
Submitted:
25 March 2024
Posted:
26 March 2024
You are already at the latest version
Abstract
We propose that a dietary reduction of Lipopolysaccharide (LPS) could reduce risk and slow the progression of cognitive decline and Alzheimer’s disease. LPS is a component of the highly inflammatory cell membrane of Gram-negative bacteria. The highest dietary sources of LPS are processed meat and dairy products, which can sharply raise plasma LPS within one to two hours. The LPS in these foods is packaged into chylomicrons, which enter the bloodstream. Smaller amounts of LPS from colonic microbiota do not account for the sharp postprandial increase of plasma LPS. Once in the bloodstream, LPS can trigger the release of inflammatory cytokines within three hours. LPS can increase cytokines via LPS binding protein and cluster of differentiation-14, triggering toll-like receptor-4, and activating nuclear factor kappa-B. Nuclear factor kappa-B can increase cytokine levels, including interleukin-1-beta, interleukin-6, and tumor necrosis factor-alpha. LPS and inflammatory cytokines are higher in Alzheimer’s disease, both in plasma and in the central nervous system. These cytokines can enter the brain via active transport and induce microglial activation and inflammatory damage in the brain, including loss of synapses and neurons. Elevated brain inflammation from dietary LPS can contribute to cognitive impairment and increased risk of dementia.
Keywords:
Alzheimer’s disease
; cognitive impairment
; cytokines
; dairy
; dementia
; diet
; endotoxin
; inflammation
; lipopolysaccharides
; meat
1. Introduction
Alzheimer’s disease (AD) is one of the most prevalent disorders among older adults, with over 55 million people worldwide suffering from dementia [1]. Identifying dietary LPS as a contributing cause of AD and cognitive impairment can provide us with another way to reduce neuroinflammation, simply by adjusting the diet. It has been hypothesized that LPS from intestinal microbiota is the main contributor to plasma LPS [2]. There is insufficient LPS produced in the small intestine to account for the after-meal surge of plasma LPS [3]. Although LPS from colonic bacteria may contribute to plasma LPS, this is a chronic leakage and it cannot explain the spike of plasma LPS after a meal [4]. LPS from certain foods can sharply increase plasma LPS from one to four hours after a meal.
We will examine how much LPS is in certain foods and how it enters the bloodstream [5]. We will examine the mechanisms of action whereby plasma LPS increases inflammatory cytokines (Figure 1). We explore how these inflammatory cytokines enter the brain, activate microglia, and damage delicate neurons [6]. Studies where humans are injected with tiny amounts of LPS show cognitive impairment within hours [7]. Diets containing high-LPS foods, such as meat, also show this effect on cognitive impairment [8]. In conclusion, reducing dietary LPS may slow the progression of AD and cognitive impairment.
2. What Are Lipopolysaccharides?
There are three potential sources of lipopolysaccharides (LPS): from bacterial infection, from colonic microbiota, and from food. LPS represents about 80% of the cell wall of Gram-negative bacteria. The terms LPS and endotoxin are used synonymously. LPS is a compound localized on the outer membrane of bacterial cells. LPS is made up of an antigen-O specific chain, a core region of a hetero-12 oligosaccharide, and a lipid A region that is the inflammatory part of the LPS [3]. Toll-like receptor-4 (TLR-4) is a transmembrane receptor that can sense LPS and is expressed on the surface of monocytes and macrophages, dendritic cells, intestinal epithelial, and blood vessel endothelial cells [9]. TLR-4 triggers NFκB to transcriptionally activate hundreds of inflammatory genes, including proinflammatory cytokines such as tumor necrosis factor-alpha (TNFα), interleukin-6 (IL-6) and interleukin 1-beta (IL-1β). LPS can increase microglial reactive oxygen species (ROS) production, neurotoxicity, and phagocytosis of neurons [6]. LPS from different Gram-negative bacteria can induce specific immune responses, which makes comparative studies difficult. LPS concentration is usually expressed in ng/mL, or in endotoxin units (EU), where 1 ng/mL LPS is about 10 EU/mL [10].
3. LPS in Food and Diets
3.1. Cooking and LPS
LPS was found to be highly resistant to typical cooking times and temperatures [11]. LPS has a strong thermal stability, and food processing only partially removes LPS. Note that the amount of LPS triples in meat during 10 days of refrigerated storage [12]. However, LPS in meat may be reduced as much as 60% by cooking, but boiling LPS in saline for up to 1 hour resulted in limited or no reduction in the biological activity of LPS [11].
3.2. LPS in Milk
Milk was analyzed in five European countries. Milk in these countries often contained LPS in the amount of 50 ng/mL, although values varied from a low of 0.9 ng/mL to a high of 100 ng/mL. Thus, an average 8 fluid ounce cup of milk (240 mL) was found to contain 12,000 ng of LPS [13]. Another study found that urban raw milk contained 18,000 ng LPS per cup and that LPS in eggs may have 27 times more endotoxin than red meat [14]. For comparison, note that an injection of as little as 60 ng of LPS raised TNFα 44-fold and IL-6 69-fold in 2 hours [15].
In a milk powder survey, infant formula studies from seven countries and 31 brands found LPS levels ranging from 4 ng/g to 5500 ng/g in the milk powder formula. This is equivalent to 2400 to 3,300,000 ng LPS/day from 100 g of milk powder. The average infant intake of LPS per day from 100 g milk powder was estimated to be about 280,000 ng/day [16].
Pasteurized skim milk was tested for LPS content. The most common level of LPS in this milk was 50-100 ng/ml, or about 12,000-24,000 ng per cup—more than a thousand times more than the 10 or less ng of LPS in the entire jejunum [10].
3.3. LPS in Meat and Processed Foods
A recent study found that the largest dietary risk factor for AD was meat consumption [8]. LPS levels were found to be highest in beef, pork, and turkey. After refrigerating turkey for eight days, LPS (TLR-4 stimulants) went from 40 to 550 ng/g. An average serving of turkey is 230 g, so the LPS per serving of turkey was 9000 to 125,000 ng, averaging 67,000 ng. The ingestion of dietary LPS can result in systemic cytokine release and inflammation. Much of the dietary LPS translocates into the systemic circulation in a biologically active form [11].
A study on toll-like receptors indicate that a 400 g processed food meal, may contain an average of about 200,000 ng LPS. Examples of processed food meals are included in Table 1. By comparison, intravenous injection of as little as 8 ng LPS is able to stimulate a detectable inflammatory response in man [15]. This suggests that the quantity of LPS in a processed food meal can be up to 25,000-fold higher than is required to stimulate inflammation, if given by injection (Table 1) [17].
Lobster contained 1200 ng/g LPS after 12 hours, and a serving of 145 g was found to contain 170,000 ng of LPS [18]. Ground beef was examined for LPS content in 84 samples. The average content of LPS in the ground beef was 3100 ng/g. A typical 98 g hamburger patty was found to contain 303,000 ng of LPS. Please see Table 1 for quantities of LPS in pork, minced pork, turkey, minced turkey, minced beef, and other foods. Foods containing minced meat and cheese contained a particularly high abundance of LPS compared with the rest of the food extracts (Table 1) [11].
Increased processed meat intake was shown to be associated with a significant increase in risk of elevated inflammation and cardiovascular disease. By contrast, the Mediterranean dietary pattern is defined in part by relatively low intake of pre-packaged ‘ready meals’ and processed meat, and is likely a lower LPS diet for these reasons [19].
Common foods can contain LPS, and the greatest concentrations were present in meat-based products. Meat content of LPS increased over time in each meat examined. LPS reached the highest levels (approximately 600,000 ng in a 3-ounce serving) in meat that was minced rather than intact, and when refrigerated in air (at 5 deg. C=41 deg. F) rather than under a modified atmosphere. Unprocessed fruit, vegetables, grains, and potato are quite low in LPS (Figure 2) [11].
Restricting dietary LPS could reduce plasma LPS levels in order to significantly reduce inflammation [20]. Also those who ate more fruit had 34% less circulating LPS. Those who ate more legumes had 20% less circulating LPS. Only fruit and legumes were significantly associated with lower LPS levels in this large prospective study with 912 participants. Adherence to the Mediterranean diet was also significantly associated with lower circulating LPS when compared to a Western Diet [7,21].
3.4. LPS in Diets
Nutritional changes can lower plasma LPS [22]. It is possible to raise or lower plasma LPS with dietary modification. Placing eight healthy subjects on a Western-style diet for one month in a controlled metabolic ward induced a 71% increase in plasma levels of LPS activity, whereas a prudent-style diet reduced levels by 31% [23]. A high fat diet triggered inflammation and cytokines independently of LPS [24]. If each meal contains high levels of LPS that stimulates inflammation for over four hours, then over 12 hours per day may be spent in an inflammatory postprandial state [25].
A study of 668 participants found that healthy dietary choices, such as consumption of fresh vegetables, fruits, and berries may be associated with positive health outcomes by reducing systemic endotoxemia. Cheese highly raised plasma LPS in this study [26]. A significant association was found between adherence to the Mediterranean diet and lower rates of cognitive impairment [21].
Plasma LPS level is highest with a Western diet high in meat (beta = 0.30, p=0.009). Mediterranean and complex carbohydrate diets are moderately lower in LPS (beta = -0.12, barely significant). A “prudent” diet containing fruit and vegetables, but low in cookies, had the least plasma LPS (beta = -0.28, p=0.009) [27]. Meals with plenty of fiber and fruit do not induce inflammation, do not increase the expression of TLR-4, and do not raise plasma concentrations of LPS or LPS binding protein (LBP) [28].
Dietary patterns with high intakes of vegetables and fruits, and low consumption of meat have been correlated with a healthier microbiota in the colon, which can be beneficial for the brain by limiting LPS translocation. However, overconsumption of foods rich in simple carbohydrates, saturated and trans fatty acids, red meat, processed meat, organ meats, and chips, can have a proinflammatory influence on the AD patient’s brain [29].
3.5. LPS after a Test Meal
Serum LPS significantly increased at each postprandial time point (compared to before a meal) of thin crust cheese pizza, with peak plasma LPS 65-fold at five hours postprandial (P=0.001) [30].
