Submitted:
28 March 2024
Posted:
28 March 2024
You are already at the latest version
Abstract
The chemotactic cytokine fractalkine (FKN, chemokine CX3CL1) has unique properties resulting from the combination of chemoattractants and adhesion molecules. The soluble form (sFKN) has chemotactic properties and potently attracts T cells and monocytes. The membrane-bound form (mFKN) facilitates diapedesis and is responsible for cell-to-cell adhesion, especially by promoting the strong adhesion of leukocytes (monocytes) to activated endothelial cells with the subsequent formation of an extracellular matrix and angiogenesis. FKN signaling occurs via CX3CR1, which is the only known member of the CX3C chemokine receptor subfamily. Signaling within the FKN-CX3CR1 axis plays an important role in many processes related to inflammation and the immune response, which often occur simultaneously and overlap. FKN is strongly upregulated by hypoxia and/or inflammation-induced inflammatory cytokine secretion, and it may act locally as a key angiogenic factor in the highly hypoxic tumor microenvironment. The importance of the FKN/CX3CR1 signaling pathway in tumorigenesis and cancer metastasis results from its influence on cell adhesion, apoptosis and cell migration. This review presents the role of the FKN signaling pathway in the context of angiogenesis in inflammation and cancer. The mechanisms determining the pro- or anti-tumor effects are presented, which are the cause of the seemingly contradictory results that create confusion regarding the therapeutic goals.
Keywords:
chemokine CX3CL1
; fractalkine
; CX3CR1
; fractalkine receptor
; CX3CL1/CX3CR1 axis
; angiogenesis
; tumorigenesis
; inflammation
; inflammation-induced angiogenesis.
1. Introduction
1.1. Angiogenesis
A well-developed microcirculatory network ensuring optimal blood-target tissue interactions is essential for preserving optimal metabolism. No metabolically active tissue in the body is more than a few hundred micrometers from a blood capillary, which is formed by the process of angiogenesis [1]. Angiogenesis involves two different mechanisms of vessel formation and two types of vessels formed. Angiogenesis, in a strict sense, describes the formation of new blood vessels from preexisting functional vessels via sprouting or splitting (also known as intussusceptive angiogenesis). This process is accompanied by the migration and proliferation of endothelial cells (ECs) [2]. However, de novo vessel formation may occur as a result of the differentiation of endothelial progenitor cells (EPCs) and their subsequent integration into the vascular wall. Human EPCs are generally defined as circulating cells that express a variety of cell surface markers similar to the markers expressed by vascular ECs, adhere to the endothelium at sites of hypoxia/ischemia or vascular injury, and participate in neoangiogenesis, including tumor angiogenesis [3,4]. Many studies revealed heterogeneity in blood ECs, where some cells express endothelial and hematopoietic antigens, and others express mature or immature endothelial markers [5]. Some studies indicate that the direct source of EPCs is not the bone marrow, as was commonly assumed, but the activation of cells with EPC properties residing in tissues (e.g., heart muscle) [6,7]. In contrast to sprouting angiogenesis, splitting angiogenesis is a rapid recovery adaptation of the existing microvascular network. However, this process only relies on the proliferation of ECs to a small extent, and it primarily relies on the reorganization of ECs and the invasion of EPCs [8].
Capillary growth and proliferation are rarely observed in normal adult tissues, except during wound healing and cyclical events in the female reproductive cycle (e.g., ovulation and menstruation), but angiogenesis and neoangiogenesis may be initiated with appropriate stimuli. Angiogenesis in adults is typically initiated in response to tissue hypoxia by the release of hypoxia inducible factors (HIFs), predominantly HIF-1α, which directly increase the expression and/or release of angiogenic stimulators, including vascular endothelial growth factor (VEGF), vascular endothelial growth factor receptor-1 (VEGFR-1) and tyrosine kinase with immunoglobulin-like and EGF-like domains 2 (TIE-2) receptors [9]. Because there is extreme hypoxia in the microenvironment of tissues surrounding tumors, the proliferation of tumor masses may also be indirectly dependent on HIF-1α, which enhances angiogenesis and increases the supply of oxygen to the area of malignant cell transformation [10,11,12,13]. Therefore, a hypoxic microenvironment is clinically associated with metastasis and poor prognosis in numerous cancers [12,14,15].
During physiological and pathological angiogenesis, EC activation is the first process that occurs [16,17]. The activation of ECs leads to the degradation of the vascular basement membrane (VBM), followed by vascular sprouting or vascular splitting, depending on the dominant share of ECs or EPCs, respectively. During this sprouting/splitting phase, transmembrane receptors that help cell‒cell and cell-extracellular matrix (ECM) adhesion, the integrins avβ3 and avβ5, play key roles in EC/EPC proliferation, the migration of cells along a gradient of ECM-bound chemoattractants (i.e., haptotaxis) and survival [18,19,20].
Angiogenesis is tightly regulated by the balance between pro-angiogenic and anti-angiogenic factors, including various cytokines and cytokine profiles [21,22]. Tumor angiogenesis is a consequence of disturbances in cytokine balance, and it differs significantly from physiological angiogenesis. The abnormal structure of vessels in cancer is accompanied by altered interactions between ECs and mural cells (pericytes and vascular smooth muscle cells), altered blood flow, increased permeability and delayed maturation [23].
The inflammatory response is also an important factor affecting angiogenesis [24,25,26]. Inflammation is defined as the homeostatic response of vascularized tissue to a sub-lethal damaging agent that destroys or inactivates invading pathogens, removes waste and debris, and permits the restoration of normal function, via resolution or repair [27,28]. Analogous to angiogenesis, the initiation and resolution of the inflammatory response involves the complex and coordinated expression of many factors, including cytokines, chemokines, growth factors, proteases, oxidative stress products, and lipid mediators [29,30]. Angiogenesis initiation is often associated with increased capillary permeability, and vascular permeability is dramatically increased in acute and chronic inflammation, cancer, and wound healing [31]. This hyperpermeability is mediated by acute or chronic exposure to vascular permeabilizing agents, particularly vascular permeability factor/vascular endothelial growth factor (VPF/VEGF, VEGF-A) [32]. Inflammatory and angiogenic processes overlap, which is why inflammation promotes angiogenesis, and new vessels may enhance tissue inflammation [30]. Vascular endothelial cells are a type of innate immune cell that are dependent on pathological conditions. This interaction explains why inflammation contributes to tissue proliferation, tumorigenesis, metastatic spread, and disordered tissue perfusion [31].
1.2. Chemokines
Chemokines are small cytokines with chemotactic properties encoded by a large gene family with at least 45 members in humans, and they play important roles in inflammation and angiogenesis [33]. Chemokines are defined by their primary amino acid sequence and the arrangement of specific structurally important conserved L-cysteine (C) residues at the N-terminus within the mature protein. Variation in the precise configuration of the two cysteines closest to the N-terminus allows chemokines to be split into four subfamilies: C, CC, CXC, and CX3C [34].
Chemokines activate their target cells by signaling via seven (pass)-transmembrane G protein-coupled receptors (GPCRs), which are further divided into conventional chemokine receptors (cCKRs) and atypical chemokine receptors (ACKRs) [35,36]. The promiscuity of chemokines and chemokine receptors on the cell surface, combined with biased signaling and allosteric modulation of receptor activation, guarantees a tightly controlled recruitment and positioning (directional migration) of individual cells within the local environment at a given time [37].
Homeostatic chemokines are constitutively expressed under physiological conditions and play a role in cell migration and homing. The local secretion of inflammatory chemokines is rapid and dynamic and aims to recruit effector cells to inflamed tissues [38]. Therefore, chemokines play a key role in the development and homeostasis of the immune system and all protective and destructive immune/inflammatory responses by participating in promotion of the activation, migration, differentiation, proliferation and apoptosis of immune cells. Chemokine signaling and the associated chemotaxis of various cell populations play important roles in the tumor microenvironment (TME) [39,40,41]. This role translates into phenomena directly related to tumor immunology and angiogenesis within the TME, which undoubtedly influence tumor progression and metastasis.
The chemokine CX3CL1 (fractalkine, FKN) deserves special attention in the context of inflammatory angiogenesis and tumorigenesis. CX3CL1 is the only member of the CX3C class of chemokines, and it has well-documented roles in ECs. CX3CL1 is a unique chemokine that combines the properties of a chemoattractant and an adhesion molecule [42,43,44,45].
This review presents the role of the FKN signaling pathway in the context of angiogenesis in inflammation and cancer.
2. Chemokine CX3CL1 (Fractalkine, FKN)
Two independent teams of researchers from the United States discovered the presence of a chemokine (C-X3-C motif) ligand 1 (CX3CL1), but named this molecule fractalkine and neurotactin in 1997 [46,47]. Baran et al. detected CX3CL1 by searching for chemokine-like sequences in an expressed sequence tag database in the National Centre for Biotechnology Information (NCBI). Bazan et al. chose the name “fractalkine”, which refers to the primary structure of the CX3CL1 molecule, where repeating subunits (fractals) recapitulate the whole. Pan at al. named the CX3CL1 molecule neuroactin after its isolation via sequencing of the mouse choroid plexus, but it did not remain in common use because it was misleading and referred to a surface glycoprotein previously discovered in Drosophilia melanogaster [48]. Imai et al. (1997) identified a functional high-affinity CX3CL1 receptor, named V28, which was later renamed CX3CR1 [42]. Signaling through CX3CR1, which is the only known FKN receptor with documented function, is responsible for the adhesive and chemoattractant properties of CX3CL1 [42,43,49,50,51].
2.1. CX3CL1 Structure
Due to the unique structure of the FKN molecule, the CX3C subclass of chemokines is characterized by a 3-amino acid spacing between the first two conserved L-cysteine residues within its chemokine domain [43,52]. Synthesized as a type I transmembrane protein, FKN exists in two forms, a full-length membrane-bound form and a soluble cleaved form (sFKN), which are generated under physiological conditions via disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) [53,54,55,56]. Stress factors, such as tumor necrosis factor alpha (TNF-α) converting enzyme (TACE or ADAM17), matrix metalloprotease-2 (MMP-2) or lysosomal cathepsin S (CTS), which act locally under conditions of disturbed homeostasis, may also contribute to the formation of sFKN [57,58,59].
The precursor form of membrane-anchored FKN is a polypeptide composed of 397 amino acid residues and contains a 24-amino acid signal peptide (SP) [57]. Mature (SP-free) transmembrane FKN consists of 373 amino acids, which form an extracellular N-terminal (chemokine) domain (aa 76), a mucin-like stalk (aa 241), a transmembrane α helix (aa 19), and a short intracellular domain (aa 37) in the form of a cytoplasmic tail [53,61]. The total molecular weight of FKN is approximately 17.5 kDa, but it is 95 kDa after glycosylation [46,47]. Typically composed of 317 amino acids, sFKN consists of a chemokine domain and an extracellular mucin-like stalk, which weigh approximately 14.7 kDa and 80 kDa after glycosylation, respectively [53,62,63]. However, there are some inconsistencies in the molecular weight and amino acid content of sFKN, which is likely due to the multiple forms of soluble CX3CL1 generated by shedding from the cell surface at alternative sites [64,65]. The molecular structures of both forms of FKN are shown in Figure 1.
2.2. CX3CL1 Function
The membranous and soluble forms of CX3CL1 (mFKN and sFKN, respectively) exhibit different functions, although both signal through the CX3CR1 receptor, which is a class of GPCR within the superfamily of seven transmembrane-spanning proteins that respond to a diverse array of sensory and chemical stimuli [66].
2.2.1. Membrane-Bound CX3CL1 (mFKN) as an Adhesion Molecule
Notably, the chemokine CXCL16 and FKN are the only chemokines that bind directly to the cell membrane via the transmembrane domain and mucin-like stalk [67,68,69]. FKN synthesis occurs primarily in ECs, which means that this chemokine has direct access to leukocytes in the bloodstream. Therefore, membrane-bound FKN mediates strong adhesion via binding to CX3CR1 on leukocytes [70,71]. This strong interaction between FKN and CX3CR1-expressing blood cells may result from a very low dissociation rate of this endothelial-white blood cell adhesion that may support the transendothelial migration of leukocytes during inflammation [72,73,74].
FKN is an adhesion molecule that mediates leukocyte adhesion directly or in cooperation with other tethering proteins, such as cadherins, immunoglobulin superfamily cell adhesion molecules (e.g., cluster of differentiation 106 [CD106] or vascular cell adhesion protein 1, also known as vascular cell adhesion molecule 1 [VCAM-1]), selectins and syndecans [43,75,76,77]. The indirect effect of FKN via cell adhesion molecules was also investigated. By counteracting the shear stress forces in most vascular beds, FKN does not recruit leukocytes alone because it does not provide optimal adhesion strength [57,77]. The significant adhesion of leukocytes to FKN peaks at 2 dynes/cm2 but is minimal at 10 dynes/cm2. In contrast, VCAM-1 recruits cells from whole blood at 10 dynes/cm2. However, when acting together, FKN and VCAM-1 show synergistic effects and cause a twofold increase in the number of adherent cells compared to VCAM-1 alone, which suggests that FKN mediates adhesion at high shear when combined with a molecule that mediates leukocyte tethering [74].
FKN is a multi-domain transmembrane chemokine that triggers leukocyte adherence without rolling and migration by presenting its chemokine domain (CD) to CX3CR1 [78]. Other domains of the FKN molecule also have key functional importance, which shows that the molecular structure of FKN may be precisely adapted to capture CX3CR1 in circulating cells. For example, the mucin stalk (mucin-like domain) holds and presents the CD away from the cell membrane surface, and its stiffness is achieved via a high degree of glycosylation. Therefore, the shortening of the mucin stalk of CX3CL1 and mutation of the potentially glycosylated residues of the mucin stalk are deleterious for the adhesive potency of FKN to a greater extent than the absence of the cytosolic domain (cytoplasmic tail) [79]. The cytosolic domain ensures the robustness of adhesion via the cytoskeleton. FKN is present as a monodisperse bundle on the surface of CX3CR1-expressing cells, and its packing is driven by its transmembrane domain (transmembrane α-helical region), which generates the permanent aggregation of an adequate amount of monomers to guarantee adhesion and prevent rolling [69].
The membrane compartment of FKN mediates a special dynamic equilibrium between the plasma membrane and intracellular vesicular trafficking from the intracellular compartment. Therefore, constitutive internalization of pre-synthesized FKN molecules occurs, which prevents FKN degradation by cell surface metalloproteases and accelerates the mobility of its intracellular content [80,81]. The two-adapter protein 2 (AP2)-binding motifs that bind clathrin, which plays a major role in the formation of coated vesicles, are crucial for the endocytosis-based internalization of FKN: YQSL is located within amino acid residues 362-365 (the cytoplasmic tail), and YVLV is located at positions 392-395 (the precursor form of FKN) [80]. The spatial distribution of FKN in individual subcellular compartments is also clearly influenced by the properties of vesicle-associated v-soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins, such as syntaxin 13 (STX13) and vesicle-associated membrane protein 3 (VAMP3) [82].
2.2.2. Soluble CX3CL1 (sFKN) as a Chemoattractant
Soluble FKN is cleaved via proteolysis by metalloproteinases, and it contains a chemokine domain (CD). SFKN exhibits functions typical of conventional chemokines and chemotaxis. As in many chemotactic cells, the signal regulating motility is initiated via the binding of sFKN on the cell surface to a GPCR class receptor, to which CX3CR1 belongs [43,83,84]. The main tasks of chemokines in the body are modulation and targeting of the immune response implemented via chemotactic effects on leukocytes by creating a concentration gradient [85,86,87,88]. The versatility and diversity of the chemotactic effects of sFKN occur because CX3CR1 is expressed constitutively or is involved in inflammatory responses in various cells, including hemato- and non-hematopoietic lines. The former also includes blood cells circulating in the vascular system, such as CD4+ and CD8+ T cells, B cells (CD19+), natural killer (NK; CD56+CD3-) cells, monocytes (CD14+), thrombocytes, dendritic cells (CD11c+), mast cell (MC) progenitors, peripheral blood-derived hematopoietic stem cells (PBHSCs), and neutrophils, to a much lesser extent [89,90,91,92,93]. The weak expression of CX3CR1 in neutrophils may explain why sFKN is not a major chemoattractant for the migration of neutrophils across the microvascular endothelium [94,95]. Notably, the very weak or ineffective effect of sFKN on stimulating neutrophil migration is not determined by CX3CR1 expression per se. Imai et al. [42] showed that although 80% of CD14+ monocytes expressed CX3CR1, only 1% of the input cells, i.e., only 1.3% of the receptor-expressing cells, migrated to sFKN. T helper-1 cells, a monocytic cell line, migrated to monocyte chemoattractant protein-1 (MCP-1) but not to sFKN, although these cells expressed the FKN receptor on their surface. Therefore, membrane-bound FKN, which is present predominantly in vascular ECs, efficiently mediates the binding and adhesion of neutrophil populations [42,54].
3. CX3CR1 – the Sole Fractalkine Receptor
The direct biological actions of both forms of fractalkine, adhesive for membrane-bound FKN and chemotactic for sFKN, are the result of interaction with the dedicated CX3C motif chemokine receptor 1 (CX3CR1, previously designated V28), also known as the fractalkine receptor or G-protein coupled receptor 13 (GPR13) [42,43,49,50,51]. Noticeably, the existence of the sole receptor for CX3CL1 makes it much easier to interpret the observed biological effects related to this chemotactic cytokine with respect to the CX3CL1/CX3CR1 axis.
3.1. CX3CR1 Structure
The genome of the CX3CR1 gene in humans is located on the short arm of chromosome 3 (3p22.2). CX3CR1 is composed of six exons (only two contain coding regions), and three intronic elements and three promoters are involved in the regulation of its genomic sequence [69,99]. CX3CR1 is evolutionarily conserved and encodes identical or similar sequences (four transcript variants) in mice and rats, despite the different locations of CX3CR1 (on chromosome 9 [9qF4] in mice and on chromosome 8 [8q32] in rats) [99,100,101].
The sequence of the 355 amino acids and the spatial structure of the transmembrane protein (MW ∼ 40 kDa) constituting CX3CR1 are well known [42]. The binding of CX3CR1 to metabotropic receptors within the most numerous class A (rhodopsin-like receptors) in the GPCR family of proteins, which are composed of a monomeric protein containing an extracellular domain with a signaling ligand binding site and an intracellular domain that binds the G protein [102]. A structural diagram of CX3CR1 is shown in Figure 2.
The polypeptide chain of CXC3CR1 is composed of seven α-helical structures extending across the cell membrane (transmembrane segments or domains: TM1–TM7) and exceeding its thickness in both directions, i.e., into the extracellular space and the cytoplasm. This conformation creates eight amino acid chains on both sides of the cell membrane that connect individual TMs: three extracellular loops (ECL1–ECL3), another three intracellular loops (ICL1–ICL3), and the two linear chains forming an extracellular amino terminus (NH2) and an intracellular carboxyl terminus (COOH) at the ends of the molecule [61,110]. The CX3CR1 molecule contains a disulfide bond connecting two conserved cysteine (C) residues located at the top of the extracellular side of TM3 and within ECL2 [105].
There are binding sites within the ECLs and NH2 terminus for functional ligands, such as CX3CL1 (FKN), and CCL26 (eotaxin-3), antibodies and some pathogens, such as bacteria and viruses [111,112,113,114]. The appropriate level of tyrosine (T) sulfation at the NH2-terminus is required for maintaining the normal activity of most GPCR receptors for chemokines [57,115].
ICL2 is of key functional importance on the other side of the plasma membrane because it contains the canonical DRYLAIV motif, which is composed of a sequence of seven amino acids (with 3-letter abbreviations: Asp-Arg-Tyr-Leu-Ala-Ile-Val) [100,106]. This protein sequence motif provides a docking point that is essential for the coupling of CX3CR1 to the heterotrimeric G protein, which belongs to the Gαi family. This binding is crucial for the induction of classical signaling pathways because metabotropic receptors do not contain ion channels in their structure and only influence ion flow by activating the intermediary G protein [57,116]. The binding of an agonist to CX3CR1 causes conformational changes in the receptor with subsequent dissociation of the components of the heterotrimeric G complex, which consists of alpha (α), beta (β) and gamma (γ) subunits. The binding of guanosine diphosphate (GDP) allows the α subunit to bind to the β and γ subunits to form an inactive trimer (inactive Gα-GDP state). Binding of an extracellular signal (ligand) to CX3CR1 allows the G protein to bind to the receptor and causes GDP to be replaced with guanosine triphosphate (GTP) to create the active Gα-GTP state [110,116,117]. The COOH-terminal serine residues (S) are susceptible to G protein-coupled receptor kinase (GRK)-mediated phosphorylation and subsequent desensitization. However, more recently characterized functions of GRKs as scaffolds and signaling adapters suggest that this small family of proteins modulates CX3CR1 activity in a more complex way [118].
Similar to other chemokine receptors, CX3CR1 exhibits polymorphisms, which may account for its varying affinity for CX3CL1 and other ligands [119]. The polymorphic residues at positions 249 and 280 may be responsible for dysfunctional CX3CR1 variants, including variants identified in various cancers [108,109]. Polymorphism of CX3CR1 is also related to diseases of the cardiovascular system (e.g., atherosclerosis), nervous system (e.g., Alzheimer’s disease) and infections (e.g., systemic candidiasis) [120,121,122,123].
