
Submitted:
25 March 2024
Posted:
25 March 2024
You are already at the latest version
Abstract
Endothelial cell (EC) injury is a crucial contributor to progression of diabetic kidney disease (DKD), but the specific EC populations and mechanisms involved remain elusive. Kidney ECs (n=5,464) were collected at 3 timepoints from diabetic BTBRob/ob mice and non-diabetic littermates. Their heterogeneity, transcriptional changes, and alternative splicing during DKD progression were mapped using SmartSeq2 single-cell RNA sequencing (scRNAseq) and elucidated by pathway, network, and gene ontology enrichment analyses. We identified 13 distinct transcriptional EC phenotypes corresponding to different kidney vessel subtypes, confirmed by in-situ hybridization and immunofluorescence. EC subtypes along nephrons displayed extensive zonation related to their functions. Differential gene expression analyses in peritubular and glomerular EC in DKD underlined regulation of DKD-relevant pathways including EIF2 signaling, oxidative phosphorylation, and IGF1 signaling. Importantly, it revealed differential alteration of these pathways between the two EC subtypes and changes during disease progression. Furthermore, glomerular and peritubular EC also displayed aberrant and dynamic alteration of al-ternative splicing (AS), strongly associated with DNA repair. Strikingly, genes displaying differential transcription or alternative splicing participate in divergent biological processes. Our study reveals spatiotemporal regulation of gene transcription and AS linked to DKD progression, providing insight into pathomechanisms and clues to novel therapeutic targets for DKD treatment.
Keywords:
diabetic nephropathy
; transcriptomics
; endothelium
1. Introduction
DKD is an increasing healthcare challenge worldwide imposing a large unmet medical need. About 40% of DKD is likely progressed from the heterogenous diabetic patient population [1]. Lack of longitudinal data from patients and animal models limits the understanding of the disease driving mechanisms and hinders development of novel therapies. Specifically, EC dysfunction and capillary rarefaction are commonly observed in DKD, correlating with impaired kidney function, and predicting future development of end-stage renal disease [2].
Little is known about changes in EC gene expression accompanying DKD progression, and how such changes over time correlate with specific EC subtypes and spatial location. The molecular heterogeneity of EC has been investigated recently by scRNAseq [3,4,5,6]. These studies characterized murine renal EC at developmental and physiological stages [3] or during adaptation to dehydration [4], whereas the underlying mechanisms driving endothelial damage and DKD progression remain unresolved. Limited information is available on EC from the whole kidney or glomerular single-cell transcriptome in mouse DKD [7,8,9,10]. Notably, none of these studies distinguished transcriptional and post-transcriptional processes. In humans, approximately 95% of multiexon genes undergo AS [11], which adds another layer of gene expression regulation. Importantly, a recent scRNAseq study [12] demonstrated that splice isoform switching is critical in mesenchymal-epithelial transition in kidney development. This underscored the feasibility and importance of exploring differential AS in specific cell types during DKD progression.
In the current study, we utilized diabetic BTBRob/ob mice, a model of obesity-induced type-2 diabetes that develops early pathological features of human DKD [13,14]. We applied SmartSeq2 full-length scRNAseq of EC at 3 timepoints to investigate the specific transcriptional response and aberrant AS in different vascular compartments in DKD progression.
2. Results
2.1. Molecular Profiling and EC Population
To characterize molecular heterogeneity of renal ECs and changes during DKD progression, we analyzed kidney EC from 6/11/20-week-old diabetic BTBRob/ob and non-diabetic BTBRLean mice (Figure 1A). The resulting dataset contained transcriptomes of 5,464 cells covering different genotypes, EC subtypes, and timepoints (Suppl. Table S1).
Uniform manifold approximation and projection (UMAP) of ECs from BTBRLean mice identified 13 EC clusters (Figure 1B), expressing EC markers (Figure 1C) [15,16] but no other renal cell markers (see Methods). For EC subtype annotation (Figure 1D), we compared cluster-specific gene patterns with published renal EC scRNAseq data and protein and mRNA localization in situ [3,4,5]. This allocated the 13 clusters to arteries/afferent arterioles (AA), glomerular (GEC), efferent arteriole (EA), ascending vasa recta (AVR), descending vasa recta (DVR), peritubular capillaries (PCEC), venous (VEC), proliferating (cycling), tip cells, and lymphatic (LVEC). One unknown cluster remained.
2.2. Results from Renal EC Heterogeneity, Annotations, and Validation
As most EC populations did not manifest unique singular markers, we performed RNA-ISH and immunofluorescence (IF) against a combination of markers to determine EC identity. AA was characterized by high expression of Fbln5, Gja5, Cxcl12, Cldn5 and low expression of Plvap [3,4]. Both RNA-ISH for Cldn5 and Cldn5-GFP reporter expression showed strong signals in AA (Suppl. Figure 1A, B). RNA-ISH also indicated Cldn5 expression in podocytes, which confirms previous findings [5,17]. However, Cldn5-GFP did not display detectable GFP signal in podocytes (Suppl. Figure 1A), suggesting that relevant cis-acting promoter sequences for podocyte expression is missing in this transgenic construct. We identified Ace as a novel marker for AA, as confirmed by RNA-ISH (Suppl. Figure 1B). GEC showed weak Ace expression, as previously reported [18].
EA differed substantially from AA, and while they express Fbln5, they have very low Cldn5 and no Ace expression. Calca appears to be specific to EA (Suppl. Figure 1C), as previously reported [4]. In addition, Tspan8 and Npnt were highly expressed in EA/DVR, however, some datasets report non-EC kidney expression of these markers [19].
DVR tightly control perfusion to the outer and inner medulla and function both as exchange (capillary) and resistance (arteriolar) vessels [20]. DVR shared certain markers with AA, including Gja5 and Cldn5, but also expressed the specific marker Slc14a1 (Suppl. Figure 2A). Among ECs, DVR showed high and specific expression of Aqp1, which was otherwise, expressed primarily in epithelial cells (Suppl. Figure 2B).
GEC/PCEC/AVR endothelium all have fenestrations [21,22], but only fenestrations in PCEC/AVR have PLVAP+ diaphragms (Suppl. Figure 2C) [23], while healthy GEC lack diaphragms [24]. Immunohistochemistry staining confirmed PLVAP expression in PCEC/AVR, while Cldn5 was absent (Suppl. Figure 1A, 2C-E). We identified Il33 as a novel transcript in PCEC (Suppl. Figure 2D), with expression also in VEC/AVR, but not in GEC. Car8 was expressed in PCEC/AVR/VEC, and colocalized with Plvap in AVR, whereas DVR (Cldn5+) were Plvap negative (Suppl. Figure 2E). Car8 expression in VEC has been reported previously [4]. Igf1 was expressed in AVR/PCEC and colocalized with Plvap. DVR (Pecam1+, Plvap-) were negative for Igf1 (Suppl. Figure 2F). GEC were annotated by previously reported markers, including high expression of Gata5, Ehd3, Tbx3, Plat, Lpl, Kcnj5, Adamtsl5, and low expression of Cldn5 and Plvap [3,4,5]. RNA-ISH for Gata5 showed strong and specific staining in GEC (Suppl. Figure 2G). All data is presented in a searchable database with data output in bar plots (see Cldn5 and Plvap in Suppl. Figure 3).
Endothelial cell types in human kidney were highly conserved in the mouse kidney [6] and the gene expression profiles of GECs and PCECs in our dataset correlated well with recently described human GECs and PCECs [25] (Suppl. Figure 4A), suggesting cross-species translatability of our mouse data. Furthermore, 10 of 53 genes associated with human DKD by GWAS [26] could be mapped to EC expression in our dataset (Suppl. Figure 4C). Of these, Igfbp5 and Tspan9 showed abundant EC expression: Igfbp5 was enriched in GEC/PCEC/AVR, while Tspan9 expressed in all clusters (Suppl. Figure 4C). Comparison with the human Nephrocell whole-kidney scRNAseq database [27] confirmed enrichment of IGFBP5 in GEC/PCEC, in contrast to ubiquitous expression of TSPAN9 (Suppl. Figure 4D).
We noticed two clusters each for GEC and PCEC. One of each had relatively higher levels of immediate early gene transcripts (IEG) (GECIEG-high and PCECIEG-high, respectively). IEGs are induced within minutes in response to cell-extrinsic and cell-intrinsic signals and do not require de novo protein synthesis for their enhanced expression [28,29]. RNA in situ hybridization (RNA-ISH) for IEGs including Fos, JunB, and Atf3, did not show expression patterns specific to any anatomical location, such as cortical versus juxtamedullary glomeruli (Suppl. Figure 5). High IEG expression has been reported in a proportion of vascular cells in scRNAseq data and suggested to result from stress and hemodynamic changes during tissue dissociation and cell isolation [30,31].
2.3. Functional Zonation of the Healthy Renal Vasculature
In BTBRLean mice we identified differentially expressed genes (DEGs) in each EC cluster relative to the remaining clusters (Suppl. File S1). Ingenuity Pathway Analysis (IPA) of DEGs displayed complex zonation of pathway activation, related to immune responses, vascular tone, coagulation, and growth factor signaling (Figure 2A, Suppl. File S2).
EC communicates with immune cells through chemokines, cytokines, and adhesion molecules. Chemokines showed enriched expressions in AA/GEC, including Cxcl1, Cxcl12/Sdf1, Cxcl16, and Cx3cl1 (Figure 2B). Interleukins Il16 and Il33 displayed higher expression in DVR and VEC, respectively. Adhesion molecules Selp and Sele were expressed in a small portion of AA and AVR, respectively, while Icam1 and Vcam1 were enriched in VEC. Furthermore, MHC class-II molecules and Cd74 were enriched in PCEC, while Lbp in VEC. The anatomically defined patterns of immune genes within the EC compartments may contribute to the nephron’s immune zonation [32].
EC Weibel-Palade bodies regulate coagulation, whose components Vwf, Selp, Edn1 and CD63 were mainly expressed in AA/DVR (Figure 2C). GEC and AA expressed Plat and its inhibitor Serpin1, respectively. Coagulation Factor F8 was enriched in GEC. EC-released prostaglandins inhibit platelet aggregation: the prostaglandin synthase Ptgs1 and transporter Slco2a1 were expressed in VEC and AA, respectively. These findings suggest differential initiation of coagulation and platelet activation along nephrons.
Regulators for vasodilation and constriction were enriched in AA, suggesting EC zonation of vascular tone regulation (Figure 2D). Ca2+-calmodulin dependent Nos3 and Ca2+-activated Kcnn3 and Kcnn4 trigger hyperpolarization [33], spreading via Gja4/Gja5 to elicit vasodilation in VSMCs [34,35], Gja4/5 affected renin secretion in granular cells [36] and together with Ace, may coordinate conversion of angiotensinogen. EC Ca2+ may trigger release of Edn1 [37] in AA/EA/DVR. Furthermore, regulators for intracellular Ca2+ homeostasis were enriched in AA/EA, including Atp2a3, Trpv4, F2rl1, and Gper1 [38] and for extracellular Ca2+ homeostasis in AA/GEC/EA including Pthlh, Calca, Sost [39], and Mgp. Our study suggests that glomerulus-linked (AA/GEC/EA) and DVR zonation fine-tunes renal vascular tone.
The IGF system displays a comprehensive expression profile along nephron segments [40]. The renal EC predominantly expressed three isoforms of Igfbp: Igfbp3 (DVR), Igfbp4 (AA), and Igfbp5 (GEC/PCEC) (Figure 2E). Igf1 and Igf2 were mainly expressed by VEC and AA, respectively, and their receptors Igf1r and Igf2r in a small proportion of ECs. The zone-distinct expression likely fine-tunes segment-specific regulation of IGF signaling.
2.4. Transcriptional Responses in Progression of DKD
The diabetic phenotype for BTBRob/ob mice is shown in Suppl. Figure S6. BTBRob/ob mice showed elevated blood glucose and proteinuria with increased glomerular size already evident at 6-weeks, confirming a DKD phenotype (Suppl. Figure 6A-D). Capillary density progressively declined over time in BTBRob/ob mice (Suppl. Figure 6E-H). In agreement, FACS of PECAM1+ cells showed reduced numbers of EC in 11- and 20-week-old diabetic mice (Suppl. Figure 6I).
UMAP showed that majority of clusters for BTBRLean and BTBRob/ob mice overlapped (Figure 3A). For comparison of DEGs between non-diabetic and diabetic mice, PCEC/PCECIEG-high were pooled to PCEC, and GEC/GECIEG-high to GEC (Figure 3B, Suppl. File S3). GEC and PCEC comprised the majority of captured cells and displayed the largest number of DEGs (Suppl. Table S2), hence became the focus for further analysis. Strikingly, many overlapping DEGs changed direction of regulation at 20-week compared to 6/11-week (Figure 3). Among overlapping genes, Ucp2 and Serinc3 were regulated similarly between PCEC and GEC. Mitochondrial Ucp2 reduces oxidative stress and plasma membrane Serinc3 inhibits apoptosis [41].
