
Submitted:
27 March 2024
Posted:
28 March 2024
You are already at the latest version
Abstract
The development of multiple drug resistant (MDR) cancer all too often signals the need for alternative toxic therapy or palliative care. Our recent in vivo and in vitro studies using canine MDR lymphoma cancer cells demonstrated that the Anaphase Promoting Complex (APC) is impaired in MDR cells compared to normal canine control and drug sensitive cancer cells. Here, we sought to establish whether this phenomena is a generalizable mechanism independent of species, malignancy type or chemotherapy regime. To test the association of blunted APC activity with MDR cancer behavior, we used matched parental and MDR MCF7 human breast cancer cells, and a patient derived xenograft (PDX) model of human breast cancer. We show that APC activating mechanisms, such as APC subunit 1 (APC1) phosphorylation and CDC27/CDC20 protein associations, are reduced in MCF7 MDR cells when compared to chemosensitive matched cell lines. Consistent with impaired APC function in MDR cells, APC substrate proteins failed to be effectively degraded. Similar to our previous observations in canine MDR lymphoma cells, chemical activation of the APC using Mad2 Inhibitor-1 (M2I-1) in MCF7 MDR cells enhanced APC substrate degradation and resensitized MDR cells in vitro to the cytotoxic effects of the alkylating chemotherapeutic agent, Doxorubicin (DOX). Using cell cycle arrest/release experiments we show that chemo-sensitive and -resistant MCF7 cells progress through the cell cycle at similar rates, but mitosis is delayed in MDR cells with elevated substrate levels. When treated with M2I-1, MDR cells progress through mitosis at a faster rate that coincides with reduced levels of APC substrates. In our PDX model, mice growing a clinically-MDR human breast cancer tumor show significantly reduced tumor growth when treated with M2I-1, with evidence of increased DNA damage and apoptosis. Thus, our results strongly support the hypothesis that APC impairment is a driver of aggressive tumor development and that targeting the APC for activation has the potential for meaningful clinical benefits in treating recurrent cases of MDR malignancy.
Keywords:
Multiple Drug Resistant breast cancer
; Anaphase Promoting Complex
; cell culture
; PDX mouse model
Introduction
Cancer is the leading cause of death in Canada, accounting for 28% of all deaths [1,2]. It is estimated that 2 in 5 will develop cancer in their lifetimes with 1 in 4 dying from cancer. The 5-year survival rate for all cancers was at 64% from 2015-17, but it varies, with lung cancer survival at 22% and breast cancer survival at 89%. Cancer recurrence is a major issue with almost all cancers. Initial treatment benefits can be met in later life with recurrence of the cancer. The rates differ, with glioblastoma experiencing almost 100% recurrence and estrogen receptor positive breast cancers experiencing 5-9% recurrence [3]. Breast cancers (BC) are heterogenous in nature and are typically described as Luminal A (estrogen/progesterone receptor (ER/PR)+, HER2-, Ki-67-), Luminal B (ER/PR+, HER2-, Ki-67+), Luminal HER2 (ER/PR+, HER2+), HER2 enriched (ER/PR-, HER2+), Basal-like (ER/PR-, HER2-, EGFR+) and triple negative (ER/PR-, HER -, EGFR-) [4]. Luminal A has the best prognosis and lowest rate of recurrence, while the triple negative BC subtype is the most aggressive and most likely to relapse. ER+ tumors are the most common, at ~80% [5,6], with the recurrence of these tumors presenting a significant clinical problem globally. After a 5-year survival period, patients with low grade ER+ tumors experienced recurrence rates of 10%, whereas those with high grade tumors had a recurrence rate of 17%, after 5-20 years [7]. Recurrent drug resistant cancers occur for a number of reasons, with a variety of hallmark responses described: decreased expression of drug targets, increased expression of drug pumps and drug detoxification mechanisms, reduced apoptosis capacity, increased DNA repair, and altered proliferation [8]. Although we are gaining advanced knowledge of the variety of resistance mechanisms displayed, we still do not have a clear understanding of how these mechanisms function, why they are induced, nor how to impede them.
An additional mechanism driving MDR that has come to light in recent years is impairment of Anaphase Promoting Complex (APC) activity that is associated with drug resistant cancer [9,10]. Numerous studies have observed that APC inhibition is linked with aggressive cancer development in vitro and in vivo [11,12,13,14,15]. Decreased APC activity impairs and slows mitotic progression, permitting further mutagenesis through mitotic delays, increasing aneuploidy and subsequent mitotic slippage [16,17,18]. Decreased APC activity via APC subunit mutation, co-activator (CDH1) mutations [11,19,20,21] or impaired upstream signaling is associated with genomic instability and MDR onset [22,23,24,25,26].
Our recent work supports observations that the APC is inhibited in aggressive cancer cells and that APC activation can reverse drug resistance [9]. We demonstrated that metformin treatment, when combined with CHOP chemotherapy (Cyclphosphamide, Doxorubicin (DOX), Vincristine, and Prednisone), reversed MDR lymphoma in canines in vivo, as it did in vitro [27]; all dogs tested showed reduced expression of markers of MDR and one canine went into remission [9]. We found that tumor samples derived from the canines expressed high levels of all 33 different APC substrate mRNAs that were present on the canine microarray, and that metformin treatment reduced all levels to normal, indicating that metformin induced APC activity. Using OSW canine lymphoma cells selected for MDR, we demonstrated that activation of the APC reduced RNA and protein levels of all APC substrates tested and resensitized the canine MDR cells to chemotherapy. Accumulation of APC substrates and presumed APC inactivation was previously described during cancer progression towards more aggressive behaviors and treatment nonresponsiveness [20]. Indeed, the accumulation of mitotic specific proteins in G1 is associated with aggressive cancer progression in patient samples [28]. This supports targeting the APC to increase its activity to manage MDR malignancies potentially through a mechanism enabling prompt (not delayed) entry into mitosis; driving cancer cells rapidly through anaphase while carrying heavy mutational loads results in chromosome instability and appears to be unsustainable, causing mitotic catastrophe and cell death [29,30,31].
The study described here used human MCF7 breast cancer cells selected for resistance to DOX or Tamoxifen (TAM). We observed that APC activity is impaired in both DOX- or TAM-resistant human MCF7 cells. The doubling time appeared similar in parental and selected cells, but MDR cells required more time to progress through mitosis. Furthermore, we also found that APC mitotic substrates take longer to degrade in mitosis, and begin to accumulate faster as the cell cycle progressed into G1, indicating a strong uncoupling between cell cycle passage into and out of mitosis, and APC E3 activity in this MDR cell population. We also demonstrate that in vitro activation of the APC in MDR-selected cells i) enhanced the turnover of APC targets in synchronous and asynchronous cells, ii) recoupled APC activity with cell cycle progression through mitosis, and iii) synergized with DOX to increase cell killing. Our observations are consistent with the critical nature of the APC in protecting cells from aggressive cancer behavior, and confirms that its influence extends beyond a single cancer type, a single species, or a single chemotherapy class.
Results
APC Activity Is Impaired in Drug Resistant MCF7 Human Breast Cancer Cells
To assess whether the restoration of chemosensitivity in MDR cells following APC activation was independent of species and cancer type, human MCF7 ER+ breast cancer cell populations were selected for resistance to TAM according to our published methods [27,32]. As shown in Figure 1A, cells selected for resistance to TAM alone were more resistant to both 5 mM TAM and 1 mM DOX than matched parental cell populations. To assess APC activity in these cell populations, we began by measuring APC1 phosphorylated at serine 355, a key marker of APC activation [33,34]. APC1 must be phosphorylated in order for CDC20 co-activator recruitment to the APC at mitosis. We show, using APC1 serine 355 (APC1S355ph) specific antibodies, that APC1 phosphorylation is reduced in MCF7 cells selected for resistance to TAM (Figures 1B, 1C). Notably, these MCF7 parental and resistant cells treated with an APC chemical activator (M2I-1; [35]) or an APC chemical inhibitor (APCIN; [36]) demonstrated that APC1S355ph was indeed downregulated when treated with APCIN and elevated when treated with M2I-1 (Figures 1B, 1C). In Figure 1C, the phosphorylated versus total APC1 (APC1S355ph:APC1tot ) ratio was determined after all bands from 3 separate experiments were scanned, normalized to the GAPDH load control, and then plotted. It is clear that the inherent level of APC1 phosphorylation in MCF7 chemoresistant cells is similar to that in unselected parental cells treated with APCIN, and that the level in resistant cells normalizes up to parental levels when treated with M2I-1. This is consistent with our findings that M2I-1 exposure reduced APC substrate protein levels, a marker of APC activation, in canine OSW lymphoma cells selected for resistance to DOX [9]. This also suggests that the APC defect is fully reversible, which is clinically important when considering this as a potential treatment target.
Next, we measured the degree of recruitment of the CDC20 coactivator to the APC in parental versus MDR cell populations; CDC20 interacts with the APC subunit CDC27 upon APC1 phosphorylation at mitosis and contributes to APC activation [37]. First, we immunoprecipitated CDC27 from MCF7 sensitive and resistant cells and measured the relative amount of CDC20 that was co-immunoprecipitated (Figure 1D). As a control we used antibodies against CDC27 to show that similar amounts of CDC27 were immunoprecipitated from both resistant and parental cells. Antibodies against CDC20 demonstrate that markedly less CDC20 was found associated with CDC27 in the resistant cells. We next used antibodies against a second APC co-activator, CDH1. Similar to CDC20, there is less CDH1 associated with CDC27 in TAM selected cells. Figure 1E shows the quantification of the CDC20-bound lanes in two repeats of the experiment shown in Figure 1D. The reciprocal co-immunoprecipitation experiment was performed (Figure 1F) with consistent results, again showing decreased CDC27 associations with CDC20 specifically in resistant cells. Furthermore, we used antibodies against protein markers of MDR (BCRP, MDR-1 and TFPI) [9,27,32,38] and DNA damage (gH2AX) [39] to confirm that the TAM selected cells are indeed multiple drug resistant and experiencing higher levels of DNA damage (Figures 2A, 2B; two separate experiments done independently are shown in A and B). Our data shown here indicates that the reduction of CDC20 and CDH1 interactions with CDC27 are not cell cycle specific and reflect an impairment of both APCCDC20 and APCCDH1.
We then compared the relative abundance of APC degradation-substrates in MCF7 parental and chemoresistant cell populations. If the APC is specifically impaired in MCF7 resistant cells, then APC substrates should accumulate compared to parental cells. Consistent with this, multiple APC substrate proteins (CDC20, Cyclin B1, HURP and Securin) were elevated in TAM selected cells. Next, we assessed MDR and APC substrate protein levels in DOX selected MCF7 cells to ensure that it was not a drug-specific effect, as TAM and DOX are unrelated first line therapeutics for breast cancer. As anticipated, higher levels of MDR protein markers (TFPI) were observed (Figure 2C), as shown previously [27,32,38]. Furthermore, higher APC substrate protein levels (CDC20 and Cyclin B1) were present. Taken together, our observations strongly support the hypothesis that APC activity is impaired in human MCF7 breast cancer cells selected for resistance to unrelated chemotherapeutic agents. This suggests that our observation that APC activity is reduced in canine MDR cancer cells in vitro and in vivo [9] is not canine specific, but potentially a common or recurrent feature of drug resistant cancer cells.
