
Submitted:
26 March 2024
Posted:
26 March 2024
You are already at the latest version
Abstract
DNA Topoisomerase IIa (Top2A) is a nuclear enzyme that is a cancer drug target, and there is interest in identifying novel sites on the enzyme to inhibit cancer cells more selectively and to reduce off-target toxicity. The C-terminal domain (CTD) is one potential target, but it is an intrinsically disordered domain, which prevents structural analysis. Therefore, we set out to analyze the sequence of Top2A from 105 species using bioinformatic analysis including the PSICalc algorithm, entropy analysis, and other approaches. Our results demonstrate that large (10th-order) interdependent clusters are found including non-proximal positions across the major domains of Top2A. Further, CTD-specific clusters of 3rd, 4th, and 5th-order were identified that included positions that had been previously analyzed via mutation and biochemical assays. Some of these clusters coincided with positions that when mutated either increased or decreased relaxation activity. Finally, sites of low entropy (i.e., high information content) were identified and mapped as key positions in the CTD. Included in the low entropy sites are phosphorylation sites and charged positions. Together, these results help to build a clearer picture of the critical positions in the CTD and provide potential sites/regions for further analysis.
Keywords:
topoisomerase II
; cancer
; bioinformatics
; intrinsically-disordered domain
; interdependency
; DNA
; topology
; protein structure.
1. Introduction
Protein sequence analysis has been in use for decades and is an important tool in biochemistry to explore protein structure and function and clarify enzyme mechanisms [1,2,3,4,5]. One form of protein sequence analysis is to look for interdependencies within protein sequences using multiple sequence alignments [2,3,6]. Interdependencies can help identify positions and regions that may interact either proximally or by way of a long-distance relationships [2,6]. Previously, we reported the application of a derivation of the K modes clustering algorithm to the question of protein sequence interdependency using the PSICalc algorithm [6]. This study aims to utilize an updated version of PSICalc and other bioinformatic tools to make discoveries on a critical target of chemotherapy: topoisomerase II.
Humans express two type II topoisomerases known as topoisomerase IIα and IIβ (Top2A and Top2B, respectively) [7,8,9]. These enzymes are found in the nucleus of all cells. Type II topoisomerases in eukaryotes are homodimers that unknot and alter the supercoiling state of DNA through a double-strand DNA cleavage and strand passage mechanism where the enzyme makes a temporary break in one double helix and passes another double helix through the break (Figure 1) [10,11]. Top2A expression fluctuates with the cell cycle and is most highly expressed in S-phase and M-phase consistent with its involvement in replication and mitosis [12,13,14,15,16]. Top2B is more involved in chromatin modulation and remodeling during transcription [12,13,14,16,17]. Type II topoisomerases have been the targets of anticancer drugs for decades because disruption of topoisomerase activity leads to DNA damage and consequently cell death [11,18]. Top2 inhibitors block catalytic activity without increasing cleaved DNA while Top2 poisons tend to lead to increased levels of DNA strand breaks [11,18].
Unfortunately, topoisomerase-targeted agents lack specificity, and both Top2A and Top2B are affected by the common Top2 inhibitors and poisons [11,19]. Most of the clinically-used Top2 drugs are poisons such as the anthracyclines and etoposide, which target the active site during the catalytic cycle of the enzyme. The active sites of Top2A and Top2B are very similar, which makes it hard for one to be targeted without hitting the other. One consequence of this is that there are some severe adverse events associated with Top2 poisons such as treatment-induced leukemia with etoposide and cardiotoxicity of the anthracyclines like doxorubicin [11,19,20,21]. There is evidence that these adverse events are mediated by Top2B [21,22,23,24,25]. Therefore, there is interest in designing specific inhibitors of Top2A since this isoform is very active in dividing cells (like cancer cells) and is reduced in expression in many differentiated tissues [13,14].
One challenge of designing selective inhibitors of Top2A is that Top2A and Top2B share a high degree of sequence identity through the ATPase and cleavage/ligation domain of the protein: ~81% identity (overall 69% identity for whole sequence). As depicted in Figure 1, the structure of Top2 includes an N-terminal ATPase domain followed by a transducer domain that enables communication between the ATPase domain and the core of the protein [26,27,28,29]. After the transducer domain, there is the TOPRIM domain that is a metal-binding domain found in topoisomerases and primases [30,31]. The TOPRIM domain is followed by the active site and DNA binding regions that coordinate with the TOPRIM domain during DNA cleavage and ligation [10]. The next portion is referred to as the C-terminal gate (or C-Gate), which is involved in releasing transported DNA segments from the enzyme [32]. A long α-helix leading up the side of the C-Gate leads to a large intrinsically disordered region (IDR) comprised of ~400 amino acids known as the C-terminal domain [33,34,35,36,37,38,39,40,41,42]. This region shares only ~42% identity between Top2A and Top2B. The CTD is important in localization, substrate selection, and regulating the activity of Top2 [34,35,36,41,42,43,44]. The CTD appears to interact with other proteins including histone 3A [38,40,45]. In addition, the CTD contributes to regulating the isoform specific localization and functions of Top2A and Top2B [34,35,36,37,46,47].
Recent studies have demonstrated that the IDR of the CTD in eukaryotic Top2 (including S. cerevisiae Top2 and human Top2A and Top2B) is involved in liquid-liquid phase separation and can form phase condensates with Top2 and DNA [42]. Consistent with previous studies of the CTD, these results validate the role of the CTD of Top2 in complex interactions that appear to regulate biochemical function of Top2 [39,40,41].
Over the last several years, we have aimed to characterize various regions of the CTD and understand the roles of the CTD in biochemical function [40,41]. Using a series of mutants, we analyzed function and identified regions that influence catalytic activity [41,48]. In addition, we have employed a bioinformatic tool called PSICalc to analyze the sequence and identify interdependencies in the sequence and develop additional mutants based upon this data [6].
In the present study, we report on the analysis of a previously-published Top2A-specific multiple-sequence alignment (MSA) with 105 species [49]. We have examined the MSA using an updated version of PSICalc, and we have identified various significant clusters within the protein and have analyzed them with a focus on interdomain interdependencies and clustering order. Our data will highlight clusters between separate domains and within the CTD. Further, we bring key observations together into a model for how PSICalc can be used for protein analysis and what can be learned from this tool about Top2A.
2. Materials and Methods
2.1. Top2A MSA Dataset
An MSA of Top2A from 105 organisms was obtained from the authors of Pereia, et. al [49]. The dataset was modified to place Homo sapiens Top2A at the top of the MSA before running the sequences through the PSICalc algorithm. Full MSA is available as a CSV file in Supplementary Materials.