2.5. Pathway and Network Analyses of EC Responces in DKD
We next performed IPA on PCEC and GEC DEGs to identify significantly enriched and regulated canonical pathways. In PCEC, multiple pathways were inactivated in 11-week BTBRob/ob mice contrary to 6-week (Figure 4A, Suppl. File S2), in conjunction with capillary rarefaction (Suppl. Figure S6), and most pathways remained inactivated at 20-week. In contrast, GEC showed transient activation of multiple pathways at 6-week (Figure 5A, Suppl. File S2), in conjunction with increased glomerular area (Suppl. Figure S6).
Oxidative phosphorylation (OXPHOS) and EIF2 signaling were among the top regulated pathways in GEC and PCEC. Inactivation of OXPHOS in PCEC started at 11-week in diabetic mice and was evident in both PCEC and GEC at 20-week (Figure 5A). Identified DEGs (Suppl. File S4) at 20-week are represented in Suppl. Figure S7. EIF2 signaling is central to eukaryotic translation initiation, and its dysregulation was manifested with myriad DEGs (Suppl. File S5) and regulated functional complexes (Suppl. Figure S8-S9). PCEC and GEC displayed opposite regulation of EIF2 signaling and upstream growth factor signaling at 6-week. In contrast, EIF2 signaling was inactivated in both PCEC and GEC at 20-week, concurrent with increased downstream apoptosis (Figure 4A, Figure 5A).
The IPA summary illustrates important pathways, effects and regulators in PCEC (Figure 4) and GEC (Figure 5), with IGF signaling as a central factor. Intriguingly, while IGF1 pathway was activated in GECs at both 6/20-week, it was inactivated in PCEC at 11-week. Analysis showed that Igf1 and several Igfbp isoforms were altered in GEC and PCEC from diabetic mice (Suppl. Figure S10A-B). Igfbp5 was significantly regulated in GEC (Suppl. Figure S10A). Igfbp5 RNA in situ hybridization showed increased expression in GEC and mesangial cells (Suppl. Figure S10C-D).
2.6. Alternative Splicing in Progression of DKD
The full-length sequencing data generated by SmartSeq2 allowed analysis of differential splicing events (DSE). Analysis distinguished five basic modes of splicing events (Figure 6A) and calculated inclusion levels of exons or introns for each event. The median inclusion level difference for splicing events (FDR<0.05) was ~0.4 between BTBRLean and BTBRob/ob (GEC/PCEC) at each timepoint (Suppl. Figure S11A). We identified 903 (PCEC 11-wk) to 1111 (GEC 6-wk) significant DSEs (Figure 6B, DSE details and links to Sashimi plot in Suppl. File S6). The dominant AS mode was exon skipping, accounting for 60% of overall DSEs and about 29% of DSE genes involved multiple DSEs. (Suppl. Figure S11B).
For GEC and PCEC, 109 and 96 genes, respectively, involved DSEs at all timepoints (Figure 6C, Suppl. File S6). As examples, Lias, Chek2, and Tbc1d31 (Figure 6D-E) showed no differential gene expression, but significant DSEs at all timepoints either at the same (Lias and Chek2/GEC) or different (Tbc1d31/PCEC) splice junctions. Lias, essential for mitochondrial energy metabolism and redox regulation in DKD [42,43], displayed exon skipping in BTBRLean but not in BTBRob/ob GEC. In contrast, Chek2, involved in DNA repair and cell cycle arrest upon DNA damage, displayed induced exon skipping in BTBRob/ob GEC (Figure 6D). Tbc1d31, essential for ciliogenesis, showed reduced exon skipping at different splice junctions between 6- and 11/20-week BTBRob/ob mice. Overall, the distinct and overlapping DSEs and involvement of different AS modes or junctions suggest intricate regulation of AS in DKD progression.
2.7. DEG and DSE Linked to Divergent Biological Processes
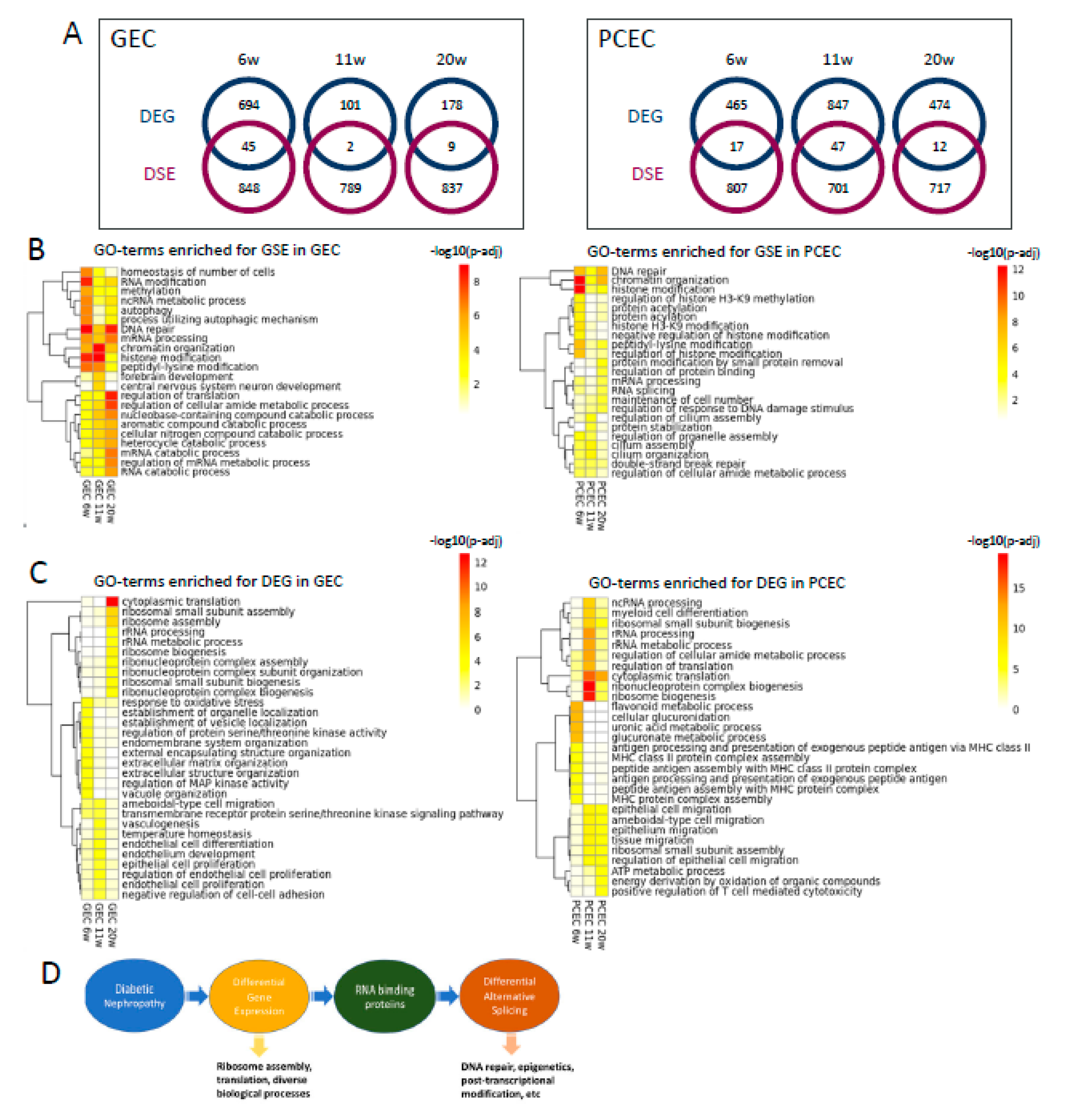
To explore the relationship between differential transcription and splicing, we compared DEGs and DSE genes and found little overlap between these two regulatory mechanisms (Figure 7A). For DSE genes, enriched gene ontology biological processes (GOBP) were predominantly related to DNA repair, epigenetics, and post-transcriptional modification, initiated already at 6-week (Figure 7B). Additionally, post-transcriptional processes including RNA modification, processing, and splicing, were enriched in both EC subtypes across timepoints. Furthermore, GEC and PCEC displayed diversely enriched GOBP, e.g. cilium assembly in PCEC and autophagy in GEC.
In contrast to DSE, the top enriched GOBPs in DEGs were related to ribosomal complex and protein translation, more prominent at 11/20-week (Figure 7C). In comparison with DSE genes, DEGs were involved in more diverse biological processes. DEG-enriched GOBPs showed a more dynamic pattern, in accordance with the fewer overlapping DEGs between timepoints (Figure 3C-D). RNA-binding proteins (RBP) regulate AS [44]. We identified 91 RBPs that were differentially expressed in GEC/PCEC for at least one timepoint. Most RBPs showed differential expression at 6-week in GEC; but at 11-week in PCEC (Suppl. Figure S12). Some of these RBPs are known to regulate splicing, e.g., Rbm4b [44] at 6-week, Son [45] at 11/20-week, and Igf2bp2 [46] at 6/20-week.
3. Discussion
To determine vascular heterogeneity in healthy kidneys and in progression of DKD, we employed scRNAseq using the SmartSeq2 protocol [47,48]. In healthy kidneys, our data pinpoint EC populations similar to those described previously[3,4] but more accurately distinguished them, including AVR and VEC [3] as well as tip and proliferating EC [4]. This is likely thanks to the superior mRNA capture and sequencing depth of SmartSeq2 compared to droplet-based methods. Our scRNAseq data identifies on average >4400 genes/cell, compared with <2500 genes/cell in previous studies [3,4], thereby permitting identification of novel EC-population specific markers, such as Il33 in VEC/PCEC/AVR and Ace in AA. Pathway analysis of EC subtypes revealed extensive zonation of the immune system, coagulation system, vascular tone, and calcium homeostasis. We also observed EC zonation of IGF signaling, underscored by previous studies [3]. These pathways may all account for EC functions in DKD progression, and our data could facilitate therapeutic design and targeting of specific EC compartments.
To elucidate longitudinal changes in EC during DKD progression we studied three timepoints in the BTBRob/ob mouse model that resembles early human DKD [14,49,50]. Capillary rarefaction was initiated as early as 6-week in BTBRob/ob mice. Our data suggests opposite regulation of many genes over time in diabetes. A similar pattern has been observed in whole kidney transcriptomics from patient biopsies, suggesting compensatory and adaptive mechanisms [51].
IPA on DEGs for PCEC and GEC in our study resolve significantly enriched and regulated canonical pathways in DKD progression. Inactivation of OXPHOS in PCEC and GEC at 20-week is likely associated with GEC dysfunction and PCEC loss, as OXPHOS and mitochondrial dysfunction are linked to DKD and vascular rarefaction [52,53,54]. The activation of EIF2 in GEC is likely caused by compensatory glomerular growth factor signaling in DKD [55]. Inactivation in PCEC possibly represents an adaptive response to cellular stress through inhibition of global translation [56]. Moreover, persistent inactivation of EIF2 signaling may be associated with apoptosis, which is common in both EC populations at late timepoints.
IGF1 signaling was a central factor altered in diabetic mice. Interestingly, GEC and PCEC showed contrariwise changes, activation in GEC and inactivation in PCEC. The IGFBP family fine-tune IGF1 signaling displaying distinct spatial expression and functions [57]. Igfbp5 was expressed in both GEC/PCEC and was significantly regulated in GEC in diabetes, as shown in human DKD [26], and enriched in GEC/PCEC in human kidney [25,27]. These observations implicate IGFBP5/IGF1 in the pathogenesis of DKD, and thus emphasize the functional significance of our observation of IGF1-related DEGs.
We performed the first comprehensive profiling of AS landscape in renal EC during DKD progression, which may shed light on intricate regulation of kidney disease evolution. Aberrant AS has been implicated in chronic kidney diseases [44], diabetes [58], and diabetic vasculopathy [59,60], where multiple splice isoforms exhibited functional significance during disease progression. Notably, AS can serve as a therapeutic target [44], given that the protective effects of SGLT2 inhibitor are partially mediated by regulation of AS in the proximal tubule in DKD [9]. The DSE profile in this study provides a comprehensive data source for interrogating splice isoform switching in early DKD and their biological relevance. DNA repair emerged as a key biological process associated with aberrant AS. DNA damage and repair are implicated in the pathogenesis of DKD, as manifested in urine-derived cells in diabetic patients [61] and in podocytes in DKD model [62].
Our study has certain limitations. First, we cannot rule out the possible loss of Pecam1 protein in individual EC during DKD progression, which could lower their representation in the scRNAseq data. Second, GOBP was used to compare DEG and DSE, as limited understanding of splice isoforms excluded analysis by IPA. Third, quantitative validation of DEGs and splice isoform switching in small EC subsets remain technically challenging. Although RNA-seq reliably identifies DEGs in contrast to microarray [63,64,65], technical developments in this field are warranted to further validate our data.
In conclusion, we have built a single-cell renal EC transcriptomic atlas and identified novel EC markers that characterize different EC populations along the intrarenal microvasculature. We discovered differential regulation of GEC/PCEC during progression of murine DKD. Moreover, the involvement of differential splicing and transcription in divergent biological processes is a novel finding and adds much information to our atlas. Our findings revealed novel spatiotemporal gene expression regulation in DKD, which might allow more precise targeting of the intrarenal microcirculation during disease progression and in turn discovery of therapeutic targets.
