Z.; Barrett, E.J. Metformin Improves Skeletal Muscle Microvascular Insulin Resistance in Metabolic Syndrome. Am J Physiol Endocrinol Metab 2022, 322, E173–E180. [Google Scholar] [CrossRef]
- Barroso, E.; Montori-Grau, M.; Wahli, W.; Palomer, X.; Vázquez-Carrera, M. Striking a Gut–Liver Balance for the Antidiabetic Effects of Metformin. Trends Pharmacol Sci 2023, 44, 457–473. [Google Scholar] [CrossRef]
- Schäfer, G. Site-Specific Uncoupling and Inhibition of Oxidative Phosphorylation by Biguanides. II. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1969, 172, 334–337. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front Endocrinol (Lausanne) 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- LaMoia, T.E.; Butrico, G.M.; Kalpage, H.A.; Goedeke, L.; Hubbard, B.T.; Vatner, D.F.; Gaspar, R.C.; Zhang, X.M.; Cline, G.W.; Nakahara, K.; et al. Metformin, Phenformin, and Galegine Inhibit Complex IV Activity and Reduce Glycerol-Derived Gluconeogenesis. Proc Natl Acad Sci U S A 2022, 119, e2122287119. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin Suppresses Gluconeogenesis by Inhibiting Mitochondrial Glycerophosphate Dehydrogenase. Nature 2014 510:7506 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on Mechanisms of Action and Repurposing Potential. Nature Reviews Endocrinology 2023 19:8 2023, 19, 460–476. [Google Scholar] [CrossRef] [PubMed]
- Lamoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr Rev 2021, 42, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Cejuela, M.; Martin-Castillo, B.; Menendez, J.A.; Pernas, S. Metformin and Breast Cancer: Where Are We Now? Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Chi Thent, Z.; Hannim Zaidun, N.; Fairuz Azmi, M.; Senin, M.; Haslan, H.; Salehuddin, R. Is Metformin a Therapeutic Paradigm for Colorectal Cancer: Insight into the Molecular Pathway? Curr Drug Targets 2016, 18, 734–750. [Google Scholar] [CrossRef]
- Ng, C.A.W.; Jiang, A.A.; Toh, E.M.S.; Ng, C.H.; Ong, Z.H.; Peng, S.; Tham, H.Y.; Sundar, R.; Chong, C.S.; Khoo, C.M. Metformin and Colorectal Cancer: A Systematic Review, Meta-Analysis and Meta-Regression. Int J Colorectal Dis 2020, 35, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Siegel, P.M.; St-Pierre, J. Metabolic Profiles Associated With Metformin Efficacy in Cancer. Front Endocrinol (Lausanne) 2018, 9, 372. [Google Scholar] [CrossRef]
- Col, N.F.; Ochs, L.; Springmann, V.; Aragaki, A.K.; Chlebowski, R.T. Metformin and Breast Cancer Risk: A Meta-Analysis and Critical Literature Review. Breast Cancer Res Treat 2012, 135, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.H.; Satkunam, M.; Pond, G.R.; Steinberg, G.R.; Blandino, G.; Schunemann, H.J.; Muti, P. Association of Metformin with Breast Cancer Incidence and Mortality in Patients with Type II Diabetes: A GRADE-Assessed Systematic Review and Meta-Analysis. Cancer Epidemiol Biomarkers Prev 2018, 27, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence That Metformin Exerts Its Anti-Diabetic Effects through Inhibition of Complex 1 of the Mitochondrial Respiratory Chain. Biochemical Journal 2000, 348, 607. [Google Scholar] [CrossRef]
- Jensen, J.B.; Sundelin, E.I.; Jakobsen, S.; Gormsen, L.C.; Munk, O.L.; Frøkiær, J.; Jessen, N. [11C]-Labeled Metformin Distribution in the Liver and Small Intestine Using Dynamic Positron Emission Tomography in Mice Demonstrates Tissue-Specific Transporter Dependency. Diabetes 2016, 65, 1724–1730. [Google Scholar] [CrossRef]
- Liang, X.; Giacomini, K.M. Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J Pharm Sci 2017, 106, 2245–2250. [Google Scholar] [CrossRef]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical Pharmacokinetics of Metformin. Clin Pharmacokinet 2011, 50, 81–98. [Google Scholar] [CrossRef]
- Glossmann, H.H.; Lutz, O.M.D. Pharmacology of Metformin - An Update. Eur J Pharmacol 2019, 865. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dzierlenga, A.L.; Nelson, N.R.; Li, H.; Werts, S.; Goedken, M.J.; Cherrington, N.J. Mechanism of Altered Metformin Distribution in Nonalcoholic Steatohepatitis. Diabetes 2015, 64, 3305–3313. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Sundelin, E.I.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Christensen, M.M.H.; Brøsen, K.; Frøkiær, J.; Jessen, N. In Vivo Imaging of Human 11C-Metformin in Peripheral Organs: Dosimetry, Biodistribution, and Kinetic Analyses. J Nucl Med 2016, 57, 1920–1926. [Google Scholar] [CrossRef]
- Alshawi, A.; Agius, L. Low Metformin Causes a More Oxidized Mitochondrial NADH/NAD Redox State in Hepatocytes and Inhibits Gluconeogenesis by a Redox-Independent Mechanism. J Biol Chem 2019, 294, 2839–2853. [Google Scholar] [CrossRef]
- He, L.; Wondisford, F.E. Metformin Action: Concentrations Matter. Cell Metab 2015, 21, 159–162. [Google Scholar] [CrossRef]
- Wilcock, C.; Bailey, C.J. Accumulation of Metformin by Tissues of the Normal and Diabetic Mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin Pathways: Pharmacokinetics and Pharmacodynamics. Pharmacogenet Genomics 2012, 22, 820–827. [Google Scholar] [CrossRef]
- Sakellakis, M. Why Metformin Should Not Be Used as an Oxidative Phosphorylation Inhibitor in Cancer Patients. Chemotherapy 2023, 68, 185–189. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J Biol Chem 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, M. Inhibitors of NADH-Ubiquinone Reductase: An Overview. Biochim Biophys Acta 1998, 1364, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of Metformin and Other Biguanides on Oxidative Phosphorylation in Mitochondria. Biochem J 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Bridges, H.R.; Blaza, J.N.; Yin, Z.; Chung, I.; Pollak, M.N.; Hirst, J. Structural Basis of Mammalian Respiratory Complex I Inhibition by Medicinal Biguanides. Science 2023, 379, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin Inhibits Hepatic Gluconeogenesis in Mice Independently of the LKB1/AMPK Pathway via a Decrease in Hepatic Energy State. J Clin Invest 2010, 120, 2355–2369. [Google Scholar] [CrossRef]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin Reduces Liver Glucose Production by Inhibition of Fructose-1-6-Bisphosphatase. Nat Med 2018, 24, 1395–1406. [Google Scholar] [CrossRef]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides Suppress Hepatic Glucagon Signalling by Decreasing Production of Cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat Rev Mol Cell Biol 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Scott, J.W.; Kemp, B.E. AMPK Functions as an Adenylate Charge-Regulated Protein Kinase. Trends Endocrinol Metab 2012, 23, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Seo, W.-Y.; Song, K.-H.; Chanda, D.; Kim, Y.D.; Kim, D.-K.; Lee, M.-W.; Ryu, D.; Kim, Y.-H.; Noh, J.-R.; et al. AMPK-Dependent Repression of Hepatic Gluconeogenesis via Disruption of CREB.CRTC2 Complex by Orphan Nuclear Receptor Small Heterodimer Partner. J Biol Chem 2010, 285, 32182–32191. [Google Scholar] [CrossRef]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front Endocrinol (Lausanne) 2019, 10, 442419. [Google Scholar] [CrossRef]
