
Submitted:
26 March 2024
Posted:
27 March 2024
You are already at the latest version
Abstract
Plant transcriptomes are an extremely dynamic entities shaped spatially and temporally by many intracellular and environmental cues. In this review, we first summarize the complexity and diversity of plant genomes and transcriptomes as a start point for the multitude of transcriptomic responses. Numerous alterations within various tissue and organ-specific transcriptomes as well as the most relevant transcriptomic responses associated with plant acclimation to selected abiotic and biotic stress conditions, from the current studies employing high-throughput transcriptomic analysis are widely discussed. Understanding changes within plant transcriptomes, revealed by in silico functional analysis, allows for the characterization of stress-affected genes and stress acclimatory mechanisms, as well as allows to perform plant metabolic engineering. The latter allow cultivars to produce more secondary metabolites in the future, which are often desirable substances in the biomedical industry. Accordingly, in this review special attention was also paid to characterize the potential of transcriptomic analyses of medicinal species, particularly to search for new cultivars. Extensive characterization of transcriptomic responses in stress would also result in the development of new cultivars that display physiological and molecular mechanisms that allow them to cope with adverse environmental conditions more adequately.
Keywords:
differentially expressed genes
; medicinal species
; plant organs
; secondary metabolites
; plant transcriptome
; stress
1. Introduction
Higher plants, known as vascular or telome plants (Thelomophyta), appeared during plant evolution back in the Palaeophytic era. They are characterized by the presence of organs, the development of tissues distributing water, mineral compounds, and photosynthesis products and the dominance of the sporophyte over the gametophyte [1,2]. Due to the sessile life cycle, higher plants were evolutionarily forced to develop multiple adaptations that allowed them to respond to stress [3], including transcriptomic alterations. To better understand how plants respond to environment, detailed transcriptomic analyses are still necessary. The development of high-throughput RNA-seq platforms with a subsequent decrease in sequencing costs, as well as data meta-analyses, simplified the transcriptomic studies. They allow for the analysis of the gene expression profile manifested by a given plant cell, tissue, or organ. Seasonal, environmental, or developmental transcriptomic variations can be also observed [4].
In recent years, plant transcriptomic reviews focused mostly on methodology issues [5,6,7]. Excellent studies with species-specific omics analyses, including those on stress response and focused on the relevance of transcription factors (TFs), hormones, translational reprogramming, and the epigenetic level, as well as phenotypic and physiological analyses were also published [8,9,10,11,12,13]. However, they did not always refer exhaustively to the transcriptomic data, and they were sometimes limited to the study of noncoding RNA in acquiring stress tolerance [14]. Owing to those factors, in this review the diversity of transcriptomic responses among diverse organs in the developmental context, and responses to various stressors, both abiotic (including UV radiation, chemical treatments, drought, cold and heat stress) as well as biotic (fungal and bacterial infections) stressors, will be presented. The cited studies coming from recent high-throughput analyses, will be discussed in the context of the complexity of plant genome and transcriptome structure as a basis for a variety of transcriptomic alterations. We will also reveal the applicative aspects of the recent searches for useful new medicinal plant cultivars resistant to stress.
2. The Diversity of Higher Plant Genomes and Transcriptomes as a Basis for the Complexity of Transcriptomic Responsiveness
Genome is composed of a complete set of all DNA molecules in the cell containing the information necessary for an individual to function and develop. In plants, three genomes are recognized: nuclear, chloroplast (the plastome) as well as mitochondrial genome (the mitogenome) [15].
Plant nuclear genome size is not correlated with the size of organellar genomes. Of all the major taxonomic groups, land plants contain the most conserved size of the nuclear genome. For example, Genlisea aurea is a carnivorous plant species with one of the smallest nuclear genomes, which spans 43.3 Mbps, while one of the largest known nuclear genomes (up to 150 Gbps) is known in Paris japonica (Table S1). Currently, two main factors are thought to be responsible for the large-scale variation of genome size among land plants: the recurrent cases of whole genome duplication (WGD), i.e., polyploidy, and the dynamics of the loss and gain of transposable elements. Even though duplication of chromosome number is an obvious reason for the increasing genome size, transposon gain-and-loss results in the greatest variation in the genome size. In some nuclear genomes, transposons comprise about 3% of their size; in other ones, they can occupy even 85% of the genome. Interestingly, the relationship between plant nuclear genome size and transposon content is generally linear [16]. Numerous evidence suggests that plant species with small nuclear genomes are characterized by broader phenotypic and ecological features, while species with larger genomes live only under conditions with more relaxed pressure of natural selection. Interestingly, plant nuclear genome correlates with the nutrient availability. Under nitrogen or phosphorus deficiency, species with larger genomes were unable to compete and dominate in ecosystems [17].
According to Margulis [18] concept, plastids and mitochondria originated from the engulfing and maintenance of single-celled organisms in the early eukaryotic cell under endosymbiosis. The proof of such an event would be the presence of their own genetic material, prokaryotic ribosomes, double membraned envelope as well as size [19]. The mitochondrial ancestors were probably γ-proteobacteria, while the plastid ancestors were organisms similar to cyanobacteria. Most organellar genes underwent an evolutionary transfer to the nucleus, and still organellar DNA insertions can be found in nuclear genomes [20]. Genome-containing organelles thus exhibit a particularly complex pattern of their biogenesis, depending on the concerted regulated expression of nuclear and organellar genes by anterograde and retrograde signaling [21,22].
Although nuclear genomes can reach 119.1 Mbps in Arabidopsis to 150 Gbps in size in Paris japonica, the plastome is relatively conserved in length, gene composition and organization, and in angiosperms typically ranges in size from 120 to 180 kbps [23,24]. The length of the Arabidopsis plastome (with nuclear genome of 119.1 Mbps) is 154,478 bps, when P. japonica, the owner of the largest land plant nuclear genome (150 Gbps) has a plastome of 155,957 bp. The difference in sizes between Arabidopsis and P. japonica plastomes is therefore about only ~1,500 bps (Table S1). Most plastomes of land plant encode a limited number of proteins involved primarily in photosynthesis, transcription, translation, and plastid signaling. Similarly to mitogenomes, plastomes also encode rRNAs, tRNAs, and various ncRNAs [25,26]. Multiple factors of nuclear origin regulate and control the expression of plastid proteins at different stages, including transcription, RNA editing, post-transcriptional modification, splicing, and translation [27]. Metabolites synthesized in plastids are often signaling molecules for distant communication pathways and play an important role in the response of plants to stress [25,28]. Therefore, knowledge on interactions between the nucleus and the plastids might contribute to understanding the plant functioning under stress.
The size of copies of the given organellar genomes can vary in the same species, e.g., the mitogenome of potato (Solanum tuberosum) is composed of few molecules from 49,171 bps, 49,230 bps, and 112,800 bps to 247,843 bps and 297,014 bps (Table S1) [29]. In general, the size of plant mitogenomes ranges from 208 kbps for white mustard (Brassica hirta), 366 kbps for Arabidopsis, up to 2,500 kbps for musk melon (Cucumis melo); even larger, fast evolving mitogenomes (ca. 11.3 Mbps) occurs in some Silene species (Table S1), while the gene number (ca. 60 genes) remain relatively stable [30,31,32]. Mitogenomes of higher plants are thus particularly large and possess a complex structure, which could arise during plant terrestrialization and optimize seed germination in novel land ecosystems. Both the structure and the expression pattern of the plant mitogenomes improved the regulation of organellar functions and mitochondrial biogenesis in seed development [21].
While the genome is set of all DNA molecules in cells, the transcriptome is set of various RNA molecules (mRNA, rRNA, tRNA, as well as numerus ncRNAs, e.g., miRNAs, siRNAs, etc.) at a given spatial and temporal step. According to Imadi et al. [33], the transcriptome can be seen as a translated fraction of genome information required by the organism to function and respond to its surrounding environment. For instance, with the comparison to maize (Zea mays) nuclear genome sizing of approximately 2,2 Gbps, the estimated transcriptome capacity spans only 97 Mbps, which consists of ca. 4% of the nuclear genome size [34].
To date, Best et al. [35] have published preliminary maps of Arabidopsis and cauliflower (Brassica oleracea var. botrytis) mitochondrial transcriptomes. Arabidopsis and cauliflower mitogenomes encode 28 and 33 protein-coding genes, 3 and 3 rRNAs, 22 and 18 tRNAs, and cover approximately. 85 and 35 ORFs of >100 aminoacid residues, respectively. In addition to rRNAs, numerous tRNAs, and ncRNA molecules [26], plant mitogenomes encode OXPHOS apparatus (respiratory chain complexes), ATP synthase subunits, mitoribosomal proteins, few proteins indispensable for cyt. c biogenesis, and the translocase of the twin arginine protein. Up today, at least 42 and 33 transcription units were found by RNA-seq of Arabidopsis and cauliflower mitogenomes, respectively. The expression of several Arabidopsis mitogenes leads to the formation of mono- or bicistronic transcripts; for example, OXPHOS and ribosomal protein genes are often expressed at various levels. Various open reading frames within the same polycistronic transcript could be diversely expressed within the post-transcriptional RNA processing, which involves few key steps, including cis- and trans-splicing and RNA editing; the latter one arose from embryophytes separated from ancestral algal lineages and is therefore believed to be absent in green algae [35,36].
It is worth noting that the transcriptome composition within the same organism depends not only on the tissue, but also on the sampling procedure [6]. Transcriptomic studies allow us not only to know what proteins are synthesized in the cell; factors that regulate the transcriptional activity of a given biological system may be also characterized [37]. Tissue-specific transcriptomes illustrate the distinctness within gene expression patterns across various plant tissues. These transcriptomes offer valuable information on the underlying molecular processes that affect tissue-specific functions, such as photosynthesis in leaves, nutrient uptake in roots, or reproductive processes in flowers. Furthermore, by deciphering tissue-specific transcriptomes, specific genes and regulatory mechanisms that display unique attributes and roles in diverse plant tissues can be described, thus propelling advancements in plant biology and agriculture [38]. The transcriptome composition also changes dynamically under various environmental cues. Tissue-specific transcriptomes play a crucial role in understanding how plants respond to these conditions. This knowledge is instrumental in the development of strategies to improve plant acclimation under challenging environmental conditions [39].
3. Tissue-Specific and Organo-Specific Alterations in Plant Transcriptomes
3.1. Xylem and Phloem Transcriptomes as Examples of Tissue-Specific Transcriptomes
Well-investigated examples of tissue-specific transcriptomes are transcriptomes of vascular elements. During the primary growth of the stems and roots, procambium derived from the apical meristems divides and differentiates into xylem and phloem. Precursor xylem cells form xylem crumb cells or fibers. During unaffected angiosperm development, in the vascular bundle, xylem, procambium and phloem cells show dorsoventral polarization: xylem is located on the dorsal (adaxial) side, phloem occurs on the ventral (axial) side, whereas procambium is located between the phloem and xylem (Figure 1a) [40,41]. Current transcriptomic studies indicate an antagonistic influence of TFs from HD-ZIP III (e.g., ATHB8, ATHB15/CNA, PHV, PHB, REV/IFL1 TFs) and the KANADI families (e.g., KAN1, KAN2, and KAN3) in the differentiation of procambium cells to xylem and phloem, respectively. Mutations in REV, PHB, and PHV genes for specific TFs have been shown to affect the organization of vascular tissue that contains the phloem, which surrounds the xylem (Figure 1b) [42]. However, in the case of mutations within KANADI genes, xylem starts to surround phloem elements (Figure 1c) [43,44]. Roszak et al. [45] employed single-cell RNA-seq approach to study the development of protophloem from the progenitor cell. This study underlined the relevance of other TFs, namely PEAR proteins that initiate differentiation (lineage bifurcation) program, controlled by PLETHORA TFs. PEAR TFs mediate early asymmetric divisions during phloem cell biogenesis and in laterally adjacent cells of procambium.
Transcriptomic analyses of the biogenesis of plant vascular elements showed that more differentially expressed genes (DEGs) appeared to be upregulated in phloem than in xylem ([46]; Table S2). Among DEGs upregulated in phloem, genes for RNA polymerase II subunits B2, B7, B9, ABC3 and ABC5, and RNA polymerase III subunits C1 and C2 as well as genes involved in galactose metabolism and polysaccharide synthesis were enriched. On the contrary, genes involved in fatty acid biogenesis (ACOX1, ACOX3, ACADM, PAAF, and ECHS1) were upregulated in xylem, indicating the demand for high-energy compounds. Furthermore, the elevated expression of the mentioned above genes accompanied the activity of genes for the synthesis of fibrous elements from the phloem [46,47,48].
Nearly 26,898 DEGs in potato (Solanum tuberosum) leaf petioles of the 39,000 genes identified in the potato genome were identified. However, only a limited number of genes (mostly downregulated) differentiated all studied tissues (Table S2). The expression patterns between the tissues studied were enriched with DEGs involved in signaling pathways, plant response to light, hormonal response, flowering, or RNA binding. Despite tissue sampling, gene expression varied depending on where the plant material was taken [49].
An interesting study showing the seasonal variation of changes in the transcriptome during plant development over months was carried out by Mishima et al. [50], who sequenced transcriptomes of tissues of the meristem cambium and derivative cells of Japanese cedar (Cryptomeria japonica) from a few developmental stages. Almost 55,051 DEGs were found, with 10,380 DEGs involved in xylem formation. More genes were expressed during inhibition of cell divisions than during peak xylem biogenesis (xylogenesis) at spring reactivation (Table S2). During xylogenesis, genes associated with primary cell wall biosynthesis, secondary cell wall deposition, and lignification were markedly upregulated. The expression of genes, coding for enzymes involved in the lignification process, e.g., PAL, 4CL, C4H, HCT, CCOAOMT, and CCR in the period from March to August also increased but decreased since September. Therefore, this study confirms environmental analyses, indicating that several cambium cells increase rapidly between March and June. Such results suggest that the genes involved in lignification belong to the main genes involved in the biogenesis of Japanese cedar xylem [50].
3.2. Selected Organo-Specific Transcriptomes
The specialization of individual plant organs to perform specific functions is a consequence of differences in the expression levels of numerous gene families. For example, distinct gene expression patterns are presented in root tissues and leaves or in generative organs [51]. Details on affected genes from developmental and organo-specific studies were summarized in Table S2.
3.2.1. Root and Leaf Transcriptomes
The main function of roots is to store energy in the form of high-energy compounds, i.e., complex carbohydrates, while leaves convert solar energy into chemical energy during photosynthesis [52,53].
A high-resolution single-cell RNA-seq expression atlas of Arabidopsis root was constructed by Denyer et al. [54]. It allows in unprecedented manner to retrieve expression features of all major cell types within roots (with finely resolved marker genes defining unique cluster identities and gene expression waves) and to characterize key developmental regulators and genes governing root morphogenesis. Among them, expression profiles of at least 239 TFs were distinctive, including the ones for root hair biogenesis. In addition, Shulse et al. [55] profiled Arabidopsis root transcriptome by single-cell RNA-seq across major tissues and developmental stages (in particular, during endodermis histogenesis, which resulted in the identification of at least 800 dynamically responding genes). Those authors also investigated the impact of sucrose supplementation on the root transcriptome under its development. In the earlier study, Efroni et al. [56] employed single-cell RNA-seq to study root cells regeneration after excision of root tip. Authors concluded that the transcriptome of regenerating cells prior to stem cell activation resembles that of an embryonic root progenitor cell. Plant root regeneration follows the developmental stages of embryonic patterning and is governed by spatial information provided by complementary hormone domains.
The pattern of the lateral roots and cell-specific transcriptomic alterations were summarized in Kortz et al. [57] review. In tissue-specific transcriptomes in Arabidopsis leaves, the number of DEGs decreased from the leaf vasculature (the highest one) to the epidermis and mesophyll, indicating the variable complexity of these tissues and their decreased sensitivity to transcriptional responsiveness. Interestingly, both the epidermis and mesophyll expression profiles were mainly enriched with downregulated DEGs. Genes specific for epidermal biogenesis coded proteins for lipid metabolism, stomatal development, auxin signaling, and cuticle wax synthesis. Genes active specifically in mesophyll coded proteins that participate in hormone signaling, oxidative stress response, defense, and photosynthesis (a few genes only for the latter GO: term) as well as various DNA binding proteins (notably, ERF and WRKY TFs). In contrast, vasculature-specific genes were indispensable for the biogenesis of vascular elements, the cell wall, genes responsible for the protein fate, plant development, and morphogenesis as well as for the accumulation of TFs containing MYB and NAC domains [39].
Recently, Tenorio Berrío et al. [58] investigated transcriptomes of over 1,800 Arabidopsis leaf cells by single-cell RNA-seq. This study characterized at least 14 diverse cell populations (not only from the core tissues) in young leaves and detected at least 19,000 various messengers focusing on the cellular diversity of developing leaves intertwined by developmental, metabolic, or stress-response routes. Main leaf tissues were well-identified by transcriptomic analysis (which indicated for thew high sensitivity of mesophyll transcriptome to low water potential) and developmental gradients in leaf epidermis were analyzed. Results indicated also for the diversity of cells building the vascular architecture. This study provided also valuable set of genetic markers for distinct cell populations. In contrast, during 3-week-long senescence, the transcriptome of Arabidopsis leaves became notably different from the one of mature leaves [59]. Several different TFs active in different stages of development were identified. DEGs active between 29 and 33 days after sowing were grouped into two clusters. In general, during Arabidopsis leaf senescence DEGs related with photosynthesis, chlorophyll biogenesis, cytokinin signaling, ribosome biogenesis, aminoacid metabolism, the utilization of carbon skeletons and cell cycle appeared declined in expression level, while genes for jasmonic acid (JA) and ethylene signaling, response to stimulus, caspase and pectinoesterase activity, lipid degradation, catalytic activity, cytoskeleton, metal binding and transport were upregulated. Those regulated genes perform protective functions that the plant takes in stress acclimation. Novel groups of co-regulated genes were also discovered among upregulated DEGs. Moreover, Chrobok et al. [60] performed the meta-analysis of Breeze et al. [59] data, that allowed for the identification of more than 1,000 genes for mitochondrial proteins active at least at a single timepoint; those alterations were especially visible at the initial stages of senescence. Few clusters were proposed based on expression values of the mitochondrial cluster, particularly enriched in genes for OXPHOS functions, auxin signaling, reaction to nutrient and light depletion, plastid functions, stress response, protein fate, transport, and assembly. Those results indicate for the relevance of mitochondrial biogenesis during plant senescence.