4. Materials and Methods
Kidney ECs were collected from diabetic BTBRob/ob mice and non-diabetic littermates at different timepoints. Their heterogeneity and changes in transcription and alternative splicing during DKD progression were mapped using SmartSeq2 scRNAseq and elucidated by pathway, network, and gene ontology enrichment analyses. Identified EC populations were validated by immunohistochemistry and in situ hybridization.
4.1. Animals
All animal experiments were carried out in accordance with Swedish legislation and local guidelines and regulation for animal welfare and were approved by the Regional Laboratory Animal Ethics Committee of Gothenburg, Sweden, (ID 38-2015) and the Regional Laboratory Animal Ethics Committee of Uppsala, Sweden, (ID C110-12 and 5.8.18-04862-220). In this study we used several mouse strains, all maintained as breeding colonies at the local animal facility. All animals were housed in 12 h light-12 h dark cycle and had ad libitum access to water and chow. The housing temperature was kept at 20±2°C and the relative humidity at 50±5%. Blood glucose and urinary albumin creatinine ratio (UACR) were measured at 11 weeks of age. BTBRV(B6)-Lepob/WiscJ stock no. 004824 (Jackson Laboratory), female diabetic BTBRob/ob mice, a model of diabetic nephropathy, and their non-diabetic littermate heterozygous or wildtype mice (BTBRLean) were euthanized, and kidneys harvested at 6-, 11-, and 20-week of age (Figure 1a). For validation of EC clusters, we used 6-, 11-, and 20-week-old C57Bl6/J (Jackson Laboratory) mice, and reporter mice with Cldn5GFP (Tg(Cldn5-GFP)Cbet/U) or Cdh5-TdTomato. Cdh5-TdTomato mice were inducible Cdh5-CreERT2 mice [66] bred to a reporter mouse, Ai14-TdTomato [67] induced with three doses of tamoxifen (2 mg) in peanut oil by oral gavage at 4 weeks of age.
4.2. Isolation of Renal Single Cells
Mice were euthanized by cervical dislocation, and the kidneys immediately harvested and placed into ambient phosphate-buffered saline (PBS) solution (DPBS, Thermo Fisher Scientific). The papilla was removed, and the tissue then diced and incubated in dissociation buffer, 0.13U/ml Liberase TL (5401020001, Merck) in RPMI 1640 (Thermo Fisher Scientific) at 37°C water bath with horizontal shaking at 500-800 rpm for 20-30 minutes. Thereafter the cell suspension was sequentially passed through 70 µm and 40 µm cell strainers. The 70 µm cell strainer was additionally washed with 5 ml RPMI 1640. Cells were pelleted by centrifugation at 300×g for 15 min at 4°C, the supernatant removed, and the pellet resuspended in ACK lysing buffer (A1049201, Thermo Fisher Scientific) for 5 min. After lysing red blood cells, cells were pelleted by centrifugation at 300×g for 10 min at 4°C, supernatant removed, and pellet resuspended in FACS buffer (DMEM supplemented with 1% fetal calf serum and 5mM EDTA). For labelling of EC and live cells, the cell suspension was incubated with APC-conjugated anti-CD31 antibody (1:100, 551262, BD Biosciences) and Calcein AM (2µM, C3099, Thermo-Fisher Scientific) for 20 min at room temperature (RT), then centrifugated as above, supernatant removed, and the cell pellet resuspended in FACS buffer and placed on ice.
4.3. Fluorescent Activated Cells Sorting (FACS)
Antibody-stained cell suspensions were analyzed using Beckton Dickinson FACSAria III cell sorter equipped with a 100 µm nozzle. Single cells meeting the selection criteria as described below were deposited into 384-well plates containing 2.3µl lysis buffer (0.2% Triton X-100, 2 U/ml RNase inhibitor, 2mM dNTPs, 1µM Smart-dT30VN, ERCC 1:4x104 dilution). For single cell sorting, first a gate for forward scatter-area/side scatter-area (FCS-A/SSC-A, linear scale) was set generously around present events only excluding events with low values (cell debris and red blood cells). Second, doublet discrimination was implemented using FCS-A/FSC-height and SSC-A/SCC-height. Third, selected events were then analyzed for live cell marker and positive fluorescent signal, unstained cells were used to ensure correct gating and as negative controls. Plates were briefly centrifugated prior to sorting, while correct deposition of the droplet into the 384-well plate was controlled by test-spotting of beads onto the seal of the respective plate, and if necessary, the plate position was adjusted for each new plate placed in the machine. The sample and plate holder of the machine were kept at 4°C, and the plates were placed on dry-ice immediately after the sorting was completed and subsequently stored at -80°C until downstream processing.
4.4. SmartSeq2 Library Preparation and Sequencing
Single-cell cDNA libraries were prepared according to a previously described SmartSeq2 protocol [68]. In brief, mRNA was transcribed into cDNA using oligo(dT) primer and Superscript II reverse transcriptase (Thermo Fisher Scientific). Second strand cDNA was synthesized using a template switching oligo. The synthesized cDNA was then amplified by polymerase chain reaction (PCR) for 24 cycles. Purified cDNA was quality controlled (QC) by analysis on a 2100 Bioanalyzer with a DNA high sensitivity chip (Agilent Biotechnologies). When the sample passed the QC, the cDNA was fragmented and tagged (tagmented) using Tn5 transposase, and each single cell uniquely indexed using the Illumina Nextera XT index kits (Set A-D). Thereafter, the uniquely indexed cDNA libraries from one 384-well plate were pooled into one sample to be sequenced on one lane of a HiSeq3000 sequencer (Illumina), using a sequencing strategy of dual indexing and single 50 base-pair reads.
4.5. Sequence Data Processing
Pooled single cell cDNA library samples were sequenced as described above. Demultiplexing into single cell fastq files was performed with standard parameters of the Illumina pipeline (bcl2fastq) using Nextera index adapters. The demultiplexed individual fastq files were then aligned to the mouse reference genome GRCm38 (mm10), using TopHat (version 2.1.1) [69,70], and the adapter sequences in each read removed using trim galore before read mapping. The alignment BAM files were sorted according to the mapping position and the duplicated reads removed using Samtools software (version 0.1.18). The raw read counts for each gene were calculated using featureCounts from the Subread package (version 1.4.6-p5) [71]. Cells with total reads counts less than 50,000 were removed. Cells that displayed a clearly contaminated transcriptome with, e.g., epithelial cell-specific gene signature (Lrp2, Pax8, Slc12a1, and Slc12a3) were excluded. This resulted in a final dataset for analysis composed of cells from 3 lean and 3 obese female mice from most timepoints (6, 11, and 20 weeks), with exception of the 6-week lean mice (n=2, one sample removed due to poor quality). After sequencing and quality control, a dataset of 5464 cells was constructed and used for downstream bioinformatic analyses.
The dataset was imported in the R-Seurat package for clustering, visualization and marker gene analysis (version: 3.1.1) [72]. For visualization of all single cells, the dimension reduction method UMAP was applied (UMAP: uniform manifold approximation and projection) [73] The counts values were normalized to 500,000 counts library size per cell for visualization in bar plots. Differentially expressed gene (DEG) analysis was performed using the FindMarkers function in the Seurat package. Single cell EC from non-diabetic (BTBRLean) mice were used to identify DEGs between EC compartments in the dataset, using cutoff values of foldchange >1.5 and an adjusted p-value <0.05. DEGs were identified in the same way as above to compare diabetic EC (BTBRob/ob) and non-diabetic EC (BTBRLean) for peritubular capillary endothelium (PCEC) and glomerular endothelium (GEC) at the different time points. For this analysis, cells were pooled regardless of their immediate early gene transcripts (IEG): PCEC and PCECIEG-high were pooled to PCEC, and GEC and GECIEG-high pooled to GEC. While all mice were female (confirmed by external genitalia), 2 mice showed expression of X- chromosome inactivation genes (Tsix and Xist). These genes were removed from visualization in heatmaps but included in calculations of DEGs (Suppl. File 3).
A published single-cell dataset [25] was used for comparison to human GEC and PCEC. In this data, we annotated cluster N4 (GATA5+, EHD3+) as GEC and cluster N2 as PCEC, as previously described [3,4], which were then compared with our mouse GEC and PCEC, respectively.
GWAS (genome-wide association studies) genes associated with CKD [26] were tested for expression in our dataset and presented in a heatmap for EC populations.
4.6. Ingenuity Pathway Analysis
Ingenuity Pathway Analysis (IPA, Qiagen) was utilized to describe pathways and their regulation by DEGs in our dataset. All analysis in IPA had a criterion of p<0.05 and z score >2. Based on the function and expression of DEGs, IPA also assigned an activation score to each enriched pathway, which were further compared between all EC clusters using IPA comparison analysis. In addition, IPA predicts which upstream regulators are activated or inhibited to explain the up-regulated and down-regulated genes observed in the dataset, while downstream effects analysis enables visualization of biological trends in the experiment and predicts the effect of molecular changes observed in the dataset on biological processes and disease or on toxicological functions. The IPA graphical summary illustrates the most important pathways, effects, and regulators.
4.7. Alternative Splicing Analysis
Raw reads were aligned to the mouse reference genome GRCm38 (mm10) using STARSolo (STAR v2.7.5c) [74,75]. Cells of each group of interest were combined into pseudo-bulk samples by merging bam files of the individual cells into one bam file per group using Samtools (v1.15.1) [76]. Differential alternative splicing analysis between different pairwise group comparisons was performed with rMATS (v4.1.2) [77] using the merged bam files as input. rMATS distinguishes five basic modes of splicing events and calculate inclusion levels of exons or introns for each event. This generated five result tables, one for every splicing event mode (SE, MXE, RI, A5SS, A3SS), containing p-value, FDR and inclusion level difference for each comparison. For further analysis, the result files based on both junction counts and exon counts were used. Sashimi plots for selected splicing events were generated using rmats2sashimiplot (v2.0.4).
4.8. Gene Ontology Enrichment Analysis
GO-term enrichment analysis was run using the enrichGO function of the R package ClusterProfiler (v4.2.0) [78]. For GO-term enrichment of DSE, genes involving significant differential splicing events (FDR<0.05) and an absolute inclusion level difference >0.4 were used as the input gene list [78]. For GO-term enrichment of DEG, significantly (adjusted p <0.05) differentially expressed genes were used as the input gene list. Heatmaps were made for the top 10 GO-terms for each comparison using the R package pheatmap (v1.0.12) (https://CRAN.R-project.org/package=pheatmap).
4.9. Identification of RNA Binding Proteins
DEGs were compared with a compiled list of mouse RBPs from 3 databases, rMAPS (http://rmaps.cecsresearch.org), RBPDB (http://rbpdb.ccbr.utoronto.ca), and Splicesome (http://spliceosomedb.ucsc.edu). The list consisted of 659 unique RBPs in total.
4.10. Validation of EC Cluster Markers with RNA In Situ Hybridization (RNA-ISH)
For RNA-ISH, the RNAscope® system (Advanced Cell Technologies) was applied according to the manufacturer’s protocol. In brief, tissue sections were prepared as described below (Immunofluorescence staining). After dehydration, HRP was quenched with Bloxall (Sp-6000, Vector Technologies) blocking solution for 10 min at RT followed by Pretreat III solution for 30 min at RT. Then, RNAscope®probes (Suppl. Table 3) were hybridized on the sections for 2 h at 40°C, followed by RNAscope® multiplex fluorescent Reagent kit v2 assay according to the manufacturer’s instructions. Opal 520, Opal 570, and Opal 690 (Akoya Biosciences) were diluted 1:1000. Sections were mounted with ProLong®Gold mounting medium and images obtained using a confocal microscope (Leica Microsystems Sp8) at 400x.
4.11. Immunofluorescence Staining
Standard methods for immunostaining were used. In brief, harvested kidneys were fixated in 4% formaldehyde for 4 h at RT, followed by immersion in 30% sucrose/PBS solution for at least 24h at 4°C. Tissues were then embedded for cryo-sectioning in Tissue-Tek OCT and sectioned at 14 µm on a Cryostat NX70 (Thermo Fisher Scientific). Sections were stored at -80°C. For staining, sections were blocked >1 h at RT with blocking-buffer (X0909, Dako, Agilent) supplemented with 0.25% Triton X-100 (Sigma Aldrich). Thereafter, sections were sequentially incubated with primary antibodies overnight at 4°C and corresponding fluorescently conjugated secondary antibodies (Suppl. Table 4) and Hoechst 33342 (Dako) for 2 h at RT. Sections were mounted with ProLong®Gold mounting medium (Thermo Fisher Scientific). Images were analyzed utilizing ImageJ/Fiji software [79]. For quantification of capillary density, the endothelial marker Pecam1 was quantified by immunohistochemistry and light microscopy in paraffin sections of kidney from 5 BTBRob/ob and 5 BTBR Lean 11-week-old mice for different regions of the kidney: cortex, outer stripe of medulla (OSOM), inner stripe of medulla (ISOM), and inner medulla (IM). Large arteries and glomeruli were excluded. To document changes in capillary density over time we performed immunofluorescence and confocal imaging of renal cortex with additional endothelial markers, endomucin and podocalyxin (3 BTBRob/ob and 3 BTBR Lean mice for each timepoint). Stained area relative to total area was calculated using Otsu thresholding from 5 images from the cortex of each mouse.