- Wilcock, C.; Wyre, N.D.; Bailey, C.J. Subcellular Distribution of Metformin in Rat Liver. J Pharm Pharmacol 1991, 43, 442–444. [Google Scholar] [CrossRef]
- Zhang, C.-S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.-W.; Wu, Y.-Q.; Lin, S.-Y.; Lin, S.-C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab 2016, 24, 521–522. [Google Scholar] [CrossRef]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal V-ATPase-Ragulator Complex Is a Common Activator for AMPK and MTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab 2014, 20, 526–540. [Google Scholar] [CrossRef]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-Dose Metformin Targets the Lysosomal AMPK Pathway through PEN2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.-M.; Zhang, D.; Camporez, J.-P.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin Inhibits Gluconeogenesis via a Redox-Dependent Mechanism in Vivo. Nat Med 2018, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Brisson, D.; Vohl, M.C.; St-Pierre, J.; Hudson, T.J.; Gaudet, D. Glycerol: A Neglected Variable in Metabolic Processes? Bioessays 2001, 23, 534–542. [Google Scholar] [CrossRef]
- Lin, E.C. Glycerol Utilization and Its Regulation in Mammals. Annu Rev Biochem 1977, 46, 765–795. [Google Scholar] [CrossRef] [PubMed]
- Saheki, T.; Iijima, M.; Li, M.X.; Kobayashi, K.; Horiuchi, M.; Ushikai, M.; Okumura, F.; Meng, X.J.; Inoue, I.; Tajima, A.; et al. Citrin/Mitochondrial Glycerol-3-Phosphate Dehydrogenase Double Knock-out Mice Recapitulate Features of Human Citrin Deficiency. J Biol Chem 2007, 282, 25041–25052. [Google Scholar] [CrossRef] [PubMed]
- Migoya, E.M.; Bergeron, R.; Miller, J.L.; Snyder, R.N.K.; Tanen, M.; Hilliard, D.; Weiss, B.; Larson, P.; Gutierrez, M.; Jiang, G.; et al. Dipeptidyl Peptidase-4 Inhibitors Administered in Combination with Metformin Result in an Additive Increase in the Plasma Concentration of Active GLP-1. Clin Pharmacol Ther 2010, 88, 801–808. [Google Scholar] [CrossRef]
- Logie, L.; Harthill, J.; Patel, K.; Bacon, S.; Hamilton, D.L.; Macrae, K.; McDougall, G.; Wang, H.-H.; Xue, L.; Jiang, H.; et al. Cellular Responses to the Metal-Binding Properties of Metformin. Diabetes 2012, 61, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vancura, A. Activation of AMP-Activated Protein Kinase by Metformin Induces Protein Acetylation in Prostate and Ovarian Cancer Cells. J Biol Chem 2016, 291, 25154–25166. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic Effects of Metformin: From Molecular Mechanisms to Clinical Implications. Diabetes Obes Metab 2018, 20, 1553–1562. [Google Scholar] [CrossRef]
- Ji, X.; Wang, S.; Tang, H.; Zhang, Y.; Zhou, F.; Zhang, L.; Zhu, Q.; Zhu, K.; Liu, Q.; Liu, Y.; et al. PPP1R3C Mediates Metformin-Inhibited Hepatic Gluconeogenesis. Metabolism 2019, 98, 62–75. [Google Scholar] [CrossRef]
- Cuyàs, E.; Fernández-Arroyo, S.; Joven, J.; Menendez, J.A. Metformin Targets Histone Acetylation in Cancer-Prone Epithelial Cells. Cell Cycle 2016, 15, 3355–3361. [Google Scholar] [CrossRef] [PubMed]
- Verdone, L. Histone Acetylation in Gene Regulation. Brief Funct Genomic Proteomic 2006, 5, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Verdura, S.; Llorach-Parés, L.; Fernández-Arroyo, S.; Joven, J.; Martin-Castillo, B.; Bosch-Barrera, J.; Brunet, J.; Nonell-Canals, A.; Sanchez-Martinez, M.; et al. Metformin Is a Direct SIRT1-Activating Compound: Computational Modeling and Experimental Validation. Front Endocrinol (Lausanne) 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Nayuni, N.K.; Kieswich, J.; Khan, N.Q.; Yaqoob, M.M.; Corder, R. Metformin Suppresses Hepatic Gluconeogenesis through Induction of SIRT1 and GCN5. J Endocrinol 2010, 205, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Bian, Y.; Zhang, Y.; Ren, G.; Li, G. Metformin Activates AMPK/SIRT1/NF-ΚB Pathway and Induces Mitochondrial Dysfunction to Drive Caspase3/GSDME-Mediated Cancer Cell Pyroptosis. Cell Cycle 2020, 19, 1089–1104. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.-R.; Liu, B. Mitochondrial Sirtuin 3: New Emerging Biological Function and Therapeutic Target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef] [PubMed]
- Li, X. SIRT1 and Energy Metabolism. Acta Biochim Biophys Sin (Shanghai) 2013, 45, 51–60. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 Modulation of the Acetylation Status, Cytosolic Localization, and Activity of LKB1. Journal of Biological Chemistry 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Stelmaszyk, A.; Mikołajczak, P.; Dworacka, M. Sirtuin 1 as the Mechanism of Action of Agents Used in the Diabetes Mellitus Pharmacotherapy. Eur J Pharmacol 2021, 907, 174289. [Google Scholar] [CrossRef]
- Sun, Z.; Li, J.; Luo, G.; Liu, W.; He, Y.; Wang, F.; Qian, Y.; Fan, C. Pharmacological Activation of SIRT1 by Metformin Prevented Trauma-Induced Heterotopic Ossification through Inhibiting Macrophage Mediated Inflammation. Eur J Pharmacol 2021, 909, 174386. [Google Scholar] [CrossRef]
- Ren, H.; Shao, Y.; Wu, C.; Ma, X.; Lv, C.; Wang, Q. Metformin Alleviates Oxidative Stress and Enhances Autophagy in Diabetic Kidney Disease via AMPK/SIRT1-FoxO1 Pathway. Mol Cell Endocrinol 2020, 500, 110628. [Google Scholar] [CrossRef] [PubMed]
- Felgueiras, R.; Neto, A.C.; Rodrigues, A.R.; Gouveia, A.M.; Almeida, H.; Neves, D. Anti-Oxidant Effect of Metformin through AMPK/SIRT1/PGC-1α/SIRT3– Independent GPx1 Expression in the Heart of Mice with Endometriosis. Horm Mol Biol Clin Investig 2022, 43, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Kim, S.G.; Kang, Y.J.; Choi, H.C. Metformin Mitigates Stress-Induced Premature Senescence by Upregulating AMPKα at Ser485 Phosphorylation Induced SIRT3 Expression and Inactivating Mitochondrial Oxidants. Mech Ageing Dev 2022, 206, 111708. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; He, J.; Xu, Y.; Zuo, Y.; Zhou, W.; Yue, Z.; Shao, X.; Cheng, J.; Wang, T.; Mou, S. AMPK Activation Coupling SENP1-Sirt3 Axis Protects against Acute Kidney Injury. Molecular Therapy 2023, 31, 3052–3066. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, Z.; Zhang, C.; Bian, Y.; Zhang, X.; Liu, X.; Chen, W.; Zhao, Y. Metformin Promotes in Vitro Maturation of Oocytes from Aged Mice by Attenuating Mitochondrial Oxidative Stress via SIRT3-Dependent SOD2ac. Front Cell Dev Biol 2022, 10. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, W.-N.; Xue, Y.-N.; Zhang, L.-C.; Zhang, J.-J.; Lu, S.-Y.; Yan, X.-Y.; Yu, H.-M.; Su, J.; Sun, L.-K. SIRT3 Aggravates Metformin-Induced Energy Stress and Apoptosis in Ovarian Cancer Cells. Exp Cell Res 2018, 367, 137–149. [Google Scholar] [CrossRef]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.-F.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin Regulates Global DNA Methylation via Mitochondrial One-Carbon Metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin Alters DNA Methylation Genome-Wide via the H19/SAHH Axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef]