In the sugarcane (Saccharum spp. hybrids) transcriptome, more organ specific DEGs were expressed in leaves, contrary to roots; during analyses 77,359 genes were identified [61]. In the leaf transcriptome, messengers for photosynthetic proteins were upregulated. On the contrary, the root transcriptome was enriched with transcripts encoding proteins for polysaccharide biogenesis, aminoacid metabolism, or involved in high-energy compound catabolism. Although organospecific differences resulted from various functions of both organs, some general characteristics of expression profiles remained unchanged [51,61].
The maize (Zea mays) leaf transcriptome showed well-noted differences in gene expression consistent with leaf axial symmetry [62]. In basal tissues, genes for cell wall biosynthesis, DNA synthesis, cell cycle regulation, auxin biosynthesis, and cellular transport were upregulated. In contrast, in the apical part of the leaves, photosynthetic genes appeared visibly upregulated, especially genes encoding enzymes for the biosynthesis of isoprenoids (e.g., carotenoids), Calvin cycle, redox homeostasis, starch, and sucrose metabolism. These data show that global changes in the level of gene expression also occur in such a seemingly ‘uniform’ organ as the plant leaf. During analysis, Li et al. [62] identified almost 25,800 maize unigenes.
3.2.2. Transcriptomes of Generative Organs
Sexual reproduction of higher plants is performed by generative organs- flowers. Their morphology is species-specific and depends mostly on the pollination strategy. Despite the differences between the formation of generative organs, the transition from the vegetative to the generative phase is undoubtedly the most important step in plant development and thus genetically controlled [63]. Multiple studies have shown that flowering depends on the photoperiod, vernalization, activity of plant hormones (including GA), thermosensing, and a variety of ageing-associated processes [64,65,66]. Both the photoperiod and the vernalization control the initiation of flowering, which protect the plant from the energy-consuming flower development in unfavorable conditions [67,68].
An analysis of the Annona squamosa transcriptome showed elevated expression levels of genes involved in vernalization and photoperiod induction [68]. More than 71,948 A. squamosa genes were identified, of which about a third were differentially expressed. Genes involved in the photoperiod pathway embraced some phytochrome (PHY) and cryptochrome (CRY) genes as well as early flowering genes (EF1, EF3). Distinct DEGs appeared to be homologous to genes involved in vernalization, including FIE and VIN3 genes. Another study showed that MADS protein genes have a large morphogenetic effect on flower formation. MADS belong to transcriptionally important regulators [69], for example, SOC1 and FUL factors. Disturbances in the expression pattern of these genes contributed to delayed flower formation, despite the unaffected photoperiod [64].
At the stage of colorful petal formation, genes responsible for encoding enzymes for pigment synthesis, genes involved in the synthesis of secondary metabolites in tissue, such as carotenoids or anthocyanins, were significantly upregulated in Achimenes [70]. Other gene families were involved during the development of broccoli (Brassica oleracea var. italica) floral buds [71]. DEGs involved in broccoli curd heterosis active in light and H2O2-mediated signaling appeared among newly discovered functional classes that control crop yield, hormone signaling stress response, and flowering. Overall, expressed genes (especially the upregulated ones) seemed to prevail in hybrid lines rather than in parental lines. Numerous DEGs from this group encoded proteins important for growth and development, fatty acid and carbohydrate metabolism, protein synthesis and modifications.
3.2.3. Seed Transcriptomes
Seeds perform a reproductive function among angiosperms and allow the development of a new plant generation, resulting from sexual reproduction, as well as protect the embryo from adverse conditions. The seed endosperm and embryo are developing after a double fertilization. In contrast, the seed cover is formed from embryo stem cells. Seeds often contain high-energy compounds, allowing for the germination. Analysis of the seed transcriptome appears thus extremely important for the characterization of genes active in seed biogenesis and germination [72,73].
Recently, a spatial transcriptomic profiling of germinating barley (Hordeum vulgare) seeds was exhaustively studied by Peirats-Llobet et al. [74]. Such methodology (Visium Gene Expression slides, 10x Genomics) allowed for a complete study of gene expression profiles across individual cell types within the plant seeds. For sequencing, 8 μm- thick longitudinal sections of the barley grains were prepared. The study allowed to obtain spatially resolved cellular map for barley germination and identifies specific functional genomics targets to characterize better cellular processes during germination. Results of Peirats-Llobet et al. [74] study extends previous attempts of using single-cell RNA-seq for deciphering of root and other organ transcriptomes [55,75], as time-series spatial transcriptomic analysis of transcriptome allows far more complex output from all cell types in the investigated tissue/ organ with preserved information on their spatial location. Visium methodology allowed also for detection of 83-90% transcripts reported from the previous, tissue-specific studies [76], however only a portion of seed tissue was subjected to RNA-seq. This approach allowed clear distinguishing of tissues and tissue regions both by their physical location and gene co-expression patterns. Various functional gene categories were revealed among over 14,000 DEGs at the initial 24 h after seed imbibition (at 0 [dry], 1, 3, 6 and 24 hour-timepoints). Selected gene families, including aquaporins (highly expressed in mesocotyl, scutellum and coleorhiza), thionins (in endosperm and aleurone), starch catabolism enzymes (in various seed tissues, including aleurone and endosperm), cell wall modification proteins (in radicle and scutellum), transporters (including mitochondrial ones in coleorhiza, embryo, and scutellum during early imbibition), ribosomal proteins, RNA-binding proteins and elongation factors (the last three categories in embryo tissues) and TFs (including bZIP, bHLH, and DREB proteins) displayed a spatially temporal expression pattern. Distinct gene clusters were observed in coleorhiza, radicle and scutellum, and embryo was divided into 3 expression clusters. Most genes encoding transcription and translation functions were expressed in the embryo. The spatial gene expression patterns suggested that the ability of germination to proceed without transcription is a specific embryo function. Genes for mitochondrial biogenesis were not expressed at a very high levels until 24 h after imbibition, however, genes for mitochondrial expression machinery were initially expressed and genes for glycolytic enzymes appeared even more active. LEA genes were mostly expressed at early imbibition stages. Endosperm and aleurone transcriptomes were distinct.
A valuable analysis of the Arabidopsis seed transcriptome at multiple developmental stages allowed the identification of active genes before, during and after seed ripening. The highly expressed genes for seed development were detected in the globular embryo and the developed cotyledon. A lower, but noticeable, expression level of genes specific for seed development was observed in the mature and post-mature stages of the green embryo, associated with the transition of the seed into dormancy. Of the genes for 48 diverse TFs, LEC1, LEC1-LIKE, LEC2, FUS3, MEDEA and PEI1 were active. Mutations that occurred in these genes resulted in embryo defects, which confirmed their key role in seed development [77]. Further studies of the ttranscriptional reprogramming in Arabidopsis germinating seeds resulted in the first dynamic TF network model of seed germination. Numerous already known as well as novel TFs were found. After seed stratification genes for bZIP and AP2/EREBP TFs appeared downregulated. The DEGs affected by seed germination count for significantly novel data in the field. Among the functional terms related with DEGs in seeds, the functions of RNA splicing, and histone prevailed, unlike the functions related to light and roots. Genes encoding proteins that interact with nucleic acids and metabolic enzymes were also enriched. Moreover, genetic isoforms varied between dry and post-imbibed seeds [78].
An analysis of the Polygonatum cyrtonema seed transcriptome revealed upregulated genes for the synthesis of secondary metabolites; the other genes for proteins active in the synthesis of lignin in the germinated seed stage were downregulated. In accordance with previous analyses, upregulation of genes involved in starch catabolism, which can provide the energy for Polygonatum seedlings, were evident [79].
3.3. Transcriptomic Analyses of Plant Species with Medicinal Applications
Transcriptomic studies have found applications in medicinal plant research, e.g., for the functional gene characterization and the identification of key metabolic pathways of pharmaceutical importance. Although most medicinal species do not belong to the model ones, transcriptomic data from various plant studies allow a comparison of newly discovered genes with existing data, helping to determine the potential function of gene expression product and to find genes responsible for the synthesis of bioactive components [80,81]. For instance, understanding tissues responsible for the increased synthesis of given compounds may allow optimization of the production of medicinal substances produced by aloe (Aloe vera), which can be used, for example, in the pharmaceutical or cosmetic industries. Analysis of aloe root and leaf transcriptomes identified in total 113,063 and 141,310 unigenes, respectively. DEGs in leaves (but not in roots) were mainly downregulated. Some DEGs were involved in lignin, saponin, and alloin biosynthesis [82]. In contrast, analysis of Trillium govanianum root, leaf, fruit, and stem transcriptomes revealed the highest number of DEGs between leaves and roots [83]. Among them, several genes involved in the biosynthesis of steroid saponins and other secondary metabolites, including brassinosteroids, carotenoids, terpenoids, flavonoids, phenylpropanoids, and steroids, were enriched. Almost 141 DEGs were involved in the metabolic pathways of these metabolites and 78 genes appeared tissue specific. Genes for the flavonoid synthesis were upregulated primarily in fruits and stems. Contrary to that, genes involved in the synthesis of terpenoid had increased expression in leaves, while genes responsible for the synthesis of steroids appeared to be upregulated in roots. Those results illustrate the fact, how diverse compounds with the pharmaceutical potential may be synthesized in tissue- or organ-specific manner. In general, the study by Singh et al. [83] may allow the production and extraction of medicinal substances from T. govanianum.
Transcriptomic analyses of the roots, stems, and leaves of Entada phaseolides allowed the identification of 61,046 DEGs in total in these three organs. At least 26 genes for cytochrome P450 biogenesis and 17 genes for uridine diphosphate glycosyltransferase isoforms involved in the biosynthesis of triterpenoid saponins were upregulated in stems compared to roots and leaves. The elevated expression level of these genes in stems corresponds to the fact that the content of triterpenoid saponins in this plant organ is higher than in roots and leaves [84].
Transcriptomic studies of medicinal species also cover DEGs for anthocyanin biogenesis. Analysis of the leaf transcriptome of Tetrastigma hemsleyanum, a species from which an important extract with antibiotic properties is obtained, allowed for identification of almost 100,540 unigenes and DEGs involved mainly in anthocyanin biogenesis (e.g., CHS, CHI, F3H, DFR, AS, UF3GT). The results of this study also suggested the differential participation of secondary metabolite biogenesis pathways in leaf metabolism depended on the developmental stage [85].
Due to its broad pharmacological activities, Dendrobium huoshanense stems belong to the widely used medical herb among health care products. However, the biosynthetic pathways of bioactive substances and the molecular regulation of stem development are often unclear in few points. The study on transcriptomes of leaves, stems, and roots of D. huoshanense revealed candidate genes involved in stem development and polysaccharide biosynthesis (at least 103 genes). The collected data was enriched in 27 genes related to fructose and mannose metabolism as well as 74 genes for glycosyltransferases and 15 genes involved in the biosynthesis of flavonoids. For photoperiod control and maintenance, genes coding for specific TFs, including CO, MADS-box, and AP2-like ones, were notable. Other genes coded products related to hormone biogenesis [86]. In general, those data may provide an important resource for future studies, in particular for the investigation of molecular processes engaged in the active compound biogenesis and the organogenesis. For additional analysis of genes necessary for polysaccharide biosynthesis among medicinal plants, transcriptomes of leaf, root, and rhizome of Polygonatum cyrtonema, an edible plant used in traditional Chinese medicine, were investigated. In this study, 164,573 genes in total were identified, while 59,271, 52,184 and 45,893 genes were expressed in leaves, roots, and rhizomes, respectively. Upregulated genes involved in polysaccharide metabolism were notably enriched in rhizomes. Furthermore, 1,131 genes for candidate TFs governing expression of genes for polysaccharide biosynthesis were found, e.g., for MYB, AP2-EREBP, WRKY, bHLH, zinc finger C3H and C2H2, and NAC TFs [87].
Another species containing secondary metabolites used in medicine is Polygonum cuspidatum. It contains resveratrol and bioactive anthraquinones, emodin and physicon, displaying antimicrobe and anticancer activities [88]. Due to the lack of availability of P. cuspidatum whole genomic data, functional studies are hampered. To address genetic insights into the metabolic pathways of natural products, a comprehensive analysis of P. cuspidatum root, stem, leaf, flower, and fruit transcriptomes revealed 27, 18 and 20 genes that encode enzymes for the mevalonate, methylerythritol phosphate, and shikimate biosynthesis pathways, respectively. Few genes coding important enzymes involved in resveratrol synthesis (PAL, C4H, 4CL, as well as STS/CHS synthases for the last step of resveratrol condensation) were identified. Some STS/CHS genes were upregulated in root tissues of P. cuspidatum, suggesting that they could be specifically involved in the resveratrol and flavonoid metabolism [89].
Based on the gene inventory involved in the processes occurring in various plant organs, it was possible to select genes coding proteins participating in given metabolic pathways, and thus it became possible to optimize the occurrence of a chosen process, or to manipulate a given mechanism of specific metabolite biogenesis by genetic engineering. Furthermore, analysis of the transcriptome of medicinal species allowed us to understand the metabolic pathways of secondary metabolites, which play an important role in the adaptation of plants to environmental conditions and have therapeutic effects on various plant organs at different developmental stages. Data obtained from these studies can provide information on the accumulation of secondary metabolites and the efficient production of active ingredients in different parts of medicinal plants, as well as can be used to develop novel genotypes with enhanced stress resistance [81]. Further analysis of metabolic pathways will help to understand how natural products are synthesized and provide resources for engineering large-scale production of natural products and generating novel chemicals in future synthetic biology applications.
Results of transcriptomic analyses of medicinal species were summarized in Table S2.
4. Plant Transcriptomic Response under Stress Acclimation
Stress can be defined as internal and external factors that affect the efficiency of physiological, metabolic, and molecular plant processes that lead to a reduction in the efficiency of energy-to-biomass conversion. These stressors can be divided into abiotic (Section 4.1) and biotic (Section 4.2) ones [90]. Due to the sedentary lifestyle, plants have developed adaptive abilities at multiple levels to cope with adverse conditions. Plants ‘sense’ external cues, which in turn generate a cellular response transmitting stimuli from the cell surface or cytoplasmic receptors to the transcription machinery. This links specific ‘cellular sensitivity’ to environmental factors. Studies on plant transcriptomes, which participate in such response may thus allow for better understanding how plants adapt to changing environmental conditions [91]. Figure 2 shows the widely used methodological pipeline for studying the transcriptomic response under stress in plant species. It combines both experimental and in silico analyses to identify systematically stress-responsive genes in cultivars varying with the stress resistance. Below discussed are the most interesting alterations that occur in gene expression profiles under diverse stress conditions from selected basic and applied studies.
4.1. Alterations in Plant Transcriptomes during Abiotic Stress – the Selected Examples
Abiotic stress (e.g., drought, suboptimal temperature including cold and heat, salinity, heavy metals, radiation, nutrient deficiency, and mechanical damage) results from the action of multiple physical or chemical stimuli [80,92]. During acclimation to abiotic stress, plant transcriptomes respond particularly dynamically. Details on genes affected under abiotic stress from high-throughput transcriptomic studies are given in Tables S2 and S3 and the summary of the cellular functions governed by diverse stressors affecting gene expression profiles in various plant species was depicted in Figure 3. In public transcriptomic repositories, e.g., Expression Atlas [93], relatively complete Arabidopsis transcriptomic data is available. The g:Profiler analysis [94] of functional GO: terms related to Arabidopsis stress-responsive genes collected from Expression Atlas showed the huge variability of gene response under those conditions, even between similar treatments. However, stress response involves not only distinct, but also similar gene families, especially across diverse duration of the given treatment, although depending on stressor quality and dosage (Figure 4, Table S3). Regarding organ-specific response, leaves are particularly affected by various stress conditions. They belong to plant organs involved in carbon skeleton metabolism and photosynthetic energy capture; however, studies regarding the impact of stress on leaf tissues, contrary to roots, may be still underrepresented in certain aspects [39].
4.1.1. Chemical Treatment and UV Radiation
Data on the influence of chemical stimuli on plant transcriptomes come frequently from leaf studies. A variability of Arabidopsis leaf tissue transcriptomic responses under UV radiation, as well as under chemical treatments (antimycin A, 3-amino-1,2,4-triazole, methyl viologen and salicylic acid [SA]) was characterized by Berkowitz et al. [39]. The epidermis and mesophyll cells had a higher number of poorly expressed genes than the vasculature after all treatments (compare with Section 3.2.1 data). In those conditions, the tissue-specific genes ranged between 20 and 80% in number. The response patterns of those tissues to stress were complex and highly specific for each treatment. For example, antimycin A resulted in similar responses in all tissues studied; however, UV-affected genes were expressed mainly in the vasculature and epidermis. Genes for photosynthetic proteins were downregulated in all Arabidopsis tissues after 3-amino-1,2,4-triazole and SA treatments, while only the genes that encode the components of PSI and PSII were upregulated by methyl viologen, and UV radiation downregulated photosynthetic genes in the epidermis and upregulated them in the mesophyll. In Arabidopsis, UV can also affect the expression level of genes for proteins necessary for chlorophyll biogenesis, protein folding, oxidoreductase and ligase genes, and genes for glyceraldehyde-3-phosphate dehydrogenase in diverse extent between UV-A and UV-B treatments; however, participation of TFs in both radiation responses is notable. Tissues studied also have distinct mitochondrial responses to antimycin A [39], which affect the expression pattern of respiratory genes (including alternative oxidase), general oxidoreductase activity genes, glutathione transferase, as well as genes related to Ser/Thr kinase activity and export across the plasma membrane. On the contrary, SA treatment resulted in genes affected for polysaccharide and heme binding, lactoperoxidase activity, H2O2 scattering, Ca2+ binding and genes for ER response proteins (Figure 4, Table S3).
