
Submitted:
25 March 2024
Posted:
27 March 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
This study investigated the influence of single nucleotide polymorphisms (SNPs) in genes asso-ciated with the interferon pathway (IFNAR2 rs2236757), antiviral response (OAS1 rs10774671, OAS3 rs10735079), and viral entry (ACE2 rs2074192) on COVID-19 severity and their association with non-alcoholic fatty liver disease (MAFLD). We did not observe a significant association between the investigated SNPs and COVID-19 severity. While the IFNAR2 rs2236757 A allele correlated with higher creatinine levels upon admission and the G allele with lower band neu-trophils upon discharge, these findings require further investigation. The distribution of OAS gene polymorphisms (rs10774671, rs10735079 did not differ between MAFLD and non-MAFLD patients. Our study population's distribution of ACE2 rs2074192 genotypes and alleles differed from the European reference population. Overall, our findings suggest that these specific SNPs may not be major contributors to COVID-19 severity in our patient population, highlighting the potential role of other genetic factors and environmental influences.
Keywords:
MAFLD
; liver disease
; SNP
; COVID-19
; SARS-CoV-2
; interferon
1. Introduction
COVID-19 (Coronavirus Disease 2019) is a respiratory illness resulting from infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). This highly pathogenic coronavirus exhibits significant contagiousness and the potential to induce severe respiratory complications.
The vast majority of COVID-19 cases (approximately 85%) manifest with mild symptomatology. However, a subset of infected individuals (roughly 5%) progresses to severe illness, characterized by acute respiratory distress syndrome (ARDS) and damage to multiple organs [1]. The emergence of COVID-19 has resulted in a significant global pandemic, with substantial morbidity and mortality. As of this manuscript's writing, the worldwide death toll attributable to COVID-19 has surpassed 6.8 million [2]. A critical distinction between SARS-CoV-2 and earlier coronavirus strains lies in the heightened virulence of SARS-CoV-2, which translates to more severe clinical presentations [3].
Recent research suggests that tiny variations in our genes, called SNPs (single nucleotide polymorphism), might affect how likely we are to get COVID-19 and how severe it could be [4,5,6,7]. These SNPs are likely to exert their effects through various biological pathways implicated in the disease process. The intricate interplay between an individual's genetic architecture, characterized by a unique constellation of single nucleotide polymorphisms (SNPs), is hypothesized to significantly influence the pathogenesis and clinical course of COVID-19.
Coronaviruses, including SARS-CoV-2, utilize spike (S) glycoproteins to bind to host cell receptors, facilitating viral entry and membrane fusion [8]. Angiotensin-converting enzyme 2 (ACE2), expressed on the cell surface, serves as the primary receptor for SARS-CoV-2 attachment [9]. Higher levels of ACE2 expression translate to increased viral binding sites on host cells, potentially rendering individuals more susceptible to infection [10]. Notably, the ACE2 rs2074192 has been associated with an increased risk of arterial hypertension in obese males [11]. Additionally, this polymorphism has been implicated in the pathogenesis of both type 2 diabetes mellitus (T2DM) and cardiovascular disease [12]. A variation in the ACE2 gene, called rs2074192, might affect how effectively the instructions for building the ACE2 protein are followed. This variation is found in a non-coding region, but it could still influence how the ACE2 gene is processed. This disruption could potentially change how strongly the virus that causes COVID-19 binds to human cells. [13]. A study by Sienko et al. supports this idea, finding a link between the rs2074192 variation and how severe COVID-19 gets in adults [14].
Innate immunity represents a critical line of defense against SARS-CoV-2 infection. Interferons (IFNs) are essential mediators of the innate immune response against viral infections, displaying both anti-proliferative and immune-regulatory properties [15,16,17]. Polymorphisms within IFN genes or their receptors have been implicated in heightened susceptibility to COVID-19 and more severe clinical outcomes [6,18,19]. A large genetic study (GWAS) by Pairo-Castineira et al. (2020) found a strong connection between a specific variation (rs2236757) in the IFNAR2 gene and worse COVID-19 symptoms [6]. This link between IFNAR2 variations and severity was further supported by Fricke-Galindo et al. (2022) who discovered that several variations were associated with a higher risk of death in COVID-19 patients. Higher levels of soluble IFNAR2 (sIFNAR2) were observed in survivors, suggesting enhanced antiviral activity facilitated by sIFNAR2 stability [18].
Another critical genetic mechanism involves three specific genes, OAS1, OAS2, and OAS3, that code for enzymes called OAS enzymes that have antiviral properties [20]. These enzymes work by activating another molecule, RNase L (latent form of ribonuclease L), which helps fight off viruses [21,22]. The RNase L pathway plays a significant role in the host immune response against SARS-CoV-2 infection [6].
A recent study by Banday et al. (2022) showed a strong connection between a variation (rs10774671) in the OAS1 gene and how much of the OAS1 protein is produced [23]. This OAS1 protein is an important weapon our bodies use to fight viruses, including SARS-CoV-2. Their study suggests that this polymorphism's functional impact on OAS1 protein abundance contributes to the association with COVID-19 hospitalization outcomes [23]. Another study looking at genes involved in the antiviral response, Pairo-Castineira et al. (2020), found a connection between a variation (rs10735079) in the OAS3 gene and an increased risk of severe COVID-19 illness [6]. This suggests that variations in these genes, like OAS1 and OAS3, might influence our susceptibility to severe COVID-19.
Deciphering the genetic basis of COVID-19 severity is a formidable task due to the remarkable complexity of human genetics. Numerous genes interact in intricate pathways, making it challenging to isolate the precise genetic factors at play.
Metabolic-associated fatty liver disease (MAFLD) represents the most prevalent cause of chronic liver disease globally, affecting a substantial proportion of the population [24,25]. This spectrum of liver conditions encompasses a range from simple steatosis (fat accumulation) to more severe presentations characterized by inflammation and potential progression to cirrhosis [26,27]. Notably, individuals diagnosed with MAFLD exhibit elevated levels of interleukin-6 (IL-6) [28]. IL-6 is a critical cytokine implicated in the inflammatory cytokine storm observed in severe COVID-19 cases [29,30,31]. This finding suggests that MAFLD may exacerbate COVID-19 outcomes by further amplifying this inflammatory response.
Chronic inflammation, a well-established contributor to the pathogenesis of fatty liver disease, is orchestrated by hepatic and adipose tissue macrophages through the secretion of cytokines and adipokines [32]. Notably, MAFLD appears to exacerbate the overreaction of the immune system seen in severe COVID-19. This happens because MAFLD is linked to the release of a flood of inflammatory molecules, including interleukin-6 (IL-6), which contribute to the cytokine storm [28]. Recent studies have highlighted the potential importance of interferon regulatory factors (IRFs) in MAFLD, with these molecules playing a critical role in the induction of interferon (IFN) transcription [33].
2. Materials and Methods
2.1. Research Approach and Participants
This prospective cohort study was undertaken at the I. Horbachevsky Ternopil National Medical University (TNMU) in Ternopil, Ukraine. The study population included individuals of European ancestry (Ukrainian ethnicity) and ranged in age from 23 to 86 years.
The study enrolled a total of 72 adult participants who tested positive for SARS-CoV-2 via nasopharyngeal swab samples analyzed using real-time polymerase chain reaction (RT-PCR) [31]. All participants were admitted to the hospital between October 2022 and May 2023. A control group of 24 patients without COVID-19 and MAFLD was also included.
The inclusion criteria: adults who were diagnosed with COVID-19 and needed to be hospitalized. The severity of their illness was categorized based on guidelines set by the National Institutes of Health (NIH) as moderate, severe, or critical [36]; control group patients with similar baseline characteristics and confirmed absence of COVID-19.
The exclusion criteria were: people who died within 48 hours of being admitted to the hospital; pre-enrollment use of corticosteroids; patients in palliative care; people who had a serious bacterial infection when they were admitted; people with cirrhosis or chronic liver disease; pregnant women; alcohol dependence; weakened immune systems; HIV infection.
All participants were checked for signs of metabolic syndrome. MAFLD diagnosis was then established based on current criteria. The identification of steatosis usually consists of using various modalities (imaging, blood biomarkers, or histology) in conjunction with the next criteria: T2DM, overweight/obesity or demonstrable evidence of metabolic abnormalities [37,38]. In order to assess the presence of hepatic steatosis, we employed the hepatic steatosis index (HSI). This scoring system considers factors like body mass index, liver enzymes, and presence of diabetes to estimate the likelihood of fat accumulation in the liver [23].
Following the National Institutes of Health (NIH) guidelines [36], participants were stratified into three subgroups based on COVID-19 severity: moderate – characterized by bilateral pneumonia with oxygen saturation (SpO2) ≥ 94% on room air; severe – defined by the presence of dyspnea (difficulty breathing) and/or tachypnea (respiratory rate > 24 breaths/minute) and/or SpO2 < 94%.; critical – requiring intensive care unit (ICU) admission, fulfilling criteria for Acute Respiratory Distress Syndrome (ARDS), or necessitating advanced respiratory support using high-flow nasal cannula (HFNC), non-invasive ventilation, or invasive mechanical ventilation.
Following enrollment and application of inclusion/exclusion criteria, the final study sample comprised 33 participants diagnosed with COVID-1 and MAFLD, 39 participants COVID-19 without MAFLD, and a control group of 24 individuals. All participants were naïve to prior research participation and provided written informed consent. The study was approved by a special committee at I. Horbachevsky Ternopil National Medical University that oversees research ethics (protocol No. 74).
2.2. Laboratory and Clinical Data
As part of the standard diagnostic workup, a comprehensive laboratory analysis was conducted. This included hematological indices (white blood cell count with differential, erythrocyte sedimentation rate, hematocrit, platelet count), coagulation parameters (international normalized ratio, prothrombin time, activated partial thromboplastin time, fibrinogen), liver function tests (total bilirubin, alanine aminotransferase, aspartate aminotransferase), renal function (creatinine), markers of cholestasis (gamma-glutamyl transferase), and inflammatory markers (C-reactive protein). Additionally, blood glucose levels were measured. Body mass index (BMI) was recorded for all participants.
Genomic DNA isolation: Peripheral blood leukocytes were used to isolate genomic DNA for subsequent genotyping analysis. This procedure employed a commercially available kit (Thermo Scientific™ GeneJET™ Whole Blood Genomic DNA Purification Mini Kit). Briefly, 200 µL of whole blood from each participant was digested with proteinase K followed by addition of lysis buffer. The subsequent steps involved washing and elution of purified DNA.
Genotyping analysis: Real-time polymerase chain reaction (RT-PCR) was employed to analyze polymorphisms in four genes: ACE2 (rs2074192), IFNAR2 (rs2236757), OAS1 (rs10774671), and OAS3 (rs10735079). The CFX96™ Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., USA) was used for this purpose. Specific TaqMan™ SNP Genotyping Assays were utilized for each targeted SNP. Amplification of the DNA was achieved using TaqMan™ Universal Master Mix II.