- García-Calzón, S.; Schrader, S.; Perfilyev, A.; Martinell, M.; Ahlqvist, E.; Ling, C. DNA Methylation Partially Mediates Antidiabetic Effects of Metformin on HbA1c Levels in Individuals with Type 2 Diabetes. Diabetes Res Clin Pract 2023, 202, 110807. [Google Scholar] [CrossRef]
- García-Calzón, S.; Perfilyev, A.; Männistö, V.; de Mello, V.D.; Nilsson, E.; Pihlajamäki, J.; Ling, C. Diabetes Medication Associates with DNA Methylation of Metformin Transporter Genes in the Human Liver. Clin Epigenetics 2017, 9, 102. [Google Scholar] [CrossRef]
- Hooten, N.N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; Cabo, R.; Evans, M.K. Metformin-mediated Increase in DICER1 Regulates MicroRNA Expression and Cellular Senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef]
- Demirsoy, İ.H.; Ertural, D.Y.; Balci, Ş.; Çınkır, Ü.; Sezer, K.; Tamer, L.; Aras, N. Profiles of Circulating MiRNAs Following Metformin Treatment in Patients with Type 2 Diabetes. J Med Biochem 2018, 37, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.J.; Mercader, J.M.; Moreno-Navarrete, J.M.; Rovira, O.; Guerra, E.; Esteve, E.; Xifra, G.; Martínez, C.; Ricart, W.; Rieusset, J.; et al. Profiling of Circulating MicroRNAs Reveals Common MicroRNAs Linked to Type 2 Diabetes That Change with Insulin Sensitization. Diabetes Care 2014, 37, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.C.K.; Than, W.H.; Kwan, B.C.H.; Lai, K.B.; Chan, R.C.K.; Teoh, J.Y.C.; Ng, J.K.C.; Chow, K.M.; Cheng, P.M.S.; Law, M.C.; et al. Adipose and Plasma MicroRNAs MiR-221 and 222 Associate with Obesity, Insulin Resistance, and New Onset Diabetes after Peritoneal Dialysis. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Xie, D.; Chen, F.; Zhang, Y.; Shi, B.; Song, J.; Chaudhari, K.; Yang, S.-H.; Zhang, G.J.; Sun, X.; Taylor, H.S.; et al. Let-7 Underlies Metformin-Induced Inhibition of Hepatic Glucose Production. Proc Natl Acad Sci U S A 2022, 119, e2122217119. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ye, Y.; Wang, B.; Zhao, S. MiR-140-5p Aggravates Insulin Resistance via Directly Targeting GYS1 and PPP1CC in Insulin-Resistant HepG2 Cells. Diabetes Metab Syndr Obes 2021, 14, 2515–2524. [Google Scholar] [CrossRef]
- Alimoradi, N.; Firouzabadi, N.; Fatehi, R. How Metformin Affects Various Malignancies by Means of MicroRNAs: A Brief Review. Cancer Cell Int 2021, 21, 207. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.G.; Stratigou, T.; Tsagarakis, S. Metformin and Gut Microbiota: Their Interactions and Their Impact on Diabetes. Hormones 2019, 18, 141–144. [Google Scholar] [CrossRef]
- Petakh, P.; Kamyshna, I.; Kamyshnyi, A. Effects of Metformin on the Gut Microbiota: A Systematic Review. Mol Metab 2023, 77, 101805. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, J.; Feng, S.; Huang, C.; Wang, H.; Huo, F.; Liu, H. Akkermansia Muciniphila, Which Is Enriched in the Gut Microbiota by Metformin, Improves Cognitive Function in Aged Mice by Reducing the Proinflammatory Cytokine Interleukin-6. Microbiome 2023, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lou, X.; Jiang, C.; Ji, X.; Tao, X.; Sun, J.; Bao, Z. Gut Microbiota Is Correlated with Gastrointestinal Adverse Events of Metformin in Patients with Type 2 Diabetes. Front Endocrinol (Lausanne) 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Silamiķele, L.; Silamiķelis, I.; Ustinova, M.; Kalniņa, Z.; Elbere, I.; Petrovska, R.; Kalniņa, I.; Kloviņš, J. Metformin Strongly Affects Gut Microbiome Composition in High-Fat Diet-Induced Type 2 Diabetes Mouse Model of Both Sexes. Front Endocrinol (Lausanne) 2021, 12, 626359. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.T.; Differding, M.K.; Zhang, M.; Maruthur, N.M.; Juraschek, S.P.; Miller, E.R.; Appel, L.J.; Yeh, H.-C. Metformin Affects Gut Microbiome Composition and Function and Circulating Short-Chain Fatty Acids: A Randomized Trial. Diabetes Care 2021, 44, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Perdigones, C.M.; Muñoz-Garach, A.; Álvarez-Bermúdez, M.D.; Moreno-Indias, I.; Tinahones, F.J. Gut Microbiota of Patients with Type 2 Diabetes and Gastrointestinal Intolerance to Metformin Differs in Composition and Functionality from Tolerant Patients. Biomedicine & Pharmacotherapy 2022, 145, 112448. [Google Scholar] [CrossRef]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut Microbiota and Intestinal FXR Mediate the Clinical Benefits of Metformin. Nat Med 2018, 24, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive Impact of Non-Antibiotic Drugs on Human Gut Bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef]
- Morales, D.R.; Morris, A.D. Metformin in Cancer Treatment and Prevention. Annu Rev Med 2015, 66, 17–29. [Google Scholar] [CrossRef]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global Burden of Colorectal Cancer in 2020 and 2040: Incidence and Mortality Estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef]
- Cancer Today Available online:. Available online: https://gco.iarc.fr/today/en (accessed on 4 March 2024).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Garczorz, W.; Kosowska, A.; Francuz, T. Antidiabetic Drugs in Breast Cancer Patients. Cancers (Basel) 2024, 16. [Google Scholar] [CrossRef]
- Lu, Y.; Hajjar, A.; Cryns, V.L.; Trentham-Dietz, A.; Gangnon, R.E.; Heckman-Stoddard, B.M.; Alagoz, O. Breast Cancer Risk for Women with Diabetes and the Impact of Metformin: A Meta-Analysis. Cancer Med 2023, 12, 11703–11718. [Google Scholar] [CrossRef]
- Bellerba, F.; Chatziioannou, A.C.; Jasbi, P.; Robinot, N.; Keski-Rahkonen, P.; Trolat, A.; Vozar, B.; Hartman, S.J.; Scalbert, A.; Bonanni, B.; et al. Metabolomic Profiles of Metformin in Breast Cancer Survivors: A Pooled Analysis of Plasmas from Two Randomized Placebo-Controlled Trials. J Transl Med 2022, 20, 1–16. [Google Scholar] [CrossRef]
- Chikermane, S.G.; Sharma, M.; Abughosh, S.M.; Aparasu, R.R.; Trivedi, M. V.; Johnson, M.L. Dose-Dependent Relation between Metformin and the Risk of Hormone Receptor-Positive, Her2-Negative Breast Cancer among Postmenopausal Women with Type-2 Diabetes. Breast Cancer Res Treat 2022, 195, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Löfling, L.L.; Støer, N.C.; Andreassen, B.K.; Ursin, G.; Botteri, E. Low-Dose Aspirin, Statins, and Metformin and Survival in Patients with Breast Cancers: A Norwegian Population-Based Cohort Study. Breast Cancer Research 2023, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, H.; Cao, L.; Yin, Y.; Shen, Y.; Zhu, W. Prognostic Value of Metformin in Cancers: An Updated Meta-Analysis Based on 80 Cohort Studies. Medicine 2022, 101, E31799. [Google Scholar] [CrossRef] [PubMed]
- Najafi, F.; Rajati, F.; Sarokhani, D.; Bavandpour, M.; Moradinazar, M. The Relationship between Metformin Consumption and Cancer Risk: An Updated Umbrella Review of Systematic Reviews and Meta-Analyses. Int J Prev Med 2023, 14. [Google Scholar] [CrossRef]