To protect themselves against the harmful action of UV-B on DNA, plants accumulate various compounds, including flavonol, anthocyanin, and proanthocyanidin. Flavonoids are economically important substances in fruits that are also beneficial for human health. Accordingly, Song et al. [95] observed that among the upregulated DEGs in V. corymbosum, genes for the phenylpropanoid and flavonoid biosynthetic pathways prevailed. Genes involved in plant hormone signal transduction were significantly enriched after 1 h, followed by genes involved in phenylpropanoid biosynthesis after 3 h, and by genes involved in the flavonoid anthocyanin pathway after 6 h of UV-B exposure. These results suggest that phytohormone-related genes may constitute the primary response to UV-B radiation. The altered expression of several genes involved in flavonoid biosynthesis was characterized. Genes involved in proanthocyanidin and flavanol biosynthesis, including the Pal1, 4CL2, CHS, CHI3, VcFSL and VcUFGT genes (for phenylalanine ammonia lyase, 4-coumarate CoA ligase, chalcone synthase, chalcone isomerase, flavanol synthase, and anthocyanidin 3-O-glucosyltransferase, respectively) were all upregulated under radiation, and their expression level lasted high after the 24h treatment. Arabidopsis genes essential for oxidoreductase activity, porphyrin metabolism, plastid organization, and carbohydrate metabolism regulation also responded to UV-B (Figure 4, Table S3), however, according to Dong et al. [96] transcriptional analysis, in UV-B response of Pachycladon cheesemanii, a native New Zealand species close to Arabidopsis, multiple genes active in the wound healing, regeneration, flavonoid biosynthesis are specifically enriched and the production of anthocyanins belong to the most important strategy of acquiring of tolerance to radiation. Nevertheless, common genes for UV-B response in Arabidopsis and P. cheesemanii included the ones for the aminoacid, vitamin, pigment, and secondary compound metabolism [96]. To explore the molecular mechanism underlying the increase in flavonoid biosynthesis under UV-B radiation, Song et al. [95] investigated the transcriptomes of Vaccinium corymbosum exposed to various dosages (1- 24 h) of UV-B radiation. Comparative analysis of the data obtained detected 16,899 DEGs between different treatments, while 806 common genes were differentially expressed in all samples tested. The highest number of DEGs appeared among plants exposed to UV-B treatment for 24 h.
Lettuce (Lactuca sativa) grown in greenhouse conditions usually contains a lover level of ascorbic acid (ASC) compared to plants grown in field conditions. ASC is an antioxidative nutrient for human growth, reproduction, and health. As the accumulation level of ASC is correlated with light intensity, lettuce plants were treated with low, intermediate, and high dose of UV-B to investigate its effect on the ASC level in plants. The subsequent transcriptomic analysis resulted in the identification of numerous DEGs within the low-dose and the high-dose radiation treated groups. Furthermore, the comparison of these two groups revealed 809 DEGs that overlapped between them, of which 351 genes were upregulated and 458 genes appeared downregulated; two MIOX genes (for myo-inositol oxygenase, important enzyme in myo-inositol pathway) genes were significantly upregulated within the high-dose UV-B treated variant. The expression of the MIOX gene was elevated 2-3 times by ASC in Arabidopsis. Genes such as APX and MDHAR that are related to the recycling of ASC, were also significantly upregulated in this variant [97,98]. In general, results of those studies suggest that expression of the MIOX, APX and MDHAR genes may contribute to the regulation of the ASC level induced by UV-B radiation. In this model, UV-B might not directly increase ASC synthesis, but rather indirectly - by affecting other biological pathways that lead to an increase in the ASC level in plant tissues.
Figure 4.
In silico functional profiling of differentially expressed Arabidopsis genes under various stress treatments. Counts of affected genes, experiment accession numbers, links to the Expression Atlas data and experiment descriptors (italicized, in quotes) were all shown (above each table). The experiment descriptors carry the information on the compared variants (treatment vs control conditions). For in silico analysis, treatment identifiers (‘UV-A radiation’, ‘UV-B radiation’, ‘antimycine A', ‘salicylic acid’, ‘drought’, ‘cold’, ‘heat’) were used for browsing of the Expression Atlas data under 'Biological conditions' option. The complete Arabidopsis data were taken from the Expression Atlas (https://www.ebi.ac.uk/gxa/home; [93]) and placed in Table S1. Next, the retrieved Arabidopsis Genome Initiative (AGI) locus codes, representing affected gene sets from the given experiment with wild-type Arabidopsis, were used as queries (organism: Arabidopsis thaliana) for the search of the most relevant driver functional terms (the functional profiling) in g:Profiler (https://biit.cs.ut.ee/gprofiler/gost; [94]). During GO: term enrichment analysis, terms were extracted and depicted in tables, together with their IDs and the source (BP- biological process; MF- molecular function; CC- cellular component). The counts for the unique and the common differentially expressed genes from various experiments (indicated by diverse colors) with plants treated with diverse UV-A, UV-B, antimycin A, salicylic acid and cold dosages were compared on DeepVenn diagrams (at the bottom or next to the g:Profiler tables). DeepVenn diagrams (circle area proportional to gene quantity) were generated using an online tool from the http://www.deepvenn.com/ Web page [99]. Colors on diagrams are the same as within compared datasets. All links were accessed January 9, 2024. (a) The data for UV and chemical/ hormone response; (b) The data for drought, heat, and cold stress response.
Figure 4.
In silico functional profiling of differentially expressed Arabidopsis genes under various stress treatments. Counts of affected genes, experiment accession numbers, links to the Expression Atlas data and experiment descriptors (italicized, in quotes) were all shown (above each table). The experiment descriptors carry the information on the compared variants (treatment vs control conditions). For in silico analysis, treatment identifiers (‘UV-A radiation’, ‘UV-B radiation’, ‘antimycine A', ‘salicylic acid’, ‘drought’, ‘cold’, ‘heat’) were used for browsing of the Expression Atlas data under 'Biological conditions' option. The complete Arabidopsis data were taken from the Expression Atlas (https://www.ebi.ac.uk/gxa/home; [93]) and placed in Table S1. Next, the retrieved Arabidopsis Genome Initiative (AGI) locus codes, representing affected gene sets from the given experiment with wild-type Arabidopsis, were used as queries (organism: Arabidopsis thaliana) for the search of the most relevant driver functional terms (the functional profiling) in g:Profiler (https://biit.cs.ut.ee/gprofiler/gost; [94]). During GO: term enrichment analysis, terms were extracted and depicted in tables, together with their IDs and the source (BP- biological process; MF- molecular function; CC- cellular component). The counts for the unique and the common differentially expressed genes from various experiments (indicated by diverse colors) with plants treated with diverse UV-A, UV-B, antimycin A, salicylic acid and cold dosages were compared on DeepVenn diagrams (at the bottom or next to the g:Profiler tables). DeepVenn diagrams (circle area proportional to gene quantity) were generated using an online tool from the http://www.deepvenn.com/ Web page [99]. Colors on diagrams are the same as within compared datasets. All links were accessed January 9, 2024. (a) The data for UV and chemical/ hormone response; (b) The data for drought, heat, and cold stress response.


4.1.2. Water Deficiency (drought)
To acclimate to water deficiencies, plants undergo several biochemical, physiological, and molecular (ABA-dependent or independent) responses, including, for example, stomatal limitation of gas exchange due to stomatal closure, which is a consequence of reduced osmotic pressure and decreased cell turgor [100] as well as multiple alterations in gene expression profiles. As drought belongs to environmental factors that affect most crop productivity, the characterization of those adjustments by omics studies would reveal drought mechanisms in crops that result in rational development of stress resistant cultivars [101]. Characterization of sweet potato (Ipomoea batatas) transcriptome in drought allowed for the detection of almost 73,636 unigenes and various genes for ABA (IbZEP, IbNCED, IbABA2 and IbAAO2), ethylene (IbACS, IbACO) and JA (IbLOX, IbAOS, IbOPR, IbACOX1I, IbACOX3, IbMFP2) biosynthesis, which appeared all upregulated. Those results indicate for the involvement of hormonal signaling under water deficit. Interestingly, those analyses also showed that genes associated with SA synthesis were not affected while among the main genes affected in drought, genes for some enzymes such as ABI phosphatase or Ca2+-ATPase were identified [102].
Analysis of the transcriptome of two drought-resistant chickpea (Cicer arietinum) cultivars under drought revealed a very few DEGs which were common for those lines. Interestingly, downregulations in the transcriptomes of both genotypes prevailed, showing detrimental drought impact on the transcriptomic level. Multiple genes for AP2-EREBP, bHLH, bZIP, C3H, MYB, WRKY or MADS TFs were involved in drought acclimation. Those TFs governed signaling regulation, secondary metabolism, or transition to the generative phase [103]. The transcriptomic response of Phoebe bournei, a Chinese wood species, to drought employed also numerous genes for TFs from 25 families, including WRKY, AP2, HLH, bZIP, CCAAT-binding, GATA zinc finger, SBP domain, TCP, Dof, GRF zinc finger, HD, and NAM TFs. Interestingly, AP2 domain containing TFs as well as some NAM TFs were preferentially expressed at 30 h and 45 h-long stress. Moreover, POD, SOD, and CAT genes were upregulated and genes necessary for plant-hormone signal transduction, MAPK signaling, phenylpropanoid biosynthesis, flavonoid biosynthesis, and starch and sucrose metabolism were significantly affected, especially after 15 d-long drought. In addition, genes for two light-harvesting complex I chlorophyll a/b binding proteins (LHCA1 and LHCA2), light-harvesting complex II chlorophyll a/b binding proteins (LHCB1, LHCB2, LHCB4) and porphyrin biosynthesis proteins were differentially expressed across treatments [104].
Another study [105] included a differential analysis of transcriptomes from two wheat (Triticum aestivum) varieties with contrasting drought sensitivities. During the comparative analysis of both transcriptomes, a significant difference was shown in gene expression in the drought resistant cultivar, which involved genes for the synthesis of secondary metabolites. Genes necessary for the drought acclimation included, among others, those coding important TFs (e.g., asMT, FT, AP2, ABA2, ARF6, WRKY6, AOS, LOX2). Furthermore, the recent investigation of genes and pathways involved in transcriptomic response of two rice (Oryza sativa) cultivars (stress-susceptible and resistant) in terminal drought revealed the general prevalence of downregulated genes across tested lines and organs [106]. Notably, the number of affected DEGs decreased in stress-resistant line. The differential expression of genes for NAC and ZIP TFs, ABA signaling, LEA proteins and proteins related with redox homeostasis played crucial roles in achieving of stress tolerance. Tyagi et al. [106] concluded that those genes are good candidates for the genetic improvement of drought tolerance in rice.
De novo sequencing of the transcriptome of Medicago falcata seedlings subjected to drought revealed almost 4,460 DEGs in response to 2h-long water deficiency; the upregulated DEGs prevailed. M. falcata housekeeping genes were expressed at a relatively high level, although with little variation. The signaling pathways of the hormone and Nod factors (interplay with legume symbiosis) were primarily enriched among DEGs. Genes for numerous TFs (e.g., NAC family) were also enriched and genes for ABA biosynthesis were upregulated, in contrast to genes for ABA catabolism. Also, genes for ABF TFs (e.g., ABF1), JA biosynthesis, ERFs, nucleic acid helicases and diverse genes for RNA polymerases and DNA repair proteins appeared upregulated. In contrast, gibberellin biogenesis genes were antagonistically expressed comparing to ABA-related genes, except for GID1 gene [107]. For the global analysis of transcriptome of another Fabaceae member, Glycine max, in the progressing drought, Transcriptogramer tool was employed [108]. Six to sixteen diverse functional categories were enriched among DEGs at 1-, 6- and 12 h-long drought, including cell division, cell cycle, cell wall organization, stress responses, hormone signaling, signal transduction, and regulation of gene expression. The participation of genes for Ca-binding proteins in the stress response was notable. Analyses of De Oliveira-Busatto et al. [108] indicated also for the presence of most observable responses in the first hour of drought action, including downregulations in DEGs for Fe metabolism and oxidative stress response and upregulated genes for chaperone binding/protein folding, helicase activity, nucleotide-binding site, leucine-rich repeat proteins, programmed cell death (PCD), proteasome, cell cycle, DNA metabolism, protein biosynthesis and RNA regulatory mechanisms. However, most functional categories were repressed after 6 h-long drought.
A transcriptomic reprogramming under recurrent dehydration and rehydration cycles in Ceratostigma plantagineum, a resurrection species, was studied [109]. DEGs were grouped into seven functional groups. The most abundant in functional terms was cluster with genes active during stress response, hormone signaling, aminoacid catabolism, sucrose and fatty acid biogenesis, energy and respiratory metabolism, protein modification and transport, and membrane organization. In silico analyses indicated downregulation of photosynthetic genes for light reactions and Calvin cycle proteins to decrease photooxidative damage during dehydration. Notably, among the multitude of genes induced in the initial stages of dehydration, the OXPHOS genes were distinctive, allowing the recovery of respiratory metabolism recovery. Under stress, numerous genes for RNA processing and regulation were enriched and genes for the ubiquitin proteasome system were significantly upregulated. Overall, data from Xu et al. [109] indicate the flexibility of primary and secondary metabolism in water shortage and rewatering, using, among others, an alternative respiratory pathway, the C3-CAM switch, and the GABA shunt.
Drought-affected Arabidopsis genes code various proteins for cell wall biogenesis, carbohydrate and heme binding, transmembrane proteins, water channels, detoxification processes, secondary metabolism, extracellular region, cell-cell junctions, and secretory activities (Figure 4, Table S3). Transcriptomic analysis of another representative of Brassicaceae- rapeseed (Brassica napus) revealed that most significantly enriched GO terms of the upregulated DEGs were related with the response to water deprivation, ABA signaling, osmotic stress, and other abiotic stimuli and lipid metabolism as well as cutin, suberin, and wax biogenesis, fatty acid degradation and secondary compound metabolism. On the contrary, downregulated DEGs were connected mainly with photosynthetic activities, porphyrin metabolism, carbon, and nitrogen metabolism [110].
Due to the growing trend of global warming and the simultaneous severe scarcity of water resources, discussed analyses can help develop new varieties of crops that are resistant to drought and will be able to grow in areas with limited water resources in the future [80]. In addition, the usage of stress-alleviating compounds may be also beneficial. For instance, the growth regulator 5-aminolevulinic acid (ALA) has been used to alleviate abiotic stress consequences, including attempts to mitigate drought in grapevine (Vitis vinifera), by the increase of anti-oxidative responses [111]. Transcriptomic and physiological analyses allowed to establish that chlorophyll metabolism and photosynthetic apparatus are primarily affected by ALA, which uses synergistic mechanisms to alleviate drought. Under drought, in the ALA presence, alterations in the expression patten of DEGs for chlorophyll synthesis (upregulated genes) and degradation (downregulated genes), Rubisco-related genes (upregulated) and photorespiratory DEGs (attenuated) played important roles that enable ALA to maintain cell homeostasis in the water scarcity.
4.1.3. Elevated Temperature (Heat Stress)
Elevated temperature affects cereal development and productivity, especially the development of male reproductive organs and the viability and maturation of pollen grains [112,113]. Heat stress leads to the increased lipid peroxidation and degradation, membrane damage, elevated amount of reactive oxygen species (ROS), and simultaneous decrease in ROS scavenger activity, leading to nucleic acid damage and consequently to apoptosis [114,115]. Asseng et al. [116] in their model tested the impact of growing temperature on wheat yield; when the average temperature increased by 2°C during the growing season, the possibility of a decrease in yield increased by up to 50%. It is possible to alleviate the consequences of heat treatment by chemical treatment of plants, for example, by endogenous melatonin. Chrysanthemum leaf transcriptomes in heat with or without additional supplementation with melatonin were compared by Xing et al. [117]. In general, heat treatment alone downregulated more genes than upregulated others. Double treatments (melatonin + heat) significantly increased the expression level of several DEGs. Interestingly, melatonin treatment regulated the expression pattern of the HSF and HSP genes, genes for starch and sucrose metabolism, signaling, and chlorophyll, flavonoid, carotenoid biosynthesis., and the level of various TFs, including MADS, MYB, NAC, TCP, WRKY, and bHLH (Figure 3).
The comparison of microspore transcriptomes in heat stress in two tomato (Solanum lycopersicum) genotypes with contrasting heat tolerance revealed among the upregulated DEGs at least 11 HSP genes, which encode cytosolic, mitochondrial, plastid, and ER HSPs. The increased expression of HSP genes suggests its key role in response to plant heat. Five APX genes (which expression products allow us to combat the increased ROS level more effective) were notably upregulated in maturing microspores [118].