3.2. FKN signaling via CX3CR1
3.2.1. Conformational Rearrangements Following FKN Binding
Metabotropic receptors, including CX3CR1, constitute the largest family of cell surface proteins involved in signaling across biological membranes, and conformational changes after ligand attachment are crucial for initiating intracellular signaling pathways [110,117]. Under physiological conditions, the sole endogenous CX3CR1 ligand, FKN, binds to the orthosteric sites of the receptor [73]. The affiliation of CX3CR1 with the A1 subfamily within the rhodopsin-like GPCRs, established on the basis of phylogenetic analysis, indicates that the receptor molecule resembles the structure of rhodopsin and transduces extracellular signals via interaction with guanine nucleotide-binding (G) proteins [124,125]. On the basis of the predicted structure, conservation of a few amino acids in the region critical for G protein activation, and activation by a small ligand, rhodopsin is a model compound for the assessment of conformational changes associated with the activation of subclass A GPCRs [126,127]. Conformational changes in CX3CR1 after ligand binding were also examined indirectly using US28, which is a virus-encoded GPCR showing 29% sequence identity with CX3CR1 [128,129]. US28 also binds to CX3CL1 and acts during human cytomegalovirus infection [130].
The term “seven-transmembrane (7TM) receptors” is often used interchangeably with GPCRs and reflects their seven membrane-embedded helices and additional signaling independent of G proteins [131,132].
Despite resolving the spatial structure of many complexes of chemokines in classes other than FKN with proper receptors, knowledge of specific chemokine recognition mechanisms in the CX3C subfamily remains incomplete [129,133]. Creating models of the crystal structure and the use of cryo-electron microscopy (cryo-EM) to study US28-FKN and US28-engineered FKN (chemokine CX3CL1.35) complexes was insufficient to explain the conformational changes in CX3CR1 that occur with its activation in humans [134,135]. However, cryo-electron microscopy data indicate the involvement of cholesterol in the regulation of CX3CR1 activation, which translates into conformational changes in the CX3CR1-Gαi complex observed in ligand-free and CX3CL1-bound states at 2.8- and 3.4-Ångström (Å) resolution, respectively [110,117]. Comparison of the overall structures of the CX3CR1-Gα and CX3CR1-CX3CL1-Gαi complexes revealed that although these two complexes exhibit similar conformations with a receptor Cα (residues T31 to Y305) root mean square deviation (RMSD) of 1.4 Å, the superposition of CX3CR1 in the two states reveals obvious differences in the extracellular region of the receptor. Compared to the ligand-free complex, the N-terminus of the activated receptor moves closer to the center axis of the helical bundle, and extracellular loop 2 (ECL2) is pushed away upon CX3CL1 binding. [117]. The movement outside the cell, shown by helix VI and treated as a specific “conformational marker” of activation within the previously structurally solved representatives of class A GPCRs, is clearly smaller for the CX3CR1-CX3CL1-Gαi complex [136]. Initially, ligand-free CX3CR1 shows a more central helix VI location. For example, it shows only 2.3-Å outward movement of the intracellular end of helix VI, while the range of this movement is 8.2 and 6.7 Å in the active structures of C-C chemokine receptor type 5 (CCR5)-Gαi and US28-Gαi, respectively. Therefore, the different conformations of CX3CR1 cause its Gαi-coupling surface area to be 900 Å2, which is much larger than the CCR-Gαi (826 Å2) and US28-Gαi (790 Å2) complexes. Therefore, the distinct conformation of CX3CR1 and the Gαi coupling interface suggest different activation mechanisms of CX3CR1 and provide some insight into the diversity of G protein-dependent intracellular signaling of FKN [117,137,138]. This activity is complemented by the fact that activated CX3CR1 exhibits conformational rearrangements of key conserved activation motifs in class A GPCRs, including the canonical DRYLAIV motif, which provides a docking point for protein G [38,139,140].
Natural variants of CX3CR1 may differ conformationally during activation and conformational changes in CX3CR1 accompanying activation may be disrupted as a result of mutations and may be associated with various diseases, including changes in the risk of several cancers [110,141,142,143,144,145,146]. Glutaminyl cyclase (QC)-catalyzed N-terminus pyroglutamate (pGlu) formation of the ligand (FKN) may determine the stability or interaction with CX3CR1, and it is therefore essential for the full biological activity of FKN [147].
3.2.2. Main FKN/CX3CR1 Signaling Pathways
The binding of membrane-bound FKN and the soluble form of cleaved FKN to CX3CR1 at its extracellular determinants localized within the amino terminus and the third extracellular loop (ECL3; see Figure 2) independently contributes and is a necessary condition for proper conformational rearrangements preceding the activation of heterotrimeric G proteins associated with CX3CR1 [152]. This binding of the agonist to the receptor involves two steps. Step one mediates high-affinity FKN binding involving Tyr14, Asp25, and Glu254. This initial interaction then triggers the engagement of Glu13, Asp16, and Asp266 (step two) [152,153]. On the other side of the cell membrane (inside the cell), the Gα subunit disassociates from the membrane and interacts with G protein regulatory (GPR) domain-containing proteins. RIC8 (synembryn), which is a non-receptor guanine-nucleotide exchange factor for Gα subunits, facilitates the exchange of GTP for GDP [154,155]. The presence of active GTP-bound Gα in the G protein complex leads to its dissociation into Gαi-GTP and a GβGγ dimer. Activated Gαi interacts with downstream effectors [156].
The resulting FKN-CX3CR1 axis transduces several well-characterized signaling pathways that lead to the activation of several transcription factors (e.g., signal transducer and activator of transcription protein [STAT], nuclear factor kappa-light-chain-enhancer of activated B cells [NF-κβ], and cAMP/Ca2+ response element binding protein [CREB]) and the inhibition of other factors (e.g., members of the class O forkhead box transcription factor [FOXO]) [44,102,157]. Most of these CX3CR1 signaling pathways are shared with other chemokine receptors, including:
❶ stimulation of calcium mobilization from intracellular stores via the phospholipase C (PLC)/protein kinase C (PKC) pathway [158,159],
activation of the respective kinases, resulting in subsequent downstream signaling within
❷ the Janus kinase (JAK)/STAT pathway, ❸ phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/IkappaBeta (Iκβ) kinase (IKK)/Iκβ/NF-κβ pathway, ❹ Ras kinases (Ras)/Raf kinases (Raf)/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) and ❺ MEK kinase (MEKK)/cJun NH(2)-terminal kinase (JNK)/CREB or MEKK/mitogen-activated protein kinases (P38)/CREB pathways [96,160,161,162].
The most important signaling pathways within the FKN/CX3CR1 axis are presented in Figure 3.
Both forms of FKN, the membrane-bound and – resulting from membrane shedding by lysosomal protease cathepsin S (CTS) and/or metalloproteases (a disintegrin and metalloproteinase domain-containing protein 10 [ADAM10], a disintegrin and metalloprotease 17 [ADAM17], also called tumor necrosis factor alpha converting enzyme, TACE], and matrix metalloprotease-2 [MMP-2]) – soluble FKN, activate the same signaling pathways promoting adhesion or chemotaxis, respectively [152,163]. The presence of an active, guanosine triphosphate (GTP)-bound Gα in the G protein complex, leads to the dissociation into Gαi-GTP and a GβGγ dimer. Once activated, Gαi can go on to interact with downstream effectors [156]. The effect on gene transcription is achieved by activating the signal transducer and activator of transcription protein (STAT), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ) and cAMP/Ca2+ response element binding protein (CREB), while inhibiting the members of the class O of forkhead box transcription factors (FOXO) [44,102,157]. The involvement of the phospholipase C (PLC)/ protein kinase C (PKC) pathway in intracellular divalent calcium cations (Ca2+) mobilization that may influence chemotaxis is well documented [158,159]. By inhibiting the expression of pro-apoptotic proteins and FOXO activity, signaling pathways via phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/IkappaBeta (Iκβ) kinase (IKK)/Iκβ/NF-κβ pathway, Ras kinases (Ras)/Raf kinases (Raf)/mitogen-activated protein kinase kinase(MEK)/ extracellular signal-regulated kinase (ERK) and MEK kinase (MEKK)/cJun NH(2)-terminal kinase (JNK)/CREB or MEKK/ mitogen-activated protein kinases (P38)/CREB pathways can increase the survival of cells, including cancer cells [164,165,166].
Other abbreviations: AC – adenylyl cyclase; cAMP – cyclic adenosine monophosphate; Gα, Gβ, Gγ – subunits of the heterotrimeric G-proteins (G protein complex); Gαi – activated Gα subunit of the G protein complex; GDP – guanosine diphosphate; IP3 – inositol 1,4,5-trisphosphate; JAK – Janus kinase; PIP2 – Phosphatidylinositol 4,5-biphosphate; RIC8 – a non-receptor guanine-nucleotide exchange factor for Gα subunits (also known as synembryn)
One of the most commonly understood signals generated by the FKN/CX3CR1 pathway is the prevention of apoptosis, primarily in monocytes. FKN/CX3CR1 signaling induces the expression of anti-apoptotic genes, primarily BCL2 and BCL-xL (B-cell lymphoma extra-large) [167]. Reduced expression of pro-apoptotic proteins and FOXO promotes cell survival and cancer cells, in certain conditions [164,165,166].
Metabotropic CX3CR1 is not isolated within the biological membrane. Therefore, many signaling pathways activated by FKN depend on the functional state of other receptors, including epidermal growth factor receptor (EGFR), a member of the family closely related to receptor tyrosine kinases ErbB-1 (EGFR) and ErbB-2 (HER2/neu) [168,169]. EGFR is involved in cell signaling pathways that control cell division and survival [170,171,172], whereas CX3CR1-dependent intracellular signaling cascades are responsible for the processes of migration and proliferation based on increased cell survival [96,156,157,158]. Mutations of the EFGR gene located at the short arm of chromosome 7 (7p11. 2) affect its expression or activity and may contribute to the development of many cancers, with CX3CR1 acting as one of the transactivators of ErbB-1 and ErbB-2 (see Chapter 5). FKN/CX3CR1 axis and tumorigenesis) [98,173,174,175,176,177]. Transactivation, which is well characterized for EGFR, represents the process whereby GPCRs activate receptor tyrosine kinases (RTKs), which are receptors for several neurotrophic factors and growth factors, with subsequent downstream signaling, such as mitogen-activated protein kinase (MAPK) [178,179].
A significant advance in CX3CR1 research was the demonstration that FKN exhibits an autoregulatory function and may induce its own expression via activation of pertussis toxin-sensitive G proteins, PI3K, phosphoinositide-dependent kinase 1 (PDK1), Akt, NIK, IKK and NF-κβ. Tumor necrosis factor alpha (TNFα) plays a key role in this autoregulatory loop because it induces the expression of FKN and CX3CR1 in an NF-κβ-dependent manner [43,180]. CX3CR1 autoregulation occurs via the use of NF-κβ inhibitors, and a reduced FKN concentration is accompanied by increased CX3CR1 expression [181].
4. FKN/CX3CR1 Axis and Inflammation
As mentioned in subsection 2.2.2. CX3CR1 is a chemoattractant of soluble CX3CL1 (sFKN) that is constitutively expressed or induced by inflammation in many hemato- and non-hematopoietic lines [89,90,91,92,93]. The differential degree of CX3CR1 expression throughout the body applies to immune and non-immune system cells, primarily in a cell type-specific manner [70,182].
Activation of the FKN/CX3CR1 axis suggests the occurrence of immune cell chemotaxis, which is determined by ligand concentration gradients [49,109]. Because cells expressing CX3CR1 are involved in inflammatory and anti-inflammatory responses, the final effect of this chemotaxis is dependent primarily on local environmental conditions. This effect constitutes a specific duality of action of FKN, which may facilitate the maintenance of homeostasis, but FKN may play a key role in the pathomechanism of inflammation in pathological conditions [73,123,183,184]. However, there is growing evidence that the impact of CX3CR1 may be tissue- and disease-specific [70]. Notably, in addition to inducing the adhesion and chemotaxis of leukocytes, FKN has anti-apoptotic effects and increases the survival of multiple cell types during homeostasis and inflammation [185,186,187].
The involvement of FKN and FKN/CX3CR1 signaling under physiological conditions and the pathological inflammatory response in selected tissues/organs/systems are summarized inTable 1.
4.1. FKN/CX3CR1 Axis and Inflammation-Induced Angiogenesis
4.1.1. Interdependence of Inflammation and Angiogenesis
Angiogenesis, i.e., the formation of new blood vessels from pre-existing vessels by sprouting or splitting, is fundamentally important in body development and tissue regeneration [265,266]. Phenomena accompanying angiogenesis, such as the migration, growth and differentiation of endothelial cells, are tightly controlled primarily by cytokines that use multiple signaling pathways. Notably, angiogenesis after the completion of individual development primarily occurs in conjunction with a chronic inflammatory response [267]. Chronic inflammation and hypoxia, which are the principal physiological stimuli that induce angiogenesis, clearly coincide [268,269]. Therefore, inflammation and angiogenesis, including tumor angiogenesis, are two interdependent processes that may enhance each other [266,269]. Most angiogenic factors have a functional duality, consisting of pro-inflammatory and pro-angiogenic effects [220,271,272]. The process of initiating vessel formation itself is most often associated with an initial increase in the permeability of the microcirculatory vessel wall, which allows angiogenic factors contained in the plasma to penetrate into the interstitial compartment. After destabilization of the endothelial cell monolayer, directional motility (haptotaxis) of these cells occurs toward angiogenic stimuli within the extravascular space [273]. Integrins (avβ3 and avβ5) play important roles in this process and determine adhesion to matrix proteins, with concomitant proliferation of ECs lining the vessel wall to replace previously migrated cells. Neuropilins (NRP-1 and NRP-2), which are highly conserved type I membrane glycoproteins that act as co-receptors for vascular endothelial growth factors (VEGFs) together with VEGF receptors (VEGFRs), also play important roles in this stage of angiogenesis. High levels of NRP1 are detected on the arterial endothelium, but NRP2 expression is restricted to lymphatic and venous endothelial cells [274,275]. For example, NRP-1 binding to several VEGF-A isoforms promotes the interaction of growth factors with VEGFR-2, which increases receptor phosphorylation. NRP-1 is required for EC adhesion to soluble VEGFR-1 [276]. NRP-2 is an important angiogenic player in the promotion of EC migration and adhesion by regulating integrin alpha 5 (ITGA5/CD49e) recycling [277]. NRP-2 may also act as an inflammation-sensing protein and is rapidly and dramatically induced in myeloid cells, especially macrophages, under inflammatory conditions [278]. In response to hypoxia, there is a local increase in hypoxia-inducible factor-1 alpha (HIF-1α) production, which likely controls angiogenesis and metabolism by upregulating hypoxia-induced genes, such as the interleukin-33 (IL-33) gene and VEGF gene [279].
The migrating and proliferating ECs form cord-like structures in target tissues that later canalize to form functional vessels, which are further stabilized by surrounding pericytes [280]. Tight cell–cell adhesion results from the expression and function of different adhesion molecules, such as platelet-endothelial adhesion molecule-1 (PECAM-1, CD31) and vascular-endothelial cadherin (VE-cadherin, CD144) [281,282]. Newly formed vessel network stabilization requires remodeling, hierarchization, and quiescence [273,283].
The proper course of angiogenesis requires precise coordination at the level of many signaling pathways, which allows for a change in the dynamic balance between angiogenesis inhibitors and stimulators (anti- and pro-angiogenic factors) toward the latter [16,284]. Clinical observations and histopathological data confirm that the angiogenesis that accompanies chronic inflammation tends to prolong and intensify the inflammatory response [282,285]. Numerous pathological conditions, such as inflammatory bowel disease (IBD), cancer growth and tumor metastasis, arthritis, diabetic retinopathy and ischemic cardiovascular diseases (including stroke), are associated with abnormal inflammatory angiogenesis [286,287,288,289,290].
4.1.2. Proangiogenic Effects of FKN/CX3CR1 Signaling on the Inflammatory Response
Coincidental inflammation and hypoxia are generally found in chronic inflammation. Both of these conditions contribute to the activation and/or potentiation of the NFĸB gene regulator. This effect is important for the activity of the FKN/CX3CR1 axis because NF-κB is a main controller of FKN expression [291]. An increase in NF-ĸB activity in chronic inflammation is induced by the action of many pro-inflammatory cytokines, including TNFα and IL-1 [292,293]. Stimulation of cluster of differentiation 40 (CD40, also known as TNFRSF5 – tumor necrosis factor receptor superfamily member 5), which is a costimulatory protein expressed on antigen-presenting cells (APCs), by CD40 ligand (CD40L) expressed on CD4 T lymphocytes (helper T cells) shortly after activation induces the classical and alternative NF-κB pathways in endothelial cells and promotes CX3CL1 expression [294,295]. The intracellular signaling pathways leading to increased FKN expression via increased NF-ĸB synthesis include the activation of phosphoinositide-3-kinase (PI3K). An autoregulatory mechanism in which FKN controls its own expression in an inflammatory environment also involves the PI3K/Akt/IKK/IKβ/NF-ĸB signaling pathway [180]. Akt signaling-related activation of the vasodilation pathway via eNOS activation and increased NO production in the endothelium are also closely associated with angiogenesis [296,297].
In addition to the PI3K/Akt/IKK/IKβ/NFĸB pathway, the proangiogenic effects of FKN/CX3CR1 axis activation by vascular ECs are mediated via the PKC/Ras/Raf/MEK-ERK or PKC/MEKK/MEK/ERK signaling pathways [164]. A two-step sequence of events then occurs: HIF-1α is upregulated and phosphorylated in hypoxia via an ERK-dependent pathway, with subsequent enhancement of VEGF-A gene transcription in monocytes [298,299,300]. VEGF-mediated angiogenesis requires NO production from activated endothelial NO synthase (eNOS) because sprouting angiogenesis requires vasodilation and increased vascular permeability [301,302].
The proangiogenic effects of FKN/CX3CR1 signaling on the inflammatory response are shown in Figure 4.
Membrane-bound FKN (mFKN), which determines adhesion, and soluble FKN (sFKN) associated with chemotaxis, are involved in the activation of the CX3CR1 signaling pathway [152,163]. Once activated, CXCR1 interacts with downstream effectors, ultimately leading to angiogenesis accompanied by vasodilation and resistance to apoptosis [156]. The pro-angiogenic effects of activation of the FKN/CX3CR1 axis are mediated by phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/IkappaBeta (Iκβ) kinase (IKK)/Iκβ/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ), as well as protein kinase C (PKC) /Ras kinases (Ras)/Raf kinases (Raf)/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) or PKC/MEK kinase (MEKK)/MEK/ERK signal pathways [164]. Activation of PI3K intracellular downstream signaling is linked to increased NF-ĸB synthesis and, the secondary, increased FKN expression, whereas PKC … ERK signaling is responsible for upregulation of hypoxia-inducible factor 1-alpha (HIF-1α). Next, HIF-1α affects its target genes, which results in, among others, increasing VEGF-A transcription in monocytes [298,299]. Vasodilation is related to Akt signaling with subsequent endothelial nitric oxide synthase (eNOS) activation and increased nitric oxide (NO) production [296,297]. Increased cell survival due to resistance to apoptosis is a consequence of Akt and ERK signaling with inhibition of pro-apoptotic proteins, and in the case of Akt pathway, also the members of the class O of forkhead box transcription factors (FOXO) inhibition and increased expression of the (anti-apoptotic) B-cell lymphoma (Bcl-2) protein. The involvement of the phospholipase C (PLC)/ protein kinase C (PKC) pathway in intracellular divalent calcium cations (Ca2+) mobilization that may influence chemotaxis is well documented [158,159].
5. FKN/CX3CR1 Axis and Tumorigenesis
5.1. Hypoxia and Angiogenesis in the Tumor Microenvironment (TME)
The main difference between cancer cells and normal cells is that cancer cells grow uncontrollably beyond the control of the immune system. Because chemotaxis is one of the key phenomena during cancer development, invasion/progression and metastasis, the presence of chemokines in the tumor microenvironment, especially chemokines with angiogenic properties, is essential [303]. Another characteristic feature of cancer is disturbed cell adhesion processes with significant changes in the function and quantitative profile of cell adhesion molecules, which lead to the loss of cell-to-cell adhesion [304].
The essential role of angiogenesis in tumor growth and the spread and establishment of metastases has been well documented [305,306]. Cancer tissue cells show spheroidal growth in vitro have an upper volume limit determined by the nutrients and oxygen diffusion distance (from the culture medium to the spheroid core) [307,308]. The expansion of tumors growing in tissue in vivo is limited by the nutrient diffusion distance from the nearest capillary, which is 100–500 microns [309,310,311]. Therefore, a further increase in the size of the tumor requires increased vascularization within the tumor tissue, and the process by which tumor-associated neovessels sprout from existing blood vessels is called “tumor angiogenesis” after Folkman [309].
Compared to normal vessels, new vessels produced by most types of cancer cells during tumor angiogenesis have abnormal morphology and function (e.g., increased permeability), which lead to limited blood supply efficiency [312]. Paradoxically, abnormal blood and lymphatic vessels that create a hostile tumor microenvironment (TME) that is characterized by hypoxia, lowered pH, and elevated interstitial fluid pressure, which noticeably maintain the malignancy of the tumor [313]. Hypoperfusion-related hypoxia makes cancer cells more aggressive, and “leaking vessels” allow these cells to travel to distant sites to metastasize [314]. Hypoxia creates an abnormal TME in which angiogenesis is constantly upregulated and immune system functions are limited (e.g., recruitment of tumor-infiltrating lymphocytes – TILs), which lead to tumor-mediated immunosuppression [315]. Hypoxia contributes to the progression of cancer and resistance to photodynamic therapy, chemotherapy and radiotherapy [316,317,318].