Genotyping assays were conducted on all specimens utilizing TaqMan® probes and TaqMan® Genotyping Master Mix (4371355) in conjunction with the CFX96™ Real-Time PCR Detection System. The PCR protocol strictly adhered to the manufacturer's instructions (Applied Biosystems, USA). TaqMan® Genotyping Master Mix includes AmpliTaq Gold® DNA polymerase, dNTPs, ROX™ reference dye, and optimized reaction buffers. TaqMan® probes are allele-specific oligonucleotides with reporter dyes (VIC® for Allele 1 and 6-FAM™ for Allele 2) attached to the 5' end and a non-fluorescent quencher (NFQ) at the 3' end. Genomic DNA (10 μL) was amplified in a reaction mixture containing primers, probes, Master Mix, and the target DNA. Allele discrimination based on relative fluorescence units (RFU) was employed for genotyping using CFX-Manager™ software.
The PCR cycling conditions were as follows: Initial denaturation: 95 °C for 10 minutes; Amplification cycles (49 cycles); Denaturation: 95 °C for 15 seconds; Annealing: 60 °C for 1:10 minutes; Final melting curve analysis: Temperature ramp to 95 °C. Genotype determination was performed based on the melting curve analysis using the CFX96™ Real-Time PCR Basic Software (Bio-Rad Laboratories, Inc., USA).
2.3. Statistical Analysis
In this study, a comprehensive statistical analysis was performed to evaluate the collected data. Demographic information, clinical characteristics, and laboratory parameters were meticulously assessed and presented using descriptive statistics. To describe the data, we reported how often things occurred (frequencies) and the middle value (medians) along with the spread of the data (interquartile ranges).
For investigating group-wise differences in categorical variables, appropriate statistical tests were chosen based on sample size and table dimensions. Frequency tables with 2x3 dimensions were analyzed using the chi-square test (χ2), while 2x2 tables employed the two-tailed Fisher exact test. The significance level (p-value) was calculated for each test. Genotype frequencies were assessed for conformance to Hardy-Weinberg equilibrium (HWE) to ensure the study population reflected the expected distribution of alleles in the target population (p > 0.05 in the chi-square test).
To identify key predictors of COVID-19 severity within the study population, logistic regression analysis was performed. Statistical significance was set at a p-value of less than 0.05. Furthermore, GeneMANIA network data was utilized to evaluate potential interactions between the investigated genes [39]. A priori sample size calculations and post-hoc power analysis were performed using “G*Power 3.1.9.7”.
For comparisons between two independent groups, the non-parametric Mann–Whitney U test was employed. In the case of three or more groups, statistical comparisons were performed using the Kruskal–Wallis test, a non-parametric alternative to one-way ANOVA. Dunn's multiple comparisons test was subsequently employed for pairwise comparisons between groups. All statistical tests were two-tailed, with a significance level of p-value less than 0.05. The point-biserial correlation coefficient was utilized to assess the relationship between binary and continuous data within a correlation matrix. To evaluate the performance of the binary logistic regression model, ROC analysis was conducted. The strength of association between variables was expressed as an odds ratio (OR) along with its corresponding 95% confidence interval (CI). Statistical software programs including GraphPad Prism (version 8.4.3), IBM SPSS Statistics (version 25), and Jamovi (version 2.4.11) were used for all statistical analyses.
3. Results
3.1. Sample Characteristics
A total of 96 patients were included in the study and categorized into three groups: COVID-19 with MAFLD (n=33, 64% males, median age 66 years, interquartile range [IQR] 50–72 years), COVID-19 without MAFLD (n=39, 56% males, median age 65 years, IQR 41–72 years), and control (n=39, 71% males, median age 50 years, IQR 38.75–58.5 years)(Table 1).
Patients with COVID-19 and MAFLD were significantly older than those in the control group (median age 66 years vs. 50 years, p=0.011). The COVID-19 with MAFLD group had a significantly higher BMI compared to both the non-MAFLD group (30.8 vs. 24.0, p<0.001) and control group (30.8 vs. 24.1, p<0.001). The prevalence of type 2 diabetes mellitus (T2DM), arterial hypertension, and coronary heart disease was significantly higher in the COVID-19 with MAFLD group compared to the other two groups (all p-values <0.001).
Sex distribution (χ2= 1.346, p=0.510) and presence of chronic obstructive pulmonary disease (COPD) (χ2= 2.364, p=0.510) did not show statistically significant differences between the groups.
The distribution of genotypes for ACE2 rs2074192, IFNAR2 rs2236757, OAS1 rs10774671, and OAS3 rs10735079 polymorphisms conformed to Hardy-Weinberg equilibrium (HWE) in both the COVID-19 and control groups (p > 0.05). Detailed data on genotype frequencies are presented in Table 2.
3.2. Genotype and Allele Distribution in the Studied Groups
No statistically significant differences in genotype frequencies were observed among the investigated groups (COVID-19 with MAFLD, COVID-19 without MAFLD, and control) for the four polymorphisms analyzed: IFNAR2 rs2236757 (χ2 = 0.496, df = 4, p = 0.974), ACE2 rs2074192 (χ2 = 1.310, df = 4, p = 0.8597), OAS1 rs10774671 (χ2 = 2.432, df = 4, p = 0.6569), and OAS3 rs10735079 (χ2 = 2.314, df = 4, p = 0.6783). These findings suggest that genetic variations within these specific genes do not appear to be associated with MAFLD or COVID-19 susceptibility in the studied population.
Figure 1.
Genotype distribution in patients with COVID-19 with MAFLD vs. without MAFLD vs. control. IFNAR2—interferon alpha and beta receptor subunit; ACE2—angiotensin converting enzyme 2; OAS1—2′-5′-oligoadenylate synthetase 1; OAS3—2′-5′-oligoadenylate synthetase 3.
Figure 1.
Genotype distribution in patients with COVID-19 with MAFLD vs. without MAFLD vs. control. IFNAR2—interferon alpha and beta receptor subunit; ACE2—angiotensin converting enzyme 2; OAS1—2′-5′-oligoadenylate synthetase 1; OAS3—2′-5′-oligoadenylate synthetase 3.
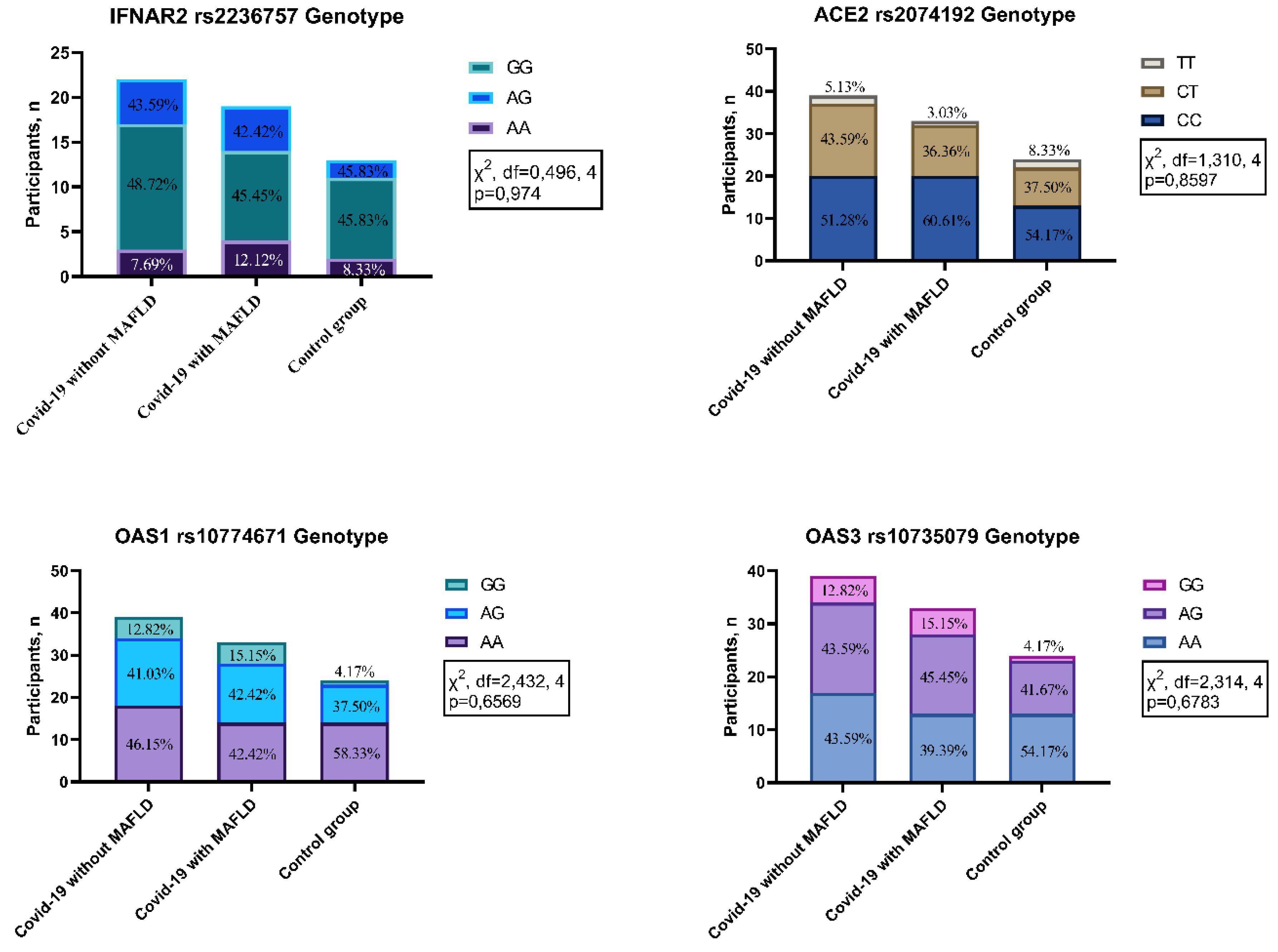
Analysis revealed statistically significant differences in the genotype and allele frequencies of the ACE2 rs2074192 polymorphism between the study groups (COVID-19 with MAFLD, COVID-19 without MAFLD, and control) and the European reference population (χ2 = 16.86, df = 4, p < 0.001 for genotype; Fisher's exact test, p < 0.001 for allele). Notably, the difference in allele frequency was also observed when comparing the control group to the European population. In contrast, no significant differences in genotype or allele frequencies were observed for the other investigated polymorphisms (IFNAR2 rs2236757, OAS1 rs10774671, and OAS3 rs10735079) when comparing the study groups with the European population (Table 3).
3.3. Risk alleles and Genotype in COVID-19 Patients
Analysis of the investigated polymorphisms (details provided in Table 4) revealed no statistically significant differences in allele frequencies among the three groups: COVID-19 with moderate severity, COVID-19 with severe/critical illness, and the control group. These findings suggest that the specific genetic variations analyzed do not appear to be associated with the degree of severity in COVID-19 patients.