2.2. Data Analysis
The interdependency data was collected utilizing PSICalc version 0.5.1. Blue Book for Mac OS available on Github (https://github.com/jdeweeselab/psicalc-package). The data was run selecting first row mapping and a percentage of non-insertion data set to 56%. The spread of 1 was selected to compare each column with each other column. The entropy threshold was set at 0.1 (Supplementary Materials: Data S2) and an additional run was completed at 0.11 (Supplementary Materials: Data S3) for comparison. Analysis in this paper focuses on the dataset from the 0.1 entropy threshold run. Data were output as Excel files available in the Supplementary Materials. Structure images were generated using Pymol 2.5.2 from crystallographic and Alpha-Fold structures. Entropy values isolated from PSICalc were plotted using Graphpad Prism 10. Amino acid frequency logo figure was generated using Weblogo (https://weblogo.berkeley.edu/).
3. Results
3.1. Improvements Made to the PSICalc Software Tool and Entropy Filtering
PSICalc utilizes a derivation of k-modes clustering where Normalized Mutual Information is the metric/distance measure used to compare amino acid changes in a column of an MSA with other columns of the MSA and to discover patterns between columns of the MSA (see Figure 2 for color-coded example). The algorithm discovers relationships between pairs of columns (where each column represents an aligned site in the MSA), and then clusters additional sites making 3rd, 4th, 5th and higher order clusters [6]. This pattern discovery approach does not require structural information, which allows it to be employed on both structured and disordered regions. As such, this tool can be applied to the CTD of Top2A and other proteins with IDRs. While the clusters may imply proximal interactions between amino acid positions, these may also imply long-distance interdependencies that may not be obvious based upon structural information.
In analyzing data from the previous version of PSICalc, it was recognized that some clusters identified in the analysis included one or two high entropy MSA columns (multiple amino acids found in the column) grouping with clusters of low entropy columns (little change in amino acids within a column) [see Supplementary Materials: Data S1, Anomalous Clusters]. It was determined that columns with an entropy close to 0 (i.e. columns with little variation in amino acids throughout the MSA) were causing a potential issue in the clustering algorithm. Since these columns were nearly invariant (e.g., varying ~1-2 species across the MSA), their significance is recognized in the overall protein structure. In other words, amino acid positions that are shared across all or nearly all species in the alignment are clearly critical to the protein. However, they offer little additional information regarding interdependencies within the protein since there are no clear patterns of association within the MSA. Therefore, we added a feature to the PSICalc software tool to be able to filter out low entropy columns using a sliding scale from 0-0.25 where the values are a measure of entropy, based upon the calculation used in the software (see Supplementary Materials file for entropy calculation). The calculation ignores gaps/insertions in sequence data and calculates based upon actual amino acid variations. In addition, the software tool now outputs the full column data for amino acids in each column of a cluster, for pairwise up to 10th-order clusters (Figure 2 and full data in Supplementary Materials: Data S2). We re-analyzed the Top2 MSA dataset from our previous paper and have presented clusters for selected positions in the Supplementary Materials (Data S1, Updated Clusters).
Previous work by Periea and colleagues on examining Top2A and Top2B generated a dataset of 105 sequences for Top2A that we have analyzed using an updated version PSICalc with entropy filtering [6]. We rearranged the sequences to place human Top2A as the first sequence and used this sequence for generating the position numbers for clusters from PSICalc. Within PSICalc, we adjusted the percentage of non-insertion data until the sequence length matched the length of human Top2A (1531 amino acids). The data were run with a spread of 1, which enables the comparison of each position. In addition, the entropy cut-off was set to 0.1, which removed positions that changed only once or not at all throughout this MSA. Full dataset output is available in the Supplementary Information (Data S2) along with the MSA used in the analysis (MSA File). As seen in the sample shown in Figure 2, PSICalc clusters positions from across the protein by identifying the patterns of amino acids in a given column compared with other columns.
The addition of the entropy cut-off removed a significant number of columns from the dataset under study. Over 700 columns (732 out of 1531 in Top2A) were found to have an entropy at or below 0.1 and were removed (see Supporting Information for output file). The remaining positions were clustered. As seen in Figure 3, mapping the entropy of each amino acid position across the protein demonstrates that the most variable region of the protein is the intrinsically disordered CTD (see Supplementary Materials File for entropy calculation. For example, of the 732 columns with low entropy, only 22 are found between 1175-1531. Thus, only 6.2% (22/356) of the columns in the CTD are low entropy compared to 60.3% (709/1175) of the N-terminal portion and 47.8% of the whole (732/1531). Interestingly, a review of the positions that were below the entropy cutoff demonstrates that domains involved with the catalytic cycle directly such as the ATPase, TOPRIM, active site, and structured DNA binding domains are highly invariant among the 105 species examined.
3.2. PSICalc Identifies Complex Interdomain Clusters
Upon analysis of the Top2A data, complex clusters spanning multiple domains were identified and examined. As seen in Figure 2, a portion of a 10th-order cluster is shown with positions representing multiple domains (see Supplementary Materials: Data S2 for full output file). Three clusters were selected from the 10th-order cluster set as outlined in Table 1.
As seen in Table 1, a strong interdependency is found among clusters of residues spread across the protein. In each case, positions from the N-terminus, core domains, and C-terminus cluster together (Figure 4). The strength of these intedependency relationships is quantified by the statistical redundancy mode (SRMode) value (max value of 0.2 for a 10th-order cluster), which has been defined previously [6]. Visual examination of the columns also shows the interdependence among the amino acid positions (Figure 2; Data S2). It is worth noting that every cluster examined at the 10th-order level included positions from across the domains of the protein (see Data S2).
3.3. PSICalc Identifies C-Terminal Domain Clusters
While clusters at the 10th order represented interdomain groupings, smaller clusters were identified that represented groupings within specific domains. Therefore, clusters that were exclusive to the C-terminal domain were examined. Table 2 contains the top CTD clusters of the 3rd, 4th, and 5th-orders from the analysis. At least one cluster represents sites that are adjacent: 1394, 1397, 1399. The remaining clusters are represented by sites spread across the CTD.
Included in several clusters in Table 2 are amino acid positions that were either mutated or are adjacent to positions that have previously been mutated to analyze the CTD of Top2A [41,48]. Some of the mutations had significant impact on catalytic activity, which likely resulted from altered binding/association properties. While the PSICalc clusters do not directly match up with the positions in the mutants, these previous results at least serve as a basis to consider the biochemical effect of changes to the regions under consideration.
3.4. Entropy Filtering Identifies Invariant Sites in the CTD
While the PSICalc algorithm identifies interdependencies among amino acid positions based upon the patterns of intrinsically linked variation, mutual information, it is also recognized that invariant positions have a critical role in protein structure and/or function. Interestingly, while the CTD is the most variable domain, there are 22 sites in the CTD that were filtered out due to low entropy (below 0.1 as calculated by PSICalc), which suggests these sites may have critical roles that are shared across various species (Figure 5, Table 3).
