
Submitted:
25 March 2024
Posted:
26 March 2024
You are already at the latest version
Abstract
Hypertensive disorders of pregnancy (HDP), including preeclampsia (PE) and gestational hypertension (GH), are major causes of maternal and fetal morbidity and mortality. This review elucidates the role of regulatory T cells (Tregs) in the immunological aspects of HDP and explores their therapeutic potential. Tregs, known for maintaining immune tolerance, are crucial in pregnancy to prevent immune-mediated rejection of the fetus. In HDP, Treg numbers and function imbalance are observed, correlating with disease severity.
The review highlights that Tregs contribute to immunological adaptation in normal pregnancy, ensuring fetal acceptance and placental development. In contrast, HDP is associated with Treg dysfunction, marked by decreased numbers and impaired regulatory capacity, leading to inadequate immune tolerance and abnormal placental development. This dysfunction is particularly evident in PE, where Tregs fail to adequately modulate the maternal immune response against fetal antigens, contributing to the pathophysiology of the disorder.
Therapeutic interventions aiming to modulate Treg activity represent a promising avenue for HDP management. Studies in animal models and limited clinical trials suggest that enhancing Treg functionality could mitigate HDP symptoms and improve pregnancy outcomes. However, the review acknowledges the complexity of translating these findings into effective clinical therapies, given the multifactorial nature of HDP and the intricate regulatory mechanisms of Tregs.
In conclusion, while the precise role of Tregs in HDP is still being unravelled, their central role in immune regulation during pregnancy is indisputable. Further research is needed to fully understand the mechanisms by which Tregs contribute to HDP and to develop targeted therapies that can safely and effectively harness their regulatory potential for treating hypertensive diseases of pregnancy.
Keywords:
Tregs
; preeclampsia
; hypertensive disorders
; immune tolerance
; semi allogenic fetus
1. Introduction
Hypertensive disease in pregnancy (HDP) is broadly divided into chronic hypertension (CH), gestational hypertension (GH) and preeclampsia (PE). (1) Overall, HDP complicates 5-10% of pregnancies, with PE affecting 3-5% [1,2] and accounts for 47,000 maternal and 500,000 fetal/neonatal deaths worldwide annually [3,4]. GH is a systolic blood pressure ≥140 mmHg and diastolic blood pressure ≥90 mmHg. PE has traditionally been diagnosed at ≥20 weeks’ gestation by the combination of hypertension and significant proteinuria (Urinary Protein: Creatinine ratio >30mmol) through an updated definition diagnoses PE based on GH or chronic hypertension (CH), and subsequent acute kidney injury, liver dysfunction, neurological symptoms, haemolysis or thrombocytopenia or fetal growth restriction [4,5,6,7,8].
Offspring exposed to PE and hypertension have a significant increase in all-cause mortality of 29 and 12%, respectively, in addition to increased risks of endocrine, nutritional, metabolic and cardiovascular disease [9]. According to the 2018-20 MBRRACE report, 8 patients died from PE and eclampsia – a figure essentially unchanged over the last decade. Various figures exist for the cost to healthcare systems of PE; in the U.K., a conservative figure is an average of around £3000 per patient [10]; therefore, efforts to better understand the mechanisms of HDP are important from a public health perspective, particularly given the potential for targeted therapies and interventions [4,5].
Risk factors associated with an incidence of HDP include increased maternal age and weight, black and South Asian ethnicity, family and personal history of PE, conception by IVF, prolonged interpregnancy interval, diabetes mellitus, chronic hypertension, chronic kidney disease and autoimmune disease [5,11,12].
Growing evidence shows that abnormal immune function is central to developing adverse pregnancy outcomes. A fetus is only a semi-allograft of its host with fetal alloantigens encoded by paternally inherited genes able to provoke a maternal immune response leading to adverse pregnancy outcomes, including PE and GH [1,12]; this presents a complex immunological problem for the continuation of any pregnancy. For a fetus to develop, a host of immunological mechanisms are at play to minimise rejection of the fetus.
The immune system is broadly divided into innate and adaptive branches; the adaptive immune system acts as an immunological memory using T and B lymphocytes, both of which affect various cell-mediated immune responses to antigens [13]. Regulatory T cells, or Tregs, are a specialised subpopulation of T cells that suppress an individual’s immune response [14]. There are several subsets of Tregs, with FoxP3 being the most specific marker for these cells. Tregs are identifiable as expressing high levels of interleukin 2 (IL-2) receptor alpha chain (CD25) and the transcription factor FOXP3 while expressing low levels of IL-7 receptor (CD127). Tregs can be broadly divided into thymus-derived (tTregs) peripherally derived (pTregs) [15]. With reduced numbers of Tregs or dysfunctional Tregs, there is an increased incidence of adverse pregnancy outcomes [16]. Recent evidence suggests Tregs are central to inducing immunological tolerance to fetal and placental antigens and have a specific role in the remodelling of uterine spiral arteries, a process widely accepted to be important in developing PE [17].
This paper brings together current evidence comparing the role of Tregs in PE and GH with that in normal pregnancies.
2. The Immune System in Pregnancy; A Focus on Regulatory T Cells (Tregs)
T cells are derived from haematopoietic stem cells in the bone marrow and are characterised by T cell receptors on their surface and a CD3 protein complex [18]. T cells differentiate into subgroups, identifiable by the specific chemokines and cytokines they produce to exert their effects, in addition to numerous other functional molecules [18,19]
CD4+ lymphocytes are the predominant cell population in the cell-mediated immune response. Depending on the microenvironment, these differentiate into either CD4+CD25+ regulatory T cells (Tregs) or T helper type 1, 2, 3, 9, 17, 22 and follicular T helper cells (Th1/2/3/9/17/22/Tfh) [1]. The cytokines that induce the differentiation of T cells into Th1, Th2 and Th17 and the cytokines they subsequently produce are outlined in Figure 1.
CD4+ T cell differentiation is under the careful control of transcription factors that determine their cell surface markers and the cytokines they produce [20], with T-bet and GATA3 being particularly important. T-bet promotes the Th1 phenotype, while GATA3 promotes Th2 and Th17 lineages. Loss of T-bet in mouse models causes differentiation of CD4+ T cells into Th2 and Th7 lineages. Conversely, GATA-3 prevents differentiation into Th2 lineage. GATA3 and Tbet are antagonistic; Tbet represses GATA3 binding to a promoter, IL5, one of several sites GATA3 binds to activate expression of the Th2 cytokine locus [21]. Differentiation into Th1 and Th2 is, therefore, mutually exclusive.
Th1 cells target intracellular pathogens and support a pro-inflammatory response [13], while Th2 cells promote an anti-inflammatory response through the production of IL-4, 5, 6 10 and 13. Th 17 cells play a specific pro-inflammatory role in endothelial defence by producing IL-17 and 22 to protect against microorganisms [13]. The various cytokines and markers can be used as proxy markers of T cell function.
Tregs are a specialised subpopulation of T cells that work to suppress an individual’s immune response [14]. The transcription factor Forkhead Box P3 (FOXP3) is the master gene for Treg differentiation, and it’s stable expression is characteristic of Tregs. Tregs can be divided into thymus-derived (tTregs), in vitro-induced (iTregs) or peripherally derived (pTregs) [15]. tTregs and pTregs are not distinguishable in humans but express different levels of Neuropilin 1 (Nrp1) in mice [17]. Thymic Tregs (tTregs) are generated in the thymus due to intermediate and high-affinity interactions with self-antigens presented to T cells by MHC class II molecules. pTregs, or peripherally generated Tregs are, however, induced in the periphery in response to specific antigens and in the presence of specific cytokines. While these are seldom differentiated in literature, there is evidence pTregs are particularly important in maternal-fetal tolerance [22]. TGF-b plays a role in inducing FOXP3 expression in naïve T cells in vitro, so-called iTregs; though likely closely related, iTregs do not replicate the suppressive capacity of pTregs [14].
Figure 1.
Cytokines inducing differentiation of naïve CD4 cells and subsequent production of cytokines by subset. Cytokines induce the differentiation of naive CD4+ T cells into Th1, Th2, Th17, and Tregs, with the downstream production of cytokines by each subset. The Tregs illustrated here represent pTregs [23].