5.2. FKN in the TME
As a unique chemokine expressed in many cell types that exhibits chemoattractant (sFKN) and pro-angiogenic effects and the typical features of an adhesion molecule (mFKN), FKN has been intensively studied in the context of carcinogenesis and metastasis [220,246,300]. The presence of a signaling pathway associated with only the metabotropic receptor CX3CR1 facilitates the interpretation of the observed relationships and narrows potential therapeutic targets [44,194,207].
Because each cancer shows considerable heterogeneity (including DNA methylation heterogeneity) depending on the type and cell-specific variability within the same type, receptor expression, chemotaxis and cellular adhesion are not fully predictable [319,320,321]. This relationship also applies to the function of the FKN/CX3CR1 axis within the TME. It is crucial to precisely define whether FKN/CX3CR1 signaling may be a therapeutic target in a given oncological case and determine whether its inhibition or stimulation is beneficial [84].
5.2.1. FKN/CX3CR1 Signaling May Promote Tumorigenesis
Most publications demonstrated a pro-tumorigenic and pro-metastatic role of FKN/CX3CR1 signaling across multiple blood and solid malignancies [322]. The promotion of tumor growth and metastatic spread is generally associated with the production of new vessels in response to hypoxia within the TME and, less often, with chemotaxis of tumor cells. Recruitment of circulating CX3CR1+ monocytes is critical for tumor angiogenesis [323].
For example, CX3CR1 signaling enhances the accumulation of tumor-associated microglia/macrophages and angiogenesis during the progression and malignant transformation of low-grade gliomas. Notably, one study revealed increased survival in patients (n = 45) with the presence of a common CX3CR1 V249I polymorphism, which correlated with reduced tumor vessel density and reduced M2 macrophage infiltration [145].
Studies in hepatocellular carcinoma (HCC), the course of which is inherently associated with the inflammatory process and the up-regulation of cytokines, have shown that FKN knockout inhibits the in vitro and in vivo angiogenesis of HCC HepG2 cells [324].
CX3CR1 was expressed in a histological grade- and stage-dependent manner in histopathological samples obtained from patients with colorectal cancer. CX3CR1 upregulation correlated with poor prognosis due to increased survival of angiogenic macrophages in the TME, which contributed to tumor metastasis [325]. High CX3CR1 expression or high expression of the CX3CL1/CX3CR1 axis were also independent negative prognostic factors in pancreatic ductal adenocarcinoma. CX3CL1 and CX3CR1 expression (77.1 and 66.7%, respectively) was clearly greater in malignant areas than in peritumoral areas [326].
mFKN promotes cell‒cell adhesion for communication between tumor cells and vascular ECs and, consequently, angiogenesis. This finding was confirmed by the result that small interfering RNA-mediated knockdown of the FKN gene inhibited melanoma B16-F0 cell growth in vivo, which correlated with decreased angiogenesis around the tumor [327].
The FKN/CX3CR1 interaction may be crucial in the development of prostate cancer metastases to bone tissue. FKN promotes the adhesion of human prostate cancer cells to bone marrow endothelial cells and their migration toward human osteoblasts in vitro [328]. This effect occurs because osteoblasts and stromal and mesenchymal cells derived from human bone marrow express mFKN, whereas sFKN is present in bone marrow supernatants [329]. After malignant transformation, endothelial cells overexpress CX3CR1, but CX3CR1 occurs in minimal quantities in the endothelium of the normal prostate gland. Androgens may promote the extravasation of CX3CR1-bearing cancer cells on an FKN concentration gradient but their ability to adhere to the bone marrow endothelium is not altered [330]. In vivo animal models showed that the overexpression of CX3CR1 induced the spinal metastasis of prostate cancer via the FKN/steroid receptor coactivator (Src)/focal adhesion kinase (FAK) signaling pathway [331]. Exposure to FKN may result in epithelial-to-mesenchymal transition (EMT) with enhanced CX3CR1+ cell migration, which is symptomatic of increased invasive and metastatic potential during prostate cancer progression. This mechanism of FKN-dependent EMT involves activation of tumor necrosis factor-α converting enzyme (TACE)/transforming growth factor-α (TGF-α)/epidermal growth factor receptor (EGFR)] signaling [98].
CX3CR1 was more highly expressed in spinal metastases than para-tumor tissue in breast cancer. Despite ambiguous results on the concentration of FKN in various breast cancer tissue samples, in vitro studies demonstrated the influence of FKN on the migration and invasion abilities of cancer cells mediated via the Src family kinase (Src)/focal adhesion kinase (FAK) signaling pathway following EGFR activation by ADAMs. Given the relatively high expression of FKN in spinal cancellous bone, CX3CR1-expressing metastatic tumor cells may be attracted [332]. An in vitro study demonstrated that the Src/FAK signaling pathway played a vital role in the FKN-dependent promotion of lung cancer cell migration and invasion [333].
5.2.2. FKN/CX3CR1 Signaling May Be a Good Prognostic Factor in Cancer
However, publications indicating the adverse effects of the FKN/CX3CR1 axis in promoting the growth and spread of various cancers are accompanied by an increasing number of contradictory reports. These cancers include frequently diagnosed cancers, such as colorectal cancer, breast cancer and lung cancer, in which the absence or low expression of FKN/CX3CR1 in tumor tissue is a poor prognostic factor associated with an increased risk of metastatic progression. The explanation for these observations may be clearly related to the activity of the immune response toward the tumor, which is determined by the chemotaxis of CX3XR1+ cells [334].
For example, the CX3CR1 gene has been identified as a hub gene in colorectal cancer, i.e., a gene that interacts with many other genes in the gene network and commonly plays a critical role in biological processes and gene regulation in the course of the disease [335,336,337]. Using the Tumor IMmune Estimation Resource (TIMER) database and CIBERSORT analysis, correlations between CX3CR1 and tumor-infiltrating immune cells were estimated in Yue et al. [338]. A co-culture of the human monocytic cell line THP-1-derived macrophages with the human colon carcinoma cell line HCT8 with low CX3CR1 expression was established in which immune marker expression, cell viability, and migration were investigated. Patients with low immune marker expression scores had significantly shorter survival than patients with high immune marker expression scores. CX3CR1 may act as a prognostic biomarker in colorectal cancer because its expression was associated with immune marker expression, immune cell infiltration levels and macrophage polarization. CX3CR1 contributes to the recruitment and regulation of immune-infiltrating cells and macrophage polarization in colorectal cancer. Therefore, silencing of CX3CR1 may promote the proliferation and migration of colorectal cancer cells [338].
Another study showed that co-expression of FKN and CX3CR1 in colorectal cancer cells (FKN-CX3CR1 axis-positive tumors) was associated with a significantly longer period of disease-free and disease-specific survival [339]. Therefore, the appropriate level of FKN-CX3CR1 axis activity in tumor cells acts as a retention factor, which likely increases homotypic cell adhesion and limits tumor spreading to metastatic sites. Conversely, no or low expression of FKN-CX3CR1 in axis-negative colorectal cancer cells poses an increased risk of tumor relapse and an increased likelihood of metachronous metastasis [339].
FKN overexpression may be a predictive biomarker for identifying antibody-dependent cellular cytotoxicity (ADCC)-based therapy responders [340]. FKN overexpression attracted tumor-suppressive lymphocytes, including NK cells, and inhibited tumor growth and lung metastasis in a syngeneic 4T1 cell line in a mouse model of breast cancer. Increased NK cell–mediated cytotoxicity acted synergistically with trastuzumab, a humanized anti-HER2 oncogene monoclonal antibody, for the treatment of breast cancer [340]. FKN overexpression in humanized tumor mice, which show human tumors and the human immune system, resulted in enhanced efficacy of trastuzumab treatment, especially in preventing lung metastases composed of FKN-overexpressing breast cancer cells [341].
The tumor growth-inhibiting effect of FKN, which is a derivative of increased NK cell activity, was also observed in an orthotopic implantation of a lung cancer model in vivo [342]. Analysis of lung cancer data from the Gene Expression Omnibus database and The Cancer Genome Atlas revealed that increased FKN mRNA expression in tumor tissues from lung adenocarcinoma patients was associated with improved overall survival (OS) and was thus a positive prognostic factor [343].
5.2.3. Possible Reasons for the Contradictory Results of FKN/CX3CR1 Signaling in Cancer
As cited in subsections 5.2.1. and 5.2.2., the results on the role of the FKN/CX3CR1 axis in common cancers remain clearly contradictory [84]. Although we notice the fragmentary nature of these reports, these results indicate that signaling involving this peculiar chemokine with adhesion molecule properties is more complex [346].
In addition to the heterogeneity of tumors mentioned in this chapter, the explanation for this surprising discrepancy in the interpretation of the impact of FKN (favorable vs. unfavorable) on the course of cancer may be that the pro-inflammatory, anti-apoptotic and pro-angiogenic effects mediated by the FKN/CX3CR1 axis are in opposition to the impact of this signaling pathway on immune system functions. CX3CR1-positive leukocytes, including CD4+ and CD8+ T cells, monocytes, B cells, neutrophils, natural killer (NK) cells and dendritic cells (DCs), are subject to sFKN-related chemotaxis [347]. Therefore, FKN is an important tumor-infiltrating lymphocyte (TIL)-recruiting chemokine and a key regulator of cytotoxic T-cell-mediated immunity. There is also growing evidence that the FKN pathway is involved in the maintenance of effector memory cytotoxic T-cell populations responsible for anti-viral and anti-tumor immunity [84].
The explanation for the efficacy of the immune response related to the FKN/CX3CR1 axis in various types of cancers (anti-tumor response) or lead to disease progression (pro-tumor response) is related to the presence of two forms of FKN: membrane-bound and soluble. Upregulated expression of FKN leads to increased accumulation of CX3CR1+ immune system cells in tumor tissue and significantly increases local CX3CR1 density with the possibility of CX3CR1 induction in tumor cells [146,338]. Simultaneous co-expression of mFKN and CX3CR1 in cancer cells leads to cell adhesion, which significantly impedes cellular migration and tumor spread [339].
However, this beneficial effect of FKN/CX3CR1 signaling may not occur when mFKN is cleaved by proteinases, such as ADAM10 or TACE/ADAM17, and the mFKN/sFKN balance is shifted in favor of the soluble form. Under these conditions, tumor cells no longer adhere to each other or adhere much weaker because the dominant CX3CR1 ligand becomes sFKN, which promotes chemotaxis. An increase in CX3CR1 expression increases sFKN-induced cancer cell migration [348]. The source of sFKN may be the cancer cells themselves and other TME components, e.g., fibroblasts [349,350]. Therefore, simultaneous increases in sFKN and CX3CR1 expression may be responsible for the pro-tumor effect of the FKN/CX3CR1 axis, which increases the risk of metastasis [326].
The transient or tumor-specific activity of ADAM10 or TACE/ADAM17 may play a key role because changing the mFKN:sFKN ratio modifies the signaling of the FKN/CX3CR1 axis to reveal anti- or pro-tumor effects [351,352].
Overall, uncontrolled proliferation and apoptosis resistance of cancer cells do not necessarily depend directly on CX3CR1 but rather on EFGR signaling [98]. This effect may be difficult to separate because some downstream signaling pathways related to FKN, including the PI3K/Akt/IKK/Iκβ/NF-κβ pathways, are also activated by EGFR stimulation [176,185].
The mechanisms determining the pro-tumor or anti-tumor effects of FKN/CX3CR1 signaling in cancer are summarized in Figure 5.
6. Concluding Remarks
The FKN/CX3CR1 signaling pathway plays an important role in angiogenesis, especially in the chronic inflammatory response. This role also applies to angiogenesis in the TME, where typical stimuli are hypoxia and inflammation, which modulate cell polarization and induce cell plasticity to promote tumorigenesis. Beneficial and detrimental effects are observed in angiogenesis accompanying inflammation, and angiogenesis within the TME is always related to tumor growth and metastasis. Therefore, there is an important need to determine how the FKN-CX3CR1 axis influences the course of cancer for therapeutic reasons. The difficulty is that the effect of FKN on the functioning of the immune system in the TME is complex and depends on the type of cancer and the heterogeneity of a specific tumor, which affects the expression of FKN and CX3CR1 in cells. The variable proportions of two forms of FKN, membrane-bound (mFKN) and soluble (sFKN), are responsible for adhesion and chemotaxis, respectively, and may determine the occurrence of antitumor or protumor effects. Therefore, FKN/CX3CR1 may be a favorable or unfavorable prognostic factor, with the evidence for antitumor functions primarily coming from prognostic studies, and the evidence for protumor function tends to be from tumor development studies. This paradoxical situation suggests that the FKN-CX3CR1 axis is a double-edged sword in cancer biology, which reveals a conflict between therapeutic goals. The essence of the development of new treatment methods must be to increase the FKN-dependent recruitment of CX3CR1+ immune cells, such as NK cells, DCs, and CD4+ and CD8+ T cells, to the TME. The homing of these cells to the TME may be enhanced by increasing CX3CR1 expression and/or increasing sFKN expression in the tumor. However, the increased expression of CX3CR1 in cancer cells that do not express mFKN and the migration of these cells under the influence of the chemotactic effect of sFKN should be prevented because it promotes tumor growth via angiogenesis and increases the risk of metastasis. Limiting treatment only to the FKN/CX3CR1 signaling pathway will not be fully effective due to the numerous processes involved in tumors, which result in cancer immune evasion.
Abbreviations
Å – Ångström, unit of length, equal to 10−10 m, or 0.1 nm
AC – adenylyl cyclase
ACKRs – atypical chemokine receptors
ADAM10 – a disintegrin and metalloproteinase domain-containing protein 10
ADAM17 – a disintegrin and metalloproteinase domain-containing protein 17, also known as tumor necrosis factor alpha (TNF-α) converting enzyme (TACE)
ADCC – antibody-dependent cellular cytotoxicity
Akt – protein kinase B
ALS – amyotrophic lateral sclerosis
AMI – acute myocardial infarction
AP – angina pectoris
AP2 – adapter protein 2
APC – antigen presenting cells
BCL2 – B-cell lymphoma 2 gene
BCL-xL – B-cell lymphoma extra-large gene
BM – bone marrow
BMSCs – bone marrow-derived mesenchymal stem cells
cAMP – cyclic adenosine monophosphate
cCKRs – conventional chemokine receptors
CCL26 – chemokine eotaxin-3
CCR2 – C-C chemokine receptor 2
CCR5 – C-C chemokine receptor type 5
CD – chemokine domain
CD106 – cluster of differentiation 106, an adhesion molecule
CD40 – cluster of differentiation 40 also known as – tumor necrosis factor receptor superfamily member 5 (TNFRSF5)
CD40L – cluster of differentiation 40 (CD40) ligand
CNS – central nervous system
CREB – cyclic adenosine monophosphate(cAMP)/Ca2+ response element binding protein
Cryo-EM – cryo-electron microscopy
CTCs – circulating tumor cells
CTS – cathepsin S
CX3CL1 – chemokine (C-X3-C motif) ligand 1, also known as fractalkine (FKN)
CX3CL1.35 – chemokine, US28-engineered fractalkine
CX3CR1 – high-affinity fractalkine (FKN) receptor or chemokine (C-X3-C motif) ligand 1 (CX3CL1) receptor, also known as G-protein coupled receptor 13 (GPR13), previously known as V28
DC – dendritic cell
ECM – extracellular matrix
EC – endothelial cell
ECL1–ECL3 – three extracellular loops within G protein-coupled receptor (GPCR)
EGFR – epidermal growth factor receptor, a member of the family closely related to receptor tyrosine kinases ErbB-1 (EGFR) and ErbB-2 (HER2/neu)
EMT – epithelial-to-mesenchymal transition
eNOS – endothelial nitric oxide synthase
EPCs – endothelial progenitor cells
ERK – extracellular signal-regulated kinase
FAK – focal adhesion kinase
FGR – fetal growth restriction
FKN – fractalkine also known as chemokine (C-X3-C motif) ligand 1 (CX3CL1)
FLSs – fibroblast-like synoviocytes
FOXO – member of the class O of forkhead box transcription factors
Gα, Gβ, Gγ – subunits of the heterotrimeric G-proteins (G protein complex)
Gαi – activated Gα subunit of the G protein complex
GDP – guanosine diphosphate
GPCR – G protein-coupled receptor
GPR domain – G-protein regulatory domain containing specific protein
GTP – guanosine triphosphate
HCC – hepatocellular carcinoma
HIF – hypoxia inducible factor
HIF-1α – hypoxia inducible factor 1 alpha
HIMECs – human intestinal microvascular endothelial cell
IBD – inflammatory bowel disease
ICL1–ICL3 – three intracellular loops within G protein-coupled receptor (GPCR)
IDD – intervertebral disc degeneration
IKK – IkappaBeta (Iκβ) kinase
IL-1, IL-1β, IL-6, IL-33 – interleukin-1, -1β, -6 and -33, respectively
IP3 – inositol 1,4,5-trisphosphate
ITGA5 – integrin alpha 5 also known as anti-CD49e antigen
JAK – Janus kinase
JNK – cJun NH(2)-terminal kinase
(KLRG1)+ NK cells – (killer cell lectin-like receptor subfamily G member 1)+ natural killer cells, a subset considered terminally differentiated
LPS – lipopolysaccharide
MACEs – major adverse cardiovascular events
MAPK – mitogen-activated protein kinase
MC – mast cells
MCP-1 – monocyte chemoattractant protein-1
MEK – mitogen-activated protein kinase kinase
MEKK – mitogen-activated protein kinase kinase (MEK) kinase
mFKN – membrane-bound form of fractalkine
MLN – mesenteric lymph node
MM – multiple myeloma
MMP-2 – matrix metalloprotease-2
MVO – microvascular obstruction
NAFLD – nonalcoholic fatty liver disease
NASH – nonalcoholic steatohepatitis
NCBI – National Centre for Biotechnology Information
NF-κβ – nuclear factor kappa-light-chain-enhancer of activated B cells
NK cell – natural killer cell
NO – nitric oxide
NRP-1, NRP-2 – neuropilin 1, neuropilin 2
NTx – N-terminal telopeptide of type I collagen
OA – osteoarthritis
OCPs – osteoclast precursors
P38 – mitogen-activated protein kinases
PAH – pulmonary arterial hypertension
PBHSC – peripheral blood-derived hematopoietic stem cells
PD – Parkinson’s disease
PDK1 – phosphoinositide-dependent kinase 1
PE – preeclampsia
PECAMN-1 – platelet-endothelial adhesion molecule-1 also known as CD31
pGlu – pyroglutamate
PI3K – phosphoinositide 3-kinase
PIP2 – phosphatidylinositol 4,5-biphosphate
PKC – protein kinase C
PLC – phospholipase C
QC – glutaminyl cyclase
RA – rheumatoid arthritis
Raf – Raf kinases
Ras – Ras kinases
RIC8 – a non-receptor guanine-nucleotide exchange factor for Gα subunits (also known as synembryn)
RMSD – root mean square deviation
RTKs – receptor tyrosine kinases
sFKN – soluble form of fractalkine
SNARE – vesicle-associated v-soluble N-ethylmaleimide-sensitive factor attachment protein receptor
SP – signal peptide
Src – steroid receptor coactivator
STAT – signal transducer and activator of transcription protein
STEMI – ST(segment)-elevation myocardial infarction
STX13 – protein syntaxin 13
TACE – tumor necrosis factor alpha (TNF-α) converting enzyme, also known as a disintegrin and metalloproteinase domain-containing protein 17
TGF-α – transforming growth factor alpha
TIE-2 – tyrosine kinase with immunoglobulin-like and EGF-like domains 2
TIL – tumor-infiltrating lymphocytes
TIMER – Tumor IMmune Estimation Resource database
TM1–TM7 – seven hydrophobic α-helical transmembrane segments or domains within G protein-coupled receptor (GPCR)
TME – tumor microenvironment
TNFα – tumor necrosis factor alpha
TNFRSF5 – tumor necrosis factor receptor superfamily member 5 also known as cluster of differentiation 40 (CD40)
TRACP-5b – tartrate-resistant acid phosphatase 5b
US28-engineered FKN – chemokine CX3CL1.35
VAMP3 – vesicle-associated membrane protein 3
VCAM-1 – vascular cell adhesion molecule 1 also known as vascular cell adhesion protein 1
VE-cadherin – vascular-endothelial cadherin also known as CD144
VEGF – vascular endothelial growth factor
VEGFR – vascular endothelial growth factor (VEGF) receptor
VEGFR-1, VEGFR-2 – vascular endothelial growth factor (VEGF) receptor type 1 and 2, respectively
VEGF-A – vascular endothelial growth factor (VEGF) isoform A
V/EVTI – vascular/extravascular tissue index
VPF – vascular permeability factor
VSMCs – vascular smooth muscle cells
Funding Statement
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created. Instead, the data are quoted from the available cited literature.
Conflicts of Interest
The author declares no conflict of interest.
References
- Adair TH, Montani JP. Angiogenesis. San Rafael (CA): Morgan & Claypool Life Sciences; 2010. Chapter 1, Overview of Angiogenesis. Available from: https://www.ncbi.nlm.nih.gov/books/NBK53238.