Meals with a high LPS and fat content have been implicated as a trigger of postprandial inflammation through the activation of NFkB [31,32]. One study used a healthy American Heart Association meal compared to an equicaloric high-fat meal with egg sausage muffin with hash browns. The high-fat meal induced inflammation through LPS and TLR-4, increased TNFα, IL-6, and increased oxidative stress for two to three hours after the meal. Plasma LPS went up 45% and NFkB increased 72% with the high fat meal only. The healthier meal did not raise LPS, TLR-4, or cytokines [28].
One study compared isocaloric meals (300 kcal) from a glucose drink, orange juice, or dairy cream among 42 healthy adults. Only dairy cream consumption increased plasma LPS levels, suggesting that this dairy cream intake was responsible for postprandial endotoxemia [7].
Ingestion of LPS and saturated fat in 100 ml heavy whipping cream promoted LPS-mediated inflammation. Two hours after the whipping cream, plasma LPS increased 83%. 100 mL of heavy cream also raised TNFα 11% after two hours. LPS area under the curve (AUC) went up 70-270 in obese individuals. TNFα plasma LPS levels went up 23-40 incremental AUC [33].
It has been calculated that a cup of milk may contain 12,000 ng LPS (Table 1). Milk fat, 40 g, with breakfast, more than doubled plasma LPS. AUC plasma IL-6 levels doubled when consuming 40 g compared to 10 g milk fat with breakfast. Plasma LPS went up three to five-fold after three hours in the high milk fat group. In obese subjects, plasma LPS went from 1 to 17.5 ng/mL (1.8-1.9 ng/mL is normal) [34]. Another study looked at people with a low-fat diet (largely excluding high-fat meat and cheese) and serum LPS levels were much lower, ranging from 0 to 0.2 ng/mL [2].
Circulating LPS after a high fat meal was raised at least 150%, from 0.4-0.6 ng/mL up to 0.6-1.4 ng/mL after four hours [35]. Participant serum LPS concentration was increased during the postprandial period (2-3 hours after eating) after the consumption of a saturated fat meal (including 16 g coconut oil). Serum LPS increased by 60% following the consumption of foods high in saturated fatty acids, lasting for 3 hours post meal [36].
4. Dietary LPS Enters the Bloodstream and Triggers Inflammation
LPS in food can trigger systemic cytokine release and inflammation. Some of the LPS in food gets into systemic circulation in a biologically active form [11].
LPS absorption is largely a postprandial phenomenon, but can also be from chronic dysbiosis (imbalanced gut bacteria) or oral infection [37]. Once LPS-laden chylomicrons are in the bloodstream, LPS are taken up by LPS binding protein (LBP). LBP is produced by the liver and present in the blood at concentrations of approximately 2-20 µg/mL. LBP is transferred to the receptor cluster of differentiation-14 (CD14). CD14 is expressed on the plasma membrane of various cell types, such as monocytes, macrophages, and human intestinal epithelial cell lines. Besides this membrane-bound state, CD14 is also found in a circulating soluble form (sCD14). sCD14 is involved in the bioactivity of circulating LPS and can be considered as an accurate marker of LPS in plasma. Both membrane-bound and soluble forms of CD14 can bind the LPS-LBP complex and mediate signal transduction, including the activation of the transcription factor nuclear factor-kappa-B (NFқB) via TLR-4. This signaling cascade results in the release of pro-inflammatory cytokines such as Interleukin IL-1β, IL-6 and TNFα. Intestinal cells are able to produce, express, and release molecules of LBP, CD14, and TLR-4 [3,37].
Pro-inflammatory LPS co-absorbs with dietary lipids. This explains how consumption of milk fat allows intestinal absorption of LPS. The LPS are then incorporated into chylomicrons, enter the bloodstream, and consequently contribute to postprandial endotoxemia and inflammation. LPS levels in chylomicrons increased from near zero up to half of a nanogram per milliliter, spiking at three hours after the high milk fat (40 g) meal. Emulsified milk fat triggered an early and sharp increase in chylomicron-endotoxemia at 60 min after ingestion [5]. Plasma LPS resulted in a robust cytokine response [38].
4.1. Colonic Sources of Plasma LPS
High-fat diets are associated with a reduction in intestinal bacterial diversity, increased colonic membrane permeability, increased numbers of Gram-negative bacteria, and increased LPS translocation to plasma. This can lead to the production of low-intensity systemic inflammation. In contrast to the immediate (1-4 hours) raised serum LPS from LPS in food, this colonic dysbiosis represents a chronic condition [39]. LPS from periodontitis can also enter the gut and become absorbed into the bloodstream. Even saliva of healthy subjects has been measured to contain a median of 7 ng/ml of LPS [40].
Exposure to a high-fat diet increases systemic LPS concentration. LPS leakage from the colon is determined by the integrity of the colonic epithelial barrier, which may also be less effective with a high-fat diet [31]. Five days of a high saturated fat diet in healthy participants resulted in an approximately two-fold increase in fasting serum LPS concentrations [41]. Another high-fat diet study found that bile acid synthesis was increased 10-fold and that these bile acids induced intestinal hyperpermeability. Note that bile acids do not reach the colon fast enough to impact the release of LPS from the colon in one to four hours [2].
A high-fat diet increased colonic LPS biosynthesis. The high-fat diet was also associated with raised colonic inflammatory arachidonic acid levels, leading to elevated plasma proinflammatory factors such as leukotriene-4 and prostaglandin-2 [42].
Residual dietary fatty acids (estimated as 5% of total fat eaten) reach the colon where they can interact with the gut microbiota and intestinal cells. Saturated fats, but not unsaturated fats, may lead to damage to the colonic mucus barrier [43]. Normally, the intestinal epithelium acts as a continuous barrier to avoid LPS translocation into the plasma [44]. When high fat meals are eaten regularly, this may increase LPS production in the large intestine and damage the intestinal barrier to allow more LPS to enter the bloodstream [7]. However, this is a slower and longer-term process compared to food-borne LPS absorption. It is unlikely that food ingested could have an impact on colonic LPS absorption in only a few hours. The fastest intestinal transit time recorded in 1000 tests was 14 hours with 24-48 hours being much more common [45]. Hence, the postprandial elevation of plasma LPS is unlikely to come from colonic sources.
4.2. Small Intestine Microbiota Versus LPS in Food
The small intestine is likely to be the primary site of LPS absorption after a fatty meal. Only low levels of endogenous LPS are present in the small intestine due to the very limited Gram-negative bacteria present in this part of the gut [46].
LPS is fat-soluble and is absorbed in a manner similar to that of the other fat-soluble nutrients via incorporation into chylomicrons with dietary fat in the small intestine (but not the large intestine). LPS leaking from the large intestine enters the portal venous system, proceeding directly to the liver, which removes LPS from the blood with great efficiency. By contrast, dietary LPS co-absorbed with dietary fat passes first into the lymphatic system and then through the thoracic duct and into the blood, thereby avoiding the first-pass clearance by the liver. Taken together, these considerations suggest that the small intestine, rather than the large intestine, is likely to be the major site of postprandial LPS absorption in the body [40].
It has been estimated that the entire digestive tract may contain one gram or more of LPS [3,39,47]. The large intestine has a high density of bacteria with between 1010 to 1012 colony-forming units (CFU)/mL [40,48].
The small intestine is a harsh environment for microbial life, owing to the short transit time, the oxygen-rich environment, the presence of antimicrobial peptides produced by Paneth cells, the influx of digestive enzymes, bile, and food substrates [48]. The jejunum is the upper part of the small intestine where chylomicrons containing food-borne LPS are formed. It is estimated that the jejunum has a bacterial density of 100-4 CFU/mL [3,40]. Unlike that of the large intestine, the microbiota of the jejunum consists mainly of Gram-positive bacteria, and the numbers of Gram-negative bacteria are low. The total LPS content of the microbiota of the small intestine seems likely to be low and is estimated to be only 1-10 nanograms of LPS. The low numbers of bacteria in the small intestine therefore suggest that the majority of the LPS present in the small intestine is likely to be derived from food and possibly from oral microbiota, rather than from products of bacteria endogenous to the small intestine [46].
With this uneven distribution of bacteria and LPS in the gut, we estimate that the entire jejunum contains only 1-10 nanograms of LPS. Even if much of the LPS were absorbed, it would barely affect blood LPS or inflammation. The LPS content of several commonly consumed processed foods can reach concentrations that are thousands of fold-higher than those normally present in the entire healthy small intestine [40]. For comparison, about 48 ng of LPS were injected in humans to provoke increased cytokines and decreases in memory performance [49,50]. Pasteurized skim milk was tested for LPS content. The most common level of LPS in this milk was 12,000-24,000 ng per cup—more than a thousand times more than the 10 or less ng of bacterial LPS in the entire jejunum [16].
LPS was present mainly in meat products and processed foods, but were minimal or undetectable in fresh fruit and vegetables [46]. Although LPS are generally not detectable in fresh, whole plant foods, they can be abundant in a number of processed foods common to the Western diet. In particular, processed foods derived from meat and dairy products contained LPS, reaching levels as high as 11,000 ng/g. A 3-ounce serving may contain 940,000 ng of LPS. These concentrations therefore far exceed those normally encountered in the small intestine [40].
The duodenum and jejunum are tasked with facilitating most of the nutrient assimilation and absorption [48]. We have given several examples of high levels of LPS and resulting cytokines in blood 1-4 hours after eating certain high fat meals. LPS can be internalized by intestinal epithelial cells through TLR-4 recognition, and transported to the Golgi compartment, where newly assembled chylomicrons are located prior to their secretion. A high-fat diet increases chylomicron formation. Chylomicrons have high affinity for LPS and transport postprandial fat along with significant amounts of concomitantly absorbed gut LPS [47,51].
The bulk of the fats and LPS in a meal will be absorbed in the jejunum and delivered to the bloodstream via chylomicron formation and transport. There is a transient increase of LPS blood levels following ingestion of high-fat meals [43]. As we have seen, there is not enough microbiota-produced LPS present in the jejunum to elevate LPS and cytokines to the extent observed. One to four hours after a meal is not enough time for the fatty food to reach the colon and release the LPS therein. Therefore, the postprandial efflux of LPS into the bloodstream must come from the food eaten (with the addition of periodontal-derived LPS).