Figure 1.
Cytokines inducing differentiation of naïve CD4 cells and subsequent production of cytokines by subset. Cytokines induce the differentiation of naive CD4+ T cells into Th1, Th2, Th17, and Tregs, with the downstream production of cytokines by each subset. The Tregs illustrated here represent pTregs [23].
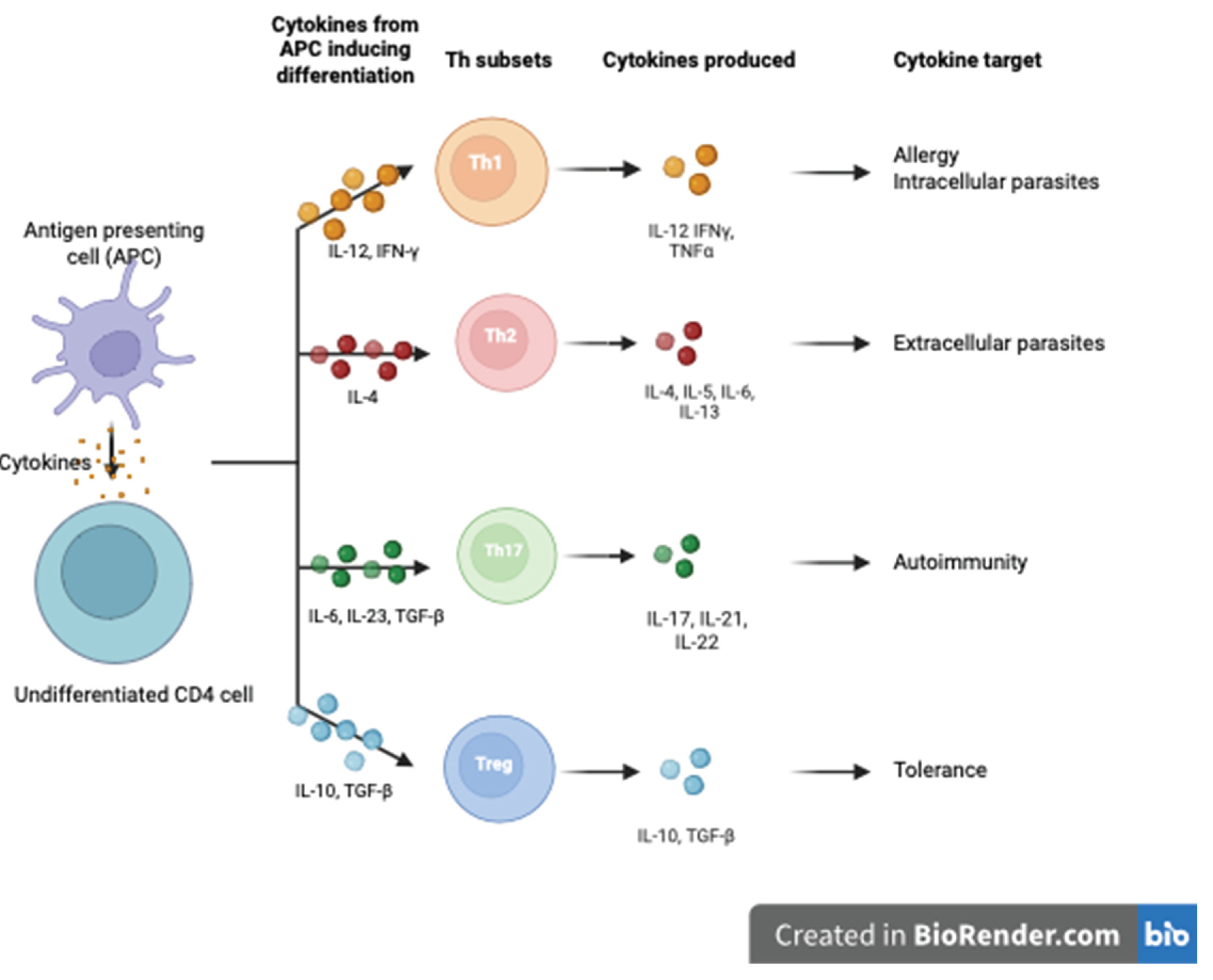
tTregs are the most studied population of CD4+ T regs, and are identified as expressing high levels of interleukin 2 (IL-2) receptor alpha chain (CD25), the co-inhibitory receptor cytotoxic T lymphocyte Ag 4 (CTLA4) and the transcription factor FOXP3 while expressing low levels of IL-7 receptor (CD127) [14]. Differentiating tTregs from pTregs is difficult in humans; there is a lack of clear markers to distinguish the two. There is a differential expression of Helios, an Ikaros family transcription factor, with this being highly expressed in tTregs but less so in the periphery, though there is debate as to whether this can be upregulated in pTregs and iTregs [24]. Nevertheless, its intracellular localisation limits its usefulness as a marker for their purification. In mouse models, tTregs also express high levels of Nrp-1, potentially before they mature in the thymus, though pTregs can upregulate Nrp-1 in inflammation, demonstrating considerable plasticity [24,25].
Mechanisms of [15,17] Treg suppression of the immune response can be broadly divided into cell-cell contact and secretion of soluble factors. Tregs exert this suppressive effect on a variety of cell types, including B cells, NK cells, monocytes, dendritic cells and conventional T cells (Tconv). Considering cell-cell contact, murine models have demonstrated that upon contact with Tconvs, Tregs inhibit TCR-induced proliferation and IL-2 transcription. Furthermore, once activated, T regs appear to carry this suppressive capacity independently of antigen, so-called bystander suppression [26,27]. CTLA-4, a co-inhibitory molecule constitutively expressed by Tregs, is another mechanism by which Tregs exert direct cell-cell effects [27]. CTLA-4 downregulates CD80/CD86 expression on APCs, directly inhibiting Tcon activation [28,29]. CTLA-4 possesses a unique ability, trans-endocytosis (TE), which physically captures CD80/CD86 from cells and destroys them via T-cell mediated endocytosis and lysosomal degradation [30].
LAG-3, an adhesion molecule that binds to MHC Class II molecules to suppress dendritic cell activation, has also been proposed to be involved in Tregs suppressive capacity [31,32]. However, unlike CTLA-4 knockout mice, which show fatal autoimmune disease [33], LAG-3 deficient mice do not show signs of autoimmune disease [32]. Other cell surface proteins, including CD40, a costimulatory molecule; A20, a deubiquitinase which acts to attenuate antigen-presentation; and Neuropilin-1, have also been described as playing a role in Treg cell: cell-mediated effects [27].
Soluble factors, primarily suppressive cytokines, are key players in the effector functions of Tregs [34]. Tregs can produce membrane-bound and soluble TGF-b, which suppress T cell proliferation and play a key role in lymphocyte haemostasis [27,34]. Knockout mouse models have elegantly demonstrated the importance of suppressing Th1 and Th2 and preventing autoimmunity. IL-10 exerts immunosuppressive effects on various cell types, and its role in preventing colitis is well studied [34,35]. Furthermore, IL-10 is essential to control IFN-γ production by T cells in the skin but not in lymph tissue [36].
IL-35 directly suppresses Tcon proliferation, with Tregs deficient in IL-35 demonstrating reduced suppressive capacity in a model of inflammatory bowel disease [37]. Although IL-35 is not constitutively expressed in humans, induced Treg populations preferentially expressing IL-35 (iTreg35) were highly suppressive. Furthermore, long-term activation of Tregs leads to the upregulation of IL-35, which explicitly induces iTreg35 cells [27,34,36,37].
There are a number of phenotypically distinct subgroups of Tregs, which have been widely characterised in the literature [38]. Outside of lymph tissue, T regs are localised to visceral adipose tissue, intestine, skin and muscle, where they regulate inflammation and contribute to tissue repair [39].
The suppressive ability of Tregs is vital to dampen the immune response in pregnancy, with significant autoimmunity resulting from depletion of CD25 [40]. There is increasing recognition and research into the role of Tregs in pregnancy. With reduced numbers of Tregs or dysfunctional Tregs, there is an increased incidence of adverse pregnancy outcomes [16]. Recent evidence suggests Tregs are central to inducing immunological tolerance to fetal and placental antigens and have a specific role in the remodelling of uterine spiral arteries, a process widely accepted to be important in developing PE [17].