- Szewczyk G, Maciejewski TM, Szukiewicz D. Current progress in the inflammatory background of angiogenesis in gynecological cancers. Inflamm Res. 2019;68(4):247-260. [CrossRef]
- Yoder, MC. Yoder MC. Human endothelial progenitor cells. Cold Spring Harb Perspect Med. 2012 ;2(7): a006692. [CrossRef]
- Marçola M, Rodrigues CE. Endothelial progenitor cells in tumor angiogenesis: another brick in the wall. Stem Cells Int. 2015;2015:832649. [CrossRef]
- Ng CY, Cheung C. Origins and functional differences of blood endothelial cells. Semin Cell Dev Biol. 2024;155(Pt C):23-29. [CrossRef]
- Fujisawa T, Tura-Ceide O, Hunter A, Mitchell A, Vesey A, Medine C, Gallogly S, Hadoke PWF, Keith C, Sproul A, Roddie H, McQuaker G, Wilmut I, Mills NL, Brittan M. Endothelial Progenitor Cells Do Not Originate From the Bone Marrow. Circulation. 2019;140(18):1524-1526. [CrossRef]
- Li Z, Solomonidis EG, Meloni M, Taylor RS, Duffin R, Dobie R, Magalhaes MS, Henderson BEP, Louwe PA, D’Amico G, Hodivala-Dilke KM, Shah AM, Mills NL, Simons BD, Gray GA, Henderson NC, Baker AH, Brittan M. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur Heart J. 2019;40(30):2507-2520. [CrossRef]
- Ackermann M, Houdek JP, Gibney BC, Ysasi A, Wagner W, Belle J, Schittny JC, Enzmann F, Tsuda A, Mentzer SJ, Konerding MA. Sprouting and intussusceptive angiogenesis in postpneumonectomy lung growth: mechanisms of alveolar neovascularization. Angiogenesis. 2014;17(3):541-51. [CrossRef]
- Hickey MM, Simon MC. Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Curr Top Dev Biol. 2006;76:217-57. [CrossRef]
- Lin S, Chai Y, Zheng X, Xu X. The role of HIF in angiogenesis, lymphangiogenesis, and tumor microenvironment in urological cancers. Mol Biol Rep. 2023;51(1):14. [CrossRef]
- Yuan X, Ruan W, Bobrow B, Carmeliet P, Eltzschig HK. Targeting hypoxia-inducible factors: therapeutic opportunities and challenges. Nat Rev Drug Discov. 2023. [CrossRef]
- Wang Y, Yang Y, Yang Y, Dang Y, Guo Z, Zhuang Q, Zheng X, Wang F, Cheng N, Liu X, Guo W, Zhao B. Hypoxia induces hepatocellular carcinoma metastasis via the HIF-1α/METTL16/lnc-CSMD1-7/RBFOX2 axis. iScience. 2023;26(12):108495. [CrossRef]
- Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5’-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26(14):5336-47. [CrossRef]
- Feng Y, Luo S, Fan D, Guo X, Ma S. The role of vascular endothelial cells in tumor metastasis. Acta Histochem. 2023;125(6):152070. [CrossRef]
- Ebeling S, Kowalczyk A, Perez-Vazquez D, Mattiola I. Regulation of tumor angiogenesis by the crosstalk between innate immunity and endothelial cells. Front Oncol. 2023;13:1171794. [CrossRef]
- Bisht M, Dhasmana DC, Bist SS. Angiogenesis: Future of pharmacological modulation. Indian J Pharmacol. 2010;42(1):2-8. [CrossRef]
- Wong BW, Marsch E, Treps L, Baes M, Carmeliet P. Endothelial cell metabolism in health and disease: impact of hypoxia. EMBO J. 2017;36(15):2187-2203. [CrossRef]
- Senger DR, Davis GE. Angiogenesis. Cold Spring Harb Perspect Biol. 2011;3(8):a005090. [CrossRef]
- Nan W, He Y, Wang S, Zhang Y. Molecular mechanism of VE-cadherin in regulating endothelial cell behaviour during angiogenesis. Front Physiol. 2023;14:1234104. [CrossRef]
- Wen JH, Choi O, Taylor-Weiner H, Fuhrmann A, Karpiak JV, Almutairi A, Engler AJ. Haptotaxis is cell type specific and limited by substrate adhesiveness. Cell Mol Bioeng. 2015; 8(4):530-542. [CrossRef]
- Kazerounian S, Lawler J. Integration of pro- and anti-angiogenic signals by endothelial cells. J Cell Commun Signal. 2018;12(1):171-179. [CrossRef]
- Geindreau M, Bruchard M, Vegran F. Role of Cytokines and Chemokines in Angiogenesis in a Tumor Context. Cancers (Basel). 2022;14(10):2446. [CrossRef]
- Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. 2020;77(9):1745-1770. [CrossRef]
- Balogh E, Biniecka M, Fearon U, Veale DJ, Szekanecz Z. Angiogenesis in Inflammatory Arthritis. Isr Med Assoc J. 2019;21(5):345-352.
- Lu E, Li C, Wang J, Zhang C. Inflammation and angiogenesis in the corpus luteum. J Obstet Gynaecol Res. 2019;45(10):1967-1974. [CrossRef]
- Corliss BA, Azimi MS, Munson JM, Peirce SM, Murfee WL. Macrophages: An Inflammatory Link Between Angiogenesis and Lymphangiogenesis. Microcirculation. 2016;23(2):95-121. [CrossRef]
- Walsh DA, Pearson CI. Angiogenesis in the pathogenesis of inflammatory joint and lung diseases. Arthritis Res. 2001;3(3):147-53. [CrossRef]
- Tsoupras A, Lordan R, Zabetakis I. Inflammation, not Cholesterol, Is a Cause of Chronic Disease. Nutrients. 2018 May 12;10(5):604. [CrossRef]
- Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2017;9(6):7204-7218. [CrossRef]
- Fiedler U, Augustin HG. Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol. 2006;27(12):552-8. [CrossRef]
- Jeong JH, Ojha U, Lee YM. Pathological angiogenesis and inflammation in tissues. Arch Pharm Res. 2021;44(1):1-15. [CrossRef]
- Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11(2):109-19. [CrossRef]
- Mehrad B, Keane MP, Strieter RM. Chemokines as mediators of angiogenesis. Thromb Haemost. 2007;97(5):755-62.
- Miller MC, Mayo KH. Chemokines from a Structural Perspective. Int J Mol Sci. 2017; 18(10):2088. [CrossRef]
- Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. 2018;285(16): 2944-2971. [CrossRef]
- Kufareva I, Salanga CL, Handel TM. Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol Cell Biol. 2015;93(4):372-83. [CrossRef]
- Legler DF, Thelen M. New insights in chemokine signaling. F1000Res. 2018;7:95. [CrossRef]
- Stone MJ, Hayward JA, Huang C, E Huma Z, Sanchez J. Mechanisms of Regulation of the Chemokine-Receptor Network. Int J Mol Sci. 2017;18(2):342. [CrossRef]
- Portella L, Bello AM, Scala S. CXCL12 Signaling in the Tumor Microenvironment. Adv Exp Med Biol. 2021;1302:51-70. [CrossRef]
- Nazari A, Khorramdelazad H, Hassanshahi G. Biological/pathological functions of the CXCL12/CXCR4/CXCR7 axes in the pathogenesis of bladder cancer. Int J Clin Oncol. 2017; 22(6):991-1000. [CrossRef]
- Shakir M, Tang D, Zeh HJ, Tang SW, Anderson CJ, Bahary N, Lotze MT. The chemokine receptors CXCR4/CXCR7 and their primary heterodimeric ligands CXCL12 and CXCL12/high mobility group box 1 in pancreatic cancer growth and development: finding flow. Pancreas. 2015;44(4):528-34. [CrossRef]
- Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91(4):521-30. [CrossRef]
- Wojdasiewicz P, Poniatowski LA, Kotela A, Deszczyński J, Kotela I, Szukiewicz D. The chemokine CX3CL1 (fractalkine) and its receptor CX3CR1: occurrence and potential role in osteoarthritis. Arch Immunol Ther Exp (Warsz). 2014;62(5):395-403. [CrossRef]
- Loh SX, Ekinci Y, Spray L, Jeyalan V, Olin T, Richardson G, Austin D, Alkhalil M, Spyridopoulos I. Fractalkine Signalling (CX3CL1/CX3CR1 Axis) as an Emerging Target in Coronary Artery Disease. J Clin Med. 2023;12(14):4821. [CrossRef]
- Imaizumi T, Yoshida H, Satoh K. Regulation of CX3CL1/fractalkine expression in endothelial cells. J Atheroscler Thromb. 2004;11(1):15-21. [CrossRef]
- Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385(6617):640-4. [CrossRef]
- Pan Y, Lloyd C, Zhou H, Dolich S, Deeds J, Gonzalo JA, Vath J, Gosselin M, Ma J, Dussault B, Woolf E, Alperin G, Culpepper J, Gutierrez-Ramos JC, Gearing D. Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature. 1997;387(6633):611-7. Erratum in: Nature 1997 Sep 4;389(6646):100. [CrossRef]
- Hortsch M, Patel NH, Bieber AJ, Traquina ZR, Goodman CS. Drosophila neurotactin, a surface glycoprotein with homology to serine esterases, is dynamically expressed during embryogenesis. Development. 1990;110(4):1327-40. [CrossRef]
- Zhuang Q, Cheng K, Ming Y. CX3CL1/CX3CR1 Axis, as the Therapeutic Potential in Renal Diseases: Friend or Foe? Curr Gene Ther. 2017;17(6):442-452. [CrossRef]
- D’Haese JG, Demir IE, Friess H, Ceyhan GO. Fractalkine/CX3CR1: why a single chemokine-receptor duo bears a major and unique therapeutic potential. Expert Opin Ther Targets. 2010;14(2):207-19. [CrossRef]
- Wu CY, Peng PW, Renn TY, Lee CJ, Chang TM, Wei AI, Liu JF. CX3CL1 induces cell migration and invasion through ICAM-1 expression in oral squamous cell carcinoma cells. J Cell Mol Med. 2023;27(11):1509-1522. [CrossRef]
- White GE, Greaves DR. Fractalkine: a survivor’s guide: chemokines as antiapoptotic mediators. Arterioscler Thromb Vasc Biol. 2012;32(3):589-94. [CrossRef]
- Lucas AD, Chadwick N, Warren BF, Jewell DP, Gordon S, Powrie F, Greaves DR. The transmembrane form of the CX3CL1 chemokine fractalkine is expressed predominantly by epithelial cells in vivo. Am J Pathol. 2001;158(3):855-66. [CrossRef]
- Jones BA, Beamer M, Ahmed S. Fractalkine/CX3CL1: a potential new target for inflammatory diseases. Mol Interv. 2010;10(5):263-70. [CrossRef]
- O’Sullivan SA, Gasparini F, Mir AK, Dev KK. Fractalkine shedding is mediated by p38 and the ADAM10 protease under pro-inflammatory conditions in human astrocytes. J Neuroinflammation. 2016;13(1):189. [CrossRef]
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102(4):1186-95. [CrossRef]
- Poniatowski ŁA, Wojdasiewicz P, Krawczyk M, Szukiewicz D, Gasik R, Kubaszewski Ł, Kurkowska-Jastrzębska I. Analysis of the Role of CX3CL1 (Fractalkine) and Its Receptor CX3CR1 in Traumatic Brain and Spinal Cord Injury: Insight into Recent Advances in Actions of Neurochemokine Agents. Mol Neurobiol. 2017;54(3):2167-2188. [CrossRef]
- Jones BA, Riegsecker S, Rahman A, Beamer M, Aboualaiwi W, Khuder SA, Ahmed S. Role of ADAM-17, p38 MAPK, cathepsins, and the proteasome pathway in the synthesis and shedding of fractalkine/CX₃ CL1 in rheumatoid arthritis. Arthritis Rheum. 2013;65(11):2814-25. [CrossRef]
- Bourd-Boittin K, Basset L, Bonnier D, L’helgoualc’h A, Samson M, Théret N. CX3CL1/fractalkine shedding by human hepatic stellate cells: contribution to chronic inflammation in the liver. J Cell Mol Med. 2009;13(8A):1526-35. [CrossRef]
- Uchida M, Ito T, Nakamura T, Igarashi H, Oono T, Fujimori N, Kawabe K, Suzuki K, Jensen RT, Takayanagi R. ERK pathway and sheddases play an essential role in ethanol-induced CX3CL1 release in pancreatic stellate cells. Lab Invest. 2013;93(1):41-53. [CrossRef]
- Lu, X. Lu X. Structure and Function of Ligand CX3CL1 and its Receptor CX3CR1 in Cancer. Curr Med Chem. 2022;29(41):6228-6246. [CrossRef]
- Iemmolo M, Ghersi G, Bivona G. The Cytokine CX3CL1 and ADAMs/MMPs in Concerted Cross-Talk Influencing Neurodegenerative Diseases. Int J Mol Sci. 2023;24(9):8026. [CrossRef]
- Turner SL, Mangnall D, Bird NC, Blair-Zajdel ME, Bunning RA. Effects of pro-inflammatory cytokines on the production of soluble fractalkine and ADAM17 by HepG2 cells. J Gastrointestin Liver Dis. 2010;19(3):265-71.
- Fonović UP, Jevnikar Z, Kos J. Cathepsin S generates soluble CX3CL1 (fractalkine) in vascular smooth muscle cells. Biol Chem. 2013;394(10):1349-52. [CrossRef]
- Hundhausen C, Schulte A, Schulz B, Andrzejewski MG, Schwarz N, von Hundelshausen P, Winter U, Paliga K, Reiss K, Saftig P, Weber C, Ludwig A. Regulated shedding of transmembrane chemokines by the disintegrin and metalloproteinase 10 facilitates detachment of adherent leukocytes. J Immunol. 2007;178(12):8064-72. [CrossRef]
- Scarselli M, Donaldson JG. Constitutive internalization of G protein-coupled receptors and G proteins via clathrin-independent endocytosis. J Biol Chem. 2009;284(6):3577-85. [CrossRef]
- Hattermann K, Gebhardt H, Krossa S, Ludwig A, Lucius R, Held-Feindt J, Mentlein R. Transmembrane chemokines act as receptors in a novel mechanism termed inverse signaling. Elife. 2016;5:e10820. [CrossRef]
- Shimaoka T, Nakayama T, Fukumoto N, Kume N, Takahashi S, Yamaguchi J, Minami M, Hayashida K, Kita T, Ohsumi J, Yoshie O, Yonehara S. Cell surface-anchored SR-PSOX/CXC chemokine ligand 16 mediates firm adhesion of CXC chemokine receptor 6-expressing cells. J Leukoc Biol. 2004;75(2):267-74. [CrossRef]
- Ostuni MA, Hermand P, Saindoy E, Guillou N, Guellec J, Coens A, Hattab C, Desuzinges-Mandon E, Jawhari A, Iatmanen-Harbi S, Lequin O, Fuchs P, Lacapere JJ, Combadière C, Pincet F, Deterre P. CX3CL1 homo-oligomerization drives cell-to-cell adherence. Sci Rep. 2020;10(1): 9069. [CrossRef]
- Lee M, Lee Y, Song J, Lee J, Chang SY. Tissue-specific Role of CX3CR1 Expressing Immune Cells and Their Relationships with Human Disease. Immune Netw. 2018;18(1):e5. [CrossRef]
- Ni Y, Zhuge F, Ni L, Nagata N, Yamashita T, Mukaida N, Kaneko S, Ota T, Nagashimada M. CX3CL1/CX3CR1 interaction protects against lipotoxicity-induced nonalcoholic steatohepatitis by regulating macrophage migration and M1/M2 status. Metabolism. 2022;136:155272. [CrossRef]
- Guo S, Dong L, Li J, Chen Y, Yao Y, Zeng R, Shushakova N, Haller H, Xu G, Rong S. C-X3-C motif chemokine ligand 1/receptor 1 regulates the M1 polarization and chemotaxis of macrophages after hypoxia/reoxygenation injury. Chronic Dis Transl Med. 2021;7(4):254-265. [CrossRef]
- Cormican S, Griffin MD. Fractalkine (CX3CL1) and Its Receptor CX3CR1: A Promising Therapeutic Target in Chronic Kidney Disease? Front Immunol. 2021;12:664202. [CrossRef]
- Kerfoot SM, Lord SE, Bell RB, Gill V, Robbins SM, Kubes P. Human fractalkine mediates leukocyte adhesion but not capture under physiological shear conditions; a mechanism for selective monocyte recruitment. Eur J Immunol. 2003;33(3):729-39. [CrossRef]
- Goda S, Imai T, Yoshie O, Yoneda O, Inoue H, Nagano Y, Okazaki T, Imai H, Bloom ET, Domae N, Umehara H. CX3C-chemokine, fractalkine-enhanced adhesion of THP-1 cells to endothelial cells through integrin-dependent and -independent mechanisms. J Immunol. 2000;164(8):4313-20. [CrossRef]
- Flierl U, Bauersachs J, Schäfer A. Modulation of platelet and monocyte function by the chemokine fractalkine (CX3 CL1) in cardiovascular disease. Eur J Clin Invest. 2015;45(6):624-33. [CrossRef]
- Umehara H, Imai T. Role of fractalkine in leukocyte adhesion and migration and in vascular injury. Drug News Perspect. 2001;14(8):460-4. [CrossRef]
- Hermand P, Pincet F, Carvalho S, Ansanay H, Trinquet E, Daoudi M, Combadière C, Deterre P. Functional adhesiveness of the CX3CL1 chemokine requires its aggregation. Role of the transmembrane domain. J Biol Chem. 2008;283(44):30225-34. [CrossRef]
- Ostuni MA, Guellec J, Hermand P, Durand P, Combadière C, Pincet F, Deterre P. CX3CL1, a chemokine finely tuned to adhesion: critical roles of the stalk glycosylation and the membrane domain. Biol Open. 2014;3(12):1173-82. [CrossRef]
- Huang YW, Su P, Liu GY, Crow MR, Chaukos D, Yan H, Robinson LA. Constitutive endocytosis of the chemokine CX3CL1 prevents its degradation by cell surface metalloproteases. J Biol Chem. 2009;284(43):29644-53. [CrossRef]
- Wong HS, Jaumouillé V, Heit B, Doodnauth SA, Patel S, Huang YW, Grinstein S, Robinson LA. Cytoskeletal confinement of CX3CL1 limits its susceptibility to proteolytic cleavage by ADAM10. Mol Biol Cell. 2014;25(24):3884-99. [CrossRef]
- Liu GY, Kulasingam V, Alexander RT, Touret N, Fong AM, Patel DD, Robinson LA. Recycling of the membrane-anchored chemokine, CX3CL1. J Biol Chem. 2005;280(20):19858-66. [CrossRef]
- Miller AF, Falke JJ. Chemotaxis receptors and signaling. Adv Protein Chem. 2004;68:393-444. [CrossRef]
- Conroy MJ, Lysaght J. CX3CL1 Signaling in the Tumor Microenvironment. Adv Exp Med Biol. 2020;1231:1-12. [CrossRef]
- Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36(5):705-16. [CrossRef]
- Richmond, A. Richmond A. Chemokine research moves on. Exp Cell Res. 2011;317(5):553-5. [CrossRef]
- Mortier A, Van Damme J, Proost P. Overview of the mechanisms regulating chemokine activity and availability. Immunol Lett. 2012;145(1-2):2-9. [CrossRef]
- Johnston B, Butcher EC. Chemokines in rapid leukocyte adhesion triggering and migration. Semin Immunol. 2002;14(2):83-92. [CrossRef]
- Nanki T, Imai T, Nagasaka K, Urasaki Y, Nonomura Y, Taniguchi K, Hayashida K, Hasegawa J, Yoshie O, Miyasaka N. Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheum. 2002;46(11):2878-83. [CrossRef]
- Hamann I, Unterwalder N, Cardona AE, Meisel C, Zipp F, Ransohoff RM, Infante-Duarte C. Analyses of phenotypic and functional characteristics of CX3CR1-expressing natural killer cells. Immunology. 2011 ;133(1):62-73. [CrossRef]
- Mionnet C, Buatois V, Kanda A, Milcent V, Fleury S, Lair D, Langelot M, Lacoeuille Y, Hessel E, Coffman R, Magnan A, Dombrowicz D, Glaichenhaus N, Julia V. CX3CR1 is required for airway inflammation by promoting T helper cell survival and maintenance in inflamed lung. Nat Med. 2010;16(11):1305-12. [CrossRef]
- Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. 2003;197(12):1701-7. [CrossRef]
- Papadopoulos EJ, Fitzhugh DJ, Tkaczyk C, Gilfillan AM, Sassetti C, Metcalfe DD, Hwang ST. Mast cells migrate, but do not degranulate, in response to fractalkine, a membrane-bound chemokine expressed constitutively in diverse cells of the skin. Eur J Immunol. 2000;30(8):2355-61. [CrossRef]
- Beck GCh, Ludwig F, Schulte J, van Ackern K, van der Woude FJ, Yard BA. Fractalkine is not a major chemoattractant for the migration of neutrophils across microvascular endothelium. Scand J Immunol. 2003;58(2):180-7. [CrossRef]
- Hall JD, Kurtz SL, Rigel NW, Gunn BM, Taft-Benz S, Morrison JP, Fong AM, Patel DD, Braunstein M, Kawula TH. The impact of chemokine receptor CX3CR1 deficiency during respiratory infections with Mycobacterium tuberculosis or Francisella tularensis. Clin Exp Immunol. 2009;156(2):278-84. [CrossRef]
- Volin MV, Huynh N, Klosowska K, Reyes RD, Woods JM. Fractalkine-induced endothelial cell migration requires MAP kinase signaling. Pathobiology. 2010;77(1):7-16. [CrossRef]
- Liu JF, Tsao YT, Hou CH. Fractalkine/CX3CL1 induced intercellular adhesion molecule-1-dependent tumor metastasis through the CX3CR1/PI3K/Akt/NF-κB pathway in human osteosarcoma. Oncotarget. 2016;8(33):54136-54148. [CrossRef]
- Tang J, Xiao L, Cui R, Li D, Zheng X, Zhu L, Sun H, Pan Y, Du Y, Yu X. CX3CL1 increases invasiveness and metastasis by promoting epithelial-to-mesenchymal transition through the TACE/TGF-α/EGFR pathway in hypoxic androgen-independent prostate cancer cells. Oncol Rep. 2016;35(2):1153-62. [CrossRef]
- Garin A, Pellet P, Deterre P, Debré P, Combadière C. Cloning and functional characterization of the human fractalkine receptor promoter regions. Biochem J. 2002;368(Pt 3):753-60. [CrossRef]
- Raport CJ, Schweickart VL, Eddy RL Jr, Shows TB, Gray PW. The orphan G-protein-coupled receptor-encoding gene V28 is closely related to genes for chemokine receptors and is expressed in lymphoid and neural tissues. Gene. 1995;163(2):295-9. [CrossRef]
- Combadiere C, Gao J, Tiffany HL, Murphy PM. Gene cloning, RNA distribution, and functional expression of mCX3CR1, a mouse chemotactic receptor for the CX3C chemokine fractalkine. Biochem Biophys Res Commun. 1998;253(3):728-32. [CrossRef]
- Zhuang Q, Ou J, Zhang S, Ming Y. Crosstalk between the CX3CL1/CX3CR1 Axis and Inflammatory Signaling Pathways in Tissue Injury. Curr Protein Pept Sci. 2019;20(8):844-854. [CrossRef]
- Schwarz N, Pruessmeyer J, Hess FM, Dreymueller D, Pantaler E, Koelsch A, Windoffer R, Voss M, Sarabi A, Weber C, Sechi AS, Uhlig S, Ludwig A. Requirements for leukocyte transmigration via the transmembrane chemokine CX3CL1. Cell Mol Life Sci. 2010;67(24):4233-48. [CrossRef]
- Raucci R, Costantini S, Castello G, Colonna G. An overview of the sequence features of N- and C-terminal segments of the human chemokine receptors. Cytokine. 2014;70(2):141-50. [CrossRef]
- Szpakowska M, Perez Bercoff D, Chevigné A. Closing the ring: a fourth extracellular loop in chemokine receptors. Sci Signal. 2014;7(341):pe21. [CrossRef]
- Nomiyama H, Yoshie O. Functional roles of evolutionary conserved motifs and residues in vertebrate chemokine receptors. J Leukoc Biol. 2015;97(1):39-47. [CrossRef]
- Mafi A, Kim SK, Goddard WA 3rd. The mechanism for ligand activation of the GPCR-G protein complex. Proc Natl Acad Sci U S A. 2022;119(18):e2110085119. [CrossRef]
- Tardáguila M, Mañes S. Tardáguila M, Mañes S. The complex role of chemokines in cancer: The case of the CX3CL1/CX3CR1 axis. In Oncology Theory & Practice, 1st ed.; iConcept Press Ltd.: Madrid, Spain, 2014; Chapter 8.
