
Submitted:
24 March 2024
Posted:
25 March 2024
Read the latest preprint version here
Abstract
In this study, a simple machine learning-based quantitative structure-activity relationship (QSAR) model was developed to predict the inhibitory potency (pIC50 values) of FLT3 tyrosine kinase inhibitors, pivotal in treating Acute Myeloid Leukemia (AML). Distinctively, our model leverages an extensive and diverse dataset, 14 times larger than those employed in prior studies within this field, enabling an unparalleled scope of compound analysis. This vast dataset, combined with further exploration of molecular descriptors, enabled predictions of extraordinary precision, covering a broader spectrum of FLT3 inhibitors than was previously possible. The Random Forest Regressor (RFR) algorithm, selected for its superior predictive performance, was trained with 1080 inputs and validated through comprehensive external and internal methods. It achieved an remarkable coefficient of determination (R^2) of 0.941 and a standard deviation of 0.235 on a test set of 270 compounds, highlighting the efficacy of model in predicting FLT3 inhibitory activity. Key molecular descriptors were identified, enhancing our understanding of structural requirements for inhibitor potency. Additionally, we developed a user-friendly computational tool that enables the rapid prediction of pIC50 values. Utilizing this tool, potential FLT3 inhibitors were identified through ligand-based virtual screening. This study represents a major advancement in FLT3 inhibitor discovery by utilizing a simple QSAR-machine learning model. It enables more efficient and precise identification of potential drug candidates at an early stage, promising a faster development of targeted therapies and streamlining the ligand-based drug design process.
Keywords:
FLT3 Inhibitors
; Ligand-based Drug Design
; Computer-Aided Drug Design
; QSAR Modeling
; AML Treatment
1. Introduction
Acute Myeloid Leukemia (AML) represents a formidable challenge in oncology, characterized by the uncontrolled proliferation of clonal cells within the hematopoietic system, leading to extensive tissue infiltration and disease progression. A pivotal player in the pathophysiology of AML is the Fms-like tyrosine kinase 3 (FLT3) receptor gene [1]. This gene, when mutated, particularly through internal tandem duplications (ITD), endows leukemic cells with a significant proliferative advantage. This is achieved by activating multiple signaling pathways, which are crucial in both the progression of the disease and its prognosis. FLT3 mutations are thus a key factor in the aggressive nature of AML and have been extensively studied for their role in the development and outcomes of the disease [2,3,4].
For decades, the treatment landscape for AML has predominantly relied on a conventional regimen of continuous-infusion of cytarabine combined with anthracycline drugs [5]. The effectiveness of this traditional approach is influenced by factors such as the genetic profile of the leukemia and patient age; with older patients often showing markedly lower response rates. This realization established an urgent need for novel therapeutic strategies that could improve outcomes across different patient demographics [2]. In response, the AML treatment paradigm has evolved, marked by the introduction of FLT3 inhibitors such as midostaurin and gilteritinib, targeting specific mutations, and sorafenib, quizartinib. This shift towards customized therapy, including the development of new combinations like CPX-351 and gemtuzumab ozogamicin, reflects an ongoing effort to match treatment strategies with the molecular characteristics of the disease [6]. The advent of such targeted therapies and the expansion of the treatment arsenal signify a substantial step towards more effective and tailored treatment approaches for AML, offering hope for improved outcomes in a wider range of patient groups [7,8,9].
The understanding and development of FLT3 inhibitors has greatly benefited from the implementation of Quantitative Structure-Activity Relationships (QSAR) and molecular docking [10,11,12,13,14,15,16]. The study by Sandoval et al. [10] exemplifies the use of QSAR in predicting with notable accuracy the anti-leukemic activity of compounds, employing linear discriminant and multilinear regression analyses. Similarly, Shih and Bhujbal et al. [12,14] identified key structural features and designing novel compounds with enhanced FLT3 inhibitory activity by integrating molecular docking with 3D-QSAR approaches. Ghosh et al. [16] further demonstrated the efficacy of computational modeling, including molecular dynamics and 3D-QSAR, in understanding the structure-activity relationship of FLT3 inhibitors. These methodologies, endorse by studies like those of Fernandes and Islam et al. [15,17], have provided invaluable insights into the molecular interactions and binding affinities of potential FLT3 inhibitors, emphasizing the significance of these approaches in the realm of drug discovery and development for AML.
Additionally, the integration of machine learning (ML) in drug discovery has revolutionized the identification and prediction of kinase inhibitors; including those targeting FLT3. Nasimian et al. [18] demonstrated the potential of a machine learning-based model in predicting drug sensitivity, revealing crucial insights into AXL dependency in AML. Janssen et al. [19] introduced the Drug Discovery Maps (DDM) model, employing algorithms like t-SNE to visualize and predict interactions across the kinase family, leading to the discovery of potent FLT3 inhibitors. Furthermore, Zhao et al. [20] applied ML methods to classify and analyze the structure-activity relationship of a vast number of FLT3 inhibitors, uncovering key structural features associated with high inhibitory activity. These advancements, as discussed by Eckardt et al. [21], highlight the growing importance of ML in managing AML, from diagnosis to therapy optimization. Such ML-based approaches offer a promising avenue for enhancing the efficacy and precision of FLT3 inhibitor development, signifying a paradigm shift in the treatment strategies for AML.
Despite significant strides in FLT3 inhibitor development, unresolved challenges persist, notably in the predictive accuracy of current QSAR models. These models often exhibit limited predictive performance, suggesting a need for enhanced precision. A common limitation is the reliance on a narrow range of molecular data for model training, which hampers the ability to generalize findings across a broader chemical space. The lack of molecular diversity and heterogeneity in these datasets fails to fully capture the complexity of potential FLT3 inhibitors. Furthermore, there is a notable absence of user-friendly models that provide rapid and reliable results, stressing the necessity for more practical and trustworthy methodologies in drug discovery.
In response to these challenges, our research introduces an innovative QSAR-ML model. This model is trained on a more extensive and diverse dataset, encompassing a wider range of molecules to improve robustness and generalizability. Integrating advanced machine learning techniques with sophisticated molecular descriptors, this model aims to surpass the predictive limitations of current QSAR models. Additionally, our QSAR-ML model is designed for user accessibility, offering quick and reliable outcomes. This approach promises to enhance the identification of new FLT3 inhibitors for AML treatment, setting a precedent for more efficient and accessible drug discovery tools, potentially revolutionizing the development of FLT3 inhibitors and accelerating progress towards more effective, personalized AML treatments.