Tregs and T helper cell lineages (Th1/2/17) are closely related and can convert to other lineages, so-called plasticity. The balance of T helper and Tregs appear to be important in achieving immune tolerance generally, which has obvious implications for pregnancy [41]. When this balance is disrupted, and there is under-expression of Tregs or over-expression of T helper lineages, there appears to be a disturbance to the delicate immune homeostasis of uncomplicated pregnancy [1]. These cell lineages, therefore, have important diagnostic and therapeutic potential.
3. Role of T Regs in Healthy Pregnancy
There are a number of outcomes where Tregs have been proposed to have a role; hypertensive disease in pregnancy (including pregnancy-induced hypertension and PE), gestational diabetes, and miscarriage are amongst the most closely researched [17]. Maternal immune cells are tightly regulated to moderate an immune reaction to the fetus and minimise the risk of adverse pregnancy outcomes [17]. As such, the level of Tregs is increased in the peripheral blood of pregnant people vs. non-pregnant controls [42]. The proportion of T regs as a total of T cells changes throughout pregnancy, with a significant increase in decidual Tregs later in pregnancy. This gestational variation is summarised in Figure 3.
Mouse models have elegantly demonstrated the importance of Tregs in successful implantation and maintenance of pregnancy with a significant increase in litter size with injection of CD25+ Tregs into mice prone to miscarriage and small litter size [43,44]. Conversely, depleting CD4+CD25+ Tregs during implantation increases the miscarriage rate in mice [45,46]. Depletion of Tregs mid-gestation leads to high resorption rates in mice models [47]. Treg depletion studies also support an essential role for Tregs in avoiding the development of immunity to fetal alloantigens with a subsequent Th1 and Th17 response, causing fetal loss.
Decidual development plays a central role in establishing pregnancy and, eventually, the development of hypertensive diseases in pregnancy, with Tregs being a fundamental regulator of that normal development (Figure 4). Tregs accumulate in the decidua early in pregnancy, and they make up around 30% of decidual T cells in mouse models [17]. Furthermore, Tregs are essential in maternal vascular remodelling by affecting the decidual leukocyte network. Tregs exert anti-inflammatory actions on uterine natural killer cells (uNK), macrophages and mast cells, all of which influence vascular remodelling [48]. Mouse models have demonstrated Tregs are protective against hypertensive sequelae and reverse vascular damage [49,50].
Analysis of the blood of patients with gestational diabetes mellitus (GDM) has demonstrated reduced CD4+CD25+ Treg cell numbers and increased pro-inflammatory cytokines IL-6 and TNF-a [51]. As with other adverse pregnancy outcomes, reduced quality and quantity of Tregs in GDM has been shown with reduced production of TGF-b and IL-10 [51,52].
Several mechanisms of immune tolerance are at play in pregnancy to moderate the immune responses that would otherwise threaten the pregnancy. The capacity of Tregs to reduce and resolve inflammation in embryo implantation is pivotal to promoting immune tolerance throughout gestation [17]. This is much the same as the role of Tregs throughout the body, where they suppress T effector cells responding to non-dangerous stimulants or preventing autoimmunity to self-antigens [17].
4. Role of T Regs in GH/PE
There is a wealth of evidence linking reduced Treg numbers with PE, but there are no publications on Tregs in GH. Whether GH and PE exist on a spectrum or are separate pathologies entirely remains a matter of debate within obstetrics and perhaps contributes to the lack of distinct and separate discussion of each.
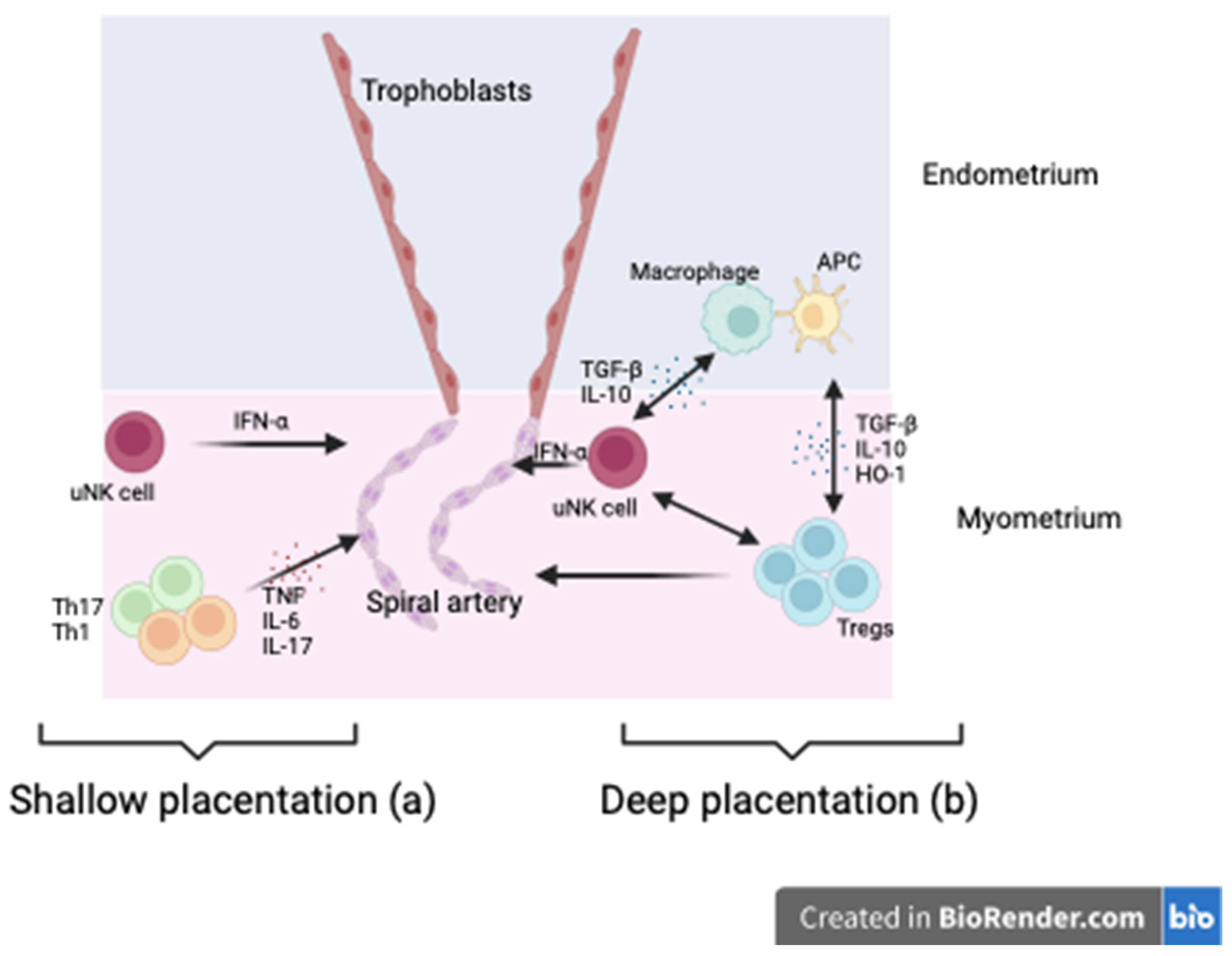
It is widely hypothesised that early pregnancy dysfunction or disturbed immune adaptation precedes placental development and underpins the emergence of adverse pregnancy outcomes, including GH and PE, later in pregnancy. There is increasing acceptance of the hypothesis that HDP results from insufficient, or shallow, placentation early in pregnancy [53,54,55] with a failure of spiral artery remodelling and trophoblast invasion compromising placental development and function. Tregs, alongside other immune cells, are potential key regulators of the decidual leucocyte network controlling implantation and placentation, with inadequate numbers of Tregs in the decidua being associated with shallow placentation [3]. Therefore, it is a reasonable assertion that Tregs are an upstream modulator of placentation and contribute to the development of hypertensive disease in pregnancy (Figure 4).
