
Submitted:
24 March 2024
Posted:
25 March 2024
You are already at the latest version
Abstract
Due to its large hole mobility, Organic Rubrene (C42H28) has attracted research questions on its applications in electronic devices. In this work, extensive first-principles calculations were made to predict some temperature and doping dependent properties of Organic Semi- conductor Rubrene. We use Density Functional Theory (DFT) to investigate the electronic structure, elastic and transport properties of the Orthorhombic phase of the Rubrene compound. The calcu- lated band structure shows the Orthorhombic phase has a direct bandgap of 1.26 eV. From the Vickers hardness (1.080 GPa), our calculations show that Orthorhombic Rubrene is not a super hard material and can find useful application as a flexible semicon- ductor. The calculated transport Inverse Effective Mass and Elec- tronic Fitness Function show that the Orthorhombic Rubrene Crystal Structure is a p-type thermoelectric material at high temperatures.
Keywords:
Electronic Structure
; DFT
; Thermoelectric
; Organic Semiconductor Rubrene
; Transport Properties
1. Introduction
Organic Semiconductors (OS) contain pi-bonded molecules which are made up of carbon and hydrogen atoms [1] and are bound together by van der Waals interactions. Therefore, OS have much lower hole mobility than inorganic semiconductors due to significantly reduced covalent coupling generated by the van der Waals contact [2]. Nevertheless, interest and research in OS have grown in recent years. They are seen as potential candidates for the future generation of electronic gadgets as they offer the possibility to be used for the manufacturing of low-cost and lightweight devices that may be huge in size and flexible [3]. The OS earliest application as a photo-conductive coating in laser printers [4] paved the way to other successful opto-electronic devices, such as Organic Solar Cells (OSCs) and Organic Light-Emitting Diodes (OLEDs) [5]. Due to their improved transport properties [6], OS may soon substitute inorganic Semiconductors as primary material for high-speed electronics [6] devices. Graphene is an excellent example, for its potential to replace Inorganic Semiconductors such as Silicon and Gallium-Arsenide in the near future.
In spite their promising future, there is still room for improvement in the characterization of OS, for their rational design and optimization of their properties. These properties are completely determined by the content and arrangements of the molecules making up the OS. This stems from the fact that these molecules are experimentally created via synthetic Chemistry. Besides, OS have an extremely low dielectric constant of approximately 3–5 [6]. This yields a large exciton binding energy and most often poor electrical characteristics. Nevertheless, recent breakthroughs have enabled to overcome many of the obstacles, providing new fundamental insight into the main factors governing the performance of OS [6].
Despite their low mobility, Organic Semiconductors still find application in OLEDs and solar cells. Organic Light-Emitting Diodes are now widely employed in commercial displays [7]. They benefit from having a high contrast, brightness, and lower power usage. As mentioned earlier, despite the limited mobility and high exciton binding energy, OS are also used in OLED televisions and smartphone displays [8]. These characteristics make OS more appropriate for emissive devices than absorptive ones. The OS are primarily, electrical insulators that can become semiconductors when charges are introduced from valence to conduction band through photo-excitation or doping [9]. In some cases, OS exist as amorphous thin films or molecular solids [10]. Due to remarkable charge carrier mobility and luminous efficiency [11], Rubrene C42H28 (5,6,11,12-tetraphenylnaphthacene) is also a typical OS material that is useful in Organic Thin-Film Transistors (OTFTs), OLEDs and OSCs. Rubrene is also commonly used in OLEDs as yellow fluorescent dye to boost luminous efficiency, stability, and durability [12].
The Rubrene structure is essentially a four-winged tetracene molecule [13] which belongs to the Polycyclic Aromatic Hydrocarbon family. Organic molecular crystals are potential semiconductor materials for LEDs, Solar Cells, and electrical applications. Organic crystals differ from inorganic solids with covalent or ionic bondings in terms of mobility and photo-excitement due to weak intermolecular interactions [14]. Rubrene has the highest carrier mobility for holes 40 cm2/(V.s) and a high photoconductivity which make it an exceptional OS [15]. These properties are employed in applications of Rubrene in luminous and flexible devices. Rubrene crystals have closed molecular packing that is oriented in a certain direction which is known as the "b" direction; this implies that transport in a Rubrene crystal is one-dimensional in nature [16]. Growing high crystalline Rubrene thin films is expensive and this impedes its practical applications [17]. Several techniques have been developed over the years to grow Rubrene thin films. Rubrene thin films can be grown in amorphous, polycrystalline, or single crystal forms utilizing various techniques. Some of the experimental techniques include pulsed laser evaporation of solidified solutions [18] and physical vapor transport [19].
In recent years, the increase in demand for new innovations in electronics has grown rapidly, hence the need to investigate the elastic, mechanical, temperature and doping dependent properties of organic semiconductor Rubrene. These findings will enable the creation of entirely new design rules with improved durability and precise parameter control.
Therefore, for the first time we attempt to calculate and investigate the temperature and doping dependent properties orthorombic Rubrene using density functional theory (DFT) calculations. In this research, we represent the structural, electronic, elastic and mechanical (elastic constants, shear, bulk and young modulus, Pugh’s ratio and Vickers hardness) properties. Also, we intend to record the values for Pugh’s ratio and Vickers hardness for the first time.
2. Method and Computational Procedure
Structural optimization of the initial atomic positions and lattice structure of Orthorhombic Rubrene were performed after obtaining initial parameters from the study of Reyes-Martinez [20]. The electronic cutoff for wavefunctions, the Monkhorst-Pack k-point grid [21] and the lattice parameters optimization were carried out with a convergence criteria of 1mRy per atom. The atomic positions were relaxed to obtain the ground state structure of Orthorhombic Rubrene. The structural optimization and electronic structure were carried out with the Perdew-Burke-Enzenhoff [22,23,24] exchange correlation functional of the density functional theory, DFT [25,26] as implemented in the Quantum Espresso Package [27,28] using a plane-wave cut-off of 45 Ry and a 2 x 6 x 4 Monkhorst-Pack grid with a Gaussian broadening of 0.01 Ry. The thermo_pw code across a strain range of -0.0075 to +0.0075 at 0.005 steps was used to calculate the elastic constants using the stress-strain technique. Ultrasoft pseudopotentials [29] as obtained from QE Pslibrary [30] were used to treat the valence and core electrons interactions. To calculate the elastic properties of Orthorhombic Rubrene via the stress-strain method, the stiffness matrix Cij corresponding to nine independent elastic constants were obtained for Rubrene crystal structure with Laue class D_2h (mmm), while ions were relaxed in each deformation. Furthermore, mechanical properties of Rubrene were obtained from the elastic constants such as, the internal strain and load-deflection [31]. The elastic constants were evaluated for small strains (∈) by applying Hooke’s law and its energy E.
In the determination of the transport properties, a plane-wave calculation was performed to relax the system followed by a non-self consistence calculation with a dense 40000 k-points. In the G-vectors, 311243 dense grids were used in the Fast Fourier Transform dimensions, while the starting wave-functions were randomized with 784 atomic wave-functions. The results obtained from the Boltztrap calculations were then used to obtain the transport effective mass, and the electronic fitness function through the TransM code [32]. These calculations help screen semiconductors as either thermoelectric or in relation to conductivity.
3. Results and Discussion
3.1. Structural Properties of Orthorhombic Rubrene
As shown in Table 1, a lattice constant of 26.79 (50.8257 a.u.) was obtained and it is in good agreement with the experimental value of Ref. [20]. Table 1 shows the optimized lattice parameters a = 26.7903 Å, b = 7.17001 Å, and c = 14.2112 Å for Orthorhombic Rubrene. These values are in close agreement with those reported in theoretical studies Ref. [33] and Ref. [34].