Some transcriptomic reports focused on the root transcriptome in heat. The dynamics of the transcriptome in root hairs was studied by Valdés-López et al. [119] in soybean (Glycine max) plants subjected to elevated temperature at diverse timepoints. The authors identified 46,366 soybean genes expressed in all variants, as well as on average 1,849 and 3,091 DEGs in heated root hairs and stripped roots per given time point. Interestingly, a limited number of DEGs was common for all time points (465 genes only) within 10 functional modules regulated by a few TFs (HSF, AP2/EREBP, MADS-box and WIRKY). In general, heat affected the expression pattern of genes for soybean root metabolism, protein folding genes, chromatin remodeling, and lipid and ATP synthesis very efficiently.
Arabidopsis leaf transcriptome under various stress conditions (salinity, osmotic stress, and heat stress) was recently investigated by Sewelam et al. [120]. Of all these treatments, the elevated temperature appeared to have the most notable effect on transcriptomic profiles (with the dominant upregulated DEGs). Interestingly, only triple treatment reached the maximum DEG number. However, eight clusters contained responsive genes and selected conditions (osmotic and heat stress) acted antagonistically, while together they largely reprogrammed the gene expression pattern. The DEGs affected by heat covered multiple genes that encoded HSPs (11 genes induced, except HSFA1E, HSF3 and HSFA5), late embryogenesis abundant (LEA) proteins, receptor-like kinases (RLKs), glutathione S-transferases and WRKY TFs. Furthermore, heat stress affected Arabidopsis genes for carbohydrate binding, UDP-galactosyltransferases, membrane transporters, and PCD (Figure 4, Table S3). Heat and its combinations with other stressors induced various mitochondrial genes, presumably as a compensative response to excessive protein degradation, which was previously suggested [121]. However, heat also repressed several cell cycle genes, ribosomal protein genes (234 genes in the triple treatment) as well as genes involved in DNA synthesis and repair [120].
The transcriptomes in the preexisted leaves of rice under heat acclimation (after a shift from 30/25oC to 40/35oC [day/night]) were also studied [122]. Notably, after the thermal shift, a transient adjustment in metabolic gene transcript level in preexisted leaves before homoeostasis was reached within 1 day, which accompanied decline in the abundance of some proteins. At least 19,308 rice genes were retained for expression analysis by RNA-seq in three timepoints after transferring of plants to the heat treatment. 1,818 and 1,465 genes in preexisted leaves appeared differentially expressed after 2 and 6 h after the thermal transfer, respectively, and multiple DEGs overlapped between those timepoints. Numerous genes for primary metabolism and responding to abiotic stimuli, and for metabolite biosynthesis were affected, including only few photosynthetic/ respiratory genes were affected (genes for complex II subunits, external NAD(P)H dehydrogenase, uncoupling proteins, alternative oxidase, and some glycolytic enzymes).
The elevated temperature often acts simultaneously with the water deficit. A few recent studies focused on investigation of the impact of heat stress, drought, and the double treatment (heat plus drought) on total plant transcriptomes. The discussed responses in barley (Hordeum vulgare) flag leaves were recently studied by Mikołajczak et al. [123]. Heat alone resulted in a notably lower number of DEGs in three timepoints and across three size groups of flag leaves, than the drought; however, the double treatment engaged almost as much DEGs as drought stress. In medium-sized leaves, heat stress (similarly to drought) affected the higher gene number, irrespectively of stress duration. However, during longer heat and drought treatments, more DEGs were also notable for large-sized leaves. Gene sets assigned to various processes underlying the drought and heat response, including photosynthesis, the abscisic acid pathway, and lipid transport were identified. Investigated stressors affected especially expression level of LEA and HSP genes. Overall, Mikołajczak et al. [123] study provided novel insight into the molecular mechanisms of barley flag leaf that determine drought and heat response, as well as their co-occurrence. Furthermore, according to the Mahalingam et al. [124] study, the number of DEGs increased in barley heads (but not in flag leaves) in stress-tolerant genotype as heat progressed. On the contrary, in stress-sensitive genotype, DEGs declined in number in those conditions. Heat response engaged transporter protein genes, genes for ABA response as well as resulted in differential expression of LEA genes in stress-sensitive genotype; in contrast, genes for non-specific lipid transfer proteins and carbonate dehydratase activity were enriched in stress-tolerant genotype. Only prolonged drought resulted in the increased number of (mostly downregulated) affected genes irrespectively from the tissue or genotype. The combined treatment (heat with drought) resulted in the notable increase of DEG number only in stress-sensitive barley line, however alterations in DEG pattern were very distinct from the single treatments. In general, multiple genes for RNA metabolism and Hsp70 chaperone components (HsP70, ClpB and Hsp70-dependent nucleotide exchange factor) appeared hub genes for heat, drought and the double treatment in co-expression network analysis that can be useful for future attempts in engineering stress tolerance. At least 900 TFs aided transcriptional reprogramming in two lines of barley across all treatments. Another study [125] pointed out to higher number of DEGs affected in double (heat and drought) treatment of Lolium temulentum, which coded for proteins necessary for the transcriptional regulation, protein folding, cell cycle, organellar biogenesis, binding, transport, oxidoreductase, and antioxidant activity, and cellular signaling. Affected genes coded also for TFs from APETALA2/Ethylene Responsive Factor, NAC, WRKY, bHLH, MYB, and GATA families as well as multiple zinc finger-type proteins.
When heat stress was combined with the elevated CO2 level, the detrimental effect of heat was alleviated only by partial deregulation of primary and secondary metabolism genes. The transcriptome of flag leaves of durum wheat (Triticum durum) in heat, elevated CO2, and the combination of those stimuli was investigated. Their data covered almost 60,209 transcripts, of which 29% of messengers specifically responded in abundance to heat. Most DEGs coded proteins involved in stress response, nucleic acid metabolism, miscellaneous enzymes, and cellular signaling. Genes upregulated by CO2 were often downregulated by heat (they coded, inter alia, photosynthetic, and OXPHOS proteins, enzymes of lipid and amino acid metabolism and glutathione-ascorbate cycle, hormone signaling, nucleic acid metabolism, and transport proteins). However, heat upregulated surprisingly Rubisco subunits [126].
Chen and Li [127] study included Brachypodium distachyon leaf transcriptome analysis, showing DEGs participating in processes such as the spliceosome and PSI, and PSII biogenesis, or in protein folding. Notably, more than 43 upregulated genes coded machinery for alternative RNA splicing. According to those analyses, the upregulation of genes involved in alternative splicing indicates an increased ability of plant cells to perform alternative splicing in response to high temperatures to create alternative forms of proteins that can alleviate physiological consequences of heat exposure.
To describe thermotolerance and protective mechanisms against thermal stress in desert species, Obaid et al. [128] studied the transcriptome of Rhizya stricta, the evergreen shrub, at elevated temperature. DEGs were grouped into 32 and 38 groups for mature and apical leaves, respectively. Massive downregulations included, among others, genes for cyclin, cytochrome p450/secologanin synthase and U-box-containing proteins. On the contrary, the upregulated genes covered those for HSPs, chaperones, UDP-glycosyltransferase, aquaporins and transparent protein testa 12, indicating that thermotolerance in R. stricta leaves is controlled mainly by improving protein folding and preventing protein degradation. TFs putatively regulating expression of HSP genes under heat included HSFA, AP2-EREBP, and WRKY TFs (Figure 3). In general, numerous metabolites, including polyols, sugars, methionine, and phenolics were involved in the development of R. stricta thermotolerance.
4.1.4. Low Temperature
Similarly, to elevated temperature, also reduced temperature (e.g., cold treatment and freeze) can contribute to plant growth and development aberrations. The cold response covers, inter alia, the direct inhibition of metabolic reactions, and because of the limited osmosis, cell dehydration and the oxidative stress occur simultaneously with other lowered temperature effects. The formation of ice crystals under conditions of reaching the freezing point is the greatest thread, which results in protein degradation and mechanical damage. Most plants, however, can adapt to the cold and gain tolerance to ice formation in their cells by gradually being exposed to reduced (non-freezing) temperatures under cold acclimation process [129].
Analysis of Amur vine (Vitis amurensis) transcriptome under cold treatment allowed for the categorization of DEG products in diverse families, including signal transduction, transcription regulation, and alternative splicing. At least 38 major families of TFs involved in the regulation of cold response in V. amurensis were detected, including novel TFs (e.g., PLATZ, LIM, EIL, NIN-like, TUB, WHIRLY, and PcG). Regarding genes for the signal transduction, CIPK8, CXIP4, CDP, and CRK2 genes, whose expression products are responsible for Ca2+ binding, appeared upregulated. Notably, results of those analyses may further allow us to elucidate the mechanisms of cold tolerance [130].
Many early cold response genes encode TFs that induce the expression of genes for the subsequent stress response [92]. Cheng et al. [131] studied effects of the overexpression of MeTCP4 (a specific TF from cassava [Manihot esculenta]) in Arabidopsis plants during cold stress. The affected genes were classified as stress-responsive in all conditions tested, to TF activity and DNA binding in control, and to oxidoreductase, peroxidase, and antioxidative activity under cold stress. Du et al. [132] analyzed the transcriptome of Agropyron mongolicum seeds at different developmental stages. 191,204 unigenes were detected, which were classified into 25 functional groups. The response to cold of A. mongolicum engaged ABA receptors and increased the level of expression of genes for bZIP and NAC TFs. This results in the downregulation of target genes as cold tolerance increases. DEGs were assigned to 136 metabolic pathways, of which the majority were enriched in carbohydrate metabolism, hormonal and phosphatidylinositol signaling as well as to biogenesis of phenylpropanoids, flavonoids, stilbenoids, diaryloheptanoids, gingerols and isoflavonoids.
The cold treatment appeared detrimental also to Medicago. falcata seedling transcriptome (downregulated transcripts prevailed). The study of M. falcata stress responses focused on phytohormone and nodulation signaling, revealing some similarities with drought responses (Section 4.1.2), however, ABF1 and GID1 transcripts decreased in abundance. Interestingly, the GH3 auxin-responsive gene was intensely upregulated in cold (similarly to DIMI1, but contrary to DIMI2 and DIMI3, all of which encode important nodulation factors). On the contrary, the AUX transcripts encoding the auxin receptor decreased in abundance. Furthermore, at least 16 genes for MYB and 12 genes for NAC TFs were induced by cold, indicating their participation in the acquisition of cold tolerance [107].
Arabidopsis data for chilling response (contrary to sub-zero cold response) covered particularly a high number of affected genes; interestingly those two responses overlapped surprisingly little (Figure 4). Chilling engaged, inter alia, secondary metabolism (e.g., flavonoid biogenesis) genes and genes for transporter proteins. Instead, during sub-zero Arabidopsis acclimation, genes for proteins participating in hormone signaling, especially JA signaling and ABA receptors, were affected. In general, Arabidopsis response to cold stress involved, inter alia, genes for PSI biogenesis, carbohydrate, and secondary compound metabolism, O-glycosyl compound hydrolases, flavonoid biosynthesis, nitrate transporter activity, and pigment metabolism (Figure 4, Table S3). Interestingly, in P. cheesemanii, Arabidopsis close relative, the cold response employs more functional gene families than in Arabidopsis leaves, including the specifically affected genes for the glycosinolate metabolism [96]. However, in both Arabidopsis and P. cheesemanii, genes for wound-like, circadian clock as well as for flavonoid, trehalose, phenylpropanoid and oxylipin metabolism become important under the early cold response.
An important stage during cold acclimation is also the regulation of metabolic processes occurring in plant cell organelles. Naydenov et al. [133] investigated mitochondrial and nuclear transcriptomes of germinated wheat (Triticum aestivum) germ. Some genes encoding mitochondrial proteins, including Mn superoxide dismutase (SOD) and alternative oxidase (AOX), appeared upregulated; however, the level of expression of nuclear genes essential for mitochondrial biogenesis visibly decreased. This indicates mutual control of gene expression between mitochondrial and nuclear transcriptomes, executed by anterograde and retrograde signaling.
4.2. Plant Transcriptomic Response under Biotic Stress
To cope with biotic stress, plants have developed several defensive responses, many of which are precisely induced by the pathogen attack [100]. Plant cells contain plasma membrane receptors, the purpose of which is to recognize the pathogen associated molecular patterns (PAMPs). When a pathogen is recognized by a receptor, PAMP-triggered immunity (PTI) is initiated, which usually stops the infection before it spreads throughout the plant. However, due to the constant combat between pathogens and their victims, pathogens can neutralize PTI by secreting special proteins called effectors into the cytosol of the plant cell. Plants, in response to the effectors secreted by pathogens, developed the ability to detect microorganisms by recognizing a specific effector, which is effector-triggered immunity (ETI). This response consists of the production by the plant of intracellular receptors designed to recognize effector molecules produced by pathogens. The interaction between the receptor and the effector triggers a complex network of cell responses aimed at determining infection resistance [134].
Higher plants do not possess specialized defense cells, and to protect against pathogens, they use 'oxidative outbreak'. Furthermore, a high level of ROS is required to initiate the hypersensitive response (HR). This reaction belongs to PCD manifestations, which is supposed to limit the access of the pathogen to water and nutrients and is aimed at limiting the spread of the pathogen [135]. Details on genes affected under biotic stress (fundal and bacterial infections) described across high-throughput transcriptomic studies were given in Table S2 and the summary of the cellular functions governed by biotic stress affected genes in various plant species was presented on Figure 3.
4.2.1. Fungal Infections
Pathogenic fungi can be divided into biotrophic and necrotrophic fungi. Biotrophic fungi feed on living host tissue. Necrotrophic fungi kill the host and then, secreting special enzymes, ‘digest’ host tissues and feed on them. Numerous species of pathogenic fungi, depending on the developmental stage, behave as biotrophic or necrotrophic ones. Depending on the stage of fungal infection, different resistance signaling pathways can be activated. SA signaling is believed to be involved in resistance to biotrophic and hemibiotrophic pathogens (pathogens that are early biotrophs and necrotrophs later). JA and ethylene signaling appeared to be indispensable for necrotrophic resistance [136].
Recently, transcriptomic analyses broadened our knowledge on plant affected genes under attack of fungal pathogens. Analysis of the transcriptome of pumpkin leaves (Cucurbita moschata) infected with powdery mildew (Blumeria graminis) 24 and 48h after the onset of infection showed a downregulation of multiple DEGs including bHLH87, WRKY21, ERF014 (coding TFs for ethylene signaling) as well as the HSF and MLO3 genes (coding powdery mildew resistance). Genes for PSI and PSII subunits, oxygen-enhancing proteins, chlorophyll-binding proteins, and magnesium chelatase were additionally regulated. Upregulation of genes associated with foliar photosynthesis after 48 h of infection indicates for untouched photosynthetic activity during infection and associates with the appearance of initial fungal hyphae. The upregulation of genes associated with the photosynthesis was also shown in the case of fungal-infected wheat (Triticum aestivum) [137].
The transcriptome of wheat leaves infected with powdery mildew (Erysiphales species) as well as with striped rust (Puccinia striiformis) were compared with the one from sample not infected with any pathogen and among themselves [138]. In total, 186,632 unigenes were detected. Comparative analysis of powdery mildew- and with striped rust-infected samples showed a deregulation of numerous genes among three variants. The response to powdery mildew infection induced stronger changes in the gene expression pattern. Genes for phenylalanine metabolism, phenylpropanoid biosynthesis, alpha-linolenic acid metabolism, flavonoid biosynthesis, and phenylalanine, tyrosine and tryptophan biosynthesis were differentially regulated under powdery mildew infection. However, in the case of the attack by striped rust, genes involved in metabolic processes such as carbon binding in the photosynthesis, carotenoid synthesis, PS antenna protein synthesis, and ubiquinone biosynthesis were all upregulated. This indicates the use of differential gene expression patterns associated with defense mechanisms in response to various fungal infections [138].
Two transcriptome datasets of fungus-infected silk of maize (Zea mays) obtained from National Centre for Biotechnology Information-Sequence Read Archive (NCBI SRA) database were compared by Kumar et al. [139]. Each dataset consisted of results of RNA-seq analysis for leaves of maize infected by different fungal species belonging to Hypocreaceae (e.g., Trichoderma atroviride), Nectriaceae (e.g., Fusarium verticillioides, F. graminearum), and Ustilaginaceae (e.g., Ustilago maydis). Set A contained data for silk samples affected by F. graminearum and U. maydis, while set B contained data from samples infected by F. verticillioides and T. atroviride (every dataset also contained the control results). Analysis of such data resulted in the reveal of 14,694 and 14,808 DEGs between control data sets of A and B, respectively. Almost 4,519 upregulated and 5,125 downregulated genes were identified, however, only 21 genes were differentially expressed in the four different variants of fungal infection. Among these genes, the genes encoding the peroxidase (POD) enzyme that controls the lengthening of germ tube to protect maize kernels from fungal disease were upregulated. Strong expression of SKIP19 genes that influence and respond to biotic and abiotic stress conditions. Differential expression pattern was found for the OSM34 (osmotin-like protein) gene, which improves host defense and immune defense against stress, and the DUF26 gene for the receptor-like protein kinase subfamily (its domain plays crucial role in stress resistance and antifungal defense); those two genes were upregulated in F. verticillioides, F. graminearum, and U. maydis infected samples, however, they appeared downregulated in samples affected by T. atroviride. Genes affected between three, two, and after infection by a single fungal species were also revealed.
Identifying genes expressed during infection by various fungal species can help to find genes essential for defense against those microbes. As the global climate is altered, important agriculturally, plant species become gradually exposed also to increasing biotic stress. The findings discussed above would help develop plant cultivars that are highly resistant to fungal infections.
4.2.2. Bacterial Infections
Analysis of the transcriptome of rice plants (Oryza indica) infected with Xanthomonas oryzae revealed multiple DEGs; the upregulated ones were classified into 10 functional groups, among which genes coding proteins involved in processes such as signal transduction, carbohydrate metabolism, and transcription regulation were enriched. On the contrary, downregulated genes were assigned to 12 functional groups, among which the groups of genes coding for TFs and proteins involved in lipid catabolism, oxidative burst, and associated with the cell cycle or metabolic pathways were the most distinctive [140].