- (a)
- Tregs modulate uterine natural killer cells (uNK) and macrophages while suppressing inflammation via TGF-βIL-10 and HO-1 release. uNK cells promote vascular remodelling via IFN-γ essential to trophoblast invasion through the decidual later of the endometrium into the myometrium. This improved blood flow to the placenta.
- (b)
The paradigm that disorders of inadequate placentation are foundational to developing particular adverse pregnancy outcomes, particularly PE, is underpinned by the peak of Tregs cell numbers coinciding with intense vascular activity, trophoblast invasion and spiral artery remodelling [46]. It is, therefore, reasonable to postulate that Tregs’ primary role is in the early phase of pregnancy and that they set the course of pregnancy.
Furthermore, Treg cells produce both TGF-b and IL-10, which contribute to their anti-inflammatory function and influence vascular activity, potentially contributing directly to blood pressure control [46]. The target receptor of IL-10, IL-10-R, is expressed on various cells at the maternal-fetal interface, including placental trophoblasts, decidual stromal cells, macrophages and uterine natural killer cells [52]. In mice deficient in IL-10, there is increased sensitivity to pro-inflammatory stimuli, including viral infections, with resultant increased systolic blood pressure and urinary protein concentrations, suggesting altered Treg function can precipitate specific adverse pregnancy outcomes [56,57]. Administration of IL-10 reversed hypoxia-induced hypertension, proteinuria and growth restriction in IL-10-depleted mice models [58].
Treg treatment in hypertensive mouse models also demonstrates maintenance of other T cell populations, demonstrating that the role of Tregs is exponential [59]. Similarly, soluble endoglin, which has been implicated in the pathogenesis of PE, does so by inhibiting TGF-b, which is a crucial anti-inflammatory cytokine produced by Tregs, further supporting the hypothesis that altered or reduced function of Tregs is instrumental in the pathogenesis of PE [59,60].
There is uncertainty as to whether immunoregulation plays a central role in vascular haemostasis in pregnancy; that is to say, immunoregulation contributes to the peripheral vasodilatation required to accommodate the significantly increased cardiac output. Understanding this has hitherto focused on natural killer cells. Still, various studies have suggested a role for Tregs in modulating heart fibrosis in hypertension and coronary arteriole endothelial dysfunction in hypertensive mice models, as well as suppressing angiotensin II-induced hypertension and subsequent vascular injury. Treatment of AngII-infused hypertensive mice with Tregs reduced both blood pressure and vascular damage [61], suggesting a direct role for Tregs in modulating vascular haemostasis and preventing hypertensive disease.
Furthermore, NOX-2 expression in Tregs has been identified as consequential for the development of Angi-II-induced hypertension and cardiac remodelling. In a mouse model with CD4-targeted NOX-2 deficient Tregs demonstrated a greater suppressive capacity and inhibition of AngII-induced hypertension with reduced infiltration of Teffs [62]. The mechanism by which Tregs can limit Ang-II-induced inflammation in the vasculature observed in hypertension has previously been unclear; though these mouse models are not specific to pregnancy, this serves as further evidence of the complex role Tregs have in vascular haemostasis.
Meta-analysis of 17 studies supported the hypothesis that pregnant people with preeclampsia have fewer T regs in overall number and functionality [63]. There is also increased T effector cell activity in these patients, particularly Th1 and Th17 [64,65,66]. Other studies suggest pregnancy-induced hypertension follows the same pattern, though the definition in such studies generally includes GH and PE [1,51]. A further meta-analysis by Green et al. suggests low Treg cell numbers may be an independent risk factor for PE but does not discuss pregnancy-induced hypertension specifically [12]
However, the Treg changes in PE are more nuanced than a simple fall in overall numbers; decidual Tregs demonstrate a reduced ability to induce peripheral Tregs [67], while peripheral HLADR neg CD45RA+ Tregs are reduced in number [68]. Over time, the number of Tregs is also central to proper immune regulation; a study using CVS at 10-12 weeks gestation identified altered expression of decidual and immune cell genes from the 1st trimester [69]. Another study using CVS identified higher placental expression of IL-6 in patients who develop PE, which is known to oppose Tregs by reducing their stability and promoting Th17 production [70].
Furthermore, aside from overall numbers, the function of Tregs is vital, as demonstrated by Han et al., who used mass cytometry to identify an association between impaired Treg function and PET [71].
The reduced uterine perfusion pressure (RUPP) mouse model, which creates conditions of placental ischaemia and oxidative stress using clips on murine vessels placed on day 14 of pregnancy, replicates PE symptoms seen in humans with a rise in pro-inflammatory cytokines, PLGF and VEGF. This model halves Treg numbers compared to controls with increased CD4+ T cells and Th17. Infusion of Tregs from healthy pregnant mice administered after the RUPP procedure mitigates PE symptoms [72].
Whether GH and PE are part of a clinical spectrum or two distinct immunological processes is beyond the scope of this review. What is clear, though, is that there is little distinction between the two in literature and a tendency to discuss PE as a catch-all for HDP. Nevertheless, Tregs play an undeniable role in preventing PE, and their dysfunction bears some responsibility for PE either directly or via downstream effectors when Treg number and function are altered.
5. Tregs as a Therapeutic Tool in Adverse Pregnancy Outcomes
There have been a number of attempts to use Treg therapy in murine models, particularly to prevent spontaneous abortion. Though there is a firth of literature on Treg treatment hypertensive disorders of pregnancy, these insights from other APOs will likely prove essential to evolving work in HDP therapy.
Wang et al. examined whether the adoptive transfer of Tregs reverse the increase in abortion rates caused by IL-17, a pro-inflammatory cytokine, in the CBA/J x BALB/c mouse model. They found that the transfer of pregnancy-induced T regs from pregnant mice 2 days before mating was protective against IL-17-induced abortion. Interestingly, Treg therapy on day 7 had no effect. They found increased TGF-β and IL-10 in those mice transfused with Tregs 2 days before mating [73].
Similarly, Idali at al. compared iTregs generated from the spleens of CBA/J mice treated with either 17 β oestradiol, progesterone or transforming growth factor-β1 plus retinoic acid with Tregs isolated from pregnant CBA/J mice mated with BALB/c mice. DBA/2 mated pregnancy CBA/I mice were treated on days 1-4 of pregnancy. Treatment with iTregs significantly reduced resorption rates, with suppression of CD4+CD25- T cells evident on a 3H thymidine incorporation assay [74]. Similar to Idali et al. Yin examined the efficacy of adoptive transfer of iTregs and freshly isolated Tregs on the incidence of abortion in CBA./J x DBA/2J mice; they found iTregs significantly reduced fetal resorption, particularly with early treatment with increased serum IL-109, TGF-b1 and IL10:IFNγ ratio [75].
Woidacki et al. also used CBA/J mice but focused on the interplay between Tregs and uterine mast cells (uMC); postulating treatment with Tregs would promote the expansion of uMCs and, therefore, promote angiogenesis. They isolated Tregs from the spleens and lymph nodes of healthy pregnancy mice and transferred these into abortion-prone mice during the day. They found that uMC numbers were corrected by adoptive transfer of Tregs with subsequent improvement in spiral artery remodelling and placental development with increased levels of soluble fms-like tyrosine kinase (sFLT-1) [76].
Whether results will be as convincing in HDP remains to be seen, but there is a great deal of promise in using adoptive transfer of Tregs, particularly iTregs, for treating AOPs.
6. Therapeutic Potential of Tregs in Hypertensive Disorders of Pregnancy
There is a shortage of literature exploring the therapeutic potential of Tregs in HDP [72,77,78]. There are broadly two schools of thought in the existing literature: using ex-vivo expanded Treg populations directly as therapy or using other medicines to increase Treg numbers/function. These other therapies include preconception strategies and pharmacological intervention [48,63,79,80,81]. Strategies to increase or maintain endogenous Tregs preconception are mainly theoretical, with little literature available. Saftlas et al. demonstrated that increased preconception priming with the partners’ seminal fluid via unprotected vaginal intercourse has the potential to decrease PE, likely due to increased priming of pTregs specific for paternal antigens that will later be expressed by the fetus and placenta [82]. In a similar vein, it has been suggested decreasing preconception inflammatory load present in conditions such as smoking, obesity, and pre-diabetes might improve Treg function [83]
Where preconception methods of optimising Treg number/function fail, there is potential to augment Treg number/function using pharmaceuticals. There is evidence that progesterone directly affects Tregs; in cord blood, progesterone drives the activation of T cells into Tregs while reducing differentiation into pro-inflammatory Th17 cells [84] in vitro. Progesterone is further postulated to increase Treg stability and function by inhibition of the mTOR pathway [85]. Similar results are demonstrated in mouse models, where progesterone increases the number and function of CD4+FOXP3+ cells, though these results specifically refer to mid-gestation. Thus far, there has been no evidence that the use of progesterone reduces the incidence of PE in clinical trials.