- Chaudhri A, Bu X, Wang Y, Gomez M, Torchia JA, Hua P, Hung SH, Davies MA, Lizee GA, von Andrian U, Hwu P, Freeman GJ. The CX3CL1-CX3CR1 chemokine axis can contribute to tumor immune evasion and blockade with a novel CX3CR1 monoclonal antibody enhances response to anti-PD-1 immunotherapy. Front Immunol. 2023;14:1237715. [CrossRef]
- Goode-Romero G, Dominguez L. Computational study of the conformational ensemble of CX3C chemokine receptor 1 (CX3CR1) and its interactions with antagonist and agonist ligands. J Mol Graph Model. 2022;117:108278. [CrossRef]
- Mizoue LS, Bazan JF, Johnson EC, Handel TM. Solution structure and dynamics of the CX3C chemokine domain of fractalkine and its interaction with an N-terminal fragment of CX3CR1. Biochemistry. 1999;38(5):1402-14. [CrossRef]
- Nakayama T, Watanabe Y, Oiso N, Higuchi T, Shigeta A, Mizuguchi N, Katou F, Hashimoto K, Kawada A, Yoshie O. Eotaxin-3/CC chemokine ligand 26 is a functional ligand for CX3CR1. J Immunol. 2010;185(11):6472-9. [CrossRef]
- Faure S, Meyer L, Costagliola D, Vaneensberghe C, Genin E, Autran B, Delfraissy JF, McDermott DH, Murphy PM, Debré P, Théodorou I, Combadière C. Rapid progression to AIDS in HIV+ individuals with a structural variant of the chemokine receptor CX3CR1. Science. 2000;287(5461):2274-7. [CrossRef]
- Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, Littman DR, Reinecker HC. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307(5707):254-8. [CrossRef]
- Ludeman JP, Stone MJ. The structural role of receptor tyrosine sulfation in chemokine recognition. Br J Pharmacol. 2014;171(5):1167-79. [CrossRef]
- Villaseca S, Romero G, Ruiz MJ, Pérez C, Leal JI, Tovar LM, Torrejón M. Gαi protein subunit: A step toward understanding its non-canonical mechanisms. Front Cell Dev Biol. 2022; 10:941870. [CrossRef]
- Lu M, Zhao W, Han S, Lin X, Xu T, Tan Q, Wang M, Yi C, Chu X, Yang W, Zhu Y, Wu B, Zhao Q. Activation of the human chemokine receptor CX3CR1 regulated by cholesterol. Sci Adv. 2022; 8(26):eabn8048. [CrossRef]
- Laganà M, Schlecht-Louf G, Bachelerie F. The G Protein-Coupled Receptor Kinases (GRKs) in Chemokine Receptor-Mediated Immune Cell Migration: From Molecular Cues to Physiopathology. Cells. 2021;10(1):75. [CrossRef]
- Niessner A, Marculescu R, Haschemi A, Endler G, Zorn G, Weyand CM, Maurer G, Mannhalter C, Wojta J, Wagner O, Huber K. Opposite effects of CX3CR1 receptor polymorphisms V249I and T280M on the development of acute coronary syndrome. A possible implication of fractalkine in inflammatory activation. Thromb Haemost. 2005;93(5):949-54. [CrossRef]
- Wu J, Yin RX, Lin QZ, Guo T, Shi GY, Sun JQ, Shen SW, Li Q. Two polymorphisms in the Fractalkine receptor CX3CR1 gene influence the development of atherosclerosis: a meta-analysis. Dis Markers. 2014;2014:913678. [CrossRef]
- Chamera K, Szuster-Głuszczak M, Basta-Kaim A. Shedding light on the role of CX3CR1 in the pathogenesis of schizophrenia. Pharmacol Rep. 2021;73(4):1063-1078. [CrossRef]
- Sakai M, Takeuchi H, Yu Z, Kikuchi Y, Ono C, Takahashi Y, Ito F, Matsuoka H, Tanabe O, Yasuda J, Taki Y, Kawashima R, Tomita H. Polymorphisms in the microglial marker molecule CX3CR1 affect the blood volume of the human brain. Psychiatry Clin Neurosci. 2018;72(6):409-422. [CrossRef]
- Imai T, Yasuda N. Therapeutic intervention of inflammatory/immune diseases by inhibition of the fractalkine (CX3CL1)-CX3CR1 pathway. Inflamm Regen. 2016;36:9. [CrossRef]
- Mirzadegan T, Benkö G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42(10):2759-67. [CrossRef]
- Joost P, Methner A. Phylogenetic analysis of 277 human G-protein-coupled receptors as a tool for the prediction of orphan receptor ligands. Genome Biol. 2002;3(11):RESEARCH0063. [CrossRef]
- Zhou XE, Melcher K, Xu HE. Structure and activation of rhodopsin. Acta Pharmacol Sin. 2012; 33(3):291-9. [CrossRef]
- Lefkowitz, RJ. Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci. 2004;25(8):413-22. [CrossRef]
- Vischer HF, Hulshof JW, de Esch IJ, Smit MJ, Leurs R. Virus-encoded G-protein-coupled receptors: constitutively active (dys)regulators of cell function and their potential as drug target. Ernst Schering Found Symp Proc. 2006;(2):187-209. [CrossRef]
- Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, Angelini A, Waghray D, Dror RO, Ploegh HL, Garcia KC. Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science. 2015;347(6226): 1113-7. [CrossRef]
- Hjortø GM, Kiilerich-Pedersen K, Selmeczi D, Kledal TN, Larsen NB. Human cytomegalovirus chemokine receptor US28 induces migration of cells on a CX3CL1-presenting surface. J Gen Virol. 2013;94(Pt 5):1111-1120. [CrossRef]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3(9):639-50. [CrossRef]
- Palczewski, K. Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743-67. [CrossRef]
- Gustavsson M, Zheng Y, Handel TM. Production of Chemokine/Chemokine Receptor Complexes for Structural Biophysical Studies. Methods Enzymol. 2016;570:233-60. [CrossRef]
- Miles TF, Spiess K, Jude KM, Tsutsumi N, Burg JS, Ingram JR, Waghray D, Hjorto GM, Larsen O, Ploegh HL, Rosenkilde MM, Garcia KC. Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. Elife. 2018;7:e35850. [CrossRef]
- Tsutsumi N, Maeda S, Qu Q, Vögele M, Jude KM, Suomivuori CM, Panova O, Waghray D, Kato HE, Velasco A, Dror RO, Skiniotis G, Kobilka BK, Garcia KC. Atypical structural snapshots of human cytomegalovirus GPCR interactions with host G proteins. Sci Adv. 2022;8(3):eabl5442. [CrossRef]
- Zhou Q, Yang D, Wu M, Guo Y, Guo W, Zhong L, Cai X, Dai A, Jang W, Shakhnovich EI, Liu ZJ, Stevens RC, Lambert NA, Babu MM, Wang MW, Zhao S. Common activation mechanism of class A GPCRs. Elife. 2019;8:e50279. [CrossRef]
- Syrovatkina V, Alegre KO, Dey R, Huang XY. Regulation, Signaling, and Physiological Functions of G-Proteins. J Mol Biol. 2016;428(19):3850-68. [CrossRef]
- Lagerström MC, Schiöth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7(4):339-57. Erratum in: Nat Rev Drug Discov. 2008;7(6):542. [CrossRef]
- Cancellieri C, Vacchini A, Locati M, Bonecchi R, Borroni EM. Atypical chemokine receptors: from silence to sound. Biochem Soc Trans. 2013;41(1):231-6. [CrossRef]
- Wingler LM, Lefkowitz RJ. Conformational Basis of G Protein-Coupled Receptor Signaling Versatility. Trends Cell Biol. 2020;30(9):736-747. [CrossRef]
- Hoffmann C, Zürn A, Bünemann M, Lohse MJ. Conformational changes in G-protein-coupled receptors-the quest for functionally selective conformations is open. Br J Pharmacol. 2008; 153 Suppl 1(Suppl 1):S358-66. [CrossRef]
- Hamza A, Samad A, Parray ZA, Ara S, Ahmed A, Almajhdi FN, Hussain T, Islam A, Parveen S. Mutation in the CX3C Motif of G Protein Disrupts Its Interaction with Heparan Sulfate: A Calorimetric, Spectroscopic, and Molecular Docking Study. Int J Mol Sci. 2022;23(4):1950. [CrossRef]
- Darbandi-Tehrani K, Hermand P, Carvalho S, Dorgham K, Couvineau A, Lacapère JJ, Combadière C, Deterre P. Subtle conformational changes between CX3CR1 genetic variants as revealed by resonance energy transfer assays. FASEB J. 2010;24(11):4585-98. [CrossRef]
- Ishizuka K, Fujita Y, Kawabata T, Kimura H, Iwayama Y, Inada T, Okahisa Y, Egawa J, Usami M, Kushima I, Uno Y, Okada T, Ikeda M, Aleksic B, Mori D, Someya T, Yoshikawa T, Iwata N, Nakamura H, Yamashita T, Ozaki N. Rare genetic variants in CX3CR1 and their contribution to the increased risk of schizophrenia and autism spectrum disorders. Transl Psychiatry. 2017;7 (8): e1184. [CrossRef]
- Lee S, Latha K, Manyam G, Yang Y, Rao A, Rao G. Role of CX3CR1 signaling in malignant transformation of gliomas. Neuro Oncol. 2020;22(10):1463-1473. [CrossRef]
- Rivas-Fuentes S, Salgado-Aguayo A, Arratia-Quijada J, Gorocica-Rosete P. Regulation and biological functions of the CX3CL1-CX3CR1 axis and its relevance in solid cancer: A mini-review. J Cancer. 2021;12(2):571-583. [CrossRef]
- Kehlen A, Haegele M, Böhme L, Cynis H, Hoffmann T, Demuth HU. N-terminal pyroglutamate formation in CX3CL1 is essential for its full biologic activity. Biosci Rep. 2017; 37(4):BSR20170712. [CrossRef]
- Maciejewski-Lenoir D, Chen S, Feng L, Maki R, Bacon KB. Characterization of fractalkine in rat brain cells: migratory and activation signals for CX3CR-1-expressing microglia. J Immunol. 1999;163(3):1628-35.
- Pallandre JR, Krzewski K, Bedel R, Ryffel B, Caignard A, Rohrlich PS, Pivot X, Tiberghien P, Zitvogel L, Strominger JL, Borg C. Dendritic cell and natural killer cell cross-talk: a pivotal role of CX3CL1 in NK cytoskeleton organization and activation. Blood. 2008;112(12):4420-4. [CrossRef]
- Foussat A, Coulomb-L’Hermine A, Gosling J, Krzysiek R, Durand-Gasselin I, Schall T, Balian A, Richard Y, Galanaud P, Emilie D. Fractalkine receptor expression by T lymphocyte subpopulations and in vivo production of fractalkine in human. Eur J Immunol. 2000;30(1): 87-97. [CrossRef]
- Chidambaram H, Das R, Chinnathambi S. Interaction of Tau with the chemokine receptor, CX3CR1 and its effect on microglial activation, migration and proliferation. Cell Biosci. 2020; 10:109. [CrossRef]
- Chen Y, Green SR, Almazan F, Quehenberger O. The amino terminus and the third extracellular loop of CX3CR1 contain determinants critical for distinct receptor functions. Mol Pharmacol. 2006;69(3):857-65. [CrossRef]
- Kharche S, Joshi M, Chattopadhyay A, Sengupta D. Conformational plasticity and dynamic interactions of the N-terminal domain of the chemokine receptor CXCR1. PLoS Comput Biol. 2021;17(5):e1008593.
- Srivastava D, Gakhar L, Artemyev NO. Structural underpinnings of Ric8A function as a G-protein α-subunit chaperone and guanine-nucleotide exchange factor. Nat Commun. 2019;10(1):3084. [CrossRef]
- Wright SJ, Inchausti R, Eaton CJ, Krystofova S, Borkovich KA. RIC8 is a guanine-nucleotide exchange factor for Galpha subunits that regulates growth and development in Neurospora crassa. Genetics. 2011;189(1):165-76. [CrossRef]
- Weis WI, Kobilka BK. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu Rev Biochem. 2018;87:897-919. [CrossRef]
- Arnoux I, Audinat E. Fractalkine Signaling and Microglia Functions in the Developing Brain. Neural Plast. 2015;2015:689404. [CrossRef]
- Lee YS, Morinaga H, Kim JJ, Lagakos W, Taylor S, Keshwani M, Perkins G, Dong H, Kayali AG, Sweet IR, Olefsky J. The fractalkine/CX3CR1 system regulates β cell function and insulin secretion. Cell. 2013;153(2):413-25. [CrossRef]
- Wang A, Yang T, Zhang L, Jia L, Wu Q, Yao S, Xu J, Yang H. IP3-Mediated Calcium Signaling Is Involved in the Mechanism of Fractalkine-Induced Hyperalgesia Response. Med Sci Monit. 2018;24:8804-8811. [CrossRef]
- Wojdasiewicz P, Turczyn P, Dobies-Krzesniak B, Frasunska J, Tarnacka B. Role of CX3CL1/CX3CR1 Signaling Axis Activity in Osteoporosis. Mediators Inflamm. 2019;2019:7570452. [CrossRef]
- Lyons A, Lynch AM, Downer EJ, Hanley R, O’Sullivan JB, Smith A, Lynch MA. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem. 2009;110(5):1547-56. [CrossRef]
- Park J, Song KH, Ha H. Fractalkine increases mesangial cell proliferation through reactive oxygen species and mitogen-activated protein kinases. Transplant Proc. 2012;44(4):1026-8. [CrossRef]
- Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J Biol Chem. 2001;276(41):37993-8001. [CrossRef]
- Lee SJ, Namkoong S, Kim YM, Kim CK, Lee H, Ha KS, Chung HT, Kwon YG, Kim YM. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am J Physiol Heart Circ Physiol. 2006;291(6):H2836-46. [CrossRef]
- Roy SK, Srivastava RK, Shankar S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J Mol Signal. 2010;5:10. [CrossRef]
- Wang H, Cai J, Du S, Guo Z, Xin B, Wang J, Wei W, Shen X. Fractalkine/CX3CR1 induces apoptosis resistance and proliferation through the activation of the AKT/NF-κB cascade in pancreatic cancer cells. Cell Biochem Funct. 2017;35(6):315-326. [CrossRef]
- Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgamer R, Shagdarsuren E, Lira SA, Weissman IL, Weber C, Jung S. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113(4):963-72. [CrossRef]
- Kim ES, Khuri FR, Herbst RS. Epidermal growth factor receptor biology (IMC-C225). Curr Opin Oncol. 2001;13(6):506-13. [CrossRef]
- Cai Z, Zhang H, Liu J, Berezov A, Murali R, Wang Q, Greene MI. Targeting erbB receptors. Semin Cell Dev Biol. 2010;21(9):961-6. [CrossRef]
- Ledonne A, Mercuri NB. Insights on the Functional Interaction between Group 1 Metabotropic Glutamate Receptors (mGluRI) and ErbB Receptors. Int J Mol Sci. 2020;21(21):7913. [CrossRef]
- Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol. 2005;1:2005.0010. [CrossRef]
- Thomas SM, Bhola NE, Zhang Q, Contrucci SC, Wentzel AL, Freilino ML, Gooding WE, Siegfried JM, Chan DC, Grandis JR. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006;66(24):11831-9. [CrossRef]
- Cantor AJ, Shah NH, Kuriyan J. Deep mutational analysis reveals functional trade-offs in the sequences of EGFR autophosphorylation sites. Proc Natl Acad Sci U S A. 2018;115(31):E7303-E7312. [CrossRef]
- Jurišić V, Obradovic J, Pavlović S, Djordjevic N. Epidermal Growth Factor Receptor Gene in Non-Small-Cell Lung Cancer: The Importance of Promoter Polymorphism Investigation. Anal Cell Pathol (Amst). 2018;2018:6192187. [CrossRef]
- Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117(8):2051-8. [CrossRef]
- Tardáguila M, Mira E, García-Cabezas MA, Feijoo AM, Quintela-Fandino M, Azcoitia I, Lira SA, Mañes S. CX3CL1 promotes breast cancer via transactivation of the EGF pathway. Cancer Res. 2013;73(14):4461-73. [CrossRef]
- Bai Q, Wang J, Zhou X. EGFR exon20 insertion mutations in non-small cell lung cancer: Clinical implications and recent advances in targeted therapies. Cancer Treat Rev. 2023; 120:102605. [CrossRef]
- Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 2001;20(13):1594-600. [CrossRef]
- Burch ML, Osman N, Getachew R, Al-Aryahi S, Poronnik P, Zheng W, Hill MA, Little PJ. G protein coupled receptor transactivation: extending the paradigm to include serine/threonine kinase receptors. Int J Biochem Cell Biol. 2012;44(5):722-7. [CrossRef]
- Chandrasekar B, Mummidi S, Perla RP, Bysani S, Dulin NO, Liu F, Melby PC. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem J. 2003;373(Pt 2):547-58. [CrossRef]
- Szukiewicz D, Wojciechowska M, Bilska A, Stangret A, Szewczyk G, Mittal TK, Watroba M, Kochanowski J. Aspirin Action in Endothelial Cells: Different Patterns of Response Between Chemokine CX3CL1/CX3CR1 and TNF-α/TNFR1 Signaling Pathways. Cardiovasc Drugs Ther. 2015;29(3):219-29. [CrossRef]
- Zwijnenburg AJ, Pokharel J, Varnaitė R, Zheng W, Hoffer E, Shryki I, Comet NR, Ehrström M, Gredmark-Russ S, Eidsmo L, Gerlach C. Graded expression of the chemokine receptor CX3CR1 marks differentiation states of human and murine T cells and enables cross-species interpretation. Immunity. 2023;56(8):1955-1974.e10. [CrossRef]
- Ren M, Zhang J, Dai S, Wang C, Chen Z, Zhang S, Xu J, Qin X, Liu F. CX3CR1 deficiency exacerbates immune-mediated hepatitis by increasing NF-κB-mediated cytokine production in macrophage and T cell. Exp Biol Med (Maywood). 2023;248(2):117-129. [CrossRef]
- Wojdasiewicz P, Poniatowski ŁA, Kotela A, Skoda M, Pyzlak M, Stangret A, Kotela I, Szukiewicz D. Comparative Analysis of the Occurrence and Role of CX3CL1 (Fractalkine) and Its Receptor CX3CR1 in Hemophilic Arthropathy and Osteoarthritis. J Immunol Res. 2020;2020:2932696. Erratum in: J Immunol Res. 2020;2020:7179283. [CrossRef]
- White GE, Tan TC, John AE, Whatling C, McPheat WL, Greaves DR. Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signalling. Cardiovasc Res. 2010;85(4):825-35. [CrossRef]
- White GE, McNeill E, Channon KM, Greaves DR. Fractalkine promotes human monocyte survival via a reduction in oxidative stress. Arterioscler Thromb Vasc Biol. 2014;34(12):2554-62. [CrossRef]
- Raei Sadigh A, Darabi M, Salmassi A, Hamdi K, Farzadi L, Ghasemzadeh A, Fattahi A, Nouri M. Fractalkine and apoptotic/anti-apoptotic markers in granulosa cells of women with polycystic ovarian syndrome. Mol Biol Rep. 2020;47(5):3593-3603. [CrossRef]
- Sheridan GK, Murphy KJ. Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol. 2013;3(12):130181. [CrossRef]
- Pawelec P, Ziemka-Nalecz M, Sypecka J, Zalewska T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells. 2020;9(10):2277. [CrossRef]
- Paolicelli RC, Bisht K, Tremblay MÈ. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front Cell Neurosci. 2014;8:129. [CrossRef]
- Camacho-Hernández NP, Peña-Ortega F. Fractalkine/CX3CR1-Dependent Modulation of Synaptic and Network Plasticity in Health and Disease. Neural Plast. 2023;2023:4637073. [CrossRef]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979(1-2):65-70. [CrossRef]
- Mecca C, Giambanco I, Donato R, Arcuri C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int J Mol Sci. 2018;19(1):318. [CrossRef]
- Subbarayan MS, Joly-Amado A, Bickford PC, Nash KR. CX3CL1/CX3CR1 signaling targets for the treatment of neurodegenerative diseases. Pharmacol Ther. 2022;231:107989. [CrossRef]
- Cipriani R, Villa P, Chece G, Lauro C, Paladini A, Micotti E, Perego C, De Simoni MG, Fredholm BB, Eusebi F, Limatola C. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci. 2011;31(45):16327-35. [CrossRef]
- Luo P, Chu SF, Zhang Z, Xia CY, Chen NH. Fractalkine/CX3CR1 is involved in the cross-talk between neuron and glia in neurological diseases. Brain Res Bull. 2019;146:12-21. [CrossRef]
- Bivona G, Iemmolo M, Ghersi G. CX3CL1 Pathway as a Molecular Target for Treatment Strategies in Alzheimer’s Disease. Int J Mol Sci. 2023;24(9):8230. [CrossRef]
- Nash KR, Moran P, Finneran DJ, Hudson C, Robinson J, Morgan D, Bickford PC. Fractalkine over expression suppresses α-synuclein-mediated neurodegeneration. Mol Ther. 2015;23(1):17-23. [CrossRef]
- Juliani J, Vassileff N, Spiers JG. Inflammatory-Mediated Neuron-Glia Communication Modulates ALS Pathophysiology. J Neurosci. 2021;41(6):1142-1144. [CrossRef]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. 2010;177(5):2549-62. [CrossRef]
- Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, Ransohoff RM, Gan L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011;286(37):32713-22. [CrossRef]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;13(4):411-3. [CrossRef]
- Eugenín J, Eugenín-von Bernhardi L, von Bernhardi R. Age-dependent changes on fractalkine forms and their contribution to neurodegenerative diseases. Front Mol Neurosci. 2023;16:1249320. [CrossRef]
- Stratoulias V, Venero JL, Tremblay MÈ, Joseph B. Microglial subtypes: diversity within the microglial community. EMBO J. 2019;38(17):e101997. [CrossRef]
- Jacquelin S, Licata F, Dorgham K, Hermand P, Poupel L, Guyon E, Deterre P, Hume DA, Combadière C, Boissonnas A. CX3CR1 reduces Ly6Chigh-monocyte motility within and release from the bone marrow after chemotherapy in mice. Blood. 2013;122(5):674-83. [CrossRef]
- Hoshino A, Ueha S, Hanada S, Imai T, Ito M, Yamamoto K, Matsushima K, Yamaguchi A, Iimura T. Roles of chemokine receptor CX3CR1 in maintaining murine bone homeostasis through the regulation of both osteoblasts and osteoclasts. J Cell Sci. 2013;126(Pt 4):1032-45. [CrossRef]
- Kuboi Y, Kuroda Y, Ohkuro M, Motoi S, Tomimori Y, Yasuda H, Yasuda N, Imai T, Matsuo K. The Fractalkine-CX3CR1 Axis Regulates Non-inflammatory Osteoclastogenesis by Enhancing Precursor Cell Survival. JBMR Plus. 2022;6(10):e10680. [CrossRef]
- Yano R, Yamamura M, Sunahori K, Takasugi K, Yamana J, Kawashima M, Makino H. Recruitment of CD16+ monocytes into synovial tissues is mediated by fractalkine and CX3CR1 in rheumatoid arthritis patients. Acta Med Okayama. 2007;61(2):89-98. [CrossRef]
- Marchica V, Toscani D, Corcione A, Bolzoni M, Storti P, Vescovini R, Ferretti E, Dalla Palma B, Vicario E, Accardi F, Mancini C, Martella E, Ribatti D, Vacca A, Pistoia V, Giuliani N. Bone Marrow CX3CL1/Fractalkine is a New Player of the Pro-Angiogenic Microenvironment in Multiple Myeloma Patients. Cancers (Basel). 2019;11(3):321. [CrossRef]
- Sciumè G, De Angelis G, Benigni G, Ponzetta A, Morrone S, Santoni A, Bernardini G. CX3CR1 expression defines 2 KLRG1+ mouse NK-cell subsets with distinct functional properties and positioning in the bone marrow. Blood. 2011;117(17):4467-75. [CrossRef]
- Zhang Y, Zheng J, Zhou Z, Zhou H, Wang Y, Gong Z, Zhu J. Fractalkine promotes chemotaxis of bone marrow-derived mesenchymal stem cells towards ischemic brain lesions through Jak2 signaling and cytoskeletal reorganization. FEBS J. 2015;282(5):891-903. [CrossRef]
- Teupser D, Pavlides S, Tan M, Gutierrez-Ramos JC, Kolbeck R, Breslow JL. Major reduction of atherosclerosis in fractalkine (CX3CL1)-deficient mice is at the brachiocephalic artery, not the aortic root. Proc Natl Acad Sci U S A. 2004;101(51):17795-800. [CrossRef]
- Ma J, Luo J, Sun Y, Zhao Z. Cytokines associated with immune response in atherosclerosis. Am J Transl Res. 2022;14(9):6424-6444.