4.3. Transport of LPS into the Blood by Saturated Fats and Chylomicrons
LPS and saturated fatty acids have been shown to be transported by chylomicrons, suggesting a mechanism through which a high fat diet may increase LPS levels [31]. Therefore, consuming a diet high in saturated fat can increase the risk of elevated plasma LPS. Saturated fat efficiently transports LPS from the gut lumen into the bloodstream [52]. An increase in postprandial plasma LPS concentration was observed in healthy adults on high saturated fat diets, in contrast to subjects who received diets high in polyunsaturated fats. Dietary LPS can be incorporated into micelles, absorbed, and added to chylomicrons to increase plasma LPS. Saturated fats along with LPS can directly stimulate TLR-4 and increase inflammation [39].
4.4. High-Fat Diets Can Increase Endotoxemia
A chronic high-fat diet can result in increased endotoxemia and inflammation due to the repeated LPS absorption from the jejunum during the digestion of lipids. Intestinal absorption of endogenous LPS from microbiota can result in increased plasma LPS and inflammation as described above, though at much lower levels of LPS and over a longer period of time. Patients submitted to a high fat meal present a large increase of IL-6. The inflammatory response to the high-fat meal is characterized by an increase of the pro-inflammatory cytokines IL-6 and TNFα in plasma. It has been reported that a high-fat meal is more able to enhance these inflammatory cytokines compared to a high carbohydrate meal [3].
High-fat diets can contribute to endotoxemia, which is defined by elevated LPS levels in blood plasma [47]. In a large sample of healthy men, a link was found between fatty food intake and plasma LPS. Fat was found to be more efficient in transporting bacterial LPS from the gut lumen into the bloodstream than was carbohydrate [53].
LPS is transported together with dietary fat from the gut after a high-fat meal, thus increasing plasma LPS concentrations postprandially. Plasma LPS increased significantly (P <0.05) by about 50% after a high-fat meal [54]. The ingestion of a single high-fat meal can mediate systemic increases of a wide range of inflammatory factors with noted activation of NFkB. LPS from a high-fat meal may trigger the inflammatory cascade within 1-4 hours [32].
5. Association of High Plasma LPS with AD and Related Dementias
Here we examine whether higher plasma LPS can be associated with neuroinflammation, AD, and cognitive impairment (Table 2). It has been noted that LPS is implicated in increasing progressive neurodegeneration in AD [8].
5.1. LPS Increases Risk of Cognitive Decline and AD
Systemic markers of innate immunity, increased by LPS, are risk factors for late-onset AD. Activation of microglia precedes cerebral atrophy in AD patients. Microglial activation can be detected in around 50% of patients with mild cognitive impairment. Prospective population-based cohort studies indicate that higher serum levels of inflammatory markers can predict dementia [60]. LPS and LPS-induced inflammation may be significant initiators of inflammatory degeneration in AD. LPS induces neuronal cell loss by greatly increasing TNFα and IL-1β [61].
Higher levels of LBP, an acute-phase reactant involved in the pro-inflammatory response to LPS, were significantly associated with a 30% higher odds ratio of developing AD over 12 years in the 3-City Cohort. There was a dose-response trend between LBP levels and increased odds of developing AD. Higher levels of circulating LBP was recently found to be associated with loss of white matter integrity and impaired cognitive performance [57].
LPS can infiltrate into the brain from the periphery and initiate the cascade of chronic neuroinflammatory reactions and neurodegenerative changes that can cause AD [29]. High levels of plasma LPS can trigger inflammatory immune activation. Recent data clearly show that immune activation in AD can have the capacity to facilitate and trigger the pathophysiology of AD [62]. LBP impacted negatively on the Digit Span Test. Higher circulating LBP is associated with brain white matter damage and poorer working memory/short-term verbal memory [63].
In humans, low-dose LPS administration impaired both immediate and delayed recall, but did not affect attention or executive functions. Administration of IL-6 impaired concentration in this study [58].
LPS activates microglia that damage neurons via nitric oxide, ROS, and cytokines, leading to phagocytosis of synapses and neurons [6]. In addition, LPS expresses its neurotoxicity in the central nervous system (CNS) in part by reducing neuron-specific neurofilaments and synaptic signaling proteins, promoting neurodegeneration [64].
Higher plasma LPS can degrade memory functioning partly due to arachidonic acid eicosanoid production, which is partially reversible with COX (cyclooxygenase) inhibitors [65]. Microglia treated with LPS produced 14 times the prostaglandin-E2 (PGE2), and PGE2 from arachidonic acid increases IL-6 inflammation [66]. Another study confirms that LPS can activate COX2 and increase production of ROS, for example, peroxides and superoxide [67].
5.2. LPS Is Elevated in AD
Recent investigations reported higher LPS levels in grey and white matter in brains from patients with AD than in those from participants free of dementia. LPS co-localized with Amyloid-β in amyloid plaques in AD brains [57]. LPS is present and associated with white matter injury in AD [60]. LPS has been reported to be higher in the AD brain and may impede the ability to repair damaged myelin [68]. Another study reported three-fold higher LPS levels in the hippocampus of four AD brains compared to two age-matched control brains. There was a two-fold higher abundance of LPS in neocortical extracts from six AD brains compared to six age-matched control brains. In some advanced AD patients, the hippocampus exhibited up to a 26-fold increase in LPS concentration [57]. Also, plasma LPS levels were three-fold higher in 18 AD participants (mean 61 ρg/mL) than in 18 healthy controls (mean 21 ρg/mL) [7]. A Norwegian study also found increased levels of LPS in the hippocampus of patients with AD, compared to healthy age-matched controls [60].
LPS was reported to be elevated in the blood and brain of AD patients [69]. Those with AD have plasma LPS levels three times higher than controls [70]. Higher plasma LPS was correlated with cognitive decline and lower cognitive function. Higher plasma LPS concentration doubled the risk (OR per 1 EU/mL =2.1) of mild cognitive impairment in participants without dementia [4].
Subjects affected by mild cognitive impairment (MCI) had double the serum LPS levels, compared to normal aging controls. Also, those with dementia had about double the serum LPS compared to MCI patients. Serum LPS was near zero in young normal people, 150 ng/mL in normal aged people, 400 ng/mL in patients affected by MCI, and 700 ng/mL in patients affected by dementia [71]. Another study confirmed that levels of LPS are increased in the serum and cerebrospinal fluid of patients with AD and in the serum of patients with MCI [4].
5.3. LPS Stimulates Brain Inflammation
LPS can stimulate macrophages to produce IL-1β, IL-6, and TNFα, which go on to affect the brain. After LPS stimulation, patients with AD produced higher levels of IL-1β, IL-6, IL-10, and TNF-α than normal controls. Note that IL-10 is a compensatory anti-inflammatory cytokine [72].
LPS can stimulate astrocytes and microglia in the CNS to secrete cytokines such as TNFα, IL-6, and interferon-gamma (IFN-γ). Microglial activation preceded the apparent neuronal degeneration [73]. Systemic inflammation evoked by LPS can induce microglial expression of IL-1β and also increase neuronal apoptosis in the brain. Thus, both central and peripheral inflammation can increase local brain inflammation and neuronal death, increasing neurodegeneration [74].
Higher circulating LPS levels increased TLR-4 protein expression two-fold (p<0.05). Higher LPS caused a significant increase in TNFα from 1 pg to 33 pg and IL-6 secretion from 2.7 ng/mL to 4.8 ng/mL. NFқB is clearly involved in this increase of cytokines because a NFқB inhibitor stopped the increase of IL-6 [75].
Increases in IL-1β have been documented after LPS challenge. IL-1β can induce microglial proliferation, stimulate microglial expression of IL-6, and activate microglia. IL-1β may promote neurodegeneration through generation of reactive nitrogen species such as peroxynitrite. IL-1β also appears to be a mediator of apoptosis. Additional neurotoxic properties of IL-1β include blood-brain barrier (BBB) damage and increased amyloid-β [72].
LPS can compromise the integrity of the BBB and contribute to early neuroinflammatory changes and AD by priming microglia and impairing amyloid clearance. Circulating LPS can activate the receptor for advanced glycation end-products (RAGE) to amplify pro-inflammatory signaling and promote chronic neuroinflammation and neurodegeneration, particularly in brain regions sensitive to AD such as the hippocampus [76].
5.4. LPS, Neuroinflammation, and Amyloid
LPS can trigger the lipopolysaccharide receptor CD14 that interacts with fibrillar, but not with aggregated, amyloid-β peptide in AD [77]. LPS, through CD14 and TLR-4, can strongly increase amyloid-β microglial activation and neurotoxicity. Thus, LPS through CD14 may significantly contribute to the overall neuroinflammatory response to amyloid peptide [78].
Pro-inflammatory cytokines are known to be centrally up-regulated in regions undergoing neurodegeneration and, in the case of AD, in regions manifesting amyloid-β deposits or neurofibrillary tangles. Once the inflammatory cascade has been stimulated, cytokines can amplify their own production [72].
One study found that neuroinflammation induced by LPS increases cognitive impairment through an increase of beta-amyloid generation [79]. Amyloid plaques are co-localized with pro-inflammatory cytokines, clusters of activated microglia, and reactive astrocytes [77]. Although the presence of amyloid-β plaque is not well correlated with reduced cognition, inflammatory plaque does show a stronger correlation with impaired cognition [72].
LPS can increase amyloid-β production and accumulation and the hyperphosphorylation of tau protein [4]. High plasma levels of LPS can increase the permeability of the blood-brain barrier, allowing toxic plasma components, including amyloid β and α-synuclein into the brain. LPS may also promote the production or aggregation of amyloid β, tau, and α-synuclein. LPS is found in amyloid plaques [6,80].
High LPS levels may compromise BBB integrity, stimulate and sustain inflammation in the CNS and facilitate the AD pathological cascade. Inflammatory cytokines may have a direct effect on amyloid aggregation or an indirect effect via endothelial dysfunction. Increased central and systemic expression of cytokines has been reported in AD patients [81].
5.5. Saturated Fatty Acids Can Raise Serum LPS and Impact Cognition
High intakes of dietary saturated fat can almost double the risk of dementia (RR = 1.9) [82]. Saturated fat intake promoted increases in circulating LPS levels and TLR-4 gene expression in women. Elevation of serum LPS due to a high-fat diet was dependent on the dietary levels of saturated fatty acids (SFAs) [2]. Women with elevated LPS performed worse on the post-meal cognitive test and had poorer attention during testing [52]. Two of the saturated fatty acids, lauric and palmitic acids, along with LPS, activated TLR-4 to increase inflammation, whereas unsaturated fatty acids did not increase the activation of TLR-4 [25]. Note that it is possible that SFAs are unable to directly stimulate TLR-signaling without the presence of LPS [11].