Analysis of the transcriptome of tomato plants (Solanum lycopersicon) infected with Clavibacter michiganensis allowed a better understanding of the response of the plant host to infection. Upregulated genes coded proteins that participate in processes such as protein phosphorylation, plant defense response, and hormonal signaling. The resistance gene analogues (RGAs) in response to C. michiganensis were upregulated, also including genes RLK genes. The expression of genes encoding TFs, such as WRKY, NAC, HSF, and CBP60, which belong to factors involved in the antibacterial plant response, was also elevated (Figure 3). In addition, an increase in the SA accumulation in tomato tissues due to bacterial infection was showed. Moreover, the use of exogenous SA resulted in the induction of genes for WRKY TFs. This suggests the appearance of gene regulation by SA in response to infection with C. michiganensis, resulting in improved quality of the plant immune response [141].
In turn, Lu et al. [142] investigated transcriptomes of rice (Oryza sativa) cultivars resistant and susceptible to infection with Xanthomonas oryzaena. This pathogen causes a cereal disease called bacterial leaf streak (BLS). Data analysis showed that, generally, more transcripts responded in abundance among the infection-susceptible cultivar. These analyses revealed elevated levels of expression of genes encoding proteins involved in phenylpropane metabolic pathways, phenylalanine metabolism, and flavonoid biosynthesis, which were closely related to resistance to plant diseases. Furthermore, the results of the study by Lu et al. [142] also suggested the participation of selected WRKY, NAC, MYB, and bHLH TFs in plant response to bacterial infection (Figure 3).
Recent research on resistant (IBL2353) and susceptible (Ohio88119) tomato (S. lycopersicum) cultivars infected by C. michiganensis presented new gene families in defense mechanisms and confirmed the important role of the WRKY family of TFs in the response to biotic stress. Comparison of transcriptomic data derived from resistant and non-resistant tomato cultivars affected by C. michiganensis revealed that 215 genes were upregulated and further 362 genes appeared downregulated among all stress variants (12 h- and 24 h-post-infection) in both genotypes, respectively. From the analyses of the GO and KEGG pathway of genes associated with resistance, almost 25 tomato genes were selected for further analysis. To compare the differences in those genes between susceptible and resistant genotypes, a heat map was created. Thanks to this, out of 25 genes coding proteins associated with the plant defense response, a WAKL20 gene (for the wall-associated receptor kinase similar to wall 20) was particularly characterized, due to its increased expression level in the IBL2353 cultivar. The WAKL20 protein is the only member of the WAKS subfamily that plays an important role in innate resistance to multiple pathogens. The virus-induced silencing of the WAKS20 gene in the IBL2353 tomato cultivar resulted in the appearance of susceptibility to C. michiganensis infection. These results seem to confirm an important role for the WAKS20 gene in the defense mechanism of plants and establish the foundation for further research into the search for new more resistant plant varieties [143].
5. Conclusions and Future Perspectives
Plant transcriptome can be dynamically shaped by many factors, functioning on diverse levels of biological diversity [81,91,93]. Understanding the composition of the transcriptome allows to recognize mechanisms and genes involved in acclimation to adverse environmental conditions to which plants have been subjected. In plant cells, in addition to the transcriptome being a product of nuclear genome expression, plastid and mitochondrial transcriptomes are also present, contributing to the overall dynamicity and complexity of plant transcriptomes [25,26,27,35,121]. Diverse nuclear and organellar genes are induced in various organs and under acclimation to numerous abiotic and biotic stressors [121,133]. In the current review, we focused mainly on such diversity of global plant transcriptomic responses to various abiotic (UV, chemical treatments, drought, heat, low temperature) and biotic (bacterial and fungal infections) stress conditions assayed by high throughput transcriptome sequencing studies. In addition, we characterized distinct tissue/organ-specific transcription patterns also analyzed by RNA-seq, including single cell RNA-seq approach and other current methodologies. For that, we focused mostly on the transcriptional biogenesis of vascular elements as well as on seed, leaf, and generative organ transcriptomes. We commented also on perspectives of application of biomedicinally important species in transcriptomic analyses.
Extensive research on the plant transcriptome is being developed at various levels. Transcriptomic data can be acquired both from differential expression studies on tissue/ organs or even from single cells, and modern RNA-seq approaches considerably extended our knowledge on transcriptome content and size. Owing Best et al. [35] study, more organellar transcriptomes for important crop species should be analyzed in the future that would allow for the better understanding of plastome and mitogenome responses in stress and their relevance in particular developmental steps. Also, more transcriptomic studies employing single cell RNA-seq coupled with microscopic analyzes are expected, in order (1) to broaden the spatio-temporal data of tissue/organ transcriptomes, (2) to explore diversity of transcriptomes in distinct cells contributing to the organ architectural integrity, and (3) to build complete expression datasets/ atlases for agriculturally important species in the future. Due to the underrepresentation of the transcriptomic data in stress for some plant organs, further studies are still expected [39]. In a future, clear distinguishing of tissues and tissue regions both by their physical location and investigating of gene co-expression patterns in transcriptomic analyses would be also necessary [76].
Higher plants, as complex organisms with specialized structures, show diverse and very dynamically changing gene expression patterns depending on the organ or conditions to which they are subjected, which leads to the metabolic flexibility [80,109]. In addition, stress response at the transcriptomic level is actively modulated depending on the stressor quality and its duration (for some stressors, like drought, affecting transcriptomic pattern mostly after a prolonged exposure [108]). In this review, transcriptomic data coming not only from Arabidopsis (Table S3), but also from diverse plant species with various potentials of agricultural applications were discussed. Notably, DEGs connected with the given stress responses in crop species varied from the Arabidopsis ones, employing diverse gene families or genes for diverse regulatory proteins; nevertheless, commonly regulated genes often covered the ones for the low-molecular compound biogenesis and for secondary metabolism enzymes [96].
Even similar environmental cues resulted in the diversity of gene responses, leading to the induction or upregulation of distinct genes. Transcriptomic stress responses engaged induction of both common genes for diverse stressors as well as specific gene modules, coding certain protein families active in selected treatments only. This was also evident in the discussed meta-analysis of Arabidopsis transcriptomic data (Figure 4), where only limited number of DEGs appeared common for diverse abiotic stress treatments; even multiple dosage of the given stressor resulted in a high variability of transcriptomic response. Some genes for the given hormone (e.g., SA) signaling were unaffected by certain treatments, for instance, by water deficit which appeared particularly detrimental also at the transcriptomic level [102,103,106]. Among affected transcripts, numerous mRNAs encoded for a plethora of transcription factors from diverse families (Figure 3). It seems that some most detrimental stressors (for instance high temperature treatments) employed particularly high number of transcription factors regulating gene expression [124].
Double stress treatments resulted in highly specific responses, in some cases increasing the number of affected DEGs [120,125] as much as the most detrimental stressor [123], indicating that still there is a need for multiple stressor analyses. Generally, stress-sensitive cultivars engage more DEGs in their stress responses, however, those patterns highly depend on actual tissue/organ stressed (Table S3) [124]. However, most of the transcriptomic data discussed here came from plant material cultivated under highly controlled laboratory conditions. Therefore, in our opinion, field experiments testing the variations of multiple stress responses (which act simultaneously on field cultivated plants) in plant nuclear and organellar transcriptomes will be continuously awaited. Additionally, plant responses on biotic stress indicated for the importance of plant-microorganism interactions which can overall influence stress tolerance [144], and this would be challenging point also for future transcriptional studies. Thus, impact of the plant microbiome on stress response on the transcriptional level should be investigated in more broadened context.
With the knowledge of individual gene profiles and mechanisms responsible, for example, for the synthesis of secondary metabolites, it would be possible to use them practically (1) during genetic engineering of medicinal plant species, (2) to investigate of processes engaged in the active compound biogenesis and the organogenesis, and (3) to understand better how metabolic pathways contribute to various stress acclimation strategies. This would also allow an exemplary manipulation of plant metabolic pathways in the future to obtain more secondary metabolites, which often depends on the developmental stage [83,85]. Last, but not least, understanding metabolic pathways and plant responses to unfavorable conditions, such as drought, reduced or elevated temperature, or pathogen infection, can contribute to the development of new more stress-resistant plant cultivars. Those pending actions will include modern methodological tools, including advanced genetic engineering and gene editing strategies to alleviate future crop losses due to suboptimal weather and the increased food demands in the future [81,145].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. this submission contains three Supplementary Tables: Table S1: Comparison of the selected, differently sized plant nuclear and organellar (plastid, mitochondrial) genomes, Table S2: Differentially expressed genes (DEGs) in studies on organ-/tissue- specific transcriptomes (lines 3-26) as well as plant transcriptomes under stress acclimation (lines 27-64), Table S3: Arabidopsis differentially expressed genes (DEGs) for various treatments and stress conditions.
Author Contributions
M.R. and M. S.: carried out the literature review; M.R.: conceptualization; M.R. and M.S.: writing—original draft preparation; M.S.: preparation of Figures 1-2 and Table S1; M.R.: preparation of Figures 3-4, and Tables S2-S3; M.R.: writing—review and editing; M.R.: supervision; M.R.: funding acquisition. All authors have read and agreed the submitted version of the manuscript. Authors agree to be personally accountable for their own contributions and for ensuring that questions related to the accuracy or integrity of any part of the work. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Excellent Initiative – Research University (ID-UB) program at the Adam Mickiewicz University, Poznań and KNOW RNA Research Center at Adam Mickiewicz University, Poznań, grant no. 01/KNOW2/2014.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Publicly available datasets (Tables S1 and S3) were analyzed in this study; those data can be found here: https://www.ncbi.nlm.nih.gov/datasets/genome/, https://www.ebi.ac.uk/gxa/home, https://biit.cs.ut.ee/gprofiler/gost,
Acknowledgments
We would like to thank colleagues from our Institute for the valuable remarks during the preparation of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| AAO | aldehyde oxidase |
| ABA | abscisic acid |
| ABF | ABSCISIC ACID RESPONSIVE ELEMENT-BINDING FACTOR |
| ACADM | medium-chain acyl-CoA dehydrogenase |
| ACO | 1-aminocyclopropane-1-carboxylic oxidase |
| ACOX | acyl-CoA oxidase |
| ACS | 1-aminocyclopropane-1-carboxylic synthase |
| ALA | 5-aminolevulinic acid |
| AOS | allene oxide synthase |
| AOX | alternative oxidase |
| AP | APETALA |
| APX | ascorbate peroxidase |
| ARF | auxin response factor |
| AS | anthocyanidin synthase |
| asMT | acetylserotonin O-methyltransferase |
| ASC | L-ascorbic acid |
| ATHB | small homeodomain-leucine zipper family |
| AUX | auxin receptor |
| bHLH | basic helix–loop–helix |
| BLS | bacterial leaf streak |
| bZIP | basic (region) leucine zipper |
| CAM | crassulacean acid metabolism |
| CAT | catalase |
| CBP | CALMODULIN-BINDING PROTEIN |
| CCOAOMT | caffeoyl-CoA O-methyltransferase |
| CDP | CAAT displacement protein (transcriptional repressor) |
| CCR | cinnamoyl-CoA:NADP reductase |
| C3H | zinc finger transcription factor |
| CHI | chalcone isomerase |
| CHS | chalcone synthase |
| C4H | 4-cinnamate hydroxylase |
| CIPK | CBL-interacting protein kinase |
| 4CL | 4-coumarate: CoA ligase |
| Clp | caseinolytic protease |
| CO | CONSTANS |
| CRK | cysteine-rich receptor-like kinase |
| CRY | cryptochrome |
| CXIP | CAX-interacting protein |
| DEGs | differentially-expressed genes |
| DFR | dihydroflavonol 4-reductase |
| DIMI | DIMINUTO |
| Dof | DNA-binding with one finger |
| DREB | dehydration-responsive element-binding |
| ECHS | short chain enoyl-CoA hydratase |
| EF | early flowering |
| EIL | ethylene insensitive-like |
| ER | endoplasmic reticulum |
| EREBP | ethylene-responsive element binding protein |
| ERF | ethylene response factor |
| ESTs | expressed sequence tags |
| ETI | effector-triggered immunity |
| F3H | flavanone-3-hydroxylase |
| FIE | fertilization-independent endospermia |
| FSL | flavonol synthase |
| FT | FLOWERING LOCUS T |
| FUL | FRUITFUL |
| FUS | fused in sarcoma |
| GABA | g- aminobutyric acid |
| GID | GA-INSENSITIVE DWARF |
| GRF | Growth Regulating Factor |
| HCT | hydroxycinnamoyl transferase |
| HD | homeodomain |
| HLH | helix-loop-helix |
| HR | hypersensitive response |
| HSF | heat shock factor |
| HSP | heat shock protein |
| IFL | INTERFASCICULAR FIBERLESS |
| JA | jasmonic acid |
| KAN | KANADI |
| LEA | late-embryogenesis abundant |
| LHC | light-harvesting complex |
| LEC | little elongation complex |
| LIM | homeobox transcription factor |
| LOX | lipoxygenase |
| MADS | MINICHROMOSOME MAINTENANCE 1/ AGAMOUS/ DEFICIENS/ SERUM RESPONSE FACTOR |
| MAPK | mitogen-assiociated protein kinase |
| MDHAR | monodehydroascorbate reductase |
| MEDEA | motif enrichment in differential elements of accessibility |
| MFP | multifunctional protein |
| MIOX | myo-inositol oxygenase |
| MLO | mildew locus o |
| MYB | myeloblastosis |
| NAC | no apical meristem/ ATAF1/ cup-shaped cotyledon |
| NAM | no apical meristem |
| NCED | 9-cis-epoxycarotenoid dioxygenase |
| NIN | nodule inception |
| OPR | 12-oxophytodienoic acid reductase |
| OSM | osmotin-like protein |
| OXPHOS | oxidative phosphorylation |
| PAAF | 23-dehydroadipyl-CoA hydratase |
| PAL | phenylalanine ammonia lyase |
| PAMP | pathogen-associated molecular pattern |
| PCD | programmed cell death |
| PcG | Polycomb group |
| PEAR | PHLOEM EARLY DNA-BINDING-WITH-ONE-FINGER |
| PEI | Cys3His zinc finger domain-containing protein |
| PHB | PHABULOSA |
| PHV | PHAVOLUTA |
| PHY | phytochrome |
| POD | peroxidase |
| PS | photosystem |
| PTI | PAMP-triggered immunity |
| PLATZ | plant AT-rich sequence and zinc-binding protein |
| REV | REVOLUTA |
| RGA | resistance gene analogue |
| RLK | receptor-like kinase |
| ROS | reactive oxidative species |
| SA | salicylic acid |
| SBP | SQUAMOSA-pROMOTER BINDING PROTEIN |
| SOC | suppressor of overexpression of constans |
| SOD | superoxide dismutase |
| STS | stachyose synthase |
| TCP | bHLH DNA-binding domain |
| TF | transcription factor |
| TUB | transcription factor family |
| UF3GT | UDP-glucose: flavonoid 3-O-glycosyltransferase |
| WAKL20 | wall-associated receptor kinase-like 2 |
| WGD | whole-genome duplication |
| WRKY | transcription factor family |
| VIN | VERNALIZATION INSENSITIVE |
| ZEP | zeaxanthin epoxidase |
| ZIP | leucine zipper |
References
- Kenrick, P.; Crane, P.R. The Origin and Early Diversification of Land Plants: A Cladistic Study; Smithsonian series in comparative evolutionary biology, 1st ed.; Smithsonian Institution Press: Washington, DC, USA, 1997. [Google Scholar]
- Forster, B.P.; Heberle-Bors, E.; Kasha, K.J.; Touraev, A. The Resurgence of Haploids in Higher Plants. Trend Plant Sci. 2007, 12, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.L.; Puttick, M.N.; Clark, J.W.; Edwards, D.; Kenrick, P.; Pressel, S.; Wellman, C.H.; Yang, Z.; Schneider, H.; Donoghue, P.C.J. The Timescale of Early Land Plant Evolution. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E2274–E2283. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; Prahalad, S.; Colbert, R. Integrative Genomics. Textbook of Pediatric Rheumatology, 7th ed.; Elsevier: Philadelphia, PA, USA, 2016; pp. 43–53.e3. [Google Scholar]
- Schliesky, S.; Gowik, U.; Weber, A.P.M.; Bräutigam, A. RNA-Seq Assembly – Are We There Yet? Front. Plant Sci. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Singh, D.; Mathur, S.; Singh, A.; Ranjan, R. Upcoming Progress of Transcriptomics Studies on Plants: An Overview. Front. Plant Sci. 2022, 13, 1030890. [Google Scholar] [CrossRef]
- Chen, C.; Ge, Y.; Lu, L. Opportunities and Challenges in the Application of Single-Cell and Spatial Transcriptomics in Plants. Front. Plant Sci. 2023, 14, 1185377. [Google Scholar] [CrossRef]
- Singh, S.; Parihar, P.; Singh, R.; Singh, V.P.; Prasad, S.M. Heavy Metal Tolerance in Plants: Role of Transcriptomics, Proteomics, Metabolomics, and Ionomics. Front. Plant Sci. 2016, 6, 1143. [Google Scholar] [CrossRef]
- Ahmad, M. Genomics and Transcriptomics to Protect Rice (Oryza sativa. L.) from Abiotic Stressors: -Pathways to Achieving Zero Hunger. Front. Plant Sci. 2022, 13, 1002596. [Google Scholar] [CrossRef]
- Bhat, K.A.; Mahajan, R.; Pakhtoon, M.M.; Urwat, U.; Bashir, Z.; Shah, A.A.; Agrawal, A.; Bhat, B.; Sofi, P.A.; Masi, A.; et al. Low Temperature Stress Tolerance: An Insight Into the Omics Approaches for Legume Crops. Front. Plant Sci. 2022, 13, 888710. [Google Scholar] [CrossRef] [PubMed]