APC Activation In Vitro Slows MDR Cancer Cell Growth and Restores APC Substrate Protein Levels to Normal
We have previously shown that APC activation in vitro reduced APC substrate protein levels in chemoresistant canine lymphoma cells, and resensitized them to DOX [9]. Here, we exposed DOX-resistant MCF7 breast cancer cells to increasing doses of the APC activator, M2I-1, and assessed APC substrate levels and changes to relative DOX resistance. Western analysis for levels of the APC substrate HURP (Figure 3A) showed that it is elevated in selected cells compared to matched parental cells, and the levels in MDR populations return to parental levels at the highest M2I-1 dose used. We also noted that M2I-1 can reduce protein levels of the APC target Cyclin B1 in MCF7 parental cells, but a higher dose of M2I-1 is required (Figure 3B). Next, we measured the viability of MCF7 parental and TAM selected cells, using MTT, following pretreatment of cells with the doses of M2I-1 shown for 18 hours, followed by 48 hours of DOX exposure at levels previously determined to differentiate between chemosensitive and chemoresistant populations (Figure 1A). Importantly, M2I-1 alone did not impact cell viability at the concentrations used, but when combined with DOX it restored chemosensitivity, as demonstrated by the reduced viability of MCF7TAM cells that was comparable to that of parental sensitive cells (Figure 3C).
In Vivo APC Activation in Tumor-Bearing Mice Stalls Tumor Growth
We have described our patient-derived MDR breast cancer tumour tissue, 4-28, that reliably grows as a xenograft (PDX) in mice (NOD/PrkdcSCID/IL2RN-/-) [27]. 4-28 tumor slices taken from a mouse xenograft tumor were implanted into new mice and monitored until palpable. Each mouse received either mock, 5, 10 or a maximum of 25 mg/kg M2I-1 via intraperitoneal injection every 2 days for 8 days (n of 3 per treatment arm).We observed that growth of the 4-28 tumor proceeded unencumbered in mock treated mice (Figure 4A), whereas there was a dose dependent decrease in tumor growth with M2I-1 exposure; notably, the highest M2I-1 dose blocked further tumor growth (Figure 4A). To confirm that APC activity was increased with M2I-1, we measured APC target degradation in excised tumor and liver tissue from both mock and 25 mg/ml M2I-1 treated mice. Western analyses with antibodies against APC substrates Cyclin B1 and CDH1 were performed. Regardless of whether MDR (tumor) or normal tissue (liver) was used, we observed a decrease in Cyclin B1 with M2I-1 treatment. The lower Cyclin B1 band is likely a cleaved Cyclin B1 band as described previously [40]. On the other hand, CDH1 decreased only in MDR tissue and not in liver tissue. In conclusion, within MDR tumors, both Cyclin B1 and CDH1 substrate levels were decreased with M2I-1 treatment, an indication that the APC was activated.
We also assessed these tumor samples for evidence of apoptosis (via PARP cleavage) [41] and DNA damage (via H3K9Ac) [42] following APC activation by M2I-1 (Figure 4B). We noted that the apoptosis present in mock treated tumor was significantly increased upon M2I treatment, in the absence of any chemotherapy, whereas liver tissue had no signal. Similarly, the DNA damage biomarker, H3K9Ac, appeared only in MDR tumor tissue after M2I-1 exposure (Figure 4B). Together, this analysis implies that the APC was activated by M2I-1 in the tumors grown in mice, leading to increased apoptosis, DNA damage, and stalled tumor growth. Importantly, while the systemic dose of M2I-1 used in this analysis was detrimental to tumors, it did not have any obvious impact on the molecular markers measured in the normal liver tissue and the mice after 8 days did not display any overt negative signs of the treatment. This is consistent with the nontoxic nature of M2I-1 in vitro in our hands (Figure 3C).
APC Substrate Degradation Is Delayed during Mitosis in Drug Resistant Cells
To determine if impaired APC function correlates with a delay in mitotic passage in MDR cell populations, as a means of permitting abnormal and/or damaged cells to repair unsustainable DNA damage [43,44,45], we compared cell cycle progression and cycle positioning over time, as well as APC activity in synchronized cells from both sensitive and resistant MCF7 cell lines. We arrested parental and TAM selected MCF7 cells in mitosis (100 nM nocodazole for 16 hours), or in S-phase (a double thymidine block), then washed the cells to remove the arresting agent and allow synchronized cell cycle re-entry, with samples taken at indicated timepoints for up to 24 hours for both flow cytometry and APC target quantification, using western blotting against multiple APC protein substrates. As can be seen in Figure 5A, the abundance of APC substrates HURP and CDC20 are highest in mitotically arrested cells, with yet higher levels observed in TAM selected cells (R), and equally low levels in S arrested cells. The shift from high to low protein levels from M to S is expected [20,46] and validates our experimental approach. The specific increase of APC target proteins in TAM selected mitotic cells, but not in S arrested cells, indicates that APCCDC20 is impaired in mitosis, and not necessarily APCCDH1 in G1/S.
We next determined if increased APC substrate accumulation in MCF7TAM cells could be partially attributed to an extension of the mitotic phase (maximal protein abundance in normal cycling cells), rather than normal timed release into G1 but with inappropriately elevated substrate protein levels. Cells were arrested in mitosis with nocodazole, then the nocodazole was washed away to allow synchronized release into the cell cycle, with samples removed every 2 hours for 24 hours. In Figure 5B, we assessed APC substrate level degradation with western analysis from samples taken every 4 hours. Overall, substrate levels were higher in MCF7TAM (R) cells when arrested with nocodazole, and remained higher for upwards of 8-12 hours for at least HURP. At later time points, HURP, CDC20 and Securin all began to accumulate in MCF7TAM cells earlier than in MCF7Sens (S) cells (see Supplemental Figure 1A for quantitation of the Figure 5B), indicating that control of substrate levels entering and exiting mitosis was consistently impaired in selected cell populations compared to parental MCF7 cell lines. The duration of a complete cell cycle progression compared between parental and selected cells was not obviously different (Figure 5B, lower panel), showing that APC substrate levels began to accumulate incorrectly in selected cells prior to complete synthesis of the genome. Flow cytometry indicated that APC substrates accumulate in MDR cells before a shift from G1 to G2/M, whereas in parental cells APC substrates have not begun to rise (a complete cell cycle flow cytometry profile over the 24 hour time course is presented in Supplemental Figure 1B). Thus, cell cycle progression and APC substrate degradation appear to be temporally uncoupled in selected cells, but are synchronous in parental cells. To confirm that APC substrate degradation is impaired while progressing through mitosis in TAM selected cells, we assessed multiple samples taken over 8 hours from the experiment shown in Figure 5B. This analysis revealed that HURP, CDC20 and Securin levels all remained highly elevated in TAM selected cells for at least 2 hours following release (Figure 5C), in contrast to their degradation in parental cells. An exended analysis of this experiment over 18 hours assessing CDC20 levels is shown in Supplemental Figure 2, confirming that CDC20 degradation occurs more rapidly in parental cells than in selected cells following release from a mitotic arrest.
APC Activation Increases Cell Cycle Progression through Mitosis in MCFTAM Cells in Coordination with Enhanced APC Substrate Degradation
To determine if APC activation using M2I-1 can reduce substrate levels during mitosis and realign the cell cycle with APC activity, we pretreated MCF7Sens and MCF7Tam cells with M2I-1 for 24 hours prior to nocodazole arrest (M2I-1 treatment continued during the nocodazole arrest), or left cells untreated, then assessed target degradation and cell cycle progression. Following the 24 hour M2I-1 pretreatment, samples from unarrested cells (cycling), nocodazole arrested cells (mitosis), and samples taken hourly for 6 hours after synchronized release back into the cell cycle were removed for western and flow cytometry analyses (Figure 6). As expected, untreated asynchronously cycling MCF7Sens and MCF7TAM cells were distributed throughout the cell cycle, and their treatment with nocodazole resulted in a strong M arrest for both (Figure 6A, rows 1 and 3). There were notable differences between MCF7Sens and MCF7TAM cells after M2I-1 pretreatment but before arrest (cycling). It was observed that M2I-1 treatment alone shifted the proportion of both cell populations towards G1, suggesting that M2I-1 promotes or accelerates passage through mitosis (Figure 6A, rows 2 and 4). The subsequent arrest of these pretreated cells also showed differences, as mitotic arrest of MCF7Sens cells was not as efficient as for MCF7TAM cells, indicating that activation of the APC apparently blunts nocodazole-dependent mitotic arrest in parental MCF7 cells, perhaps by allowing some progression into G1. This inefficiency was not observed in pre-treated MCF7TAM cells, where APC is inherently less active.
Synchronized release of untreated cells from mitotis back into the cell cycle also revealed differences between sensitive and MDR cell populations: MCF7TAM cells had a prolonged mitotic pause where it took 4 hours to begin entrance back into the cell cycle, where it then continued with high M content for the remainder of the time course. In contrast, MCF7Sens cells exited mitosis and entered G1 at the 3 hour mark with continuous cell cycle progression (compare Figure 6A, row 1 vs 3). Synchonized release of MCF7Sens M2I-1 pretreated cells from mitosis back into the cell cycle was only modestly faster than MCF7Sens cells that had not been pretreated (Figure 6A row 1 vs 2), whereas the pre-treated MCF7TAM cells did not enter G1 until the 6 hour mark (Figure 6A, row 2 vs 4), similar to untreated MCF7Sens cells.
Lastly, we determined whether M2I-1 activation of the APC in pretreated cells led to coordinated degradation of APC substrates in alignment with the observed increased rate of passage through mitosis and entrance into G1. Samples were taken from the cells grown for Figure 6A before and at mitotic arrest, and then hourly after syncrhonized release back into the cell cycle out to 6 hours. Western analyses of APC substrates was done to determine differences in target protein abundance between synchronized MCF7Sens and MCF7TAM cell populations after M2I pretreatment. Three APC targets were assessed over the 6 hour experiment: CDC20, HURP and Securin (Figure 6B). Without pretreatment, MCF7TAM cells harboured higher levels of all target substrates than in MCF7Sens at mitotic arrest (time 0) other than CDC20 (CDC20 degradation relies primarily on APCCDH1 [47,48], which is not fully functional in mitosis [20]) and all substrates clearly declined over the 6 hour timecourse. The differences in the rate of target degradation was accentuated upon M2I-1 pretreatment in both selected and parental populations. Significantly, despite the delayed entry into G1 in MCF7TAM cells, we determined that the E3-dependent target protein degradation rates in the MCF7TAM population was restored to that of MCF7Sens cells following M2I-1 treatment. The HURP western was repeated and quantified to assess the degradation rate changes following M2I-1 pretreatment in both MCF7Sens and MCF7TAM cells, demonstrating the normalization of target levels in MCF7TAM cells despite initially elevated levels (Supplemental Figure 3). A different HURP antibody was used in Figure 6B (Proteintech) compared to that used in Figure 5 (Sigma), revealing high levels of phosphorylated HURP in MCF7TAM cells. HURP phosphorylation is known to require the Aurora kinase, an APC substrate that accumulates in cancer cells [49] and occurs in cells where when the APC is inhibited [50]. This adds additional evidence that the APC is inhibited in MCF7TAM cells. In conclusion, APC activation in MCF7Res cells normalizes rates of progression through mitosis, increases the degradation of APC substrates to that of parental cell populations, and enhances cell killing by DOX to match that of parental cell populations. Given that APC substrate overabundance correlates with more aggressive cancers and less responsive therapy, the reduction of APC targets to normal holds significant potential for therapy.