Table 1.
Calculated and measured lattice constant of orthorhombic Rubrene.
| Reference | Lattice Constants | Method/Theory | ||
|---|---|---|---|---|
| a () | b () | c () | ||
| Present work | 26.79 | 7.17 | 14.21 | GGA |
| Ref. [33] | 26.86 | 7.19 | 14.43 | GGA |
| Ref. [34] | 26.660 | 7.142 | 14.025 | vdW-DFT |
| Ref. [34] | 26.965 | 7.206 | 14.442 | Experiment at 294K |
| Ref. [35] | 26.789 | 7.170 | 14.211 | Experiment at 100K |
| Ref. [36] | 14.1289 | 7.1455 | 26.7450 | GGA |
The relaxed structure of orthorhombic Rubrene (Figure 1b) shows the compound to be a polymorph with the Cmca space group. Whenever Rubrene molecules unite to form Orthorhombic crystals, the resulting molecules have a centrosymmetric structure with 2/m symmetry. The cell parameters of Orthorhombic Rubrene crystal (as reported in Table 1) shows the molecules are organized in a herringbone packing pattern with nearly complete - stacking in the b direction. This is responsible for its large charge-carrier mobility, which has been affirmed in several pieces of research [16,34]. The mobility of charge carriers in Orthorhombic monocrystals can approach 40 cm2/Vs, which is equivalent to that of amorphous silicon [37,38]. As seen in most organic crystals, Rubrene’s electronic transport is very anisotropic. High mobility values can only be obtained along the lattice’s b axis. The features obtained from Figure 1b agrees reasonably well with the results of Ref. [34] and [39]. The structural properties obtained in this work agree with previous theoretical and experimental values.
3.2. Electronic Properties
Figure 2 depict the density of state and band structure, as well as the state and behaviour of electrons in an Orthorhombic Rubrene crystal. The numerical values for the direct bandgap between the Highest Occupied Molecular Orbit (HOMO) and the Lowest Unoccupied Molecular Orbit (LUMO) are shown in Table 2. The highest valence band results from the highest occupied molecular orbital (HOMO), whereas the lowest conduction band results from the lowest unoccupied molecular orbital (LUMO). Generally, an energetic bandgap of ∼1-5eV has been recorded for OS such as Orthorhombic Rubrene [33], as they tend to have a diminishing density of states around the Fermi energy. This is proportional to the size of the electrical gap, which is fairly considerable. This reduces the density of thermally generated charge carriers in pure organic crystals, such as silicon.
Table 2.
Comparison of band gaps (HOMO - LUMO) of orthorhombic Rubrene obtained in this work, experiments and previous calculations.
Table 2.
Comparison of band gaps (HOMO - LUMO) of orthorhombic Rubrene obtained in this work, experiments and previous calculations.
| Reference | Band Gap | Theory | |
|---|---|---|---|
| Nature | Energy (ev) | ||
| Present work | Direct | 1.26 | PBE |
| Ref. [36] | Direct | 1.13 | PBE |
| Ref. [40] | Direct | 1.357 | B3LYP 6-311G |
| Ref. [41] | Direct | 2.50 | B3LYP/6-311G(d,p) |
As shown in the band structure (Figure 2), the bottom of the conduction band (CB) and the top of the valence band (VB) occur at the same momentum value. This implies that the band structure of organic semiconductor Rubrene has a direct bandgap, as can be observed with some other organic semiconductors. The direct bandgap of 1.26 eV obtained within PBE agrees with the calculation of Ref [36] using PBE also with a direct bandgap of 1.13 eV Ref. [41] and [42] found a greater disparity between HOMO and LUMO. Their calculated band gap is 2.50 eV and 2.60 eV respectively at B3LYP/6-311G(d,p) level, which differs from other calculations. So far, there is no theoretical literature report on the consequential variation in the HOMO-LUMO gap [43]. The highest VB’s structural characteristic controls hole transport behaviour. The band splitting of the VB of orthorhombic Rubrene is fairly minimal. Pressure increases intermolecular interaction, resulting in increased mobility [44]. High mobilities are only for holes, and electron mobilities are several orders of magnitude lower [42,45]. Rubrene is a p-type material, as are the vast majority of organic semiconductors [42].
The number of possible electrons (or hole) states per volume at a given energy is given by the density of states as shown in Figure 2. The density of states distribution at the top of VB is relatively smooth in the orthorhombic Rubrene.
The high-symmetry points’ reciprocal coordinates are = (0, 0, 0), X = (0, 0, 0.5), S = (0, 0.5, 0.5), Y = (0, 0.5, 0), = (0, 0, 0), Z = (-0.5, 0, 0), U = (-0.5, 0, 0.5), R = (-0.5, 0.5, 0.5), T = (-0.5, 0.5, 0), and Z = (-0.5, 0, 0). Each band in the band structures occurs in pairs because the orthorhombic Rubrene crystal structure comprises two molecules in a unit cell. Figure 2 depicts the band structure along the k-path – X – S – Y – – Z – U -R – T - Z, where -Z corresponds to the `a’ crystal axis and -Y to the `b’ axis in real space [16].
3.3. Elastic Properties
The values of the computed Cij show that the Born-Huang Stability criteria have been fully satisfied [46] which proves Rubrene to be mechanically stable. The estimated bulk modulus B, shear modulus G, Young’s modulus E, Poisson ratio, Pugh ratio, and Vickers Hardness using the Voigt-Reuss-Hill approximation are shown in Table 4 [47]. According to [48], a material is considered to be ductile if its bulk to shear modulus ratio, B/G, is more than 1.75; otherwise, it is brittle. The obtained Pugh ratio is 0.74667, which shows Rubrene to be brittle than ductile. This property implies the ability to break with minimal elastic deformation when stressed and no considerable plastic deformation. Brittle materials have more strength than ductile materials. Brittle materials are more resistant to compression. Even high-strength brittle materials absorb relatively little energy before breakage. This feature proves the ability of orthorhombic Rubrene as an organic semiconductor useful in redefining the future of flexible and stretchable electronics. Furthermore, the Poisson’s ratio can also prove the ductility of a material if the ratio is greater than 0.26 and brittleness is less than 0.26. The obtained Poisson ratio is 0.03292, which agrees with the Pugh ratio can classify orthorhombic Rubrene as brittle.
Table 3.
Comparison of independent elastic constants of orthorhombic Rubrene obtained in this work, experiment and previous calculations.
Table 3.
Comparison of independent elastic constants of orthorhombic Rubrene obtained in this work, experiment and previous calculations.
| Reference | This work | Ref. [20] | Ref. [34] | Ref. [34] |
|---|---|---|---|---|
| Method | PBE | AIREBO | vdw-DFT | Experiment |
| C11 (GPa) | 18.8 | 15.54 | 25.31 | 18.48 |
| C12 (GPa) | -8.7 | 1.08 | 6.94 | 2.63 |
| C13 (GPa) | 1.6 | 2.08 | 6.78 | 7.68 |
| C22 (GPa) | 13.6 | 17.85 | 16.99 | 13.39 |
| C23 (GPa) | 9.4 | 10.82 | 10.53 | 7.77 |
| C33 (GPa) | 14.6 | 13.29 | 13.94 | 14.32 |
| C44 (GPa) | 7.2 | 2.03 | 6.66 | 6.46 |
| C55 (GPa) | 13.2 | 1.97 | 4.41 | 2.8 |
| C66 (GPa) | 6.5 | 3.36 | 3.67 | 6.8 |
The Vickers hardness Hv is obtained as shown in Table 4. The hardness of a material measures its resistance to plastic deformation produced by applied forces. The result helps characterise the elastic and plastic properties of a solid. The predicted Vickers hardness Hv of 1.080 GPa was obtained for orthorhombic Rubrene using Chen’s model [49]. The result shows that orthorhombic Rubrene is far from hard and cannot be classified as superhard.