The 22 invariant sites are spread across the CTD, but most invariant sites are concentrated in range of 1175-1295 (Figure 5). The latter portion of this range includes a Nuclear Localization Sequence (NLS) from 1259-1296 [45,50]. Additionally, several sites are within the second NLS (1454-1497) [45,50]. Further, K1228 has evidence of being Sumoylated while S1295, S1332, and S1525 are phosphorylation sites [51]. Phosphorylation of S1525 is associated with the decatenation checkpoint [44].
3.5. Low Entropy Positions Correlate with Charged Positions
As seen in Table 3 and Figure 5, many of the sites that do not change represent charged sites, especially Arg and Lys. Interestingly, an analysis of the frequency of amino acids at each site in the CTD across the 105 species in the alignment displays some key themes and regions through the CTD. As seen in Figure 6, there is a high proportion of charged residues, especially Lys, Arg, Asp, and Glu throughout the CTD, and many of these positions are highly invariant as seen in the sequence logo. In the sequence logo, taller letters indicate the position is less variable within the MSA, which implies higher information content (bits) compared to sites that are more variable. In addition, these residues appear to come in an alternating pattern (positive – negative – positive – negative), especially closer to the end of the sequence.
From an examination of Figure 6, the 0.1 value for entropy cutoff did not catch all the low entropy positions in the CTD. Positions such as M1227, F1282, K1283, and R1313 are marginally above the threshold but are still mostly invariant. Adjusting the value to 0.11 or higher allows these sites to be removed from the clustering. Re-running that analysis at 0.11 (which included positions that change twice across the MSA such as 1517, 1518) provides a very similar set of clusters indicating that these sites were not affecting the bulk of the clustering process (See Supplementary Materials: Data S3).
4. Discussion
In this present study, we use an updated PSICalc clustering algorithm to analyze a dataset of Top2A from 105 species originally published by Moreira, et. al [49]. The original PSICalc algorithm errantly clustered one or two high entropy columns with nearly invariant (low entropy) columns [Supplementary Materials Data S1]. As a result, spurious clusters were formed. The latest version removes the invariant and nearly invariant clusters using an entropy-filtering approach. In addition, PSICalc now outputs clusters both numerically with SRMode values and as groups of columns representing clustered positions in the MSA numbered according to the first row of the MSA.
Our results here show that even with the removal of the low entropy positions, PSICalc still discovers clusters within and among various domains of the protein. Interestingly, several large clusters are identified with moderately strong SRMode values where positions from the N-terminus, core domain, and C-terminus of Top2A are all within the cluster. These groupings imply long-range interdependencies within the protein. While the biochemical details of such interdependencies have not been worked out, the results suggest that interdomain relationships may play a key role in large, multidomain proteins like Top2A. Interdomain interactions have been explored on some level but many details remain to be examined [54].
Further, CTD-specific clusters were also identified and compared with previous results of biochemical experiments characterizing CTD mutants (Table 2). Strikingly, many of the identified interdependent clusters either included one or more mutated position or were adjacent to the mutated positions in our previous study. These mutations were selected by identifying groupings of Ser and Thr residues and mutating them as a group to determine whether those mutations impacted biochemical activity [41]. As noted in Table 2, some of the mutations either increased or decreased plasmid DNA relaxation activity, which may imply that these regions influence substrate selection, substrate binding, stability of the enzyme:DNA interaction, and/or other aspects of the catalytic cycle [41,48].
Entropy filtering removed invariant and nearly invariant sites, which are certainly recognized as being critical. Given that almost half of Top2A is essentially invariant across 105 species, it appears that many positions in this enzyme are fixed. This includes 22 positions in the C-terminal domain that were identified by the entropy filtering. Interestingly, these are primarily found within one region (~1176-1295). Most of the positions are either charged or polar and several are found within the NLSs. A few are associated with known phosphorylation sites. S1295, S1332, and S1525 are all known to be phosphorylated in a cell cycle-dependent manner and all appear to impact catalytic activity [41,44,48,53,55]. Interestingly, each of these sites were mutated in our previous study looking for roles of clusters of CTD residues [41,48]. While the regions including S1295 and S1332 appeared to decrease relaxation when mutated, there was no observed effect on relaxation when S1525, but there was a slight increase in DNA cleavage levels, especially with etoposide [41].
Finally, there appears to be a clear pattern in the CTD of alternating charged residues between Asp/Glu and Arg/Lys (Figure 6). Importantly, the charged residues appear to be sites that are often less variant than positions around these sites. This implies that the charges are likely involved in the function of the CTD. While some charged positions may be explained as being a part of an NLS, other positions could be involved in DNA and/or protein interactions. McClendon, et al. proposed that patches of positively charged amino acids in the CTD are potentially involved in recognition of substrate topology [35]. Deletion of one or more of these patches appears to eliminate the ability of Top2A to differentiate between positive and negative supercoils [35]. Vanden Broeck, et al. found that a small flexible “linker” region of the Top2A CTD was visible by cryo-electron microscopy (residues 1191-1217) [54]. Of note, this region includes a series of positively charged amino acids and appears to interact with the Gate-segment of DNA [54]. Our data are consistent with their findings and indicate that this region includes relatively invariant positive charges along with some other critical positions.
Additional evidence regarding the role of the charged patches in the CTD comes from recent work by Jeong et al. which demonstrates that phase condensation of scTop2, Top2A, and Top2B is sensitive to salt concentration [42]. Increasing concentrations of salt (from 150 up to 400 mM potassium acetate) disrupted the phase condensates formed by Top2 in the presence or absence of DNA [42]. The ionic disruption of these interactions appears to indicate the charges in the CTD are likely important in the interactions between Top2 and potential binding partners including DNA and proteins. Additionally, the alteration of activity by phosphorylation and other modifications is supportive of this role as well [39,40,51,53].
Considering the above data, various strategies could be employed to identify potential sites for targeting Top2A in a selective manner. One possible strategy is to develop a way to target the ionic interactions between the CTD and various targets. This will require identification of specific binding partners that influence the activity of Top2 in the nucleus, and then characterize the binding modalities to exploit features that could lead to altered enzyme activity.