Discussion
When multiple drug resistant cancer develops, treatment may revert to the use of highly toxic second line chemotherapeutics, or palliative care. There are very few treatment options, if any, that will reverse drug resistance, and certainly no widely used therapy that benefits multiple cancer types. In this study, we show that Anaphase Promoting Complex (APC) activity is low in multiple drug resistant (MDR) MCF7 breast cancer cells and that activation of the APC using the small chemical APC activator M2I-1 restored APC activity in resistant cells, recoupled cell cycle progression with APC substrate degradation, and resensitized MCF7 cells selected for resistance to Tamoxifen (MCF7TAM) to DOX, to levels noted in unmodified parental cell populations. Further, treatment of mice growing PDX triple negative breast cancer (TNBC) cells with M2I-1 stalled tumor growth, reduced APC substrate levels, and induced PARP cleavage and histone H3 acetylation. Therefore, our prior (canine lymphoma [9]) and current observations that APC activation reverses MDR cancer behavior applies to different cell line models (breast cancer and lyphoma), across species lines (canine and humans), in vitro and in vivo (cell line, canine and PDX mouse models), and to different methods of selecting cells for resistance (DOX and TAM). We propose that APC function is a general and critical means to maintain cell health and protect against aggressive drug resistant cancer development. Taken together, our results build a strong case supporting the use of APC activation as a promising means to reverse drug resistant cancer.
There is ample evidence supporting the idea that normal APC activity protects against cancer development. Many APC subunit mutations have been identified in a variety of spontaneous human cancers [19,21,51,52], which can cause cells to survive exposure to chemotherapy (acquired resistance); mutations in at least 7 different APC subunits have been associated with resistance to spindle assembly checkpoint inhibitors [11]. It has been observed that APC impairment is associated with an extended duration of mitosis, allowing time for increased DNA repair, for suppression of chromosome segregation errors, and avoidance of mitotic catastrophe [19], thus providing a rational mechanism whereby malignant cell populations survive cytotoxic chemotherapy exposures. In alignment with this idea, we have determined that restoring APC activity in MDR cell populations results in stalled cancer cell proliferation in vitro and in vivo, and promotes DNA damage and apoptosis (Figure 3 and Figure 4) [9,35,53]. Consistent with previous literature, we found that passage through mitosis was delayed in MDR cancer cells (Figure 6A). It has been suggested that slow-growing cancer cells harboring high loads of chromosome instability use the DNA damage response pathway during mitosis as a genome protective mechanism to survive mitotic catastrophe [43,44,45]. This may in turn promote further genomic instability by linking pre-mitotic DNA damage with chromosome instabilities that are then propagated during chromosome segregation. This mechanism may moderate the amount of chromosomal damage carried, as moderate levels of chromosome instability appear to confer treatment resistance and poor prognoses, whereas high or low levels of chromosome instability are associated with better treatment responses [54,55,56]. Therefore, mutations that impair APC function create an environment that is conducive to genomic instability moderation due to slowed mitotic progression.
Another mechanism whereby impaired APC activity may contribute to cancer development, aggressive behavior, and treatment resistance may be due to the failure of pro-oncogenic APC substrates to be appropriately degraded, resulting in a cancer-promoting environment. Multiple APC substrates are known to contribute to cancer development and progression, and have repeatedly been found to be elevated in many cancers, presumably due to reduced APC function and blunting of its E3 activity to target and clear them via ubiquitin-dependent proteasomal degradation (see [20,51] and references therein). APC-targeted proteins, such as CDC20, Securin, HURP, FOXM1, PLK1, and the Aurora kinases, accumulate in multiple unrelated cancer types and are generally associated with more aggressive disease and worse clinical outcomes. In our MDR cell populations, we not only confirmed the accumulation of multiple APC substrates (Figure 2), but also noted enhanced HURP phosphorylation in mitosis (Figure 6B), which is attributable to increased Aurora kinase activity [49], although we did not directly demonstrate its protein accumulation.
These protein ‘biomarkers’ of poor prognosis have lead to the development of targeted inhibitors against many APC degradation targets, in isolation, without compelling benefits in patient survival [57]. Aurora kinase inhibitors are currently in phase I-III clinical trials with some success as monotherapy and showed promise as a combined therapy, but the inhibitors exhibit high toxicity [58]. Clinical trials using inhibitors against the APC substrate PLK1 have also met with inconsistent results [59,60]. We believe that targeting the root cause, that of normalizing the APC inhibition present in MDR populations, will be the key to reducing all pro-oncogenic APC substrates and facilitating real clinical benefits to therapy, potentially ones that may be well tolerated. We posit that the APC itself be targeted for activation to normalize the levels of the pro-oncogenic protein degradation targets en masse.
It is important to acknowledge that there is literature demonstrating that APC inhibition, not activation, leads to death of cancer cells in vitro. At its simplest, this may reflect the essential nature of this protein complex for cell survival. The APC substrate, CDC20, an APC co-activator in mitosis, is frequently highly overexpressed in different cancer cell lines and human tumors [61,62,63], leading to consideration that elevated CDC20 is an important driver of tumorigenesis, and can serve as a prognostic marker, and a therapeutic target. It is possible that the gene and protein signature of CDC20 elevations and its correlation with more aggressive or metastatic malignancies may be due to CDC20 being the most potent pro-oncogenic APC substrate. This would lead to the possibility that inhibition of just this protein, in a potential background of other elevated substrates, would be sufficient to curtail the growth of these cancer cells. Studies using inhibitors against CDC20 or knockdown of CDC20 have shown cytotoxicity in vitro [64,65,66]. Similarly, anti-mitotic agents that inhibit APCCDC20 result in SAC activation (and therefore APC inhibition), delayed or arrested mitosis, and triggered apoptosis in a Bim-dependent manner in vitro [52,67]. Two indirect APC chemical inhibitors work through altering CDC20 binding and activation of the APC: Tosyl-L-Arginine Methyl Ester (TAME) and APC inhibitor (APCIN). TAME blocks the binding of both APC-coactivators, CDC20 and CDH1, to the APC, whereas APCIN binds to CDC20, ultimately impairing the ubiquitination and degradation efficiency of APC substrates (reviewed in [52]). Both inhibitors have anti-tumoral effects [68,69,70,71], despite their different mechanisms of action, and show synergistic activity when both are used together to create a more potent anti-tumoral effect [36].
The observed anti-cancer effect of inhibiting CDC20 through gene silencing, or chemically through APCIN or TAME, can be interpreted in several ways. First, inhibition of the CDC20 oncoprotein by silencing suggests that, since it is an APC activator, the APC itself must be a critical driver of cancer development. In this case, using an APC activator in cells, such as M2I-1, would be predicted to cause uncontrolled proliferation by pushing compromised cells inappropriately through mitosis. Contrary to this notion, we and others have found that M2I-1 has antiproliferative activity on cancer cells in vitro and in vivo (Figure 3C, Figure 4A) [9,53,72]. Another explanation for why elevated CDC20 levels promote cancer progression is that CDC20 accumulation reflects compromised APC activity, and is not therefore able to target CDC20 (or its other targets) for degradation, which is consistent with the overabundance of multiple APC substrates observed in unrelated cancer tissues. While CDC20 may be pro-oncogenic, it is unlikely to act in isolation, as at least 60 of the known 69 human APC substrates are associated with multiple cancer types when they accumulate [20], and are now considered a cancer signature [73,74]. A recent series of papers found that APC substrate-mRNAs, including HURP and CDC20, are elevated in multiple cancers, and are now recognized as a hub or signature gene set predictive of poor prognosis cancer (a subset of references are included here; [75,76,77,78]). These substrate accumulations are also associated with more aggressive cancers; in 182 breast tumor samples tested from high grade TNBC, 58% of the samples stained for G1 markers, yet expressed high levels of APC substrates, a cell cycle point when substrates should instead be degraded and at their nadir levels [28]. It has been shown that mitotic slippage can cause this effect where cells bypass a block in mitosis and continue cycling, leading to more aggressive tumors [10].
While the optimal use of APC activators and inhibitors in cancer therapy remains unresolved, it is extremely important to consider that the APC is an essential component for normal cell growth, and is necessary for normal cell function. Genetic mouse models lacking either CDC20 or CDH1 are lethal [79,80,81], highlighting the neccessity of fine dose management should APC inhibitors, such as APCIN and/or TAME, be considered in the future for human cancer therapy. Conversely, we do not anticipate that APC activation will have the same limitations making dosing theoretically easier; our use of M2I-1 in vitro was not cytotoxic when used alone, yet synergized strongly with DOX to kill MDR cell populations (Figure 3C). M2I-1 use in vivo also did not obviously impact the health of mice when injected in a short-term experiment (Figure 4).
In conclusion, our work supports the hypothesis that APC activation in vitro, in aggressive cancer cells, such as cultured breast cancer cells selected for drug resistance, is sufficient to stall the growth of these cells. We observed that APC activity is reduced in drug resistant cells (Figure 1 and Figure 2), and that APC activation increases the degradation of APC substrates, and resensitizes cells to chemotherapy (Figure 3). In our mouse PDX TNBC model, APC activation in vivo, as monotherapy, was sufficient to stall tumor growth (Figure 4), demonstrating that our in vitro results with human (Figure 1, Figure 2 and Figure 3) and canine [9] cancer cells reflects our in vivo situation. A possible underlying mechanism to explain these effects may be through the restoration of mitotic progression and avoidance of mitotic slippage when APC activity is restored. We base this on our observation that, in resistant cell populations, APC substrates have delayed degradation during mitosis (Figure 5B and 5C) and that mitotic progression into G1 is slowed (Figure 6A). APC activation in MDR cells through pretreatment with M2I-1 normalized progression through mitosis with substrates degraded more rapidly, similar to non-MDR cell populations. Even though mitotic exit in resistant cells treated with M2I-1 did not fully retore it to that of sensitive cells in our hands, it was accompanied with reduced APC substrate levels. This suggests that APC activation recouples mitotic progression with substrate degradation in resistant cells, decreasing the time resistant cells have to manage chromosome instability and survive the next round of division. We suggest that restoration of APC activity may be a general means of killing aggressive cancer cells that is applicable to more than one cancer type, that spans different chemotherapy classes, and may be generalizable given that these observations were consistent across evolutionary boundaries.
Materials And Methods
Cell Lines and Materials:
MCF7 human breast cells were obtained from American Type Culture Collection (ATCC) in Manassas, VA, USA. Cells were cultured in 75 cm tissue culture flasks (Corning) in a humidified atmosphere (5% CO2) at 37°C. MCF7 cells were cultured in DMEM, high glucose, media (Gibco) with 10% FBS and penicillin-streptomycin (Gibco). Doxorubicin hydrochloride (DOX; Pfizer), Tamoxifen (TAM; Cayman Chemical), APCIN (Sigma), Mad2-Inhibitor 1 (Cayman Chemical), nocodazole (Sigma), and thymidine (Sigma-aldrich cat # t1895) were acquired from the indicated providers. All treatment compounds were reconstituted in dimethylsulfoxide (DMSO). Drug treatments were applied at the concentrations and times as indicated. Flow cytometry was performed as described previously [82].
DOX-Selection of MDR Cell Lines
MCF7 parental cells were selected for drug resistance as previously described [32] with initial selection in the presence of 1 uM DOX for 48 hours. Following this treatment, the cells were washed with sterile PBS and allowed a 3-day recovery period. Drug resistance selection pressure was then reapplied to the cells by subculturing in the presence of 100 nM DOX for 2 weeks with fresh media changes every 3 days. Following the selection period, drug resistance was verified by MDR-1 Western blot analyses and using a cell proliferation assay that relies on the reduction of the yellow MTT (3-(4,5-dimethylthiazol-2-yl) 2, diphenyl-tetrazolium bromide), to a purple MTT-formazan by mitochondrial reductases, as previously described [32]. Cancer cells were cultured in 6 well multi-well plates in phenol red-free medium to avoid interference with the analysis of the purple formazan product. The formazan product and spectrophotometric analysis was performed at 570 nm. Cells were also assessed for cell viability using the Trypan Blue assay, as previously described [27].