Elastic Constants (GPa) = Calculated elasticity tensor for orthorhombic Rubrene
Also, the calculated reduced elastic constants and anisotropy ratio (C22/C33) of orthorhombic Rubrene obtained in this work is shown in Table 5. The values of reduced constants C22, C33, and the anisotropy ratio (C22/C33) are in close agreement with the result of Ref [34] from the experimental and vdw-DFT method. These findings may be beneficial not only for studying the strain effect on carrier mobility [16], but also for Rubrene’s actual use as a flexible electrical device [34].
Its directional elastic properties need to be analysed and visualised to better understand an anisotropic material such as Rubrene. This includes the young modulus, linear compressibility, shear modulus and Poisson ratio as shown in Figure 5. The debye temperature and average Debye sound velocity recorded in this research for orthorhombic Rubrene are 331.008K and 2384.484m/s, respectively (Table 4).
Table 4.
Calculated Voigt-Reuss-Hill approximation moduli for orthorhombic Rubrene in this work
| Method | PBE |
|---|---|
| Bulk Modulus (GPa) | 4.163 |
| Shear Modulus (GPa) | 11.519 |
| Young Modulus (GPa) | 5.576 |
| Poisson Ratio | 0.03292 |
| Pugh Ratio (B/G) | 0.74667 |
| Vickers Hardness (GPa) | 1.08 |
| Average Debye sound velocity ( m/s ) | 2384.484 |
| Debye temperature (K) | 331.008 |
Table 5.
Calculated reduced elastic constants and anisotropy ratio (C22/C33) of orthorhombic Rubrene obtained in this work, experiments and previous calculations.
Table 5.
Calculated reduced elastic constants and anisotropy ratio (C22/C33) of orthorhombic Rubrene obtained in this work, experiments and previous calculations.
| Reference | Method | C22(GPa) | C33 (GPa) | Anisotropy ratio (C22/C33) |
|---|---|---|---|---|
| This work | PBE | 13.6 | 14.6 | 0.93 |
| [34] | vdw-DFT | 15.08 | 12.12 | 1.24 |
| [34] | Experiment | 13.02 | 11.13 | 1.17 |
Young modulus is a mechanical property that measures the tensile/rigidity or stiffness of a material when the force is applied. It implies the ratio of the tensile stress to the proportional deformation/tensile strain.
Where tensile stress is the force per unit area and tensile strain is extension per unit length. The young modulus reported for orthorhombic Rubrene in this work is 55.76 kbar which shows Rubrene as a non-rigid material (Figure 4a).
Linear compressibility or the bulk modulus helps describe a material’s behaviour when pressure is applied, which can be either negative or positive. The 2D and 3D surface plot (Figure 4b and Figure 5b) shows the positive values of linear compressibility plotted in green and the negative value in red. The result indicates that orthorhombic Rubrene crystal structure exhibits negative linear compressibility.
Figure 4.
(a)Young’s Modulus, (b)Linear Compressibility, (c)Shear Modulus, and (d)Poisson Ratio’s in 2D showing its directional planes
Figure 4.
(a)Young’s Modulus, (b)Linear Compressibility, (c)Shear Modulus, and (d)Poisson Ratio’s in 2D showing its directional planes
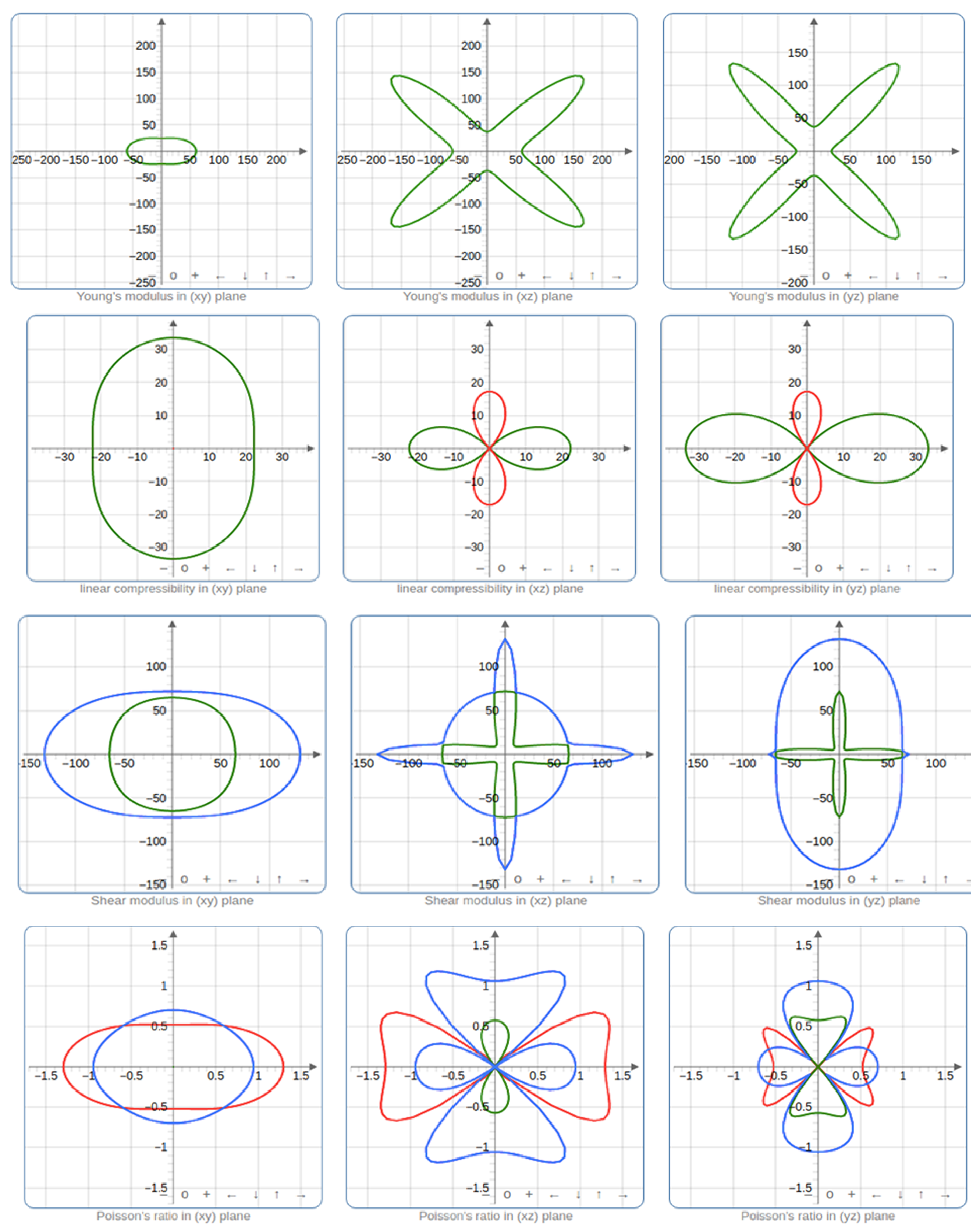
Figure 5.
(a)Young’s Modulus, (b)Linear Compressibility, (c)Shear Modulus, and (d)Poisson Ratio’s in 3D showing its directional planes
Figure 5.