- Riopel M, Vassallo M, Ehinger E, Pattison J, Bowden K, Winkels H, Wilson M, de Jong R, Patel S, Balakrishna D, Bilakovics J, Fanjul A, Plonowski A, Larson CJ, Ley K, Cabrales P, Witztum JL, Olefsky JM, Lee YS. CX3CL1-Fc treatment prevents atherosclerosis in Ldlr KO mice. Mol Metab. 2019;20:89-101. [CrossRef]
- Liu H, Jiang D. Fractalkine/CX3CR1 and atherosclerosis. Clin Chim Acta. 2011;412(13-14):1180-6. [CrossRef]
- Elliott MR, Koster KM, Murphy PS. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J Immunol. 2017;198(4):1387-1394. [CrossRef]
- Lucas AD, Bursill C, Guzik TJ, Sadowski J, Channon KM, Greaves DR. Smooth muscle cells in human atherosclerotic plaques express the fractalkine receptor CX3CR1 and undergo chemotaxis to the CX3C chemokine fractalkine (CX3CL1). Circulation. 2003;108(20):2498-504. [CrossRef]
- Harman JL, Jørgensen HF. The role of smooth muscle cells in plaque stability: Therapeutic targeting potential. Br J Pharmacol. 2019;176(19):3741-3753. [CrossRef]
- Apostolakis S, Spandidos D. Chemokines and atherosclerosis: focus on the CX3CL1/CX3CR1 pathway. Acta Pharmacol Sin. 2013;34(10):1251-6. [CrossRef]
- Skoda M, Stangret A, Szukiewicz D. Fractalkine and placental growth factor: A duet of inflammation and angiogenesis in cardiovascular disorders. Cytokine Growth Factor Rev. 2018;39:116-123. [CrossRef]
- Noels H, Weber C, Koenen RR. Chemokines as Therapeutic Targets in Cardiovascular Disease. Arterioscler Thromb Vasc Biol. 2019;39(4):583-592. [CrossRef]
- Flamant M, Mougenot N, Balse E, Le Fèvre L, Atassi F, Gautier EL, Le Goff W, Keck M, Nadaud S, Combadière C, Boissonnas A, Pavoine C. Early activation of the cardiac CX3CL1/CX3CR1 axis delays β-adrenergic-induced heart failure. Sci Rep. 2021;11(1):17982. [CrossRef]
- Njerve IU, Solheim S, Lunde K, Hoffmann P, Arnesen H, Seljeflot I. Fractalkine levels are elevated early after PCI-treated ST-elevation myocardial infarction; no influence of autologous bone marrow derived stem cell injection. Cytokine. 2014;69(1):131-5. [CrossRef]
- Yao K, Zhang S, Lu H, Hong X, Qian J, Sun A, Zou Y, Ge J. Changes in fractalkine in patients with ST-elevation myocardial infarction. Coron Artery Dis. 2015;26(6):516-20. [CrossRef]
- Xu B, Qian Y, Zhao Y, Fang Z, Tang K, Zhou N, Li D, Wang J. Prognostic value of fractalkine/CX3CL1 concentration in patients with acute myocardial infarction treated with primary percutaneous coronary intervention. Cytokine. 2019;113:365-370. [CrossRef]
- Boag SE, Das R, Shmeleva EV, Bagnall A, Egred M, Howard N, Bennaceur K, Zaman A, Keavney B, Spyridopoulos I. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J Clin Invest. 2015;125(8):3063-76. [CrossRef]
- Zhang J, Patel JM. Role of the CX3CL1-CX3CR1 axis in chronic inflammatory lung diseases. Int J Clin Exp Med. 2010;3(3):233-44.
- Balabanian K, Foussat A, Dorfmüller P, Durand-Gasselin I, Capel F, Bouchet-Delbos L, Portier A, Marfaing-Koka A, Krzysiek R, Rimaniol AC, Simonneau G, Emilie D, Humbert M. CX(3)C chemokine fractalkine in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165(10):1419-25. [CrossRef]
- Amsellem V, Abid S, Poupel L, Parpaleix A, Rodero M, Gary-Bobo G, Latiri M, Dubois-Rande JL, Lipskaia L, Combadiere C, Adnot S. Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 Chemokine Systems in Hypoxic Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2017;56(5):597-608. [CrossRef]
- Tsai WH, Chang SC, Lin YC, Hsu HC. CX3CL1(+) Microparticles-Induced MFG-E8 Enhances Apoptotic Cell Clearance by Alveolar Macrophages. Cells. 2021;10(10):2583. [CrossRef]
- El-Shazly A, Berger P, Girodet PO, Ousova O, Fayon M, Vernejoux JM, Marthan R, Tunon-de-Lara JM. Fraktalkine produced by airway smooth muscle cells contributes to mast cell recruitment in asthma. J Immunol. 2006;176(3):1860-8. [CrossRef]
- Godwin MS, Jones M, Blackburn JP, Yu Z, Matalon S, Hastie AT, Meyers DA, Steele C. The chemokine CX3CL1/fractalkine regulates immunopathogenesis during fungal-associated allergic airway inflammation. Am J Physiol Lung Cell Mol Physiol. 2021;320(3):L393-L404. [CrossRef]
- Upton N, Jackson DJ, Nikonova AA, Hingley-Wilson S, Khaitov M, Del Rosario A, Traub S, Trujillo-Torralbo MB, Habibi M, Elkin SL, Kon OM, Edwards MR, Mallia P, Footitt J, Macintyre J, Stanciu LA, Johnston SL, Sykes A. Rhinovirus induction of fractalkine (CX3CL1) in airway and peripheral blood mononuclear cells in asthma. PLoS One. 2017;12(8):e0183864. [CrossRef]
- Efsen E, Grappone C, DeFranco RM, Milani S, Romanelli RG, Bonacchi A, Caligiuri A, Failli P, Annunziato F, Pagliai G, Pinzani M, Laffi G, Gentilini P, Marra F. Up-regulated expression of fractalkine and its receptor CX3CR1 during liver injury in humans. J Hepatol. 2002;37(1):39-47. [CrossRef]
- Sutti S, Locatelli I, Bruzzì S, Jindal A, Vacchiano M, Bozzola C, Albano E. CX3CR1-expressing inflammatory dendritic cells contribute to the progression of steatohepatitis. Clin Sci (Lond). 2015; 129(9):797-808. [CrossRef]
- Nagata N, Chen G, Xu L, Ando H. An Update on the Chemokine System in the Development of NAFLD. Medicina (Kaunas). 2022;58(6):761. [CrossRef]
- Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. 2010;52(4):1390-400. [CrossRef]
- Zhang P, Wang BJ, Wang JZ, Xie XM, Tong QX. Association of CX3CL1 and CX3CR1 Expression with Liver Fibrosis in a Mouse Model of Schistosomiasis. Curr Med Sci. 2020;40(6):1121-1127. [CrossRef]
- Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, Trautwein C, Tacke F. The fractalkine receptor CX₃CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. 2010;52(5):1769-82. [CrossRef]
- Wasmuth HE, Zaldivar MM, Berres ML, Werth A, Scholten D, Hillebrandt S, Tacke F, Schmitz P, Dahl E, Wiederholt T, Hellerbrand C, Berg T, Weiskirchen R, Trautwein C, Lammert F. The fractalkine receptor CX3CR1 is involved in liver fibrosis due to chronic hepatitis C infection. J Hepatol. 2008;48(2):208-15. [CrossRef]
- Hassan GS, Flores Molina M, Shoukry NH. The multifaceted role of macrophages during acute liver injury. Front Immunol. 2023;14:1237042. [CrossRef]
- Isse K, Harada K, Zen Y, Kamihira T, Shimoda S, Harada M, Nakanuma Y. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. 2005;41(3):506-16. [CrossRef]
- Shimoda S, Harada K, Niiro H, Taketomi A, Maehara Y, Tsuneyama K, Kikuchi K, Nakanuma Y, Mackay IR, Gershwin ME, Akashi K. CX3CL1 (fractalkine): a signpost for biliary inflammation in primary biliary cirrhosis. Hepatology. 2010;51(2):567-75. [CrossRef]
- Joeris T, Müller-Luda K, Agace WW, Mowat AM. Diversity and functions of intestinal mononuclear phagocytes. Mucosal Immunol. 2017;10(4):845-864. [CrossRef]
- Bain CC, Mowat AM. Intestinal macrophages - specialised adaptation to a unique environment. Eur J Immunol. 2011;41(9):2494-8. [CrossRef]
- Ferretti E, Pistoia V, Corcione A. Role of fractalkine/CX3CL1 and its receptor in the pathogenesis of inflammatory and malignant diseases with emphasis on B cell malignancies. Mediators Inflamm. 2014;2014:480941. [CrossRef]
- Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW, Mowat AM. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6(3):498-510. [CrossRef]
- Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, Malissen B, Osborne LC, Artis D, Mowat AM. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(10):929-937. [CrossRef]
- Sans M, Danese S, de la Motte C, de Souza HS, Rivera-Reyes BM, West GA, Phillips M, Katz JA, Fiocchi C. Enhanced recruitment of CX3CR1+ T cells by mucosal endothelial cell-derived fractalkine in inflammatory bowel disease. Gastroenterology. 2007;132(1):139-53. [CrossRef]
- Siwetz M, Blaschitz A, Kremshofer J, Bilic J, Desoye G, Huppertz B, Gauster M. Metalloprotease dependent release of placenta derived fractalkine. Mediators Inflamm. 2014;2014:839290. [CrossRef]
- Siwetz M, Sundl M, Kolb D, Hiden U, Herse F, Huppertz B, Gauster M. Placental fractalkine mediates adhesion of THP-1 monocytes to villous trophoblast. Histochem Cell Biol. 2015;143(6):565-74. [CrossRef]
- Siwetz M, Dieber-Rotheneder M, Cervar-Zivkovic M, Kummer D, Kremshofer J, Weiss G, Herse F, Huppertz B, Gauster M. Placental fractalkine is up-regulated in severe early-onset preeclampsia. Am J Pathol. 2015;185(5):1334-43. [CrossRef]
- Vishnyakova P, Poltavets A, Nikitina M, Muminova K, Potapova A, Vtorushina V, Loginova N, Midiber K, Mikhaleva L, Lokhonina A, Khodzhaeva Z, Pyregov A, Elchaninov A, Fatkhudinov T, Sukhikh G. Preeclampsia: inflammatory signature of decidual cells in early manifestation of disease. Placenta. 2021;104:277-283. [CrossRef]
- Szewczyk G, Pyzlak M, Pankiewicz K, Szczerba E, Stangret A, Szukiewicz D, Skoda M, Bierła J, Cukrowska B, Fijałkowska A. The potential association between a new angiogenic marker fractalkine and a placental vascularization in preeclampsia. Arch Gynecol Obstet. 2021;304(2): 365-376. [CrossRef]
- Ullah A, Zhao J, Singla RK, Shen B. Pathophysiological impact of CXC and CX3CL1 chemokines in preeclampsia and gestational diabetes mellitus. Front Cell Dev Biol. 2023;11:1272536. [CrossRef]
- Szukiewicz D, Kochanowski J, Pyzlak M, Szewczyk G, Stangret A, Mittal TK. Fractalkine (CX3CL1) and its receptor CX3CR1 may contribute to increased angiogenesis in diabetic placenta. Mediators Inflamm. 2013;2013:437576. [CrossRef]
- Szukiewicz D, Kochanowski J, Mittal TK, Pyzlak M, Szewczyk G, Cendrowski K. CX3CL1 (fractalkine) and TNFα production by perfused human placental lobules under normoxic and hypoxic conditions in vitro: the importance of CX3CR1 signaling. Inflamm Res. 2014;63(3):179-89. [CrossRef]
- Gokce S, Herkiloglu D, Cevik O, Turan V. Role of chemokines in early pregnancy loss. Exp Ther Med. 2022;23(6):397. [CrossRef]
- Tanaka Y, Hoshino-Negishi K, Kuboi Y, Tago F, Yasuda N, Imai T. Emerging Role of Fractalkine in the Treatment of Rheumatic Diseases. Immunotargets Ther. 2020 ;9:241-253. [CrossRef]
- Koizumi K, Saitoh Y, Minami T, Takeno N, Tsuneyama K, Miyahara T, Nakayama T, Sakurai H, Takano Y, Nishimura M, Imai T, Yoshie O, Saiki I. Role of CX3CL1/fractalkine in osteoclast differentiation and bone resorption. J Immunol. 2009;183(12):7825-31. [CrossRef]
- Guo YN, Cui SJ, Tian YJ, Zhao NR, Zhang YD, Gan YH, Zhou YH, Wang XD. Chondrocyte apoptosis in temporomandibular joint osteoarthritis promotes bone resorption by enhancing chemotaxis of osteoclast precursors. Osteoarthritis Cartilage. 2022;30(8):1140-1153. [CrossRef]
- Wojdasiewicz P, Poniatowski ŁA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459. [CrossRef]
- Lu Z, Zhang A, Dai Y. CX3CL1 deficiency ameliorates inflammation, apoptosis and accelerates osteogenic differentiation, mineralization in LPS-treated MC3T3-E1 cells via its receptor CX3CR1. Ann Anat. 2023;246:152036. [CrossRef]
- Gao XW, Hu HL, Xie MH, Tang CX, Ou J, Lu ZH. CX3CL1/CX3CR1 axis alleviates inflammation and apoptosis in human nucleus pulpous cells via M2 macrophage polarization. Exp Ther Med. 2023; 26(1):359. [CrossRef]
- Folkman, J. Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1-18. [CrossRef]
- Akbarian M, Bertassoni LE, Tayebi L. Biological aspects in controlling angiogenesis: current progress. Cell Mol Life Sci. 2022;79(7):349. [CrossRef]
- Szade A, Grochot-Przeczek A, Florczyk U, Jozkowicz A, Dulak J. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life. 2015;67(3):145-59. [CrossRef]
- Narkar, VA. Narkar VA. Exercise and Ischemia-Activated Pathways in Limb Muscle Angiogenesis and Vascular Regeneration. Methodist Debakey Cardiovasc J. 2023;19(5):58-68. [CrossRef]
- Yin CL, Ma YJ. The Regulatory Mechanism of Hypoxia-inducible Factor 1 and its Clinical Significance. Curr Mol Pharmacol. 2024;17:e18761429266116. [CrossRef]
- Zhou W, Yang L, Nie L, Lin H. Unraveling the molecular mechanisms between inflammation and tumor angiogenesis. Am J Cancer Res. 2021;11(2):301-317.
- Perry BN, Arbiser JL. The duality of angiogenesis: implications for therapy of human disease. J Invest Dermatol. 2006;126(10):2160-6. [CrossRef]
- Edgar LT, Park H, Crawshaw JR, Osborne JM, Eichmann A, Bernabeu MO. Traffic Patterns of the Migrating Endothelium: How Force Transmission Regulates Vascular Malformation and Functional Shunting During Angiogenic Remodelling. Front Cell Dev Biol. 2022;10:840066. [CrossRef]
- Silvestre JS, Lévy BI, Tedgui A. Mechanisms of angiogenesis and remodelling of the microvasculature. Cardiovasc Res. 2008;78(2):201-2. [CrossRef]
- Niklason L, Dai G. Arterial Venous Differentiation for Vascular Bioengineering. Annu Rev Biomed Eng. 2018;20:431-447. [CrossRef]
- Sarabipour S, Kinghorn K, Quigley KM, Kovacs-Kasa A, Annex BH, Bautch VL, Mac Gabhann F. Trafficking dynamics of VEGFR1, VEGFR2, and NRP1 in human endothelial cells. PLoS Comput Biol. 2024;20(2):e1011798.