Western Blot Analysis
MCF7 cells were washed once with sterile PBS and harvested using a rubber cell scraper as previously described [27]. Cells were pelleted via centrifugation at 1000 rpm and resuspended in ice cold RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 1 mM EGTA, 1% NP-40) with protease and phosphatase inhibitors. The cell suspensions were then sonicated with a 70% duty pulse sonication cycle, and centrifuged to remove cell debris. The resulting cell lysates were subjected to Bradford protein analysis (BioRad) to facilitate equivalent protein loading during Western analysis. Cell lysates were treated with 5X Laemmli buffer containing ß-mercaptoethanol, boiled to reduce viscosity, resolved by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), and finally, transblotted onto nitrocellulose membranes. Transblot efficiency was verified prior to immunoprobing by nonspecific Ponceau S protein staining of the membranes. The antibodies used in this study, typically at a 1:1000/2000 dilution, included APC1tot (Abcam133397), APC1S355phos (Abcam10923), CDC20 (PA5-34775), CDH1 (Sigma), CDC27 (Abcam10538), Cyclin B1 (Sigma), HURP (Abcam70744, Proteintech), Securin (Abcam79546), MDR-1 (Sigma), BCRP (Santa Cruz Biotechnology; SCBt), TFPI (Abcam), PARP (Sigma), gH2AX (NovusBio), histone H3K9Ac (Millipore), histone H3tot (Millipore), GAPDH (Millipore), and Tubulin (Sigma). Following primary antibody incubation overnight at 4°C, the blots were probed with a 1:10000 dilution of a secondary horseradish peroxidase (HRP) secondary antibody. The antigen/antibody target signals were detected using an enhanced chemiluminescent detection kit (ECL-BioRad) and chemoluminescent detection using BioRad VersaDoc molecular imager and Software. Cells were viewed with an Olympus BX51 fluorescence microscope 100x objective equipped with an Infinity 3-1 UM camera. Images were collected using Infinity Analyse software version 5.0. A
Coimmunoprecipitation
For co-immunoprecipitation experiments, media was removed, and the dish was rinsed with 3 mL of PBS. Ice cold RIPA buffer with 0.1% Triton-X-100 and protease inhibitors was then added to the culture dish and incubated on ice for 20 minutes. Cells were then scraped into the RIPA buffer using a rubber policeman and underwent the same 70% duty pulse sonication cycle described previously. Protein concentrations were determined using a Bradford assay, with final stocks adjusted to 750 μg of protein/ml in RIPA buffer. Cell lysates were first cleared by incubation for 60 minutes with Protein A sepharose beads. The mixture was then centrifuged and the supernatant was incubated overnight at 4°C with the primary antibody of interest, either CDC27, CDC20 or CDH1. 15 μL of lysate was collected before primary antibody incubation to use as the “Input” sample. Following overnight incubation with the primary antibody, samples were incubated for 2 hours with Protein A sepharose beads. Beads were then centrifuged and the supernatant was collected as the “Unbound” sample while beads were collected as the “Bound” sample. All samples were combined with 2x electrophoresis buffer (4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromophenol blue, 0.125 M Tris HCl, pH 6.8) and boiled before being analyzed using Western Blots, as described above.
Animals
As previously described [27], 8 to 14 week-old female NOD/SCID/common gamma-chain knock-out (NSG: NOD/PrkdcSCID/IL2RN-/-) mice were obtained from Jackson Laboratory (USA). All experiments were approved by the University of Saskatchewan animal ethics office, in accord with the guidelines of the Canadian Council on Animal Care.
Murine Xenograft Experiments
We obtained written informed consent from a patient with TNBC for a tumor sample, as described previously [27], which was compliant with the Research Ethic board approved protocol at the University of Saskatchewan. We passaged a fragment from the original tumor 8 times in NSG mice prior to use in experiments. The resultant tumor was excised, chopped up into ~2 mm fragments, which were then frozen back for future use. Fragments were then grafted subcutaneously into 12 separate NSG mice. Once palpable, mice were injected using intraperitoneal (i.p.) injections with 0, 5, 10 or 25 mg/kg. This was defined as day 0. Tumor size was measured every 2 days to day 8 using calipers. Tumor sizes were normalized to day 0 and plotted. The mice were sacrificed after day 8, with tumors surgically removed and analyzed (n = 3 per treatment arm).
Cell Cycle Arrest
Cell cycle arrests of MCF7 parental and TAM resistant cells were performed in the presence or absence of M2I-1. Cells were cultured to 40% confluence before arresting in 100 nM nocodazole for 16 hours. TAM resistant cells were cultured in the absence of TAM pressure for one week prior to arrest. Cells arrested in the presence of 20 µM M2I-1 were pretreated for 24 hours before addition of nocodazole, and M2I-1 was maintained until sampling. Samples were harvested for western analysis and flow cytometry every hour upon washing twice in PBS and releasing into nocodazole free media. Cells were arrested in S phase using a double thymidine block. Cells were treated twice with 2 mM thymidine for 18 hrs, with a 9 hour break in between, according to published methods [83].
Flow Cytometry
MCF7 cells were harvested from 6-well plates via dissociation with 0.25% Trypsin-EDTA (Gibco). An equal volume of culture media was added to inactivate the trypsin, and cells were fixed by the addition of 1/10 volume 37% formaldehyde solution (Sigma) with gentle agitation at room temperature for 10 minutes. Cells were pelleted by centrifugation at 300 X G for 5 minutes, and washed by resuspension in distilled water passing through a twenty-gauge needle 5 times to eliminate clumping. The washing process was repeated 3 times. Finally, cells were resuspended in 70% ethanol for storage. Immediately prior to analysis MCF7 cells were stained in the dark for 30 minutes using Vybrant DyeCycle Violet Stain (Invitrogen). Once cells were strained, they were pelleted and resuspended in distilled water before analysis using a Beckman Coulter Cytoflex flow cytometer.
Statistical Analysis
Statistical analysis was performed using a paired t-test. Statistically significant differences are noted within their respective figure legends. Error bars define the standard error of the mean.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org
Author Contributions
Conceptualization, T.G.A. and T.A.A.H.; methodology, Z.B. and G.F.D.; validation, M.L., C.V., S.V. and G.F.D.; formal analysis, M.L., C.V., S.V., Z.B. and G.F.D.; resources, T.G.A. and T.A.A.H.; data curation, T.A.A.H.; writing—original draft preparation, T.A.A.H.; writing—review and editing, T.G.A. and T.A.A.H.; supervision, T.G.A. and T.A.A.H.; project administration, T.A.A.H.; and funding acquisition, T.G.A. and T.A.A.H. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
Liubov Lobanova is acknowledged for her assistance with the western shown in Figure 3A. Dr. Chris Eskiw is thanked for reading the final draft. Grants from the Canadian Cancer Society and the Canadian Foundation for Innovation were instrumental for the completion of this work.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Cancer statistics at a glance. Canadian cancer Society; 2023. Available: https://cancer.ca/en/research/cancer-statistics/cancer-statistics-at-a-glance (accessed March 6, 2024).
- Brenner D.R., Poirier A., Woods R.R., Ellison L.F, Billette J.M., Demers A.A., Zhang S.X., Yao C., Finley C., Fitzgerald N., et al. Canadian Cancer Statistics Advisory Committee. Projected estimates of cancer in Canada in 2022. CMAJ. 2022;194:E601-E607. [CrossRef] [PubMed] [PubMed Central]
- https://www.cancertherapyadvisor.com/home/tools/fact-sheets/cancer-recurrence-statistics/divide (ER/PR positive, HER2 negative ER/PR positive, HER2 negative negative) subtypes.
- Voduc K.D., Cheang M.C., Tyldesley S., Gelmon K., Nielsen T.O., Kennecke H. Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol. 2010;28:1684-91. [CrossRef] [PubMed]
- Sleightholm R., Neilsen B.K., Elkhatib S., Flores L., Dukkipati S., Zhao R., Choudhury S., Gardner B., Carmichael J., Smith L., et al. Percentage of Hormone Receptor Positivity in Breast Cancer Provides Prognostic Value: A Single-Institute Study. J Clin Med Res. 2021;13:9-19. [CrossRef] [PubMed] [PubMed Central]
- Yi M., Huo L., Koenig K.B., Mittendorf E.A., Meric-Bernstam F., Kuerer H.M., Bedrosian I., Buzdar A.U., Symmans W.F., Crow J.R., et al. Which threshold for ER positivity? a retrospective study based on 9639 patients. Ann Oncol. 2014;25:1004-11. [CrossRef] [PubMed] [PubMed Central]
- Pan H., Gray R., Braybrooke J., Davies C., Taylor C., McGale P., Peto R., Pritchard K.I., Bergh J., Dowsett M., et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N Engl J Med. 2017;377:1836-1846. [CrossRef] [PubMed] [PubMed Central]
- Cree, I.A., Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer. 2017;17:10. [CrossRef] [PubMed] [PubMed Central]
- Arnason T.G., MacDonald-Dickinson V., Gaunt M.C., Davies G.F., Lobanova L., Trost B., Gillespie Z.E., Waldner M., Baldwin P., Borrowman D., et al. Activation of the Anaphase Promoting Complex Reverses Multiple Drug Resistant Cancer in a Canine Model of Multiple Drug Resistant Lymphoma. Cancers. 2022;14:4215. [CrossRef] [PubMed] [PubMed Central]
- Sinha D., Duijf P.H.G., Khanna K.K. Mitotic slippage: an old tale with a new twist. Cell Cycle. 2019;18:7-15. [CrossRef] [PubMed] [PubMed Central]