(a)Young’s Modulus, (b)Linear Compressibility, (c)Shear Modulus, and (d)Poisson Ratio’s in 3D showing its directional planes
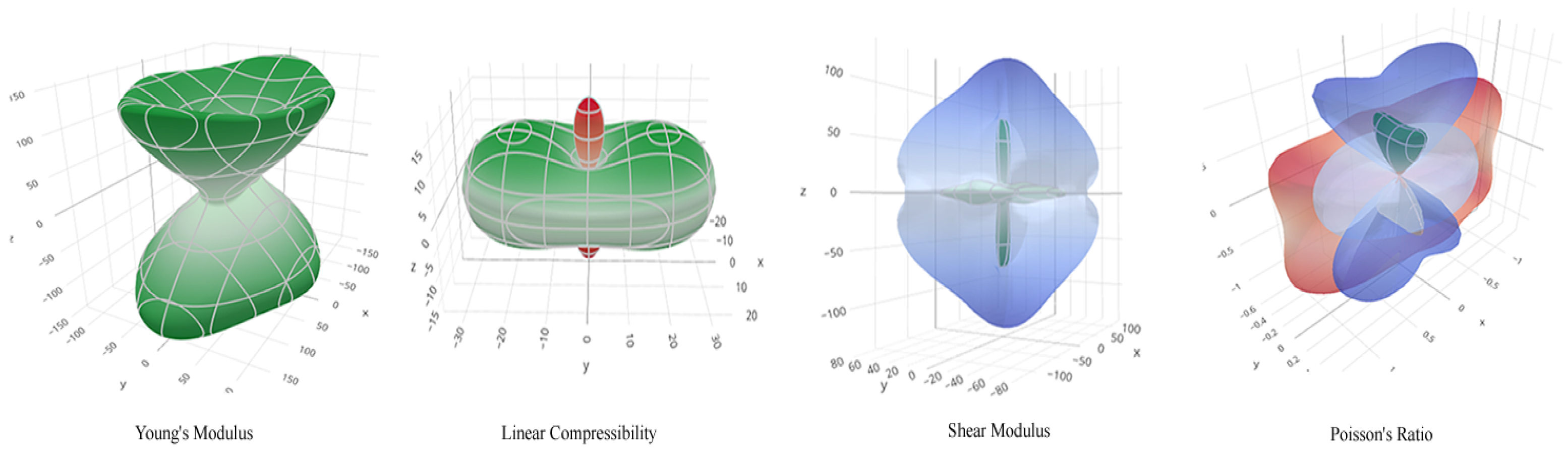
Shear modulus is the ratio of shear stress to the shear strain. It gives information on how resistant a material is to deformations as shown in Figure 4c and Figure 5c. The 2D and 3D plots of the Poisson ratio Figure 4d and Figure 5d showed the lateral strain and the longitudinal strain on orthorhombic Rubrene. Poisson ratio for elastic materials is the ratio of the lateral strain and longitudinal strain, which gives information on how materials deform under loading. However, the calculated Poisson ratio value is 0.03292, which lies between -1 to 0.5. The result showed the pristine form of orthorhombic Rubrene as almost perfectly incompressible as there is little or no transverse deformation when axial strain is applied.
3.4. Temperature and Doping Dependent Properties
To address the thermoelectric properties, the electronic fitness test is crucial as it evaluates the thermopower for arbitrary band structures and conductivity [32]. Suitable thermoelectric materials, in general, have intricate electronic structures that aren’t described by a simple parabolic band [32]. Therefore, electronic fitness function calculation is necessary to identify such materials and address the (Conductivity) and S (Seebeck coefficient) conflict.
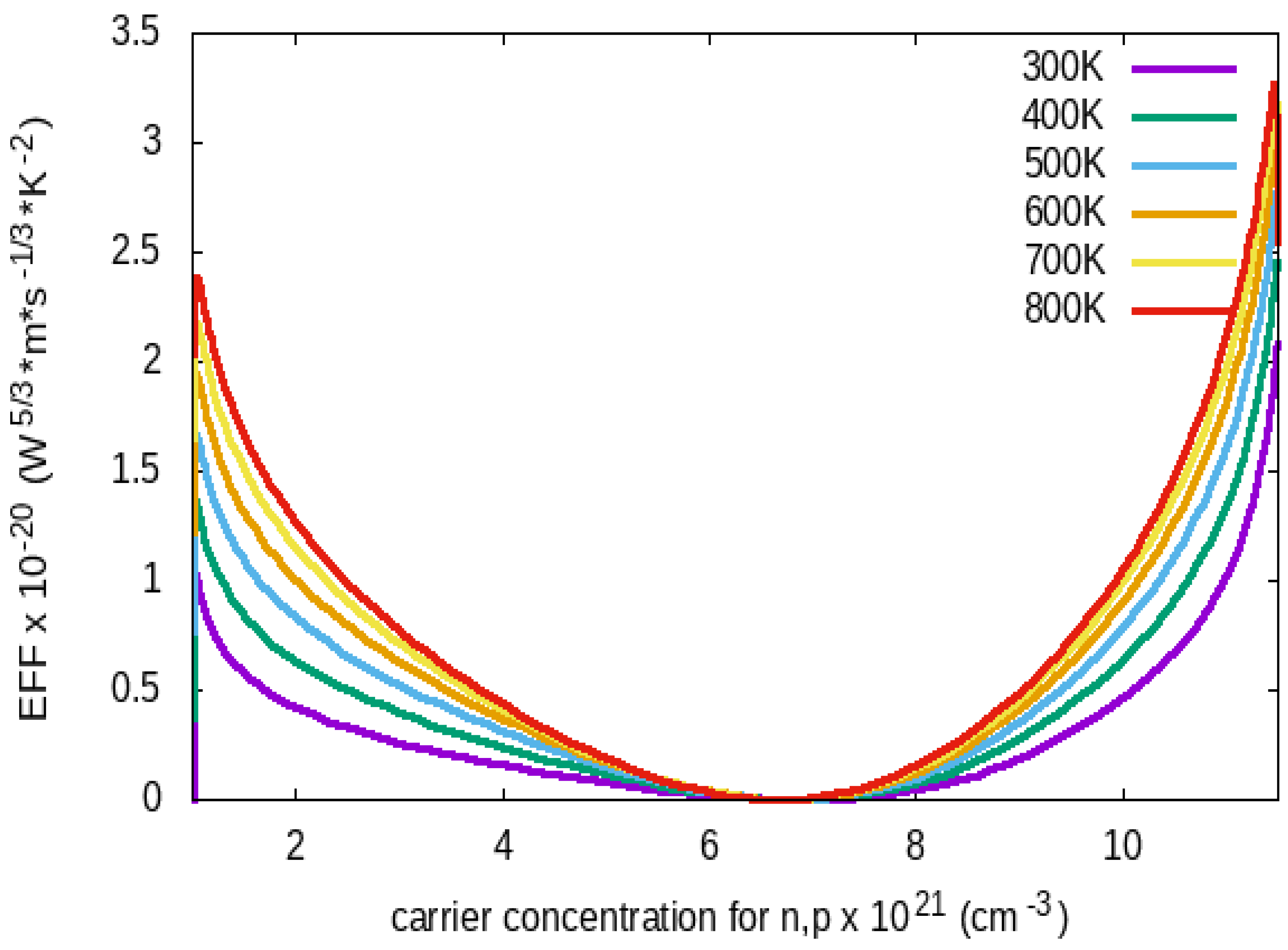
The Electronic Fitness Function (EFF) displayed in Figure 6 shows an increase with temperature for both concentrations, which is caused by the rising temperature and which improves its thermoelectric performance. There is a dip at carrier concentration 6.5 x 1021 for the hole concentration and reaches peak at 2 x 1021 and 1.1 x 1022. This work also predicted that orthorhombic Rubrene has a high electronic fitness function at about 500K. Organic semiconductors like Rubrene are mostly p-type semiconductors.