Analysis of laboratory parameters at admission revealed associations between specific genotypes and certain markers (detailed data presented in Table 5). Patients carrying the A allele of IFNAR2 rs2236757 exhibited significantly higher creatinine levels compared to those without the A allele (88.5 mmol/L vs. 50 mmol/L, IQRs: 50–72 vs. 38.75–58.5 mmol/L, p = 0.021).
For ACE2 rs2074192, the presence of the C allele was associated with both lower band neutrophil levels (9 mmol/L vs. 3 mmol/L, IQRs: 6–13.5 vs. 2–7 mmol/L, p = 0.046) and higher total bilirubin levels (12.4 mmol/L vs. 21.9 mmol/L, IQRs: 10.8–14.9 vs. 20.5–107 mmol/L, p = 0.004). Additionally, the G allele was linked to lower eosinophil levels (1% vs. 1%, IQRs: 0–1 vs. 0–2%, p = 0.036).
For OAS1 rs10774671, the G allele was associated with elevated fibrinogen (3.99 g/L vs. 3.52 g/L, IQRs: 3.55–5.49 vs. 2.88–4.83 g/L, p = 0.033) and total protein levels (70.4 g/L vs. 66.9 g/L, IQRs: 64.5–75.1 vs. 60.9–70.4 g/L, p = 0.032).
Lastly, the G allele of OAS3 rs10735079 was linked to higher total protein levels (70.4 g/L vs. 66.2 g/L, IQRs: 64.8–74.8 vs. 60.2–70.7 g/L, p = 0.011).
Further analysis of laboratory parameters upon hospital discharge revealed additional associations between specific genotypes and certain markers (detailed data in Table 6). Patients carrying the C allele of ACE2 rs2074192 exhibited significantly lower International Normalized Ratio (INR) (1.01 vs. 1.09, IQRs: 0.92–1.07 vs. 1.07–1.17, p = 0.046) and Prothrombin Time (PT) (12.6 s vs. 13.6 s, IQRs: 11.85–13.4 vs. 13.4–14.4 s, p = 0.045) compared to those without the C allele.
The presence of the G allele of OAS1 rs10774671 was associated with both elevated hematocrit (37.91% vs. 34.65%, IQRs: 32.95–42.99 vs. 30.65–39.55%, p = 0.044) and higher total protein levels (66.6 g/L vs. 63.4 g/L, IQRs: 62.45–72.15 vs. 58.35–68.78 g/L, p = 0.011) upon discharge.
Similarly, the G allele of OAS3 rs10735079 was linked to increased monocyte count (5% vs. 4%, IQRs: 4–8 vs. 2–6.5%, p = 0.049), hematocrit (37.91% vs. 33.94%, IQRs: 32.77–43 vs. 30.35–38.12%, p = 0.024), and total protein levels (66.6 g/L vs. 63.5 g/L, IQRs: 61.98–72.23 vs. 58.08–68.73 g/L, p = 0.016) at discharge.
Upon admission, patients with the ACE2 rs2074192 CC genotype exhibited significantly lower total bilirubin levels compared to those with the TT genotype (median 12.2 mmol/L, interquartile range [IQR] 10.7-14.2 mmol/L vs. median 21.9 mmol/L, IQR 20.5-107 mmol/L, p = 0.024). Additionally, the presence of the OAS3 rs10735079 AA genotype was associated with higher general protein levels compared to the AG genotype (median 66.2 g/L, IQR 60.2-70.7 g/L vs. median 70.4 g/L, IQR 65.6-75.1 g/L, p = 0.019).
At discharge, patients with the IFNAR2 rs2236757 AA genotype displayed a significantly higher band neutrophil level compared to those with the AG genotype (median 5 mmol/L, IQR 4-7 mmol/L vs. median 2.5 mmol/L, IQR 1.75-4 mmol/L, p = 0.021). Conversely, the OAS1 rs10774671 AA genotype was associated with a lower level (median 63.5 g/L, IQR 58.1-68.7 g/L vs. median 66.3 g/L, IQR 62-72.3 g/L, p = 0.047). Detailed data visualizations are presented in Figure 2 (medians with 95% CI), while Table 7 provides the IQR values.
The Kruskal-Wallis test revealed a statistically significant difference in INR levels among patients categorized by their ACE2 rs2074192 genotype (p=0.020). However, Dunn's multiple comparisons test, employed to identify specific pairwise differences, did not detect any significant variations in INR between individual genotypes (Figure 2).
3.4. Correlation Analysis
To investigate potential associations, we analyzed the studied SNPs, their corresponding genotypes, and various laboratory outcomes measured upon admission (Figure 3) and discharge (Figure 4).
Admission Findings.
IFNAR2 rs2236757: The presence of the A allele positively correlated with creatinine levels (r = 0.27, p = 0.021), while the GG genotype exhibited a negative correlation (r = -0.26, p = 0.027).
ACE2 rs2074192: The C allele displayed a positive correlation with band neutrophils (r = 0.23, p = 0.047) and a negative correlation with total bilirubin (r = -0.30, p = 0.010). Conversely, the T allele negatively correlated with eosinophils (r = -0.25, p = 0.035).
OAS3 rs10735079: Both the G allele (r = 0.30, p = 0.011) and GG genotype (r = 0.23, p = 0.047) positively correlated with general protein levels. Additionally, the GG genotype negatively correlated with blood glucose (r = -0.25, p = 0.036).
OAS1 rs10774671: The G allele positively correlated with fibrinogen (r = 0.27, p = 0.033), general protein (r = 0.25, p = 0.032), and negatively correlated with blood glucose (r = -0.23, p = 0.050). Similarly, the GG genotype exhibited a negative correlation with blood glucose (r = -0.26, p = 0.030).
Discharge Findings.
IFNAR2 rs2236757: The G allele displayed a negative correlation with band neutrophil levels (r = -0.30, p = 0.012), while the GG genotype showed a positive correlation with albumin (r = 0.24, p = 0.038).
ACE2 rs2074192: The TT genotype negatively correlated with leukocytes (r = -0.24, p = 0.041), and the C allele positively correlated with prothrombin time (PT) (r = -0.23, p = 0.048).
OAS3 rs10735079: The G allele positively correlated with monocytes (r = 0.23, p = 0.048), hematocrit (r = 0.27, p = 0.023), and general protein (r = 0.29, p = 0.015). The GG genotype also positively correlated with hematocrit (r = 0.28, p = 0.019) and general protein (r = 0.26, p = 0.025).
OAS1 rs10774671: Both the GG genotype and G allele positively correlated with hematocrit (r = 0.25, p = 0.033 and r = 0.24, p = 0.043 respectively) and general protein (r = 0.28, p = 0.019 and r = 0.30, p = 0.010 respectively).
Notably, the correlations observed between the variables were statistically weak.
3.5. Regression Analysis
We created a simple logistic regression for predicting COVID-19 severity. This predictive model has developed conditioning on IFNAR2 rs2236757 allele G, ACE2 rs2074192 allele T, OAS1 rs10774671 allele G, SpO2 level (admission) and segmented neutrophils level on admission.

Table 8.
Identification of risk factors for COVID-19 severity using logistic regression analysis.
| B (ORa) | S.E. | Wald | df | Sig. (p-value) | Exp(B) | |
| IFNAR2 rs2236757 allele G | -1.679 | 1.683 | 0.995 | 1 | 0.318 | 0.186 |
| ACE2 rs2074192 allele T | 2.024 | 1.055 | 3.678 | 1 | 0.055 | 7.569 |
| OAS1 rs10774671 allele G | 2.437 | 1.252 | 3.789 | 1 | 0.052 | 11.442 |
| SpO2 (admission) | -1.384 | 0.389 | 12.663 | 1 | 0.000 | 0.250 |
| Segmented neutrophils (%, admission) | 0.095 | 0.040 | 5.782 | 1 | 0.016 | 1.100 |
| Constant | 124.716 | 35.574 | 12.291 | 1 | 0.000 | 1.457E+54 |
Variable(s) entered on step 1: IFNAR2 rs2236757 allele G, ACE2 rs2074192 allele T, OAS1 rs10774671 allele G, SpO2 (admission), segmented neutrophils (%, admission); aodds ratio.
The resulting logistic regression model demonstrated statistical significance (p < 0.001), indicating a strong association between the selected predictors and COVID-19 severity. Additionally, the Nagelkerke R-squared of 0.763 indicates that the independent variables in the model explain a substantial proportion (76.3%) of the variation in COVID-19 severity.
The developed model achieved a high level of accuracy (90.3%), signifying its ability to accurately classify cases into severity categories. Receiver Operating Characteristic (ROC) analysis confirmed the model's effectiveness. The ROC curve analysis yielded an area under the curve (AUC) of 0.96 (95% CI: 0.92-1.00), demonstrating excellent ability to discriminate between patients with severe and non-severe COVID-19 (Figure 5).
The model exhibited a specificity of 92.9% and a sensitivity of 86.7%. Specificity refers to the model's ability to correctly identify individuals without severe COVID-19, while sensitivity reflects its accuracy in detecting severe cases.
The cut-off value for the logistic function (p) was established at 0.5. This threshold can be used to categorize patients into high-risk and low-risk groups based on the predicted probability of severe COVID-19
3.6. Genmania Interactiom
An analysis using GeneMANIA network data [39] revealed interconnectedness between the investigated SNPs and others polymorphisms, including: CLTRN (collectrin, amino acid transport regulator), SLC6A19 (solute carrier family 6 member 19), MME (membrane metalloendopeptidase), ACE (angiotensin I converting enzyme), CTSZ (cathepsin Z), CMA1 (chymase 1), IFNA1 (interferon alpha 1), IFNA5 (interferon alpha 5), IFNAR1 (interferon alpha and beta receptor subunit 1), JAK1 (janus kinase 1), TMPRSS2 (transmembrane serine protease 2), IFNB1 (interferon beta 1), ISG15 (ubiquitin like modifier), STAT1 )signal transducer and activator of transcription 1), STAT2 (signal transducer and activator of transcription 2), IRF9 (interferon regulatory factor 9), OASL (2'-5'-oligoadenylate synthetase like), USP18 (ubiquitin specific peptidase 18), IFNA2 (interferon alpha 2), OAS2 (2'-5'-oligoadenylate synthetase 2) (Table 9).
The analysis revealed that physical interactions between genes in the proposed network were documented in a high percentage of cases (77.64%), co-expression as found in a smaller proportion – 8.01%, predicted functional relationships between genes in 5.37%, co-localization in 3.63%, genetic interactions in 2.87%, pathway in 1.88% and shared protein domains in 0.60%.
4. Discussion
This study investigated the potential influence of SNPs in genes associated with the interferon pathway (IFNAR2 rs2236757), antiviral response (OAS1 rs10774671, OAS3 rs10735079), and viral entry (ACE2 rs2074192) on COVID-19 severity and their association with MAFLD.
Patients with MAFLD have increased inflammatory mediators levels [40]. Previous research linked the interferon pathway to MAFLD pathogenesis [33] and COVID-19 severity [41].