- Thu K.L., Silvester J., Elliott M.J., Ba-Alawi W., Duncan M.H., Elia A.C., Mer A.S., Smirnov P., Safikhani Z., Haibe-Kains B., et al. Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer. Proc Natl Acad Sci U S A. 2018;115:E1570-E1577. [CrossRef] [PubMed] [PubMed Central]
- De K., Grubb T.M., Zalenski A.A., Pfaff K.E., Pal D., Majumder S., Summers M.K., Venere M. Hyperphosphorylation of CDH1 in Glioblastoma Cancer Stem Cells Attenuates APC/CCDH1 Activity and Pharmacologic Inhibition of APC/CCDH1/CDC20 Compromises Viability. Mol Cancer Res. 2019;17:1519-1530. [CrossRef] [PubMed] [PubMed Central]
- Zhang Y., Li J., Yi K., Feng J., Cong Z., Wang Z., Wei Y., Wu F., Cheng W., Samo A.A., et al. Elevated signature of a gene module coexpressed with CDC20 marks genomic instability in glioma. Proc Natl Acad Sci U S A. 2019;116:6975-6984. Epub 2019 Mar 15. Erratum in: Proc Natl Acad Sci U S A. 2020 Jan 14;117(2):1234. [CrossRef] [PubMed] [PubMed Central]
- Murphy J.M., Jeong K., Ahn E.E., Lim S.S. Nuclear focal adhesion kinase induces APC/C activator protein CDH1-mediated cyclin-dependent kinase 4/6 degradation and inhibits melanoma proliferation. J Biol Chem. 2022;298:102013. [CrossRef] [PubMed] [PubMed Central]
- Gong R.H., Chen M., Huang C., Wong H.L.X., Kwan H.Y., Bian Z. Combination of artesunate and WNT974 induces KRAS protein degradation by upregulating E3 ligase ANACP2 and β-TrCP in the ubiquitin-proteasome pathway. Cell Commun Signal. 2022;20:34. [CrossRef] [PubMed] [PubMed Central]
- Russell P., Hennessy B.T., Li J., Carey M.S., Bast R.C, Venkitaraman A.R. Cyclin G1 regulates the outcome of taxane-induced mitotic checkpoint arrest. Oncogene. 2012;31:2450-60. [CrossRef] [PubMed] [PubMed Central]
- Simonetti G., Bruno S., Padella A., Tenti E., Martinelli G. Aneuploidy: Cancer strength or vulnerability? Int J Cancer. 2019;144:8-25. [CrossRef] [PubMed] [PubMed Central]
- Pernicone N., Peretz L., Grinshpon S., Listovsky T. MDA-MB-157 Cell Line Presents High Levels of MAD2L2 and Dysregulated Mitosis. Anticancer Res. 2020;40:5471-5480. [CrossRef] [PubMed]
- Sansregret L., Patterson J.O., Dewhurst S., López-García C., Koch A., McGranahan N., Chao W.C.H., Barry D.J., Rowan A., Instrell R., et al. APC/C Dysfunction Limits Excessive Cancer Chromosomal Instability. Cancer Discov. 2017;7:218-233. [CrossRef] [PubMed] [PubMed Central]
- VanGenderen C., Harkness T.A.A., Arnason T.G. The role of Anaphase Promoting Complex activation, inhibition and substrates in cancer development and progression. Aging. 2020;12:15818-15855. [CrossRef] [PubMed] [PubMed Central]
- Melloy P.G. The anaphase-promoting complex: A key mitotic regulator associated with somatic mutations occurring in cancer. Genes Chromosomes Cancer. 2020;59:189-202. [CrossRef] [PubMed]
- Kotani S., Tugendreich S., Fujii M., Jorgensen P.M., Watanabe N., Hoog C., Hieter P., Todokoro K. PKA and MPF activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol Cell. 1998;1:371-80. [CrossRef] [PubMed]
- Zhang S., Chang L., Alfieri C., Zhang Z., Yang J., Maslen S., Skehel M., Barford D. Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature. 2016;533:260-264. [CrossRef] [PubMed] [PubMed Central]
- Gao D., Inuzuka H., Tseng A., Chin R.Y., Toker A., Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol. 2009;11:397-408. [CrossRef] [PubMed] [PubMed Central]
- Song M.S., Carracedo A., Salmena L., Song S.J., Egia A., Malumbres M., Pandolfi P.P. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. 2011;144:187-99. [CrossRef] [PubMed] [PubMed Central]
- Choppara S., Malonia S.K., Sankaran G., Green M.R., Santra M.K. Degradation of FBXO31 by APC/C is regulated by AKT- and ATM-mediated phosphorylation. Proc Natl Acad Sci USA. 2018115:998-1003. [CrossRef] [PubMed] [PubMed Central]
- Davies G., Lobanova L., Dawicki W., Groot G., Gordon J.R., Bowen M., Harkness T., Arnason T. Metformin inhibits the development, and promotes the resensitization, of treatment-resistant breast cancer. PLoS One. 2017;12(12):e0187191. [CrossRef] [PubMed] [PubMed Central]
- Loddo M., Kingsbury S.R., Rashid M., Proctor I., Holt C., Young J., El-Sheikh S., Falzon M., Eward K.L., Prevost T., et al. Cell-cycle-phase progression analysis identifies unique phenotypes of major prognostic and predictive significance in breast cancer. Br. J. Cancer. 2009;100(6):959-70. [CrossRef] [PubMed] [PubMed Central]
- Bakhoum S.F., Kabeche L., Compton D.A., Powell S.N., Bastians H. Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities. Trends Cancer. 2017;3:225-234. [CrossRef] [PubMed] [PubMed Central]
- Levine M.S., Holland A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018;32:620-638. [CrossRef] [PubMed] [PubMed Central]
- Sarwar S., Morozov V.M., Purayil H., Daaka Y., Ishov A.M. Inhibition of Mps1 kinase enhances taxanes efficacy in castration resistant prostate cancer. Cell Death Dis. 2022;13:868. [CrossRef] [PubMed] [PubMed Central]
- Davies G.F., Berg A., Postnikoff S.D., Wilson H.L., Arnason T.G., Kusalik A., Harkness T.A. TFPI1 mediates resistance to doxorubicin in breast cancer cells by inducing a hypoxic-like response. PLoS One. 2014;9:e84611. [CrossRef] [PubMed] [PubMed Central]
- Kraft C., Herzog F., Gieffers C., Mechtler K., Hagting A., Pines J., Peters J.M. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003;22:6598-609. [CrossRef] [PubMed] [PubMed Central]
- Qiao R., Weissmann F., Yamaguchi M., Brown N.G., VanderLinden R., Imre R., Jarvis M.A., Brunner M.R., Davidson I.F., Litos G., et al. Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc Natl Acad Sci U S A. 2016;113:E2570-8. [CrossRef] [PubMed] [PubMed Central]
- Kastl J., Braun J., Prestel A., Möller H.M., Huhn T., Mayer T.U. Mad2 Inhibitor-1 (M2I-1): A Small Molecule Protein-Protein Interaction Inhibitor Targeting the Mitotic Spindle Assembly Checkpoint. ACS Chem Biol. 2015;10:1661-6. [CrossRef] [PubMed]
- Sackton K.L., Dimova N., Zeng X., Tian W., Zhang M., Sackton T.B., Meaders J., Pfaff K.L., Sigoillot F., Yu H., et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature. 2014;514:646–649. [CrossRef] [PubMed] [PubMed Central]
- Garrido D., Bourouh M., Bonneil É., Thibault P., Swan A., Archambault V. Cyclin B3 activates the Anaphase-Promoting Complex/Cyclosome in meiosis and mitosis. PLoS Genet. 2020;16:e1009184. [CrossRef] [PubMed] [PubMed Central]
- Davies G.F., Juurlink B.H., Harkness T.A. Troglitazone reverses the multiple drug resistance phenotype in cancer cells. Drug Des Devel Ther. 2009;3:79-88. [CrossRef] [PubMed] [PubMed Central]
- Georgoulis A., Vorgias C.E., Chrousos G.P., Rogakou E.P. Genome Instability and γH2AX. Int J Mol Sci. 2017;18:1979. [CrossRef] [PubMed] [PubMed Central]
- Tokumoto T., Yamashita M., Tokumoto M., Katsu Y., Horiguchi R., Kajiura H., Nagahama Y. Initiation of cyclin B degradation by the 26S proteasome upon egg activation. J Cell Biol. 1997;138:1313-22. [CrossRef] [PubMed] [PubMed Central]
- Soldani C., Scovassi A.I. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7:321-8. [CrossRef] [PubMed]
- Switonski P.M., Delaney J.R., Bartelt L.C., Niu C., Ramos-Zapatero M., Spann N.J., Alaghatta A., Chen T., Griffin E.N., Bapat J., et al. Altered H3 histone acetylation impairs high-fidelity DNA repair to promote cerebellar degeneration in spinocerebellar ataxia type 7. Cell Rep. 2021;37:110062. [CrossRef] [PubMed] [PubMed Central]
- Bakhoum S.F., Kabeche L., Murnane J.P., Zaki B.I., Compton D.A. DNA-Damage Response during Mitosis Induces Whole-Chromosome Missegregation. Cancer Discovery. 2014;4:1281–1289. [CrossRef] [PubMed] [PubMed Central]
- Burrell R.A., McClelland S.E., Endesfelder D., Groth P., Weller M.C., Shaikh N., Domingo E., Kanu N., Dewhurst S.M., Gronroos E. et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. [CrossRef]
- Bakhoum S.F., Kabeche L., Compton D.A., Powell S.N., Bastians H. Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities. Trends Cancer. 2017;3:225-234. [CrossRef] [PubMed] [PubMed Central]
- Bansal S., Tiwari S. Mechanisms for the temporal regulation of substrate ubiquitination by the anaphase-promoting complex/cyclosome. Cell Div. 2019;14:14. [CrossRef] [PubMed] [PubMed Central]
- Pfleger C.M., Kirschner M.W. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000;14:655-65. [PubMed] [PubMed Central]
- Robbins J.A., Cross F.R. Regulated degradation of the APC coactivator Cdc20. Cell Div. 2010;5:23. [CrossRef] [PubMed] [PubMed Central]
- Wu J.M., Chen C.T., Coumar M.S., Lin W.H., Chen Z.J., Hsu J.T., Peng Y.H., Shiao H.Y., Lin W.H., Chu C.Y., et al. Aurora kinase inhibitors reveal mechanisms of HURP in nucleation of centrosomal and kinetochore microtubules. Proc Natl Acad Sci U S A. 2013;110:E1779-87. [CrossRef] [PubMed] [PubMed Central]
- Song L., Rape M. Regulated degradation of spindle assembly factors by the anaphase-promoting complex. Mol Cell. 2010;38:369-82. [CrossRef] [PubMed] [PubMed Central]
- Hu X., Jin X., Cao X., Liu B. The Anaphase-Promoting Complex/Cyclosome Is a Cellular Ageing Regulator. Int J Mol Sci. 2022;23:15327. [CrossRef] [PubMed] [PubMed Central]
- Schrock M.S., Stromberg B.R., Scarberry L., Summers M.K. APC/C ubiquitin ligase: Functions and mechanisms in tumorigenesis. Semin Cancer Biol. 2020;67:80-91. [CrossRef] [PubMed] [PubMed Central]
- Li J., Dang N., Martinez-Lopez N., Jowsey P.A., Huang D., Lightowlers R.N., Gao F., Huang J.Y. M2I-1 disrupts the in vivo interaction between CDC20 and MAD2 and increases the sensitivities of cancer cell lines to anti-mitotic drugs via MCL-1s. Cell Div. 2019;14:5. [CrossRef] [PubMed] [PubMed Central]