The consumption of a high saturated-fat diet has been associated with increased postprandial levels of LPS and increased circulating levels of pro-inflammatory markers. LPS was increased by 60% following the consumption of a high SFA meal and the high levels of LPS lasted longer (three hours versus one hour following a low-fat meal) [7]. High intake of dietary cholesterol and SFAs increased the risk of impaired cognitive function and the development of dementia, including AD [59].
5.6. Injecting LPS Impairs Cognition
It has been reported that two hours after an injection of LPS (0.8 ng/kg of body weight), TNFα was raised 32-fold (5-160 pg/mL) and IL-6 was raised 68-fold (2-135 pg/mL), and this increase of inflammatory markers was associated with impairment of social cognitive processing [55].
After injection, LPS can be measured in plasma within a few minutes. LPS is then transported to the liver for clearance. Within the first hour after LPS administration, TNFα, IFN-γ, and IL-6 appear in plasma. TNFα peaks after 90 minutes, whereas IL-6 and IFN-γ peak after 120 minutes. IL-1β peaks between 90 and 120 minutes. Compensatory, anti-inflammatory IL-10 peaks at 3 hours post injection [9].
In one human study, injections of LPS were either 0.4 ng/kg of body weight or 0.8 ng/kg of body weight, approximately 24 ng or 48 ng, respectively. Pro-inflammatory cytokines, such as IL-6 (up 180 pg/ml) and TNFα (up 110 pg/ml), were greatly increased by injected LPS within two hours. These two cytokines play a pivotal role in mediating cognitive impairment by transferring peripheral inflammation to the brain. Long-term memory performance was impaired selectively for emotional stimuli after administration of the lower, but not of the higher, dose of LPS. The impact on cognition may vary with type of bacterial LPS and the timing of the testing. We suggest that there may be compensatory changes, such as increased anti-inflammatory IL-10, triggered by the LPS injection [56].
In a human study, LPS was injected in the small amount of 0.2 ng/kg (14 ng for a 70 kg person). TNFα was doubled and IL-6 raised 700%, peaking at three hours post injection. Performance in declarative memory was worse with increases in IL-6. Subjects with small increases in IL-6 performed better than subjects with larger increases [57,83].
LPS-induced cytokine secretion has been correlated with impairments in verbal and nonverbal declarative memory functions. LPS can decrease immediate and delayed recall [50]. Another study reported a significant impairment in declarative memory until 10 hours after the injection of LPS. Additionally, word-list learning performance worsened between 4.5 hours and six hours after LPS injection [7].
5.7. Injecting LPS Increases Neuroinflammation
Injection of 28 ng LPS resulted in marked activation of microglia in the brain for 3 hours as shown by PET scan [84]. It is well established that intravenous injection of as little as eight ng LPS can be sufficient to stimulate a detectable inflammatory response in humans. Injection of 60 ng evoked a 44-fold increase in TNFα from 1 to 44 pg/mL in two hours. IL-6 went up 69-fold from 2 mg/mL at baseline to 138 mg/mL in 2 hours. C-reactive protein (CRP) was elevated 52-fold from 0.34 to 18 mg/L with strong increases continuing up to 24 hours or more [15].
LPS administration (1.0 ng/kg, i.v.), 60 ng for a 60 kg person, increased blood levels of inflammatory cytokines. The LPS injection also significantly increased microglial activation throughout the brain, revealed by PET scan [85].
6. Association of High Cytokine Levels with AD and Cognitive Impairment
Under normal conditions, low levels of brain cytokines can help hippocampal memory and neurogenesis. When activated, microglia and astrocytes can secrete high levels of pro-inflammatory cytokines, producing detrimental effects on memory, neuronal plasticity, and neurogenesis. Administration of LPS was found to impair learning and memory. In AD subjects, higher levels of IL-6 have been correlated with the severity of the dementia [65]. The hippocampus is vulnerable to high levels of IL-1β, IL-6, and TNFα, and these cytokines may reduce synaptic plasticity and may inhibit neurogenesis [59].
High levels of inflammatory plasma IL-6 and TNFα have been significantly related to poorer memory. LPS administration produced a global decrease in memory functions, reflected by decreased immediate recall of story items, reduced delayed story recall, impaired immediate and delayed recall of figure items, and decreased performance in Word List Learning. With low dose LPS injection (.2 ng/kg), declarative memory performance was worse when there were LPS-induced increases in circulating IL-6 levels [65].
6.1. Excess Inflammation Increases Cognitive Decline
It has been demonstrated that higher plasma LPS can increase inflammatory cytokines. Studies have shown that excess immune activation can contribute to and drive AD pathogenesis. Inflammation in AD primarily concerns the innate immune system. Inflammatory IL-1β, IL-6, and TNFα are up-regulated in the brains and cerebrospinal fluid of humans with AD [62].
In three hundred community-dwelling subjects with mild to severe Alzheimer disease, high levels of TNFα were associated with a four-fold increase in the rate of cognitive decline. In contrast, subjects who had low levels of serum TNFα throughout the study showed no cognitive decline over a six-month period. It can be hypothesized that in AD, acute systemic inflammation associated with increased TNFα can lead to increased cognitive decline [86].
There is a growing appreciation of the role of cytokine-mediated inflammation in neurodegenerative diseases such as AD and vascular dementia. Peripheral cytokines enter the blood-brain barrier directly via active transport. IL-1β, IL-6, and TNFα are typically considered proinflammatory, whereas IL-4, IL-10, and IL-13 are typically considered anti-inflammatory. There is abundant evidence that inflammatory mechanisms within the central nervous system contribute to cognitive impairment via cytokine-mediated interactions between neurons and glial cells [72]. Higher serum IL-6 Level can predict the development of disability in older persons and can increase their risk of dementia [87].
High levels of serum IL-6 have been correlated with lower cognitive functioning and can raise the risk of subsequent cognitive decline [72]. Clinical observations in humans suggest that there is a cytokine-mediated decrease in memory functions as a consequence of acute peripheral inflammation. Mean reaction time was significantly reduced by 27.6 milliseconds after endotoxin administration exclusively in the high-dose group [56].
Acute systemic inflammation can elicit a negative impact on central nervous system function, inducing neuropathological changes which can accelerate the onset of neurodegenerative disease. In a cohort of 275 AD patients across a six month period, elevated serum TNFα was significantly correlated with accelerated cognitive decline. Conversely, those patients with low serum TNFα showed stable cognitive function during this period. Elevated serum IL-1β was significantly associated with increased cognitive decline across a two-month period [88].
Plasma levels of inflammatory proteins have been found to be increased before the clinical onset of dementia, indicating that they may be a contributing factor. When IL-6 was higher in the Rotterdam Study, risk of AD and dementia was elevated by 28% and risk of vascular dementia was elevated by 26%, per standard deviation of IL-6 [89].
Cytokine-mediated inflammation is likely to exacerbate progression of AD in association with factors such as reactive oxygen damage. Subjects with late-stage dementia have been shown to have higher levels of IL-6 in the entorhinal cortex and superior temporal gyrus at autopsy [72]. Systemic and CNS inflammation, including heightened IL-1β, TNFα, and interferon-beta, can induce cognitive changes and accelerate neurodegenerative disease [90].
Cytokines can activate glial cells and, conversely, glial cells can produce cytokines when activated. Specific combinations of cytokines can induce dose-dependent neuronal injury, so cytokines can act as direct contributors to neuronal injury upon release by activated microglia or astrocytes. In addition, cytokines can activate microglia to produce free radicals. The debris created by neuronal damage may further activate microglia and astrocytes, leading to a vicious cycle of damage [72].
6.2. Increased IL-6 and Activities of Daily Living
Higher circulating levels of IL-6 predict disability onset in older persons. Participants in the highest IL-6 tertile were 1.76 times more likely to develop mobility-disability and 1.62 times more likely to develop mobility plus activities of daily living (ADL)-disability compared with the lowest IL-6 tertile [87].
Cross-sectional studies have reported an association between increases in IL-6 and functional status as indicated by ADL or instrumental activities of daily living (IADL) measures. IL-6 levels are correlated with subsequent development of disability in ADLs/IADLs [91]. Interleukin-6 and C-reactive protein play an important role in the pathogenesis of dementia. Higher levels of IL-6 predict a decline in the activities of daily living [87] Higher levels of LPS almost doubled the impairment of IADL from 27% to 50% [4].
6.3. Autopsy Findings of Higher Cytokines in AD
Activated microglia and inflammation-related mediators have been found in the cerebral neocortex of autopsied patients with early AD pathology [77]. Increased levels of IL-1β, IL-6, or TNFα have been found in peripheral blood or autopsy specimens of patients with mild to moderate late-onset AD [72].
A study on autopsied AD brains showed the presence of LPS in brain lysates from the hippocampus and superior temporal lobe neocortex. LPS levels in AD brains varied from two-fold increases in the neocortex to three-fold increases in the hippocampus, compared to age-matched control brains. Some hippocampal samples from advanced AD cases exhibited up to a 26-fold increase in LPS over age-matched controls. The hippocampus has been found to be the area of the brain that developed the earliest and most profound neuropathology in AD [92].
6.4. Cytokine Therapy Can Impair Cognition
Normal individuals who receive systemic cytokine therapy such as interferon-alpha (IFN-α), IL-2, and TNFα at therapeutic doses frequently describe impaired thinking processes [93]. Chronic peripheral administration of IL-2 has been associated with hippocampal neurodegeneration and suppression of hippocampal long-term potentiation, leading to impaired memory performance. Therapeutic administration of IFN-gamma in humans can result in an array of CNS side effects including confusion, psychomotor slowing, thought blockade, subjective memory loss, impaired motor and executive functioning, and even sustained slowing of reaction times at higher doses [72].
7. LPS Can Represent a Risk Factor for Atherosclerosis and Reduced Brain Perfusion
Cytokines have prothrombotic effects and can influence many steps of the atherogenic process. IL-1β, IFNγ, and TNFα can induce smooth-muscle degeneration by apoptosis, whereas TNFα, IL-1β, or IL-6 may promote cellular adherence to endothelial cells [94].