- Colotti G, Failla CM, Lacal PM, Ungarelli M, Ruffini F, Di Micco P, Orecchia A, Morea V. Neuropilin-1 is required for endothelial cell adhesion to soluble vascular endothelial growth factor receptor 1. FEBS J. 2022;289(1):183-198. [CrossRef]
- Alghamdi AAA, Benwell CJ, Atkinson SJ, Lambert J, Johnson RT, Robinson SD. NRP2 as an Emerging Angiogenic Player; Promoting Endothelial Cell Adhesion and Migration by Regulating Recycling of α5 Integrin. Front Cell Dev Biol. 2020;8:395. [CrossRef]
- Li T, Ran J, Miao Z, Yang M, Mou D, Jiang Y, Xu X, Xie Q, Jin K. Deficiency of inflammation-sensing protein neuropilin-2 in myeloid-derived macrophages exacerbates colitis via NF-κB activation. J Pathol. 2024;262(2):175-188. [CrossRef]
- Rahane D, Dhingra T, Chalavady G, Datta A, Ghosh B, Rana N, Borah A, Saraf S, Bhattacharya P. Hypoxia and its effect on the cellular system. Cell Biochem Funct. 2024;42(2):e3940. [CrossRef]
- Cui Y, Lin H, Zhao YH, Ma JX, Li JX. Tube Formation Capability and Chemotaxis of Skin Pericytes. Discov Med. 2024;36(181):308-322. [CrossRef]
- Yang S, Graham J, Kahn JW, Schwartz EA, Gerritsen ME. Functional roles for PECAM-1 (CD31) and VE-cadherin (CD144) in tube assembly and lumen formation in three-dimensional collagen gels. Am J Pathol. 1999;155(3):887-95. [CrossRef]
- Granger DN, Senchenkova E. Inflammation and the Microcirculation. San Rafael (CA): Morgan & Claypool Life Sciences; 2010. Chapter 6, Angiogenesis. Available from: https://www.ncbi.nlm.nih.gov/books/NBK53377/.
- Ouarné M, Pena A, Franco CA. From remodeling to quiescence: The transformation of the vascular network. Cells Dev. 2021;168:203735. [CrossRef]
- Pollina EA, Legesse-Miller A, Haley EM, Goodpaster T, Randolph-Habecker J, Coller HA. Regulating the angiogenic balance in tissues. Cell Cycle. 2008;7(13):2056-70. [CrossRef]
- Aguilar-Cazares D, Chavez-Dominguez R, Carlos-Reyes A, Lopez-Camarillo C, Hernadez de la Cruz ON, Lopez-Gonzalez JS. Contribution of Angiogenesis to Inflammation and Cancer. Front Oncol. 2019;9:1399. [CrossRef]
- Britzen-Laurent N, Weidinger C, Stürzl M. Contribution of Blood Vessel Activation, Remodeling and Barrier Function to Inflammatory Bowel Diseases. Int J Mol Sci. 2023;24(6):5517. [CrossRef]
- Reyes ME, Pulgar V, Vivallo C, Ili CG, Mora-Lagos B, Brebi P. Epigenetic modulation of cytokine expression in gastric cancer: influence on angiogenesis, metastasis and chemoresistance. Front Immunol. 2024;15:1347530. [CrossRef]
- Lee YE, Lee SH, Kim WU. Cytokines, Vascular Endothelial Growth Factors, and PlGF in Autoimmunity: Insights From Rheumatoid Arthritis to Multiple Sclerosis. Immune Netw. 2024 ;24(1):e10. [CrossRef]
- Vofo BN, Chowers I. Suppressing Inflammation for the Treatment of Diabetic Retinopathy and Age-Related Macular Degeneration: Dazdotuftide as a Potential New Multitarget Therapeutic Candidate. Biomedicines. 2023;11(6):1562. [CrossRef]
- Fu J, Liang H, Yuan P, Wei Z, Zhong P. Brain pericyte biology: from physiopathological mechanisms to potential therapeutic applications in ischemic stroke. Front Cell Neurosci. 2023;17:1267785. [CrossRef]
- Bhavsar PK, Sukkar MB, Khorasani N, Lee KY, Chung KF. Glucocorticoid suppression of CX3CL1 (fractalkine) by reduced gene promoter recruitment of NF-kappaB. FASEB J. 2008;22(6): 1807-16. [CrossRef]
- Hayden MS, Ghosh S. Regulation of NF-κB by TNF family cytokines. Semin Immunol. 2014; 26(3):253-66. [CrossRef]
- Diep S, Maddukuri M, Yamauchi S, Geshow G, Delk NA. Interleukin-1 and Nuclear Factor Kappa B Signaling Promote Breast Cancer Progression and Treatment Resistance. Cells. 2022; 11(10):1673. [CrossRef]
- Chen D, Ireland SJ, Remington G, Alvarez E, Racke MK, Greenberg B, Frohman EM, Monson NL. CD40-Mediated NF-κB Activation in B Cells Is Increased in Multiple Sclerosis and Modulated by Therapeutics. J Immunol. 2016;197(11):4257-4265. [CrossRef]
- Seigner J, Basilio J, Resch U, de Martin R. CD40L and TNF both activate the classical NF-κB pathway, which is not required for the CD40L induced alternative pathway in endothelial cells. Biochem Biophys Res Commun. 2018;495(1):1389-1394.
- Yang C, Hwang HH, Jeong S, Seo D, Jeong Y, Lee DY, Lee K. Inducing angiogenesis with the controlled release of nitric oxide from biodegradable and biocompatible copolymeric nanoparticles. Int J Nanomedicine. 2018;13:6517-6530. [CrossRef]
- Heydari P, Kharaziha M, Varshosaz J, Kharazi AZ, Javanmard SH. Co-release of nitric oxide and L-arginine from poly (β-amino ester)-based adhesive reprogram macrophages for accelerated wound healing and angiogenesis in vitro and in vivo. Biomater Adv. 2024;158:213762. [CrossRef]
- Minet E, Arnould T, Michel G, Roland I, Mottet D, Raes M, Remacle J, Michiels C. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett. 2000;468(1):53-8. [CrossRef]
- Ryu J, Lee CW, Hong KH, Shin JA, Lim SH, Park CS, Shim J, Nam KB, Choi KJ, Kim YH, Han KH. Activation of fractalkine/CX3CR1 by vascular endothelial cells induces angiogenesis through VEGF-A/KDR and reverses hindlimb ischaemia. Cardiovasc Res. 2008;78(2):333-40. [CrossRef]
- Park Y, Lee J, Kwak JY, Noh K, Yim E, Kim HK, Kim YJ, Broxmeyer HE, Kim JA. Fractalkine induces angiogenic potential in CX3CR1-expressing monocytes. J Leukoc Biol. 2018;103(1): 53-66. [CrossRef]
- Smith TL, Oubaha M, Cagnone G, Boscher C, Kim JS, El Bakkouri Y, Zhang Y, Chidiac R, Corriveau J, Delisle C, Andelfinger GU, Sapieha P, Joyal JS, Gratton JP. eNOS controls angiogenic sprouting and retinal neovascularization through the regulation of endothelial cell polarity. Cell Mol Life Sci. 2021;79(1):37. [CrossRef]
- Li B, Zhang Y, Yin R, Zhong W, Chen R, Yan J. Activating CD137 Signaling Promotes Sprouting Angiogenesis via Increased VEGFA Secretion and the VEGFR2/Akt/eNOS Pathway. Mediators Inflamm. 2020;2020:1649453. [CrossRef]
- Zarychta E, Ruszkowska-Ciastek B. Cooperation between Angiogenesis, Vasculogenesis, Chemotaxis, and Coagulation in Breast Cancer Metastases Development: Pathophysiological Point of View. Biomedicines. 2022;10(2):300. [CrossRef]
- Janiszewska M, Primi MC, Izard T. Cell adhesion in cancer: Beyond the migration of single cells. J Biol Chem. 2020;295(8):2495-2505. [CrossRef]
- Folkman, J. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29(6 Suppl 16):15-8. [CrossRef]
- Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213-9. [CrossRef]
- Tannock, IF. Tannock IF. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br J Cancer. 1968;22(2):258-73. [CrossRef]
- Place TL, Domann FE, Case AJ. Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free Radic Biol Med. 2017; 113:311-322. Erratum in: Free Radic Biol Med. 2021 Jan;162:180. [CrossRef]
- Folkman, J. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182-6. [CrossRef]
- Welter M, Bartha K, Rieger H. Vascular remodelling of an arterio-venous blood vessel network during solid tumour growth. J Theor Biol. 2009;259(3):405-22. [CrossRef]
- Gerlee P, Anderson AR. Diffusion-limited tumour growth: simulations and analysis. Math Biosci Eng. 2010;7(2):385-400. [CrossRef]
- Martin JD, Seano G, Jain RK. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu Rev Physiol. 2019;81:505-534. [CrossRef]
- Lefler DS, Manobianco SA, Bashir B. Immunotherapy resistance in solid tumors: mechanisms and potential solutions. Cancer Biol Ther. 2024;25(1):2315655. [CrossRef]
- Bielenberg DR, Zetter BR. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015;21(4):267-73. [CrossRef]
- Schlößer HA, Theurich S, Shimabukuro-Vornhagen A, Holtick U, Stippel DL, von Bergwelt-Baildon M. Overcoming tumor-mediated immunosuppression. Immunotherapy. 2014;6(9):973-88. [CrossRef]
- Wan Y, Fu LH, Li C, Lin J, Huang P. Conquering the Hypoxia Limitation for Photodynamic Therapy. Adv Mater. 2021;33(48):e2103978. [CrossRef]
- Mohamed OAA, Tesen HS, Hany M, Sherif A, Abdelwahab MM, Elnaggar MH. The role of hypoxia on prostate cancer progression and metastasis. Mol Biol Rep. 2023;50(4):3873-3884. [CrossRef]
- Liu Z, Liu X, Zhang W, Gao R, Wei H, Yu CY. Current advances in modulating tumor hypoxia for enhanced therapeutic efficacy. Acta Biomater. 2024;176:1-27. [CrossRef]
- Ma S, Pan X, Gan J, Guo X, He J, Hu H, Wang Y, Ning S, Zhi H. DNA methylation heterogeneity attributable to a complex tumor immune microenvironment prompts prognostic risk in glioma. Epigenetics. 2024;19(1):2318506. [CrossRef]
- Ge R, Wang Z, Cheng L. Tumor microenvironment heterogeneity an important mediator of prostate cancer progression and therapeutic resistance. NPJ Precis Oncol. 2022;6(1):31. [CrossRef]
- Jia Q, Wang A, Yuan Y, Zhu B, Long H. Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp Hematol Oncol. 2022;11(1):24. [CrossRef]
- Sidibe A, Ropraz P, Jemelin S, Emre Y, Poittevin M, Pocard M, Bradfield PF, Imhof BA. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat Commun. 2018;9(1):355. [CrossRef]
- Richards DM, Hettinger J, Feuerer M. Monocytes and macrophages in cancer: development and functions. Cancer Microenviron. 2013;6(2):179-91. [CrossRef]
- Li F, Wang Z, Liu Y, Li J. Down-regulation of fractalkine inhibits the in vitro and in vivo angiogenesis of the hepatocellular carcinoma HepG2 cells. Oncol Rep. 2010;24(3):669-75.
- Zheng J, Yang M, Shao J, Miao Y, Han J, Du J. Chemokine receptor CX3CR1 contributes to macrophage survival in tumor metastasis. Mol Cancer. 2013;12(1):141. [CrossRef]
- Xu X, Wang Y, Chen J, Ma H, Shao Z, Chen H, Jin G. High expression of CX3CL1/CX3CR1 axis predicts a poor prognosis of pancreatic ductal adenocarcinoma. J Gastrointest Surg. 2012; 16(8):1493-8. [CrossRef]
- Ren T, Chen Q, Tian Z, Wei H. Down-regulation of surface fractalkine by RNA interference in B16 melanoma reduced tumor growth in mice. Biochem Biophys Res Commun. 2007;364(4): 978-84. [CrossRef]
- Shulby SA, Dolloff NG, Stearns ME, Meucci O, Fatatis A. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 2004;64(14):4693-8. [CrossRef]
- Goto Y, Aoyama M, Sekiya T, Kakita H, Waguri-Nagaya Y, Miyazawa K, Asai K, Goto S. CXCR4+ CD45- Cells are Niche Forming for Osteoclastogenesis via the SDF-1, CXCL7, and CX3CL1 Signaling Pathways in Bone Marrow. Stem Cells. 2016;34(11):2733-2743. [CrossRef]
- Jamieson WL, Shimizu S, D’Ambrosio JA, Meucci O, Fatatis A. CX3CR1 is expressed by prostate epithelial cells and androgens regulate the levels of CX3CL1/fractalkine in the bone marrow: potential role in prostate cancer bone tropism. Cancer Res. 2008;68(6):1715-22. [CrossRef]
- Liu P, Liang Y, Jiang L, Wang H, Wang S, Dong J. CX3CL1/fractalkine enhances prostate cancer spinal metastasis by activating the Src/FAK pathway. Int J Oncol. 2018;53(4):1544-1556. [CrossRef]
- Liang Y, Yi L, Liu P, Jiang L, Wang H, Hu A, Sun C, Dong J. CX3CL1 involves in breast cancer metastasizing to the spine via the Src/FAK signaling pathway. J Cancer. 2018 ;9(19):3603-3612. [CrossRef]
- Liu W, Liang Y, Chan Q, Jiang L, Dong J. CX3CL1 promotes lung cancer cell migration and invasion via the Src/focal adhesion kinase signaling pathway. Oncol Rep. 2019;41(3):1911-1917. [CrossRef]
- Guo J, Chen T, Wang B, Zhang M, An H, Guo Z, Yu Y, Qin Z, Cao X. Chemoattraction, adhesion and activation of natural killer cells are involved in the antitumor immune response induced by fractalkine/CX3CL1. Immunol Lett. 2003;89(1):1-7. [CrossRef]
- Chen, CK. Chen CK. Inference of gene networks using gene expression data with applications. Heliyon. 2024;10(5):e26065. [CrossRef]
- Yu D, Lim J, Wang X, Liang F, Xiao G. Enhanced construction of gene regulatory networks using hub gene information. BMC Bioinformatics. 2017;18(1):186. [CrossRef]
- Liu Y, Liu C, Huang D, Ge C, Chen L, Fu J, Du J. Identification and prognostic analysis of candidate biomarkers for lung metastasis in colorectal cancer. Medicine (Baltimore). 2024; 103(11):e37484. [CrossRef]
- Yue Y, Zhang Q, Sun Z. CX3CR1 Acts as a Protective Biomarker in the Tumor Microenvironment of Colorectal Cancer. Front Immunol. 2022;12:758040. [CrossRef]
- Erreni M, Siddiqui I, Marelli G, Grizzi F, Bianchi P, Morone D, Marchesi F, Celesti G, Pesce S, Doni A, Rumio C, Roncalli MG, Laghi L, Mantovani A, Allavena P. The Fractalkine-Receptor Axis Improves Human Colorectal Cancer Prognosis by Limiting Tumor Metastatic Dissemination. J Immunol. 2016;196(2):902-14. [CrossRef]
- Dreyer TF, Kuhn S, Stange C, Heithorst N, Schilling D, Jelsma J, Sievert W, Seitz S, Stangl S, Hapfelmeier A, Noske A, Wege AK, Weichert W, Ruland J, Schmitt M, Dorn J, Kiechle M, Reuning U, Magdolen V, Multhoff G, Bronger H. The Chemokine CX3CL1 Improves Trastuzumab Efficacy in HER2 Low-Expressing Cancer In Vitro and In Vivo. Cancer Immunol Res. 2021;9(7):779-789. [CrossRef]
- Wege AK, Dreyer TF, Teoman A, Ortmann O, Brockhoff G, Bronger H. CX3CL1 Overexpression Prevents the Formation of Lung Metastases in Trastuzumab-Treated MDA-MB-453-Based Humanized Tumor Mice (HTM). Cancers (Basel). 2021;13(10):2459. [CrossRef]
- Kee JY, Arita Y, Shinohara K, Ohashi Y, Sakurai H, Saiki I, Koizumi K. Antitumor immune activity by chemokine CX3CL1 in an orthotopic implantation of lung cancer model in vivo. Mol Clin Oncol. 2013;1(1):35-40. [CrossRef]
- Liu J, Li Y, Zhu X, Li Q, Liang X, Xie J, Hu S, Peng W, Li C. Increased CX3CL1 mRNA expression level is a positive prognostic factor in patients with lung adenocarcinoma. Oncol Lett. 2019; 17(6):4877-4890. [CrossRef]
- Matsubara T, Ono T, Yamanoi A, Tachibana M, Nagasue N. Fractalkine-CX3CR1 axis regulates tumor cell cycle and deteriorates prognosis after radical resection for hepatocellular carcinoma. J Surg Oncol. 2007;95(3):241-9. [CrossRef]
- Hyakudomi M, Matsubara T, Hyakudomi R, Yamamoto T, Kinugasa S, Yamanoi A, Maruyama R, Tanaka T. Increased expression of fractalkine is correlated with a better prognosis and an increased number of both CD8+ T cells and natural killer cells in gastric adenocarcinoma. Ann Surg Oncol. 2008;15(6):1775-82. [CrossRef]
- Korbecki J, Simińska D, Kojder K, Grochans S, Gutowska I, Chlubek D, Baranowska-Bosiacka I. Fractalkine/CX3CL1 in Neoplastic Processes. Int J Mol Sci. 2020;21(10):3723. [CrossRef]
- Shao D, Zhou H, Yu H, Zhu X. CX3CR1 is a potential biomarker of immune microenvironment and prognosis in epithelial ovarian cancer. Medicine (Baltimore). 2024;103(3):e36891. [CrossRef]
- Yao X, Qi L, Chen X, Du J, Zhang Z, Liu S. Expression of CX3CR1 associates with cellular migration, metastasis, and prognosis in human clear cell renal cell carcinoma. Urol Oncol. 2014;32(2):162-70. [CrossRef]
- Sun C, Hu A, Wang S, Tian B, Jiang L, Liang Y, Wang H, Dong J. ADAM17-regulated CX3CL1 expression produced by bone marrow endothelial cells promotes spinal metastasis from hepatocellular carcinoma. Int J Oncol. 2020;57(1):249-263. [CrossRef]
- Castellana D, Zobairi F, Martinez MC, Panaro MA, Mitolo V, Freyssinet JM, Kunzelmann C. Membrane microvesicles as actors in the establishment of a favorable prostatic tumoral niche: a role for activated fibroblasts and CX3CL1-CX3CR1 axis. Cancer Res. 2009;69(3): 785-93. [CrossRef]
- Smith TM Jr, Tharakan A, Martin RK. Targeting ADAM10 in Cancer and Autoimmunity. Front Immunol. 2020;11:499. [CrossRef]
- Wang K, Xuan Z, Liu X, Zheng M, Yang C, Wang H. Immunomodulatory role of metalloproteinase ADAM17 in tumor development. Front Immunol. 2022;13:1059376. [CrossRef]
Figure 1.
Schematic structure of the fractalkine (FKN, chemokine CX3CL1) molecule as a transmembrane protein, including the soluble form (sFKN) cleaved by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), tumor necrosis factor alpha (TNF-α) converting enzyme (TACE or ADAM17), matrix metalloproteinase-2 (MMP-2) or cathepsins (CTS).
Figure 1.
Schematic structure of the fractalkine (FKN, chemokine CX3CL1) molecule as a transmembrane protein, including the soluble form (sFKN) cleaved by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), tumor necrosis factor alpha (TNF-α) converting enzyme (TACE or ADAM17), matrix metalloproteinase-2 (MMP-2) or cathepsins (CTS).