Several lines of investigation, including genome-wide association studies (GWAS), transcriptomic analyses, and single-cell studies, have converged on IFNAR2 (rs2236757) as a potential genetic risk factor for severe COVID-19 [6]. Supporting evidence emerges from studies focused on genes associated with COVID-19 severity, where a specific variant within the IFNAR2 gene (rs2236757) has been linked to poorer clinical outcomes [6,18]. Notably, this variant (rs2236757) has been associated with an increased risk of critical illness and mortality from COVID-19 infection [18]. Interestingly, research suggests an inverse correlation between soluble IFNAR2 protein (sIFNAR2) levels and COVID-19 outcomes, with lower levels observed in deceased patients and higher levels in survivors [18].
Dieter K. et al. (2022) further strengthened the connection between the IFNAR2 rs2236757 genotype and severe COVID-19 by demonstrating an association with increased ICU admission rates and mortality [19]. However, our own investigation did not identify a significant association between the rs2236757 polymorphism and COVID-19 severity in our patient population. Additionally, this SNP did not exhibit a relationship with MAFLD.
While our findings did not reveal a substantial impact of the IFNAR2 rs2236757 genotype on COVID-19 severity or clinical outcomes, an interesting observation emerged. Patients with the A allele of rs2236757 presented with higher creatinine levels upon admission compared to those without the A allele. Conversely, upon discharge, patients with the G allele displayed a negative correlation with band neutrophils, potentially indicating lower neutrophil levels. The distribution of the IFNAR2 rs2236757 genotype also did not differ significantly between MAFLD and non-MAFLD patients in our study.
2′-5′ oligoadenylate synthetases (OAS) are a family of interferon-stimulated antiviral enzymes crucial for the innate immune response [42]. These enzymes identify viral double-stranded RNA (dsRNA) and trigger RNA destabilization via RNase L activation within infected cells. Humans possess four OAS genes (OAS1, OAS2, OAS3, and OASL) located on chromosomes 12q24.1 (OAS1-3) and 12q24.2 (OASL). OAS1, a dsRNA-activated enzyme, plays a central role in cellular antiviral defense [43]. OAS3 encodes a single transcript that results in an enzyme activating latent RNase L, leading to the degradation of both viral and cellular RNA [43].
Previous studies have explored the potential association between OAS polymorphisms and COVID-19 severity. Banday et al. (2022) investigated the influence of OAS1 rs10774671 on COVID-19 hospitalization outcomes, suggesting a functional impact on disease severity [23]. Similarly, the GenOMICC study by Pairo-Castineira et al. identified a potential role for OAS3 rs10735079 in the progression to critical illness in COVID-19 patients [6].
Our study population's genotype and allele distribution for the investigated SNPs (rs10774671 and rs10735079) did not differ significantly from the European population. Furthermore, we did not observe statistically significant differences in COVID-19 severity between patients with either OAS1 rs10774671 or OAS3 rs10735079. Interestingly, Carrying two copies of the G allele (homozygous genotype) for both polymorphisms was linked to variations in certain clinical parameters. In rs10774671 carriers, the G allele correlated with higher protein and hematocrit levels, as well as increased fibrinogen. Patients with rs10735079 and the G allele exhibited higher monocyte levels. Importantly, no significant differences in the distribution of these polymorphisms were found when comparing patients with and without MAFLD. The G allele OAS1 rs10774671 was used in logistic regression as a predictor of COVID-19 severity (p=0.052).
The 2′-5′ oligoadenylate synthetases (OAS) are interferon-induced antiviral enzymes that recognize virally produced dsRNA and initiate RNA destabilization through activation of RNase L within infected cells [42]. The human genome encodes four 2',5'-oligoadenylate synthetase (OAS) genes: OAS1, OAS2, OAS3, and OASL. These genes map to distinct chromosomal locations, with OAS1-3 clustered on chromosome 12q24.1 and OASL situated on chromosome 12q24.2. Notably, OAS1 functions as a dsRNA-activated antiviral enzyme, playing a crucial role in the innate immune response against viral infection. OAS3 encodes a single transcript that produces enzyme that activate latent RNase L, leading to degradation of both viral and endogenous RNA [43].
Banday et al. (2022) showed the influence of OAS1 rs10774671 on the hospitalization outcomes for COVID-19 patients and purposed the functional impact on COVID-19 severity [23]. Pairo-Castineira et al., in GenOMICC study discovered OAS3 rs10735079 role in progressing critical illness in COVID-19 patients [6]. The studied population did not differ from the European population in terms of SNPs genotype or alleles. No statistically significant differences in COVID-19 severity were found between patients with OAS1 rs10774671 and OAS3 rs10735079 in our study. The presence of the G allele was associated with higher protein and hematocrit levels, higher fibrinogen level in rs10774671 patients and higher monocytes level in rs10735079 patients. When comparing patients with MAFLD and non-MAFLD, no difference in the distribution of these polymorphisms was found.
The ACE2 receptor and transmembrane serine protease 2 (TMPRSS2) are established factors facilitating SARS-CoV-2 entry into human cells [44]. Viral entry begins with the binding of the viral spike (S) protein to ACE2, followed by S protein cleavage mediated by TMPRSS2. This cleavage allows for the fusion of the viral membrane with the host cell membrane, enabling viral replication and spread within the target cells [44]. Genetic polymorphisms that influence the expression of these genes could potentially impact the course of SARS-CoV-2 infection [45]. Studies have shown an association between the ACE2 rs2074192 TT genotype and increased COVID-19 mortality [45].
Furthermore, the severity of COVID-19 has been linked to pre-existing conditions such as cardiovascular disease, retinopathy in hypertensive individuals, type 2 diabetes, and hypertensive left ventricular hypertrophy, which are more prevalent in carriers of the T allele of the rs2074192 polymorphism [12,46]. These conditions, including MAFLD, are often components of metabolic syndrome.
Our investigation focused on MAFLD and did not identify any significant differences in the distribution of ACE2 rs2074192 genotypes or alleles between MAFLD and non-MAFLD groups. Additionally, the presence of the ACE2 rs2074192 polymorphism did not seem to influence COVID-19 severity in our study population. It is important to note that our population exhibited a distinct distribution of genotypes and alleles compared to the European reference population.
Our findings do not support a significant association between the investigated genes (IFNAR2 rs2236757, ACE2 rs2074192, OAS1 rs10774671, and OAS3 rs10735079) and the course of SARS-CoV-2 infection. Furthermore, we did not observe an increased prevalence of these polymorphisms in patients with MAFLD. Additionally, no interaction was identified between these polymorphisms and MAFLD that could potentially worsen the course of COVID-19 in this patient population.
5. Conclusions
This study contributes to the ongoing investigation of genetic factors influencing COVID-19 severity. We did not identify a strong association between the studied SNPs and disease severity. The distinct distribution of ACE2 rs2074192 genotypes in our population compared to the reference group suggests the need for further research considering ethnicity and geographic factors. Future studies could explore functional analyses of these polymorphisms and investigate interactions with other genetic and environmental variables to gain a more comprehensive understanding of the complex interplay of factors influencing COVID-19 outcomes.
Author Contributions
Conceptualization and writing—original draft preparation, M.B. and O.K.; writing—review and editing, O.B., V.O., and I.K.; supervision, O.K., and V.O.; project administration, V.O. and O.K.; visualization, I.K.; funding acquisition, M.B. and V.O. All authors have read and agreed to the published version of the manuscript.
Funding
RECOOP Grant #36—CSMC Senior Scientists (RCSS) “Comprehensive Analysis of Genetic Predictors for MAFLD Development in Patients with COVID-19”.
Institutional Review Board Statement
The study protocol met the requirements for biomedical research and was approved by the Local Ethics Committee of the I. Horbachevsky Ternopil National Medical University as protocol N74, dated 13 October2023.
Informed Consent Statement
All patients signed an informed consent for the study.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Pfortmueller, C.A.; Spinetti, T.; Urman, R.D.; Luedi, M.M.; Schefold, J.C. COVID-19-Associated Acute Respiratory Distress Syndrome (CARDS): Current Knowledge on Pathophysiology and ICU Treatment – A Narrative Review. Best Pract. Res. Clin. Anaesthesiol. 2021, 35, 351. [CrossRef]
- Mbbs, L.R.; D, L.B.S.M.; Neurosurgery, M.S. Coronavirus Disease ( COVID-19 ) Spreads Situation Reports. Who 2020, 75, 95–97.