2. Results and Discussion
We explore the performance of various machine learning (ML) models in predicting pIC50 values of 1350 FLT3 tyrosine kinase inhibitor compounds, based on 1269 descriptors. The models compared include Random Forest Regressor (RFR) [22], Gradient Boosting Regressor (GBR) [23], Support Vector Machine (SVM) [24], Kernel Ridge Regression (KRR) [24], Gaussian Process Regressor (GPR) [25], Bagging with Random Forest (BRF) [26], and two Artificial Neural Network (ANN) architectures implemented using Keras (ANN-K) [27] and PyTorch (ANN-P) [28].
In Table 1, we present a comprehensive comparison of the machine learning models in predicting pIC50 values of FLT3 tyrosine kinase inhibitor compounds across various metrics including R² (coefficient of determination), MAE (mean absolute error), SD (standard deviation), and RMSE (root mean square error) for both the training and testing datasets.
2.1. Model Performance Overview
2.1.1. Training Performance
The training performance was evaluated using R², MAE, SD, and RMSE metrics (see Table 1). RFR and ANN-K exhibited exceptional performance with R² values close to 1 (0.988 for both), indicating a near-perfect prediction on the training set. GBR also showed high efficacy with an R² of 0.973. In contrast, SVM model performed poorly with an R² of 0.014, suggesting almost no predictive capability. The other models displayed varying degrees of effectiveness, with KRR and GPR showing moderate to low R² values, as reflected in Table 1.
2.1.2. Testing Performance
When evaluating the models’ ability to generalize to unseen data (see Table 1), GBR and RFR showed the highest R² values on the test set (0.939 and 0.936, respectively), closely followed by BRF model (R² = 0.931). These results suggest that ensemble methods, particularly those based on decision trees, are more adept at predicting pIC50 values for FLT3 inhibitors. In contrast, SVM and GPR showed poor performance on the test set, registering negative R² values (-0.012 for SVM and -0.228 for GPR), which expose their limitations in this context.
2.1.3. Error Metrics
The MAE and RMSE values further support these findings. RFR, GBR, and BRF show lower errors on both the training and test sets (see Table 1), implying higher accuracy in their predictions. Although ANN models, particularly the one implemented with Keras (ANN-K), showed promising results on the training set, lower R² and higher error metrics compared to RFR and GBR indicate a drop-in performance on the test set.
In summary, the ensemble methods, especially Random Forest and Gradient Boosting, have proven to be highly effective in modeling the intricate data related to pIC50 values of FLT3 tyrosine kinase inhibitors. These methods are adept at capturing the nonlinear relationships between descriptors and pIC50 values and demonstrate strong generalization capabilities. The underwhelming performance of SVM and GPR underscores the complexity of parameter tuning and kernel function selection for high-dimensional data. The high training performance but reduced testing efficacy of ANN-K suggests overfitting, highlighting the need for improved regularization techniques or model architectures. The slight performance variation between ANN-K and ANN-P might reflect differences in their respective framework’s optimization and regularization processes. As supported by the data presented in Table 1, RFR stands out as the most effective model for predicting pIC50 values, showcasing its potential in handling complex, high-dimensional datasets in drug discovery applications.
2.2. Model Optimization
This section presents a comprehensive analysis of the component optimization through feature selection, focusing on the efficacy of RFR as the chosen method for predicting FLT3 tyrosine kinase inhibitor activity. Our findings, illustrated in Figure 1, stresses the paramount importance of the initial five molecular descriptors. These descriptors exhibit a combined R² test score of 0.893, indicating their critical role in model accuracy and interpretability (see below “Model Interpretation”). Subsequent inclusion of descriptors up to the eleventh markedly improves the R² test to 0.932, with a significant but diminishing return on predictive performance with each additional descriptor. Beyond the incorporation of 49 descriptors, the R² test plateaus at 0.941, suggesting that further addition of descriptors does not substantially enhance the model’s predictive capability. This observation emphasizes the effectiveness of RFR in capturing the complex nonlinear relationships between a manageable number of descriptors and pIC50 values, thus optimizing the balance between model simplicity and predictive accuracy. This outcome reaffirms the superiority of ensemble methods like RFR in handling high-dimensional data [29] while underlining the importance of a judicious feature selection process in the development of efficient and reliable predictive models for drug discovery applications.
The results presented in Table 2 demonstrate the predictive capability of the RFR model for FLT3 tyrosine kinase inhibitor compounds when optimized with 49 descriptors. With a remarkable R² value of 0.989 for training and 0.941 for testing, the model showcases exceptional accuracy and robustness in capturing the complex relationships between the descriptors and the pIC50 values. The error metrics, including MAE, SD, and RMSE, further affirm the model’s precision across both training and test datasets. The Q2LOO value of 0.926 indicates strong predictive reliability through leave-one-out cross-validation, emphasizing consistency in the model.
2.3. Comparative Analysis: QSAR Modeling
The comparative analysis provided in Table 3 reveals the significant advancements made in this work over prior QSAR studies focusing on FLT3 tyrosine kinase inhibitor compounds. Notably, the dataset size in the current study is at least 14 times larger than those used in previous research efforts, such as those by Kar et al. [11], Shih et al. [12], Abutayeh et al. [13], Bhujbal et al. [14], Fernandes et al. [15], and Ghosh et al. [16]. This substantial increase in dataset size to 1350 compounds, with a training set of 1080 and a testing set of 270, bolsters the statistical power of the study and provides a more comprehensive understanding of the molecular descriptors’ impact on pIC50 values.
The R² values obtained in this study by the RFR model with 49 descriptors, both during training (0.989) and testing phases (0.941), surpass those reported in earlier studies (see Table 3). Such high R² values suggest that the model can capture the complex non-linear relationships between the molecular descriptors and the pIC50 values, which is essential for reliable prediction of FLT3 tyrosine kinase inhibition.