The structure for the EFF in Figure 6 shows larger p-type EFF values at high doping levels. This is attributed to the secondary conduction band contribution in Rubrene. Furthermore, as the temperature rises, it exhibits improved p-type performance. Valley anisotropy, higher band degeneracy, and multiband contributions in valence bands at higher energies, particularly at high doping levels, result in larger EFF in p-type materials. The features observed from these results show orthorhombic rubrene as a promising organic material for thermoelectric applications.
Figure 7 depicts the transport inverse effective mass as a function of carrier concentrations ranging from 300K to 800K. The result shows it exhibits a light, effective mass of holes. This agrees with the experiments performed in Ref [50].
Figure 7.
Inverse Effective Mass as a function of the Carrier Concentrations from 300K to 800K
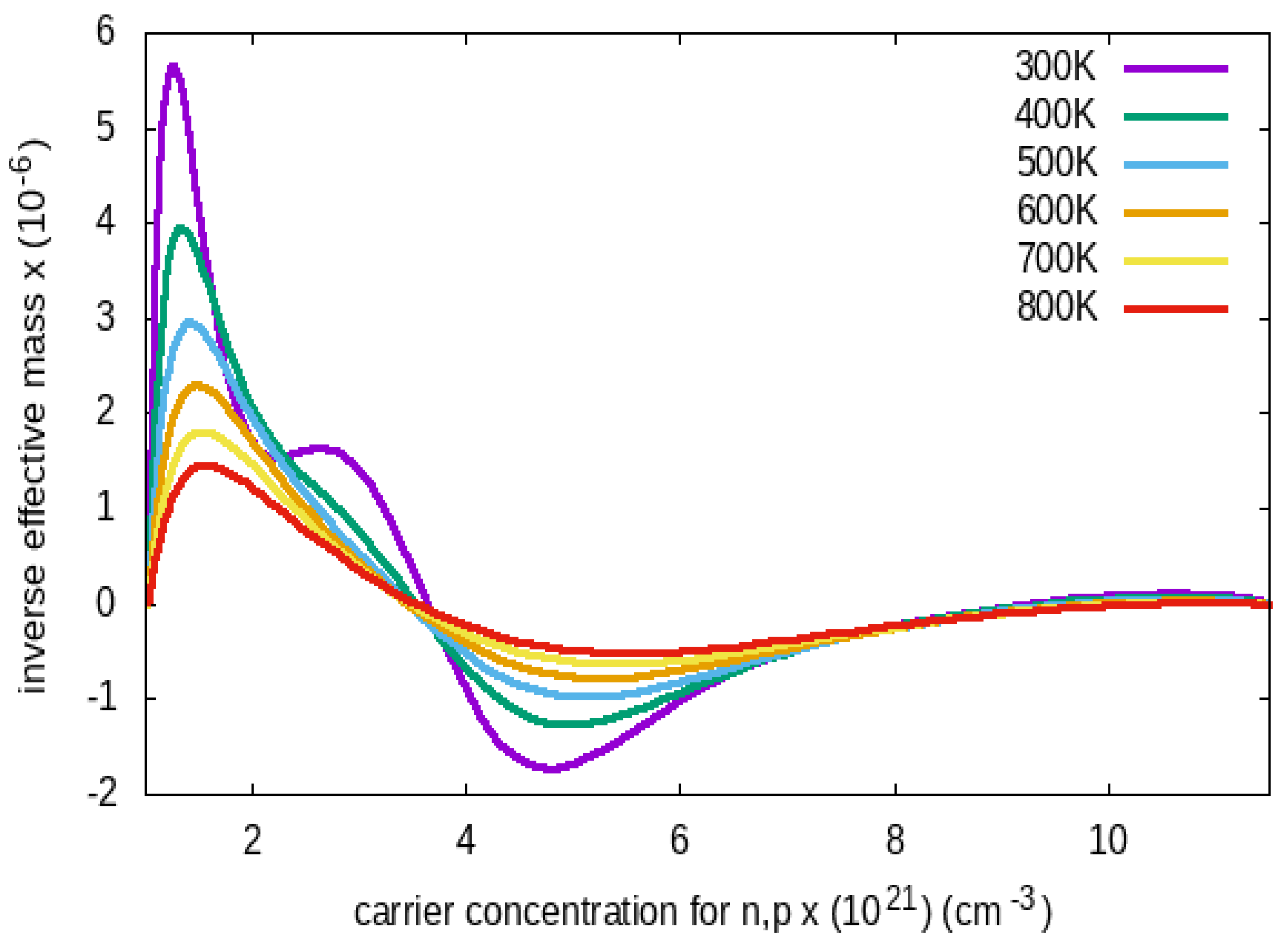
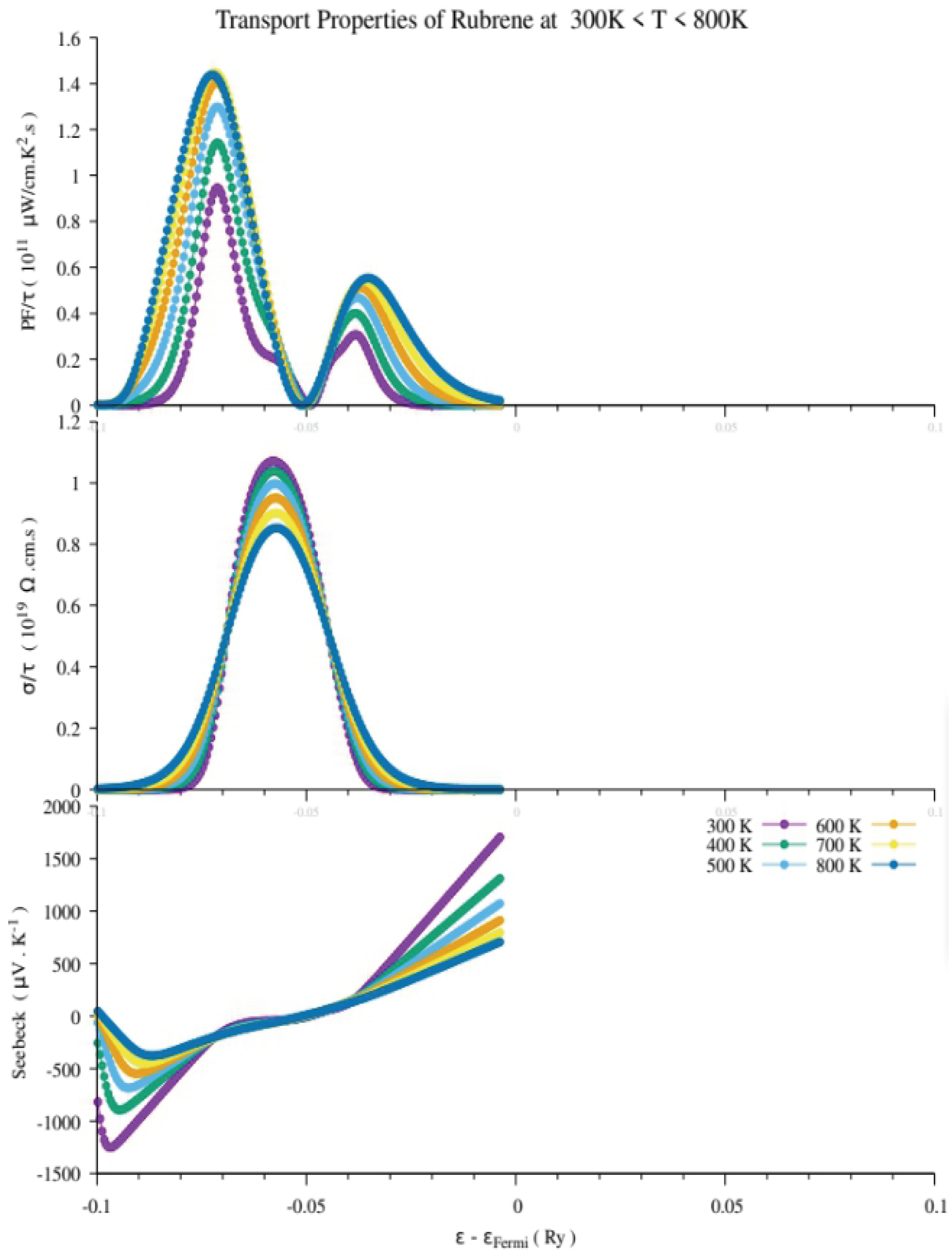
Figure 8.