The atherogenic and prothrombotic effects of cytokines may directly influence vascular dementia risk. By influencing the response to ischemia, cytokines may determine the point at which multiple serial ischemic insults overcome an individual’s cognitive reserve threshold. In addition, IL-1β and TNFα are known to modulate endothelial functions that govern the formation and stability of blood clots. Cytokines can directly influence the coagulation cascade. IL-1β, IL-2, IL-6, and TNFα can increase thrombosis [72].
7.1. Brain Perfusion
Brain perfusion of blood is vital to supply glucose and oxygen to brain neurons. High cytokine levels can exacerbate the cerebral hypoperfusion and blood–brain barrier leakiness that are associated with AD and vascular dementia [95]. Ninety minutes after LPS injection at a dose of 2 ng/kg body weight in healthy young volunteers, it has been reported that cerebral blood flow was reduced by 24% [96].
7.2. Atheroslerosis, LPS, and LBP
Upon stimulation with LPS, macrophages and neutrophils release thromboxane A2, which can constrict blood vessels and thicken blood. The presence of LPS and proinflammatory cytokines can change endothelial cells to a procoagulant profile. This may increase atherosclerosis and reduce brain perfusion [9]. High plasma LPS levels are associated with major adverse cardiovascular events, possibly by increasing platelet activation [21].
When college students ate a high-fat meal (thin crust cheese pizza), in addition to raising LPS 65 times, there were increases in oxidized LDL uptake and adhesion molecule expression in monocytes. Consumption of a high-fat meal also increased the potential of monocytes to become foam cells. Monocyte-derived foam cells are the predominant component of arterial plaques in the early stages of atherosclerosis [30].
Serum LPS was near zero in young normal people and median LPS levels in hundreds of men and women ages 50-79 was 14.3 pg/mL. Risk of atherosclerosis rose with higher LPS levels [97]. Plasma levels of LPS in patients affected by dementia was 700 ng/mL [71].
LPS and LBP Damage Cerebral Arteries
LPS binds to transmembrane receptors within the toll-like receptor family, initiating a cascade of cytokines and other inflammatory mediators, followed by vascular injury [98]. LPS-induced inflammation via TLR-4 (which is expressed in atherosclerotic plaque) has been shown to play an important role in the pathogenesis of atherosclerosis [28]. TNFα and IL-1β activate blood coagulation and stimulate the expression of adhesion molecules on endothelial cells, possibly reducing brain perfusion [9].
LBP can be used as a marker to indicate activation of an inflammatory response due to plasma exposure to LPS. LBP was reported as a factor related to atherosclerosis. Systemic inflammation, triggered by dietary LPS, has been associated with increased plasma concentrations of pro-inflammatory cytokines, and is a risk factor for atherosclerosis [37].
There was a strong correlation between serum LBP and coronary artery disease. The proinflammatory action of LBP bound to LPS might be an important contributor to the progression of cardiovascular incidents and associated mortality. In those with more severe coronary artery disease, circulating LBP was higher. Circulating LBP was found to be increased in people with elevated carotid plaque. Interestingly, LBP deposits were found inside atherosclerotic plaque. There was a consistent association between serum LBP and the carotid intima media thickness, a widely used atherosclerosis marker [99].
7.3. BBB Damage and Cytokines
The binding of cytokines to endothelial receptors in the brain vessels may stimulate the release of other mediators, such as endothelial cell adhesion molecules, chemokines, nitric oxide, and prostaglandins, which may lead to impairment of BBB integrity [72].
High levels of TNFα and IL-1β can promote the recruitment of inflammatory cells, their adherence to brain endothelial cells, and enhanced vascular permeability of the BBB. Cytokines can increase atherosclerosis and smooth muscle apoptosis of endothelial cells [94]. The barrier function of the BBB decreases with age, in AD, and in response to elevated levels of TNFα, IL-1β, and IL-6 [83].
8. Conclusions
Reducing or eliminating foods with high levels of LPS, such as meat, cheese, and milk, has the potential to reduce neuroinflammation. This can represent an alternative, novel technique to reduce the risk of cognitive impairment and AD progression. If brain inflammation is contributing to cognitive impairment, we hypothesize that cognition may improve or stabilize through a reduction of neuroinflammation. We have shown that, although colonic LPS may increase plasma LPS, the large increase postprandially is due to consuming dietary LPS in food. Dietary LPS in certain foods is absorbed into enterocytes and packaged into chylomicrons, which enter the bloodstream. Once in the bloodstream, LPS triggers the release of inflammatory cytokines within three hours. LPS increases cytokines via LBP, which binds to CD14, which triggers TLR-4, which in turn activates NFқB. NFқB increases cytokines, including interleukin-1-beta (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNFα). Increases in certain cytokines of over 60-fold have been measured after consuming LPS-rich food. These cytokines can enter the brain via active transport and induce microglial activation and a cascade of inflammatory damage in the brain, including loss of synapses and neurons. Therefore, elevated inflammation in the brain can contribute to cognitive impairment and increases the risk of cognitive damage. LPS and inflammatory cytokines are higher in Alzheimer’s disease (AD), both in plasma and in the central nervous system (CNS). We propose that a dietary reduction of LPS could both reduce risk and slow the progression of cognitive decline including Alzheimer’s disease. More studies are needed to determine the benefits of a low LPS diet on those with cognitive impairment, mixed dementia, and AD.
Author Contributions
Conceptualization, S.M.B.; writing—original draft preparation, S.M.B and C.P.B.; writing—review and editing, C.P.B. L.B. M.H. T.H. and P.P; investigation, W.B.G.; supervision, W.B.G.;. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dementia. Availabe online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on March 17, 2024).
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv Nutr 2020, 11, 77-91. [CrossRef]
- Laugerette, F.; Vors, C.; Peretti, N.; Michalski, M.C. Complex links between dietary lipids, endogenous endotoxins and metabolic inflammation. Biochimie 2011, 93, 39-45. [CrossRef]
- Saji, N.; Saito, Y.; Yamashita, T.; Murotani, K.; Tsuduki, T.; Hisada, T.; Sugimoto, T.; Niida, S.; Toba, K.; Sakurai, T. Relationship Between Plasma Lipopolysaccharides, Gut Microbiota, and Dementia: A Cross-Sectional Study. J Alzheimers Dis 2022, 86, 1947-1957. [CrossRef]
- Vors, C.; Drai, J.; Pineau, G.; Laville, M.; Vidal, H.; Laugerette, F.; Michalski, M.C. Emulsifying dietary fat modulates postprandial endotoxemia associated with chylomicronemia in obese men: a pilot randomized crossover study. Lipids Health Dis 2017, 16, 97. [CrossRef]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J Neuroinflammation 2019, 16, 180. [CrossRef]
- Andre, P.; Laugerette, F.; Feart, C. Metabolic Endotoxemia: A Potential Underlying Mechanism of the Relationship between Dietary Fat Intake and Risk for Cognitive Impairments in Humans? Nutrients 2019, 11. [CrossRef]
- Grant, W.B.; Blake, S.M. Diet’s Role in Modifying Risk of Alzheimer’s Disease: History and Present Understanding. J Alzheimers Dis 2023, 96, 1353-1382. [CrossRef]
- Andreasen, A.S.; Krabbe, K.S.; Krogh-Madsen, R.; Taudorf, S.; Pedersen, B.K.; Moller, K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem 2008, 15, 1697-1705. [CrossRef]
- Wu, H.; Wang, Y.; Li, H.; Meng, L.; Zheng, N.; Wang, J. Effect of Food Endotoxin on Infant Health. Toxins (Basel) 2021, 13. [CrossRef]
- Erridge, C. Accumulation of stimulants of Toll-like receptor (TLR)-2 and TLR4 in meat products stored at 5 degrees C. J Food Sci 2011, 76, H72-79. [CrossRef]
- Jay, J.M.; Margitic, S.; Shereda, A.L.; Covington, H.V. Determining endotoxin content of ground beef by the Limulus amoebocyte lysate test as a rapid indicator of microbial quality. Appl Environ Microbiol 1979, 38, 885-890. [CrossRef]
- Gehring, U.; Spithoven, J.; Schmid, S.; Bitter, S.; Braun-Fahrlander, C.; Dalphin, J.C.; Hyvarinen, A.; Pekkanen, J.; Riedler, J.; Weiland, S.K. et al. Endotoxin levels in cow’s milk samples from farming and non-farming families - the PASTURE study. Environ Int 2008, 34, 1132-1136. [CrossRef]
- Venter, P. Endotoxin residues in food: a review. Interim: Interdisciplinary J. 2010, 9, 106-126.
- Ferguson, J.F.; Patel, P.N.; Shah, R.Y.; Mulvey, C.K.; Gadi, R.; Nijjar, P.S.; Usman, H.M.; Mehta, N.N.; Shah, R.; Master, S.R. et al. Race and gender variation in response to evoked inflammation. J Transl Med 2013, 11, 63. [CrossRef]
- Hansen, K.; Mikkelsen, T.; Moller-Madsen, A. Use of the Limulus test to determine the hygienic status of milk products as characterized by levels of gram-negative bacterial lipopolysaccharide present. J Dairy Res 1982, 49, 323-328. [CrossRef]
- Faraj, T.A. Regulation of cardiometabolic risk factors by dietary Toll-like receptor stimulants. PhD, University of Leicester, Leicester, 2017.
- Prester, L.; Orct, T.; Macan, J.; Vukusic, J.; Kipcic, D. Determination of biogenic amines and endotoxin in squid, musky octopus, Norway lobster, and mussel stored at room temperature. Arh Hig Rada Toksikol 2010, 61, 389-397. [CrossRef]
- Herieka, M.; Faraj, T.A.; Erridge, C. Reduced dietary intake of pro-inflammatory Toll-like receptor stimulants favourably modifies markers of cardiometabolic risk in healthy men. Nutr Metab Cardiovasc Dis 2016, 26, 194-200. [CrossRef]
- Brown, B.I. Nutritional Management of Metabolic Endotoxemia: A Clinical Review. Altern Ther Health Med 2017, 23, 42-54.