Moreover, the SD in test results significantly improved, achieving a value of 0.235, markedly lower than previous studies (see Table 3). This reduction in SD attests to the model’s accuracy in predicting pIC50 values across a diverse range of compounds, reducing variability and increasing prediction certainty. Furthermore, the Q2LOO value of 0.926 supports the model’s consistent performance across various data subsets, indicating that its predictive accuracy is not overly dependent on specific data points or features.
2.4. Model Interpretation
The model’s interpretability can be enhanced by conceptually analyzing the five most influential descriptors on its performance. The five descriptors, detailed in Table 4, are recognized for their paramount importance: SHBd, MLFER_S, nBase, MaxsssN, and MLFER_BH.
2.4.1. SHBd
The relationship between SHBd values and pIC50 scores, as depicted in Figure 2A, reveals the nuanced interplay crucial for the design of FLT3 tyrosine kinase inhibitors. A specific configuration, encompassing both the quantity and the electronic and topological quality of hydrogen bond donors, is crucial for effective FLT3 inhibition. Achieving optimal inhibitory activity requires SHBd values to be within the range of 1 to 1.5. Deviations from this range lead to diminished efficacy, indicating that both insufficient and excessive hydrogen bonding capabilities can adversely affect the performance of inhibitors. This observation emphasizes the importance of molecular descriptors in guiding the optimization of therapeutic compounds, where adjusting the hydrogen bond donor capacity to a targeted range can significantly enhance the potency and selectivity of inhibitors.
2.4.2. MLFER_S
An analysis of Figure 2B reveals the optimal MLFER_S range for FLT3 tyrosine kinase inhibitors to be between 3.1 and 4.5. Within this specific interval, compounds exhibit peak inhibitory efficacy, while values outside this range result in decreased inhibitor performance. This observation emphasizes the importance of solvophobic energy contributions in determining the effectiveness of these compounds, indicating a pivotal role of solvatophilic interactions in the design of optimal inhibitors.
2.4.3. nBase, MaxsssN and MLFER_BH
The observed trends in nBase, MaxssN, and MLFER_BH, as depicted in Figure 2C–E, collectively underscore the intricate relationship between molecular structure and FLT3 inhibitory activity. A peak un inhibitory activity is observed when molecules contain 2 basic groups (see Figure 2C). This would suggest a role of basic nitrogenous groups in enhancing interactions with FLT3, possibly through a combination of electrostatic attractions and hydrogen bonding. This is further supported by the findings related to MaxssN, where compounds exhibiting values greater than 1.5 show enhanced activity, with a more pronounced effect observed beyond 2.2. This indicates that not only the presence of nitrogen atoms, but their specific electronic configuration, characterized by three single bonds, is crucial for optimal FLT3 binding and inhibition. Meanwhile, MLFER_BH extends the discussion beyond nitrogenous groups to encompass the overall hydrogen bond acceptor capacity of the molecule. The observation that compounds with MLFER_BH values greater than 3.1 exhibit the best inhibitory activity underline the universal importance of hydrogen bond interactions in the activity of FLT3 inhibitors. Although nBase and MaxssN focus on the role of nitrogenous groups, the inclusion of MLFER_BH broadens the scope to include all potential hydrogen bond acceptors, suggesting that the ability to engage in hydrogen bonding, irrespective of the atom involved, is fundamental to the inhibitory mechanism.
2.5. Novel FLT3 Inhibitors Identified by Ligand-Based Screening
After applying Ligand-Based Virtual Screening (LBVS) using our customized cheminformatics model, we identified a series of promising compounds with potential inhibitory effects against FLT3 tyrosine kinase, with the top 5 presented in Table 5. This approach, which utilizes molecular fingerprints and the Tanimoto coefficient to assess structural similarities and predict pIC50 values, has enabled the selection of candidates that exhibit significant affinity for FLT3; approaching the potency of Gilteritinib, the next-generation inhibitor [35]. This methodology accentuates the utility of LBVS in the efficient identification of compounds with desired biological activity without the need for direct physical interactions with the biological target. Additionally, the methodology also highlights the accuracy of our model in predicting inhibitory activity based on the chemical structure of the compounds. The identification of these pyrazinecarboxamide derivatives with pIC50 values close to that of Gilteritinib (9.39) [36] emphasizes the potential of this computational approach in the discovery and development of new FLT3 inhibitors for the treatment of AML with FLT3 mutations. These findings expand our understanding of the structure-activity relationships of FLT3 inhibitors and provide a solid foundation for the future experimental validation of these compounds.
2.6. Script-Like Tool Description
To enhance the user experience with our model, we have created a script-based tool that automates the prediction of pIC50 and IC50 values for any compound using its SMILES code. The reliability of model is framed by the structural similarity of the target compound to the structures used in its training. Access the tool via the following link: https://github.com/Jacksonalcazar/Prediction-of-FLT3-Inhibitory-Activity. This tool is user-friendly, delivering results swiftly within seconds.
3. Materials and Methods
3.1. Data Curation
Data on FLT3 inhibitor compounds with published IC50 values were systematically extracted from the PubChem database [37,38] using the Requests library [39] and subsequently organized in a tabular format with the Pandas library [40] in Python 3. The dataset underwent rigorous cleaning where duplicate entries were removed. For compounds with multiple IC50 values from different assays, an average value was calculated to obtain a more representative measure. We focused on compounds with an IC50 value under 10 µM to prioritize higher potency for our analysis. Finally, the cleaned data were formatted to align with the requirements of our machine learning algorithms, ensuring both data integrity and compatibility for effective modeling.
3.2. Molecular Descriptor Calculation
Initially, 1511 molecular descriptors were computed using PaDEL-Descriptor 2.21 [41] and RDKIT [42]. The dataset was curated to exclude descriptors either incompatible with all compounds or constant across the dataset, reducing the number of descriptors to 1269. This curation was essential to ensure data quality and relevance for the machine learning model, focusing on meaningful descriptors for interpretability and accuracy.
3.3. Benchmarking Machine Learning Methods with External Validation
The dataset, which includes 1350 compounds and 1269 descriptors, was imported using Python 3 in conjunction with the Pandas library [40]. The experimental pIC50 values served as our target variable. To ensure a balanced representation of the dataset, we split it into training and testing sets in an 80:20 ratio using ‘train_test_split’ function of Scikit-learn library (sklearn), with the random_state parameter set to 11 for reproducibility.