Transport properties of Rubrene showing the power factor, electrical conductivity and Seebeck coefficient, respectively.
Figure 8.
Transport properties of Rubrene showing the power factor, electrical conductivity and Seebeck coefficient, respectively.
4. Conclusion
Using Density Functional Theory via First-Principles calculations, the structural, electrical, lattice dynamics, elastic, temperature, and doping-dependent characteristics of orthorhombic Rubrene were unveiled. Other properties, such as the Pugh ratio, Vickers hardness, electronic fitness function, and transport effective mass of the orthorhombic Rubrene crystal structure, are predicted in the present work.
The relaxed structure of orthorhombic Rubrene showed that it has a high charge carrier mobility. The band structure calculations show that orthorhombic Rubrene has a direct bandgap of 1.26 eV. This property makes it suitable for use in high-power electronics. The small bulk modulus (4.163 GPa) and Vickers hardness (1.080 GPa) reflect the orthorhombic Rubrene crystal structure as not super hard. The obtained Pugh ratio is 0.74667, which shows rubrene to be brittle rather than ductile. The calculated transport inverse effective mass and electronic fitness function confirm the orthorhombic Rubrene crystal structure to be a thermoelectric material of p-type. Furthermore, the results of the transport effective mass and the electronic fitness function obtained showed that Rubrene is a promising thermoelectric material that performs best at high temperatures of about 500 K.
Acknowledgments
The Authors acknowledge computational time support from South Africa’s Centre for High Performance Computing, CHPC, through collaboration with ATR.
References
- Jung, J.; Ulański, J. , Charge Carrier Transport in Organic Semiconductor Composites – Models and Experimental Techniques. In Solution-Processable Components for Organic Electronic Devices; John Wiley and Sons, Ltd, 2019; chapter 6, pp. 309–363, [https://onlinelibrary.wiley.com/doi/pdf/10.1002/9783527813872.ch6]. [CrossRef]
- Ahmad, S. Organic semiconductors for device applications: current trends and future prospects. Journal of Polymer Engineering 2014, 34, 279–338. [Google Scholar] [CrossRef]
- Dey, A.; Singh, A.; Das, D.; Iyer, P. Organic Semiconductors: A New Future of Nanodevices and Applications 2015. pp. 97–128. [CrossRef]
- Diemer, P.J.; Harper, A.F.; Niazi, M.R.; Petty II, A.J.; Anthony, J.E.; Amassian, A.; Jurchescu, O.D. Laser-Printed Organic Thin-Film Transistors. Advanced Materials Technologies 2017, 2, 1700167. [Google Scholar] [CrossRef]
- Kim, J.T.; Lee, J.; Jang, S.; Yu, Z.; Park, J.; Jung, E.; Lee, S.; Song, M.H.; Whang, D.R.; Wu, S.; Park, S.H.; Chang, D.; Lee, B.R. Solution Processable Small Molecules as Efficient Electron Transport Layers in Organic Optoelectronic Devices. Journal of Materials Chemistry A 2020, 8. [Google Scholar] [CrossRef]
- Riede, M.; Lüssem, B.; Leo, K. Organic Semiconductors. Comprehensive Semiconductor Science and Technology, p. 448–507. [CrossRef]
- Walker, A.B. Multiscale Modeling of Charge and Energy Transport in Organic Light-Emitting Diodes and Photovoltaics. Proceedings of the IEEE 2009, 97, 1587–1596. [Google Scholar] [CrossRef]
- Chen, F.C. Organic Semiconductors. Encyclopedia of Modern Optics, p. 220–231. [CrossRef]
- El-Saba, M. , Carrier Transport in Organic Semiconductors and Insulators; 2017. [CrossRef]
- Kim, J.; Yasuda, T.; Yang, Y.; Adachi, C. Advanced Materials 2013, 25, 2666–2671. [CrossRef]
- Choi, M.; Lee, H.N. Light-Emission and Electricity-Generation Properties of Photovoltaic Organic Light-Emitting Diodes with Rubrene/DBP Light-Emission and Electron-Donating Layers. International Journal of Photoenergy 2014, 2014, 361861. [Google Scholar] [CrossRef]
- Saxena, K.; Mehta, D.; Rai, V.K.; Srivastava, R.; Chauhan, G.; Kamalasanan, M.; Jain, V. Studies on organic light-emitting diodes based on rubrene-doped zinc quinolate. Physica Status Solidi (a) 2009, 206, 1660–1663. [Google Scholar] [CrossRef]
- Weinberg-Wolf, J.R.; McNeil, L.E.; Liu, S.; Kloc, C. Evidence of low intermolecular coupling in rubrene single crystals by Raman scattering. Journal of Physics: Condensed Matter 2007, 19, 276204. [Google Scholar] [CrossRef]
- Sai, N.; Tiago, M.; Chelikowsky, J.; Reboredo, F. Optical spectra and exchange-correlation effects in molecular crystals. Physical Review B, 77. [CrossRef]
- Wikipedia contributors. Rubrene — Wikipedia, The Free Encyclopedia, 2021. [Online; accessed 11-February-2022].
- Reyes-Martinez, M.; Crosby, A.; Briseno, A. Nat. Commun, 6, 6948.
- Lin, K.; Wang, Y.; Chen, K.; Ho, C.; Yang, C.; Shen, J.; Chiu, K. Scientific Reports 2017, 7. Funding Information: We gratefully acknowledge financial supports from Chung Yuan Christian University (Grant number CYCU-107011022) and from the Ministry of Science and Technology of ROC (Grant numbers MOST 104-2112-M-033-005 and MOST 105-2112-M-033-008). Publisher Copyright: © The Author(s) 2017. [CrossRef]
- Majewska, N.; Gazda, M.; Jendrzejewski, R.; Majumdar, S.; Sawczak, M.; Śliwiński, G. Organic semiconductor rubrene thin films deposited by pulsed laser evaporation of solidified solutions. Proc. SPIE 10453, Third International Conference on Applications of Optics and Photonics, Vol. 104532H. [CrossRef]
- Zeng, X.; Zhang, D.; Duan, L.; Wang, L.; Dong, G.; Qiu, Y. Morphology and fluorescence spectra of rubrene single crystals grown by physical vapor transport. Applied Surface Science, 253, 6047–6051. [CrossRef]
- Reyes-Martinez, M.; Ramasubramaniam, A.; Briseno, A.; Crosby, A. The Intrinsic Mechanical Properties of Rubrene Single Crystals. Advanced Materials, 24, 5548–5552. [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.; Cococcioni, M.; Dabo, I. Quantum Espresso: a modular and open-source software project for quantum simulations of materials. Journal of physics: Condensed matter, 21, 395502.