- Yu, O.H.Y.; Suissa, S. Metformin and Cancer: Solutions to a Real-World Evidence Failure. Diabetes Care 2023, 46, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Essa, N.M.; Salem, H.F.; Elgendy, M.O.; Gabr, A.; Omran, M.M.; Hassan, N.A.; Tashkandi, H.M.; Harakeh, S.; Boshra, M.S. Efficacy of Metformin as Adjuvant Therapy in Metastatic Breast Cancer Treatment. Journal of Clinical Medicine 2022, Vol. 11, Page 5505 2022, 11, 5505. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Amirabadizadeh, A.; Aramjoo, H.; Llorens, S.; Roshanravan, B.; Saeedi, F.; Talebi, M.; Shakibaei, M.; Samarghandian, S. Impact of Metformin on Cancer Biomarkers in Non-Diabetic Cancer Patients: A Systematic Review and Meta-Analysis of Clinical Trials. Current Oncology 2021, 28, 1412. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Bellerba, F.; Macis, D.; Bertelsen, B.E.; Guerrieri-Gonzaga, A.; Aristarco, V.; Viste, K.; Mellgren, G.; Di Cola, G.; Costantino, J.; et al. Effect of Metformin and Lifestyle Intervention on Adipokines and Hormones in Breast Cancer Survivors: A Pooled Analysis from Two Randomized Controlled Trials. Breast Cancer Res Treat 2024. [Google Scholar] [CrossRef]
- Serageldin, M.A.; Kassem, A.B.; El-Kerm, Y.; Helmy, M.W.; El-Mas, M.M.; El-Bassiouny, N.A. The Effect of Metformin on Chemotherapy-Induced Toxicities in Non-Diabetic Breast Cancer Patients: A Randomised Controlled Study. Drug Saf 2023, 46, 587. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Coleman, N.; Piha-Paul, S.A.; Tsimberidou, A.M.; Janku, F.; Rodon, J.; Pant, S.; Dumbrava, E.E.I.; Fu, S.; Hong, D.S.; et al. Phase I Study of MTORC1/2 Inhibitor Sapanisertib (CB-228/TAK-228) in Combination with Metformin in Patients with MTOR/AKT/PI3K Pathway Alterations and Advanced Solid Malignancies. Cancer Research Communications 2024, 4, 378. [Google Scholar] [CrossRef]
- Bashraheel, S.S.; Kheraldine, H.; Khalaf, S.; Moustafa, A.E. Al Metformin and HER2-Positive Breast Cancer: Mechanisms and Therapeutic Implications. Biomedicine & Pharmacotherapy 2023, 162, 114676. [Google Scholar] [CrossRef]
- Huang, J.; Tong, Y.; Hong, J.; Huang, O.; Wu, J.; He, J.; Chen, W.; Li, Y.; Chen, X.; Shen, K. Neoadjuvant Docetaxel, Epirubicin, and Cyclophosphamide with or without Metformin in Breast Cancer Patients with Metabolic Abnormality: Results from the Randomized Phase II NeoMET Trial. Breast Cancer Res Treat 2023, 197, 525–533. [Google Scholar] [CrossRef]
- K, S.; A, T.; A, G.; L, H.; H, G.; DD, S.; LG, E.; L, K.; E, K. The Combined Treatment with Ketogenic Diet and Metformin Slows Tumor Growth in Two Mouse Models of Triple Negative Breast Cancer. Res Sq 2023. [Google Scholar] [CrossRef]
- García-Castillo, V.; López-Urrutia, E.; Villanueva-Sánchez, O.; Ávila-Rodríguez, M.A.; Zentella-Dehesa, A.; Cortés-González, C.; López-Camarillo, C.; Jacobo-Herrera, N.J.; Pérez-Plasencia, C. Targeting Metabolic Remodeling in Triple Negative Breast Cancer in a Murine Model. J Cancer 2017, 8, 178. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Zhao, Y.; Shi, F.; Zhou, Q.; Wu, J.; Lyu, S.; Song, Q. Metformin Inducing the Change of Functional and Exhausted Phenotypic Tumor-Infiltrated Lymphocytes and the Correlation with JNK Signal Pathway in Triple-Negative Breast Cancer. Breast Cancer : Targets and Therapy 2022, 14, 391. [Google Scholar] [CrossRef]
- Jiang, H.; Suo, H.; Gao, L.; Liu, Y.; Chen, B.; Lu, S.; Jin, F.; Cao, Y. Metformin Plays an Antitumor Role by Downregulating Inhibitory Cells and Immune Checkpoint Molecules While Activating Protective Immune Responses in Breast Cancer. Int Immunopharmacol 2023, 118, 110038. [Google Scholar] [CrossRef] [PubMed]
- Zilinyi, R.; Czompa, A.; Czegledi, A.; Gajtko, A.; Pituk, D.; Lekli, I.; Tosaki, A. The Cardioprotective Effect of Metformin in Doxorubicin-Induced Cardiotoxicity: The Role of Autophagy. Molecules : A Journal of Synthetic Chemistry and Natural Product Chemistry 2018, 23. [Google Scholar] [CrossRef]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lamb, R.; Hulit, J.; Howell, A.; Gandara, R.; Sartini, M.; Rubin, E.; Lisanti, M.P.; Sotgia, F. Mitochondrial Dysfunction in Breast Cancer Cells Prevents Tumor Growth: Understanding Chemoprevention with Metformin. Cell Cycle 2013, 12, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Nurzhan, S.; Bekezhankyzy, Z.; Ding, H.; Berdigaliyev, N.; Sergazy, S.; Gulyayev, A.; Shulgau, Z.; Triggle, C.R.; Aljofan, M. The Effect of Different Glucose Concentrations on the Antiproliferative Activity of Metformin in MCF-7 Breast Cancer Cells. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab 2017, 26, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and Phenformin Deplete Tricarboxylic Acid Cycle and Glycolytic Intermediates during Cell Transformation and NTPs in Cancer Stem Cells. Proc Natl Acad Sci U S A 2014, 111, 10574–10579. [Google Scholar] [CrossRef]
- Gao, Z.Y.; Liu, Z.; Bi, M.H.; Zhang, J.J.; Han, Z.Q.; Han, X.; Wang, H.Y.; Sun, G.P.; Liu, H. Metformin Induces Apoptosis via a Mitochondria-Mediated Pathway in Human Breast Cancer Cells in Vitro. Exp Ther Med 2016, 11, 1700. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, S. Metformin Inhibits Human Breast Cancer Cell Growth by Promoting Apoptosis via a ROS-Independent Pathway Involving Mitochondrial Dysfunction: Pivotal Role of Superoxide Dismutase (SOD). Cellular Oncology 2018, 41, 637–650. [Google Scholar] [CrossRef]
- Min, W.L.; Wang, B.F.; Liang, B.B.; Zhang, L.; Pan, J.Y.; Huang, Y.; Zhao, Y.; Lin, S.; Zhao, Y.H.; Zhang, S.Q.; et al. A ROS/Akt/NF-ΚB Signaling Cascade Mediates Epidermal Growth Factor-Induced Epithelial-Mesenchymal Transition and Invasion in Human Breast Cancer Cells. World J Oncol 2022, 13, 289–298. [Google Scholar] [CrossRef]
- Shi, P.; Liu, W.; Tala, T.; Wang, H.; Li, F.; Zhang, H.; Wu, Y.; Kong, Y.; Zhou, Z.; Wang, C.; et al. Metformin Suppresses Triple-Negative Breast Cancer Stem Cells by Targeting KLF5 for Degradation. Cell Discovery 2017 3:1 2017, 3, 1–13. [Google Scholar] [CrossRef]
- Safari, F.; Momeni, A.; Ramezani, M.; Ansari, Y.; Moghbelinejad, S. Metformin Caused Radiosensitivity of Breast Cancer Cells through the Expression Modulation of MiR-21-5p/SESN1axis. Asian Pac J Cancer Prev 2023, 24, 3715. [Google Scholar] [CrossRef]
- Zhuang, Y.; Haugrud, A.B.; Schaefer, M.A.; Messerli, S.M.; Miskimins, W.K. Ability of Metformin to Deplete NAD+ Contributes to Cancer Cell Susceptibility to Metformin Cytotoxicity and Is Dependent on NAMPT Expression. Front Oncol 2023, 13, 1225220. [Google Scholar] [CrossRef]
- Yu, G.H.; Li, S.F.; Wei, R.; Jiang, Z. Diabetes and Colorectal Cancer Risk: Clinical and Therapeutic Implications. J Diabetes Res 2022, 2022. [Google Scholar] [CrossRef] [PubMed]
- Lawler, T.; Walts, Z.L.; Steinwandel, M.; Lipworth, L.; Murff, H.J.; Zheng, W.; Warren Andersen, S. Type 2 Diabetes and Colorectal Cancer Risk. JAMA Netw Open 2023, 6, e2343333–e2343333. [Google Scholar] [CrossRef] [PubMed]
- Berster, J.M.; Göke, B. Type 2 Diabetes Mellitus as Risk Factor for Colorectal Cancer. Arch Physiol Biochem 2008, 114, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yan, L.; Wang, Z.; Lu, Y.; Chu, Y.; Li, X.; Liu, Y.; Rui, D.; Nie, S.; Xiang, H. Metformin Therapy and Risk of Colorectal Adenomas and Colorectal Cancer in Type 2 Diabetes Mellitus Patients: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 16017. [Google Scholar] [CrossRef] [PubMed]