4.12. Blood and Urine Analysis
Non-anesthetized plasma glucose levels were measured in tail vein blood at 11 weeks of age using a portable glucometer (Accu-Chek mobile®) following manufacturer’s instructions. Urinary albumin creatinine ratio (UACR) was measured in spot urine collected on a tray. The urine albumin concentration was measured with ELISA (Albuwell-M) and the urine creatinine was measured colorimetric/fluorometric (ab65340 Creatinine Assay Kit, Abcam) following manufacturer’s instructions.
4.13. Statistical Analysis
For capillary density measurements, data are expressed as mean ± SD. Means between groups were compared using 2-tailed unpaired Student’s t-test or one-way ANOVA with Bonferroni’s multiple comparisons were appropriate using GraphPad Prism version-8 (GraphPad Software Inc). All data was tested for uneven distribution and in the case of uneven distribution logarithmic values were used. A p<0.05 is considered statistically significant.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Figures and Tables file containing: Supplemental Figure S1. Validation of vascular segments I.Supplemental Figure S2. Validation of vascular segments II. Supplemental Figure S3. Searchable database. Supplemental Figure S4. Mouse and human expression correlation. Supplemental Figure S5. Immediate early gene (IEG) expression. Supplemental Figure S6. Validation of DKD. Supplemental Figure S7. Statistics of the top enriched pathways and oxidative phosphorylation. Supplemental Figure S8. EIF2 signaling in diabetic PCEC at 6 and 20 weeks. Supplemental Figure S9. EIF2 signaling in diabetic GEC at 6 and 20 weeks. Supplemental Figure S10. The IGF system in DKD. Supplemental Figure S11. Characterization of DSEs. Supplemental Figure S12. Overlapping DEGs with a compiled list of mouse RBPs. Supplemental Table S1. Number of sequenced cells per cluster and timepoint. Supplemental Table S2. Differentially expressed genes comparing Lean and ob/ob mice. Supplemental Table 3S. RNAscope probes and reagents. Supplemental Table S4. Antibodies. Supplemental File S1. DEGs in EC populations. Supplemental File S2. IPA analysis all data. Supplemental File S3. DEGs lean vs obese. Supplemental File S4. Oxidative phosphorylation. Supplemental File S5. EIF2 signaling. Supplemental File S6. Significant DSE with Sachimi plots.
Author Contributions
A.X.Z. contributed to single cell isolation, FACS sorting, data analysis, and writing; M. J. to IHC and ISH, data analysis, and writing; L.H., L.W., and L.M. to data analysis; P.T. and A.B.G. to BTBR model and characterization; R.T. and L.C. to analysis; M.U. to FACS sorting; J.L. to sequencing; L.O.L., C.B., and P.B. L.H. to data analysis and writing.
Funding
This research was funded by AstraZeneca.
Institutional Review Board Statement
All animal experiments were carried out in accordance with Swedish legislation and local guidelines and regulation for animal welfare and were approved by the Regional Laboratory Animal Ethics Committee of Gothenburg, Sweden, (ID 38-2015) and the Regional Laboratory Animal Ethics Committee of Uppsala, Sweden, (ID C110-12 and 5.8.18-04862-220).
Data Availability Statement
ScRNA-seq data can be accessed from NCBI’s Gene Expression Omnibus database, accession number GSE192687. All data to support the findings of this study are included in the paper, the Supplementary Information, and available in a searchable database at https://betsholtzlab.org/Publications/MouseKidenyOB/database.html (login: reviewer; password: reviewer).
Acknowledgments
We appreciate discussions with Julie M. Williams, Robert Menzies, and Santhosh V. Kumar and experimental supports from Charlotte Wennberg Huldt and Henrik Palmgren at AstraZeneca.
Conflicts of Interest
A.X.Z., P.T., L.W, A.B.G.,P.T. M.U. R.T., L.C., and P.B.L.H. are employed by AstraZeneca Gothenburg, Sweden, and L.O.L is an advisor. The funders had no role in study design, data collection and analysis,decision to publish, or preparation of the manuscript.
References
- Alicic, R. Z., M. T. Rooney, and K. R. Tuttle. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin J Am Soc Nephrol 2017, 12, 2032-45. [Google Scholar] [CrossRef]
- Kida, Y. Peritubular Capillary Rarefaction: An Underappreciated Regulator of Ckd Progression. Int J Mol Sci 2020, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Barry, D. M., E. A. McMillan, B. Kunar, R. Lis, T. Zhang, T. Lu, E. Daniel, M. Yokoyama, J. M. Gomez-Salinero, A. Sureshbabu, O. Cleaver, A. Di Lorenzo, M. E. Choi, J. Xiang, D. Redmond, S. Y. Rabbany, T. Muthukumar, and S. Rafii. "Molecular Determinants of Nephron Vascular Specialization in the Kidney." Nat Commun 10, no. 1 (2019): 5705.
- Dumas, S. J., E. Meta, M. Borri, J. Goveia, K. Rohlenova, N. V. Conchinha, K. Falkenberg, L. A. Teuwen, L. de Rooij, J. Kalucka, R. Chen, S. Khan, F. Taverna, W. Lu, M. Parys, C. De Legher, S. Vinckier, T. K. Karakach, L. Schoonjans, L. Lin, L. Bolund, M. Dewerchin, G. Eelen, T. J. Rabelink, X. Li, Y. Luo, and P. Carmeliet. "Single-Cell Rna Sequencing Reveals Renal Endothelium Heterogeneity and Metabolic Adaptation to Water Deprivation." J Am Soc Nephrol 31, no. 1 (2020): 118-38.
- He, B., P. Chen, S. Zambrano, D. Dabaghie, Y. Hu, K. Moller-Hackbarth, D. Unnersjo-Jess, G. G. Korkut, E. Charrin, M. Jeansson, M. Bintanel-Morcillo, A. Witasp, L. Wennberg, A. Wernerson, B. Schermer, T. Benzing, P. Ernfors, C. Betsholtz, M. Lal, R. Sandberg, and J. Patrakka. "Single-Cell Rna Sequencing Reveals the Mesangial Identity and Species Diversity of Glomerular Cell Transcriptomes." Nat Commun 12, no. 1 (2021): 2141.
- Zhang, K., H. Kan, A. Mao, F. Yu, L. Geng, T. Zhou, L. Feng, and X. Ma. "Integrated Single-Cell Transcriptomic Atlas of Human Kidney Endothelial Cells." J Am Soc Nephrol (2024).
- Chung, J. J., L. Goldstein, Y. J. Chen, J. Lee, J. D. Webster, M. Roose-Girma, S. C. Paudyal, Z. Modrusan, A. Dey, and A. S. Shaw. "Single-Cell Transcriptome Profiling of the Kidney Glomerulus Identifies Key Cell Types and Reactions to Injury." J Am Soc Nephrol 31, no. 10 (2020): 2341-54.
- Subramanian, Ayshwarya, Katherine Vernon, Yiming Zhou, Jamie L. Marshall, Maria Alimova, Fan Zhang, Michal Slyper, Julia Waldman, Monica S. Montesinos, Danielle Dionne, Lan T. Nguyen, Michael S. Cuoco, Dan Dubinsky, Jason Purnell, Keith Heller, Samuel H. Sturner, Elizabeth Grinkevich, Ayan Ghoshal, Astrid Weins, Alexandra-Chloe Villani, Steven L. Chang, Orit Rosenblatt-Rosen, Jillian L. Shaw, Aviv Regev, and Anna Greka. "Obesity-Instructed Trem2(High) Macrophages Identified by Comparative Analysis of Diabetic Mouse and Human Kidney at Single Cell Resolution." (2021): 2021.05.30.446342.
- Wu, H., R. Gonzalez Villalobos, X. Yao, D. Reilly, T. Chen, M. Rankin, E. Myshkin, M. D. Breyer, and B. D. Humphreys. "Mapping the Single-Cell Transcriptomic Response of Murine Diabetic Kidney Disease to Therapies." Cell Metab 34, no. 7 (2022): 1064-78 e6.
- Wilson, P. C., H. Wu, Y. Kirita, K. Uchimura, N. Ledru, H. G. Rennke, P. A. Welling, S. S. Waikar, and B. D. Humphreys. "The Single-Cell Transcriptomic Landscape of Early Human Diabetic Nephropathy." Proc Natl Acad Sci U S A 116, no. 39 (2019): 19619-25.
- Pan, Q., O. Shai, L. J. Lee, B. J. Frey, and B. J. Blencowe. "Deep Surveying of Alternative Splicing Complexity in the Human Transcriptome by High-Throughput Sequencing." Nat Genet 40, no. 12 (2008): 1413-5.
- Wineberg, Y., T. H. Bar-Lev, A. Futorian, N. Ben-Haim, L. Armon, D. Ickowicz, S. Oriel, E. Bucris, Y. Yehuda, N. Pode-Shakked, S. Gilad, S. Benjamin, P. Hohenstein, B. Dekel, A. Urbach, and T. Kalisky. "Single-Cell Rna Sequencing Reveals Mrna Splice Isoform Switching During Kidney Development." J Am Soc Nephrol 31, no. 10 (2020): 2278-91.
- Stoehr, J. P., S. T. Nadler, K. L. Schueler, M. E. Rabaglia, B. S. Yandell, S. A. Metz, and A. D. Attie. "Genetic Obesity Unmasks Nonlinear Interactions between Murine Type 2 Diabetes Susceptibility Loci." Diabetes 49, no. 11 (2000): 1946-54.
- Hudkins, K. L., W. Pichaiwong, T. Wietecha, J. Kowalewska, M. C. Banas, M. W. Spencer, A. Muhlfeld, M. Koelling, J. W. Pippin, S. J. Shankland, B. Askari, M. E. Rabaglia, M. P. Keller, A. D. Attie, and C. E. Alpers. "Btbr Ob/Ob Mutant Mice Model Progressive Diabetic Nephropathy." J Am Soc Nephrol 21, no. 9 (2010): 1533-42.
- Goncharov, N. V., A. D. Nadeev, R. O. Jenkins, and P. V. Avdonin. "Markers and Biomarkers of Endothelium: When Something Is Rotten in the State." Oxid Med Cell Longev (2017): 9759735.
- Vestweber, D. "Ve-Cadherin: The Major Endothelial Adhesion Molecule Controlling Cellular Junctions and Blood Vessel Formation." Arterioscler Thromb Vasc Biol 28, no. 2 (2008): 223-32.
- Park, J., R. Shrestha, C. Qiu, A. Kondo, S. Huang, M. Werth, M. Li, J. Barasch, and K. Susztak. "Single-Cell Transcriptomics of the Mouse Kidney Reveals Potential Cellular Targets of Kidney Disease." Science 360, no. 6390 (2018): 758-63.
- Ye, M., J. Wysocki, J. William, M. J. Soler, I. Cokic, and D. Batlle. "Glomerular Localization and Expression of Angiotensin-Converting Enzyme 2 and Angiotensin-Converting Enzyme: Implications for Albuminuria in Diabetes." J Am Soc Nephrol 17, no. 11 (2006): 3067-75.
- Tabula Muris, Consortium, coordination Overall, coordination Logistical, collection Organ, processing, preparation Library, sequencing, analysis Computational data, annotation Cell type, group Writing, group Supplemental text writing, and investigators Principal. "Single-Cell Transcriptomics of 20 Mouse Organs Creates a Tabula Muris." Nature 562, no. 7727 (2018): 367-72.
- Moffat, D. B. "The Fine Structure of the Blood Vessels of the Renal Medulla with Particular Reference to the Control of the Medullary Circulation." J Ultrastruct Res 19, no. 5 (1967): 532-45.
- Kriz, W. "Fenestrated Glomerular Capillaries Are Unique." J Am Soc Nephrol 19, no. 8 (2008): 1439-40.
- Stolz, D. B., and S. Sims-Lucas. "Unwrapping the Origins and Roles of the Renal Endothelium." Pediatr Nephrol 30, no. 6 (2015): 865-72.
- Stan, R. V., M. Kubitza, and G. E. Palade. "Pv-1 Is a Component of the Fenestral and Stomatal Diaphragms in Fenestrated Endothelia." Proc Natl Acad Sci U S A 96, no. 23 (1999): 13203-7.
- Yamamoto, I., S. Horita, T. Takahashi, K. Tanabe, S. Fuchinoue, S. Teraoka, M. Hattori, and Y. Yamaguchi. "Glomerular Expression of Plasmalemmal Vesicle-Associated Protein-1 in Patients with Transplant Glomerulopathy." Am J Transplant 7, no. 8 (2007): 1954-60.
- Young, M. D., T. J. Mitchell, F. A. Vieira Braga, M. G. B. Tran, B. J. Stewart, J. R. Ferdinand, G. Collord, R. A. Botting, D. M. Popescu, K. W. Loudon, R. Vento-Tormo, E. Stephenson, A. Cagan, S. J. Farndon, M. Del Castillo Velasco-Herrera, C. Guzzo, N. Richoz, L. Mamanova, T. Aho, J. N. Armitage, A. C. P. Riddick, I. Mushtaq, S. Farrell, D. Rampling, J. Nicholson, A. Filby, J. Burge, S. Lisgo, P. H. Maxwell, S. Lindsay, A. Y. Warren, G. D. Stewart, N. Sebire, N. Coleman, M. Haniffa, S. A. Teichmann, M. Clatworthy, and S. Behjati. "Single-Cell Transcriptomes from Human Kidneys Reveal the Cellular Identity of Renal Tumors." Science 361, no. 6402 (2018): 594-99.