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M. Advanced capabilities for materials modelling with Quantum ESPRESSO. Journal of Physics: Condensed Matter, 29, 465901.
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Baroni, S.; De Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Reviews of Modern Physics, 73, 515.
- Louail, L.; Maouche, D.; Roumili, A.; Sahraoui, F. Calculation of elastic constants of 4d transition metals. Mater. Lett, 58, 2975–2978.
- Xing, G.; Sun, J.; Li, Y.; Fan, X.; Zheng, W.; Singh, D. The thermoelectric fitness function. Phys. Rev. Mater, 1, 065405, 079901.
- Wang, D.; Tang, L.; Long, M.; Shuai, Z. First-principles investigation of organic semiconductors for thermoelectric applications. The Journal of Chemical Physics, 131, 224704. [CrossRef]
- Zhang, Y.; Manke, D.; Sharifzadeh, S.; Briseno, A.; Ramasubramaniam, A.; Koski, K. The elastic constants of rubrene determined by Brillouin scattering and density functional theory. Applied Physics Letters, 110, 071903. [CrossRef]
- Jurchescu, O.; Meetsma, A.; Palstra, T.; Crystallogr, A. Sect. B: Struct. Sci, 62, 330.
- Yanagisawa, S.; Morikawa, Y.; Schindlmayr, A. HOMO band dispersion of crystalline rubrene: Effects of self-energy corrections within the GW approximation. Physical Review B, 88. [CrossRef]
- Podzorov, V.; Menard, E.; Borissov, A.; Kiryukhin, V.; Rogers, J.; Gershenson, M. Intrinsic charge transport on the surface of organic semiconductors. Phys. Rev. Lett, 93.
- Yamagishi, M.; Takeya, J.; Tominari, Y.; Nakazawa, Y.; Kuroda, T.; Ikehata, S.; Uno, M.; Nishikawa, T.; Kawase, T. High-mobility double-gate organic single-crystal transistors with organic crystal gate insulators. Appl. Phys. Lett, 90, 182117.
- Fumagalli, E.M. Growth and physical properties of crystalline rubrene. Università degli Studi di Milano-Bicocca. Doctorate in Materials Science.
- Musa, A.; Gidado, A.; Mohammed, L.; Yunusa, K.; Suleiman, A. Molecular and Electronic Properties of Rubrene and Its Cyanide Derivative Using Density Functional Theory (DFT.
- Zhang, M.; Hua, Z.; Liu, W.; Liu, H.; He, S.; Zhu, C.; Zhu, Y. A DFT study on the photoelectric properties of rubrene and its derivatives. Journal of Molecular Modeling, 21. [CrossRef]
- Missaoui, A.; Khabthani, J.; Laissardière, G.; Mayou, D. Two-Dimensional Electronic Transport in Rubrene: The Impact of Inter-Chain Coupling. Entropy, 26. [CrossRef]
- Mukherjee, T.; Sinha, S.; Mukherjee, M. Electronic structure of twisted and planar rubrene molecules: a density functional study. Physical Chemistry Chemical Physics, 20, 18623–18629. [CrossRef]
- Rang, Z.; Nathan, M.; Ruden, P.; Podzorov, V. ; Gershenson.
- Bisri, S.; Takenobu, T.; Takahashi, T.; Iwasa, Y. Electron transport in rubrene single-crystal transistors. Appl. Phys. Lett, 96, 183304.
- Fan, Q.; Wei, Q.; Yan, H.; Zhang, M.; Zhang, D.; Zhang, J. A New Potential Superhard Phase of OsN 2. Acta Physica Polonica, A, 126.
- Hill, R. Proceedings of the Physical Society. Section A, A65, 349. [CrossRef]
- Pugh, S. XCII. Relations between the Elastic Moduli and the Plastic Properties of Polycrystalline Pure Metals. The London, Edinburgh and Dublin Philosophical Magazine and Journal of Science, 45, 823–843.
- Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [CrossRef]
- Machida, S.; Nakayama, Y.; Duhm, S.; Xin, Q.; Funakoshi, A.; Ogawa, N.; Ishii, H. Highest-Occupied-Molecular-Orbital Band Dispersion of Rubrene Single Crystals as Observed by Angle-Resolved Ultraviolet Photoelectron Spectroscopy. Physical Review Letters, 104. [CrossRef]
Figure 1.
Molecular and Crystal Structure of Organic Semiconductor Rubrene
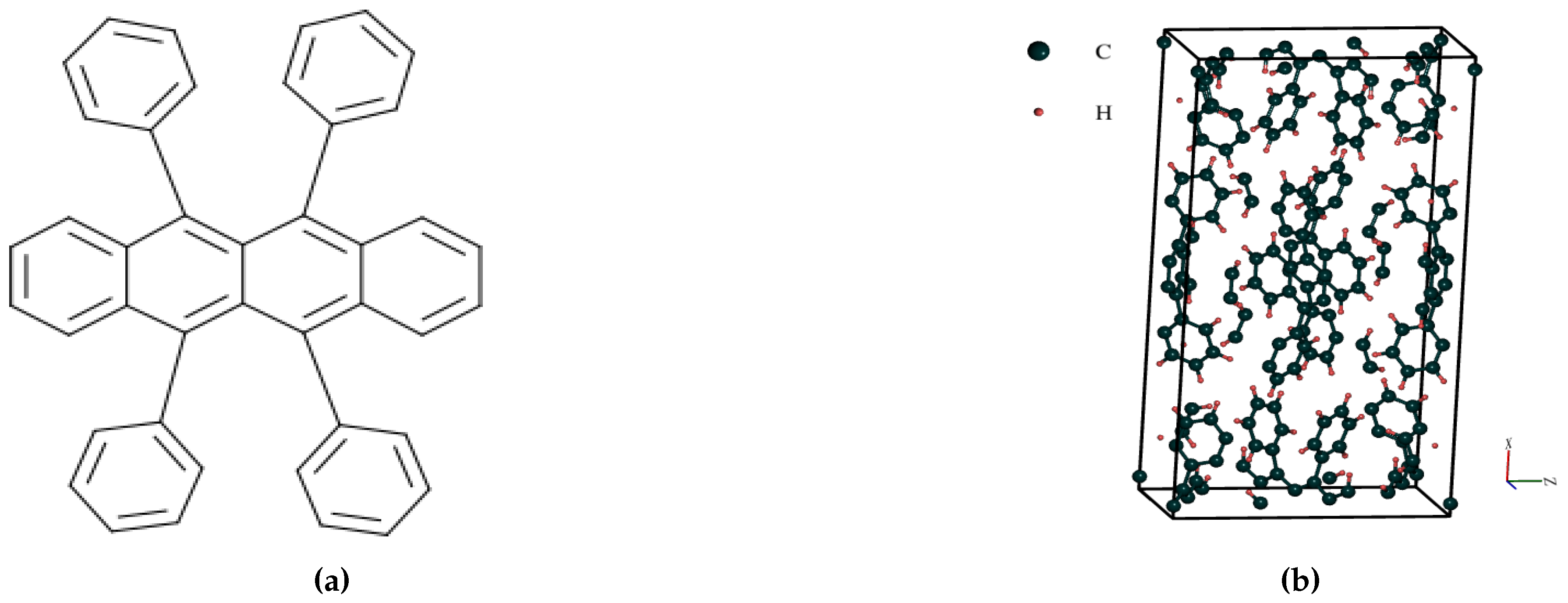
Figure 2.