- Jamal-Hanjani M., A’Hern R., Birkbak N.J., Gorman P., Grönroos E., Ngang S., Nicola P., Rahman L., Thanopoulou E., Kelly G., et al. Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: a prospective validation cohort study from the TACT trial. Ann Oncol. 2015;26:1340-6. [CrossRef] [PubMed]
- Roylance R., Endesfelder D., Gorman P., Burrell R.A., Sander J., Tomlinson I., Hanby A.M., Speirs V., Richardson A.L., Birkbak N.J., et al. Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol Biomarkers Prev. 2011;20:2183-94. [CrossRef] [PubMed] [PubMed Central]
- Birkbak N.J., Eklund A.C., Li Q., McClelland S.E., Endesfelder D., Tan P., Tan I.B., Richardson A.L., Szallasi Z., Swanton C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011;71:3447-52. [CrossRef] [PubMed] [PubMed Central]
- Komlodi-Pasztor E., Sackett D.L., Fojo A.T. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res. 2012;18(1):51-63. [CrossRef] [PubMed]
- Du R., Huang C., Liu K., Li X., Dong Z. Targeting AURKA in Cancer: molecular mechanisms and opportunities for Cancer therapy. Mol Cancer. 2021;20(1):15. [CrossRef] [PubMed] [PubMed Central]
- Su S., Chhabra G., Singh C.K., Ndiaye M.A., Ahmad N. PLK1 inhibition-based combination therapies for cancer management. Transl Oncol. 2022;16:101332. [CrossRef] [PubMed] [PubMed Central]
- Zhang J., Zhang L., Wang J., Ouyang L., Wang Y. Polo-like Kinase 1 Inhibitors in Human Cancer Therapy: Development and Therapeutic Potential. J Med Chem. 2022;65:10133-10160. [CrossRef] [PubMed]
- Kidokoro T., Tanikawa C., Furukawa Y., Katagiri T., Nakamura Y., Matsuda K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene. 2008;27:1562–1571. [CrossRef] [PubMed]
- Jiang J., Jedinak A., Sliva D. Ganodermanontriol (GDNT) exerts its effect on growth and invasiveness of breast cancer cells through the down-regulation of CDC20 and uPA. Biochem Biophys Res Commun. 2011;415:325–329. [CrossRef] [PubMed]
- Kato T., Daigo Y., Aragaki M., Ishikawa K., Sato M., Kaji M. Overexpression of CDC20 predicts poor prognosis in primary non-small cell lung cancer patients. J Surg Oncol. 2012;106:423–430. [CrossRef] [PubMed]
- Xian F., Zhao C., Huang C., Bie J., Xu G. The potential role of CDC20 in tumorigenesis, cancer progression and therapy: A narrative review. Medicine. 2023;102:e35038. [CrossRef] [PubMed] [PubMed Central]
- Zhao S.F., Leng J.F., Xie S.S., Zhu L.Q., Zhang M.Y., Kong L.Y., Yin Y. Design, synthesis and biological evaluation of CDC20 inhibitors for treatment of triple-negative breast cancer. Eur J Med Chem. 2024;268:116204. [CrossRef] [PubMed]
- Cheevapruk K., Ueno M., Sungwan P., Sittithumcharee G., Kariya R., Sampattavanich S., Okada S. Novel Midkine Inhibitor Induces Cell Cycle Arrest and Apoptosis in Multiple Myeloma. Anticancer Res. 2024;44:1023-1031. [CrossRef] [PubMed]
- Wan L., Tan M., Yang J., Inuzuka H., Dai X., Wu T., Liu J., Shaik S., Chen G., Deng J., et al. APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev Cell. 2014;29:377-91. [CrossRef] [PubMed] [PubMed Central]
- Bhuniya R., Yuan X., Bai L., Howie K.L., Wang R., Li W., Park F., Yang C.Y. Design, Synthesis, and Biological Evaluation of Apcin-Based CDC20 Inhibitors. ACS Med Chem Lett. 2022;13:188-195. [CrossRef] [PubMed] [PubMed Central]
- Song C., Lowe V.J., Lee S. Inhibition of Cdc20 suppresses the metastasis in triple negative breast cancer (TNBC). Breast Cancer. 2021;28:1073-1086. [CrossRef] [PubMed]
- Gao Y., Guo C., Fu S., Cheng Y., Song C. Downregulation of CDC20 suppressed cell proliferation, induced apoptosis, triggered cell cycle arrest in osteosarcoma cells, and enhanced chemosensitivity to cisplatin. Neoplasma. 2021;68:382-390. [CrossRef] [PubMed]
- Maes A., Maes K., De Raeve H., De Smedt E., Vlummens P., Szablewski V., Devin J., Faict S., De Veirman K., Menu E., et al. The anaphase-promoting complex/cyclosome: a new promising target in diffuse large B-cell lymphoma and mantle cell lymphoma. Br J Cancer. 2019;120:1137-1146. [CrossRef] [PubMed] [PubMed Central]
- Hu Q., Liu Q., Zhao Y., Zhang L., Li L. SGOL2 is a novel prognostic marker and fosters disease progression via a MAD2-mediated pathway in hepatocellular carcinoma. Biomark Res. 2022;10:82. [CrossRef] [PubMed] [PubMed Central]
- Zhan S.J., Liu B., Linghu H. Identifying genes as potential prognostic indicators in patients with serous ovarian cancer resistant to carboplatin using integrated bioinformatics analysis. Oncol. Rep. 2018;39:2653–2663. [CrossRef] [PubMed] [PubMed Central]
- Vriend J., Thanasupawat T., Sinha N., Klonisch T. Ubiquitin Proteasome Gene Signatures in Ependymoma Molecular Subtypes. Int J Mol Sci. 2022;23:12330. [CrossRef] [PubMed] [PubMed Central]
- Weng Y., Liang W., Ji Y., Li Z., Jia R., Liang Y., Ning P., Xu Y. Key Genes and Prognostic Analysis in HER2+ Breast Cancer. Technol Cancer Res Treat. 2021;20:1533033820983298. [CrossRef] [PubMed] [PubMed Central]
- Zhang Q., Wang Y., Xue F. ASPM, CDC20, DLGAP5, BUB1B, CDCA8, and NCAPG May Serve as Diagnostic and Prognostic Biomarkers in Endometrial Carcinoma. Genet Res. 2022;2022:3217248. [CrossRef] [PubMed] [PubMed Central]
- Zheng W., Zhao Y., Wang T., Zhao X., Tan Z. Identification of hub genes associated with bladder cancer using bioinformatic analyses. Transl Cancer Res. 2022;11:1330-1343. [CrossRef] [PubMed] [PubMed Central]
- Hu Z., Chen H., Li H., Xu S., Mu Y., Pan Q., Tong J., Xu G. Lysosome-related genes: A new prognostic marker for lung adenocarcinoma. Medicine. 2023;102:e34844. [CrossRef] [PubMed] [PubMed Central]
- Li M., York J.P., and Zhang P. Loss of Cdc20 Causes a Securin-Dependent Metaphase Arrest in Two-Cell Mouse Embryos. Molecular and Cellular Biology. 2007;27:3481–3488. [CrossRef] [PubMed] [PubMed Central]
- Li M., Shin Y.H., Hou L., Huang X., Wei Z., Klann E., Zhang P. The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat Cell Biol. 2008;10:1083-9. [CrossRef] [PubMed] [PubMed Central]
- García-Higuera I., Manchado E., Dubus P., Cañamero M., Méndez J., Moreno S., Malumbres M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008;10:802-11. [CrossRef] [PubMed]
- Davies G.F., Roesler W.J., Juurlink B.H., Harkness T.A. Troglitazone overcomes doxorubicin-resistance in resistant K562 leukemia cells. Leuk Lymphoma. 2005;46:1199-206. [CrossRef] [PubMed]
- Chen G., Deng X. Cell Synchronization by Double Thymidine Block. Bio Protoc. 2018;8:e2994. [CrossRef] [PubMed] [PubMed Central]
Figure 1.
The Anaphase Promoting Complex is compromised in MCF7TAM resistant cells. A) MCF7 cells selected for resistance to Tamoxifen (TAM) are resistant to Doxorubicin (DOX), compared to parental MCF7 cells. B) Westerns of MCF7Sens and MCF7TAM cells were performed using 30 µg of protein for each sample and antibodies against either APC1S355ph or APC1tot, with antibodies against GAPDH used as a load control. Cells were treated with 5 μM of M2I-1 or 10 μM of APCIN for 18 hours. C) The bands from 3 repeats of the the experiment shown in B) were imaged using a VersaDoc. All bands were normalized using GAPDH for each western, with the APC1ph/APC1tot ratio determined and plotted. Standard error of the mean is shown. Statistical analyses performed using a paired t-test shows a statistically significant decrease (p<0.05) in APC1S355ph in MCF7TAM cells when compared to MCF7sens cells. D) CDC27 was immunoprecipitated (IPed) from MCF7Sens and MCF7TAM cells, with resultant coimmunoprecipitated (CoIPed) proteins assessed using western blots (WB) with antibodies against CDC27, CDC20 or CDH1. The experiment was repeated 2 times. E) Protein bands from the 2 experiments performed in D) were imaged using a VersaDoc. Bound samples were normalized to input samples and plotted. Standard error of the mean is shown. Statistical analyses performed using a paired t-test shows a statistically significant decrease (p=0.002) of bound CDC20 in MCF7TAM cells compared to MCF7Sens cells. F) The reciprocal experiment was performed by coimmunoprecipating CDC27 using antibodies against CDC20.
Figure 1.
The Anaphase Promoting Complex is compromised in MCF7TAM resistant cells. A) MCF7 cells selected for resistance to Tamoxifen (TAM) are resistant to Doxorubicin (DOX), compared to parental MCF7 cells. B) Westerns of MCF7Sens and MCF7TAM cells were performed using 30 µg of protein for each sample and antibodies against either APC1S355ph or APC1tot, with antibodies against GAPDH used as a load control. Cells were treated with 5 μM of M2I-1 or 10 μM of APCIN for 18 hours. C) The bands from 3 repeats of the the experiment shown in B) were imaged using a VersaDoc. All bands were normalized using GAPDH for each western, with the APC1ph/APC1tot ratio determined and plotted. Standard error of the mean is shown. Statistical analyses performed using a paired t-test shows a statistically significant decrease (p<0.05) in APC1S355ph in MCF7TAM cells when compared to MCF7sens cells. D) CDC27 was immunoprecipitated (IPed) from MCF7Sens and MCF7TAM cells, with resultant coimmunoprecipitated (CoIPed) proteins assessed using western blots (WB) with antibodies against CDC27, CDC20 or CDH1. The experiment was repeated 2 times. E) Protein bands from the 2 experiments performed in D) were imaged using a VersaDoc. Bound samples were normalized to input samples and plotted. Standard error of the mean is shown. Statistical analyses performed using a paired t-test shows a statistically significant decrease (p=0.002) of bound CDC20 in MCF7TAM cells compared to MCF7Sens cells. F) The reciprocal experiment was performed by coimmunoprecipating CDC27 using antibodies against CDC20.
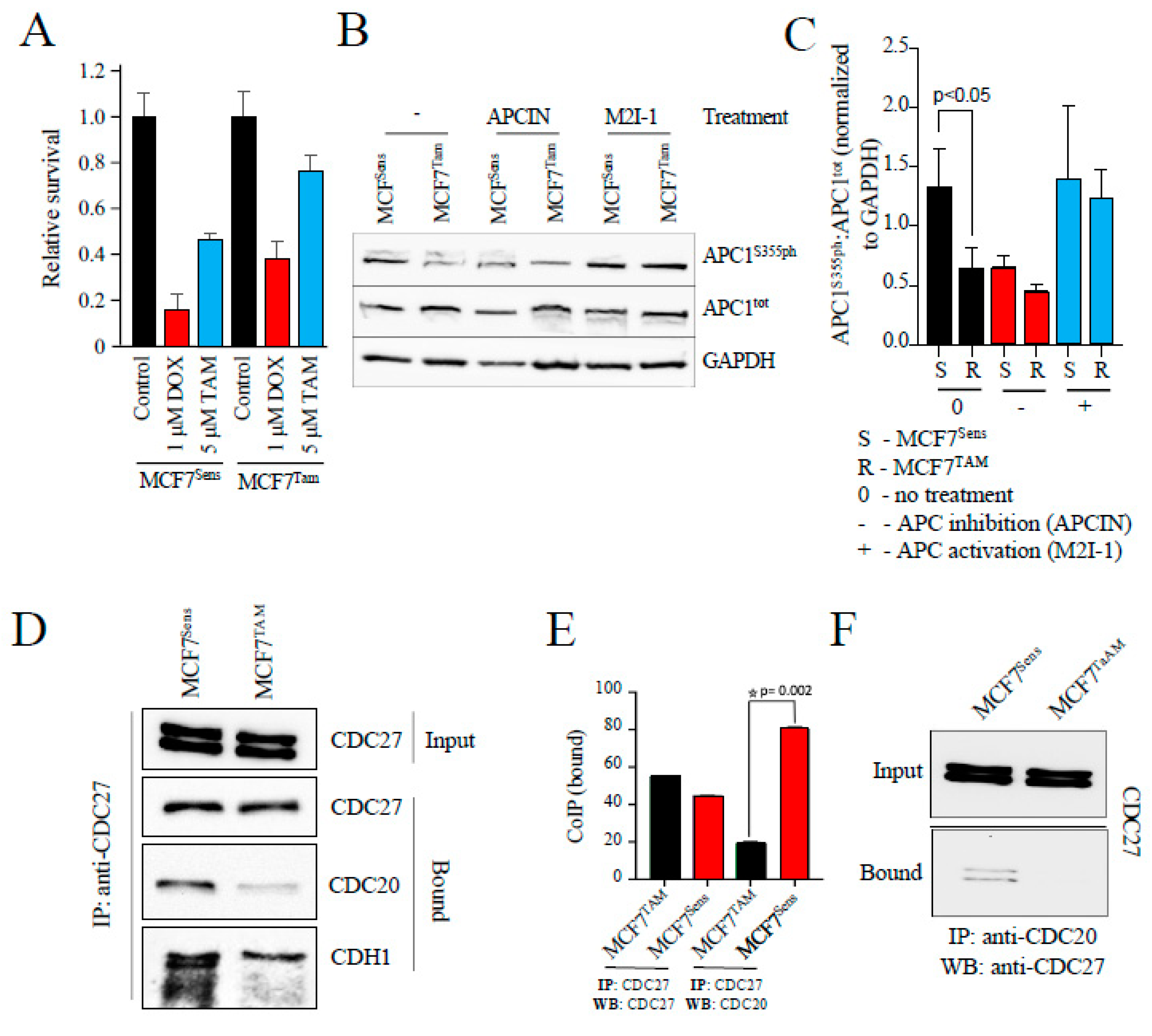
Figure 2.