- Pattaro, C., A. Teumer, M. Gorski, A. Y. Chu, M. Li, V. Mijatovic, M. Garnaas, A. Tin, R. Sorice, Y. Li, D. Taliun, M. Olden, M. Foster, Q. Yang, M. H. Chen, T. H. Pers, A. D. Johnson, Y. A. Ko, C. Fuchsberger, B. Tayo, M. Nalls, M. F. Feitosa, A. Isaacs, A. Dehghan, P. d'Adamo, A. Adeyemo, A. K. Dieffenbach, A. B. Zonderman, I. M. Nolte, P. J. van der Most, A. F. Wright, A. R. Shuldiner, A. C. Morrison, A. Hofman, A. V. Smith, A. W. Dreisbach, A. Franke, A. G. Uitterlinden, A. Metspalu, A. Tonjes, A. Lupo, A. Robino, A. Johansson, A. Demirkan, B. Kollerits, B. I. Freedman, B. Ponte, B. A. Oostra, B. Paulweber, B. K. Kramer, B. D. Mitchell, B. M. Buckley, C. A. Peralta, C. Hayward, C. Helmer, C. N. Rotimi, C. M. Shaffer, C. Muller, C. Sala, C. M. van Duijn, A. Saint-Pierre, D. Ackermann, D. Shriner, D. Ruggiero, D. Toniolo, Y. Lu, D. Cusi, D. Czamara, D. Ellinghaus, D. S. Siscovick, D. Ruderfer, C. Gieger, H. Grallert, E. Rochtchina, E. J. Atkinson, E. G. Holliday, E. Boerwinkle, E. Salvi, E. P. Bottinger, F. Murgia, F. Rivadeneira, F. Ernst, F. Kronenberg, F. B. Hu, G. J. Navis, G. C. Curhan, G. B. Ehret, G. Homuth, S. Coassin, G. A. Thun, G. Pistis, G. Gambaro, G. Malerba, G. W. Montgomery, G. Eiriksdottir, G. Jacobs, G. Li, H. E. Wichmann, H. Campbell, H. Schmidt, H. Wallaschofski, H. Volzke, H. Brenner, H. K. Kroemer, H. Kramer, H. Lin, I. M. Leach, I. Ford, I. Guessous, I. Rudan, I. Prokopenko, I. Borecki, I. M. Heid, I. Kolcic, I. Persico, J. W. Jukema, J. F. Wilson, J. F. Felix, J. Divers, J. C. Lambert, J. M. Stafford, J. M. Gaspoz, J. A. Smith, J. D. Faul, J. J. Wang, J. Ding, J. N. Hirschhorn, J. Attia, J. B. Whitfield, J. Chalmers, J. Viikari, J. Coresh, J. C. Denny, J. Karjalainen, J. K. Fernandes, K. Endlich, K. Butterbach, K. L. Keene, K. Lohman, L. Portas, L. J. Launer, L. P. Lyytikainen, L. Yengo, L. Franke, L. Ferrucci, L. M. Rose, L. Kedenko, M. Rao, M. Struchalin, M. E. Kleber, M. Cavalieri, M. Haun, M. C. Cornelis, M. Ciullo, M. Pirastu, M. de Andrade, M. A. McEvoy, M. Woodward, M. Adam, M. Cocca, M. Nauck, M. Imboden, M. Waldenberger, M. Pruijm, M. Metzger, M. Stumvoll, M. K. Evans, M. M. Sale, M. Kahonen, M. Boban, M. Bochud, M. Rheinberger, N. Verweij, N. Bouatia-Naji, N. G. Martin, N. Hastie, N. Probst-Hensch, N. Soranzo, O. Devuyst, O. Raitakari, O. Gottesman, O. H. Franco, O. Polasek, P. Gasparini, P. B. Munroe, P. M. Ridker, P. Mitchell, P. Muntner, C. Meisinger, J. H. Smit, Icbp Consortium, Agen Consortium, Cardiogram, C. HARGe-Heart Failure Group, E. CHOGen Consortium, P. Kovacs, P. S. Wild, P. Froguel, R. Rettig, R. Magi, R. Biffar, R. Schmidt, R. P. Middelberg, R. J. Carroll, B. W. Penninx, R. J. Scott, R. Katz, S. Sedaghat, S. H. Wild, S. L. Kardia, S. Ulivi, S. J. Hwang, S. Enroth, S. Kloiber, S. Trompet, B. Stengel, S. J. Hancock, S. T. Turner, S. E. Rosas, S. Stracke, T. B. Harris, T. Zeller, T. Zemunik, T. Lehtimaki, T. Illig, T. Aspelund, T. Nikopensius, T. Esko, T. Tanaka, U. Gyllensten, U. Volker, V. Emilsson, V. Vitart, V. Aalto, V. Gudnason, V. Chouraki, W. M. Chen, W. Igl, W. Marz, W. Koenig, W. Lieb, R. J. Loos, Y. Liu, H. Snieder, P. P. Pramstaller, A. Parsa, J. R. O'Connell, K. Susztak, P. Hamet, J. Tremblay, I. H. de Boer, C. A. Boger, W. Goessling, D. I. Chasman, A. Kottgen, W. H. Kao, and C. S. Fox. "Genetic Associations at 53 Loci Highlight Cell Types and Biological Pathways Relevant for Kidney Function." Nat Commun 7 (2016): 10023.
- Menon, R., E. A. Otto, P. Hoover, S. Eddy, L. Mariani, B. Godfrey, C. C. Berthier, F. Eichinger, L. Subramanian, J. Harder, W. Ju, V. Nair, M. Larkina, A. S. Naik, J. Luo, S. Jain, R. Sealfon, O. Troyanskaya, N. Hacohen, J. B. Hodgin, M. Kretzler, Kpmp Kpmp, and Network Nephrotic Syndrome Study. "Single Cell Transcriptomics Identifies Focal Segmental Glomerulosclerosis Remission Endothelial Biomarker." JCI Insight 5, no. 6 (2020).
- Herschman, H. R. "Primary Response Genes Induced by Growth Factors and Tumor Promoters." Annu Rev Biochem 60 (1991): 281-319.
- Fowler, T., R. Sen, and A. L. Roy. "Regulation of Primary Response Genes." Mol Cell 44, no. 3 (2011): 348-60.
- Vanlandewijck, M., and C. Betsholtz. "Single-Cell Mrna Sequencing of the Mouse Brain Vasculature." Methods Mol Biol 1846 (2018): 309-24.
- Kalucka, J., Lpmh de Rooij, J. Goveia, K. Rohlenova, S. J. Dumas, E. Meta, N. V. Conchinha, F. Taverna, L. A. Teuwen, K. Veys, M. Garcia-Caballero, S. Khan, V. Geldhof, L. Sokol, R. Chen, L. Treps, M. Borri, P. de Zeeuw, C. Dubois, T. K. Karakach, K. D. Falkenberg, M. Parys, X. Yin, S. Vinckier, Y. Du, R. A. Fenton, L. Schoonjans, M. Dewerchin, G. Eelen, B. Thienpont, L. Lin, L. Bolund, X. Li, Y. Luo, and P. Carmeliet. "Single-Cell Transcriptome Atlas of Murine Endothelial Cells." Cell 180, no. 4 (2020): 764-79 e20.
- Stewart, B. J., J. R. Ferdinand, M. D. Young, T. J. Mitchell, K. W. Loudon, A. M. Riding, N. Richoz, G. L. Frazer, J. U. L. Staniforth, F. A. Vieira Braga, R. A. Botting, D. M. Popescu, R. Vento-Tormo, E. Stephenson, A. Cagan, S. J. Farndon, K. Polanski, M. Efremova, K. Green, M. Del Castillo Velasco-Herrera, C. Guzzo, G. Collord, L. Mamanova, T. Aho, J. N. Armitage, A. C. P. Riddick, I. Mushtaq, S. Farrell, D. Rampling, J. Nicholson, A. Filby, J. Burge, S. Lisgo, S. Lindsay, M. Bajenoff, A. Y. Warren, G. D. Stewart, N. Sebire, N. Coleman, M. Haniffa, S. A. Teichmann, S. Behjati, and M. R. Clatworthy. "Spatiotemporal Immune Zonation of the Human Kidney." Science 365, no. 6460 (2019): 1461-66.
- Taylor, M. S., and M. Francis. "Decoding Dynamic Ca(2+) Signaling in the Vascular Endothelium." Front Physiol 5 (2014): 447.
- Coleman, H. A., M. Tare, and H. C. Parkington. "Endothelial Potassium Channels, Endothelium-Dependent Hyperpolarization and the Regulation of Vascular Tone in Health and Disease." Clin Exp Pharmacol Physiol 31, no. 9 (2004): 641-9.
- Schmidt, K., and C. de Wit. "Endothelium-Derived Hyperpolarizing Factor and Myoendothelial Coupling: The in Vivo Perspective." Front Physiol 11 (2020): 602930.
- Wagner, C., and A. Kurtz. "Distribution and Functional Relevance of Connexins in Renin-Producing Cells." Pflugers Arch 465, no. 1 (2013): 71-7.
- Marsen, T. A., M. S. Simonson, and M. J. Dunn. "Roles of Calcium and Kinases in Regulation of Thrombin-Stimulated Preproendothelin-1 Transcription." Am J Physiol 271, no. 5 Pt 2 (1996): H1918-25.
- Altmann, J. B., G. Yan, J. F. Meeks, M. E. Abood, E. Brailoiu, and G. C. Brailoiu. "G Protein-Coupled Estrogen Receptor-Mediated Effects on Cytosolic Calcium and Nanomechanics in Brain Microvascular Endothelial Cells." J Neurochem 133, no. 5 (2015): 629-39.
- Kumar, R., and V. Vallon. "Reduced Renal Calcium Excretion in the Absence of Sclerostin Expression: Evidence for a Novel Calcium-Regulating Bone Kidney Axis." J Am Soc Nephrol 25, no. 10 (2014): 2159-68.
- Bach, L. A. "The Insulin-Like Growth Factor System in Kidney Disease and Hypertension." Curr Opin Nephrol Hypertens 21, no. 1 (2012): 86-91.
- Bossolasco, M., F. Veillette, R. Bertrand, and A. M. Mes-Masson. "Human Tde1, a Tde1/Tms Family Member, Inhibits Apoptosis in Vitro and Stimulates in Vivo Tumorigenesis." Oncogene 25, no. 33 (2006): 4549-58.
- Yi, X., L. Xu, S. Hiller, H. S. Kim, V. Nickeleit, L. R. James, and N. Maeda. "Reduced Expression of Lipoic Acid Synthase Accelerates Diabetic Nephropathy." J Am Soc Nephrol 23, no. 1 (2012): 103-11.
- Zhao, Y., T. Yan, C. Xiong, M. Chang, Q. Gao, S. Yao, W. Wu, X. Yi, and G. Xu. "Overexpression of Lipoic Acid Synthase Gene Alleviates Diabetic Nephropathy of Lepr(Db/Db) Mice." BMJ Open Diabetes Res Care 9, no. 1 (2021).
- Stevens, M., and S. Oltean. "Alternative Splicing in Ckd." J Am Soc Nephrol 27, no. 6 (2016): 1596-603.
- Kim, J. H., K. Jeong, J. Li, J. M. Murphy, L. Vukadin, J. K. Stone, A. Richard, J. Tran, G. Y. Gillespie, E. K. Flemington, R. W. Sobol, S. S. Lim, and E. E. Ahn. "Son Drives Oncogenic Rna Splicing in Glioblastoma by Regulating Ptbp1/Ptbp2 Switching and Rbfox2 Activity." Nat Commun 12, no. 1 (2021): 5551.
- Zhao, F., L. Wu, Q. Wang, X. Zhao, T. Chen, C. Yin, L. Yan, and X. Yang. "Insulin-Like Growth Factor 2 Mrna-Binding Protein 2-Regulated Alternative Splicing of Nuclear Factor 1 C-Type Causes Excessive Granulosa Cell Proliferation in Polycystic Ovary Syndrome." Cell Prolif 55, no. 4 (2022): e13216.
- Muhl, L., G. Genove, S. Leptidis, J. Liu, L. He, G. Mocci, Y. Sun, S. Gustafsson, B. Buyandelger, I. V. Chivukula, A. Segerstolpe, E. Raschperger, E. M. Hansson, J. L. M. Bjorkegren, X. R. Peng, M. Vanlandewijck, U. Lendahl, and C. Betsholtz. "Single-Cell Analysis Uncovers Fibroblast Heterogeneity and Criteria for Fibroblast and Mural Cell Identification and Discrimination." Nat Commun 11, no. 1 (2020): 3953.
- Vanlandewijck, M., L. He, M. A. Mae, J. Andrae, K. Ando, F. Del Gaudio, K. Nahar, T. Lebouvier, B. Lavina, L. Gouveia, Y. Sun, E. Raschperger, M. Rasanen, Y. Zarb, N. Mochizuki, A. Keller, U. Lendahl, and C. Betsholtz. "A Molecular Atlas of Cell Types and Zonation in the Brain Vasculature." Nature 554, no. 7693 (2018): 475-80.