The machine learning models deployed in this study, implemented using sklearn [43], included the Random Forest Regressor (RFR) [22], Gradient Boosting Regressor (GBR) [23], Support Vector Machine (SVM) [24], Kernel Ridge Regression (KRR) [24], Gaussian Process Regressor (GPR) [25], Bagging with Random Forest (BRF) [26]. Additionally, two Artificial Neural Network (ANN) architectures were implemented using Keras (ANN-K) [27] and PyTorch (ANN-P) [28]. Consistency in random state settings was maintained across the applicable models. Specific hyperparameters were carefully chosen to optimize model performance: the number of base estimators in BRF was set to 10; the alpha parameter in KRR and GPR was set to 100 and 1x10-10, respectively.
In the case of the ANNs, the ANN-K was designed as a Sequential model, consisting of three Dense layers: a first layer with 500 neurons to handle the large number of features; a smaller intermediate layer with 5 neurons for abstract data representations; and a final single-neuron output layer, which was used for pIC50 value regression. The ReLU activation function was used in the first two layers, with linear activation in the output layer, and weights initialized using the HeNormal initializer. This architecture was mirrored in the PyTorch implementation (ANN-P), which also featured an input layer with 500 neurons, an intermediate layer with 5 neurons, and a single-neuron output layer, utilizing ReLU activation and PyTorch’s default initializers. Both models underwent data normalization using sklearn’s StandardScaler and were trained for 100 epochs to balance learning and prevent overfitting. They were compiled using the Adam optimizer and the ‘mean_squared_error’ loss function.
Model performance was evaluated using coefficient of determination (R²), Mean Absolute Error (MAE), Standard Deviation (SD), and Root Mean Squared Error (RMSE) on both training and testing datasets. These metrics were derived using the sklearn.metrics module, which offers robust tools for model evaluation. The testing datasets were specifically used for external validation, providing a comprehensive view of each model’s predictive accuracy and error characteristics. Ensuring reproducibility was a fundamental aspect of this methodology. This was achieved by using random seeds (set to 11) consistently across numpy, TensorFlow-Keras/PyTorch, and sklearn models, thereby maintaining a reliable and consistent assessment of model performance.
3.4. Component Optimization through Feature Selection
3.4.1. Individual Descriptor Evaluation
To ascertain the influence of each molecular descriptor on predicting FLT3 inhibitor activity, we analyzed them within the established framework (80:20 training-to-test split, random_state = 11). Utilizing the machine learning model identified as most effective in our earlier benchmarking, we examined each descriptor. The evaluation centered on the coefficient of determination within the test set (R² test). This metric was crucial as it quantitatively reflected the descriptor’s relevance, directly linking its presence to the precision of the model’s predictions.
3.4.2. Analysis and Feature Selection Process
The next step entailed analyzing the top 100 descriptors using the R² test metric to understand their correlation with FLT3 inhibitory activity. This analysis led to a selective inclusion of descriptors, starting with the most correlated and progressively adding less correlated ones. This process aimed to find an optimal balance between model complexity and predictive accuracy.
3.5. Internal Validation
After the benchmarking phase and component optimization, the optimal model underwent internal validation through a Leave-One-Out Cross-Validation (LOOCV) technique. In this method, implemented using the LeaveOneOut class from Python’s ‘sklearn.model_selection’ module, the model is trained on all data points except for one, which is reserved for testing. This process is systematically repeated for each data point in the dataset. During this comprehensive validation process, the model’s prediction accuracy was quantified using the R² metric (Q²LOO). This key parameter enables for the comparison of our selected method’s performance with that of previous studies, ensuring its robustness beyond a fixed dataset.
3.6. Ligand-Based Virtual Screening
In the quest to predict pIC50 values for FLT3 tyrosine kinase inhibitor compounds, we procured from PubChem dataset of over 10.2 million molecules, analyzed using 881-bit molecular fingerprints (PubChem finderprints) [44]. We employed the Tanimoto coefficient [45] to identify structural similarities, setting a 90% similarity threshold against the top 100 most active compounds. This rigorous filtering led to the exclusion of known FLT3 inhibitors, focusing on novel compounds with unknown activity.
These selected compounds were then processed through the model chosen in our earlier benchmarking, specifically tailored for predicting FLT3 inhibitory activity. By predicting the pIC50 values, the model facilitated the prioritization of the five most promising compounds, streamlining the path towards experimental validation and accelerating the discovery of potent FLT3 inhibitors.
4. Conclusions
This study has successfully demonstrated the applicability and efficacy of a QSAR-ML hybrid model in predicting the pIC50 values of FLT3 tyrosine kinase inhibitors, marking a significant advancement in the field of drug discovery for AML. The innovative integration of machine learning techniques with quantitative structure-activity relationship models has not only overcome the limitations of conventional QSAR models but has also introduced a novel approach that enhances predictive accuracy and generalizability across a broad spectrum of FLT3 inhibitors.
The comprehensive dataset, extensive molecular descriptor analysis, and meticulous benchmarking of various machine learning algorithms have culminated in a model that showcases superior predictive capabilities. Notably, the Random Forest Regressor emerged as the most effective model, validated through rigorous external and internal validation methods. This model is a reliable and highly accurate tool for the identification of potent FTL3 inhibitor, as evidenced by its exceptional R² of 0.941 in predicting the pIC50 values of 270 FLT3 tyrosine kinase inhibitor compounds and a SD of 0.235.
Moreover, our component optimization and feature selection process have highlighted the critical importance of specific molecular descriptors in FLT3 inhibitor efficacy, providing valuable insights into the structural features that govern inhibitor activity. This understanding facilitates the rational design of new FLT3 inhibitors, consequently streamlining the drug discovery process by focusing on compounds that exhibit these key structural characteristics.
The identification of novel FLT3 inhibitors through ligand-based virtual screening further illustrates the practical application of our QSAR-ML model in accelerating the discovery of effective treatments for AML. These promising compounds, predicted to possess high inhibitory potency, pave the way for experimental validation and potential clinical development.
Furthermore, the development of a user-friendly script-like tool for the prediction of pIC50 values represents a significant contribution to the cheminformatics toolbox, offering researchers a practical and efficient means of evaluating the FLT3 inhibitory potential of new compounds.