Robertson et al. suggest advancing new treatments targeting Tregs in HDP prevention and treatment requires further research; specifically, they suggest developing appropriate diagnostics and validating preconception interventions that improve Treg numbers and function before carefully applying robust methodology to apply Treg therapies where lower intervention approaches are unsuccessful [48]. There is currently no clinically available test for Tregs preconception or antenatally. A wide range of markers is used to predict HDP in research, reflected in clinical practice [63]. Progressing in this area of research will require a consensus on which Treg markers to measure and their minimum levels; tools such as those developed by Prins et al. [79] will be central to standardising the clinical and immune outcomes measured and compared. Extensive cytometry marker panels that consider standard Treg and emerging markers will likely need to be developed. Such assays will need to consider how to measure the suppressive function of Tregs effectively and, therefore, include emerging markers of Treg suppressive potential such as CD154 in addition to the gold standard FOXP3 methylation status [80,81]
The potential of Tregs as a form of cell therapy in pregnancy has not been explored. In theory, the advantage of directed cell therapy is the targeted immune system augmentation without provoking systemic immune responses. Robertson suggests that Tregs’ capacity to suppress the immune response in an antigen-non-specific manner and ability to induce tolerance presents a significant advantage; Tregs could reasonably be expected to react to one paternally inherited fetal alloantigen to suppress the immune response to a wide range of placental and fetal antigens.
Outside of pregnancy, the primary clinical application of Treg therapies that has been explored is in solid organ transplant, where the success of the procedure is limited by the failure of immunosuppressive mechanisms leading to graft rejection and loss and by the severe side effects of long term immunosuppression. The ONE study isolated Tregs from patients undergoing living donor kidney transplantation and expanded these before re-infusion, demonstrating this can be done safely [86]. Similarly, the ThRIL study has revealed a similar safety profile for Treg therapy in liver transplantation, pacing the way for further trials [87].
A phase IIb trial, the TWO trial is underway to assess whether Treg infusion might offer a way to reduce the need for post-transplantation immunosuppression to avoid rejection. The study will specifically assess organ rejection via biopsy at 18 months post-transplantation in those receiving a standard immunosuppression regiment compared to those receiving an autologous polyclonal ex vivo expanded T regs 5 days post-transplantation [86,88].
Disappointingly, other trials have encountered issues with rejection [48] or manufacturing. Despite this, there is an enormous potential for Treg therapy in a plethora of clinical settings; there have been considerable strides in examining the utility of Treg therapies in AOPs, with much work needed to further explore their use in HDP.
7. Limitations of This Review Article
There is a great deal of heterogeneity in the existing literature exploring the role of Tregs in adverse pregnancy outcomes, which limits the understanding of the underlying immunology and potential therapeutic targets. The various studies considered in this review have seldom standardised characteristics that are known to impact immunological function; these include ethnicity and BMI. Much of this is unavoidable and a product of research being conducted globally, but it has implications for the interpretation of data. Furthermore, studies examine the number, function, and dysfunction of Tregs at different stages of pregnancy; this makes it difficult to draw firm conclusions from these data, given the variation in Treg concentration and function throughout pregnancy.
There is no standardised use of upstream or downstream cytokines/chemokines to characterise Tregs and no standard units of measurement, making it difficult to compare the various studies considered in this review. Promisingly, there are a number of commercially available antibody panels which will likely change the research landscape in the future.
The role of Tregs in HDP has not long been researched; a complete understanding of the immunological mechanisms underpinning the role of Tregs in HDP remains elusive, and the lack of longitudinal studies makes this review a thorough but incomplete summary of the role of Tregs in HDP. Further limitations of existing research are the lack of robust data and RCTs exploring the therapeutic potential of Tregs. As with other adverse pregnancy outcomes, there is no consensus on whether the specific T cell profiles explored are a consequence of HDP or consequential of HDP.
Further research could focus on characterising the T cell profile separately throughout pregnancy and postnatally in GH and PE using a longitudinal approach. This would allow a better understanding of Tregs using one set of cytokines throughout pregnancy. As with traditional risk factors for HDP, there is likely to be variation in these profiles dependent on specific characteristics such as ethnicity and BMI. In future studies collecting PBMCs for Treg analysis, it would be worthwhile to consider collecting demographic data alongside BMI and co-morbidities to properly establish any variation in results that might be attributable to these factors.
Once their predictive value has been explored, In-vitro and mouse models will need to be utilised to establish a therapeutic potential for Tregs in HDP. This may involve establishing the effect of current therapeutic strategies on Treg profiles in addition to treatment with Tregs or their inducers.
8. Conclusions
It appears clear that the Treg profile differs in healthy pregnancies and those affected by PE and GH. Enough data is available to conclude the effects of deficient or reduced numbers of Tregs on the delicate equilibrium of the immune system during pregnancy. There is, however, a lack of clinical and longitudinal studies exploring Tregs profiles and their consequences in vivo. Alongside this, the lack of a common approach to categorising the role of Tregs in HDP makes it challenging to be sure about the part of Tregs in this disease.
It also appears clear that Tregs have a great deal of potential as a therapy for HDP; a combination of strategies, including targeting Tregs preconception and Treg therapies, may have the capacity to transform the incidence and course of HDP.
Future research should focus on longitudinal studies before assessing the therapeutic benefit of targeting targeted Tregs. The public health burden of HDP is clear, and although unlikely to be a panacea, Tregs hold the potential to alleviate the burden of the disease significantly.
Author Contributions
Conceptualisation: KH, PS; methodology: KH, VJ, VM; investigation: KH, VJ, VM; data curation: KH; writing—original draft preparation; KH, VJ, VM; writing—review and editing; KH, CS, GL, KHN, PS; visualisation: KH; supervision: PS, CS, GL, KHN. All authors have read and agreed to the published version of the manuscript.
Funding
PS is supported by a Fetal Medicine Foundation Senior Clinical Lectureship and grants from the Fetal Medicine Foundation. KH is supported by a Fetal Medicine Foundation PhD studentship, KHN is supported by the Fetal Medicine Foundation.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Jiang, L.; Tang, C.; Gong, Y.; Liu, Y.; Rao, J.; Chen, S.; et al. PD-1/PD-L1 regulates Treg differentiation in pregnancy-induced hypertension. Brazilian Journal of Medical and Biological Research. 2018, 51. [Google Scholar] [CrossRef] [PubMed]
- Lisonkova, S.; Joseph, K.S. Incidence of preeclampsia: risk factors and outcomes associated with early- versus late-onset disease. Am J Obstet Gynecol. 2013, 209, 544–e1. [Google Scholar] [CrossRef] [PubMed]
- Vatish, M.; Powys, V.R.; Cerdeira, A.S. Novel therapeutic and diagnostic approaches for preeclampsia. Curr Opin Nephrol Hypertens. 2023, 32, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Magee, L.A.; Nicolaides, K.H.; von Dadelszen, P. Preeclampsia. N Engl J Med. 2022, 386, 1817–1832. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.; Wright, A.; Nicolaides, K.H. The competing risk approach for prediction of preeclampsia. Am J Obstet Gynecol. 2020, 223, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; et al. Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Vol. 72, Hypertension. Lippincott Williams and Wilkins; 2018. p. 24–43.
- Lambert, G.; Brichant, J.F.; Hartstein, G.; Bonhomme, V.; Dewandre, P.Y. Preeclampsia: an update. Acta Anaesthesiol Belg. 2014, 65, 137–149. [Google Scholar] [PubMed]
- Gathiram, P.; Moodley, J. Preeclampsia: Its pathogenesis and pathophysiolgy. Vol. 27, Cardiovascular Journal of Africa. Clinics Cardive Publishing (PTY)Ltd; 2016. p. 71–8.