- Pastori, D.; Carnevale, R.; Nocella, C.; Novo, M.; Santulli, M.; Cammisotto, V.; Menichelli, D.; Pignatelli, P.; Violi, F. Gut-Derived Serum Lipopolysaccharide is Associated With Enhanced Risk of Major Adverse Cardiovascular Events in Atrial Fibrillation: Effect of Adherence to Mediterranean Diet. J Am Heart Assoc 2017, 6. [CrossRef]
- Estadella, D.; da Penha Oller do Nascimento, C.M.; Oyama, L.M.; Ribeiro, E.B.; Damaso, A.R.; de Piano, A. Lipotoxicity: effects of dietary saturated and transfatty acids. Mediators Inflamm 2013, 2013, 137579. [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100-1101 e1102. [CrossRef]
- Mo, Z.; Huang, S.; Burnett, D.J.; Rutledge, J.C.; Hwang, D.H. Endotoxin May Not Be the Major Cause of Postprandial Inflammation in Adults Who Consume a Single High-Fat or Moderately High-Fat Meal. J Nutr 2020, 150, 1303-1312. [CrossRef]
- Meessen, E.C.E.; Warmbrunn, M.V.; Nieuwdorp, M.; Soeters, M.R. Human Postprandial Nutrient Metabolism and Low-Grade Inflammation: A Narrative Review. Nutrients 2019, 11. [CrossRef]
- Ahola, A.J.; Lassenius, M.I.; Forsblom, C.; Harjutsalo, V.; Lehto, M.; Groop, P.H. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci Rep 2017, 7, 6511. [CrossRef]
- Andre, P.; Pais de Barros, J.P.; Mj Merle, B.; Samieri, C.; Helmer, C.; Delcourt, C.; Feart, C. Mediterranean diet and prudent diet are both associated with low circulating esterified 3-hydroxy fatty acids, a proxy of LPS burden, among older adults. Am J Clin Nutr 2021, 114, 1080-1091. [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care 2009, 32, 2281-2287. [CrossRef]
- Szczechowiak, K.; Diniz, B.S.; Leszek, J. Diet and Alzheimer’s dementia - Nutritional approach to modulate inflammation. Pharmacol Biochem Behav 2019, 184, 172743. [CrossRef]
- Henning, A.L.; Venable, A.S.; Vingren, J.L.; Hill, D.W.; McFarlin, B.K. Consumption of a high-fat meal was associated with an increase in monocyte adhesion molecules, scavenger receptors, and Propensity to Form Foam Cells. Cytometry B Clin Cytom 2018, 94, 606-612. [CrossRef]
- Hawkesworth, S.; Moore, S.E.; Fulford, A.J.; Barclay, G.R.; Darboe, A.A.; Mark, H.; Nyan, O.A.; Prentice, A.M. Evidence for metabolic endotoxemia in obese and diabetic Gambian women. Nutr Diabetes 2013, 3, e83. [CrossRef]
- Al-Disi, D.A.; Al-Daghri, N.M.; Khan, N.; Alfadda, A.A.; Sallam, R.M.; Alsaif, M.; Sabico, S.; Tripathi, G.; McTernan, P.G. Postprandial Effect of a High-Fat Meal on Endotoxemia in Arab Women with and without Insulin-Resistance-Related Diseases. Nutrients 2015, 7, 6375-6389. [CrossRef]
- Gonzalez, F.; Considine, R.V.; Abdelhadi, O.A.; Acton, A.J. Saturated Fat Ingestion Promotes Lipopolysaccharide-Mediated Inflammation and Insulin Resistance in Polycystic Ovary Syndrome. J Clin Endocrinol Metab 2019, 104, 934-946. [CrossRef]
- Vors, C.; Pineau, G.; Drai, J.; Meugnier, E.; Pesenti, S.; Laville, M.; Laugerette, F.; Malpuech-Brugere, C.; Vidal, H.; Michalski, M.C. Postprandial Endotoxemia Linked With Chylomicrons and Lipopolysaccharides Handling in Obese Versus Lean Men: A Lipid Dose-Effect Trial. J Clin Endocrinol Metab 2015, 100, 3427-3435. [CrossRef]
- Harte, A.L.; Varma, M.C.; Tripathi, G.; McGee, K.C.; Al-Daghri, N.M.; Al-Attas, O.S.; Sabico, S.; O’Hare, J.P.; Ceriello, A.; Saravanan, P. et al. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care 2012, 35, 375-382. [CrossRef]
- Lyte, J.M.; Gabler, N.K.; Hollis, J.H. Postprandial serum endotoxin in healthy humans is modulated by dietary fat in a randomized, controlled, cross-over study. Lipids Health Dis 2016, 15, 186. [CrossRef]
- Laugerette, F.; Alligier, M.; Bastard, J.P.; Drai, J.; Chanseaume, E.; Lambert-Porcheron, S.; Laville, M.; Morio, B.; Vidal, H.; Michalski, M.C. Overfeeding increases postprandial endotoxemia in men: Inflammatory outcome may depend on LPS transporters LBP and sCD14. Mol Nutr Food Res 2014, 58, 1513-1518. [CrossRef]
- White, A.J.; Wijeyekoon, R.S.; Scott, K.M.; Gunawardana, N.P.; Hayat, S.; Solim, I.H.; McMahon, H.T.; Barker, R.A.; Williams-Gray, C.H. The Peripheral Inflammatory Response to Alpha-Synuclein and Endotoxin in Parkinson’s Disease. Front Neurol 2018, 9, 946. [CrossRef]
- Netto Candido, T.L.; Bressan, J.; Alfenas, R.C.G. Dysbiosis and metabolic endotoxemia induced by high-fat diet. Nutr Hosp 2018, 35, 1432-1440. [CrossRef]
- Erridge, C. Diet, commensals and the intestine as sources of pathogen-associated molecular patterns in atherosclerosis, type 2 diabetes and non-alcoholic fatty liver disease. Atherosclerosis 2011, 216, 1-6. [CrossRef]
- Bowser, S.M.; McMillan, R.P.; Boutagy, N.E.; Tarpey, M.D.; Smithson, A.T.; Osterberg, K.L.; Neilson, A.P.; Davy, B.M.; Davy, K.P.; Hulver, M.W. Serum endotoxin, gut permeability and skeletal muscle metabolic adaptations following a short term high fat diet in humans. Metabolism 2020, 103, 154041. [CrossRef]
- Wan, Y.; Wang, F.; Yuan, J.; Li, J.; Jiang, D.; Zhang, J.; Li, H.; Wang, R.; Tang, J.; Huang, T. et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: a 6-month randomised controlled-feeding trial. Gut 2019, 68, 1417-1429. [CrossRef]
- Michalski, M.C.; Le Barz, M.; vors, C. Metabolic impact of dietary lipids: towards a role of unabsorbed lipid residues? OCL 2021, 28, 9. [CrossRef]
- Lakhan, S.E.; Kirchgessner, A. Gut inflammation in chronic fatigue syndrome. Nutr Metab (Lond) 2010, 7, 79. [CrossRef]
- Burkitt, D.P.; Walker, A.R.; Painter, N.S. Effect of dietary fibre on stools and the transit-times, and its role in the causation of disease. Lancet 1972, 2, 1408-1412. [CrossRef]
- Erridge, C. The capacity of foodstuffs to induce innate immune activation of human monocytes in vitro is dependent on food content of stimulants of Toll-like receptors 2 and 4. Br J Nutr 2011, 105, 15-23. [CrossRef]
- Moreira, A.P.; Texeira, T.F.; Ferreira, A.B.; Peluzio Mdo, C.; Alfenas Rde, C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr 2012, 108, 801-809. [CrossRef]
- Kastl, A.J. Jr.; Terry, N.A.; Wu, G.D.; Albenberg, L.G. The Structure and Function of the Human Small Intestinal Microbiota: Current Understanding and Future Directions. Cell Mol Gastroenterol Hepatol 2020, 9, 33-45. [CrossRef]
- Reichenberg, A.; Yirmiya, R.; Schuld, A.; Kraus, T.; Haack, M.; Morag, A.; Pollmacher, T. Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiatry 2001, 58, 445-452. [CrossRef]
- Cohen, O.; Reichenberg, A.; Perry, C.; Ginzberg, D.; Pollmacher, T.; Soreq, H.; Yirmiya, R. Endotoxin-induced changes in human working and declarative memory associate with cleavage of plasma “readthrough” acetylcholinesterase. J Mol Neurosci 2003, 21, 199-212. [CrossRef]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res 2009, 50, 90-97. [CrossRef]
- Madison, A.A.; Belury, M.A.; Andridge, R.; Shrout, M.R.; Renna, M.E.; Malarkey, W.B.; Bailey, M.T.; Kiecolt-Glaser, J.K. Afternoon distraction: a high-saturated-fat meal and endotoxemia impact postmeal attention in a randomized crossover trial. Am J Clin Nutr 2020, 111, 1150-1158. [CrossRef]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferrieres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr 2008, 87, 1219-1223. [CrossRef]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr 2007, 86, 1286-1292. [CrossRef]
- Moieni, M.; Irwin, M.R.; Jevtic, I.; Breen, E.C.; Eisenberger, N.I. Inflammation impairs social cognitive processing: A randomized controlled trial of endotoxin. Brain Behav Immun 2015, 48, 132-138. [CrossRef]
- Grigoleit, J.S.; Kullmann, J.S.; Wolf, O.T.; Hammes, F.; Wegner, A.; Jablonowski, S.; Engler, H.; Gizewski, E.; Oberbeck, R.; Schedlowski, M. Dose-dependent effects of endotoxin on neurobehavioral functions in humans. PLoS One 2011, 6, e28330. [CrossRef]
- Andre, P.; Samieri, C.; Buisson, C.; Dartigues, J.F.; Helmer, C.; Laugerette, F.; Feart, C. Lipopolysaccharide-Binding Protein, Soluble CD14, and the Long-Term Risk of Alzheimer’s Disease: A Nested Case-Control Pilot Study of Older Community Dwellers from the Three-City Cohort. J Alzheimers Dis 2019, 71, 751-761. [CrossRef]
- DellaGioia, N.; Hannestad, J. A critical review of human endotoxin administration as an experimental paradigm of depression. Neurosci Biobehav Rev 2010, 34, 130-143. [CrossRef]
- Melo, H.M.; Santos, L.E.; Ferreira, S.T. Diet-Derived Fatty Acids, Brain Inflammation, and Mental Health. Front Neurosci 2019, 13, 265. [CrossRef]
- Arnesen, P.M.; Wettergreen, M.; You, P.; Fladby, T. Alzheimer’s disease risk genes are differentially expressed upon Lipopolysaccharide stimulation in a myelogenic cell model. Alzheimer’s and Dementia 2023, 19, e080112.