In summary, our study provides a simple model for predicting the pIC50 values of FLT3 tyrosine kinase inhibitors and sets a new benchmark in the integration of machine learning and QSAR methodologies for drug discovery. By addressing the challenges associated with the predictive accuracy of conventional models and introducing a more accessible tool for compound evaluation, this work significantly advances the pursuit of personalized and effective therapies for AML, offering hope for improved patient outcomes in the face of this challenging disease.
Author Contributions
Conceptualization, J.J.A.; methodology, J.J.A.; software, J.J.A; validation, J.J.A. and P.R.C., formal analysis, J.J.A.; investigation, J.J.A., I.S., C.M., B.M., G.S., D.M., F.D.; resources, P.R.C.; data curation, J.J.A., I.S., C.M., B.M., G.S., D.M., F.D.; writing—original draft preparation, J.J.A., I.S., C.M., B.M., G.S., D.M., F.D.; writing—review and editing, J.J.A.; visualization, J.J.A.; supervision, J.J.A.; project administration, J.J.A.; funding acquisition, P.R.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by FONDEQUIP EQM150093, which provided essential computational resources.
Data Availability Statement
We have made the prediction model publicly available. You can access the script at the following GitHub repository: https://github.com/Jacksonalcazar/Prediction-of-FLT3-Inhibitory-Activity.
Acknowledgments
J.J.A. and P.R.C. thank the Vicerectoria de Investigación y Doctorado (VRID) and Instituto de Ciencias e Innovación en Medicina (ICIM) at the Universidad del Desarrollo for their invaluable support. Special appreciation is extended to Alessandra Misad for proofreading the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Birg, F.; Courcoul, M.; Rosnet, O.; Bardin, F.; Pébusque, M.J.; Marchetto, S.; Tabilio, A.; Mannoni, P.; Birnbaum, D. Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood 1992, 80, 2584–2593. [Google Scholar] [CrossRef]
- Small, D. FLT3 mutations: biology and treatment. Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program 2006, 2006, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Barley, K.; Navada, S.C. Acute myeloid leukemia. Oncology 2019, 373, 308–318. [Google Scholar] [CrossRef]
- Kazi, J.U.; Rönnstrand, L. FMS-like tyrosine kinase 3/FLT3: From basic science to clinical implications. Physiological Reviews 2019, 99, 1433–1466. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Short, N.J.; Fathi, A.T.; Marcucci, G.; Ravandi, F.; Tallman, M.; Wang, E.S.; Wei, A.H. Acute Myeloid Leukemia: Historical Perspective and Progress in Research and Therapy Over 5 Decades. Clinical Lymphoma, Myeloma and Leukemia 2021, 21, 580–597. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Tiong, I.S. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood 2017, 130, 2469–2474. [Google Scholar] [CrossRef]
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New directions for emerging therapies in acute myeloid leukemia: the next chapter. Blood Cancer Journal 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: current progress and future directions. Blood Cancer Journal 2021, 11, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, S.; Schlenk, R.F. Update on current treatments for adult acute myeloid leukemia: To treat acute myeloid leukemia intensively or non-intensively? That is the question. Haematologica 2023, 108, 342–352. [Google Scholar] [CrossRef]
- Kumar Kar, R.; Suryadevara, P.; Roushan, R.; Chandra Sahoo, G.; Ranjan Dikhit, M.; Das, P. Quantifying the Structural Requirements for Designing Newer FLT3 Inhibitors. Medicinal Chemistry 2012, 8, 913–927. [Google Scholar] [CrossRef]
- Shih, K.C.; Lin, C.Y.; Chi, H.C.; Hwang, C.S.; Chen, T.S.; Tang, C.Y.; Hsiao, N.W. Design of novel FLT-3 inhibitors based on dual-layer 3D-QSAR model and fragment-based compounds in silico. Journal of Chemical Information and Modeling 2012, 52, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Abutayeh, R.F.; Taha, M.O. Discovery of novel FLT3 inhibitory chemotypes through extensive ligand-based and new structure- based pharmacophore modelling methods. Journal of Molecular Graphics and Modelling 2019, 88, 128–151. [Google Scholar] [CrossRef] [PubMed]
- Bhujbal, S.P.; Keretsu, S.; Cho, S.J. Design of New Therapeutic Agents Targeting FLT3 Receptor Tyrosine Kinase Using Molecular Docking and 3D-QSAR Approach. Letters in Drug Design& Discovery 2019, 17, 585–596. [Google Scholar] [CrossRef]
- Fernandes, Í.A.; Resende, D.B.; Ramalho, T.C.; Kuca, K.; Da Cunha, E.F.F. Theoretical studies aimed at finding FLT3 inhibitors and a promising compound and molecular pattern with dual aurora B/FLT3 activity. Molecules 2020, 25, 1726. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Keretsu, S.; Cho, S.J. Molecular modeling studies of n-phenylpyrimidine-4-amine derivatives for inhibiting FMS-like tyrosine kinase-3. International Journal of Molecular Sciences 2021, 22, 12511. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, C.; Torrens, F.; Godoy, K.; Reyes, C.; Farías, J. Application of Quantitative Structure-Activity Relationships in the Prediction of New Compounds with Anti-Leukemic Activity. International Journal of Molecular Sciences 2023, 24, 12258. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Osman, O.I.; Hassan, W.M. Identifying novel therapeutic inhibitors to target FMS-like tyrosine kinase-3 (FLT3) against acute myeloid leukemia: a molecular docking, molecular dynamics, and DFT study. Journal of Biomolecular Structure and Dynamics 2023. [CrossRef] [PubMed]
- Nasimian, A.; Al Ashiri, L.; Ahmed, M.; Duan, H.; Zhang, X.; Rönnstrand, L.; Kazi, J.U. A Receptor Tyrosine Kinase Inhibitor Sensitivity Prediction Model Identifies AXL Dependency in Leukemia. International Journal of Molecular Sciences 2023, 24, 3830. [Google Scholar] [CrossRef]
- Janssen, A.P.; Grimm, S.H.; Wijdeven, R.H.; Lenselink, E.B.; Neefjes, J.; Van Boeckel, C.A.; Van Westen, G.J.; Van Der Stelt, M. Drug Discovery Maps, a Machine Learning Model That Visualizes and Predicts Kinome-Inhibitor Interaction Landscapes. Journal of Chemical Information and Modeling 2019, 59, 1221–1229. [Google Scholar] [CrossRef]
- Zhao, Y.; Tian, Y.; Pang, X.; Li, G.; Shi, S.; Yan, A. Classification of FLT3 inhibitors and SAR analysis by machine learning methods. Molecular Diversity 2023, 1, 1–17. [Google Scholar] [CrossRef]
- Eckardt, J.N.; Bornhäuser, M.; Wendt, K.; Middeke, J.M. Application of machine learning in the management of acute myeloid leukemia: Current practice and future prospects. Blood Advances 2020, 4, 6077–6085. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Machine Learning 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Annals of Statistics 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Hearst, M.A.; Dumais, S.T.; Osuna, E.; Platt, J.; Scholkopf, B. Support vector machines. IEEE Intelligent Systems and their applications 1998, 13, 18–28. [Google Scholar] [CrossRef]
- Williams, C.; Rasmussen, C. Gaussian processes for regression. Advances in neural information processing systems 1995, 8. [Google Scholar]
- Altman, N.; Krzywinski, M. Ensemble methods: bagging and random forests. Nature pubchemds 2017, 14, 933–935. [Google Scholar] [CrossRef]
- Chollet, F. Keras, 2015. In: Github Repos. https://github.com/fchollet/keras.