- Huang, C.; Wei, K.; Lee, P.M.Y.; Qin, G.; Yu, Y.; Li, J. Maternal hypertensive disorder of pregnancy and mortality in offspring from birth to young adulthood: national population based cohort study. BMJ. 2022, 379, e072157. [Google Scholar] [CrossRef]
- Zakiyah, N.; Tuytten, R.; Baker, P.N.; Kenny, L.C.; Postma, M.J.; van Asselt, A.D.I. Early cost-effectiveness analysis of screening for preeclampsia in nulliparous women: A modelling approach in European high-income settings. PLoS One. 2022, 17. [Google Scholar] [CrossRef] [PubMed]
- NICE Hypertension in pregnancy: diagnosis and management. NICE Guideline 133. 2019.
- Green, S.; Politis, M.; Rallis, K.S.; Saenz de Villaverde Cortabarria, A.; Efthymiou, A.; Mureanu, N.; et al. Regulatory T Cells in Pregnancy Adverse Outcomes: A Systematic Review and Meta-Analysis. Vol. 12, Frontiers in Immunology. Frontiers Media S.A.; 2021.
- McElwain, C.; McCarthy, F.; McCarthy, C. Gestational Diabetes Mellitus and Maternal Immune Dysregulation: What We Know So Far. Int J Mol Sci. 2021, 22, 4261. [Google Scholar] [CrossRef]
- Pellerin, L.; Jenks, J.A.; Bégin, P.; Bacchetta, R.; Nadeau, K.C. Regulatory T cells and their roles in immune dysregulation and allergy. Vol. 58, Immunologic Research. Humana Press Inc.; 2014. p. 358–68.
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061, J Immunol. 2017 Feb 1;198(3):981–5. [Google Scholar] [CrossRef] [PubMed]
- Saito, S. Reconsideration of the Role of Regulatory T Cells During Pregnancy: Differential Characteristics of Regulatory T Cells Between the Maternal-Fetal Interface And Peripheral Sites and Between Early and Late Pregnancy. Medical Principles and Practice. 2022 Oct 4.
- Robertson, S.A.; Care, A.S.; Moldenhauer, L.M. Regulatory T cells in embryo implantation and the immune response to pregnancy. J Clin Invest. 2018, 128, 4224–4235. [Google Scholar] [CrossRef] [PubMed]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4 + T Cells: Differentiation and Functions. Clin Dev Immunol. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jurewicz, M.M.; Stern, L.J. Class II MHC antigen processing in immune tolerance and inflammation. Immunogenetics. 2019, 71, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Giganti, G.; Atif, M.; Mohseni, Y.; Mastronicola, D.; Grageda, N.; Povoleri, G.A.; et al. Treg cell therapy: How cell heterogeneity can make the difference. Eur J Immunol. 2021, 51, 39–55. [Google Scholar] [CrossRef]
- Kanhere, A.; Hertweck, A.; Bhatia, U.; Gökmen, M.R.; Perucha, E.; Jackson, I.; et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun. 2012, 3, 1268. [Google Scholar] [CrossRef] [PubMed]
- Dhamne, C.; Chung, Y.; Alousi, A.M.; Cooper, L.J.N.; Tran, D.Q. Peripheral and thymic foxp3(+) regulatory T cells in search of origin, distinction, and function. Front Immunol. 2013, 4, 253. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, A.S.; Schumacher, A. The T helper type 17/regulatory T cell paradigm in pregnancy. Immunology 2016, 148, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; Thornton, A.M. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. 2014, 259, 88–102. [Google Scholar] [CrossRef]
- Weiss, J.M.; Bilate, A.M.; Gobert, M.; Ding, Y.; Curotto de Lafaille, M.A.; Parkhurst, C.N.; et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. 2012, 209, 1723–1742. [Google Scholar] [CrossRef]
- Karim, M.; Feng, G.; Wood, K.J.; Bushell, A.R. CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood 2005, 105, 4871–4877. [Google Scholar]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Oderup, C.; Cederbom, L.; Makowska, A.; Cilio, C.M.; Ivars, F. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology. 2006, 118, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Onishi, Y.; Fehervari, Z.; Yamaguchi, T.; Sakaguchi, S. Foxp3 + natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proceedings of the National Academy of Sciences. 2008, 105, 10113–10118. [Google Scholar] [CrossRef]
- Kennedy, A.; Waters, E.; Rowshanravan, B.; Hinze, C.; Williams, C.; Janman, D.; et al. Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat Immunol. 2022, 23, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Workman, C.; Lee, J.; Chew, C.; Dale, B.M.; Colonna, L.; et al. Regulatory TCells Inhibit Dendritic Cells by Lymphocyte Activation Gene-3 Engagement of MHCClass, I. I. The Journal of Immunology. 2008, 180, 5916–5926. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; et al. Role of LAG-3 in Regulatory T Cells. Immunity. 2004, 21, 503–513. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; et al. CTLA-4 Control over Foxp3 + Regulatory T Cell Function. Science (1979). 2008, 322, 271–275. [Google Scholar]
- Sojka, D.K.; Huang, Y.H.; Fowell, D.J. Mechanisms of regulatory T-cell suppression – a diverse arsenal for a moving target. Immunology. 2008, 124, 13–22. [Google Scholar] [CrossRef]
- Kamanaka, M.; Huber, S.; Zenewicz, L.A.; Gagliani, N.; Rathinam, C.; O’Connor, W.; et al. Memory/effector (CD45RBlo) CD4 T cells are controlled directly by IL-10 and cause IL-22–dependent intestinal pathology. Journal of Experimental Medicine. 2011, 208, 1027–1040. [Google Scholar] [CrossRef]
- Sojka, D.K.; Fowell, D.J. Regulatory T cells inhibit acute IFN-γ synthesis without blocking T-helper cell type 1 (Th1) differentiation via a compartmentalised requirement for IL-10. Proceedings of the National Academy of Sciences. 2011, 108, 18336–18341. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007, 450, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Rocamora-Reverte, L.; Melzer, F.L.; Würzner, R.; Weinberger, B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front Immunol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Lui, P.P.; Cho, I.; Ali, N. Tissue regulatory T cells. Immunology. 2020, 161, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell. 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Kim, J.Y.; Hur, S.E.; Kim, C.J.; Na, B.J.; Lee, M.; et al. An imbalance in interleukin-17-producing T and Foxp3+ regulatory T cells in women with idiopathic recurrent pregnancy loss. Hum Reprod. 2011, 26, 2964–2971. [Google Scholar] [CrossRef] [PubMed]
- Krop, J.; Heidt, S.; Claas, F.H.J.; Eikmans, M. Regulatory T Cells in Pregnancy: It Is Not All About FoxP3. Vol. 11, Frontiers in Immunology. Frontiers Media S.A.; 2020.