- Sehdev, A.; Shih, Y.C.T.; Vekhter, B.; Bissonnette, M.B.; Olopade, O.I.; Polite, B.N. Metformin for Primary Colorectal Cancer Prevention in Patients with Diabetes: A Case-Control Study in a US Population. Cancer 2015, 121, 1071–1078. [Google Scholar] [CrossRef]
- Berkovic, M.C.; Mikulic, D.; Bilic-Curcic, I.; Mrzljak, A. How Far along Are We in Revealing the Connection between Metformin and Colorectal Cancer? World J Gastroenterol 2021, 27, 1362. [Google Scholar] [CrossRef]
- Bradley, M.C.; Ferrara, A.; Achacoso, N.; Ehrlich, S.F.; Quesenberry, C.P.; Habel, L.A. A Cohort Study of Metformin and Colorectal Cancer Risk among Patients with Diabetes Mellitus. Cancer Epidemiol Biomarkers Prev 2018, 27, 525–530. [Google Scholar] [CrossRef]
- Jung, Y.S.; Park, C.H.; Eun, C.S.; Park, D. Il; Han, D.S. Metformin Use and the Risk of Colorectal Adenoma: A Systematic Review and Meta-Analysis. J Gastroenterol Hepatol 2017, 32, 957–965. [Google Scholar] [CrossRef]
- Skinner, H.; Hu, C.; Tsakiridis, T.; Santana-Davila, R.; Lu, B.; Erasmus, J.J.; Doemer, A.J.; Videtic, G.M.M.; Coster, J.; Yang, A.X.; et al. Addition of Metformin to Concurrent Chemoradiation in Patients With Locally Advanced Non-Small Cell Lung Cancer: The NRG-LU001 Phase 2 Randomized Clinical Trial. JAMA Oncol 2021, 7, 1324–1332. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Chen, B.E.; Gelmon, K.A.; Whelan, T.J.; Ennis, M.; Lemieux, J.; Ligibel, J.A.; Hershman, D.L.; Mayer, I.A.; Hobday, T.J.; et al. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients With Breast Cancer: The MA.32 Randomized Clinical Trial. JAMA 2022, 327, 1963–1973. [Google Scholar] [CrossRef]
- Pujalte Martin, M.; Borchiellini, D.; Thamphya, B.; Guillot, A.; Paoli, J.B.; Besson, D.; Hilgers, W.; Priou, F.; El Kouri, C.; Hoch, B.; et al. TAXOMET: A French Prospective Multicentric Randomized Phase II Study of Docetaxel Plus Metformin Versus Docetaxel Plus Placebo in Metastatic Castration-Resistant Prostate Cancer. Clin Genitourin Cancer 2021, 19, 501–509. [Google Scholar] [CrossRef]
- Akce, M.; Farran, B.; Switchenko, J.M.; Rupji, M.; Kang, S.; Khalil, L.; Ruggieri-Joyce, A.; Olson, B.; Shaib, W.L.; Wu, C.; et al. Phase II Trial of Nivolumab and Metformin in Patients with Treatment-Refractory Microsatellite Stable Metastatic Colorectal Cancer. J Immunother Cancer 2023, 11. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, M.; Zheng, Z.; Zhang, Q.; Gao, F.; Xue, Y. Effects of Metformin on CD133+ Colorectal Cancer Cells in Diabetic Patients. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Bragagnoli, A.C.; Araujo, R.L.C.; Ferraz, M.W.; dos Santos, L.V.; Abdalla, K.C.; Comar, F.; Santos, F.A.; Oliveira, M.A.; Carvalheira, J.B.C.; Cárcano, F.M.; et al. Metformin plus Lrinotecan in Patients with Refractory Colorectal Cancer: A Phase 2 Clinical Trial. British Journal of Cancer 2021 124:6 2021, 124, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Miranda, V.C.; Braghiroli, M.I.; Faria, L.D.; Bariani, G.; Alex, A.; Bezerra Neto, J.E.; Capareli, F.C.; Sabbaga, J.; Lobo dos Santos, J.F.; Hoff, P.M.; et al. Phase 2 Trial of Metformin Combined With 5-Fluorouracil in Patients With Refractory Metastatic Colorectal Cancer. Clin Colorectal Cancer 2016, 15, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.J.; Seo, Y.; Kwon, J.H.; Yoon, J.P.; Kang, J.Y.; Lee, H.J.; Park, S.J.; Hong, S.P.; Cheon, J.H.; et al. Effects of Metformin on Colorectal Cancer Stem Cells Depend on Alterations in Glutamine Metabolism. Scientific Reports 2018 8:1 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Durán, R. V.; Mackenzie, E.D.; Boulahbel, H.; Frezza, C.; Heiserich, L.; Tardito, S.; Bussolati, O.; Rocha, S.; Hall, M.N.; Gottlieb, E. HIF-Independent Role of Prolyl Hydroxylases in the Cellular Response to Amino Acids. Oncogene 2013, 32, 4549–4556. [Google Scholar] [CrossRef]
- Bozzi, F.; Mogavero, A.; Varinelli, L.; Belfiore, A.; Manenti, G.; Caccia, C.; Volpi, C.C.; Beznoussenko, G. V.; Milione, M.; Leoni, V.; et al. MIF/CD74 Axis Is a Target for Novel Therapies in Colon Carcinomatosis. Journal of Experimental and Clinical Cancer Research 2017, 36, 1–15. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The Mechanisms of Action of Metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Hosono, K.; Endo, H.; Takahashi, H.; Sugiyama, M.; Uchiyama, T.; Suzuki, K.; Nozaki, Y.; Yoneda, K.; Fujita, K.; Yoneda, M.; et al. Metformin Suppresses Azoxymethane-Induced Colorectal Aberrant Crypt Foci by Activating AMP-Activated Protein Kinase. Mol Carcinog 2010, 49, 662–671. [Google Scholar] [CrossRef]
- Tomimoto, A.; Endo, H.; Sugiyama, M.; Fujisawa, T.; Hosono, K.; Takahashi, H.; Nakajima, N.; Nagashima, Y.; Wada, K.; Nakagama, H.; et al. Metformin Suppresses Intestinal Polyp Growth in ApcMin/+ Mice. Cancer Sci 2008, 99, 2136–2141. [Google Scholar] [CrossRef]
- Zaafar, D.K.; Zaitone, S.A.; Moustafa, Y.M. Role of Metformin in Suppressing 1,2-Dimethylhydrazine-Induced Colon Cancer in Diabetic and Non-Diabetic Mice: Effect on Tumor Angiogenesis and Cell Proliferation. PLoS One 2014, 9. [Google Scholar] [CrossRef]
- Mohamed Suhaimi, N.A.; Phyo, W.M.; Yap, H.Y.; Choy, S.H.Y.; Wei, X.; Choudhury, Y.; Tan, W.J.; Tan, L.A.P.Y.; Foo, R.S.Y.; Tan, S.H.S.; et al. Metformin Inhibits Cellular Proliferation and Bioenergetics in Colorectal Cancer Patient-Derived Xenografts. Mol Cancer Ther 2017, 16, 2035–2044. [Google Scholar] [CrossRef]
- Nangia-Makker, P.; Yu, Y.; Vasudevan, A.; Farhana, L.; Rajendra, S.G.; Levi, E.; Majumdar, A.P.N. Metformin: A Potential Therapeutic Agent for Recurrent Colon Cancer. PLoS One 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Okabayashi, K.; Seishima, R.; Ishida, T.; Shigeta, K.; Tsuruta, M.; Hasegawa, H.; Kitagawa, Y. Metformin Inhibits the Development and Metastasis of Colorectal Cancer. Med Oncol 2022, 39. [Google Scholar] [CrossRef] [PubMed]
- Duraiyarasan, S.; Adefuye, M.; Manjunatha, N.; Ganduri, V.; Rajasekaran, K. Colon Cancer and Obesity: A Narrative Review. Cureus 2022, 14. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.J.O.; Sonenberg, N.; Pollak, M.N. The Effects of Adiponectin and Metformin on Prostate and Colon Neoplasia Involve Activation of AMP-Activated Protein Kinase. Cancer Prev Res (Phila) 2008, 1, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Algire, C.; Moiseeva, O.; Deschênes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin Reduces Endogenous Reactive Oxygen Species and Associated DNA Damage. Cancer Prev Res (Phila) 2012, 5, 536–543. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A. V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res 2017, 5, 9–16. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Saigal, A.; Szamosi, J.C.; Hammill, J.A.; Bezverbnaya, K.; Wang, D.; Gautam, J.; Tsakiridis, E.E.; Di Pastena, F.; McNicol, J.; et al. Metformin-Induced Reductions in Tumor Growth Involves Modulation of the Gut Microbiome. Mol Metab 2022, 61. [Google Scholar] [CrossRef]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297. [Google Scholar] [CrossRef]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin Transiently Inhibits Colorectal Cancer Cell Proliferation as a Result of Either AMPK Activation or Increased ROS Production. Sci Rep 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.A.; Van Wickle, J.; Hill, R.B.; Marchese, A.; Kalyanaraman, B.; Dwinell, M.B. Mitochondria-Targeted Drugs Stimulate Mitophagy and Abrogate Colon Cancer Cell Proliferation. Journal of Biological Chemistry 2018, 293, 14891–14904. [Google Scholar] [CrossRef] [PubMed]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.L.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.A.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-Fluorouracil Resistant Colon Cancer Cells Are Addicted to OXPHOS to Survive and Enhance Stem-like Traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef]