- Gayle, D.A.; Ling, Z.; Tong, C.; Landers, T.; Lipton, J.W.; Carvey, P.M. Lipopolysaccharide (LPS)-induced dopamine cell loss in culture: roles of tumor necrosis factor-alpha, interleukin-1beta, and nitric oxide. Brain Res Dev Brain Res 2002, 133, 27-35. [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 2015, 16, 358-372. [CrossRef]
- Moreno-Navarrete, J.M.; Blasco, G.; Puig, J.; Biarnes, C.; Rivero, M.; Gich, J.; Fernandez-Aranda, F.; Garre-Olmo, J.; Ramio-Torrenta, L.; Alberich-Bayarri, A. et al. Neuroinflammation in obesity: circulating lipopolysaccharide-binding protein associates with brain structure and cognitive performance. Int J Obes (Lond) 2017, 41, 1627-1635. [CrossRef]
- Zhao, Y.; Sharfman, N.M.; Jaber, V.R.; Lukiw, W.J. Down-Regulation of Essential Synaptic Components by GI-Tract Microbiome-Derived Lipopolysaccharide (LPS) in LPS-Treated Human Neuronal-Glial (HNG) Cells in Primary Culture: Relevance to Alzheimer’s Disease (AD). Front Cell Neurosci 2019, 13, 314. [CrossRef]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 2011, 25, 181-213. [CrossRef]
- Fiebich, B.L.; Hull, M.; Lieb, K.; Schumann, G.; Berger, M.; Bauer, J. Potential link between interleukin-6 and arachidonic acid metabolism in Alzheimer’s disease. J Neural Transm Suppl 1998, 54, 268-278.
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L. et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med 2013, 11, 200. [CrossRef]
- Zhan, X.; Hakoupian, M.; Jin, L.W.; Sharp, F.R. Lipopolysaccharide, Identified Using an Antibody and by PAS Staining, Is Associated With Corpora amylacea and White Matter Injury in Alzheimer’s Disease and Aging Brain. Front Aging Neurosci 2021, 13, 705594. [CrossRef]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Gastrointestinal Tract Microbiome-Derived Pro-inflammatory Neurotoxins in Alzheimer’s Disease. J Aging Sci 2021, 9.
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol 2009, 206, 121-124. [CrossRef]
- Sanchez-Tapia, M.; Mimenza-Alvarado, A.; Granados-Dominguez, L.; Flores-Lopez, A.; Lopez-Barradas, A.; Ortiz, V.; Perez-Cruz, C.; Sanchez-Vidal, H.; Hernandez-Acosta, J.; Avila-Funes, J.A. et al. The Gut Microbiota-Brain Axis during Aging, Mild Cognitive Impairment and Dementia: Role of Tau Protein, beta-Amyloid and LPS in Serum and Curli Protein in Stool. Nutrients 2023, 15. [CrossRef]
- Wilson, C.J.; Finch, C.E.; Cohen, H.J. Cytokines and cognition--the case for a head-to-toe inflammatory paradigm. J Am Geriatr Soc 2002, 50, 2041-2056. [CrossRef]
- Niehaus, I.; Lange, J.H. Endotoxin: is it an environmental factor in the cause of Parkinson’s disease? Occup Environ Med 2003, 60, 378. [CrossRef]
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 2005, 25, 9275-9284. [CrossRef]
- Creely, S.J.; McTernan, P.G.; Kusminski, C.M.; Fisher f, M.; Da Silva, N.F.; Khanolkar, M.; Evans, M.; Harte, A.L.; Kumar, S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab 2007, 292, E740-747. [CrossRef]
- McGrattan, A.M.; McGuinness, B.; McKinley, M.C.; Kee, F.; Passmore, P.; Woodside, J.V.; McEvoy, C.T. Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease. Curr Nutr Rep 2019, 8, 53-65. [CrossRef]
- Eikelenboom, P.; van Exel, E.; Hoozemans, J.J.; Veerhuis, R.; Rozemuller, A.J.; van Gool, W.A. Neuroinflammation - an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener Dis 2010, 7, 38-41. [CrossRef]
- Fassbender, K.; Walter, S.; Kuhl, S.; Landmann, R.; Ishii, K.; Bertsch, T.; Stalder, A.K.; Muehlhauser, F.; Liu, Y.; Ulmer, A.J. et al. The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J 2004, 18, 203-205. [CrossRef]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation 2008, 5, 37. [CrossRef]
- Zhan, X.; Stamova, B.; Jin, L.W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324-2332. [CrossRef]
- Marizzoni, M.; Cattaneo, A.; Mirabelli, P.; Festari, C.; Lopizzo, N.; Nicolosi, V.; Mombelli, E.; Mazzelli, M.; Luongo, D.; Naviglio, D. et al. Short-Chain Fatty Acids and Lipopolysaccharide as Mediators Between Gut Dysbiosis and Amyloid Pathology in Alzheimer’s Disease. J Alzheimers Dis 2020, 78, 683-697. [CrossRef]
- Kalmijn, S.; Launer, L.J.; Ott, A.; Witteman, J.C.; Hofman, A.; Breteler, M.M. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol 1997, 42, 776-782. [CrossRef]
- Krabbe, K.S.; Reichenberg, A.; Yirmiya, R.; Smed, A.; Pedersen, B.K.; Bruunsgaard, H. Low-dose endotoxemia and human neuropsychological functions. Brain Behav Immun 2005, 19, 453-460. [CrossRef]
- Salardini, A.; Hillmer, A.T.; AP, M.; A, H.-A.; E, L.; SE, S.; RS, O.D.; JE, H.; TA, G.; NB, N. et al. PBR28 brain PET imaging with lipopolysaccharide challenge for the study of microglia function in Alzheimer’s disease. In Proceedings of 2020 Alzheimer’s Association International Conference July 28, 2020.
- Sandiego, C.M.; Gallezot, J.D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.F.; Matuskey, D.; Lee, J.Y.; O’Connor, K.C.; Huang, Y. et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci U S A 2015, 112, 12468-12473. [CrossRef]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768-774. [CrossRef]
- Ferrucci, L.; Harris, T.B.; Guralnik, J.M.; Tracy, R.P.; Corti, M.C.; Cohen, H.J.; Penninx, B.; Pahor, M.; Wallace, R.; Havlik, R.J. Serum IL-6 level and the development of disability in older persons. J Am Geriatr Soc 1999, 47, 639-646. [CrossRef]
- Cunningham, C. Microglia and neurodegeneration: the role of systemic inflammation. Glia 2013, 61, 71-90. [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Meijer, J.; Kiliaan, A.; Ruitenberg, A.; van Swieten, J.C.; Stijnen, T.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol 2004, 61, 668-672. [CrossRef]
- Cunningham, C.; Campion, S.; Lunnon, K.; Murray, C.L.; Woods, J.F.; Deacon, R.M.; Rawlins, J.N.; Perry, V.H. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry 2009, 65, 304-312. [CrossRef]
- Cohen, H.J.; Pieper, C.F.; Harris, T.; Rao, K.M.; Currie, M.S. The association of plasma IL-6 levels with functional disability in community-dwelling elderly. J Gerontol A Biol Sci Med Sci 1997, 52, M201-208. [CrossRef]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Secretory Products of the Human GI Tract Microbiome and Their Potential Impact on Alzheimer’s Disease (AD): Detection of Lipopolysaccharide (LPS) in AD Hippocampus. Front Cell Infect Microbiol 2017, 7, 318. [CrossRef]
- Licinio, J.; Kling, M.A.; Hauser, P. Cytokines and brain function: relevance to interferon-alpha-induced mood and cognitive changes. Semin Oncol 1998, 25, 30-38.
- Arvin, B.; Neville, L.F.; Barone, F.C.; Feuerstein, G.Z. The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev 1996, 20, 445-452. [CrossRef]
- Asby, D.; Boche, D.; Allan, S.; Love, S.; Miners, J.S. Systemic infection exacerbates cerebrovascular dysfunction in Alzheimer’s disease. Brain 2021, 144, 1869-1883. [CrossRef]
- Moller, K.; Strauss, G.I.; Qvist, J.; Fonsmark, L.; Knudsen, G.M.; Larsen, F.S.; Krabbe, K.S.; Skinhoj, P.; Pedersen, B.K. Cerebral blood flow and oxidative metabolism during human endotoxemia. J Cereb Blood Flow Metab 2002, 22, 1262-1270. [CrossRef]
- Wiedermann, C.J.; Kiechl, S.; Dunzendorfer, S.; Schratzberger, P.; Egger, G.; Oberhollenzer, F.; Willeit, J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. J Am Coll Cardiol 1999, 34, 1975-1981. [CrossRef]
- Funari, E.; Testai, E. Human health risk assessment related to cyanotoxins exposure. Crit Rev Toxicol 2008, 38, 97-125. [CrossRef]
- Serrano, M.; Moreno-Navarrete, J.M.; Puig, J.; Moreno, M.; Guerra, E.; Ortega, F.; Xifra, G.; Ricart, W.; Fernandez-Real, J.M. Serum lipopolysaccharide-binding protein as a marker of atherosclerosis. Atherosclerosis 2013, 230, 223-227. [CrossRef]
Figure 1.
The proposed mechanisms leading to an improvement of cognition by reducing dietary LPS. LPS=lipopolysaccharides, LBP=LPS binding protein, CD14=cluster of differentiation-14, IL-1β=interleukin-1-beta, IL-6= interleukin-6, TNFα=tumor necrosis factor-alpha.
Figure 1.
The proposed mechanisms leading to an improvement of cognition by reducing dietary LPS. LPS=lipopolysaccharides, LBP=LPS binding protein, CD14=cluster of differentiation-14, IL-1β=interleukin-1-beta, IL-6= interleukin-6, TNFα=tumor necrosis factor-alpha.