Another strategy for identifying a region of interest is to compare the amino acid positions involved in CTD-only clusters and determine whether those positions are also found in Top2B. Of the CTD-only clusters in Table 2, those with the highest SRMode values include unique amino acid residues as well as those that are identical to analogous positions in Top2B (e.g., see Top2A/Top2B alignment in Supporting Information Figure S1 and Table S1). For instance, the top 5th-order cluster includes two positions that are identical in Top2B (D1304/D1345, D1344/D1387 where the positions are denoted Top2A/Top2B), one position that is similar (Q1217/R1240), and two positions that are different (L1364/K1434 and V1482/T1564). Another 5th-order cluster shows a similar pattern where E1189/V1207, E1232/S1254, and V1513/G1601 differ while T1272/T1312 and A1321/A1364 are identical. Of note, this latter cluster includes positions near the invariant region of Top2A. While additional analysis will be needed to determine whether this same region is invariant in Top2B, it is reasonable to consider this region for further studies.
5. Conclusions
Using an updated PSICalc algorithm with entropy filtering combined with clustering outputs allowed for rapid identification of significant clusters through both SRMode value ranking and visual inspection of the clustered sites across the MSA. Analysis of a 105 species MSA of Top2A sequences demonstrated that nearly half of Top2A is highly constrained across species. Further, clusters were identified that spread across the domains of Top2A. Biochemical and other analyses are needed to determine the significance of such interdomain clusters. It is possible that some of these clusters represent control and communication mechanisms. However, the significance of the low entropy clusters must also be maintained in these analyses. In addition, it will be critical going forward to identify and more fully characterize the nature of interactions of Top2 CTD with protein binding partners.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Data S1: Anomalous and Updated Cluster Examples from Previous TOP2 MSA Dataset; Data S2: Top2A 105 sp PSICalc Data 0.1 Entropy Filter; Data S3: Top2A 105 sp PSICalc Data 0.11 Entropy Filter; MSA File: Top2 105 sp; Supplemenatary Materials File: Entropy Calculation; Figure S1: Top2A and Top2B CTD alignment; Table S1: Example Clusters Compared between Top2A and Top2B CTD.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, J.E.D.; methodology, J.E.D., K.K.D. T.D.T.; software, T.D.T, K.K.D.; investigation, C.E.E., K.A.M., J.E.D.; data curation, C.E.E., K.A.M., J.E.D., T.D.T.; writing—original draft preparation, C.E.E., K.A.M., J.E.D.; writing—review and editing, C.E.E., K.A.M., J.E.D., T.D.T., K.K.D.; visualization, C.E.E., K.A.M., J.E.D.; project administration, J.E.D.; funding acquisition, J.E.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Freed-Hardeman University, Jobe Center of Excellence in Biological Sciences, FHU Undergraduate Research, Center for Science and Culture, and Biological, Physical, and Human Sciences Department (FHU).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Acknowledgments
We would like to thank Dr. Filipe Pereira for providing the alignment datasets from a previous study for us to analyze in this current work. We would like to thank Dr. Timothy L. Wallace for helpful discussions on the advancement of the software tool.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wong, A.K.; Liu, T.S.; Wang, C.C. Statistical analysis of residue variability in cytochrome c. J. Mol. Biol. 1976, 102, 287–295. [Google Scholar] [CrossRef]
- Durston, K.K.; Chiu, D.K.; Wong, A.K.; Li, G.C. Statistical discovery of site inter-dependencies in sub-molecular hierarchical protein structuring. EURASIP J. Bioinform. Syst. Biol. 2012, 2012. [Google Scholar] [CrossRef]
- Schmidt, M.; Hamacher, K. hoDCA: higher order direct-coupling analysis. BMC Bioinformatics 2018, 19, 546. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Nartey, C.M.; Koo, H.J.; Laurendon, C.; Shaik, H.Z.; O'Maille, P.; Noel, J.P.; Morcos, F. Coevolutionary Information Captures Catalytic Functions and Reveals Divergent Roles of Terpene Synthase Interdomain Connections. Biochemistry 2024, 63, 355–366. [Google Scholar] [CrossRef]
- Townsley, T.D.; Wilson, J.T.; Akers, H.; Bryant, T.; Cordova, S.; Wallace, T.L.; Durston, K.K.; Deweese, J.E. PSICalc: a novel approach to identifying and ranking critical non-proximal interdependencies within the overall protein structure. Bioinform Adv 2022, 2, vbac058. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- McKie, S.J.; Neuman, K.C.; Maxwell, A. DNA topoisomerases: Advances in understanding of cellular roles and multi-protein complexes via structure-function analysis. Bioessays 2021, e2000286. [Google Scholar] [CrossRef]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: wolf in sheep's clothing. Nucleic Acids Res. 2009, 37, 738–749. [Google Scholar] [CrossRef]
- Murphy, M.B.; Mercer, S.L.; Deweese, J.E. Inhibitors and Poisons of Mammalian Type II Topoisomerases. In Advances in Molecular Toxicology, Fishbein, J.C., Heilman, J., Eds.; Academic Press: Cambridge, MA, 2017; Volume 11, pp. 203–240. [Google Scholar]
- Capranico, G.; Tinelli, S.; Austin, C.A.; Fisher, M.L.; Zunino, F. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim. Biophys. Acta 1992, 1132, 43–48. [Google Scholar] [CrossRef]
- Prosperi, E.; Sala, E.; Negri, C.; Oliani, C.; Supino, R.; Astraldi Ricotti, G.B.; Bottiroli, G. Topoisomerase II alpha and beta in human tumor cells grown in vitro and in vivo. Anticancer Res. 1992, 12, 2093–2099. [Google Scholar]
- Christensen, M.O.; Larsen, M.K.; Barthelmes, H.U.; Hock, R.; Andersen, C.L.; Kjeldsen, E.; Knudsen, B.R.; Westergaard, O.; Boege, F.; Mielke, C. Dynamics of human DNA topoisomerases IIα and IIβ in living cells. J. Cell. Biol. 2002, 157, 31–44. [Google Scholar] [CrossRef]
- Carpenter, A.J.; Porter, A.C. Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIalpha mutant human cell line. Mol. Biol. Cell 2004, 15, 5700–5711. [Google Scholar] [CrossRef]
- Sakaguchi, A.; Kikuchi, A. Functional compatibility between isoform alpha and beta of type II DNA topoisomerase. J. Cell Sci. 2004, 117, 1047–1054. [Google Scholar] [CrossRef]