- Ericsson, A., P. Tonelius, M. Lal, A. Sabirsh, G. Bottcher, L. William-Olsson, M. Stromstedt, C. Johansson, G. Hyberg, S. Tapani, A. C. Jonsson-Rylander, and R. Unwin. "The Effects of Dual Pparalpha/Gamma Agonism Compared with Ace Inhibition in the Btbrob/Ob Mouse Model of Diabetes and Diabetic Nephropathy." Physiol Rep 5, no. 5 (2017).
- Bjornson Granqvist, A., A. Ericsson, J. Sanchez, P. Tonelius, L. William-Olsson, U. Dahlqvist, A. K. Andersson, T. Tesan Tomic, K. Hudkins, C. E. Alpers, G. Pellegrini, and M. Soderberg. "High-Protein Diet Accelerates Diabetes and Kidney Disease in the Btbrob/Ob Mouse." Am J Physiol Renal Physiol 318, no. 3 (2020): F763-F71.
- Fan, Y., Z. Yi, V. D. D'Agati, Z. Sun, F. Zhong, W. Zhang, J. Wen, T. Zhou, Z. Li, L. He, Q. Zhang, K. Lee, J. C. He, and N. Wang. "Comparison of Kidney Transcriptomic Profiles of Early and Advanced Diabetic Nephropathy Reveals Potential New Mechanisms for Disease Progression." Diabetes 68, no. 12 (2019): 2301-14.
- Huang, C., Y. Kim, M. L. Caramori, J. H. Moore, S. S. Rich, J. C. Mychaleckyj, P. C. Walker, and M. Mauer. "Diabetic Nephropathy Is Associated with Gene Expression Levels of Oxidative Phosphorylation and Related Pathways." Diabetes 55, no. 6 (2006): 1826-31.
- Forbes, J. M., and D. R. Thorburn. "Mitochondrial Dysfunction in Diabetic Kidney Disease." Nature reviews. Nephrology 14, no. 5 (2018): 291-312.
- Yu, B. B., H. Zhi, X. Y. Zhang, J. W. Liang, J. He, C. Su, W. H. Xia, G. X. Zhang, and J. Tao. "Mitochondrial Dysfunction-Mediated Decline in Angiogenic Capacity of Endothelial Progenitor Cells Is Associated with Capillary Rarefaction in Patients with Hypertension Via Downregulation of Cxcr4/Jak2/Sirt5 Signaling." EBioMedicine 42 (2019): 64-75.
- Kasinath, B. S., M. M. Mariappan, K. Sataranatarajan, M. J. Lee, G. Ghosh Choudhury, and D. Feliers. "Novel Mechanisms of Protein Synthesis in Diabetic Nephropathy--Role of Mrna Translation." Rev Endocr Metab Disord 9, no. 4 (2008): 255-66.
- Wek, R. C. "Role of Eif2alpha Kinases in Translational Control and Adaptation to Cellular Stress." Cold Spring Harb Perspect Biol 10, no. 7 (2018).
- Allard, J. B., and C. Duan. "Igf-Binding Proteins: Why Do They Exist and Why Are There So Many?" Front Endocrinol (Lausanne) 9 (2018): 117.
- Dlamini, Z., F. Mokoena, and R. Hull. "Abnormalities in Alternative Splicing in Diabetes: Therapeutic Targets." Journal of molecular endocrinology 59, no. 2 (2017): R93-R107.
- Cornelius, V. A., J. R. Fulton, and A. Margariti. "Alternative Splicing: A Key Mediator of Diabetic Vasculopathy." Genes (Basel) 12, no. 9 (2021).
- Zhao, H., H. Kong, B. Wang, S. Wu, T. Chen, and Y. Cui. "Rna-Binding Proteins and Alternative Splicing Genes Are Coregulated in Human Retinal Endothelial Cells Treated with High Glucose." J Diabetes Res 2022 (2022): 7680513.
- Hishikawa, A., K. Hayashi, N. Yoshimoto, R. Nakamichi, K. Homma, and H. Itoh. "DNA Damage and Expression of DNA Methylation Modulators in Urine-Derived Cells of Patients with Hypertension and Diabetes." Sci Rep 10, no. 1 (2020): 3377.
- Hishikawa, A., K. Hayashi, T. Abe, M. Kaneko, H. Yokoi, T. Azegami, M. Nakamura, N. Yoshimoto, T. Kanda, Y. Sakamaki, and H. Itoh. "Decreased Kat5 Expression Impairs DNA Repair and Induces Altered DNA Methylation in Kidney Podocytes." Cell Rep 26, no. 5 (2019): 1318-32 e4.
- Everaert, C., M. Luypaert, J. L. V. Maag, Q. X. Cheng, M. E. Dinger, J. Hellemans, and P. Mestdagh. "Benchmarking of Rna-Sequencing Analysis Workflows Using Whole-Transcriptome Rt-Qpcr Expression Data." Sci Rep 7, no. 1 (2017): 1559.
- Zhang, L., S. J. Yoder, and S. A. Enkemann. "Identical Probes on Different High-Density Oligonucleotide Microarrays Can Produce Different Measurements of Gene Expression." BMC Genomics 7 (2006): 153.
- Balazsi, G., and Z. N. Oltvai. "A Pitfall in Series of Microarrays: The Position of Probes Affects the Cross-Correlation of Gene Expression Profiles." Methods Mol Biol 377 (2007): 153-62.
- Pitulescu, M. E., I. Schmidt, R. Benedito, and R. H. Adams. "Inducible Gene Targeting in the Neonatal Vasculature and Analysis of Retinal Angiogenesis in Mice." Nat Protoc 5, no. 9 (2010): 1518-34.
- Madisen, L., T. A. Zwingman, S. M. Sunkin, S. W. Oh, H. A. Zariwala, H. Gu, L. L. Ng, R. D. Palmiter, M. J. Hawrylycz, A. R. Jones, E. S. Lein, and H. Zeng. "A Robust and High-Throughput Cre Reporting and Characterization System for the Whole Mouse Brain." Nature neuroscience 13, no. 1 (2010): 133-40.
- Picelli, S., O. R. Faridani, A. K. Bjorklund, G. Winberg, S. Sagasser, and R. Sandberg. "Full-Length Rna-Seq from Single Cells Using Smart-Seq2." Nat Protoc 9, no. 1 (2014): 171-81.
- Kim, D., G. Pertea, C. Trapnell, H. Pimentel, R. Kelley, and S. L. Salzberg. "Tophat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions." Genome Biol 14, no. 4 (2013): R36.
- Langmead, B., and S. L. Salzberg. "Fast Gapped-Read Alignment with Bowtie 2." Nat Methods 9, no. 4 (2012): 357-9.
- Liao, Y., G. K. Smyth, and W. Shi. "Featurecounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features." Bioinformatics 30, no. 7 (2014): 923-30.
- Butler, A., P. Hoffman, P. Smibert, E. Papalexi, and R. Satija. "Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species." Nat Biotechnol 36, no. 5 (2018): 411-20.
- Becht, E., L. McInnes, J. Healy, C. A. Dutertre, I. W. H. Kwok, L. G. Ng, F. Ginhoux, and E. W. Newell. "Dimensionality Reduction for Visualizing Single-Cell Data Using Umap." Nat Biotechnol (2018).
- Dobin, A., C. A. Davis, F. Schlesinger, J. Drenkow, C. Zaleski, S. Jha, P. Batut, M. Chaisson, and T. R. Gingeras. "Star: Ultrafast Universal Rna-Seq Aligner." Bioinformatics 29, no. 1 (2013): 15-21.
- Kaminow, Benjamin, Dinar Yunusov, and Alexander Dobin. "Starsolo: Accurate, Fast and Versatile Mapping/Quantification of Single-Cell and Single-Nucleus Rna-Seq Data." In BioRxiv, 2021.
- Danecek, P., J. K. Bonfield, J. Liddle, J. Marshall, V. Ohan, M. O. Pollard, A. Whitwham, T. Keane, S. A. McCarthy, R. M. Davies, and H. Li. "Twelve Years of Samtools and Bcftools." Gigascience 10, no. 2 (2021).
- Shen, S., J. W. Park, Z. X. Lu, L. Lin, M. D. Henry, Y. N. Wu, Q. Zhou, and Y. Xing. "Rmats: Robust and Flexible Detection of Differential Alternative Splicing from Replicate Rna-Seq Data." Proc Natl Acad Sci U S A 111, no. 51 (2014): E5593-601.
- Wu, T., E. Hu, S. Xu, M. Chen, P. Guo, Z. Dai, T. Feng, L. Zhou, W. Tang, L. Zhan, X. Fu, S. Liu, X. Bo, and G. Yu. "Clusterprofiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data." Innovation (Camb) 2, no. 3 (2021): 100141.
- Schindelin, J., I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak, and A. Cardona. "Fiji: An Open-Source Platform for Biological-Image Analysis." Nat Methods 9, no. 7 (2012): 676-82.
Figure 1.
Experimental design and EC populations. (A) Kidneys were harvested and dissociated into single-cell suspension before labelling with CD31 antibody to sort endothelial cells into 384-well plates. Single-cell sequencing utilized SmartSeq2 technology and data was subjected to bioinformatic analysis. (B) UMAP of endothelial cells from non-diabetic (Lean) mice were used to identify 13 separate clusters of EC cells annotated as afferent arteriole/arterioles (AA), ascending vasa recta (AVR), proliferating (cycling), descending vasa recta (DVR), efferent arteriole (EA), glomerulus (GEC), lymphatics (LVEC), peritubular capillaries (PCEC), tip cells (tip), and veins (VEC). GEC and PCEC each had two clusters with one cluster displaying high immediate early gene expression (GECIEG-high and PCECIEG-high). (C) UMAPs showing pan EC markers including Pecam1, Cdh5, Tek, Kdr, and Notch4. (D) Dot blot of markers for each EC population.
Figure 1.
Experimental design and EC populations. (A) Kidneys were harvested and dissociated into single-cell suspension before labelling with CD31 antibody to sort endothelial cells into 384-well plates. Single-cell sequencing utilized SmartSeq2 technology and data was subjected to bioinformatic analysis. (B) UMAP of endothelial cells from non-diabetic (Lean) mice were used to identify 13 separate clusters of EC cells annotated as afferent arteriole/arterioles (AA), ascending vasa recta (AVR), proliferating (cycling), descending vasa recta (DVR), efferent arteriole (EA), glomerulus (GEC), lymphatics (LVEC), peritubular capillaries (PCEC), tip cells (tip), and veins (VEC). GEC and PCEC each had two clusters with one cluster displaying high immediate early gene expression (GECIEG-high and PCECIEG-high). (C) UMAPs showing pan EC markers including Pecam1, Cdh5, Tek, Kdr, and Notch4. (D) Dot blot of markers for each EC population.
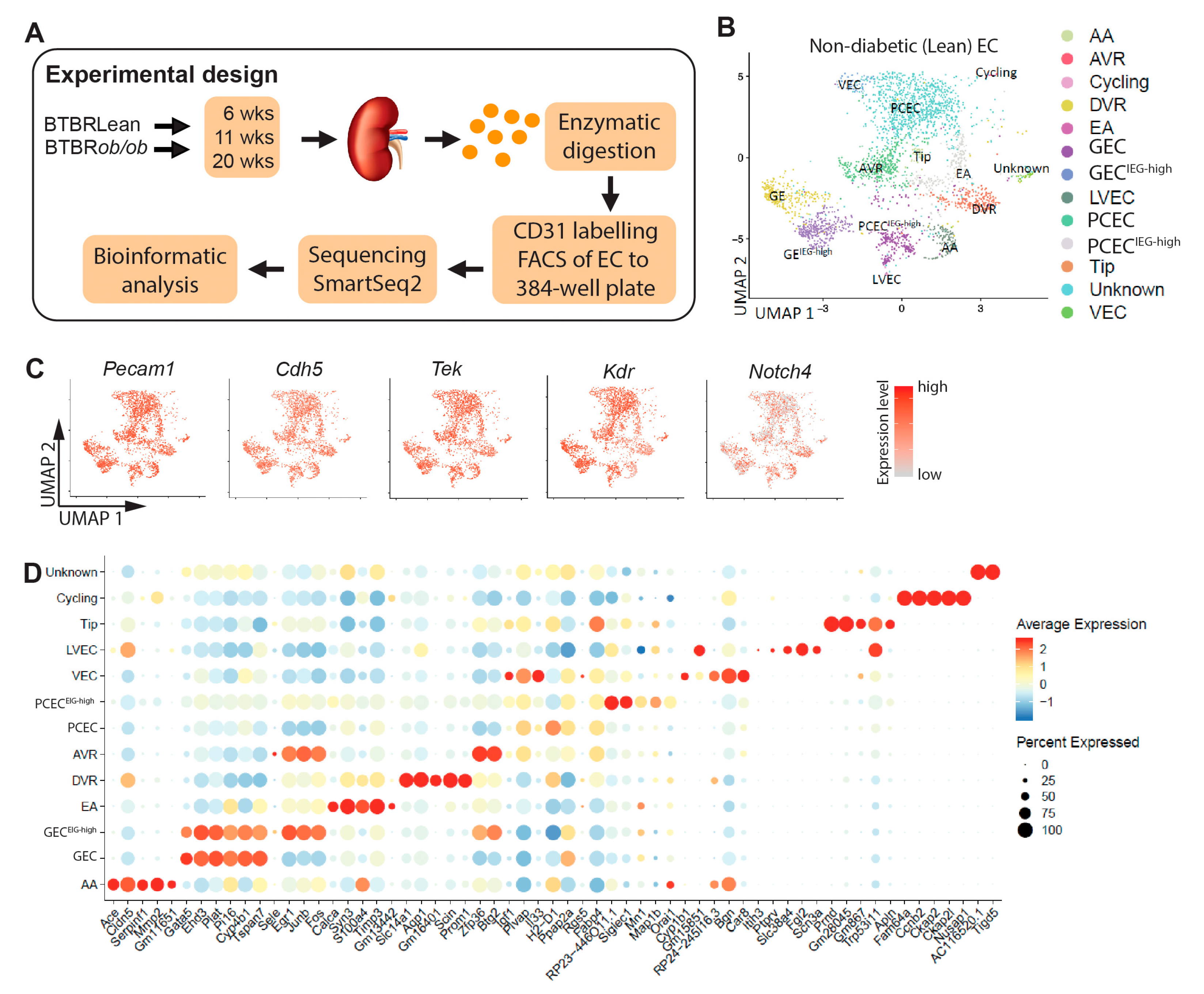
Figure 2.