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. Pytorch: An imperative style, high-performance deep learning library. Advances in neural information processing systems 2019, 32. [Google Scholar]
- Marino, S.; Zhao, Y.; Zhou, N.; Zhou, Y.; Toga, A.W.; Zhao, L.; Jian, Y.; Yang, Y.; Chen, Y.; Wu, Q.; et al. Compressive Big Data Analytics: An ensemble meta-algorithm for high-dimensional multisource datasets. Plos one 2020, 15, e0228520. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.H.; Kier, L.B. Electrotopological State Indices for Atom Types: A Novel Combination of Electronic, Topological, and Valence State Information. Journal of Chemical Information and Computer Sciences 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Euldji, I.; Si-Moussa, C.; Hamadache, M.; Benkortbi, O. QSPR Modelling of the Solubility of Drug and Drug-like Compounds in Supercritical Carbon Dioxide. Molecular Informatics 2022, 41, 2200026. [Google Scholar] [CrossRef]
- Platts, J.A.; Butina, D.; Abraham, M.H.; Hersey, A. Estimation of molecular linear free energy relation descriptors using a group contribution approach. Journal of Chemical Information and Computer Sciences 1999, 39, 835–845. [Google Scholar] [CrossRef]
- Lin, C.; Xiaoxiao, Z. Optimizing Drug Screening with Machine Learning. 2022 19th International Computer Conference on Wavelet Active Media Technology and Information Processing, ICCWAMTIP 2022 2022. [CrossRef]
- Ibrahim, Z.Y.; Uzairu, A.; Shallangwa, G.; Abechi, S. QSAR and molecular docking based design of some indolyl-3-ethanone-α- thioethers derivatives as Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitors. SN Applied Sciences 2020, 2, 1–12. [Google Scholar] [CrossRef]
- Lee, L.Y.; Hernandez, D.; Rajkhowa, T.; Smith, S.C.; Raman, J.R.; Nguyen, B.; Small, D.; Levis, M. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood 2017, 129, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.; Kurosawa, K.; Matsuya, T.; Iikubo, K.; Kondoh, Y.; Kamikawa, A.; Tomiyama, H.; Iwai, Y. Patent US8969336, 2015. Available at: https://patents.google.com/patent/US8969336B2.
- PubChem Substructure Fingerprint, 2023. [Accessed December 10, 2023].
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic acids research 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Kenneth Reitz. Requests: HTTP for Humans™— Requests 2.26.0 documentation, 2021. Available at: https://docs.python-requests.org/en/latest/.
- McKinney, W.; Team, P.D. Pandas - Powerful Python Data Analysis Toolkit. https://pandas.pydata.org, 2015.
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. Journal of Computational Chemistry 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. RDKit: Open-source cheminformatics 2022_9_5 (Q3 2022). http://www.rdkit.org, 2023. [CrossRef]
- Fabian, Pedregosa.; Gaël, Varoquaux.; Alexandre, Gramfort.; Vincent, Michel.; Bertrand, Thirion.; Olivier, Grisel.; Mathieu, Blondel.; Peter, Prettenhofer.; Ron, Weiss.; Vincent, Dubourg.; et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 2011, 12, 2825–2830. [Google Scholar]
- PubChem, 2023. [Accessed November 28, 2023].
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? Journal of Cheminformatics 2015, 7, 1–13. [Google Scholar] [CrossRef]
Figure 1.
Variation in R² test values as a function of the number of descriptors, ranked from most to least significant.
Figure 1.
Variation in R² test values as a function of the number of descriptors, ranked from most to least significant.