- Zenclussen, A.C.; Gerlof, K.; Zenclussen, M.L.; Sollwedel, A.; Bertoja, A.Z.; Ritter, T.; et al. Abnormal T-Cell Reactivity against Paternal Antigens in Spontaneous Abortion. Am J Pathol. 2005, 166, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Salvany-Celades, M.; van der Zwan, A.; Benner, M.; Setrajcic-Dragos, V.; Bougleux Gomes, H.A.; Iyer, V.; et al. Three Types of Functional Regulatory T Cells Control T Cell Responses at the Human Maternal-Fetal Interface. Cell Rep. 2019, 27, 2537–2547. [Google Scholar] [CrossRef] [PubMed]
- Shima, T.; Sasaki, Y.; Itoh, M.; Nakashima, A.; Ishii, N.; Sugamura, K.; et al. Regulatory T cells are necessary for implantation and maintenance of early pregnancy but not late pregnancy in allogeneic mice. J Reprod Immunol. 2010, 85, 121–129. [Google Scholar] [CrossRef]
- Nevers, T.; Kalkunte, S.; Sharma, S. Uterine Regulatory T cells, IL-10 and Hypertension. American Journal of Reproductive Immunology 2011, 66, 88–92. [Google Scholar] [CrossRef]
- Rowe, J.H.; Ertelt, J.M.; Aguilera, M.N.; Farrar, M.A.; Way, S.S. Foxp3+ Regulatory T Cell Expansion Required for Sustaining Pregnancy Compromises Host Defense against Prenatal Bacterial Pathogens. Cell Host Microbe. 2011, 10, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.A.; Green, E.S.; Care, A.S.; Moldenhauer, L.M.; Prins, J.R.; Hull, M.L.; et al. Therapeutic Potential of Regulatory T Cells in Preeclampsia-Opportunities and Challenges. Front Immunol. 2019, 10, 478. [Google Scholar] [CrossRef] [PubMed]
- Maganto-García, E.; Bu, D.X.; Tarrio, M.L.; Alcaide, P.; Newton, G.; Griffin, G.K.; et al. Foxp3+-Inducible Regulatory T Cells Suppress Endothelial Activation and Leukocyte Recruitment. The Journal of Immunology. 2011, 187, 3521–3529. [Google Scholar]
- Matrougui, K.; Zakaria, A.E.; Kassan, M.; Choi, S.; Nair, D.; Gonzalez-Villalobos, R.A.; et al. Natural Regulatory T Cells Control Coronary Arteriolar Endothelial Dysfunction in Hypertensive Mice. Am J Pathol. 2011, 178, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, L.; Liu , B.; Li, Q.; Wang, Z.; Fan, S.; et al. Functional Defects of Regulatory T Cell Through Interleukin 10 Mediated Mechanism in the Induction of Gestational Diabetes Mellitus. DNA Cell Biol. 2018, 37, 278–285. [Google Scholar]
- Bennett, W.A.; Lagoo-Deenadayalan, S.; Whitworth, N.S.; Brackin, M.N.; Hale, E.; Cowan, B.D. Expression and production of interleukin-10 by human trophoblast: relationship to pregnancy immunotolerance. Early Pregnancy. 1997, 3, 190–198. [Google Scholar] [PubMed]
- Kwak-Kim, J.; Bao, S.; Lee, S.K.; Kim, J.W.; Gilman-Sachs, A. Immunological Modes of Pregnancy Loss: Inflammation, Immune Effectors, and Stress. American Journal of Reproductive Immunology. 2014, 72, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Kusanovic, J.P.; Chaiworapongsa, T.; Hassan, S.S. Placental bed disorders in preterm labor, preterm PROM, spontaneous abortion and abruptio placentae. Best Pract Res Clin Obstet Gynaecol. 2011, 25, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am J Obstet Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef]
- Tinsley, J.H.; Chiasson, V.L.; Mahajan, A.; Young, K.J.; Mitchell, B.M. Toll-like receptor 3 activation during pregnancy elicits preeclampsia-like symptoms in rats. Am J Hypertens. 2009, 22, 1314–1319. [Google Scholar] [CrossRef]
- Thaxton, J.E.; Romero, R.; Sharma, S. TLR9 activation coupled to IL-10 deficiency induces adverse pregnancy outcomes. J Immunol. 2009, 183, 1144–1154. [Google Scholar]
- Lai, Z.; Kalkunte, S.; Sharma, S. A critical role of interleukin-10 in modulating hypoxia-induced preeclampsia-like disease in mice. Hypertension. 2011, 57, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, R.; Mutter, W.P.; Wolf, M.; Levine, R.J.; Taylor, R.N.; Sukhatme, V.P.; et al. First trimester placental growth factor and soluble fms-like tyrosine kinase 1 and risk for preeclampsia. J Clin Endocrinol Metab. 2004, 89, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai , J.i.; Mammoto, T.; Kim, Y.M.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006, 12, 642–649. [Google Scholar]
- Barhoumi, T.; Kasal, D.A.; Li, M.W.; Shbat, L.; Laurant, P.; Neves, M.F.; et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011, 57, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, A.; Trevelin, S.C.; Mongue-Din, H.; Becker, P.D.; Ortiz, C.; Smyth, L.A.; et al. Nox2 in regulatory T cells promotes angiotensin II–induced cardiovascular remodeling. Journal of Clinical Investigation. 2018, 128, 3088–3101. [Google Scholar] [CrossRef] [PubMed]
- Rahimzadeh, M.; Norouzian, M.; Arabpour, F.; Naderi, N. Regulatory T-cells and preeclampsia: an overview of literature. Expert Rev Clin Immunol. 2016, 12, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Darmochwal-Kolarz, D.; Suzuki, D.; Sakai, M.; Ito, M.; Shima, T.; et al. Proportion of peripheral blood and decidual CD4+ CD25bright regulatory T cells in preeclampsia. Clin Exp Immunol. 2007, 149, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Santner-Nanan, B.; Peek, M.J.; Khanam, R.; Richarts, L.; Zhu, E.; Fazekas de St Groth, B.; et al. Systemic Increase in the Ratio between Foxp3+ and IL-17-Producing CD4+ T Cells in Healthy Pregnancy but Not in Preeclampsia. The Journal of Immunology. 2009, 183, 7023–7030. [Google Scholar] [CrossRef]
- Quinn, K.H.; Lacoursiere, D.Y.; Cui, L.; Bui, J.; Parast, M.M. The unique pathophysiology of early-onset severe preeclampsia: role of decidual T regulatory cells. J Reprod Immunol. 2011, 91, 76–82. [Google Scholar] [CrossRef]