- Triggle, C.R.; Bansal, D.; Ding, H.; Islam, M.M.; Farag, E.A.B.A.; Hadi, H.A.; Sultan, A.A. A Comprehensive Review of Viral Characteristics, Transmission, Pathophysiology, Immune Response, and Management of SARS-CoV-2 and COVID-19 as a Basis for Controlling the Pandemic. Front. Immunol. 2021, 12, 631139. [CrossRef]
- Buchynskyi, M.; Oksenych, V.; Kamyshna, I.; Vari, S.G.; Kamyshnyi, A. Genetic Predictors of Comorbid Course of COVID-19 and MAFLD: A Comprehensive Analysis. Viruses 2023, 15. [CrossRef]
- Fricke-Galindo, I.; Falfán-Valencia, R. Genetics Insight for COVID-19 Susceptibility and Severity: A Review. Front. Immunol. 2021, 12, 622176. [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic Mechanisms of Critical Illness in COVID-19. Nat. 2020 5917848 2020, 591, 92–98. [CrossRef]
- Negi, K.; Agarwal, M.; Pahuja, I.; Bhardwaj, B.; Rawat, M.; Bhaskar, A.; Dwivedi, V.P. Combating the Challenges of COVID-19 Pandemic: Insights into Molecular Mechanisms, Immune Responses and Therapeutics against SARS-CoV-2. Oxford open Immunol. 2023, 4, iqad001. [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [CrossRef]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [CrossRef]
- Li, F. Receptor Recognition and Cross-Species Infections of SARS Coronavirus. Antiviral Res. 2013, 100, 246–254. [CrossRef]
- Cui, N.; Tong, H.; Li, Y.; Ge, Y.; Shi, Y.; Lv, P.; Zhao, X.; Zhang, J.; Fu, G.; Zhou, Y.; et al. Role of Prealbumin in Predicting the Prognosis of Severely and Critically Ill COVID-19 Patients. Am. J. Trop. Med. Hyg. 2021, 105, 718–726. [CrossRef]
- Liu, C.; Li, Y.; Guan, T.; Lai, Y.; Shen, Y.; Zeyaweiding, A.; Zhao, H.; Li, F.; Maimaiti, T. ACE2 Polymorphisms Associated with Cardiovascular Risk in Uygurs with Type 2 Diabetes Mellitus. Cardiovasc. Diabetol. 2018, 17, 127. [CrossRef]
- Pouladi, N.; Abdolahi, S. Investigating the ACE2 Polymorphisms in COVID-19 Susceptibility: An in Silico Analysis. Mol. Genet. genomic Med. 2021, 9, e1672. [CrossRef]
- Sienko, J.; Marczak, I.; Kotowski, M.; Bogacz, A.; Tejchman, K.; Sienko, M.; Kotfis, K. Association of ACE2 Gene Variants with the Severity of COVID-19 Disease-A Prospective Observational Study. Int. J. Environ. Res. Public Health 2022, 19. [CrossRef]
- Hammad, M.O.; Alseoudy, M.M.; Borg, A.M.; El-Mesery, A.; Elgamal, M.; Abdelghany, D.A.; Elzeiny, D. IFNL1 Rs30461 Polymorphism as a Risk Factor for COVID-19 Severity: A Cross-Sectional Study. Cytokine 2024, 176, 156500. [CrossRef]
- Kamyshnyi, A.; Koval, H.; Kobevko, O.; Buchynskyi, M.; Oksenych, V.; Kainov, D.; Lyubomirskaya, K.; Kamyshna, I.; Potters, G.; Moshynets, O. Therapeutic Effectiveness of Interferon-A2b against COVID-19 with Community-Acquired Pneumonia: The Ukrainian Experience. Int. J. Mol. Sci. 2023, Vol. 24, Page 6887 2023, 24, 6887. [CrossRef]
- Buchynskyi, M.; Kamyshna, I.; Lyubomirskaya, K.; Moshynets, O.; Kobyliak, N.; Oksenych, V.; Kamyshnyi, A. Efficacy of Interferon Alpha for the Treatment of Hospitalized Patients with COVID-19: A Meta-Analysis. Front. Immunol. 2023, 14, 250. [CrossRef]
- Fricke-Galindo, I.; Martínez-Morales, A.; Chávez-Galán, L.; Ocaña-Guzmán, R.; Buendía-Roldán, I.; Pérez-Rubio, G.; Hernández-Zenteno, R. de J.; Verónica-Aguilar, A.; Alarcón-Dionet, A.; Aguilar-Duran, H.; et al. IFNAR2 Relevance in the Clinical Outcome of Individuals with Severe COVID-19. Front. Immunol. 2022, 13, 949413. [CrossRef]
- Dieter, C.; de Almeida Brondani, L.; Lemos, N.E.; Schaeffer, A.F.; Zanotto, C.; Ramos, D.T.; Girardi, E.; Pellenz, F.M.; Camargo, J.L.; Moresco, K.S.; et al. Polymorphisms in ACE1, TMPRSS2, IFIH1, IFNAR2, and TYK2 Genes Are Associated with Worse Clinical Outcomes in COVID-19. Genes (Basel). 2022, 14. [CrossRef]
- Kozak, K.; Pavlyshyn, H.; Kamyshnyi, O.; Shevchuk, O.; Korda, M.; Vari, S.G. The Relationship between COVID-19 Severity in Children and Immunoregulatory Gene Polymorphism. Viruses 2023, 15. [CrossRef]
- Sadler, A.J.; Williams, B.R.G. Interferon-Inducible Antiviral Effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [CrossRef]
- Kerr, I.M.; Brown, R.E.; Hovanessian, A.G. Nature of Inhibitor of Cell-Free Protein Synthesis Formed in Response to Interferon and Double-Stranded RNA. Nature 1977, 268, 540–542. [CrossRef]
- Banday, A.R.; Stanifer, M.L.; Florez-Vargas, O.; Onabajo, O.O.; Papenberg, B.W.; Zahoor, M.A.; Mirabello, L.; Ring, T.J.; Lee, C.-H.; Albert, P.S.; et al. Genetic Regulation of OAS1 Nonsense-Mediated Decay Underlies Association with COVID-19 Hospitalization in Patients of European and African Ancestries. Nat. Genet. 2022, 54, 1103–1116. [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [CrossRef]
- Powell, E.E.; Wong, V.W.-S.; Rinella, M. Non-Alcoholic Fatty Liver Disease. Lancet (London, England) 2021, 397, 2212–2224. [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209. [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H.; et al. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999-2014.e1. [CrossRef]
- Gao, F.; Zheng, K.I.; Yan, H.-D.; Sun, Q.-F.; Pan, K.-H.; Wang, T.-Y.; Chen, Y.-P.; Targher, G.; Byrne, C.D.; George, J.; et al. Association and Interaction Between Serum Interleukin-6 Levels and Metabolic Dysfunction-Associated Fatty Liver Disease in Patients With Severe Coronavirus Disease 2019. Front. Endocrinol. (Lausanne). 2021, 12, 604100. [CrossRef]
- Dietz, M.; Chironi, G.; Claessens, Y.-E.; Farhad, R.L.; Rouquette, I.; Serrano, B.; Nataf, V.; Hugonnet, F.; Paulmier, B.; Berthier, F.; et al. COVID-19 Pneumonia: Relationship between Inflammation Assessed by Whole-Body FDG PET/CT and Short-Term Clinical Outcome. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 260–268. [CrossRef]
- Inciardi, R.M.; Solomon, S.D.; Ridker, P.M.; Metra, M. Coronavirus 2019 Disease (COVID-19), Systemic Inflammation, and Cardiovascular Disease. J. Am. Heart Assoc. 2020, 9, e017756. [CrossRef]
- Nowroozi, A.; Momtazmanesh, S.; Rezaei, N. COVID-19 and MAFLD/NAFLD: An Updated Review. Front. Med. 2023, 10, 1126491. [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The Role of Macrophages in Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [CrossRef]
- Møhlenberg, M.; Terczynska-Dyla, E.; Thomsen, K.L.; George, J.; Eslam, M.; Grønbæk, H.; Hartmann, R. The Role of IFN in the Development of NAFLD and NASH. Cytokine 2019, 124, 154519. [CrossRef]
- Buchynskyi, M.; Kamyshna, I.; Oksenych, V.; Zavidniuk, N.; Kamyshnyi, A. The Intersection of COVID-19 and Metabolic-Associated Fatty Liver Disease: An Overview of the Current Evidence. Viruses 2023, 15. [CrossRef]
- Buchynskyi, M.; Oksenych, V.; Kamyshna, I.; Kamyshnyi, O. Exploring Paxlovid Efficacy in COVID-19 Patients with MAFLD: Insights from a Single-Center Prospective Cohort Study. Viruses 2024, 16. [CrossRef]
- Clinical Spectrum | COVID-19 Treatment Guidelines Available online: https://www.covid19treatmentguidelines.nih.gov/overview/clinical-spectrum/ (accessed on 29 October 2023).
- Fouad, Y.; Waked, I.; Bollipo, S.; Gomaa, A.; Ajlouni, Y.; Attia, D. What’s in a Name? Renaming ‘NAFLD’ to ‘MAFLD.’ Liver Int. 2020, 40, 1254–1261. [CrossRef]
- Méndez-Sánchez, N.; Bugianesi, E.; Gish, R.G.; Lammert, F.; Tilg, H.; Nguyen, M.H.; Sarin, S.K.; Fabrellas, N.; Zelber-Sagi, S.; Fan, J.G.; et al. Global Multi-Stakeholder Endorsement of the MAFLD Definition. Lancet Gastroenterol. Hepatol. 2022, 7, 388–390. [CrossRef]
- GeneMANIA Available online: https://genemania.org/ (accessed on 24 February 2024).
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines With Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298. [CrossRef]
- Bastard, P.; Gervais, A.; Le Voyer, T.; Rosain, J.; Philippot, Q.; Manry, J.; Michailidis, E.; Hoffmann, H.-H.; Eto, S.; Garcia-Prat, M.; et al. Autoantibodies Neutralizing Type I IFNs Are Present in ~4% of Uninfected Individuals over 70 Years Old and Account for ~20% of COVID-19 Deaths. Sci. Immunol. 2021, 6. [CrossRef]
- Kristiansen, H.; Gad, H.H.; Eskildsen-Larsen, S.; Despres, P.; Hartmann, R. The Oligoadenylate Synthetase Family: An Ancient Protein Family with Multiple Antiviral Activities. J. Interf. cytokine Res. Off. J. Int. Soc. Interf. Cytokine Res. 2011, 31, 41–47. [CrossRef]
- Fagone, P.; Nunnari, G.; Lazzara, F.; Longo, A.; Cambria, D.; Distefano, G.; Palumbo, M.; Nicoletti, F.; Malaguarnera, L.; Di Rosa, M. Induction of OAS Gene Family in HIV Monocyte Infected Patients with High and Low Viral Load. Antiviral Res. 2016, 131, 66–73. [CrossRef]
- Zipeto, D.; Palmeira, J. da F.; Argañaraz, G.A.; Argañaraz, E.R. ACE2/ADAM17/TMPRSS2 Interplay May Be the Main Risk Factor for COVID-19. Front. Immunol. 2020, 11, 576745. [CrossRef]
- Sheikhian, F.; Sadeghi Mofrad, S.; Tarashi, S.; Ghazanfari Jajin, M.; Sakhaee, F.; Ahmadi, I.; Anvari, E.; Sheikhpour, M.; Fateh, A. The Impact of ACE2 Polymorphisms (Rs1978124, Rs2285666, and Rs2074192) and ACE1 Rs1799752 in the Mortality Rate of COVID-19 in Different SARS-CoV-2 Variants. Hum. Genomics 2023, 17, 54. [CrossRef]
- Fan, Z.; Wu, G.; Yue, M.; Ye, J.; Chen, Y.; Xu, B.; Shu, Z.; Zhu, J.; Lu, N.; Tan, X. Hypertension and Hypertensive Left Ventricular Hypertrophy Are Associated with ACE2 Genetic Polymorphism. Life Sci. 2019, 225, 39–45. [CrossRef]
Figure 2.
Comparison of the medians of laboratory findings in patients with different genotypes on admission/discharge: IFNAR2 rs2236757, ACE2 rs2074192, OAS1 rs10774671, OAS3 rs10735079. The Kruskal-Wallis test indicated an overall difference between groups. Dunn's test was then employed to pinpoint exactly which groups differed from each other.
Figure 2.
Comparison of the medians of laboratory findings in patients with different genotypes on admission/discharge: IFNAR2 rs2236757, ACE2 rs2074192, OAS1 rs10774671, OAS3 rs10735079. The Kruskal-Wallis test indicated an overall difference between groups. Dunn's test was then employed to pinpoint exactly which groups differed from each other.