APC substrate proteins are elevated in MCF7 cells selected for resistance to drugs. A) MCF7 sensitive cells and cells selected for resistance to TAM were prepared for western analyses. Antibodies against MDR markers (BCRP and TFPI) or APC substrates (Cdc20, Cyclin B1, HURP and Securin) are shown. B) A second batch of sensitive and MCF7 cells selected for resistance to TAM were prepared for westerns using antibodies against APC substrates, MDR markers and indicators of DNA damage. This was done to demonstrate reproducibility of the method. C) MCF7 sensitive and cells selected for resistance to DOX were prepared for westerns using an MDR marker (TFPI) and APC substrate proteins (Cdc20 and Cyclin B1). GAPDH and Tubulin were used as loading controls.
Figure 2.
APC substrate proteins are elevated in MCF7 cells selected for resistance to drugs. A) MCF7 sensitive cells and cells selected for resistance to TAM were prepared for western analyses. Antibodies against MDR markers (BCRP and TFPI) or APC substrates (Cdc20, Cyclin B1, HURP and Securin) are shown. B) A second batch of sensitive and MCF7 cells selected for resistance to TAM were prepared for westerns using antibodies against APC substrates, MDR markers and indicators of DNA damage. This was done to demonstrate reproducibility of the method. C) MCF7 sensitive and cells selected for resistance to DOX were prepared for westerns using an MDR marker (TFPI) and APC substrate proteins (Cdc20 and Cyclin B1). GAPDH and Tubulin were used as loading controls.
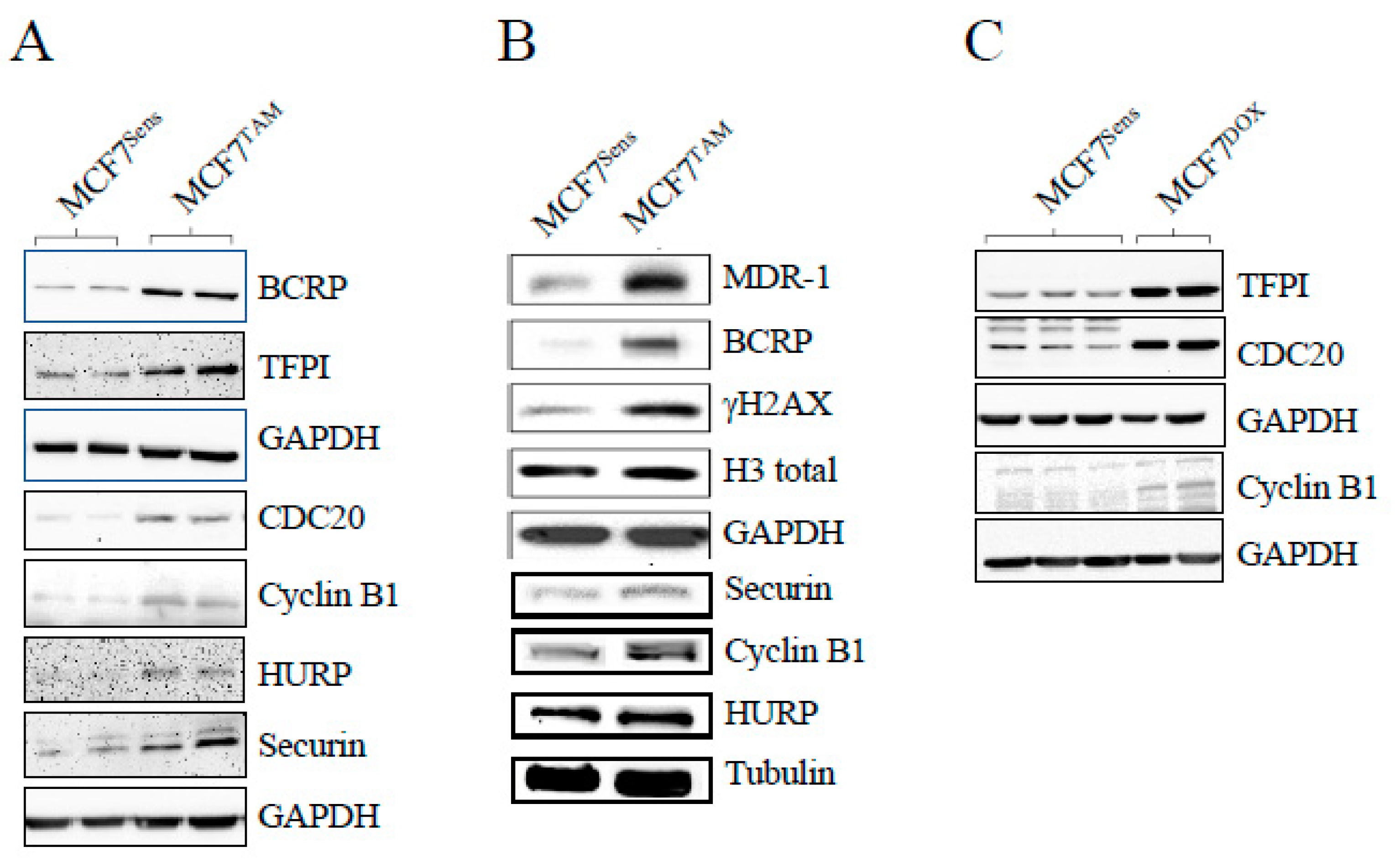
Figure 3.
APC activation reduces APC substrate levels and synergizes with DOX to kill drug resistant cells. A) MCF7DOX cells were treated with 0, 1 or 5 μM M2I-1 for 18 hours. Sensitive parental cells were left untreated as a control. Protein lysates from the selected cells and controls were prepared and analyzed using antibodies against the APC substrate HURP. Tubulin was used as a load control. B) MCF7Sens cells were treated with an increasing dose of M2I-1 as shown for 18 hours. Lysates were prepared and analyzed with antibodies against the APC substrate Cyclin B1. GAPDH was used as a loading control. C) MCF7Sens and MCF7TAM cells were pretreated with 0, 1 or 5 μM M2I-1 for 18 hours. MCF7TAM pretreated cells were then exposed to 1 μM DOX for 48 hours. Untreated MCF7Sens cells treated with 1 μM DOX for 48 hours were used as a control. Cell viability was measured using Trypan Blue. Three biological repeats were performed, with SEM shown.
Figure 3.
APC activation reduces APC substrate levels and synergizes with DOX to kill drug resistant cells. A) MCF7DOX cells were treated with 0, 1 or 5 μM M2I-1 for 18 hours. Sensitive parental cells were left untreated as a control. Protein lysates from the selected cells and controls were prepared and analyzed using antibodies against the APC substrate HURP. Tubulin was used as a load control. B) MCF7Sens cells were treated with an increasing dose of M2I-1 as shown for 18 hours. Lysates were prepared and analyzed with antibodies against the APC substrate Cyclin B1. GAPDH was used as a loading control. C) MCF7Sens and MCF7TAM cells were pretreated with 0, 1 or 5 μM M2I-1 for 18 hours. MCF7TAM pretreated cells were then exposed to 1 μM DOX for 48 hours. Untreated MCF7Sens cells treated with 1 μM DOX for 48 hours were used as a control. Cell viability was measured using Trypan Blue. Three biological repeats were performed, with SEM shown.
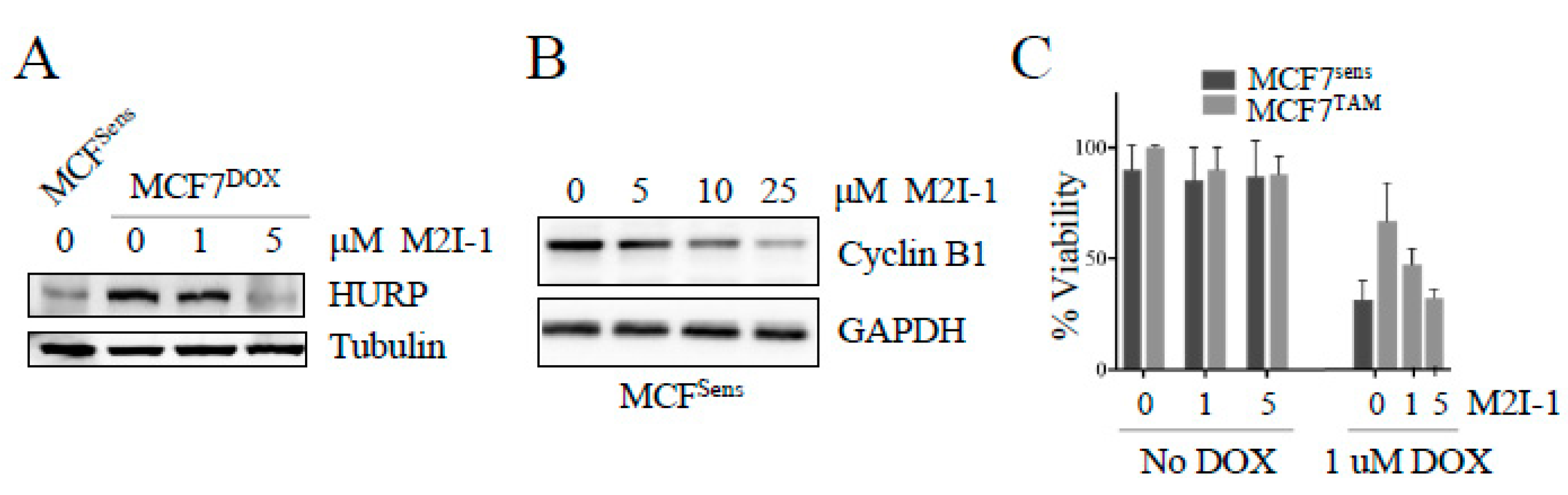
Figure 4.
Mice harboring a patient derived xenografted triple negative breast cancer (TNBC) tumor treated with M2I-1 show stalled tumor growth with protein alterations indicative of APC activation, induction of apoptosis, and cell killing. A) A patient-derived TNBC tumor sample (4-28 PDX) was engrafted into NOD/SCID mice. Intraperitoneal injections of indicated doses of M2I-1 versus mock (DMSO) were delivered once the tumors were palpable (day 0). Tumor size was measured by calipers each 2 days out to day 8. n=3 per treatment arm , SEM shown. B) Liver (control) and tumor tissue samples recovered from mock and M2I-1 treated mice were used for lysate preparation and westerns using antibodies to assess APC activity (Cyclin B1 and CDH1), apoptosis (24 kDa PARP fragment) and cell killing (inhibition of histone deacetyltransferase activity; H3K9Ac). Ponceau S was used to show equivalency of loads.