(a) Band Structure of Orthorhombic Rubrene (b) Density of States of Orthorhombic Rubrene
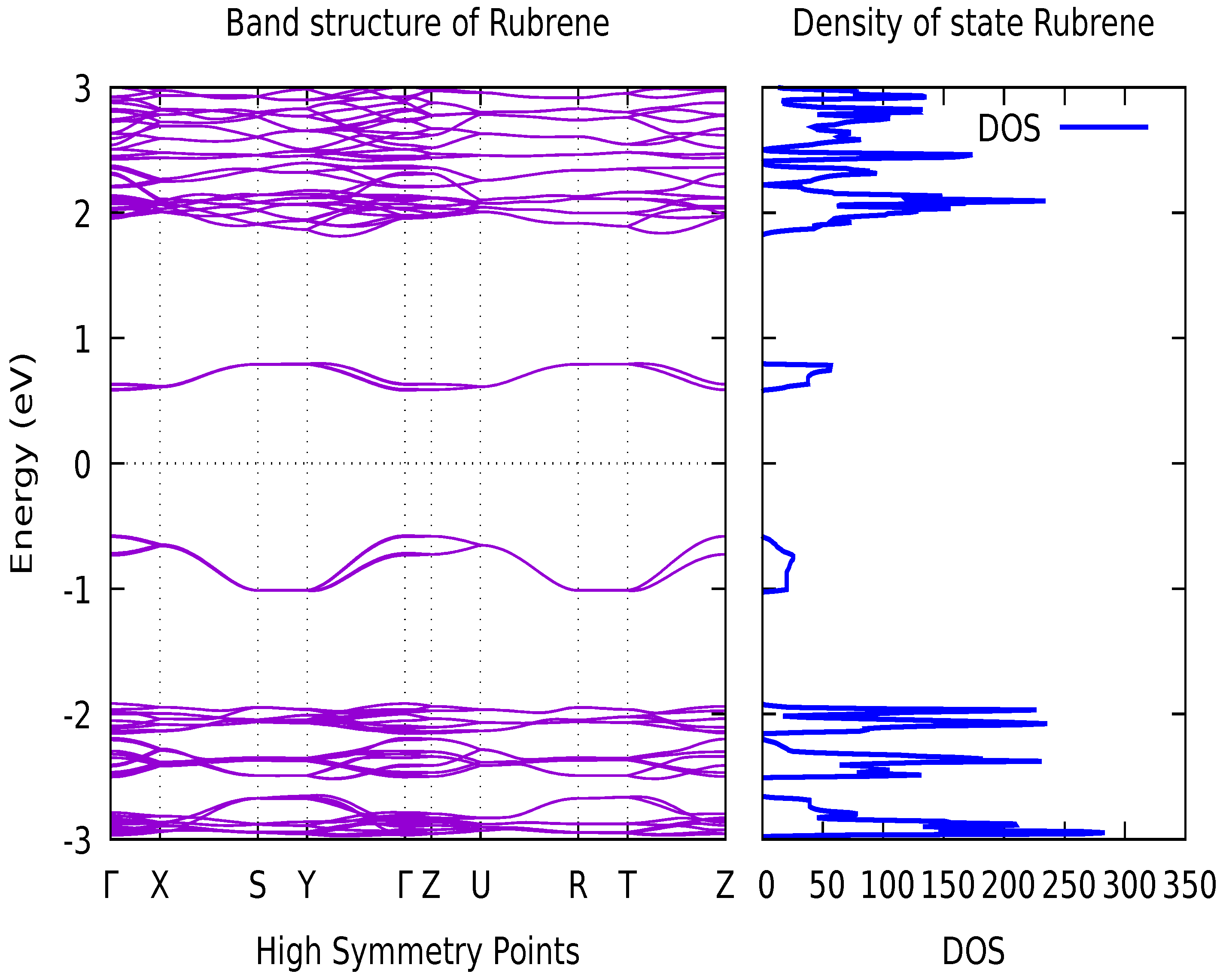
Figure 3.
Brillouin zone of pristine Orthorhombic Rubrene within the high symmetry points
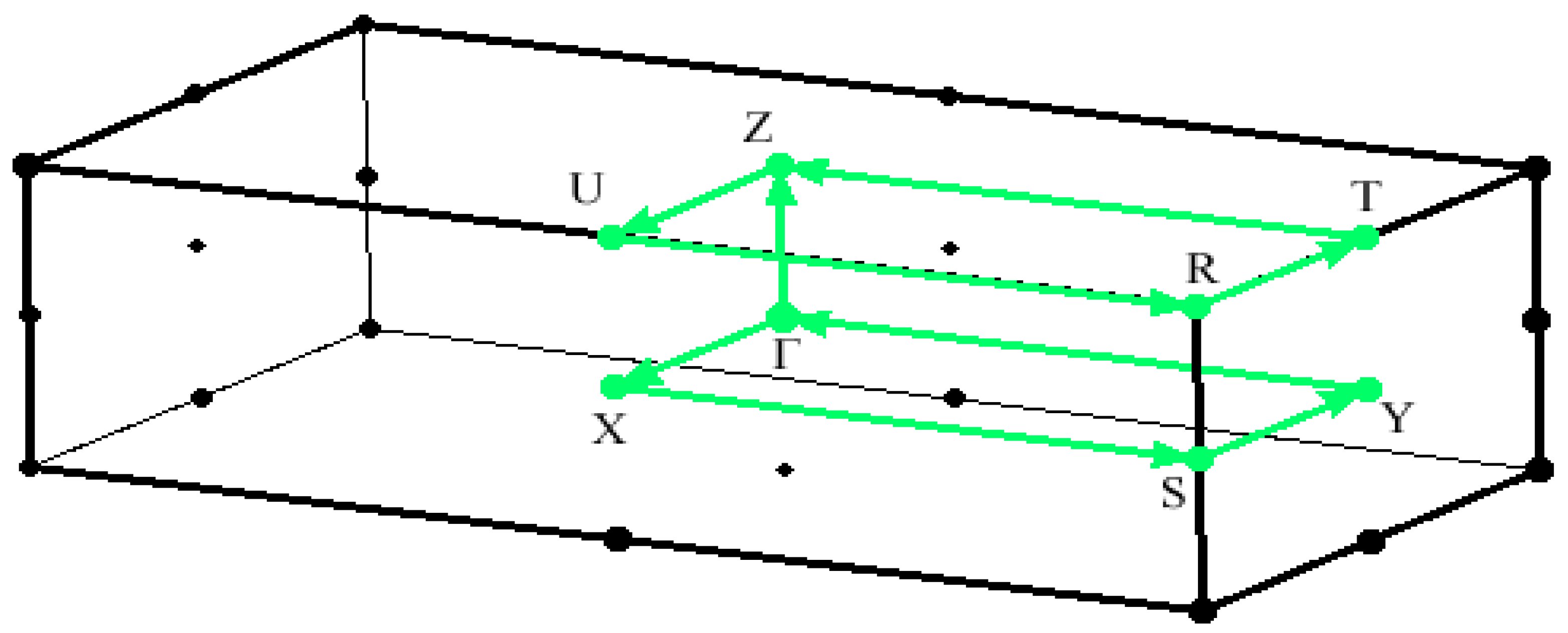
Figure 6.
Electronic Fitness Function of Rubrene against Carrier Concentrations from 300K to 800K
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