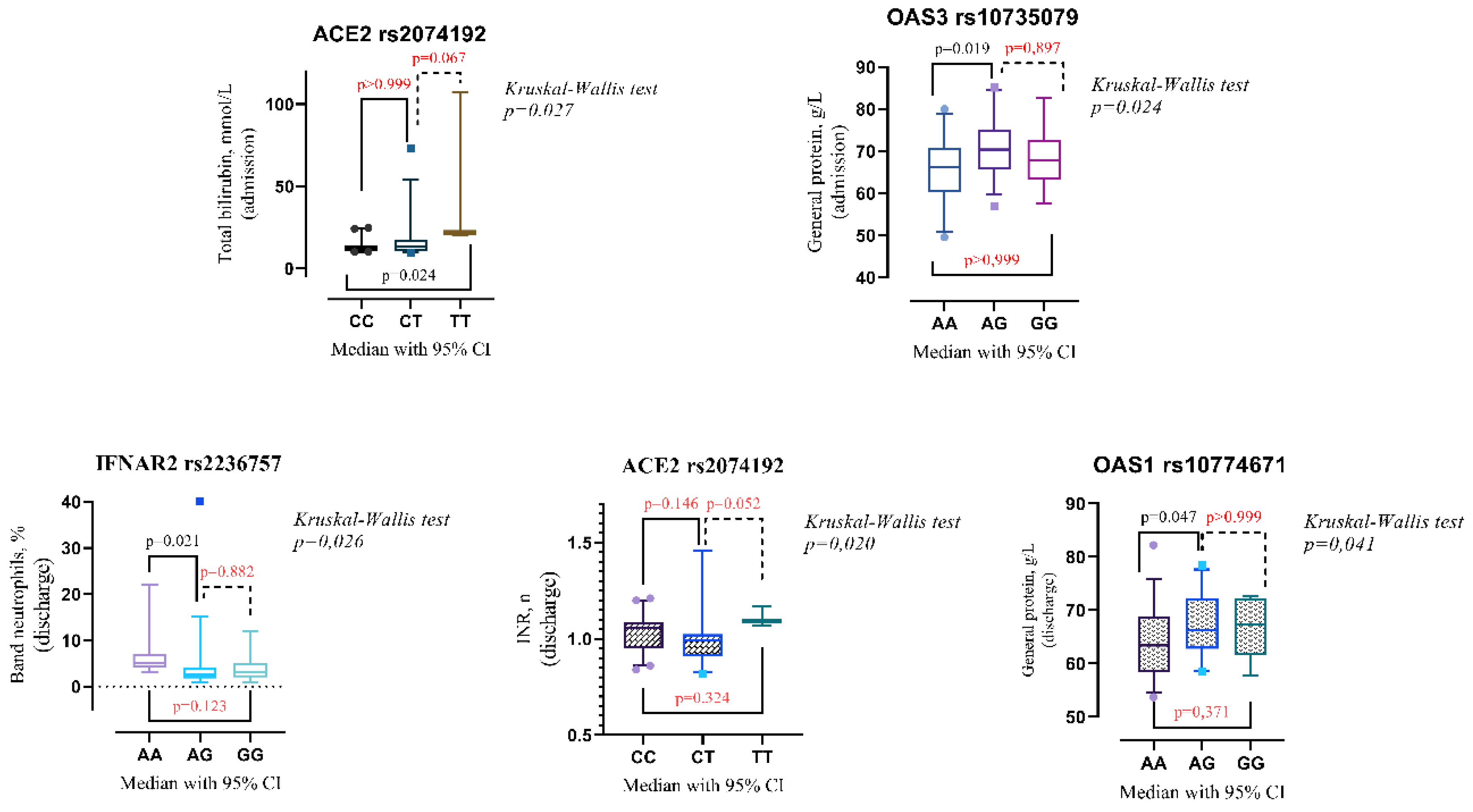
Figure 3.
Correlation correlogram. Laboratory findings on admission (likely continuous and binary data based on the mention of point-biserial correlation). Point-biserial correlation: Used for correlations between binary (yes/no) data and continuous data. Red: Strong negative correlation (r = -1.0). Blue: Strong positive correlation (r = 1.0). Only statistically significant correlations (p-value <= 0.05) are shown.
Figure 3.
Correlation correlogram. Laboratory findings on admission (likely continuous and binary data based on the mention of point-biserial correlation). Point-biserial correlation: Used for correlations between binary (yes/no) data and continuous data. Red: Strong negative correlation (r = -1.0). Blue: Strong positive correlation (r = 1.0). Only statistically significant correlations (p-value <= 0.05) are shown.
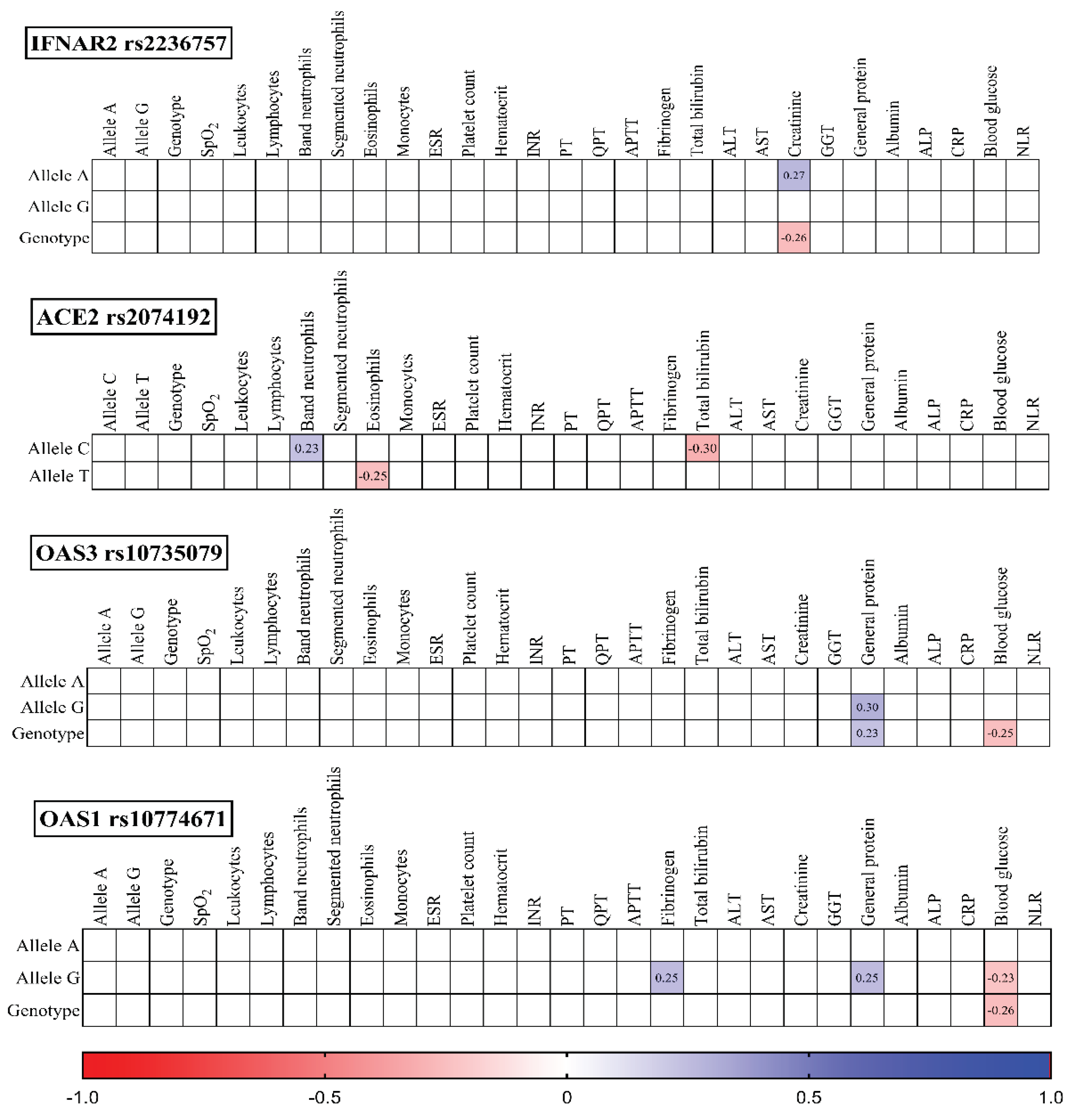
Figure 4.
Correlation correlogram. Laboratory findings on discharge (likely continuous and binary data based on the mention of point-biserial correlation). Point-biserial correlation: Used for correlations between binary (yes/no) data and continuous data. Red: Strong negative correlation (r = -1.0). Blue: Strong positive correlation (r = 1.0). Only statistically significant correlations (p-value <= 0.05) are shown.
Figure 4.
Correlation correlogram. Laboratory findings on discharge (likely continuous and binary data based on the mention of point-biserial correlation). Point-biserial correlation: Used for correlations between binary (yes/no) data and continuous data. Red: Strong negative correlation (r = -1.0). Blue: Strong positive correlation (r = 1.0). Only statistically significant correlations (p-value <= 0.05) are shown.
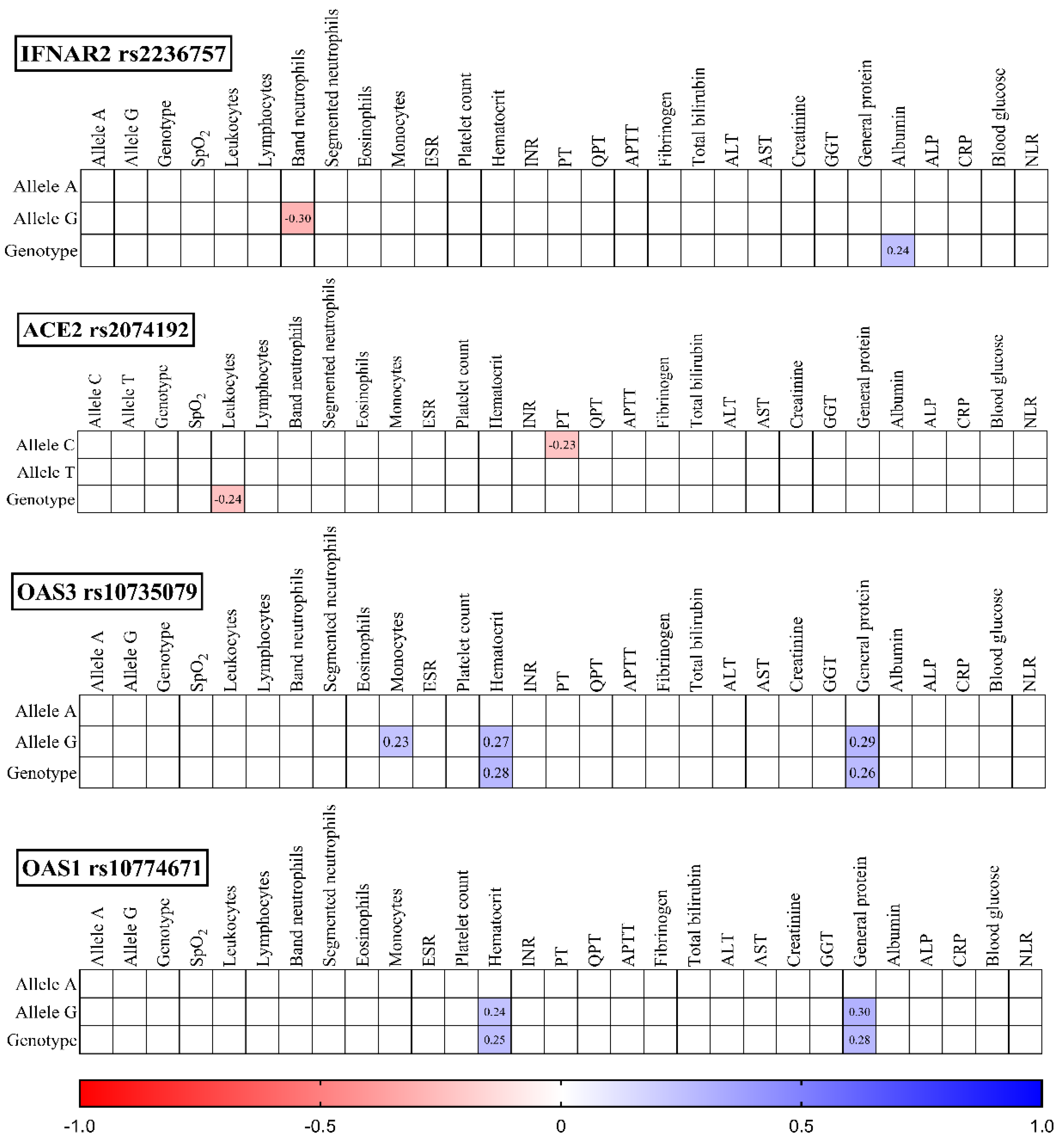
Figure 5.