- Zhang, P.; Zhao, S.; Lu, X.; Shi, Z.; Liu, H.; Zhu, B. Metformin Enhances the Sensitivity of Colorectal Cancer Cells to Cisplatin through ROS-Mediated PI3K/Akt Signaling Pathway. Gene 2020, 745. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Scott Budigner, G.R.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. Elife 2014, 3. [Google Scholar] [CrossRef]
- Khodaei, F.; Hosseini, S.M.; Omidi, M.; Hosseini, S.F.; Rezaei, M. Cytotoxicity of Metformin against HT29 Colon Cancer Cells Contributes to Mitochondrial Sirt3 Upregulation. J Biochem Mol Toxicol 2021, 35. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ung, T.T.; Li, S.; Lian, S.; Xia, Y.; Park, S.Y.; Do Jung, Y. Metformin Inhibits Lithocholic Acid-Induced Interleukin 8 Upregulation in Colorectal Cancer Cells by Suppressing ROS Production and NF-KB Activity. Sci Rep 2019, 9. [Google Scholar] [CrossRef]
- Cheng, G.; Lanza-Jacoby, S. Metformin Decreases Growth of Pancreatic Cancer Cells by Decreasing Reactive Oxygen Species: Role of NOX4. Biochem Biophys Res Commun 2015, 465, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.H.; Pu, X.X.; Huang, M.J.; Xiao, J.; Zhou, J.M.; Lin, T.Y.; Lin, E.H. 5-Fluorouracil Upregulates the Activity of Wnt Signaling Pathway in CD133-Positive Colon Cancer Stem-like Cells. Chin J Cancer 2010, 29, 810–815. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, S.C.; Ku, J.L. Metformin Increases Chemo-Sensitivity via Gene Downregulation Encoding DNA Replication Proteins in 5-Fu Resistant Colorectal Cancer Cells. Oncotarget 2017, 8, 56546–56557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Y. Metformin Attenuates Cells Stemness and Epithelial-mesenchymal Transition in Colorectal Cancer Cells by Inhibiting the Wnt3a/Β-catenin Pathway. Mol Med Rep 2019, 19, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res 2016, 76, 3904–3915. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Wadhwani, A.; Sauraj; Roychowdhury, P. ; Kang, J.H.; Ko, Y.T.; Kuppusamy, G. WZB117 Decorated Metformin-Carboxymethyl Chitosan Nanoparticles for Targeting Breast Cancer Metabolism. Polymers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Shahbazi, R.; Jafari-Gharabaghlou, D.; Mirjafary, Z.; Saeidian, H.; Zarghami, N. Design and Optimization Various Formulations of PEGylated Niosomal Nanoparticles Loaded with Phytochemical Agents: Potential Anti-Cancer Effects against Human Lung Cancer Cells. Pharmacol Rep 2023, 75, 442–455. [Google Scholar] [CrossRef]
- Kumar, V.; Sharma, K.; Sachan, R.; Alhayyani, S.; Al-abbasi, F.A.; Singh, R.; Anwar, F. Co-Drug Development of Gallic Acid and Metformin Targeting the pro-Inflammatory Cytokines for the Treatment of Breast Cancer. J Biochem Mol Toxicol 2023, 37. [Google Scholar] [CrossRef]
- Citi, V.; Barresi, E.; Piragine, E.; Spezzini, J.; Testai, L.; Da Settimo, F.; Martelli, A.; Taliani, S.; Calderone, V. Anti-Proliferative Properties of the Novel Hybrid Drug Met-ITC, Composed of the Native Drug Metformin with the Addition of an Isothiocyanate H2S Donor Moiety, in Different Cancer Cell Lines. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Solier, S.; Müller, S.; Cañeque, T.; Versini, A.; Mansart, A.; Sindikubwabo, F.; Baron, L.; Emam, L.; Gestraud, P.; Pantoș, G.D.; et al. A Druggable Copper-Signalling Pathway That Drives Inflammation. Nature 2023, 617, 386–394. [Google Scholar] [CrossRef]
- Ailuno, G.; Baldassari, S.; Balboni, A.; Pastorino, S.; Zuccari, G.; Cortese, K.; Barbieri, F.; Drava, G.; Florio, T.; Caviglioli, G. Development of Biotinylated Liposomes Encapsulating Metformin for Therapeutic Targeting of Inflammation-Based Diseases. Pharmaceutics 2024, 16, 235. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, Y.; Ding, K.; Li, S.; Liu, T. Biomimetic Nanovesicle Co-Delivery System Impairs Energy Metabolism for Cancer Treatment. J Nanobiotechnology 2023, 21. [Google Scholar] [CrossRef] [PubMed]
- Torunoglu, S.T.; Zajda, A.; Tampio, J.; Markowicz-Piasecka, M.; Huttunen, K.M. Metformin Derivatives - Researchers’ Friends or Foes? Biochem Pharmacol 2023, 215. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, Y.; Liu, Y.; Jia, B.; Liu, X.; Ma, T. Carbonized Polymer Dots Derived from Metformin and L-Arginine for Tumor Cell Membrane- and Mitochondria-Dual Targeting Therapy. Nanoscale 2023, 15, 17922–17935. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, J.; Hardy, M.; Ouari, O.; Chitambar, C.R.; Dwinell, M.B.; Kalyanaraman, B. Synergistic Inhibition of Tumor Cell Proliferation by Metformin and Mito-Metformin in the Presence of Iron Chelators. Oncotarget 2019, 10, 3518–3532. [Google Scholar] [CrossRef]
- Ji, P.; Deng, X.C.; Jin, X.K.; Zhang, S.M.; Wang, J.W.; Feng, J.; Chen, W.H.; Zhang, X.Z. Fused Cytomembrane-Camouflaged Nanoparticles for Tumor-Specific Immunotherapy. Adv Healthc Mater 2023, 12. [Google Scholar] [CrossRef]
Figure 1.
Complex I-dependent mechanism. The figure illustrates the proposed mechanism for metformin via inhibition of Complex I. Metformin inhibits complex I, causing a slight decrease in mitochondrial respiratory chain activity, resulting in an increase in the cellular redox state (NADH/NAD+) and a decrease in ATP synthesis (ATP/ADP), which leads to the inhibition of gluconeogenesis. Additionally, there is an increase in AMP levels that induces gluconeogenesis inhibition by AMPK-dependent and AMPK–independent mechanisms. Among the AMPK-independent mechanisms, increased AMP levels can inhibit the activity of gluconeogenic enzymes such AC and FBP1. AC: adenylate cyclase; FBP1: fructose-1,6-biphosphatase.
Figure 1.
Complex I-dependent mechanism. The figure illustrates the proposed mechanism for metformin via inhibition of Complex I. Metformin inhibits complex I, causing a slight decrease in mitochondrial respiratory chain activity, resulting in an increase in the cellular redox state (NADH/NAD+) and a decrease in ATP synthesis (ATP/ADP), which leads to the inhibition of gluconeogenesis. Additionally, there is an increase in AMP levels that induces gluconeogenesis inhibition by AMPK-dependent and AMPK–independent mechanisms. Among the AMPK-independent mechanisms, increased AMP levels can inhibit the activity of gluconeogenic enzymes such AC and FBP1. AC: adenylate cyclase; FBP1: fructose-1,6-biphosphatase.