- Cowell, I.G.; Casement, J.W.; Austin, C.A. To Break or Not to Break: The Role of TOP2B in Transcription. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Cowell, I.G.; Austin, C.A. Mechanism of Generation of Therapy Related Leukemia in Response to Anti-Topoisomerase II Agents. Int. J. Environ. Res. Public Health 2012, 9, 2075–2091. [Google Scholar] [CrossRef]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Frank, N.E.; Cusack, B.J.; Talley, T.T.; Walsh, G.M.; Olson, R.D. Comparative effects of doxorubicin and a doxorubicin analog, 13-deoxy, 5-iminodoxorubicin (GPX-150), on human topoisomerase IIbeta activity and cardiac function in a chronic rabbit model. Invest. New Drugs 2016, 34, 693–700. [Google Scholar] [CrossRef]
- Lyu, Y.L.; Kerrigan, J.E.; Lin, C.P.; Azarova, A.M.; Tsai, Y.C.; Ban, Y.; Liu, L.F. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef]
- Cowell, I.G.; Sondka, Z.; Smith, K.; Lee, K.C.; Manville, C.M.; Sidorczuk-Lesthuruge, M.; Rance, H.A.; Padget, K.; Jackson, G.H.; Adachi, N.; et al. Model for MLL translocations in therapy-related leukemia involving topoisomerase IIbeta-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 8989–8994. [Google Scholar] [CrossRef]
- Azarova, A.M.; Lyu, Y.L.; Lin, C.P.; Tsai, Y.C.; Lau, J.Y.; Wang, J.C.; Liu, L.F. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. USA 2007, 104, 11014–11019. [Google Scholar] [CrossRef]
- Dutta, R.; Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci 2000, 25, 24–28. [Google Scholar] [CrossRef]
- Classen, S.; Olland, S.; Berger, J.M. Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187. Proc. Natl. Acad. Sci. USA 2003, 100, 10629–10634. [Google Scholar] [CrossRef]
- Oestergaard, V.H.; Bjergbaek, L.; Skouboe, C.; Giangiacomo, L.; Knudsen, B.R.; Andersen, A.H. The transducer domain is important for clamp operation in human DNA topoisomerase IIalpha. J. Biol. Chem. 2004, 279, 1684–1691. [Google Scholar] [CrossRef]
- Bendsen, S.; Oestergaard, V.H.; Skouboe, C.; Brinch, M.; Knudsen, B.R.; Andersen, A.H. The QTK loop is essential for the communication between the N-terminal atpase domain and the central cleavage--ligation region in human topoisomerase IIalpha. Biochemistry 2009, 48, 6508–6515. [Google Scholar] [CrossRef]
- Aravind, L.; Leipe, D.D.; Koonin, E.V. Toprim--a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998, 26, 4205–4213. [Google Scholar] [CrossRef]
- Deweese, J.E.; Osheroff, N. The Use of Divalent Metal Ions by Type II Topoisomerases. Metallomics 2010, 2, 450–459. [Google Scholar] [CrossRef]
- Roca, J. The path of the DNA along the dimer interface of topoisomerase II. J. Biol. Chem. 2004, 279, 25783–25788. [Google Scholar] [CrossRef]
- Shaiu, W.L.; Hu, T.; Hsieh, T.S. The hydrophilic, protease-sensitive terminal domains of eucaryotic DNA topoisomerases have essential intracellular functions. Pac. Symp. Biocomput. 1999, 578–589. [Google Scholar] [CrossRef]
- Linka, R.M.; Porter, A.C.; Volkov, A.; Mielke, C.; Boege, F.; Christensen, M.O. C-terminal regions of topoisomerase IIα and IIβ determine isoform-specific functioning of the enzymes in vivo. Nucleic Acids Res. 2007, 35, 3810–3822. [Google Scholar] [CrossRef]
- McClendon, A.K.; Gentry, A.C.; Dickey, J.S.; Brinch, M.; Bendsen, S.; Andersen, A.H.; Osheroff, N. Bimodal recognition of DNA geometry by human topoisomerase II alpha: preferential relaxation of positively supercoiled DNA requires elements in the C-terminal domain. Biochemistry 2008, 47, 13169–13178. [Google Scholar] [CrossRef]
- Meczes, E.L.; Gilroy, K.L.; West, K.L.; Austin, C.A. The impact of the human DNA topoisomerase II C-terminal domain on activity. PLoS One 2008, 3, e1754. [Google Scholar] [CrossRef]
- Gilroy, K.L.; Austin, C.A. The impact of the C-terminal domain on the interaction of human DNA topoisomerase II alpha and beta with DNA. PLoS One 2011, 6, e14693. [Google Scholar] [CrossRef]
- Lane, A.B.; Gimenez-Abian, J.F.; Clarke, D.J. A novel chromatin tether domain controls topoisomerase IIalpha dynamics and mitotic chromosome formation. J. Cell Biol. 2013, 203, 471–486. [Google Scholar] [CrossRef]
- Clarke, D.J.; Azuma, Y. Non-Catalytic Roles of the Topoisomerase IIalpha C-Terminal Domain. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Hoang, K.G.; Menzie, R.A.; Rhoades, J.H.; Fief, C.A.; Deweese, J.E. Reviewing the Modification, Interactions, and Regulation of the C-terminal Domain of Topoisomerase IIα as a Prospect for Future Therapeutic Targeting. EC Pharmacology & Toxicology 2020, 8, 27–43. [Google Scholar]
- Dougherty, A.C.; Hawaz, M.G.; Hoang, K.G.; Trac, J.; Keck, J.M.; Ayes, C.; Deweese, J.E. Exploration of the Role of the C-Terminal Domain of Human DNA Topoisomerase IIalpha in Catalytic Activity. ACS Omega 2021, 6, 25892–25903. [Google Scholar] [CrossRef]
- Jeong, J.; Lee, J.H.; Carcamo, C.C.; Parker, M.W.; Berger, J.M. DNA-Stimulated Liquid-Liquid phase separation by eukaryotic topoisomerase ii modulates catalytic function. Elife 2022, 11. [Google Scholar] [CrossRef]
- Mirski, S.E.; Bielawski, J.C.; Cole, S.P. Identification of functional nuclear export sequences in human topoisomerase IIalpha and beta. Biochem Biophys Res Commun 2003, 306, 905–911. [Google Scholar] [CrossRef]
- Luo, K.; Yuan, J.; Chen, J.; Lou, Z. Topoisomerase IIalpha controls the decatenation checkpoint. Nat. Cell Biol. 2009, 11, 204–210. [Google Scholar] [CrossRef]
- Mirski, S.E.; Gerlach, J.H.; Cummings, H.J.; Zirngibl, R.; Greer, P.A.; Cole, S.P. Bipartite nuclear localization signals in the C terminus of human topoisomerase IIa. Exp. Cell Res. 1997, 237, 452–455. [Google Scholar] [CrossRef]
- McClendon, A.K.; Osheroff, N. The geometry of DNA supercoils modulates topoisomerase-mediated DNA cleavage and enzyme response to anticancer drugs. Biochemistry 2006, 45, 3040–3050. [Google Scholar] [CrossRef]
- Bedez, C.; Lotz, C.; Batisse, C.; Broeck, A.V.; Stote, R.H.; Howard, E.; Pradeau-Aubreton, K.; Ruff, M.; Lamour, V. Post-translational modifications in DNA topoisomerase 2alpha highlight the role of a eukaryote-specific residue in the ATPase domain. Sci. Rep. 2018, 8, 9272. [Google Scholar] [CrossRef]
- Musselman, J.R.; England, D.C.; Fielding, L.A.; Durham, C.T.; Baxter, E.; Jiang, X.; Lisic, E.C.; Deweese, J.E. Topoisomerase IIα C-terminal Domain Mutations and Catalytic Function. bioRxiv, 2007. [Google Scholar] [CrossRef]