(a) ROC Curve for COVID-19 Severity: This curve shows how well the model can distinguish between different severities of COVID-19; (b) Cut-off Plot for Optimal Prediction: This plot helps determine the best threshold for the model's predictions; (c) simple logistic regression curve.
Figure 5.
(a) ROC Curve for COVID-19 Severity: This curve shows how well the model can distinguish between different severities of COVID-19; (b) Cut-off Plot for Optimal Prediction: This plot helps determine the best threshold for the model's predictions; (c) simple logistic regression curve.

Figure 6.
GeneMANIA network analysis was employed to visualize the interaction network between the studied genes (IFNAR2, ACE2, OAS1, OAS3) and other relevant genes.
Figure 6.
GeneMANIA network analysis was employed to visualize the interaction network between the studied genes (IFNAR2, ACE2, OAS1, OAS3) and other relevant genes.
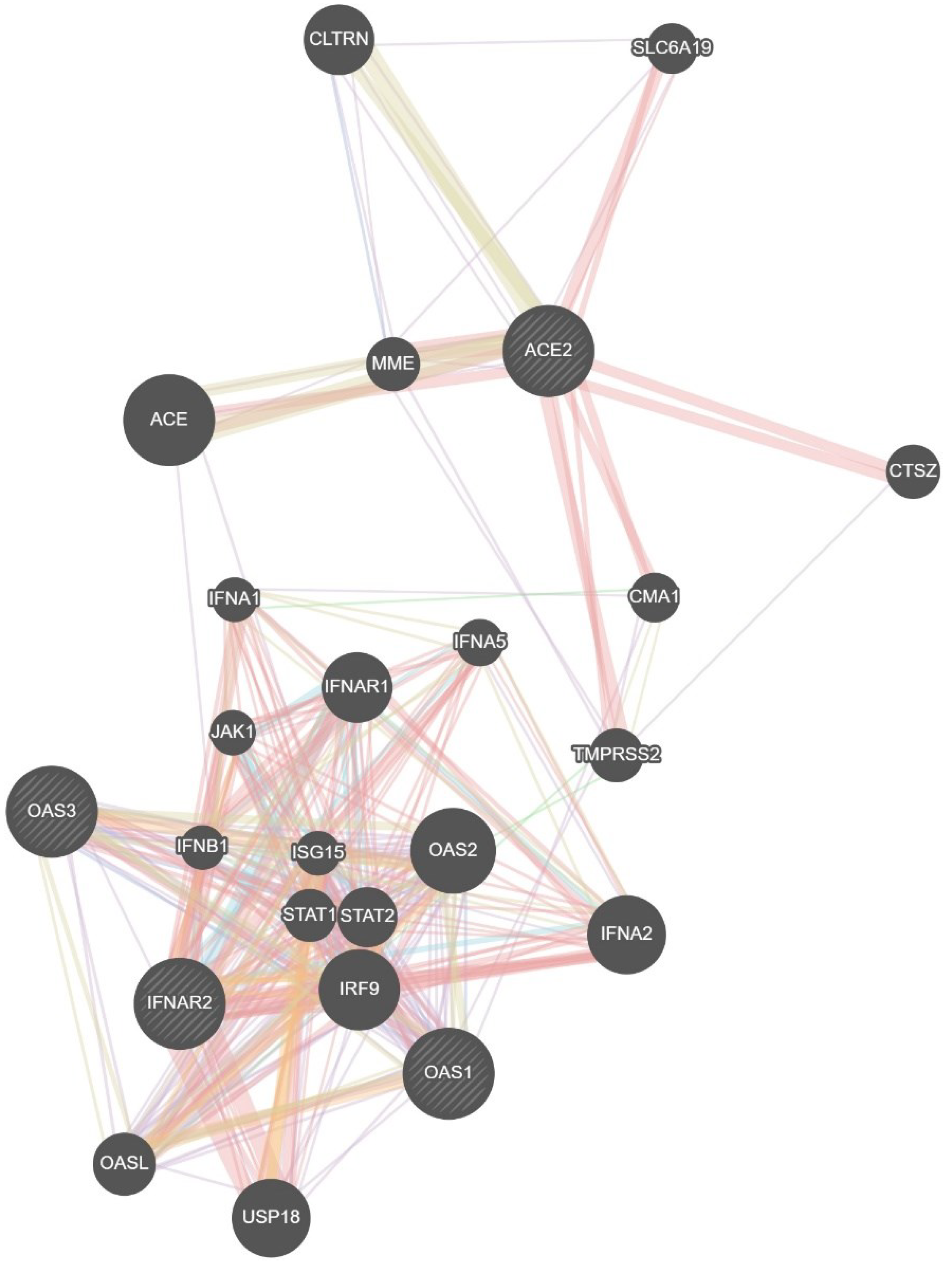
Table 1.
Baseline patients’ characteristics.
| COVID-19 with MAFLD (n=33) | COVID-19 without MAFLD (n=39) | Control (n=24) |
p-valuea | |
| Age (years), IQRb | 66 (50–72) | 65 (41–72) | 50 (38.75–58.5) | p=0.011 |
| Male, No. (%) | 21 (64%) | 22 (56%) | 17 (71%) | p=0.510 |
| BMIc, kg/m² | 30.8 (28.42-33.55) | 24 (22.4-25.35) | 24.1 (22.95-25.75) | p<0.001 |
| Comorbidities | ||||
| Diabetes mellitus | 14 (42%) | 2 (5%) | 1 (4%) | p<0.001 |
| Arterial hypertension | 25 (76%) | 18 (46%) | 5 (21%) | p<0.001 |
| Coronary heart disease | 14 (42%) | 13 (33%) | 1 (4%) | p<0.001 |
| COPDc | 3 (9%) | 1 (3%) | 0 | p=0.307 |
aChi-squared, Kruskal–Wallis test with Dunn’s multiple comparisons test; b- data are reported as medians and interquartile ranges (bIQR). cCOPD—chronic obstructive pulmonary disease. cBMI – body mass index.
Table 2.
Test for Hardy-Weinberg equilibrium for IFNAR2 rs2236757, ACE2 rs2074192, OAS1 rs10774671 and OAS3 rs10735079.
Table 2.
Test for Hardy-Weinberg equilibrium for IFNAR2 rs2236757, ACE2 rs2074192, OAS1 rs10774671 and OAS3 rs10735079.
| Group | Genotype | ACE2 rs2074192 | Genotype | IFNAR2 rs2236757 | OAS1 rs10774671 | OAS3 rs10735079 | ||||
| Expected | Expected | Expected | Observed | Expected | Observed | Expected | Observed | |||
| COVID-19 patients |
CC | 41.25 | 40 | AA | 8 | 7 | 30.68 | 32 | 29.39 | 30 |
| CT | 26.29 | 29 | AG | 32 | 34 | 32.64 | 30 | 33.22 | 32 | |
| TT | 4.25 | 3 | GG | 32 | 31 | 8.68 | 10 | 9.39 | 10 | |
| χ2= 0.645; p=0.422 | χ2=0.2813; p=0.596 | χ2= 0.471; p=0.493 | χ2= 0.097; p=0.755 | |||||||
| Control group | CC | 12.76 | 13 | AA | 2.34 | 2 | 14.26 | 14 | 13.5 | 13 |
| CT | 9.48 | 9 | AG | 10.31 | 11 | 8.48 | 9 | 9 | 10 | |
| TT | 1.76 | 2 | GG | 11.34 | 11 | 1.26 | 1 | 1.5 | 1 | |
| χ2=0.061; p=0.804 | χ2= 0.107; p=0.744 | χ2=0.091; p=0.763 | χ2=0.296; p=0.586 | |||||||
Table 3.
Analysis of allele frequencies in SARS-CoV-2 patients compared to a European population.
| Genotype/Allele Frequency | ||||||||
| Gene | Patients with COVID-19, n (%) | Control group, n (%) | European Population, n (%) |
p-valueа COVID-19-Control | p-valueа COVID-19-EUR | p-valuea Control-EUR | ||
| IFNAR2 rs2236757 | Genotype | AA | 7 (10) | 2 (8) | 105 (10) | χ2= 0,0762 p= 0.963 |
χ2=2.411 p=0.299 |
χ2=0.649 p=0.722 |
| AG | 34 (47) | 11 (46) | 190 (38) | |||||
| GG | 31 (43) | 11 (46) | 260 (52) | |||||
| Allele | A | 48 (33) | 15 (31) | 296 (29) | Fisher's exact test p= 0.860 |
Fisher's exact test p=0.332 | Fisher's exact test p=0.750 | |
| G | 96 (66) | 33 (69) | 710 (71) | |||||
| ACE2 rs2074192 | Genotype | CC | 40 (56) | 13 (54) | 126 (33) | χ2=0,641 p=0.726 |
χ2=16.86 p<0.001 |
χ2=4.831 p=0.089 |
| CT | 29 (40) | 9 (38) | 188 (49) | |||||
| TT | 3 (4) | 2 (8) | 69 (18) | |||||
| Allele | C | 109 (76) | 35 (73) | 440 (57) | Fisher's exact test p= 0.703 | Fisher's exact test p<0.001 | Fisher's exact test p=0.035 | |
| T | 35 (24) | 13 (27) | 326 (43) | |||||
| OAS1 rs10774671 | Genotype | AA | 32 (44) | 14 (54) | 209 (42) | χ2=2,145 p=0.342 |
χ2=0.648 p=0.723 |
χ2=3.121 p=0.210 |
| AG | 30 (42) | 9 (42) | 234 (46) | |||||
| GG | 10 (14) | 1 (4) | 60 (12) | |||||
| Allele | A | 94 (65) | 37 (77) | 652 (64) | Fisher's exact test p= 0.153 | Fisher's exact test p=1.000 | Fisher's exact test p=0.089 | |
| G | 50 (35) | 11 (23) | 354 (36) | |||||
| OAS3 rs10735079 | Genotype | AA | 30 (42) | 13 (59) | 201 (40) | χ2=2,286 p=0.319 |
χ2= 0.223 p=0.895 |
χ2= 2.654 p=0.265 |
| AG | 32 (44) | 10 (37) | 238 (47) | |||||
| GG | 10 (14) | 1 (4) | 64 (13) | |||||
| Allele | A | 92 (64) | 36 (75) | 640 (65) | Fisher's exact test p= 0.215 | Fisher's exact test p=1.000 | Fisher's exact test p=0.124 | |
| G | 52 (36) | 12 (25) | 366 (35) | |||||
aFisher exact and Chi-squared tests, as appropriate.