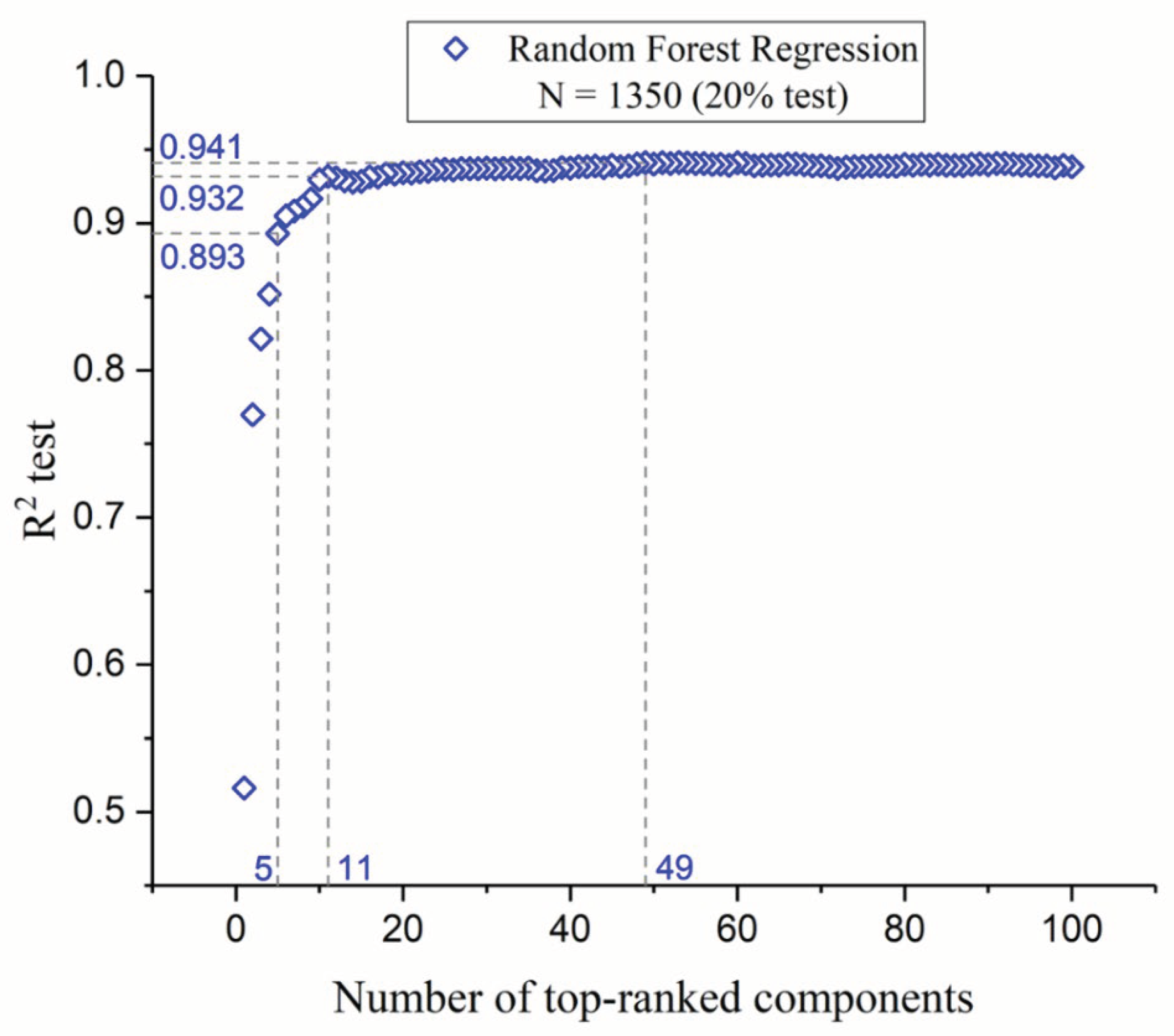
Figure 2.
Scatter plots to show the relation between the five key molecular descriptors and the FLT3 inhibitor potency of molecules in both the training set and test set. Range with higher values of potency highlighted in green in each plot.
Figure 2.
Scatter plots to show the relation between the five key molecular descriptors and the FLT3 inhibitor potency of molecules in both the training set and test set. Range with higher values of potency highlighted in green in each plot.
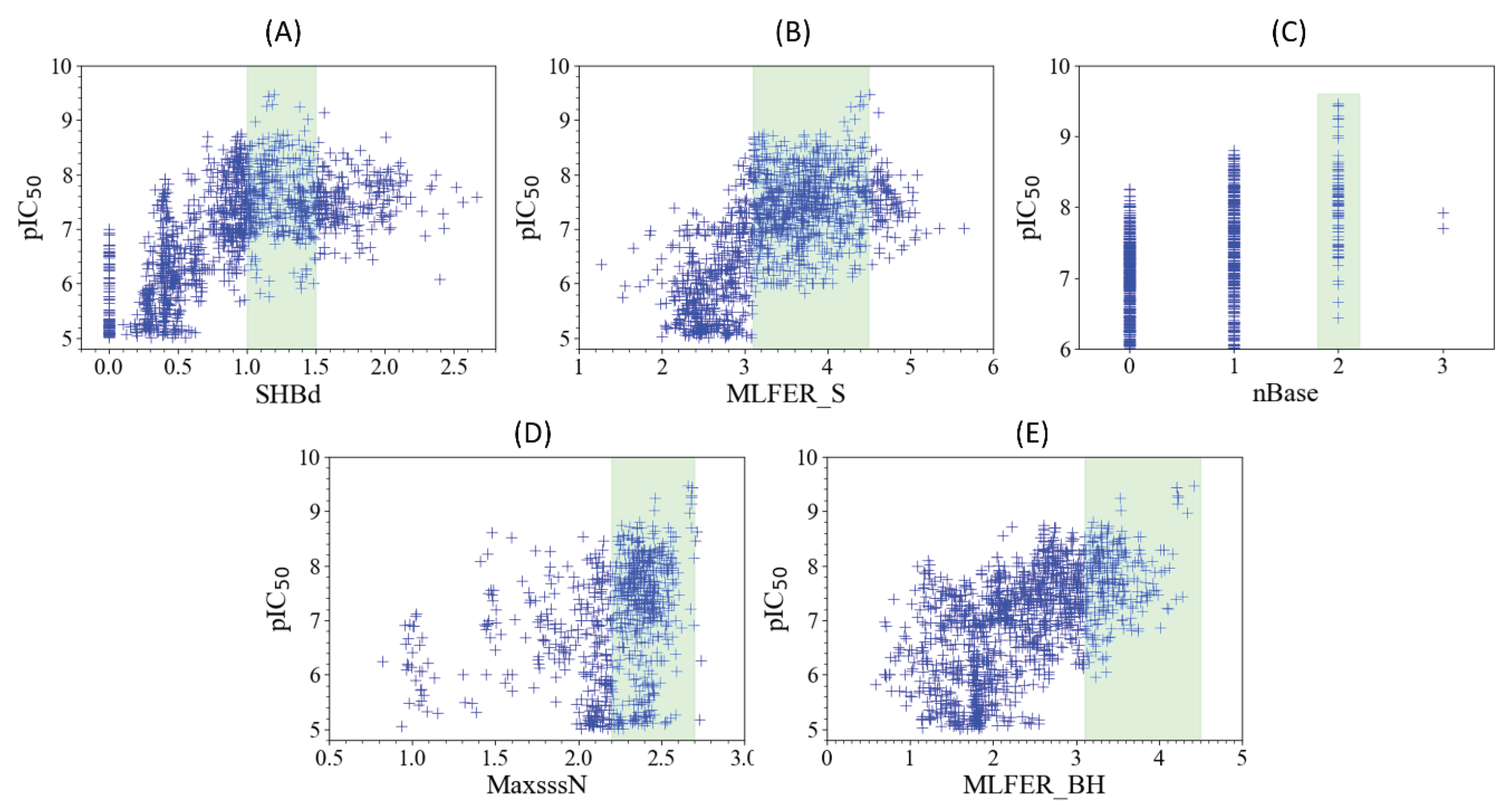

Table 1.