- Moreira, F.; Arenas, M.; Videira, A.; Pereira, F. Evolutionary History of TOPIIA Topoisomerases in Animals. J. Mol. Evol. 2022, 90, 149–165. [Google Scholar] [CrossRef]
- Mirski, S.E.; Gerlach, J.H.; Cole, S.P. Sequence determinants of nuclear localization in the alpha and beta isoforms of human topoisomerase II. Exp. Cell Res. 1999, 251, 329–339. [Google Scholar] [CrossRef]
- Lotz, C.; Lamour, V. The interplay between DNA topoisomerase 2α post-translational modifications and drug resistance. Cancer Drug Resistance 2020, 3. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–520. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Liu, X. Plk1-dependent phosphorylation regulates functions of DNA topoisomerase IIalpha in cell cycle progression. J. Biol. Chem. 2008, 283, 6209–6221. [Google Scholar] [CrossRef] [PubMed]
- Vanden Broeck, A.; Lotz, C.; Drillien, R.; Haas, L.; Bedez, C.; Lamour, V. Structural basis for allosteric regulation of Human Topoisomerase IIalpha. Nat Commun 2021, 12, 2962. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, A.; Wang, B.; Elowe, S.; Malik, R.; Zhang, F.; Bauer, M.; Schmidt, A.; Sillje, H.H.; Korner, R.; Nigg, E.A. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol. Cell. Proteomics 2011, 10, M110–004457. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Domain Structure and Catalytic Cycle of Topoisomerase II. A) Ribbon diagrams of the crystal structure of S. Cerevisiae Top2 are shown as dimers rotated 90 degrees with one monomer colored and the other in gray. Color coding matches part C. Gates are labeled at left. B) The topoisomerase II catalytic cycle is shown in six stages including 1) binding of gate-segment (G-segment); 2) binding of ATP and transport-segment (T-segment); 3) temporary cleavage of G-segment, opening of G-segment (DNA gate), hydrolysis of one ATP, transport of the T-segment; 4) closing of the G-segment, conformational change of the ATPase/transducer domains; 5) hydrolysis of the second ATP, opening of the C-gate and release of the T-segment; and 6) release of the ADP and of the G-segment. C) Domain organization of topoisomerase II. Domains are labeled and color-coded. Winged-helix domain (WHD) within the DNA binding region includes the active site tyrosine (Y805 in human Top2A). Nuclear export signals (NES), nuclear localization signals (NLS), and the Chromatin Tether domain (ChT) are also denoted. The C-terminal domain (CTD) is shown in grey and is not shown in parts A and B due to the unstructured nature of the region. Percent identity shown at the bottom based upon BLASTp comparison of P11388 Top2A with Q02880 Top2B sequences with CTD starting at 1175 (Top2A)/1193 (Top2B).
Figure 1.
Domain Structure and Catalytic Cycle of Topoisomerase II. A) Ribbon diagrams of the crystal structure of S. Cerevisiae Top2 are shown as dimers rotated 90 degrees with one monomer colored and the other in gray. Color coding matches part C. Gates are labeled at left. B) The topoisomerase II catalytic cycle is shown in six stages including 1) binding of gate-segment (G-segment); 2) binding of ATP and transport-segment (T-segment); 3) temporary cleavage of G-segment, opening of G-segment (DNA gate), hydrolysis of one ATP, transport of the T-segment; 4) closing of the G-segment, conformational change of the ATPase/transducer domains; 5) hydrolysis of the second ATP, opening of the C-gate and release of the T-segment; and 6) release of the ADP and of the G-segment. C) Domain organization of topoisomerase II. Domains are labeled and color-coded. Winged-helix domain (WHD) within the DNA binding region includes the active site tyrosine (Y805 in human Top2A). Nuclear export signals (NES), nuclear localization signals (NLS), and the Chromatin Tether domain (ChT) are also denoted. The C-terminal domain (CTD) is shown in grey and is not shown in parts A and B due to the unstructured nature of the region. Percent identity shown at the bottom based upon BLASTp comparison of P11388 Top2A with Q02880 Top2B sequences with CTD starting at 1175 (Top2A)/1193 (Top2B).
Figure 2.
Example Cluster from PSICalc Analysis with Color-Coded Amino Acids. Sites are listed across the top numbered according to human Top2A positions. Species are listed at left. Cluster truncated for display purposes. Amino acids are color coded to highlight the interdependencies. Full cluster data available in Supplementary Materials (Data S2).
Figure 2.
Example Cluster from PSICalc Analysis with Color-Coded Amino Acids. Sites are listed across the top numbered according to human Top2A positions. Species are listed at left. Cluster truncated for display purposes. Amino acids are color coded to highlight the interdependencies. Full cluster data available in Supplementary Materials (Data S2).
Figure 3.
Entropy Value for Each Amino Acid Position in the Top2A Dataset. The cutoff value of 0.1 is denoted by the dotted line. Positions below this value were not clustered.
Figure 3.
Entropy Value for Each Amino Acid Position in the Top2A Dataset. The cutoff value of 0.1 is denoted by the dotted line. Positions below this value were not clustered.
Figure 4.
Mapping of 10th-order clusters onto Alphafold Structure of Top2A Monomer. Three 10th-order clusters are highlighted in red, green, and yellow corresponding to colors in Table 1 mapped onto a monomer of Top2A with the intrinsically disordered region shown. Views are rotated 90 degrees relative to each other. Structure from AlphaFold: AF-P11388-F1. Image generated using Pymol 2.5.2.
Figure 4.
Mapping of 10th-order clusters onto Alphafold Structure of Top2A Monomer. Three 10th-order clusters are highlighted in red, green, and yellow corresponding to colors in Table 1 mapped onto a monomer of Top2A with the intrinsically disordered region shown. Views are rotated 90 degrees relative to each other. Structure from AlphaFold: AF-P11388-F1. Image generated using Pymol 2.5.2.
Figure 5.
Mapping Sites in the C-terminal Domain. Low entropy sites are shown in red. Sites that are known to be phosphorylated in association with mitosis are in blue. Purple sites indicate low entropy sites that are also known phosphorylation sites. Nuclear localization sequences (NLS) and Chromatin Tether (ChT) domains are indicated. Phosphorylation data from Phosphosite Plus. Figure generated using SnapGene.