- Hsu, P.; Santner-Nanan, B.; Dahlstrom, J.E.; Fadia, M.; Chandra, A.; Peek, M.; et al. Altered Decidual DC-SIGN+ Antigen-Presenting Cells and Impaired Regulatory T-Cell Induction in Preeclampsia. Am J Pathol. 2012, 181, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Steinborn, A.; Haensch, G.M.; Mahnke, K.; Schmitt, E.; Toermer, A.; Meuer, S.; et al. Distinct subsets of regulatory T cells during pregnancy: Is the imbalance of these subsets involved in the pathogenesis of preeclampsia? Clinical Immunology. 2008, 129, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Quinn, K.H.; Lacoursiere, D.Y.; Cui, L.; Bui, J.; Parast, M.M. The unique pathophysiology of early-onset severe preeclampsia: role of decidual T regulatory cells. J Reprod Immunol. 2011, 91, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.; Herrock, O.; Campbell, N.; Cornelius, D.; Fitzgerald, S.; Amaral, L.M.; et al. The role of immune cells and mediators in preeclampsia. Nat Rev Nephrol. 2023, 19, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ghaemi, M.S.; Ando, K.; Peterson, L.S.; Ganio, E.A.; Tsai, A.S.; et al. Differential Dynamics of the Maternal Immune System in Healthy Pregnancy and Preeclampsia. Front Immunol. 2019, 10, 1305. [Google Scholar] [CrossRef] [PubMed]
- Harmon, A.; Cornelius, D.; Amaral, L.; Paige, A.; Herse, F.; Ibrahim, T.; et al. IL-10 supplementation increases Tregs and decreases hypertension in the RUPP rat model of preeclampsia. Hypertens Pregnancy. 2015, 34, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Liu, F.J.; Xin-Liu Hao, C.F.; Bao, H.C.; Qu, Q.L.; et al. Adoptive transfer of pregnancy-induced CD4+CD25+ regulatory T cells reverses the increase in abortion rate caused by interleukin 17 in the CBA/J×BALB/c mouse model. Human Reproduction 2014, 29, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Idali, F.; Rezaii-nia, S.; Golshahi, H.; Fatemi, R.; Naderi, M.M.; Goli, L.B.; et al. Adoptive cell therapy with induced regulatory T cells normalises the abortion rate in abortion-prone mice. Reprod Fertil Dev. 2020. [CrossRef] [PubMed]
- Yin, Y.; Han, X.; Shi, Q.; Zhao, Y.; He, Y. Adoptive transfer of CD4+CD25+ regulatory T cells for prevention and treatment of spontaneous abortion. European Journal of Obstetrics & Gynecology and Reproductive Biology. 2012, 161, 177–181. [Google Scholar]
- Woidacki, K.; Meyer, N.; Schumacher, A.; Goldschmidt, A.; Maurer, M.; Zenclussen, A.C. Transfer of regulatory T cells into abortion-prone mice promotes the expansion of uterine mast cells and normalises early pregnancy angiogenesis. Sci Rep. 2015, 5, 13938. [Google Scholar] [CrossRef]
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Preeclampsia. The Lancet. 2010, 376, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Allan, S.E.; Broady, R.; Gregori, S.; Himmel, M.E.; Locke, N.; Roncarolo, M.G.; et al. CD4 + T-regulatory cells: toward therapy for human diseases. Immunol Rev. 2008, 223, 391–421. [Google Scholar] [CrossRef] [PubMed]
- Prins, J.R.; Holvast, F.; van ’t Hooft, J.; Bos, A.F.; Ganzevoort, J.W.; Scherjon, S.A.; et al. Development of a core outcome set for immunomodulation in pregnancy (COSIMPREG): a protocol for a systematic review and Delphi study. BMJ Open. 2018, 8, e021619. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, M.G.; Ronchetti, S.; Ricci, E.; Alunno, A.; Gerli, R.; Nocentini, G.; et al. GITR+ regulatory T cells in the treatment of autoimmune diseases. Autoimmun Rev. 2015, 14, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.; Eastaff-Leung, N.; Bresatz-Atkins, S.; Warner, N.; Ruitenberg, J.; Krumbiegel, D.; et al. Inhibition of activation induced CD154 on CD4 + CD25 − cells: a valid surrogate for human Treg suppressor function. Immunol Cell Biol. 2012, 90, 812–821. [Google Scholar] [CrossRef]
- Saftlas, A.F.; Rubenstein, L.; Prater, K.; Harland, K.K.; Field, E.; Triche, E.W. Cumulative exposure to paternal seminal fluid prior to conception and subsequent risk of preeclampsia. J Reprod Immunol. 2014, 101–102, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.M.; Green, E.S.; Tan, T.C.Y.; Gonzalez, M.B.; Rumbold, A.R.; Hull, M.L.; et al. Periconception onset diabetes is associated with embryopathy and fetal growth retardation, reproductive tract hyperglycosylation and impaired immune adaptation to pregnancy. Sci Rep. 2018, 8, 2114. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ulrich, B.; Cho, J.; Park, J.; Kim, C.H. Progesterone promotes differentiation of human cord blood fetal T cells into T regulatory cells but suppresses their differentiation into Th17 cells. J Immunol. 2011, 187, 1778–1787. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lydon, J.P.; Kim, C.H. Progesterone suppresses the mTOR pathway and promotes generation of induced regulatory T cells with increased stability. Eur J Immunol. 2012, 42, 2683–2696. [Google Scholar] [CrossRef]
- Sawitzki, B.; Harden, P.N.; Reinke, P.; Moreau, A.; Hutchinson, J.A.; Game, D.S.; et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. The Lancet. 2020, 395, 1627–1639. [Google Scholar] [CrossRef]
- Sánchez-Fueyo, A.; Whitehouse, G.; Grageda, N.; Cramp, M.E.; Lim, T.Y.; Romano, M.; et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. American Journal of Transplantation. 2020, 20, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Brook, M.O.; Hester, J.; Petchey, W.; Rombach, I.; Dutton, S.; Bottomley, M.J.; et al. Transplantation Without Overimmunosuppression (TWO) study protocol: a phase 2b randomised controlled single-centre trial of regulatory T cell therapy to facilitate immunosuppression reduction in living donor kidney transplant recipients. BMJ Open. 2022, 12, e061864. [Google Scholar] [CrossRef] [PubMed]
Figure 3.
Treg numbers throughout pregnancy. A schematic representation of the rising and falling number of Tregs in peripheral blood in response to a varying inflammatory load in a normal pregnancy. Implantation, labour and birth are highlighted as key events. However, a rise in inflammatory load, a Treg response during pregnancy, and a subsequent fall in inflammatory load and Treg numbers postnatally [17].
Figure 3.
Treg numbers throughout pregnancy. A schematic representation of the rising and falling number of Tregs in peripheral blood in response to a varying inflammatory load in a normal pregnancy. Implantation, labour and birth are highlighted as key events. However, a rise in inflammatory load, a Treg response during pregnancy, and a subsequent fall in inflammatory load and Treg numbers postnatally [17].
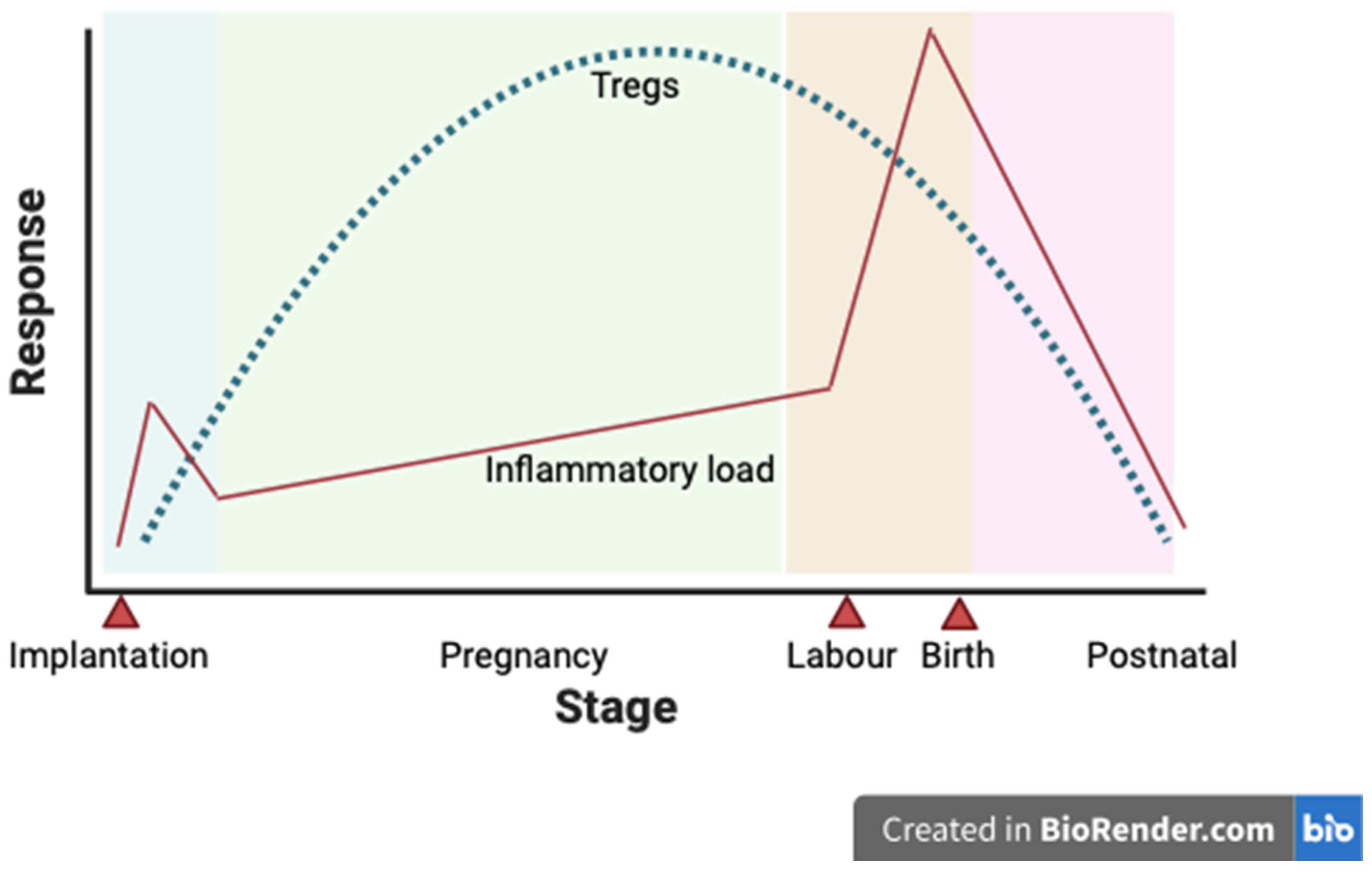
Figure 4.
Role of Tregs in promoting health placentation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