Heterogeneity of different EC compartments. (A) selection of significant canonical pathways identified by pathway analysis based on DEGs between EC populations in non-diabetic (Lean) mice (complete IPA results shown in Suppl. File S2). Dot blot and schematic representation in EC populations for genes involved in (B) immune zonation, (C) coagulation zonation, (D) vascular tone and calcium homeostasis, and (E) IGF signaling.
Figure 2.
Heterogeneity of different EC compartments. (A) selection of significant canonical pathways identified by pathway analysis based on DEGs between EC populations in non-diabetic (Lean) mice (complete IPA results shown in Suppl. File S2). Dot blot and schematic representation in EC populations for genes involved in (B) immune zonation, (C) coagulation zonation, (D) vascular tone and calcium homeostasis, and (E) IGF signaling.
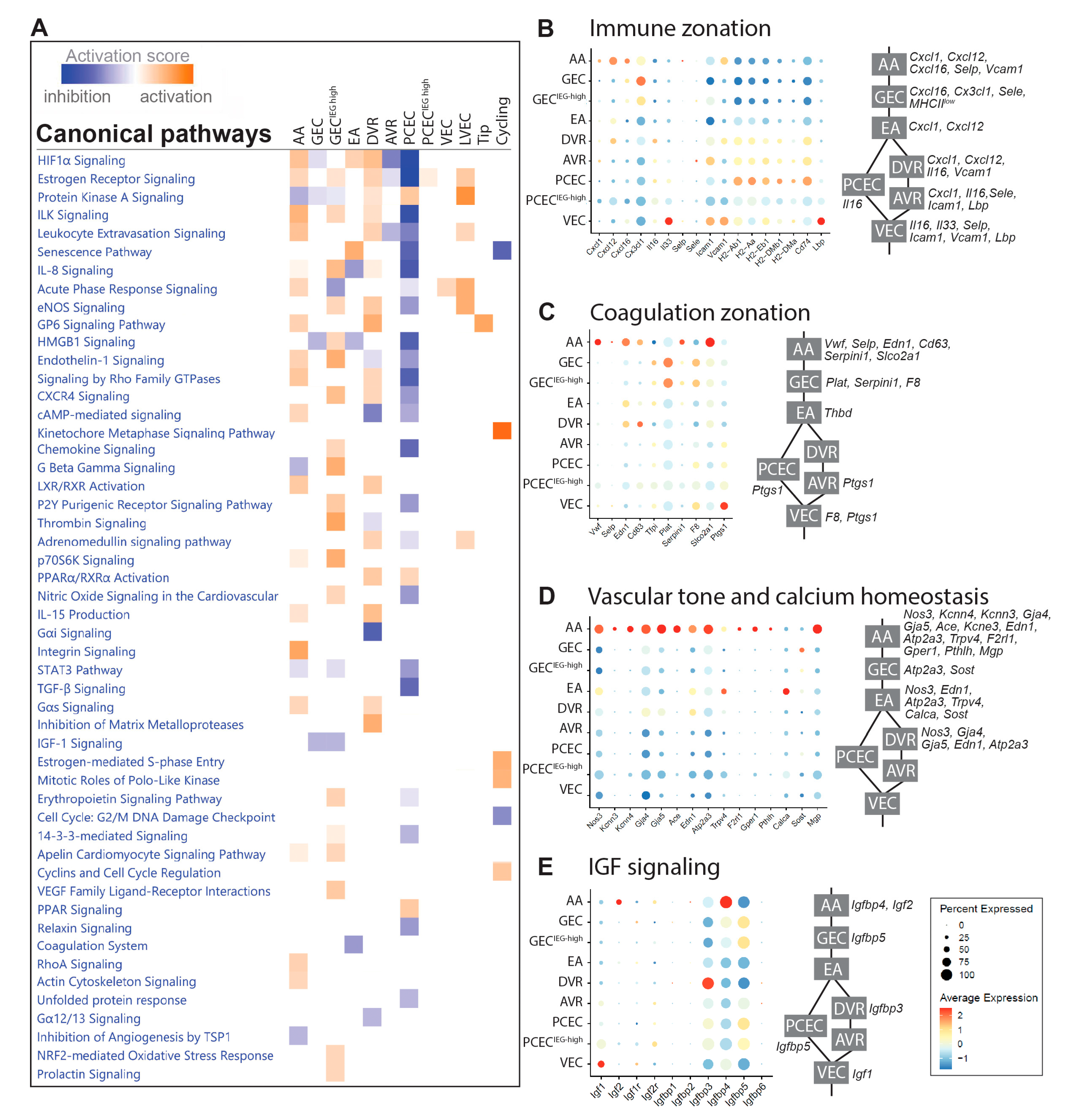
Figure 3.
Renal EC response to DKD progression. (A) UMAP of endothelial cells in non-diabetic (Lean) and diabetic (ob/ob) mice. (B) Schematic diagram of DEGs of different EC populations in non-diabetic and diabetic mice at 6, 11, and 20 weeks. (C, D) Venn diagram of DEGs for PCEC and GEC, respectively, comparing non-diabetic (Lean) with diabetic (ob/ob) mice at different timepoints. Expression heatmaps (blue, low; red, high) of the relative expression is shown for each comparison.
Figure 3.
Renal EC response to DKD progression. (A) UMAP of endothelial cells in non-diabetic (Lean) and diabetic (ob/ob) mice. (B) Schematic diagram of DEGs of different EC populations in non-diabetic and diabetic mice at 6, 11, and 20 weeks. (C, D) Venn diagram of DEGs for PCEC and GEC, respectively, comparing non-diabetic (Lean) with diabetic (ob/ob) mice at different timepoints. Expression heatmaps (blue, low; red, high) of the relative expression is shown for each comparison.
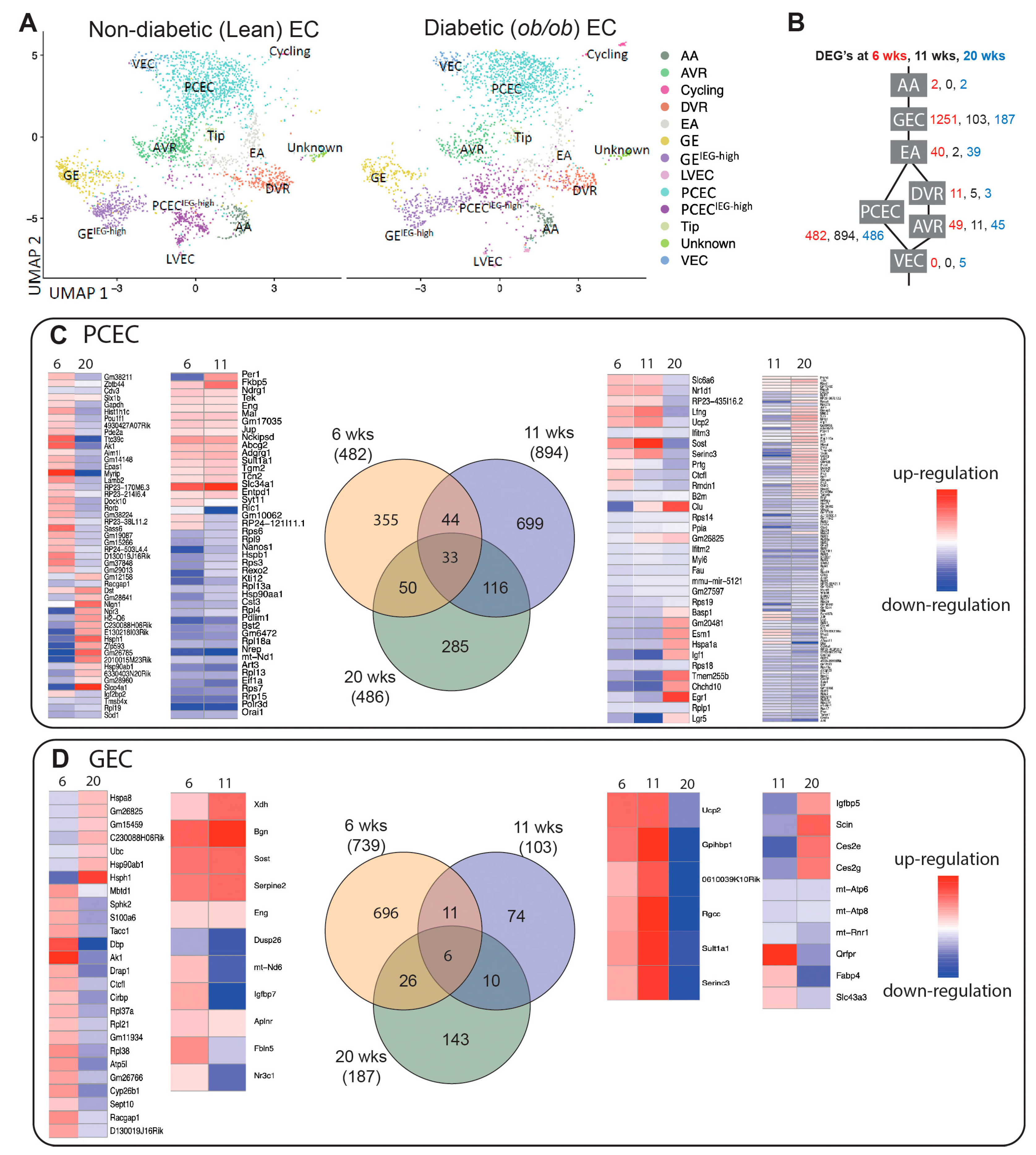
Figure 4.
Pathway analysis (IPA) of PCEC based on DEGs comparing non-diabetic (Lean) and diabetic (ob/ob) mice. (A) selection of significant canonical pathways, downstream effects, and upstream regulators identified by IPA for PCEC (the complete IPA results shown in Suppl. File S2). (B-D) Graphical summaries of pathway analysis showing pathway activation (red) or pathway inhibition (blue) in diabetic PCEC in 6-, 11-, and 20-week-old mice, respectively.
Figure 4.
Pathway analysis (IPA) of PCEC based on DEGs comparing non-diabetic (Lean) and diabetic (ob/ob) mice. (A) selection of significant canonical pathways, downstream effects, and upstream regulators identified by IPA for PCEC (the complete IPA results shown in Suppl. File S2). (B-D) Graphical summaries of pathway analysis showing pathway activation (red) or pathway inhibition (blue) in diabetic PCEC in 6-, 11-, and 20-week-old mice, respectively.