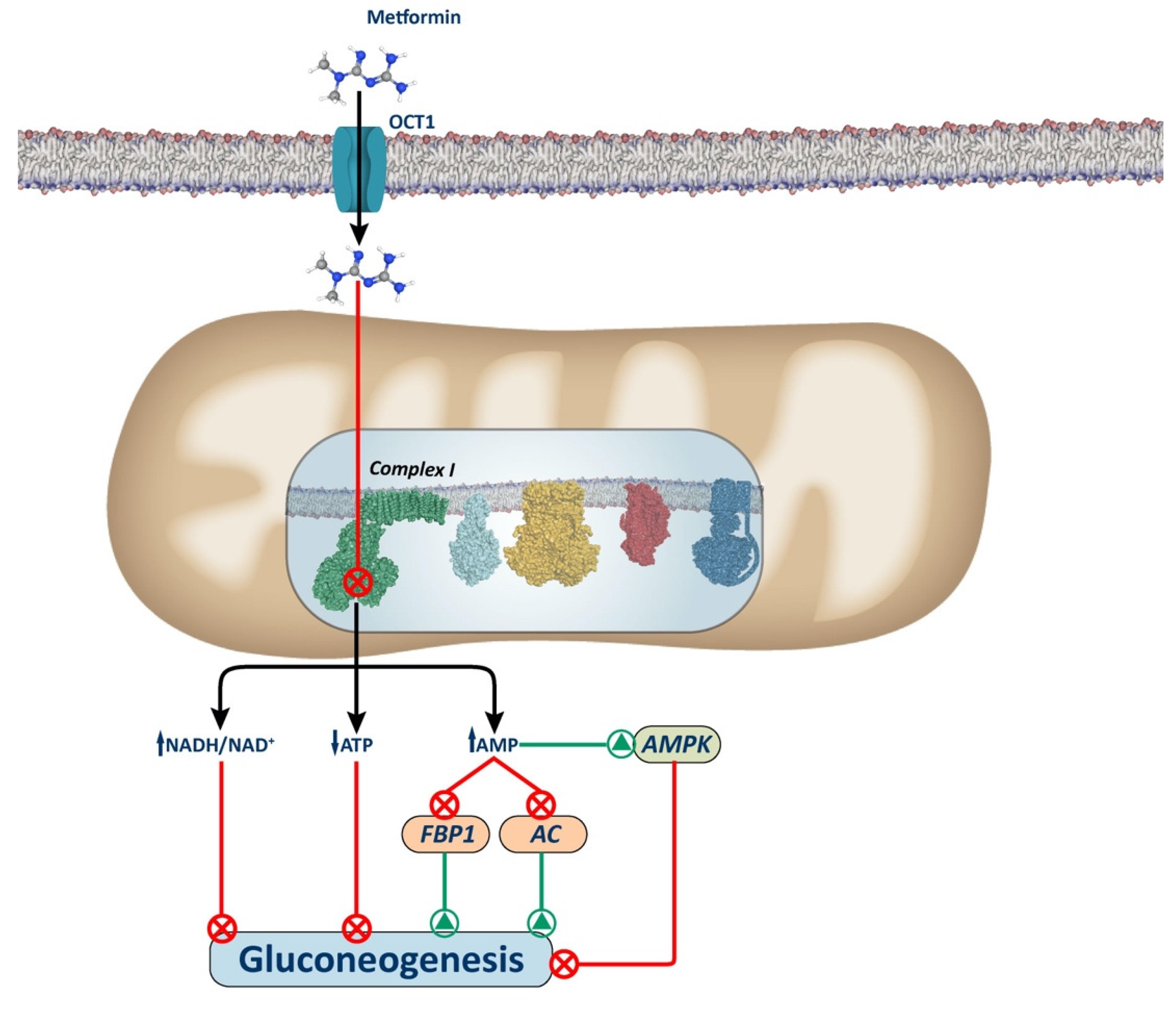
Figure 2.
Lysosomal-AMPK-dependent mechanism. The figure illustrates how metformin, through a lysosome-dependent mechanism, can activate AMPK. Metformin interacts with presenilin enhancer 2 (PEN2), which in turn is recruited by the v-ATP6P1 protein of the v-ATPase, leading to its inhibition. This inhibition promotes the translocation of AXIN-LKB1 to the surface of the lysosome, creating a docking site for AMPK activation. Studies in animals with silenced PEN2 suggest that this mechanism may play a significant role in the liver and intestine, regulating lipid metabolism and the secretion of glucagon-like peptide 1 (GLP1), respectively.
Figure 2.
Lysosomal-AMPK-dependent mechanism. The figure illustrates how metformin, through a lysosome-dependent mechanism, can activate AMPK. Metformin interacts with presenilin enhancer 2 (PEN2), which in turn is recruited by the v-ATP6P1 protein of the v-ATPase, leading to its inhibition. This inhibition promotes the translocation of AXIN-LKB1 to the surface of the lysosome, creating a docking site for AMPK activation. Studies in animals with silenced PEN2 suggest that this mechanism may play a significant role in the liver and intestine, regulating lipid metabolism and the secretion of glucagon-like peptide 1 (GLP1), respectively.
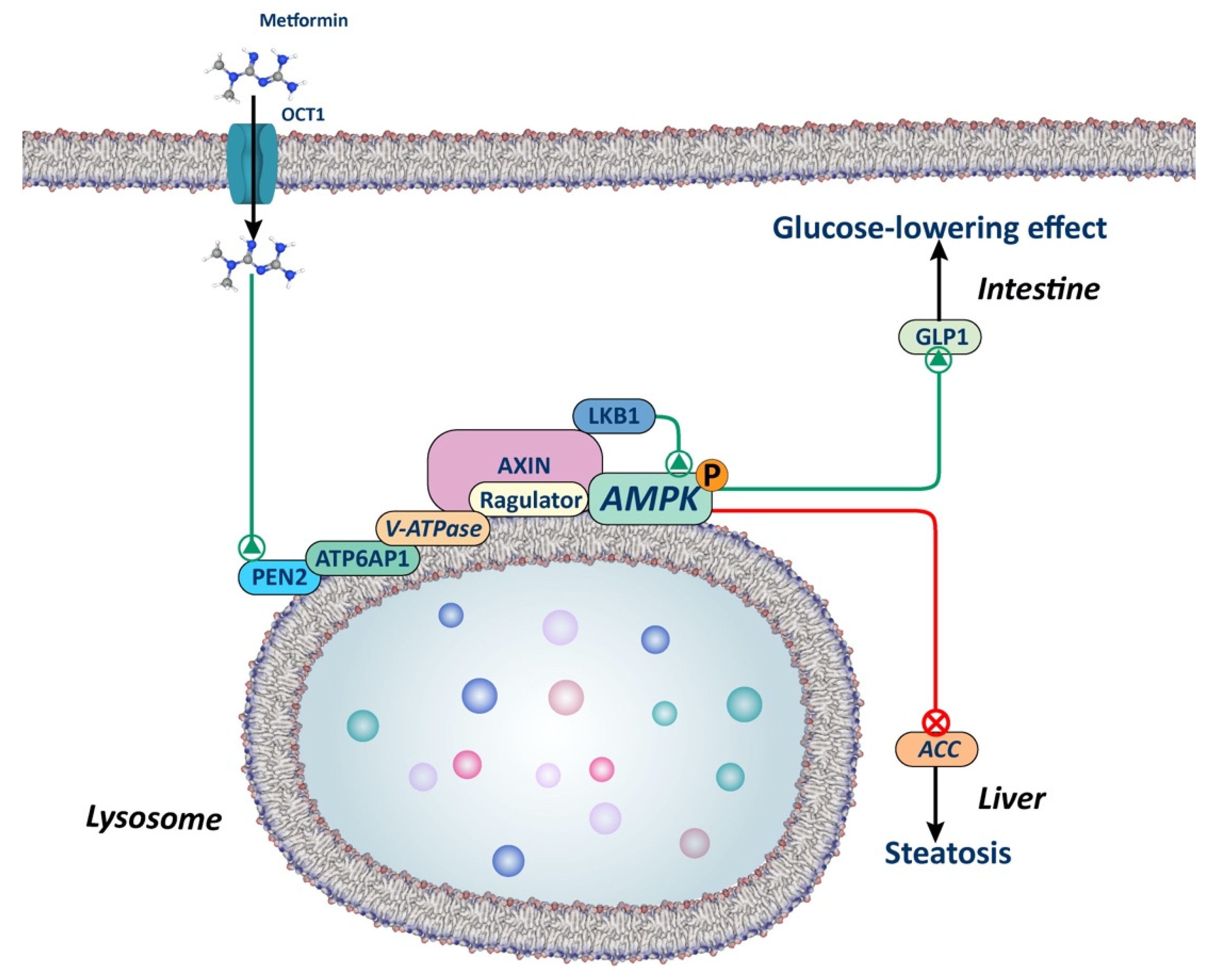
Figure 3.
Complex IV-dependent mechanism and mitochondrial GPDH-dependent mechanism. The figure illustrates the proposed mechanism for metformin via inhibition of Complex IV. Inhibition of this complex leads to a reduction in the ubiquinone pool, subsequently inhibiting mGPDH activity and thus, electron entry through the glycerol phosphate shuttle. It has been proposed that metformin may also directly inhibit this shuttle, thus contributing to an increase in the NADH/NAD+ ratio and thereby restricting the use of redox-dependent substrates such as lactate and glycerol for gluconeogenesis. Alternatively, it has also been proposed that the elevation of the NADH/NAD+ ratio induced by metformin through the inhibition of either Complex I or IV of the OXPHOS system may inhibit the aspartate-malate shuttle. In this scenario, inhibition of this shuttle would lead to compensatory activation of the glycerol phosphate shuttle, thereby decreasing glycerol-3P levels and releasing the repressive effect of this metabolite on phosphofructokinase 1. Consequently, this would enhance glycolysis as opposed to gluconeogenesis (not depicted in the figure).
Figure 3.