- Kourani, M.; Mohareb, F.; Rezwan, F.I.; Anastasiadi, M.; Hammond, J.P. Genetic and Physiological Responses to Heat Stress in Brassica napus. Front. Plant Sci. 2022, 13, 832147. [Google Scholar] [CrossRef]
- Son, S.; Park, S.R. Plant Translational Reprogramming for Stress Resilience. Front. Plant Sci. 2023, 14, 1151587. [Google Scholar] [CrossRef]
- Tu, M.; Du, C.; Yu, B.; Wang, G.; Deng, Y.; Wang, Y.; Chen, M.; Chang, J.; Yang, G.; He, G.; et al. Current Advances in the Molecular Regulation of Abiotic Stress Tolerance in Sorghum via Transcriptomic, Proteomic, and Metabolomic Approaches. Front. Plant Sci. 2023, 14, 1147328. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Wang, Z.; Li, X.; Ai, Q.; Wong, D.C.J.; Zhang, F.; Yang, J.; Zhang, N.; Si, H. Current Perspectives of lncRNAs in Abiotic and Biotic Stress Tolerance in Plants. Front. Plant Sci. 2024, 14, 1334620. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.N.; Robin Buell, C. Tapping the Promise of Genomics in Species with Complex, Nonmodel Genomes. Annu. Rev. Plant Biol. 2013, 64, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Soltis, D.E.; Kersey, P.J.; Wegrzyn, J.L.; Leebens-Mack, J.H.; Gostel, M.R.; Liu, X.; Soltis, P.S. Green Plant Genomes: What We Know in an Era of Rapidly Expanding Opportunities. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2115640118. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Margulis, L. Origin of Eukaryotic Cells: Evidence and Research Implications for a Theory of the Origin and Evolution of Microbial, Plant, and Animal Cells on the Precambrian Earth, 1st ed.; Yale Univ. Press: New Haven, U.S.A, 1970. [Google Scholar]
- McFadden, G.I. Primary and Secondary Endosymbiosis and the Origin of Plastids. J. Phycol. 2001, 37, 951–959. [Google Scholar] [CrossRef]
- Rockwell, N.C.; Lagarias, J.C.; Bhattacharya, D. Primary Endosymbiosis and the Evolution of Light and Oxygen Sensing in Photosynthetic Eukaryotes. Front. Ecol. Evol. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Best, C.; Mizrahi, R.; Ostersetzer-Biran, O. Why so Complex? The Intricacy of Genome Structure and Gene Expression, Associated with Angiosperm Mitochondria, May Relate to the Regulation of Embryo Quiescence or Dormancy—Intrinsic Blocks to Early Plant Life. Plants 2020, 9, 598. [Google Scholar] [CrossRef]
- Dobrogojski, J.; Adamiec, M.; Luciński, R. The Chloroplast Genome: A Review. Acta Physiol. Plant. 2020, 42, 98. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast Microsatellites: New Tools for Studies in Plant Ecology and Evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.L.; Aaqil Khan, M.; Muhammad Imran, Q.; Kang, S.-M.; Al-Hosni, K.; Jeong, E.J.; Lee, K.E.; Lee, I.-J. Comparative Analysis of Complete Plastid Genomes from Wild Soybean (Glycine Soja) and Nine Other Glycine Species. PLoS ONE 2017, 12, e0182281. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast Genomes: Diversity, Evolution, and Applications in Genetic Engineering. Genome Biol 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Rurek, M. Participation of Non-Coding RNAs in Plant Organelle Biogenesis. Acta Biochim. Pol. 2016, 63, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Goldschmidt-Clermont, M. Participation of Nuclear Genes in Chloroplast Gene Expression. Biochimie 2000, 82, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.J.; Sowden, R.G.; Jarvis, P. Abiotic Stress-Induced Chloroplast Proteome Remodelling: A Mechanistic Overview. J. Exp. Bot. 2018, 69, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-S.; Cho, J.-H.; Im, J.-S.; Choi, J.-G.; Park, Y.-E.; Hong, S.-Y.; Kwon, M.; Kang, J.-H.; Park, T.-H. The Complete Mitochondrial Genome Sequences of Potato (Solanum tuberosum L., Solanaceae). Mitochondrial DNA Part B 2017, 2, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Logan, D.C. Plant Mitochondrial Dynamics. Biochim. Biophys. Acta - Molecular Cell Research 2006, 1763, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid Evolution of Enormous, Multichromosomal Genomes in Flowering Plant Mitochondria with Exceptionally High Mutation Rates. PLoS Biol 2012, 10, e1001241. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Keller, S.R.; Berardi, A.E.; Sanderson, B.J.; Karpovich, J.F.; Taylor, D.R. De novo Transcriptome Assembly and Polymorphism Detection in the Flowering Plant Silene vulgaris (Caryophyllaceae). Mol. Ecol. Res. 2012, 12, 333–343. [Google Scholar] [CrossRef]
- Imadi, S.R.; Kazi, A.G.; Ahanger, M.A.; Gucel, S.; Ahmad, P. Plant Transcriptomics and Responses to Environmental Stress: An Overview. J. Genet. 2015, 94, 525–537. [Google Scholar] [CrossRef]
- Schneider, G.F.; Dekker, C. DNA Sequencing with Nanopores. Nat. Biotechnol. 2012, 30, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Best, C.; Sultan, L.D.; Murik, O.; Ostersetzer, O. Insights into the Mitochondrial Transcriptome Landscapes of Two Brassicales Plant Species, Arabidopsis thaliana (var. Col-0) and Brassica oleracea (var. botrytis). Endocyt. Cell Res. 2020, 30, 16–38. [Google Scholar] [CrossRef]
- Cahoon, A.; Nauss, J.; Stanley, C.; Qureshi, A. Deep Transcriptome Sequencing of Two Green Algae, Chara vulgaris and Chlamydomonas reinhardtii, Provides No Evidence of Organellar RNA Editing. Genes 2017, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. The Review of Transcriptome Sequencing: Principles, History and Advances. IOP Conf. Ser.: Earth Environ. Sci. 2019, 332, 042003. [Google Scholar] [CrossRef]
- Booth, M.W.; Breed, M.F.; Kendrick, G.A.; Bayer, P.E.; Severn-Ellis, A.A.; Sinclair, E.A. Tissue-Specific Transcriptome Profiles Identify Functional Differences Key to Understanding Whole Plant Response to Life in Variable Salinity. Biology Open 2022, 11, bio059147. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, O.; Xu, Y.; Liew, L.C.; Wang, Y.; Zhu, Y.; Hurgobin, B.; Lewsey, M.G.; Whelan, J. RNA-seq Analysis of Laser Microdissected Arabidopsis Thaliana Leaf Epidermis, Mesophyll and Vasculature Defines Tissue-specific Transcriptional Responses to Multiple Stress Treatments. Plant J. 2021, 107, 938–955. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Z.; Dixon, R.A. On–Off Switches for Secondary Cell Wall Biosynthesis. Mol. Plant 2012, 5, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Růžička, K.; Ursache, R.; Hejátko, J.; Helariutta, Y. Xylem Development – from the Cradle to the Grave. New Phytol. 2015, 207, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Dinneny, J.R.; Yanofsky, M.F. Vascular Patterning: Xylem or Phloem? Curr. Biol. 2004, 14, R112–R114. [Google Scholar] [CrossRef]
- Eshed, Y.; Baum, S.F.; Perea, J.V.; Bowman, J.L. Establishment of Polarity in Lateral Organs of Plants. Curr. Biol. 2001, 11, 1251–1260. [Google Scholar] [CrossRef]
- Eshed, Y.; Izhaki, A.; Baum, S.F.; Floyd, S.K.; Bowman, J.L. Asymmetric Leaf Development and Blade Expansion in Arabidopsis Are Mediated by KANADI and YABBY Activities. Development 2004, 131, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Roszak, P.; Heo, J.; Blob, B.; Toyokura, K.; Sugiyama, Y.; De Luis Balaguer, M.A.; Lau, W.W.Y.; Hamey, F.; Cirrone, J.; Madej, E.; et al. Cell-by-Cell Dissection of Phloem Development Links a Maturation Gradient to Cell Specialization. Science 2021, 374, eaba5531. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, F.; Tang, Y.; Yuan, Y.; Guo, Q. Transcriptome Sequencing and Profiling of Expressed Genes in Phloem and Xylem of Ramie (Boehmeria nivea L. Gaud). PLoS ONE 2014, 9, e110623. [Google Scholar] [CrossRef] [PubMed]
- Kalckar, H.M. Galactose Metabolism and Cell “Sociology”: Galactose, One of the Freaks of Evolution, Furnishes a Simple Illustration of the Extravagances of Nature. Science 1965, 150, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Weber, H. Fatty Acid-Derived Signals in Plants. Trend Plant Sci. 2002, 7, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Lashbrook, C.C.; Cho, S.K.; Butler, N.M.; Sharma, P.; Muppirala, U.; Severin, A.J.; Hannapel, D.J. Transcriptional Analysis of Phloem-Associated Cells of Potato. BMC Genomics 2015, 16, 665. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Fujiwara, T.; Iki, T.; Kuroda, K.; Yamashita, K.; Tamura, M.; Fujisawa, Y.; Watanabe, A. Transcriptome Sequencing and Profiling of Expressed Genes in Cambial Zone and Differentiating Xylem of Japanese Cedar (Cryptomeria japonica). BMC Genomics 2014, 15, 5920. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Huang, X.; Deng, J.; Liu, H.; Liu, Y.; Yu, K.; Huang, B. Differential Gene Expression between Leaf and Rhizome in Atractylodes Lancea: A Comparative Transcriptome Analysis. Front. Plant Sci. 2016, 7, 348. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.J.; Furtado, A.; Marquardt, A.; Hodgson-Kratky, K.; Hoang, N.V.; Botha, F.C.; Papa, G.; Mortimer, J.C.; Simmons, B.; Henry, R.J. Variation in Sugarcane Biomass Composition and Enzymatic Saccharification of Leaves, Internodes and Roots. Biotechnol. Biofuels 2020, 13, 201. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K. O2 and Other High-Energy Molecules in Photosynthesis: Why Plants Need Two Photosystems. Life 2021, 11, 1191. [Google Scholar] [CrossRef]
- Denyer, T.; Ma, X.; Klesen, S.; Scacchi, E.; Nieselt, K.; Timmermans, M.C.P. Spatiotemporal Developmental Trajectories in the Arabidopsis Root Revealed Using High-Throughput Single-Cell RNA Sequencing. Dev. Cell 2019, 48, 840–852.e5. [Google Scholar] [CrossRef] [PubMed]
- Shulse, C.N.; Cole, B.J.; Ciobanu, D.; Lin, J.; Yoshinaga, Y.; Gouran, M.; Turco, G.M.; Zhu, Y.; O’Malley, R.C.; Brady, S.M. High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types. Cell Rep. 2019, 27, 2241–2247.e4. [Google Scholar] [CrossRef] [PubMed]
- Efroni, I.; Mello, A.; Nawy, T.; Ip, P.-L.; Rahni, R.; DelRose, N.; Powers, A.; Satija, R.; Birnbaum, K.D. Root Regeneration Triggers an Embryo-like Sequence Guided by Hormonal Interactions. Cell 2016, 165, 1721–1733. [Google Scholar] [CrossRef] [PubMed]
- Kortz, A.; Hochholdinger, F.; Yu, P. Cell Type-Specific Transcriptomics of Lateral Root Formation and Plasticity. Front. Plant Sci. 2019, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Tenorio Berrío, R.; Verstaen, K.; Vandamme, N.; Pevernagie, J.; Achon, I.; Van Duyse, J.; Van Isterdael, G.; Saeys, Y.; De Veylder, L.; Inzé, D.; et al. Single-Cell Transcriptomics Sheds Light on the Identity and Metabolism of Developing Leaf Cells. Plant Physiol. 2022, 188, 898–918. [Google Scholar] [CrossRef] [PubMed]
- Breeze, E.; Harrison, E.; McHattie, S.; Hughes, L.; Hickman, R.; Hill, C.; Kiddle, S.; Kim, Y.; Penfold, C.A.; Jenkins, D.; et al. High-Resolution Temporal Profiling of Transcripts during Arabidopsis Leaf Senescence Reveals a Distinct Chronology of Processes and Regulation. Plant Cell 2011, 23, 873–894. [Google Scholar] [CrossRef] [PubMed]
- Chrobok, D.; Law, S.R.; Brouwer, B.; Lindén, P.; Ziolkowska, A.; Liebsch, D.; Narsai, R.; Szal, B.; Moritz, T.; Rouhier, N.; et al. Dissecting the Metabolic Role of Mitochondria during Developmental Leaf Senescence. Plant Physiol. 2016, 172, 2132–2153. [Google Scholar] [CrossRef]
- Mason, P.J.; Hoang, N.V.; Botha, F.C.; Furtado, A.; Marquardt, A.; Henry, R.J. Comparison of the Root, Leaf and Internode Transcriptomes in Sugarcane (Saccharum Spp. Hybrids). Curr. Res. Biotech. 2022, 4, 167–178. [Google Scholar] [CrossRef]
- Li, P.; Ponnala, L.; Gandotra, N.; Wang, L.; Si, Y.; Tausta, S.L.; Kebrom, T.H.; Provart, N.; Patel, R.; Myers, C.R.; et al. The Developmental Dynamics of the Maize Leaf Transcriptome. Nat. Genet. 2010, 42, 1060–1067. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Liu, L.-L.; Huang, J.-Q.; Wang, Z.-J.; Chen, F.-F.; Zhang, Q.-X.; Zheng, B.-S.; Chen, M. Use of Transcriptome Sequencing to Understand the Pistillate Flowering in Hickory (Carya cathayensis Sarg.). BMC Genomics 2013, 14, 691. [Google Scholar] [CrossRef]
- Melzer, S.; Lens, F.; Gennen, J.; Vanneste, S.; Rohde, A.; Beeckman, T. Flowering-Time Genes Modulate Meristem Determinacy and Growth Form in Arabidopsis thaliana. Nat. Genet. 2008, 40, 1489–1492. [Google Scholar] [CrossRef] [PubMed]
- Mutasa-Gottgens, E.; Hedden, P. Gibberellin as a Factor in Floral Regulatory Networks. J. Exp. Bot. 2009, 60, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, A.; Schmid, M. Regulation of Flowering Time: All Roads Lead to Rome. Cell. Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Doyle, M.R.; Sung, S.; Amasino, R.M. Vernalization: Winter and the Timing of Flowering in Plants. Annu. Rev. Cell Dev. Biol. 2009, 25, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Feng, S.; Pan, Y.; Zhong, J.; Chen, Y.; Yuan, C.; Li, H. Transcriptome Analysis and Identification of Genes Associated with Floral Transition and Flower Development in Sugar Apple (Annona squamosa L.). Front. Plant Sci. 2016, 7, 1695. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Hu, Q.; Zhu, Y.; Zhu, L.; Ma, T.; Zeng, H.; Zang, Q.; Li, X.; Lin, X. Comparative Transcriptomic Analysis of the Flower Induction and Development of the Lei Bamboo (Phyllostachys violascens). BMC Bioinformatics 2019, 20, 687. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.R.; Roalson, E.H. Comparative Transcriptome Analyses of Flower Development in Four Species of Achimenes (Gesneriaceae). BMC Genomics 2017, 18, 240. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yuan, J.; Wu, M.; Han, Z.; Li, L.; Jiang, H.; Jia, Y.; Han, X.; Liu, M.; Sun, D.; et al. Transcriptome and DNA Methylome Reveal Insights into Yield Heterosis in the Curds of Broccoli (Brassica oleracea L var. italica). BMC Plant Biol. 2018, 18, 168. [Google Scholar] [CrossRef] [PubMed]
- Boesewinkel, F.D.; Bouman, F. The Seed: Structure. In Embryology of Angiosperms, 1st ed.; Johri, B.M., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, Germany, 1984; pp. 567–610. [Google Scholar]
- Martín-Gómez, J.J.; Gutiérrez del Pozo, D.; Ucchesu, M.; Bacchetta, G.; Cabello Sáenz de Santamaría, F.; Tocino, Á.; Cervantes, E. Seed Morphology in the Vitaceae Based on Geometric Models. Agronomy 2020, 10, 739. [Google Scholar] [CrossRef]
- Peirats-Llobet, M.; Yi, C.; Liew, L.C.; Berkowitz, O.; Narsai, R.; Lewsey, M.G.; Whelan, J. Spatially Resolved Transcriptomic Analysis of the Germinating Barley Grain. Nucl. Acids Res. 2023, 51, 7798–7819. [Google Scholar] [CrossRef]
- Zhu, M.; Taylor, I.W.; Benfey, P.N. Single-Cell Genomics Revolutionizes Plant Development Studies across Scales. Development 2022, 149, dev200179. [Google Scholar] [CrossRef] [PubMed]
- Betts, N.S.; Berkowitz, O.; Liu, R.; Collins, H.M.; Skadhauge, B.; Dockter, C.; Burton, R.A.; Whelan, J.; Fincher, G.B. Isolation of Tissues and Preservation of RNA from Intact, Germinated Barley Grain. Plant J. 2017, 91, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Le, B.H.; Cheng, C.; Bui, A.Q.; Wagmaister, J.A.; Henry, K.F.; Pelletier, J.; Kwong, L.; Belmonte, M.; Kirkbride, R.; Horvath, S.; et al. Global Analysis of Gene Activity during Arabidopsis Seed Development and Identification of Seed-Specific Transcription Factors. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 8063–8070. [Google Scholar] [CrossRef] [PubMed]
- Narsai, R.; Gouil, Q.; Secco, D.; Srivastava, A.; Karpievitch, Y.V.; Liew, L.C.; Lister, R.; Lewsey, M.G.; Whelan, J. Extensive Transcriptomic and Epigenomic Remodelling Occurs during Arabidopsis thaliana Germination. Genome Biol. 2017, 18, 172. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lu, J.; Xing, J.; Du, M.; Wang, M.; Zhang, L.; Li, Y.; Zhang, C.; Wu, Y. Transcriptome and Metabolome Analyses Revealing the Potential Mechanism of Seed Germination in Polygonatum cyrtonema. Sci. Rep. 2021, 11, 12161. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, N.; Li, W.; Gao, X.; Cha, M.; Qin, L.; Liu, L. Advances in Transcriptomics in the Response to Stress in Plants. Global Med. Genet. 2020, 07, 030–034. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huang, Z.; Sun, J.; Cui, X.; Liu, Y. Research Progress and Future Development Trends in Medicinal Plant Transcriptomics. Front. Plant Sci. 2021, 12, 691838. [Google Scholar] [CrossRef] [PubMed]
- Choudhri, P.; Rani, M.; Sangwan, R.S.; Kumar, R.; Kumar, A.; Chhokar, V. De Novo Sequencing, Assembly and Characterisation of Aloe Vera Transcriptome and Analysis of Expression Profiles of Genes Related to Saponin and Anthraquinone Metabolism. BMC Genomics 2018, 19, 427. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Singh, G.; Bhandawat, A.; Singh, G.; Parmar, R.; Seth, R.; Sharma, R.K. Spatial Transcriptome Analysis Provides Insights of Key Gene(s) Involved in Steroidal Saponin Biosynthesis in Medicinally Important Herb Trillium govanianum. Sci. Rep. 2017, 7, 45295. [Google Scholar] [CrossRef]
- Liao, W.; Mei, Z.; Miao, L.; Liu, P.; Gao, R. Comparative Transcriptome Analysis of Root, Stem, and Leaf Tissues of Entada Phaseoloides Reveals Potential Genes Involved in Triterpenoid Saponin Biosynthesis. BMC Genomics 2020, 21, 639. [Google Scholar] [CrossRef]
- Yan, J.; Qian, L.; Zhu, W.; Qiu, J.; Lu, Q.; Wang, X.; Wu, Q.; Ruan, S.; Huang, Y. Integrated Analysis of the Transcriptome and Metabolome of Purple and Green Leaves of Tetrastigma hemsleyanum Reveals Gene Expression Patterns Involved in Anthocyanin Biosynthesis. PLoS ONE 2020, 15, e0230154. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Pu, T.; Gui, C.; Zhang, X.; Gong, L. Transcriptome Analysis Reveals Biosynthesis of Important Bioactive Constituents and Mechanism of Stem Formation of Dendrobium huoshanense. Sci. Rep. 2020, 10, 2857. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Peng, D.; Zhu, J.; Zhao, D.; Shi, Y.; Zhang, S.; Ma, K.; Wu, J.; Huang, L. Transcriptome Analysis of Polygonatum cyrtonema Hua: Identification of Genes Involved in Polysaccharide Biosynthesis. Plant Methods 2019, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Tan, H.; Li, S.; Cheung, F.; Wang, N.; Nagamatsu, T.; Feng, Y. Cancer Stem Cells: The Potential Targets of Chinese Medicines and Their Active Compounds. Int. J. Mol. Sci. 2016, 17, 893. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, C.; Li, T.; Yuan, Z.; Zhou, H.; Tamada, Y.; Zhao, Y.; Wang, J.; Zheng, Q.; Hao, X.; et al. Global Transcriptome Analysis Reveals Dynamic Gene Expression Profiling and Provides Insights into Biosynthesis of Resveratrol and Anthraquinones in a Medicinal Plant Polygonum cuspidatum. Ind. Crops and Prod. 2021, 171, 113919. [Google Scholar] [CrossRef]
- Umar, O.; Ranti, L.; Abdulbaki, A.; Bola, A.; Abdulhamid, A.; Biola, M. Stresses in Plants: Biotic and Abiotic. In Current Trends in Wheat Research; ed. Mahmood-ur-Rahman Ansari, Government College University, Faisalabad: IntechOpen 2021. [CrossRef]
- Zhu, J.-K. Salt and Drought Stress Signal Transduction in Plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Gull, A.; Ahmad Lone, A.; Ul Islam Wani, N. Biotic and Abiotic Stresses in Plants. In Abiotic and Biotic Stress in Plants, 1st ed.; Bosco de Oliveira, A., Ed.; IntechOpen, 2019.