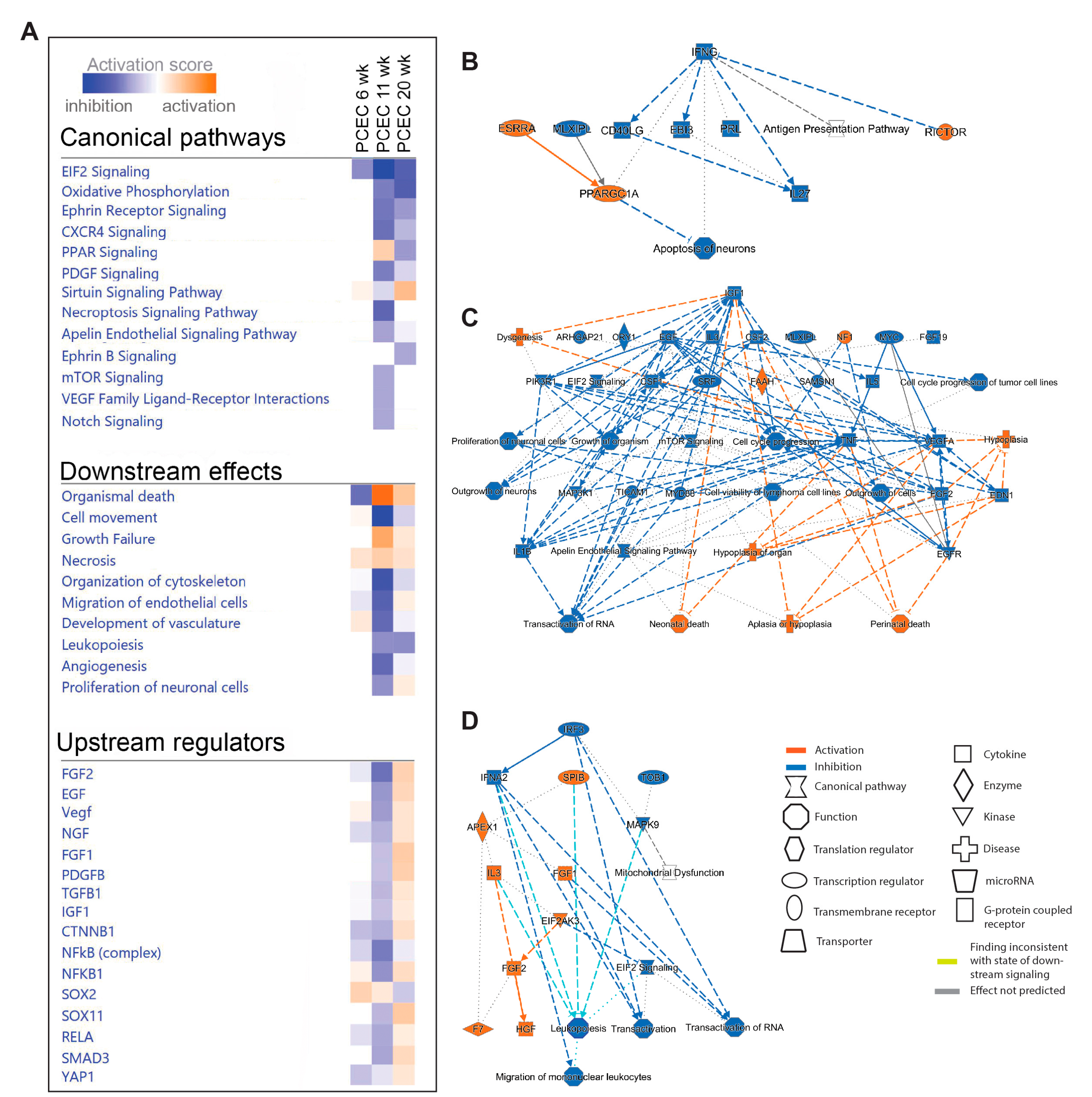
Figure 5.
Pathway analysis (IPA) of GEC based on DEGs comparing non-diabetic (Lean) and diabetic (ob/ob) mice. (A) selection of significant canonical pathways, downstream effects, and upstream regulators identified by IPA for GEC. The complete IPA results can be found in Suppl. File S2. (B, C) Graphical summaries of the pathway analysis showing activation (red) or inhibition (blue) in diabetic GEC in 6- and 20-week-old mice, respectively. No significant pathway regulation was found in 11-week-old mice.
Figure 5.
Pathway analysis (IPA) of GEC based on DEGs comparing non-diabetic (Lean) and diabetic (ob/ob) mice. (A) selection of significant canonical pathways, downstream effects, and upstream regulators identified by IPA for GEC. The complete IPA results can be found in Suppl. File S2. (B, C) Graphical summaries of the pathway analysis showing activation (red) or inhibition (blue) in diabetic GEC in 6- and 20-week-old mice, respectively. No significant pathway regulation was found in 11-week-old mice.
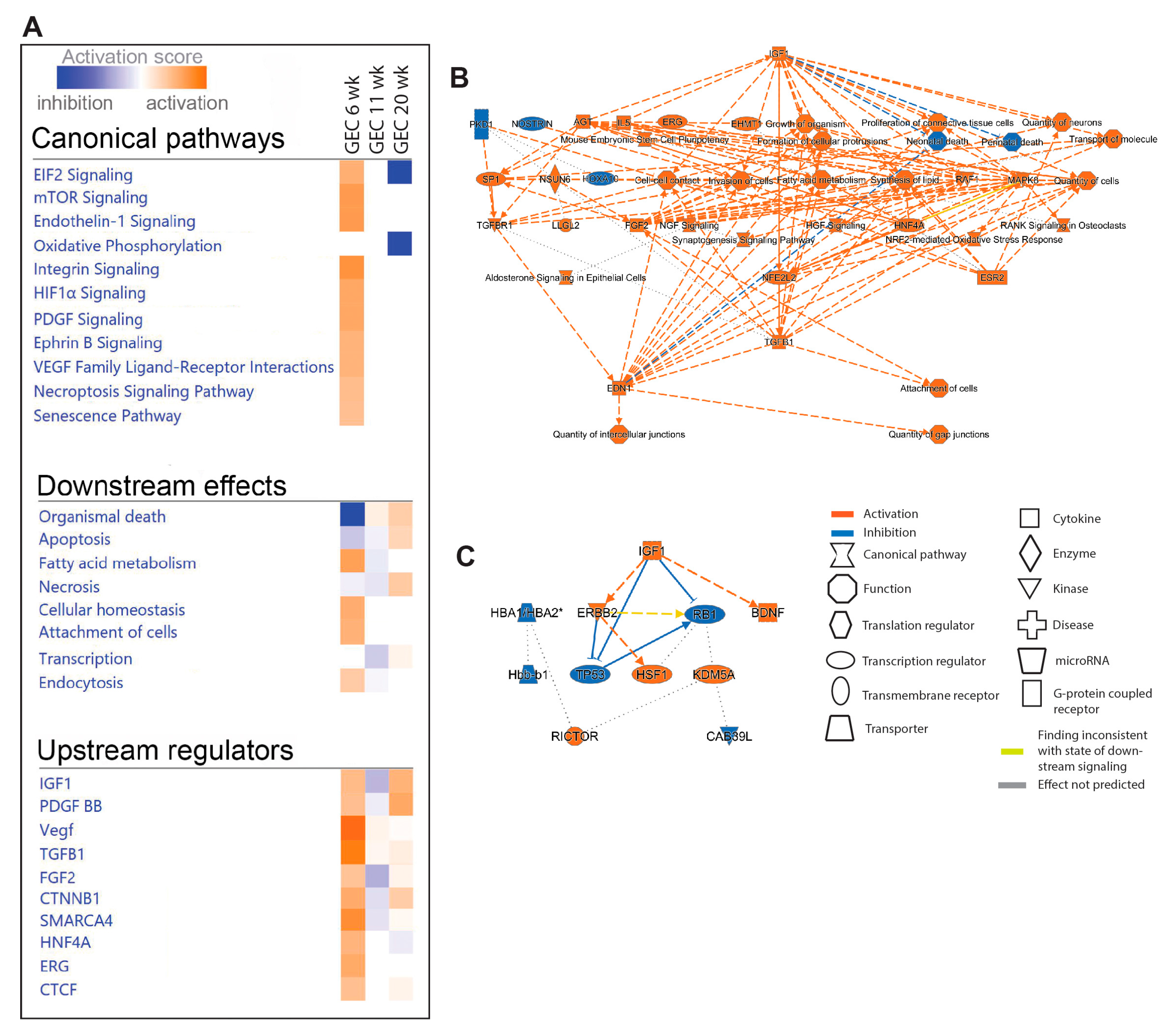
Figure 6.
Identification of DSEs in DKD progression. (A) Schematic diagram of 5 different modes of AS distinguished by rMATS. (B) Number of significant DSEs identified in comparison between BTBRLean and BTBRob/ob GEC and PCEC at different timepoints. (C) Venn diagram of DSE genes for PCEC and GEC, respectively, displaying number of overlapping or distinct DSE genes at different timepoints. Exemplary Sashimi plots visualizing read counts of splice junctions and inclusion levels of exons at selected genomic locations for Lias and Chek2 in GEC (D) and Tbc1d31 in PCEC (E) in BTBRLean and BTBRob/ob mice.
Figure 6.
Identification of DSEs in DKD progression. (A) Schematic diagram of 5 different modes of AS distinguished by rMATS. (B) Number of significant DSEs identified in comparison between BTBRLean and BTBRob/ob GEC and PCEC at different timepoints. (C) Venn diagram of DSE genes for PCEC and GEC, respectively, displaying number of overlapping or distinct DSE genes at different timepoints. Exemplary Sashimi plots visualizing read counts of splice junctions and inclusion levels of exons at selected genomic locations for Lias and Chek2 in GEC (D) and Tbc1d31 in PCEC (E) in BTBRLean and BTBRob/ob mice.
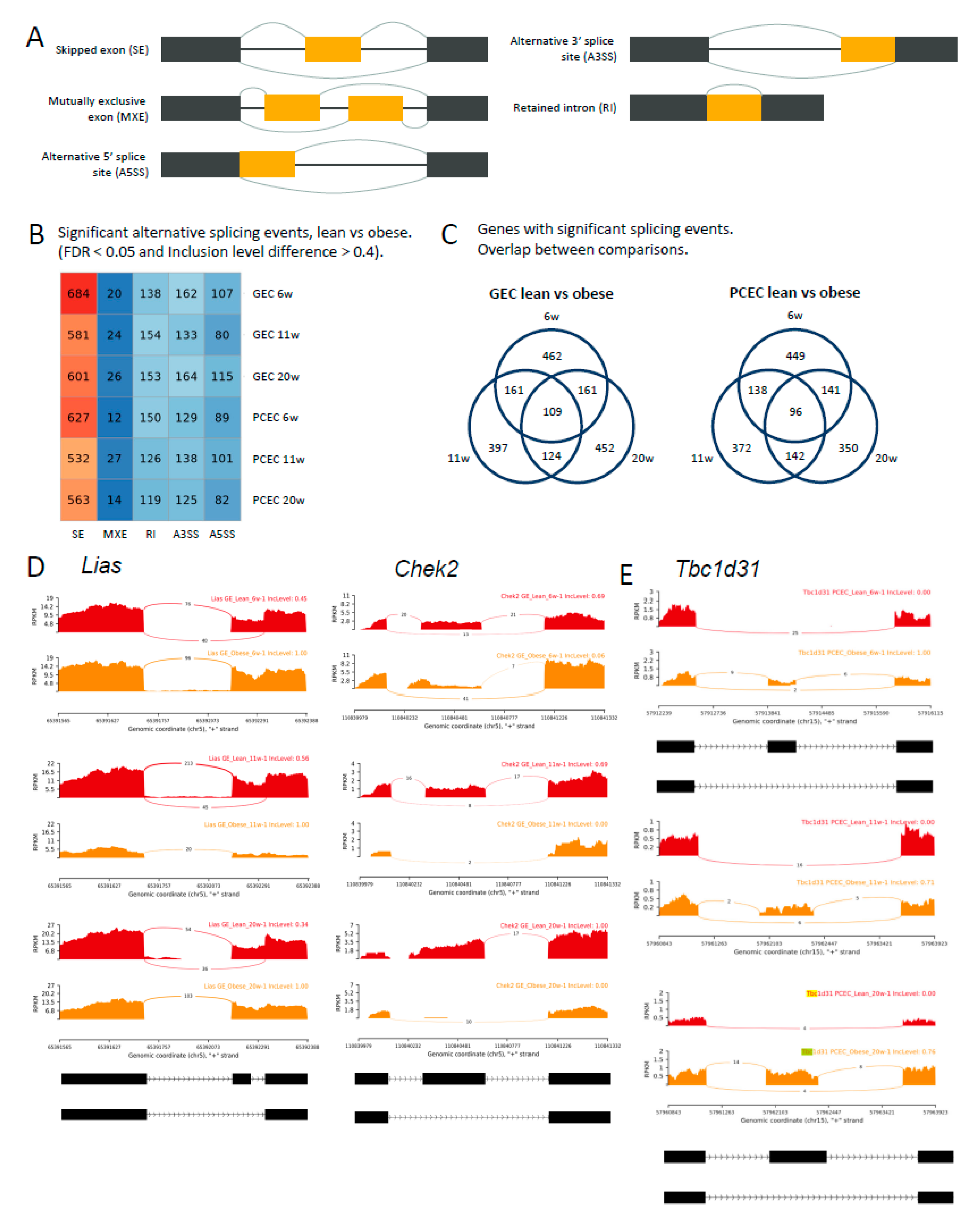
Figure 7.
Comparison between DSE genes and DEGs. (A) Venn diagram of DSE genes and DEGs for PCEC and GEC, displaying number of distinct genes and overlapping genes that are both differentially expressed and alternatively spliced. GO-term enrichment analysis for DSE genes (B) and DEGs (C) displaying the top 10 significant GOBPs at three timepoints in GEC and PCEC, respectively. Scaled bar representing -log10(adjusted p-value). (D) Proposed working model displaying the link between DEGs and DSEs and their divergently regulated biological processes.
Figure 7.
Comparison between DSE genes and DEGs. (A) Venn diagram of DSE genes and DEGs for PCEC and GEC, displaying number of distinct genes and overlapping genes that are both differentially expressed and alternatively spliced. GO-term enrichment analysis for DSE genes (B) and DEGs (C) displaying the top 10 significant GOBPs at three timepoints in GEC and PCEC, respectively. Scaled bar representing -log10(adjusted p-value). (D) Proposed working model displaying the link between DEGs and DSEs and their divergently regulated biological processes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