Complex IV-dependent mechanism and mitochondrial GPDH-dependent mechanism. The figure illustrates the proposed mechanism for metformin via inhibition of Complex IV. Inhibition of this complex leads to a reduction in the ubiquinone pool, subsequently inhibiting mGPDH activity and thus, electron entry through the glycerol phosphate shuttle. It has been proposed that metformin may also directly inhibit this shuttle, thus contributing to an increase in the NADH/NAD+ ratio and thereby restricting the use of redox-dependent substrates such as lactate and glycerol for gluconeogenesis. Alternatively, it has also been proposed that the elevation of the NADH/NAD+ ratio induced by metformin through the inhibition of either Complex I or IV of the OXPHOS system may inhibit the aspartate-malate shuttle. In this scenario, inhibition of this shuttle would lead to compensatory activation of the glycerol phosphate shuttle, thereby decreasing glycerol-3P levels and releasing the repressive effect of this metabolite on phosphofructokinase 1. Consequently, this would enhance glycolysis as opposed to gluconeogenesis (not depicted in the figure).
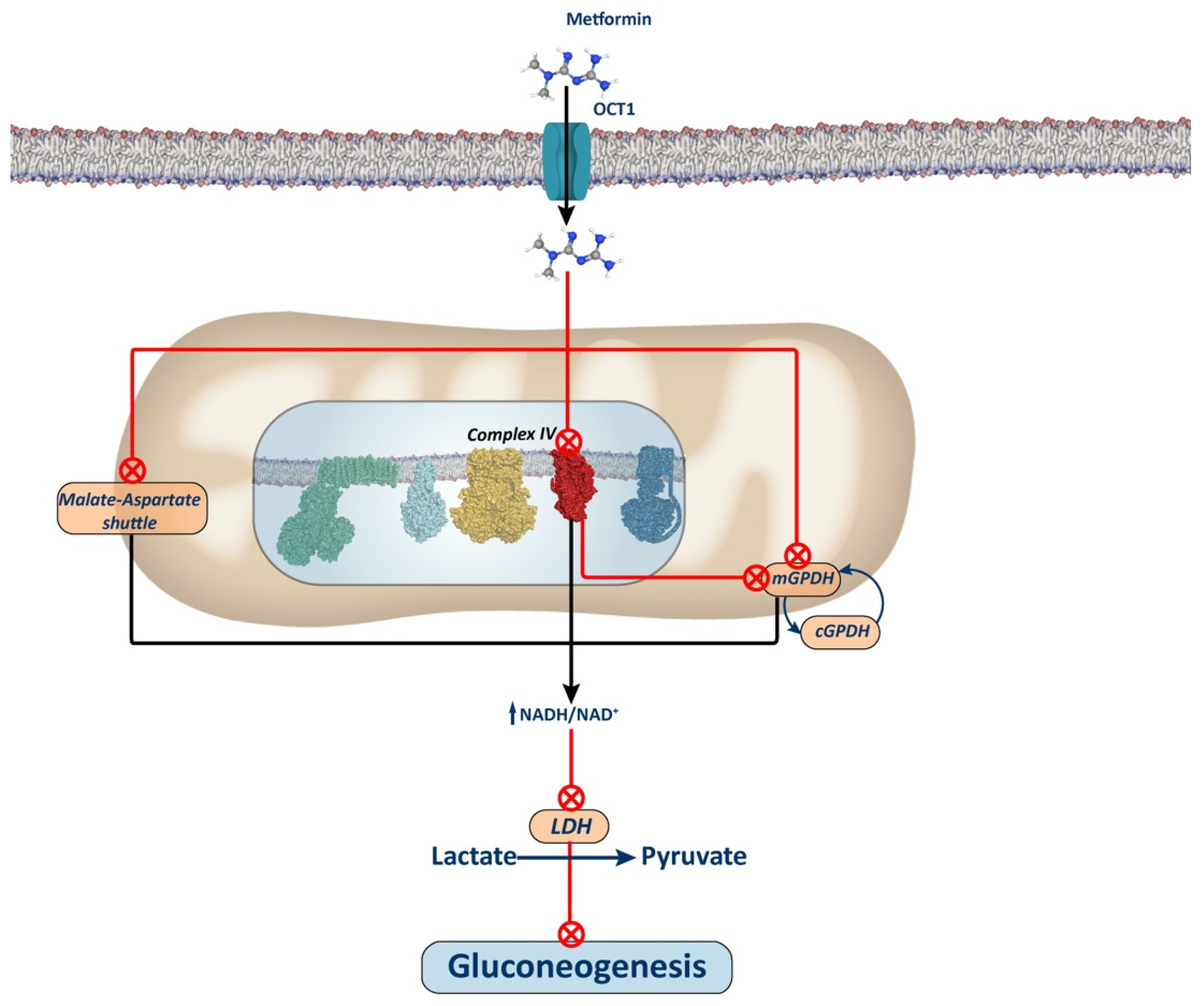
Figure 4.
Constraints and opportunities of metformin as a chemopreventive and therapeutic agent in breast and colorectal cancer management..
Figure 4.
Constraints and opportunities of metformin as a chemopreventive and therapeutic agent in breast and colorectal cancer management..
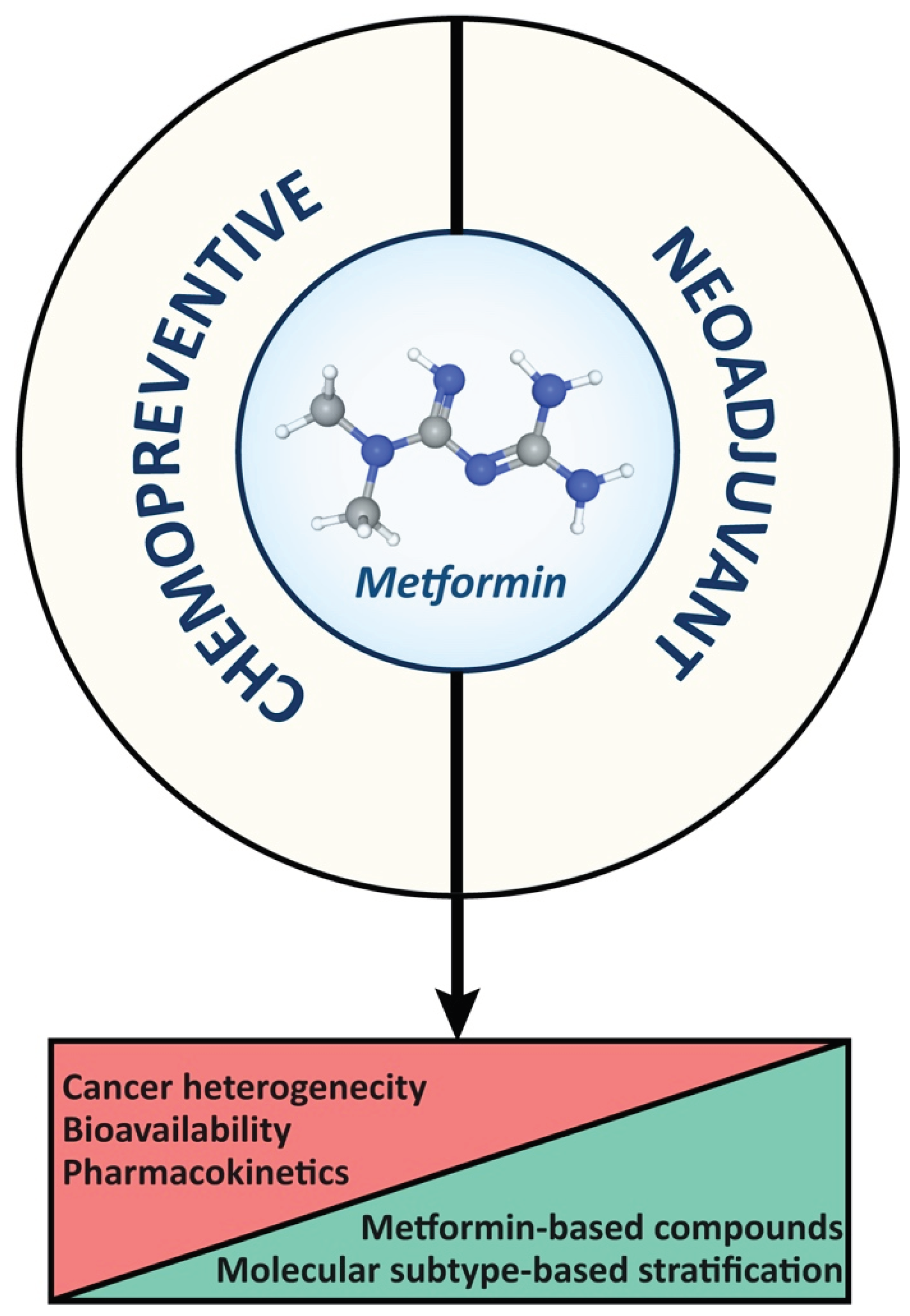
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