- EMBL-EBI. Expression Atlas. Available online: https://www.ebi.ac.uk/gxa/home (accessed on 9 January 2024). Gene expression across species and biological conditions.
- ELIXIR Recommended Interoperability Resource. g:Profiler. Available online: https://biit.cs.ut.ee/gprofiler/gost (accessed on 9 January 2024).
- Song, Y.; Ma, B.; Guo, Q.; Zhou, L.; Lv, C.; Liu, X.; Wang, J.; Zhou, X.; Zhang, C. UV-B Induces the Expression of Flavonoid Biosynthetic Pathways in Blueberry (Vaccinium corymbosum) Calli. Front. Plant Sci. 2022, 13, 1079087. [Google Scholar] [CrossRef]
- Dong, Y.; Gupta, S.; Wargent, J.J.; Putterill, J.; Macknight, R.C.; Gechev, T.S.; Mueller-Roeber, B.; Dijkwel, P.P. Comparative Transcriptomics of Multi-Stress Responses in Pachycladon cheesemanii and Arabidopsis thaliana. Int. J. Mol. Sci. 2023, 24, 11323. [Google Scholar] [CrossRef]
- Lorence, A.; Chevone, B.I.; Mendes, P.; Nessler, C.L. Myo -Inositol Oxygenase Offers a Possible Entry Point into Plant Ascorbate Biosynthesis. Plant Physiol. 2004, 134, 1200–1205. [Google Scholar] [CrossRef]
- Zhou, H.; Yu, L.; Liu, S.; Zhu, A.; Yang, Y.; Chen, C.; Yang, A.; Liu, L.; Yu, F. Transcriptome Comparison Analyses in UV-B Induced AsA Accumulation of Lactuca sativa L. BMC Genomics 2023, 24, 61. [Google Scholar] [CrossRef]
- DeepVenn - Create Area-Proportional Venn Diagrams Using the Deep Learning Framework Tensorflow. Available online: http://www.deepvenn.com/ (accessed on 8 01 2024).
- Verma, S.; Nizam, S.; Verma, P.K. Biotic and Abiotic Stress Signaling in Plants. In Stress Signaling in Plants: Genomics and Proteomics Perspective, Volume 1, 1st ed.; Sarwat, M., Ahmad, A., Abdin, M., Eds.; Springer New York: New York, NY, U.S.A, 2013; pp. 25–49. [Google Scholar]
- Shanker, A.K.; Maheswari, M.; Yadav, S.K.; Desai, S.; Bhanu, D.; Attal, N.B.; Venkateswarlu, B. Drought Stress Responses in Crops. Funct. Integr. Genomics 2014, 14, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhou, Y.; Zhai, H.; He, S.; Zhao, N.; Liu, Q. Transcriptome Profiling Reveals Insights into the Molecular Mechanism of Drought Tolerance in Sweetpotato. J. Integr. Agri. 2019, 18, 9–23. [Google Scholar] [CrossRef]
- Kumar, M.; Chauhan, A.S.; Kumar, M.; Yusuf, M.A.; Sanyal, I.; Chauhan, P.S. Transcriptome Sequencing of Chickpea (Cicer arietinum L.) Genotypes for Identification of Drought-Responsive Genes Under Drought Stress Condition. Plant Mol. Biol. Rep. 2019, 37, 186–203. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Sun, S.; Li, Y.; Jia, L.; Ye, S.; Yu, Y.; Dossa, K.; Luan, Y. Leaf-Transcriptome Profiles of Phoebe bournei Provide Insights into Temporal Drought Stress Responses. Front. Plant Sci. 2022, 13, 1010314. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Gunapati, S.; Kianian, S.F.; Singh, S.P. Comparative Analysis of Transcriptome in Two Wheat Genotypes with Contrasting Levels of Drought Tolerance. Protoplasma 2018, 255, 1487–1504. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Kumar, S.; Mohapatra, T. Biochemical, Physiological and Molecular Responses of Rice to Terminal Drought Stress: Transcriptome Profiling of Leaf and Root Reveals the Key Stress-Responsive Genes. J. Plant Biochem. Biotechnol. 2023. [Google Scholar] [CrossRef]
- Miao, Z.; Xu, W.; Li, D.; Hu, X.; Liu, J.; Zhang, R.; Tong, Z.; Dong, J.; Su, Z.; Zhang, L.; et al. De Novo Transcriptome Analysis of Medicago falcata Reveals Novel Insights about the Mechanisms Underlying Abiotic Stress-Responsive Pathway. BMC Genomics 2015, 16, 818. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira-Busatto, L.A.; De Almeida, R.M.C.; Weber, R.L.M.; Favero, D.; Bredemeier, C.; Da Silva Giordano, C.P.; Bodanese-Zanettini, M.H. The Soybean Transcriptogram Allows a Wide Genome-to-Single-Gene Analysis That Evinces Time-Dependent Drought Response. Plant Mol. Biol. Rep. 2022, 40, 1–27. [Google Scholar] [CrossRef]
- Xu, X.; Legay, S.; Sergeant, K.; Zorzan, S.; Leclercq, C.C.; Charton, S.; Giarola, V.; Liu, X.; Challabathula, D.; Renaut, J.; et al. Molecular Insights into Plant Desiccation Tolerance: Transcriptomics, Proteomics and Targeted Metabolite Profiling in Craterostigma plantagineum. Plant J. 2021, 107, 377–398. [Google Scholar] [CrossRef]
- Fang, S.; Zhao, P.; Tan, Z.; Peng, Y.; Xu, L.; Jin, Y.; Wei, F.; Guo, L.; Yao, X. Combining Physio-Biochemical Characterization and Transcriptome Analysis Reveal the Responses to Varying Degrees of Drought Stress in Brassica napus L. Int. J. Mol. Sci. 2022, 23, 8555. [Google Scholar] [CrossRef]
- Yang, Y.; Xia, J.; Fang, X.; Jia, H.; Wang, X.; Lin, Y.; Liu, S.; Ge, M.; Pu, Y.; Fang, J.; et al. Drought Stress in ‘Shine Muscat’ Grapevine: Consequences and a Novel Mitigation Strategy–5-Aminolevulinic Acid. Front. Plant Sci. 2023, 14, 1129114. [Google Scholar] [CrossRef] [PubMed]
- Young, L.W.; Wilen, R.W.; Bonham-Smith, P.C. High Temperature Stress of Brassica napus during Flowering Reduces Micro- and Megagametophyte Fertility, Induces Fruit Abortion, and Disrupts Seed Production. J. Exp. Bot. 2004, 55, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Taohua, Z.; Gui, W.; Xu, L.; Li, J.; Ding, Y. Five Pectinase Gene Expressions Highly Responding to Heat Stress in Rice Floral Organs Revealed by RNA-Seq Analysis. Biochem. Biophys. Res. Commun. 2015, 463, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Bita, C.E.; Gerats, T. Plant Tolerance to High Temperature in a Changing Environment: Scientific Fundamentals and Production of Heat Stress-Tolerant Crops. Front. Plant Sci. 2013, 4, 273. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Lu, X.; Zeng, H.; Zhang, Y.; Zhu, J. Heat Stress Induction of miR398 Triggers a Regulatory Loop That Is Critical for Thermotolerance in Arabidopsis. Plant J. 2013, 74, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Asseng, S.; Foster, I.; Turner, N.C. The Impact of Temperature Variability on Wheat Yields. Global Change Biology 2011, 17, 997–1012. [Google Scholar] [CrossRef]
- Xing, X.; Ding, Y.; Jin, J.; Song, A.; Chen, S.; Chen, F.; Fang, W.; Jiang, J. Physiological and Transcripts Analyses Reveal the Mechanism by Which Melatonin Alleviates Heat Stress in Chrysanthemum Seedlings. Front. Plant Sci. 2021, 12, 673236. [Google Scholar] [CrossRef] [PubMed]
- Frank, G.; Pressman, E.; Ophir, R.; Althan, L.; Shaked, R.; Freedman, M.; Shen, S.; Firon, N. Transcriptional Profiling of Maturing Tomato (Solanum Lycopersicum L.) Microspores Reveals the Involvement of Heat Shock Proteins, ROS Scavengers, Hormones, and Sugars in the Heat Stress Response. J. Exp.Bot. 2009, 60, 3891–3908. [Google Scholar] [CrossRef]
- Valdés-López, O.; Batek, J.; Gomez-Hernandez, N.; Nguyen, C.T.; Isidra-Arellano, M.C.; Zhang, N.; Joshi, T.; Xu, D.; Hixson, K.K.; Weitz, K.K.; et al. Soybean Roots Grown under Heat Stress Show Global Changes in Their Transcriptional and Proteomic Profiles. Front. Plant Sci. 2016, 7, 517. [Google Scholar] [CrossRef]
- Sewelam, N.; Brilhaus, D.; Bräutigam, A.; Alseekh, S.; Fernie, A.R.; Maurino, V.G. Molecular Plant Responses to Combined Abiotic Stresses Put a Spotlight on Unknown and Abundant Genes. J. Exp. Bot. 2020, 71, 5098–5112. [Google Scholar] [CrossRef]
- Rurek, M.; Czołpińska, M.; Pawłowski, T.; Krzesiński, W.; Spiżewski, T. Cold and Heat Stress Diversely Alter Both Cauliflower Respiration and Distinct Mitochondrial Proteins Including OXPHOS Components and Matrix Enzymes. Int. J. Mol. Sci. 2018, 19, 877. [Google Scholar] [CrossRef]
- Rashid, F.A.A.; Crisp, P.A.; Zhang, Y.; Berkowitz, O.; Pogson, B.J.; Day, D.A.; Masle, J.; Dewar, R.C.; Whelan, J.; Atkin, O.K.; et al. Molecular and Physiological Responses during Thermal Acclimation of Leaf Photosynthesis and Respiration in Rice. Plant Cell Environ. 2020, 43, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Mikołajczak, K.; Kuczyńska, A.; Krajewski, P.; Kempa, M.; Nuc, M. Transcriptome Profiling Disclosed the Effect of Single and Combined Drought and Heat Stress on Reprogramming of Genes Expression in Barley Flag Leaf. Front. Plant Sci. 2023, 13, 1096685. [Google Scholar] [CrossRef]
- Mahalingam, R.; Duhan, N.; Kaundal, R.; Smertenko, A.; Nazarov, T.; Bregitzer, P. Heat and Drought Induced Transcriptomic Changes in Barley Varieties with Contrasting Stress Response Phenotypes. Front. Plant Sci. 2022, 13, 1066421. [Google Scholar] [CrossRef]
- Martin, R.C.; Kronmiller, B.A.; Dombrowski, J.E. Transcriptome Analysis of Lolium temulentum Exposed to a Combination of Drought and Heat Stress. Plants 2021, 10, 2247. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Bolger, A.M.; Martínez-Carrasco, R.; Pérez, P.; Gutiérrez, E.; Usadel, B.; Morcuende, R. De Novo Transcriptome Analysis of Durum Wheat Flag Leaves Provides New Insights Into the Regulatory Response to Elevated CO2 and High Temperature. Front. Plant Sci. 2019, 10, 1605. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, H. Heat Stress Regulates the Expression of Genes at Transcriptional and Post-Transcriptional Levels, Revealed by RNA-Seq in Brachypodium distachyon. Front. Plant Sci. 2017, 7, 2067. [Google Scholar] [CrossRef]
- Obaid, A.Y.; Sabir, J.S.M.; Atef, A.; Liu, X.; Edris, S.; El-Domyati, F.M.; Mutwakil, M.Z.; Gadalla, N.O.; Hajrah, N.H.; Al-Kordy, M.A.; et al. Analysis of Transcriptional Response to Heat Stress in Rhazya stricta. BMC Plant Biol. 2016, 16, 252. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Zhu, J.; Zhu, J.-K. Cold Stress Regulation of Gene Expression in Plants. Trend Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef]
- Xu, W.; Li, R.; Zhang, N.; Ma, F.; Jiao, Y.; Wang, Z. Transcriptome Profiling of Vitis amurensis, an Extremely Cold-Tolerant Chinese Wild Vitis Species, Reveals Candidate Genes and Events That Potentially Connected to Cold Stress. Plant Mol. Biol. 2014, 86, 527–541. [Google Scholar] [CrossRef]
- Cheng, Z.; Lei, N.; Li, S.; Liao, W.; Shen, J.; Peng, M. The Regulatory Effects of MeTCP4 on Cold Stress Tolerance in Arabidopsis Thaliana: A Transcriptome Analysis. Plant Physiol. Biochem. 2019, 138, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Li, X.; Li, T.; Yu, D.; Han, B. Genome-Wide Transcriptome Profiling Provides Overwintering Mechanism of Agropyron Mongolicum. BMC Plant Biol. 2017, 17, 138. [Google Scholar] [CrossRef] [PubMed]
- Naydenov, N.G.; Khanam, S.; Siniauskaya, M.; Nakamura, C. Profiling of Mitochondrial Transcriptome in Germinating Wheat Embryos and Seedlings Subjected to Cold, Salinity and Osmotic Stresses. Genes Genet. Syst. 2010, 85, 31–42. [Google Scholar] [CrossRef] [PubMed]
- McDowell, J.M.; Dangl, J.L. Signal Transduction in the Plant Immune Response. Trend Biochem. Sci. 2000, 25, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Tsuda, K.; Wang, L.; Coller, J.; Watanabe, Y.; Glazebrook, J.; Katagiri, F. Network Modeling Reveals Prevalent Negative Regulatory Relationships between Signaling Sectors in Arabidopsis Immune Signaling. PLoS Pathog. 2010, 6, e1001011. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, C.M.J.; Leon-Reyes, A.; Van der Ent, S.; Van Wees, S.C.M. Networking by Small-Molecule Hormones in Plant Immunity. Nat. Chem. Biol. 2009, 5, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.-L.; Chen, B.-H.; Chen, X.-J.; Guo, Y.-Y.; Yang, H.-L.; Li, X.-Z.; Wang, G.-Y. Transcriptome Profiling of Pumpkin (Cucurbita moschata Duch.) Leaves Infected with Powdery Mildew. PLoS ONE 2018, 13, e0190175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Wang, C.; Liu, M.; Li, H.; Fu, Y.; Wang, Y.; Nie, Y.; Liu, X.; Ji, W. Large-Scale Transcriptome Comparison Reveals Distinct Gene Activations in Wheat Responding to Stripe Rust and Powdery Mildew. BMC Genomics 2014, 15, 898. [Google Scholar] [CrossRef]
- Kumar, A.; Kanak, K.R.; Arunachalam, A.; Dass, R.S.; Lakshmi, P.T.V. Comparative Transcriptome Profiling and Weighted Gene Co-Expression Network Analysis to Identify Core Genes in Maize (Zea mays L.) Silks Infected by Multiple Fungi. Front. Plant Sci. 2022, 13, 985396. [Google Scholar] [CrossRef]
- Kottapalli, K.R.; Rakwal, R.; Satoh, K.; Shibato, J.; Kottapalli, P.; Iwahashi, H.; Kikuchi, S. Transcriptional Profiling of Indica Rice Cultivar IET8585 (Ajaya) Infected with Bacterial Leaf Blight Pathogen Xanthomonas oryzae pv oryzae. Plant Physiol. Biochem. 2007, 45, 834–850. [Google Scholar] [CrossRef]
- Yokotani, N.; Hasegawa, Y.; Sato, M.; Hirakawa, H.; Kouzai, Y.; Nishizawa, Y.; Yamamoto, E.; Naito, Y.; Isobe, S. Transcriptome Analysis of Clavibacter michiganensis subsp. michiganensis-Infected Tomatoes: A Role of Salicylic Acid in the Host Response. BMC Plant Biol. 2021, 21, 476. [Google Scholar] [CrossRef]
- Lu, L.; Yang, D.; Tang, D.; Li, S.; Chen, Z. Transcriptome Analysis of Different Rice Cultivars Provides Novel Insights into the Rice Response to Bacterial Leaf Streak Infection. Funct. Integr. Genomics 2020, 20, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Li, Z.; Liu, X.; Yang, W.; Wang, Y. Comparative Transcriptome Analysis Reveals Potential Genes Conferring Resistance or Susceptibility to Bacterial Canker in Tomato. Horticulturae 2023, 9, 242. [Google Scholar] [CrossRef]
- Rivero, R.M.; Mittler, R.; Blumwald, E.; Zandalinas, S.I. Developing Climate-resilient Crops: Improving Plant Tolerance to Stress Combination. Plant J. 2022, 109, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Prusty, M.R.; Pandey, M.K.; Singh, P.K.; Bohra, A.; Guo, B.; Varshney, R.K. Application of CRISPR/Cas9-Mediated Gene Editing for Abiotic Stress Management in Crop Plants. Front. Plant Sci. 2023, 14, 1157678. [Google Scholar] [CrossRef]
Figure 1.