Figure 4.
Mice harboring a patient derived xenografted triple negative breast cancer (TNBC) tumor treated with M2I-1 show stalled tumor growth with protein alterations indicative of APC activation, induction of apoptosis, and cell killing. A) A patient-derived TNBC tumor sample (4-28 PDX) was engrafted into NOD/SCID mice. Intraperitoneal injections of indicated doses of M2I-1 versus mock (DMSO) were delivered once the tumors were palpable (day 0). Tumor size was measured by calipers each 2 days out to day 8. n=3 per treatment arm , SEM shown. B) Liver (control) and tumor tissue samples recovered from mock and M2I-1 treated mice were used for lysate preparation and westerns using antibodies to assess APC activity (Cyclin B1 and CDH1), apoptosis (24 kDa PARP fragment) and cell killing (inhibition of histone deacetyltransferase activity; H3K9Ac). Ponceau S was used to show equivalency of loads.
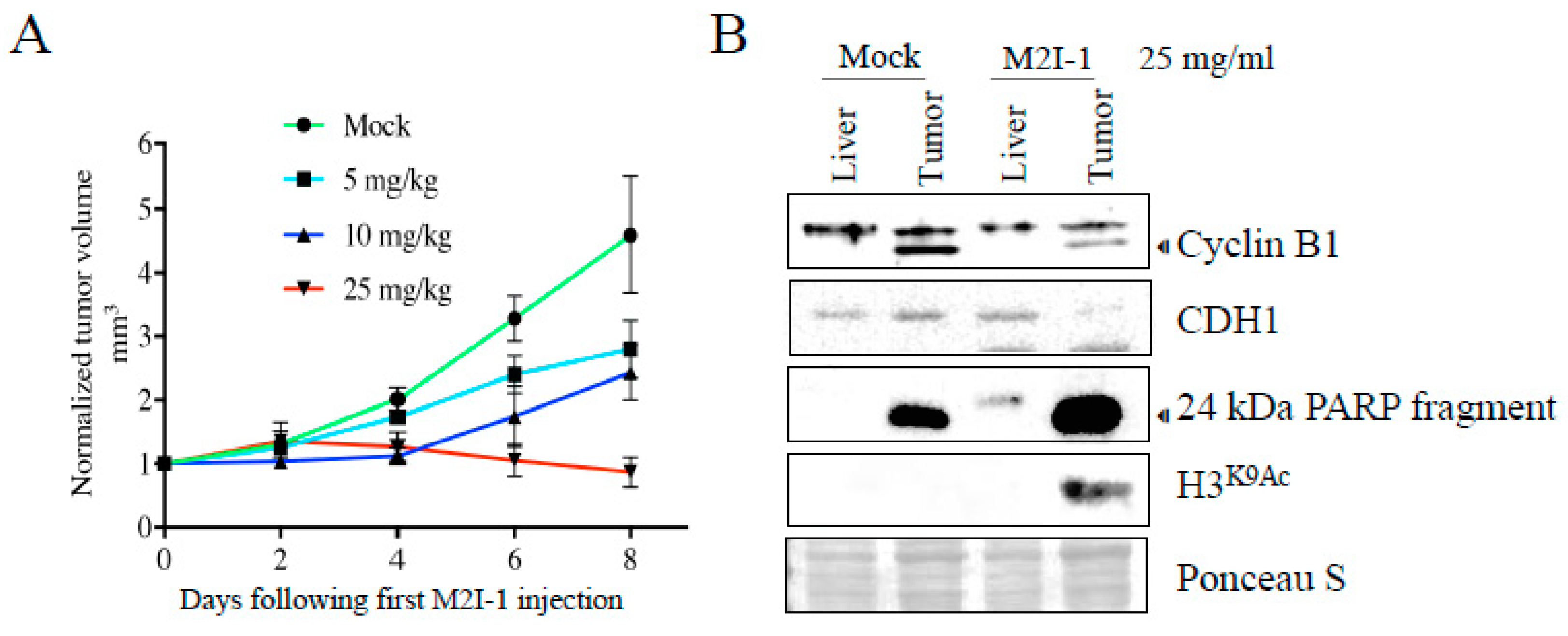
Figure 5.
APC substrate degradation is delayed in mitosis in resistant cells. A) MCF7Sens (S) and MCF7TAM (R) cells were arrested with nocodazole (NOC; 100 nM for 16 hours) at M or in S phase using a double thymidine block (two treatments of 2 mM thymidine for 18 hrs, with a 9 hour break in between). Protein lysates were prepared and assessed using antibodies against the APC substrates HURP (Sigma) and CDC20. Antibodies againt GAPDH were used as a load control. B) MCF7Sens (S) and MCF7TAM (R) cells were arrested at M with NOC. The cells were washed and realeased into fresh media and allowed to re-enter the cell cycle. Samples were removed every 2 hours for 24 hours for analyses using antibodies against the APC substrates HURP (Sigma), CDC20 and Securin. Ponceau S was used as a load control. Samples were also removed for flow cytometry to determine cell cycle progression following release of cells into fresh media. Selected time points are shown to assess the entire 24 hours. C) The early samples from the time course shown in B) are shown, as described in B).
Figure 5.
APC substrate degradation is delayed in mitosis in resistant cells. A) MCF7Sens (S) and MCF7TAM (R) cells were arrested with nocodazole (NOC; 100 nM for 16 hours) at M or in S phase using a double thymidine block (two treatments of 2 mM thymidine for 18 hrs, with a 9 hour break in between). Protein lysates were prepared and assessed using antibodies against the APC substrates HURP (Sigma) and CDC20. Antibodies againt GAPDH were used as a load control. B) MCF7Sens (S) and MCF7TAM (R) cells were arrested at M with NOC. The cells were washed and realeased into fresh media and allowed to re-enter the cell cycle. Samples were removed every 2 hours for 24 hours for analyses using antibodies against the APC substrates HURP (Sigma), CDC20 and Securin. Ponceau S was used as a load control. Samples were also removed for flow cytometry to determine cell cycle progression following release of cells into fresh media. Selected time points are shown to assess the entire 24 hours. C) The early samples from the time course shown in B) are shown, as described in B).
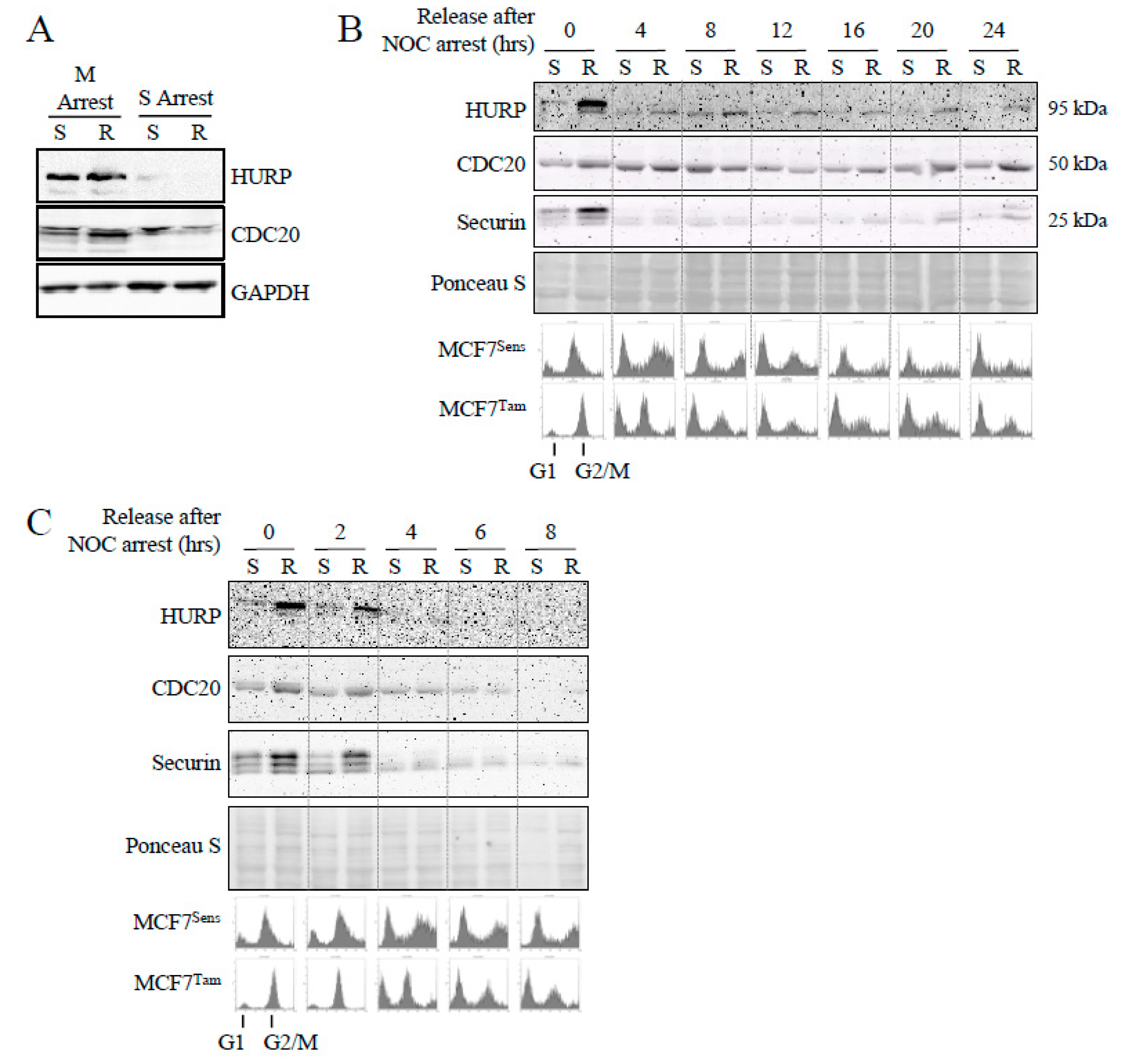
Figure 6.
APC activation increases progression through mitosis and APC substrate degradation. A) MCF7Sens (S) and MCF7TAM (R) cells were pretreated with 20 μM M2I-1 for 24 hours, or left untreated, before M arrest with 100 nM NOC for 16 hours. Cells were washed, then resuspended in fresh media to allow progression through mitosis. Samples were removed every hour for 6 hours for flow cytometry. B) Samples from the cells grown above were removed every hour following release into fresh media for western analyses using antibodies against the APC substrates HURP (Proteintech), CDC20 and Securin. GAPDH was used as a load control for HURP, whereas Ponceau S was used for CDC20 and Securin. One blot was used for the CDC20, Securin and GAPDH signals since they were of different sizes and easily separatable.
Figure 6.
APC activation increases progression through mitosis and APC substrate degradation. A) MCF7Sens (S) and MCF7TAM (R) cells were pretreated with 20 μM M2I-1 for 24 hours, or left untreated, before M arrest with 100 nM NOC for 16 hours. Cells were washed, then resuspended in fresh media to allow progression through mitosis. Samples were removed every hour for 6 hours for flow cytometry. B) Samples from the cells grown above were removed every hour following release into fresh media for western analyses using antibodies against the APC substrates HURP (Proteintech), CDC20 and Securin. GAPDH was used as a load control for HURP, whereas Ponceau S was used for CDC20 and Securin. One blot was used for the CDC20, Securin and GAPDH signals since they were of different sizes and easily separatable.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