Figure 5.
Mapping Sites in the C-terminal Domain. Low entropy sites are shown in red. Sites that are known to be phosphorylated in association with mitosis are in blue. Purple sites indicate low entropy sites that are also known phosphorylation sites. Nuclear localization sequences (NLS) and Chromatin Tether (ChT) domains are indicated. Phosphorylation data from Phosphosite Plus. Figure generated using SnapGene.
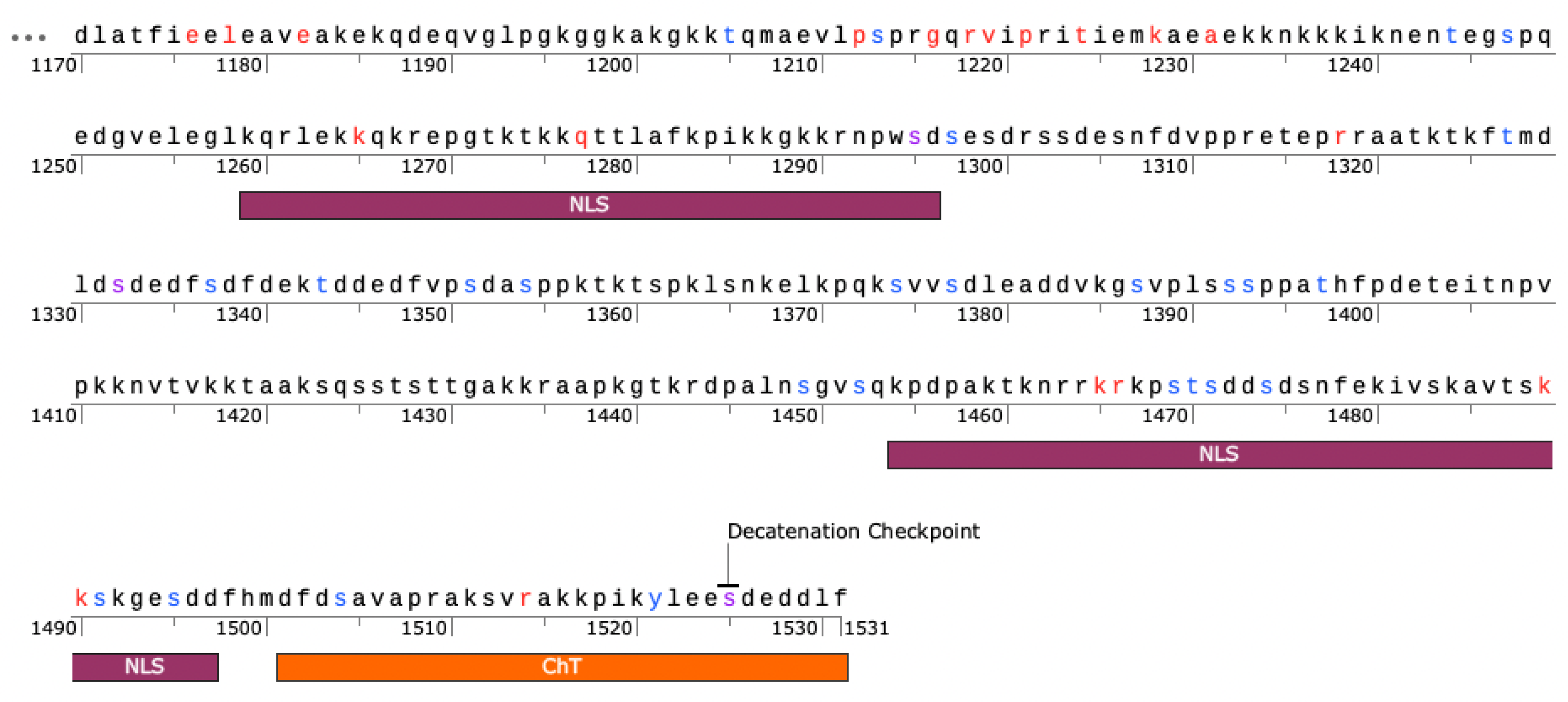
Figure 6.
Frequency Plot of Amino Acids in the CTD from the 105 Species Alignment. Taller letters indicate the frequency of the amino acid at a given position. Figure generated using WebLogo.
Figure 6.
Frequency Plot of Amino Acids in the CTD from the 105 Species Alignment. Taller letters indicate the frequency of the amino acid at a given position. Figure generated using WebLogo.
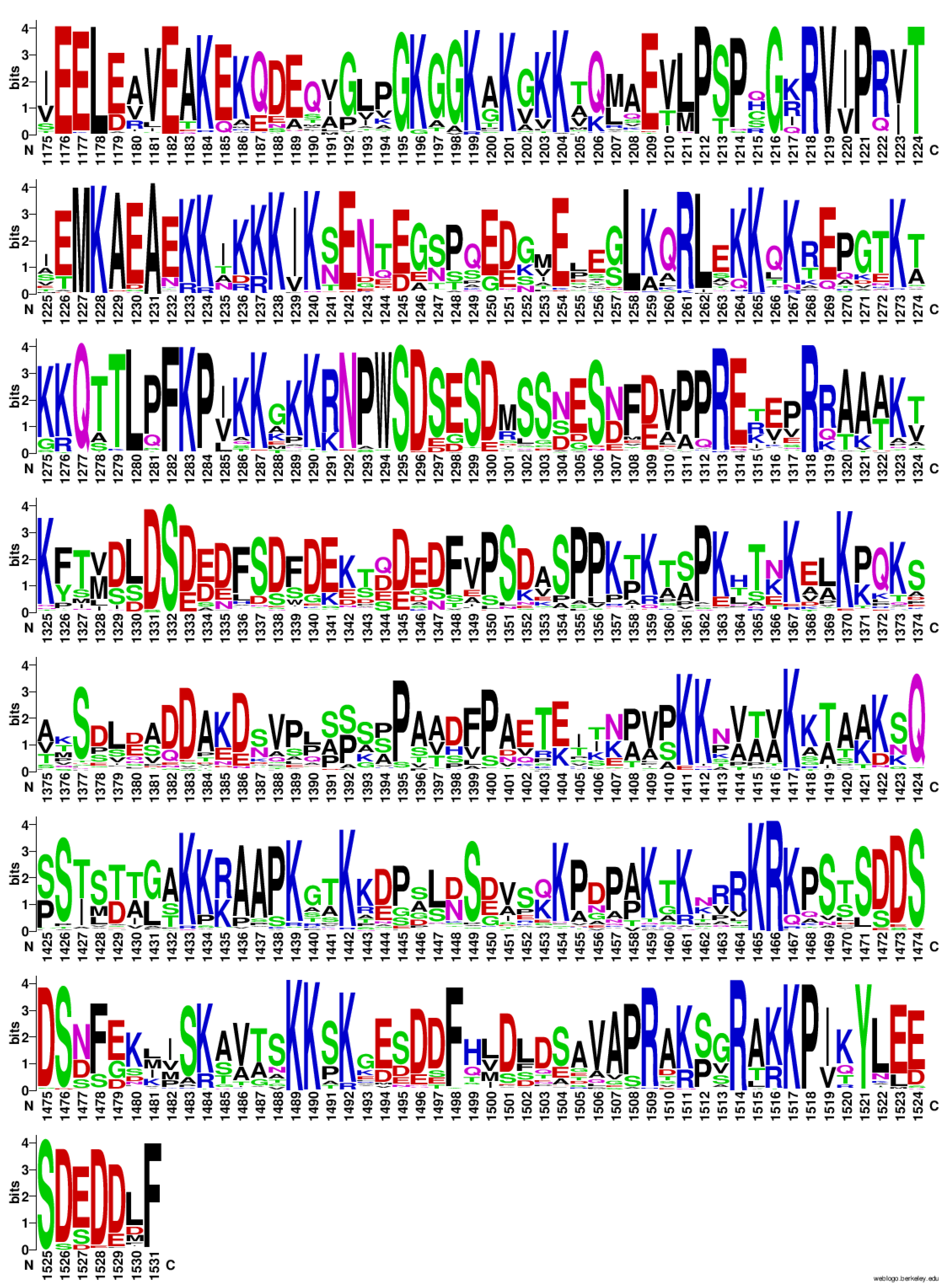

Table 1.