Models of the vascular element development in higher plants. (a) Organization of vascular elements in wild-type plants (without mutations in genes for transcription factors governing vascular biogenesis); (b) Organization of vascular elements in plant species with mutations in genes coding for REV, PHB, and PHV transcription factors; (c) Organization of vascular elements in plant species with mutation in genes coding for proteins of the KANADI family. More details in the text.
Figure 1.
Models of the vascular element development in higher plants. (a) Organization of vascular elements in wild-type plants (without mutations in genes for transcription factors governing vascular biogenesis); (b) Organization of vascular elements in plant species with mutations in genes coding for REV, PHB, and PHV transcription factors; (c) Organization of vascular elements in plant species with mutation in genes coding for proteins of the KANADI family. More details in the text.
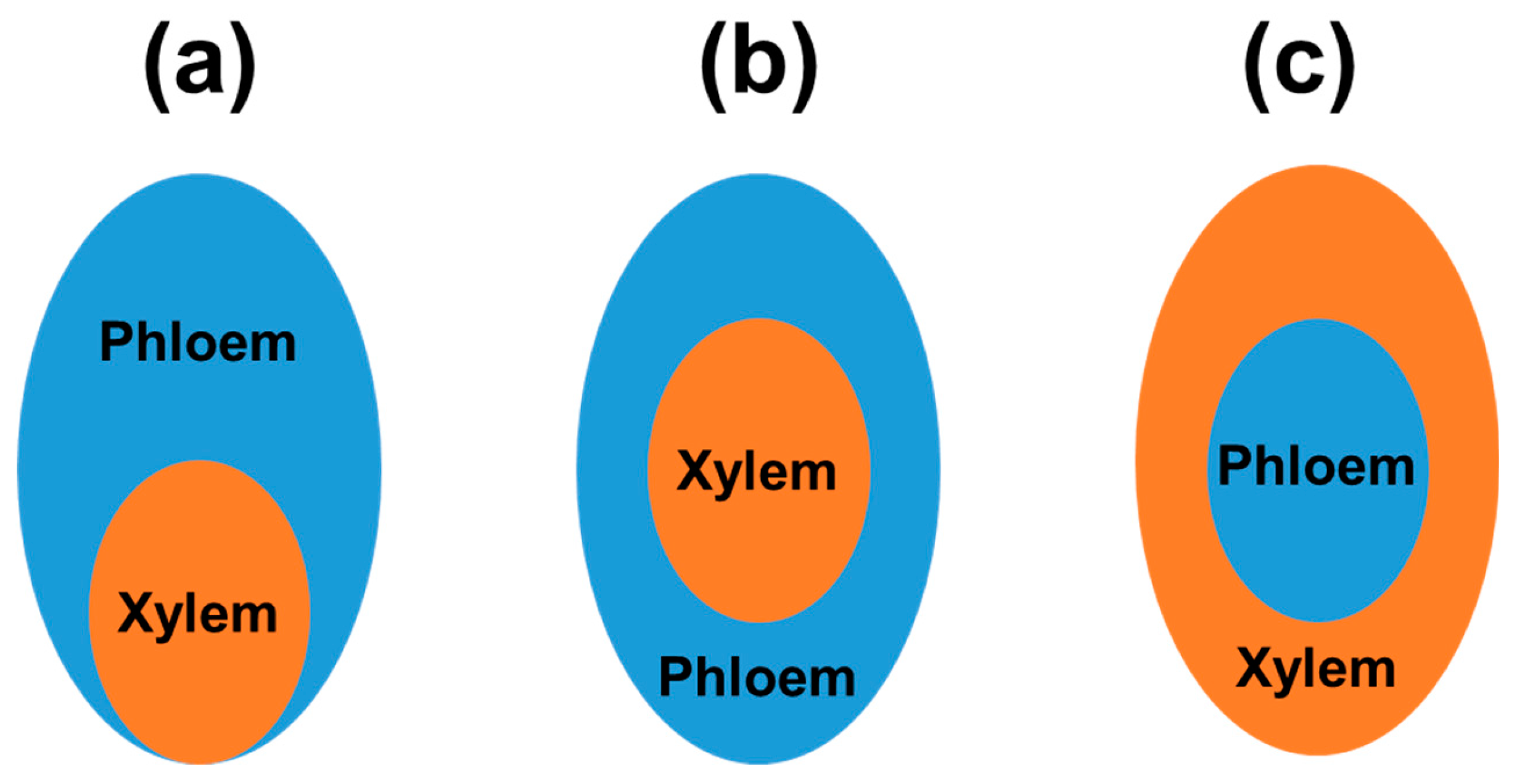
Figure 2.
The general pipeline for the high-throughput analysis of the plant transcriptome under stress acclimation between genotypes varying with stress resistance (stress susceptible and resistant lines). Key steps, from stress dosage (top) to the identification of differentially expressed gene sets (bottom) were shown. In this type of analysis, expression patterns are often compared pairwise between the stress treated and control grown plants for each genotype tested and the list of affected genes with fold change (FC) values is generated. More details in the text.
Figure 2.
The general pipeline for the high-throughput analysis of the plant transcriptome under stress acclimation between genotypes varying with stress resistance (stress susceptible and resistant lines). Key steps, from stress dosage (top) to the identification of differentially expressed gene sets (bottom) were shown. In this type of analysis, expression patterns are often compared pairwise between the stress treated and control grown plants for each genotype tested and the list of affected genes with fold change (FC) values is generated. More details in the text.
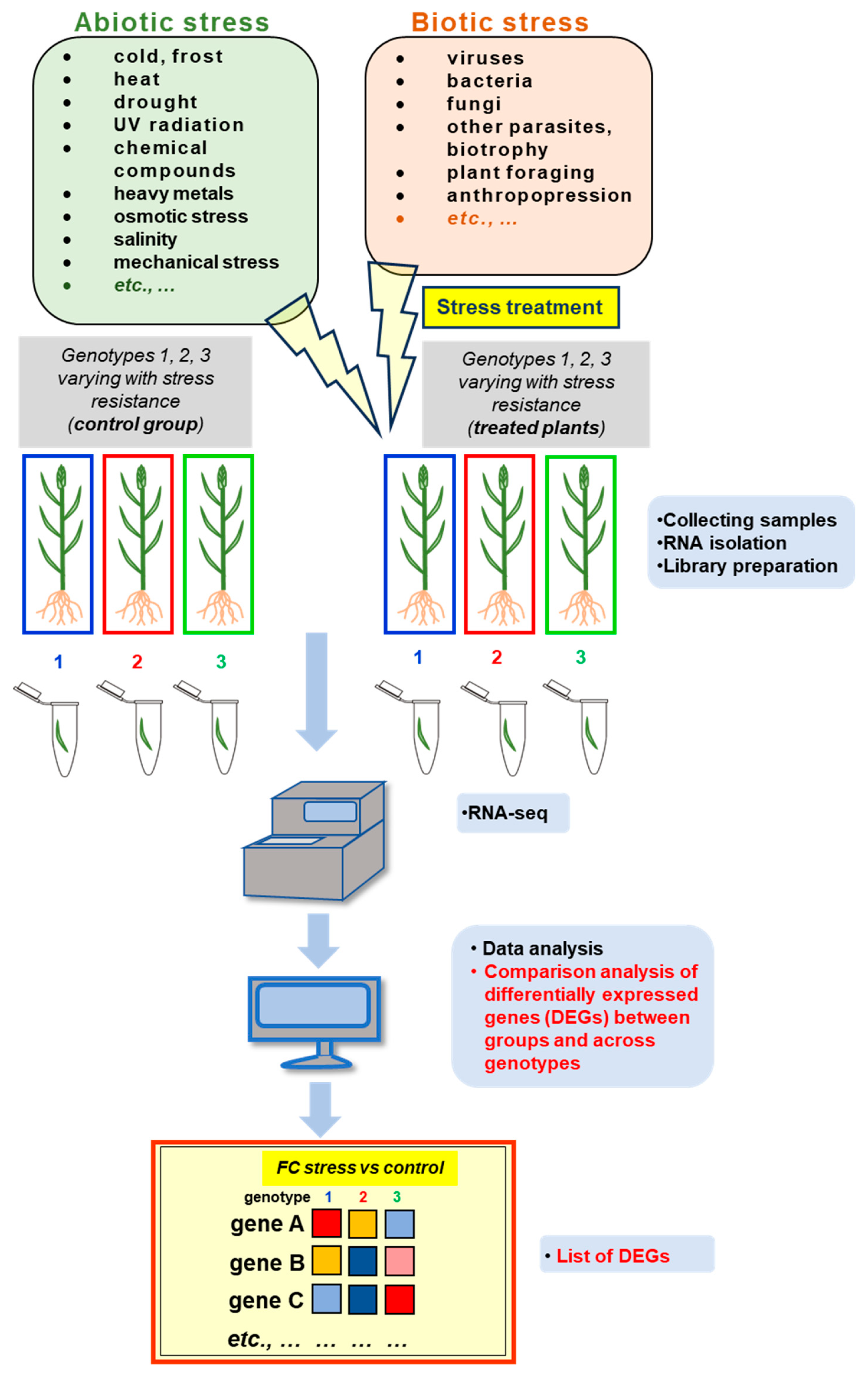
Figure 3.
Summary of the cellular functions governed by stress affected genes in various plant species. In addition, abbreviations for the key gene as well as gene families (particularly for most relevant transcription factors) were indicated next to each stressor identifier. Only most relevant, join data for both upregulated and downregulated genes were indicated (peripherally). Literature references, cited in the text, were shown (next to each stressor icons). Signaling routes between actively transcribed nuclear, plastid and mitochondrial genomes (small arrows within marked organelles) that are indispensable, for instance, for the proper organellar biogenesis under stress acclimation were also depicted (center). More details in the text.
Figure 3.
Summary of the cellular functions governed by stress affected genes in various plant species. In addition, abbreviations for the key gene as well as gene families (particularly for most relevant transcription factors) were indicated next to each stressor identifier. Only most relevant, join data for both upregulated and downregulated genes were indicated (peripherally). Literature references, cited in the text, were shown (next to each stressor icons). Signaling routes between actively transcribed nuclear, plastid and mitochondrial genomes (small arrows within marked organelles) that are indispensable, for instance, for the proper organellar biogenesis under stress acclimation were also depicted (center). More details in the text.
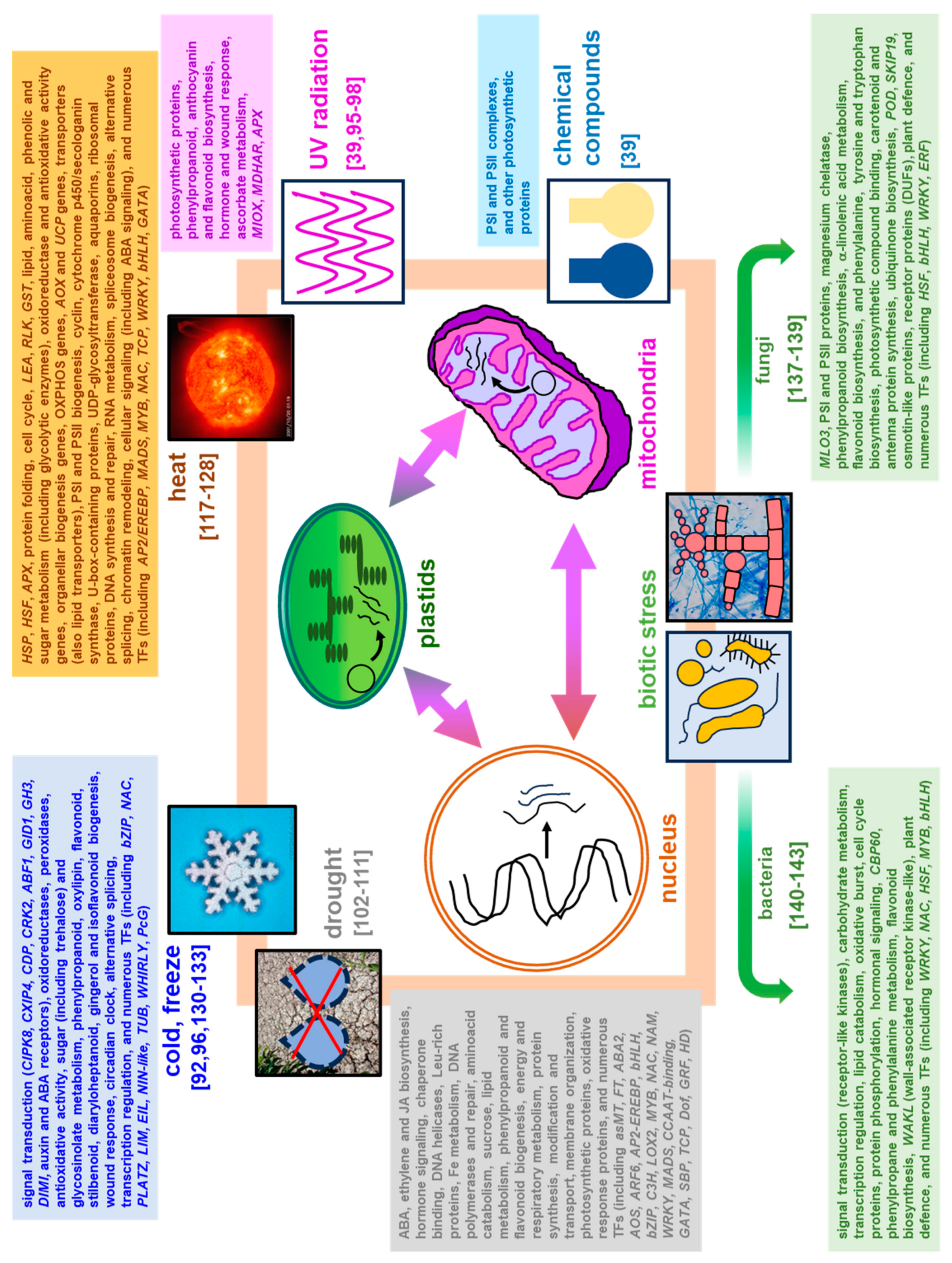
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