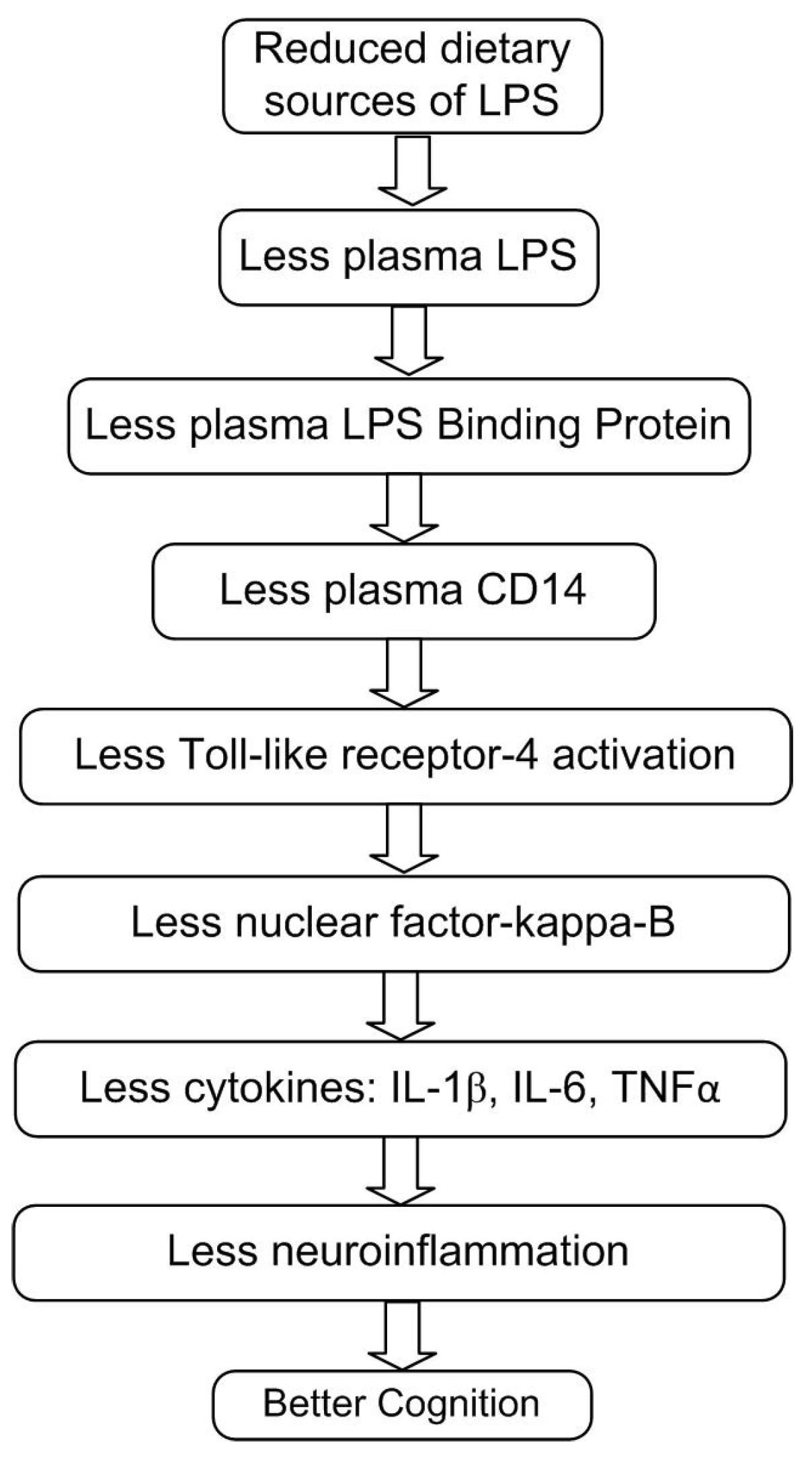
Figure 2.
LPS-triggered cytokines in food samples. Dark bars are soluble, grey insoluble (adapted from Erridge, 2011) [11].
Figure 2.
LPS-triggered cytokines in food samples. Dark bars are soluble, grey insoluble (adapted from Erridge, 2011) [11].
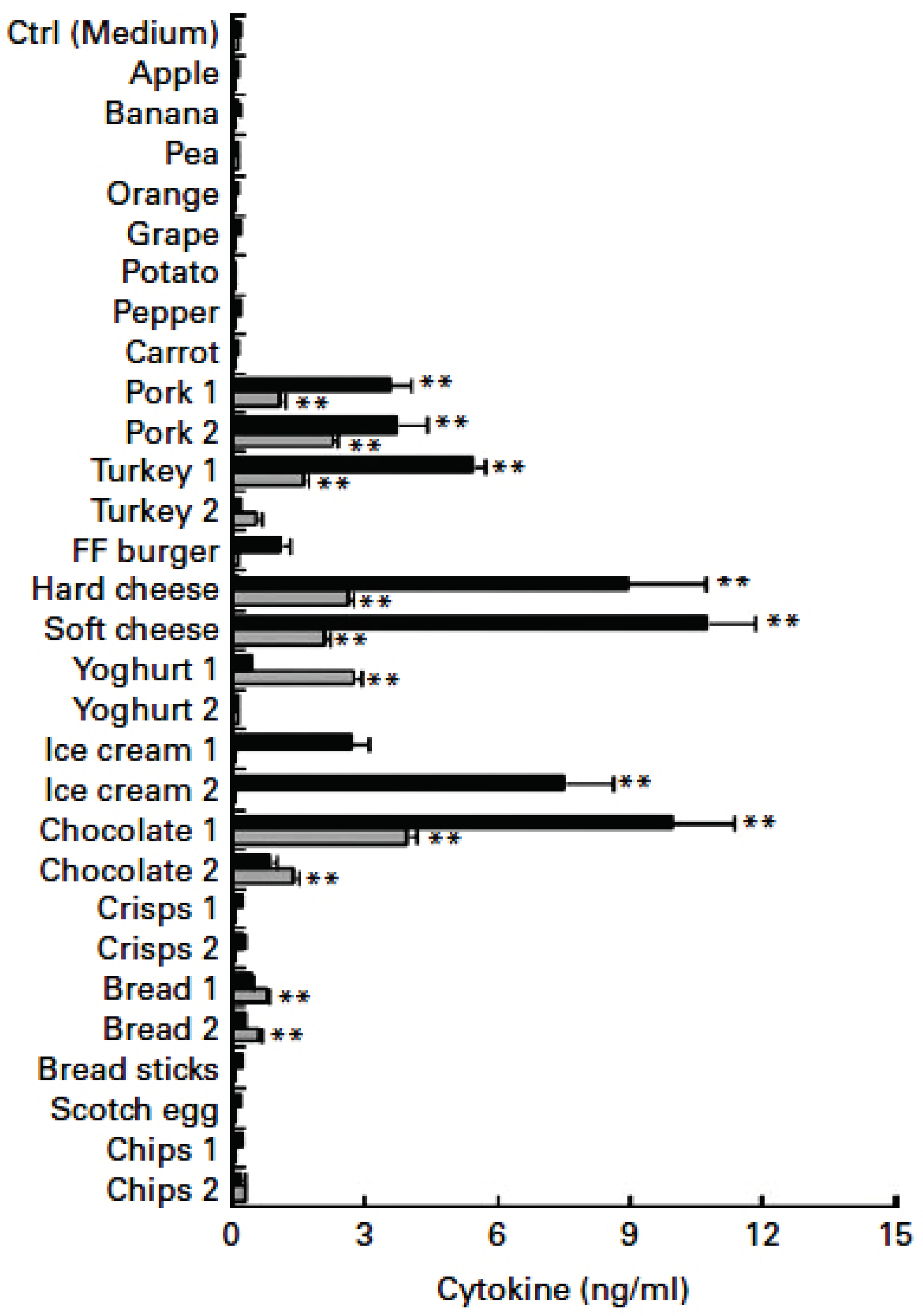

Table 1.
LPS per serving in food, references are in the text.
| Food | ng/g of LPS | Serving Size g | LPS per Serving in ng |
|---|---|---|---|
| Macaroni cheese | 6,500 | 340 | 2,200,000 |
| Minced turkey | 7800 | 230 | 1,800,000 |
| Cheese and onion Rolls | 17,000 | 74 | 1,300,000 |
| Minced pork | 10,000 | 110 | 1,100,000 |
| Minced beef | 7000 | 98 | 690,000 |
| Pork sausage rolls | 4,200 | 2 rolls 120 | 520,000 |
| Turkey | 300 | 230 | 510,000 |
| Hamburger patty | 3090 | 98 | 300,000 |
| Infant formula milk powder | 2800 | 100 | 280,000 |
| Lobster | 1200 | 145 | 170,000 |
| Pork | 1100 | 110 | 120,000 |
| Spaghetti bolognese | 220 | 400 | 90,000 |
| Minced beef and onion pie | 330 | 310 | 69,000 |
| Skim milk, one cup | 75 ng/ml | 240 | 18,000 |
| Milk, one cup | 50 | 240 | 12,000 |
Table 2.
Cognitive test results from elevated LPS, LBP, or SFAs.
| Exposure to LPS | Amount | Cognitive Test Result |
|---|---|---|
| LPS injection | 0.8 ng/kg of body weight | Impairment of social cognitive processing [55]. |
| LPS injection | 0.4 ng/kg or 0.8 ng/kg body weight | Long-term memory performance was impaired, low dose only [56]. |
| LPS injection | 0.2 ng/kg body weight | Degraded performance in declarative memory [57]. |
| LPS-induced cytokine secretion | 49 ng Injection of LPS | Impairments in verbal and nonverbal declarative memory functions and decrease immediate and delayed recall [50]. |
| LPS injection | 0.2 ng/kg body weight | Significant impairment in declarative memory and word-list learning [7]. |
| LPS injection | 0.8 ng/kg body weight | Negative effects on memory function [9]. |
| Levels of circulating LBP | Higher | Impaired cognitive performance [57]. |
| Levels of circulating LBP | Higher | Poorer working memory/short-term verbal memory and the Digit Span Test Puig, 2017}. |
| LPS administration | Low-dose | Impaired both immediate and delayed recall [58]. |
| Plasma LPS | Elevated | Performed worse on the post-meal cognitive test and had poorer attention during testing Belury, 2020}. |
| Cholesterol and SFAs | High intake | Increased the risk of impaired cognitive function and the development of dementia, including AD [59]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