Examples of Interdomain Clusters within Topoisomerase IIα Identified by PSICalc.

Table 2.
Top2A CTD Clusters with the Highest SRMode Values.
| ClusterA | SRModeB | Previously MutatedC | Biochemical EffectC |
| 3rd Order | Max: 0.66 | ||
| 1189, 1232, 1272 | 0.514864 | T1272A, T1274A, T1278A, T1279A | Inc Relaxation |
| 1244, 1256, 1374 | 0.471158 | S1374G, S1377A | Inc Relaxation |
| 1217, 1364, 1482 | 0.434758 | S1351A, S1354G, T1358A, S1361A, S1365G | Dec Relaxation; Slower Decatenation |
| 1206, 1357, 1404 | 0.43775 | ND | |
| 1247, 1249, 1338 | 0.408826 | T1324A, T1327A, S1332A, S1337A, T1343A | Dec Relaxation |
| 1317, 1358, 1499 | 0.398492 | S1351A, S1354G, T1358A, S1361A, S1365G | Dec Relaxation; Slower Decatenation |
| 1394, 1397, 1399 | 0.359155 | S1387I, S1391G, S1392A, S1393A, T1397G | Inc Relaxation; Weaker binding |
| 1390, 1405, 1456 | 0.379833 | Same as above | |
| 1209, 1214, 1411 | 0.351671 | ND | |
| 1336, 1353, 1413 | 0.344962 | S1351A, S1354G, T1358A, S1361A, S1365G | Dec Relaxation; Slower Decatenation |
| 1172, 1306, 1377 | 0.277244 | S1374G, S1377A | None |
| 1227, 1237, 1303 | 0.168657 | S1295A, S1297A, S1302G, S1303G, S1306G | Dec Relaxation |
| 1273, 1476, 1531 | 0.390435 | S1469A, T1470I, S1471A, S1474A, S1476A | Strong Dec Relaxation |
| 4th Order | Max: 0.50 | ||
| 1190, 1316, 1480, 1512 | 0.331816 | S1469A, T1470I, S1471A, S1474A, S1476A And T1487A, S1488I, S1491A, S1495I |
Strong Dec Relaxation Inc Relaxation |
| 1198, 1213, 1236, 1383 | 0.179402 | S1387I, S1391G, S1392A, S1393A, T1397G | Inc Relaxation |
| 1247, 1249, 1338, 1485 | 0.307347 | T1324A, T1327A, S1332A, S1337A, T1343A And T1487A, S1488I, S1491A, S1495I |
Dec Relaxation Inc Relaxation |
| 1200, 1227, 1237, 1303 | 0.117365 | S1295A, S1297A, S1302G, S1303G, S1306G | Dec Relaxation |
| 5th Order | Max: 0.40 | ||
| 1217, 1304, 1344, 1364, 1482 | 0.249352 | S1295A, S1297A, S1302G, S1303G, S1306G And S1351A, S1354G, T1358A, S1361A, S1365G |
Dec Relaxation Dec Relaxation; Slower Decatenation |
| 1189, 1232, 1272, 1321, 1513 | 0.259725 | T1272A, T1274A, T1278A, T1279A | Inc Relaxation |
| 1334, 1336, 1353, 1366, 1413 | 0.191835 | T1324A, T1327A, S1332A, S1337A, T1343A And S1351A, S1354G, T1358A, S1361A, S1365G |
Dec Relaxation Dec Relaxation; Slower Decatenation |
| 1323, 1390, 1405, 1453, 1456 | 0.205813 | T1324A, T1327A, S1332A, S1337A, T1343A And S1449A, S1452A |
Dec Relaxation None |
| 1200, 1215, 1227, 1237, 1303 | 0.087985 | S1295A, S1297A, S1302G, S1303G, S1306G | Dec Relaxation |
AClustered amino acid positions are numbered according to human Top2A sequence. BSRMode values calculated by PSICalc algorithm; maximum SRMode value based upon cluster number is also shown. CFrom reference: [41].
Table 3.
Low Entropy CTD Positions.
| Position in Top2A | Amino Acid | Notes | Source |
| 1176 | E | ||
| 1178 | L | ||
| 1182 | E | ||
| 1212 | P | ||
| 1216 | G | ||
| 1218 | R | ||
| 1219 | V | ||
| 1221 | P | ||
| 1224 | T | ||
| 1228 | K | SUMO Site | [52] |
| 1231 | A | ||
| 1265 | K | NLS (1259-1296) | [45,50] |
| 1277 | Q | NLS (1259-1296) | [45,50] |
| 1295 | S | PO4 Site; Mitosis Associated (PlK-1); NLS (1259-1296) | [45,50,52] |
| 1318 | R | ||
| 1332 | S | PO4 Site; Mitosis Associated (PlK-1) | [52] |
| 1465 | K | NLS (1454-1497) | [45,50] |
| 1466 | R | NLS (1454-1497) | [45,50] |
| 1489 | K | NLS (1454-1497) | [45,50] |
| 1490 | K | NLS (1454-1497) | [45,50] |
| 1514 | R | ||
| 1525 | S | PO4 Site; Decatenation checkpoint | [44,52,53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