Performance comparison of machine learning models for predicting pIC50 values of FLT3 tyrosine kinase inhibitor compounds.
Table 1.
Performance comparison of machine learning models for predicting pIC50 values of FLT3 tyrosine kinase inhibitor compounds.
| Metrics/ML | RFR | GBR | SVM | KRR | GPR | BRF | ANN-K | ANN-P |
|---|---|---|---|---|---|---|---|---|
| R² training | 0.988 | 0.973 | 0.014 | 0.546 | 0.641 | 0.967 | 0.988 | 0.983 |
| MAE training | 0.082 | 0.126 | 0.756 | 0.489 | 0.469 | 0.136 | 0.070 | 0.082 |
| SD training | 0.102 | 0.154 | 0.933 | 0.638 | 0.526 | 0.172 | 0.101 | 0.121 |
| RMSE training | 0.102 | 0.154 | 0.941 | 0.638 | 0.568 | 0.172 | 0.103 | 0.123 |
| R² test | 0.936 | 0.939 | -0.012 | 0.592 | -0.228 | 0.931 | 0.907 | 0.895 |
| MAE test | 0.197 | 0.195 | 0.786 | 0.484 | 0.876 | 0.207 | 0.235 | 0.248 |
| SD test | 0.246 | 0.237 | 0.975 | 0.619 | 0.932 | 0.255 | 0.296 | 0.313 |
| RMSE test | 0.246 | 0.239 | 0.977 | 0.620 | 1.076 | 0.256 | 0.297 | 0.315 |
Table 2.
Performance of Random Forest models for predicting pIC50 values of FLT3 tyrosine kinase inhibitor compounds based on 49 components.
Table 2.
Performance of Random Forest models for predicting pIC50 values of FLT3 tyrosine kinase inhibitor compounds based on 49 components.
| Training set | Test set | |
|---|---|---|
| Size | 1080 | 270 |
| R² | 0.989 | 0.941 |
| MAE | 0.081 | 0.190 |
| SD | 0.101 | 0.235 |
| RMSE | 0.101 | 0.236 |
| Q2LOO | 0.926 | |
Table 3.
Comparative performance of QSAR models for FLT3 inhibitors.
| Kara (2012) |
Shiha (2012) |
Abutayeha (2019) |
Bhujbala (2020) |
Fernandesa (2020) |
Ghosha (2021) |
This work |
|
|---|---|---|---|---|---|---|---|
| Dataset size | 67 | 72 | 93 | 63 | 40 | 40 | 1350 |
| Train set size | 51 | 25 | 76 | 45 | 28 | 30 | 1080 |
| Test set size | 16 | 47 | 17 | 18 | 12 | 10 | 270 |
| R² training | 0.956 | 0.98 | 0.86 | 0.956 | 0.80 | 0.983 | 0.989 |
| R² test | 0.891 | 0.76 | 0.57 | 0.707 | 0.80 | 0.698 | 0.941 |
| SD test | 0.435 | 0.66 | – | > 0.895 | 0.31 | 0.452 | 0.235 |
| Q2LOO | 0.747 | 0.58 | 0.65 | 0.57 | 0.60 | 0.802 | 0.926 |
Table 4.
Name and characterization of the five most important descriptors for the model development, ordered by priority.
Table 4.
Name and characterization of the five most important descriptors for the model development, ordered by priority.
| Priority | Descriptor | Name | Description |
|---|---|---|---|
| 1° | SHBd [30,31] |
Sum of E-States for (strong) hydrogen bond donors | The value is calculated as the sum of each atom capable of donating a hydrogen atom, weighted by its electronic environment and topological position (E-State). |
| 2° | MLFER_S [31,32] |
Molecular Linear-Free Energy Relation_S | Cumulative sum of the free energy contributions of solvatophilic groups in a molecule, calculated using previously established empirical values on their interactions with solvents. |
| 3° | nBase | Number of basic groups | Number of basic groups in the molecule, especially nitrogenous groups. |
| 4° | MaxsssN [30,33] |
Maximum atom-type E-State: >N- | Maximum electrotopological state present in nitrogen atoms with three single bonds. |
| 5° | MLFER_BH [32,34] |
Overall or summation solute hydrogen bond basicity | Total hydrogen bond basicity in a molecule calculated by summing the contributions of all possible hydrogen bond acceptor sites in the molecule. |
Table 5.
Top five candidates for FLT3 inhibitors identified by ligand-based virtual screening.
| IUPAC name | Structure | pIC50 |
|---|---|---|
| 6-Ethyl-3-[3-methoxy-4-[4-(1-methylpiperidin-4-yl)piperazin-1-yl]anilino]-5-(oxan-4-ylamino)pyrazine-2-carboxamide |  |
9.34 |
| 6-Ethyl-3-[3-methoxy-4-[4-(4-propan-2-ylpiperazin-1-yl) piperidin-1-yl]anilino]-5-(oxan-4-ylamino)pyrazine-2-carboxamide |  |
9.34 |
| 3-[4-[4-(1-Methylpiperidin-4-yl)piperazin-1-yl]anilino]-5-(oxan-4-ylamino)-6-propan-2-ylpyrazine-2-carboxamide | 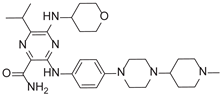 |
9.29 |
| 6-(1-Methyl-3,6-dihydro-2H-pyridin-4-yl)-3-[4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]anilino]-5-(oxan-4-ylamino)pyrazine-2-carboxamide | 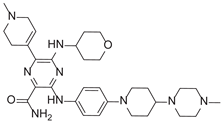 |
9.27 |
| 6-Ethyl-3-[4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]-3-propan-2-yloxyanilino]-5-(oxan-4-ylamino)pyrazine-2-carboxamide | 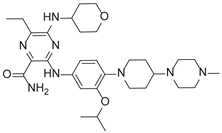 |
9.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