Table 4.
Allele influence on COVID-19 severity.
| COVID-19 severity | |||||||
| Gene | Allele | Moderate, n=42 | Severe/ critical, n=30 |
Control, n=24 | aχ2; p-value |
bp-value (moderate to severe/critical COVID-19) |
cOR (CI for OR) |
| IFNAR2 rs2236757 | A | 27 | 21 | 15 | χ2=0.200; p=0.905 |
p=0.724 | 0.880 (0.4368 to 1.812) |
| G | 57 | 39 | 33 | ||||
| ACE2 rs2074192 | C | 67 | 42 | 35 | χ2=1.927; p=0.382 |
p= 0.237 | 1.689 (0.7781 to 3.717) |
| T | 17 | 18 | 13 | ||||
| OAS3 rs10735079 | A | 52 | 40 | 36 | χ2=2.357; p=0.308 |
p= 0.601 | 0.812 (0.3955 to 1.599) |
| G | 32 | 20 | 12 | ||||
| OAS1 rs10774671 | A | 53 | 41 | 37 | χ2=2.758; p=0.252 |
p= 0.595 | 0.7923 (0.4056 to 1.579) |
| G | 31 | 19 | 11 | ||||
aChi-squared test and bFisher exact test, as appropriate; cOdds ratio.
Table 5.
Association between genotype and laboratory findings at hospital admission in COVID-19 patients.
Table 5.
Association between genotype and laboratory findings at hospital admission in COVID-19 patients.
| IFNAR2 rs2236757 | ||||||
| Allele A (n=41) | No Allele A (n=31) | p-Valuea | Allele G (n=65) | No Allele G (n=7) | p-Valuea | |
| Creatinine, mmol/L | 103 (88.5–121) | 90 (71–102) | p=0.021 | 96 (81.5–110) | 104 (80–120) | p= 0.481 |
| ACE2 rs2074192 | ||||||
| Allele С (n=69) | No Allele С (n=3) | p-Valuea | Allele T (n=32) | No Allele T (n=40) | p-Valuea | |
| Band neutrophils, % | 9 (6–13.5) | 3 (2–7) | p=0.046 | 7 (5.25–12) | 9 (6–14.8) | p=0.625 |
| Total bilirubin, mmol/L | 12.4 (10.8–14.9) | 21.9 (20.5–107) | p=0.004 | 13.8 (10.8–18.9) | 12.2 (10.7–14.2) | p=0.162 |
| Eosinophils, % | 1 (0–1.5) | 1 (0–3) | p=0.915 | 1 (0–1) | 1 (0–2) | p=0.036 |
| OAS1 rs10774671 | ||||||
| Allele A (n=62) | No Allele A (n=10) | p-Valuea | Allele G (n=40) | No Allele G (n=30) | p-Valuea | |
| Fibrinogen, g/L | 3.77 (3.11–5.16) | 3.85 (3.55–5.16) | p=0.598 | 3.99 (3.55–5.49) | 3.52 (2.88–4.83) | p=0.033 |
| General protein, g/L | 68.6 (62.2–73.7) | 68 (63.2–72.7) | p=0.907 | 70.4 (64.5–75.1) | 66.9 (60.9–70.4) | p=0.032 |
| OAS3 rs10735079 | ||||||
| Allele A (n=62) | No Allele A (n=10) | p-Valuea | Allele G (n=42) | No Allele G (n=30) | p-Valuea | |
| General protein, g/L | 68.6 (62.2–73.7) | 68 (63.2–72.7) | p=0.907 | 70.4 (64.8–74.8) | 66.2 (60.2–70.7) | p=0.011 |
aMann–Whitney U test, as appropriate.
Table 6.
Association between genotype and laboratory findings at hospital discharge in COVID-19 patients.
Table 6.
Association between genotype and laboratory findings at hospital discharge in COVID-19 patients.
| ACE2 rs2074192 | ||||||
| Allele С (n=69) | No Allele С (n=3) | p-Valuea | Allele T (n=32) | No Allele T (n=40) | p-Valuea | |
| Leukocytes, 109/L | 8.93 (6.15–11.3) | 6.99 (5.17–7.37) | p=0.299 | 7.55 (5.35–9.24) | 9.34 (6.39–12.46) | p=0.051 |
| INR*, n | 1.01 (0.92–1.07) | 1.09 (1.07–1.17) | p=0.046 | 1.01 (0.92–1.07) | 1.05 (0.95–1.09) | p=0.281 |
| PT*, sec | 12.6 (11.85–13.4) | 13.6 (13.4–14.4) | p=0.045 | 12.55 (11.7–13.4) | 12.85 (12.2–13.4) | p=0.571 |
| QPT*, % | 96.2 (82.2–104.7) | 79.3 (71.6–84.3) | p=0.053 | 98.65 (83.18–102.1) | 91.15 (81.4–106.3) | p=0.869 |
| OAS1 rs10774671 | ||||||
| Allele A (n=62) | No Allele A (n=10) | p-Valuea | Allele G (n=40) | No Allele G (n=32) | p-Valuea | |
| Hematocrit, % | 36 (31.34–42.18) | 38.36 (34.96–44.2) | p=0.179 | 37.91 (32.95–42.99) | 34.65 (30.65–39.55) | p=0.044 |
| General protein, g/L | 64.55 (60.88–70.93) | 67.25 (61.58–72.23) | p=0.444 | 66.6 (62.45–72.15) | 63.4 (58.35–68.78) | p=0.011 |
| OAS3 rs10735079 | ||||||
| Allele A (n=62) | No Allele A (n=10) | p-Valuea | Allele G (n=42) | No Allele G (n=30) | p-Valuea | |
| Monocytes, % | 5 (3–7.25) | 5 (2–9.25) | p=0.725 | 5 (4–8) | 4 (2–6.5) | p=0.049 |
| Hematocrit, % | 36 (31.34–42.18) | 38.36 (34.96–44.20) | p=0.179 | 37.91 (32.77–43) | 33.84 (30.35–38.12) | p=0.024 |
| General protein, g/L | 64.55 (60.88–70.93) | 67.25 (61.58–72.23) | p=0.444 | 66.6 (61.98–72.23) | 63.5 (58.08–68.73) | p=0.016 |
aMann–Whitney U test, as appropriate. Abbreviations*: INR—international normalized ratio; PT—prothrombint time; QPT—quick prothrombin time.
Table 7.
Difference in clinical and laboratory findings on admission/ discharge in patients with different genotype.
Table 7.
Difference in clinical and laboratory findings on admission/ discharge in patients with different genotype.
| Admission | |||||||
| ACE2 rs2074192 Genotype | |||||||
| СС (n=40) | СT (n=29) | TT (n=3) | p-Valuea | ||||
| Total bilirubin, mmol/L, IQRb | 12,2 (10,7–14,2) | 13,4 (10,8–17,4) | 21,9 (20,5–107) | p=0,027 | |||
| OAS3 rs10735079 Genotype | |||||||
| AA (n=30) | AG (n=32) | GG (n=10) | p-Valuea | ||||
| General protein, g/L | 66,2 (60,2–70,7) | 70,4 (65,6–75,1) | 68 (63,2–72,7) | p=0,024 | |||
| Discharge | |||||||
| IFNAR2 rs2236757 Genotype | |||||||
| AA (n=7) | AG (n=34) | GG (n=31) | p-Valuea | ||||
| Band neutrophils, % | 5 (4–7) | 2,5 (1,75–4) | 3 (2–5) | p=0,026 | |||
| ACE2 rs2074192 Genotype | |||||||
| СС (n=40) | СT (n=29) | TT (n=3) | p-Valuea | ||||
| INR, n | 1,06 (0,95–1,09) | 0,99 (0,91–1,03) | 1,09 (1,07–1,17) | p=0,020 | |||
| OAS1 rs10774671 Genotype | |||||||
| AA (n=32) | AG (n=30) | GG (n=10) | p-Valuea | ||||
| General protein, g/L | 63,5 (58,1–68,7) | 66,3 (62–72,3) | 67,3 (61,6–72,2) | p=0,041 | |||
aKruskal–Wallis test as appropriate. bIQR-interquartile range.
Table 9.
GenMANIA interaction.
| Physical Interactions | Co-expression | Predicted | Co-localization | Genetic Interactions | Pathway | Shared protein domains |
| ACE–ACE2 IRF9–OAS3 IRF9–OAS1 IRF9–IFNAR2 IFNA2–FNAR2 USP18–IFNAR2 IFNAR1–IFNAR2 STAT2–OAS3 STAT2–OAS1 STAT2–IFNAR2 CTSZ–ACE2 MME–ACE2 STAT1–OAS3 STAT1–IFNAR2 CMA1–ACE2 IFNA5–IFNAR2 JAK1–IFNAR2 IFNA1–IFNAR2 IFNB1–IFNAR2 ACE–ACE2 TMPRSS2–ACE2 SLC6A19–ACE2 |
OAS2 –OAS1 IRF9–OAS1 OASL–OAS1 STAT1–OAS1 OAS1–OAS3 ISG15–OAS1 USP18–OAS1 ACE–ACE2 OAS1–OAS3 OAS2–OAS3 USP18–OAS1 CLTRN–ACE2 OASL–OAS3 MME–ACE2 STAT1–OAS3 ISG15–OAS3 STAT2–ACE IFNAR1–IFNAR2 STAT1–IFNAR2 CLTRN–ACE2 USP18–OAS3 MME–ACE2 SLC6A19–ACE2 CLTRN–ACE2 TMPRSS2–OAS1 CMA1–OAS1 IFNB1–IFNAR2 STAT2–OAS1 |
OAS2– OAS3 OASL–OAS1 JAK1–IFNAR2 IRF9–IFNAR2 STAT1–IFNAR2 |
IFNB1–IFNAR2 OAS1–OAS3 STAT2–OAS3 STAT2–OAS1 STAT1–OAS3 |
TMPRSS2–OASL TMPRSS2–STAT2 JAK1–STAT2 IFNA1–CMA1 |
IRF9–IFNAR2 IFNA2–IFNAR2 IFNAR1–IFNAR2 STAT2–IFNAR2 STAT1–IFNAR2 IFNA5–IFNAR2 IFNB1–IFNAR2 IFNAR1–IFNAR2 |
OAS1–OAS3 ACE–ACE2 OAS2–OAS3 OAS2–OAS1 CLTRN–ACE2 IFNAR1–IFNAR2 OASL–OAS3 OASL–OAS1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