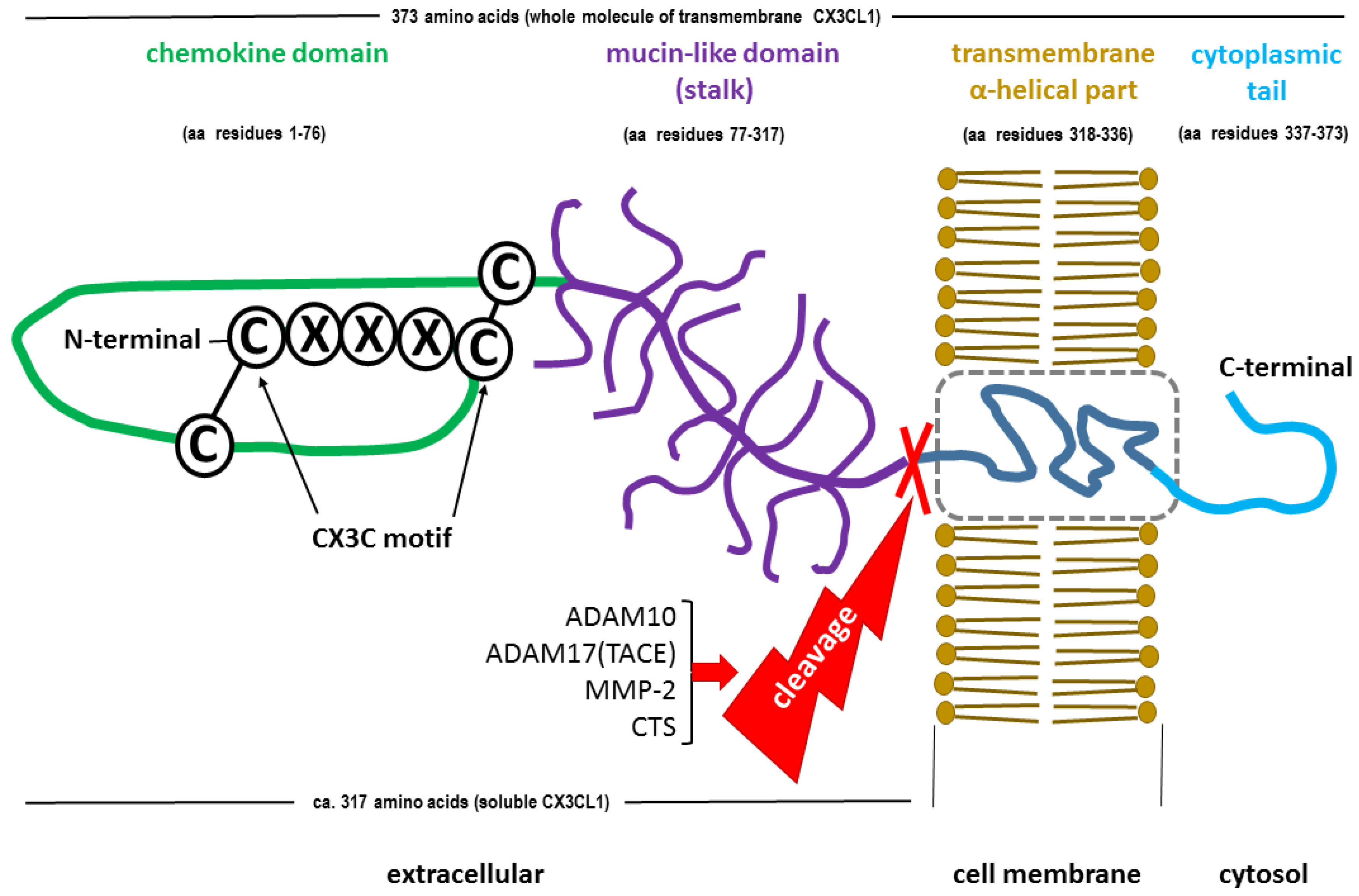
Figure 2.
Schematic diagram and amino acid sequence of CX3C motif chemokine receptor 1 (CX3CR1), the only fractalkine (FKN, chemokine CX3CL1) receptor. Belonging to the most numerous class A (rhodopsin-like receptors) in the G protein-coupled receptor (GPCR) family of proteins, CX3CR1 share a common structural signature of the polypeptide chain (355 aa; ∼ 40 kDa), containing seven hydrophobic α-helical transmembrane (TM1–TM7) segments or domains, with an extracellular amino terminus (NH2) and an intracellular carboxyl terminus (COOH). These transmembrane segments are connected to each other by three intracellular (ICL1–ICL3) and three extracellular loops (ECL1–ECL3) [103,104]. A disulfide bridge is marked, connecting two conservative cysteine (C) residues located at the top of the extracellular side of TM3 and within ECL2, respectively [105]. The ICL2 contains the canonical DRYLAIV motif, composed of a sequence of seven amino acids (in 3-letter abbreviations: Asp-Arg-Tyr-Leu-Ala-Ile-Val) forming a docking site that is essential for CX3CR1 coupling to G protein and induction of classical signaling pathways [100,106]. The binding of an agonist to CX3CR1 causes conformational changes of the receptor with subsequent dissociation of the components of the heterotrimeric G complex, consisting of alpha (α), beta (β) and gamma (γ) subunits. Binding of guanosine diphosphate (GDP) allows the α subunit to bind to the β and γ subunits to form an inactive trimer. Binding of an extracellular signal (ligand) to CX3CR1 allows the G-protein to bind to the receptor and causes GDP to be replaced with guanosine triphosphate (GTP; not shown) [102,107]. The COOH-terminal serine residues (S) are susceptible to G protein-coupled receptor kinase (GRK)-mediated phosphorylation (marked with tiny yellowish dots containing P). * The polymorphic residues at positions 249 and 280 may be responsible for dysfunctional CX3CR1 variants [108,109].
Figure 2.
Schematic diagram and amino acid sequence of CX3C motif chemokine receptor 1 (CX3CR1), the only fractalkine (FKN, chemokine CX3CL1) receptor. Belonging to the most numerous class A (rhodopsin-like receptors) in the G protein-coupled receptor (GPCR) family of proteins, CX3CR1 share a common structural signature of the polypeptide chain (355 aa; ∼ 40 kDa), containing seven hydrophobic α-helical transmembrane (TM1–TM7) segments or domains, with an extracellular amino terminus (NH2) and an intracellular carboxyl terminus (COOH). These transmembrane segments are connected to each other by three intracellular (ICL1–ICL3) and three extracellular loops (ECL1–ECL3) [103,104]. A disulfide bridge is marked, connecting two conservative cysteine (C) residues located at the top of the extracellular side of TM3 and within ECL2, respectively [105]. The ICL2 contains the canonical DRYLAIV motif, composed of a sequence of seven amino acids (in 3-letter abbreviations: Asp-Arg-Tyr-Leu-Ala-Ile-Val) forming a docking site that is essential for CX3CR1 coupling to G protein and induction of classical signaling pathways [100,106]. The binding of an agonist to CX3CR1 causes conformational changes of the receptor with subsequent dissociation of the components of the heterotrimeric G complex, consisting of alpha (α), beta (β) and gamma (γ) subunits. Binding of guanosine diphosphate (GDP) allows the α subunit to bind to the β and γ subunits to form an inactive trimer. Binding of an extracellular signal (ligand) to CX3CR1 allows the G-protein to bind to the receptor and causes GDP to be replaced with guanosine triphosphate (GTP; not shown) [102,107]. The COOH-terminal serine residues (S) are susceptible to G protein-coupled receptor kinase (GRK)-mediated phosphorylation (marked with tiny yellowish dots containing P). * The polymorphic residues at positions 249 and 280 may be responsible for dysfunctional CX3CR1 variants [108,109].
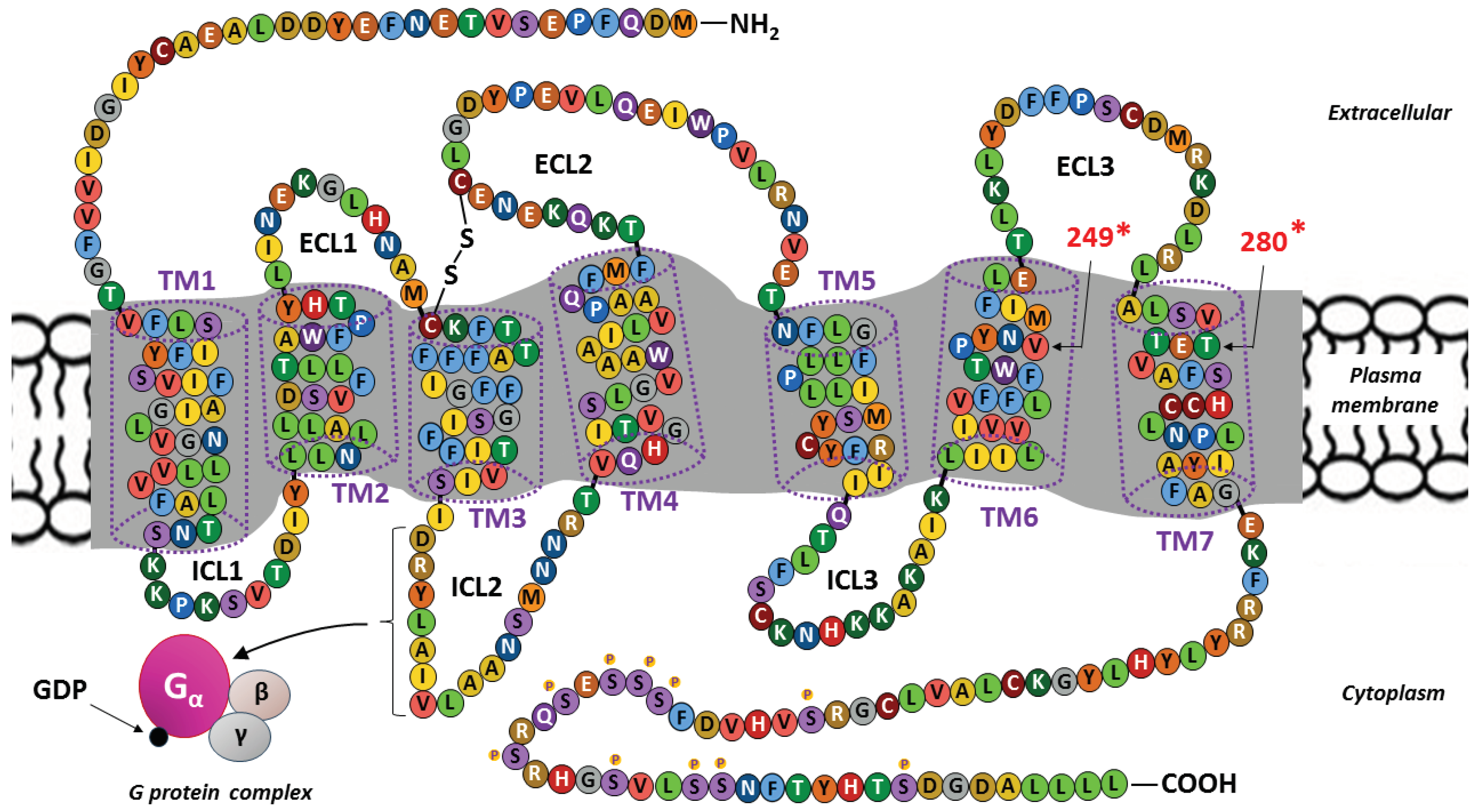
Figure 3.
Main signaling pathways initiated by activation of the CX3C motif chemokine receptor 1 (CX3CR1) receptor through the binding of its sole endogenous ligand FKN (fractalkine, chemokine CX3CL1). For the sake of clarity, interactions with other receptors have been omitted.
Figure 3.
Main signaling pathways initiated by activation of the CX3C motif chemokine receptor 1 (CX3CR1) receptor through the binding of its sole endogenous ligand FKN (fractalkine, chemokine CX3CL1). For the sake of clarity, interactions with other receptors have been omitted.
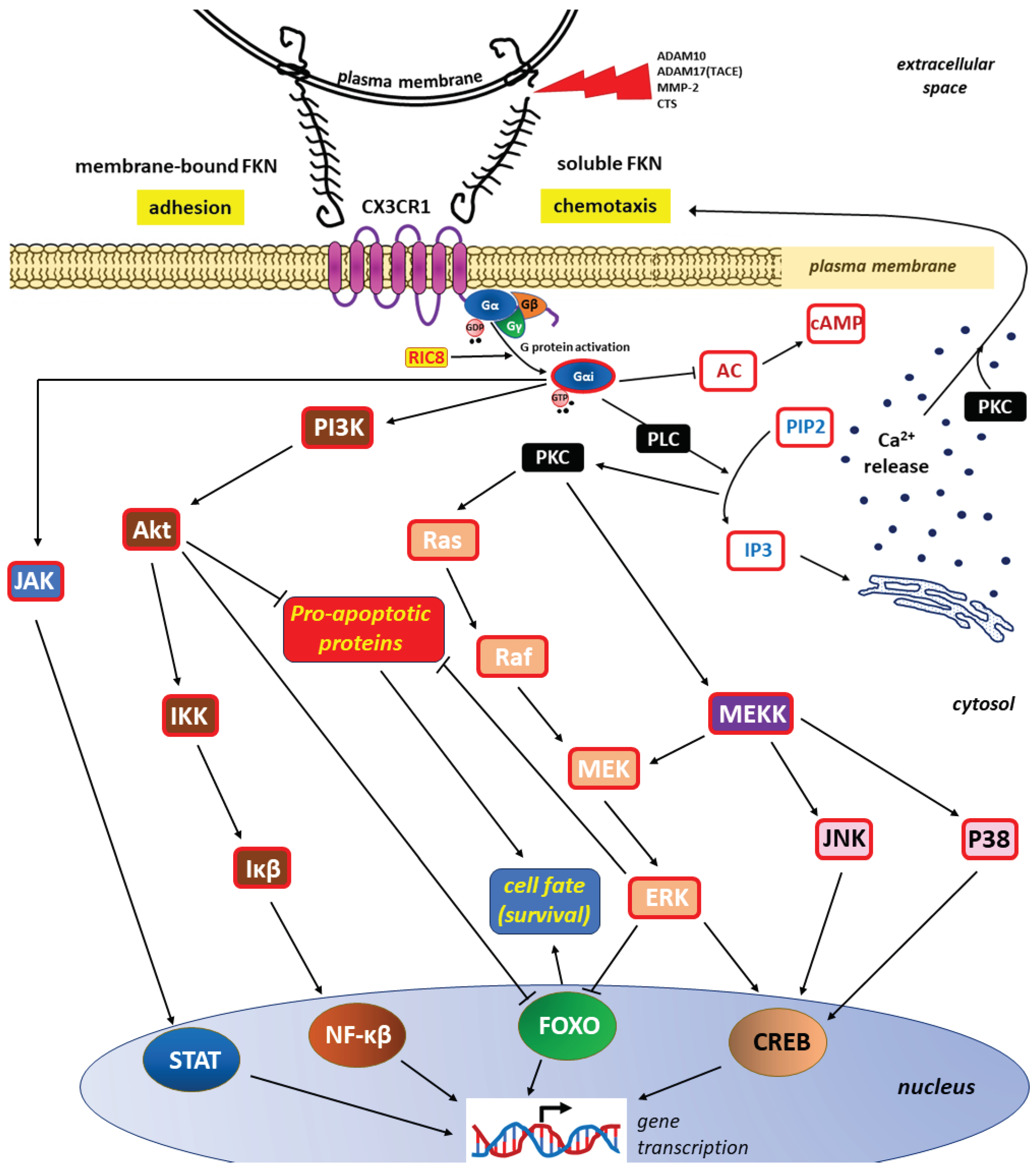
Figure 4.
Angiogenesis during (chronic) inflammatory response as a consequence of specific signaling through the FKN/CX3CR1 axis. (For the full spectrum of signaling via CX3CR1, see Figure 3.).
Figure 4.
Angiogenesis during (chronic) inflammatory response as a consequence of specific signaling through the FKN/CX3CR1 axis. (For the full spectrum of signaling via CX3CR1, see Figure 3.).
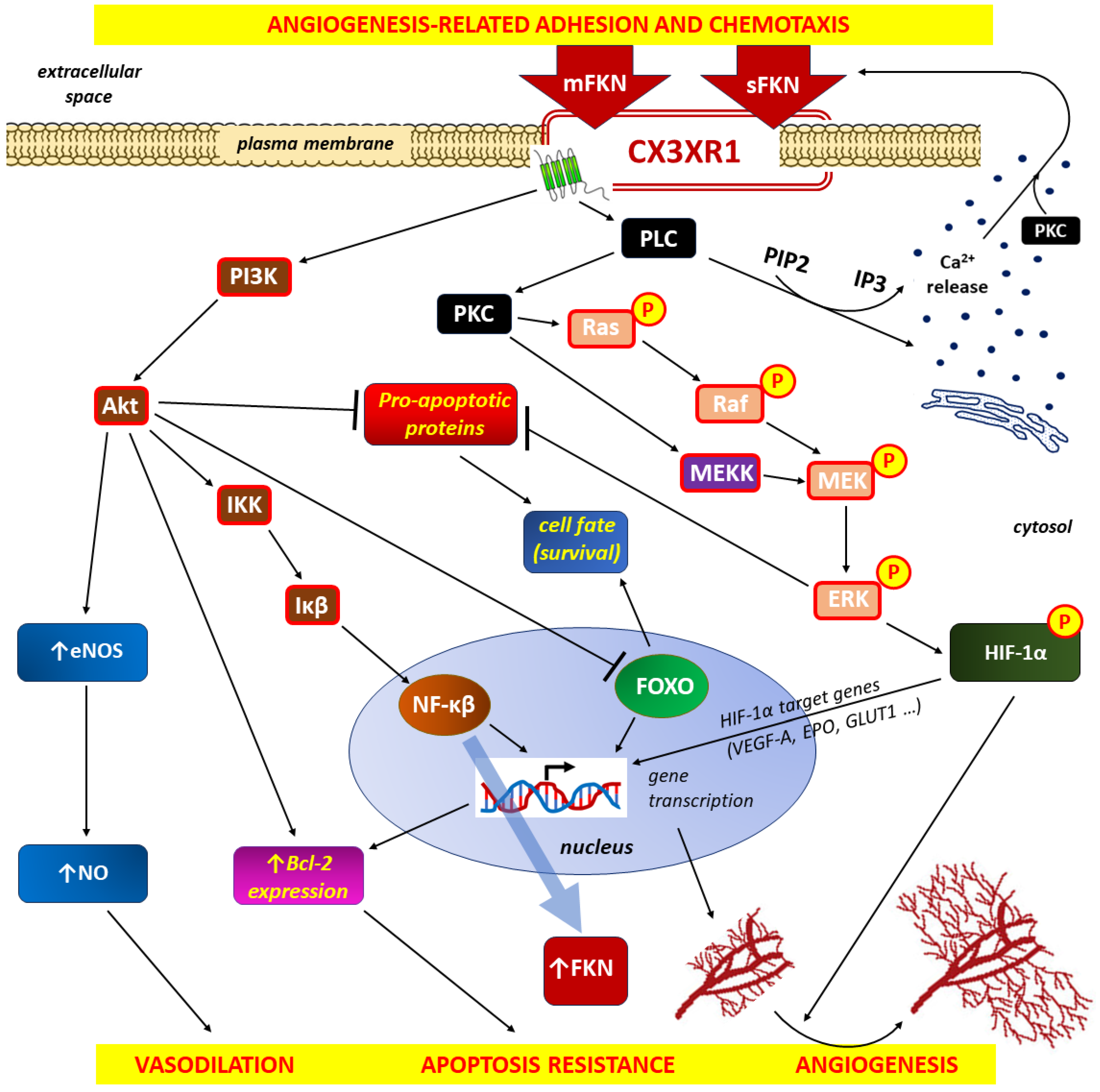
Figure 5.
The opposing actions of the FKN/CX3CR1 axis in the tumor microenvironment (TME) explain the occurrence of both anti-tumor (A.) and pro-tumor (B.) effects. A. Chronic inflammation in the tumor microenvironment (TME) upregulates both forms of fractalkine (FKN). The soluble form (sFKN) acts as a chemoattractant bringing cytotoxic and cytokine-producing cells to areas of inflammation. Thus, FKN serves as an important chemokine recruiting CX3CR1+ tumor-infiltrating leukocytes (TIL) such as CD4+ and CD8+ T cells, monocytes (M), B cells (B), neutrophils (N), natural killer (NK) and dendritic cells (DC) [347]. Enhanced cytotoxic T cell-mediated immunity is associated with anti-tumor effects. Additionally, increased CX3CR1 density in the TME, resulting from the accumulation of CX3CR1+ cells of the immune system, may induce CX3CR1 expression in tumor cells (TC) [146,338]. Simultaneous co-expression of the membrane-bound FKN (mFKN) and CX3CR1 in cancer cells activates integrins, which leads to cells adhering and sticking together (marked with anchors), which significantly impedes their migration and spread of the tumor [339]. B. Chronic inflammation in TME may activate proteinases, mainly tumor necrosis factor-alpha converting enzyme (TACE) also known as a disintegrin and metalloprotease 17 (ADAM17) TACE/ADAM17, transiently or in a tumor-specific manner [351,352]. As a result, mFKN on TC is cleaved and the mFKN/sFKN balance is shifted in favor of the soluble form. TC adhesion is significantly weakened (marked with a crossed out anchor) because sFKN, responsible for chemotaxis, has become the main CX3CR1 ligand. If this is accompanied by increased expression of CX3CR1 in TC, sFKN-induced cancer cell migration will occur [348]. Therefore, simultaneous increase in sFKN and CX3CR1 expression may cause pro-tumor effects of the FKN/CX3CR1 axis with increased risk of metastasis due to the presence of circulating tumor cells (CTCs), separated from the primary tumor and traveling through the blood stream [326]. Because TACE/ADAM17 activates epidermal growth factor receptor (EGFR), hypoxia-induced angiogenesis, uncontrolled proliferation and apoptosis resistance of cancer cells do not necessarily depend directly on CX3CR1, but on EFGR signaling [98]. Moreover, some FKN signaling pathways, including PI3K/Akt/IKK/Iκβ/NF-κβ, are also EGFR-mediated [176,185].
Figure 5.
The opposing actions of the FKN/CX3CR1 axis in the tumor microenvironment (TME) explain the occurrence of both anti-tumor (A.) and pro-tumor (B.) effects. A. Chronic inflammation in the tumor microenvironment (TME) upregulates both forms of fractalkine (FKN). The soluble form (sFKN) acts as a chemoattractant bringing cytotoxic and cytokine-producing cells to areas of inflammation. Thus, FKN serves as an important chemokine recruiting CX3CR1+ tumor-infiltrating leukocytes (TIL) such as CD4+ and CD8+ T cells, monocytes (M), B cells (B), neutrophils (N), natural killer (NK) and dendritic cells (DC) [347]. Enhanced cytotoxic T cell-mediated immunity is associated with anti-tumor effects. Additionally, increased CX3CR1 density in the TME, resulting from the accumulation of CX3CR1+ cells of the immune system, may induce CX3CR1 expression in tumor cells (TC) [146,338]. Simultaneous co-expression of the membrane-bound FKN (mFKN) and CX3CR1 in cancer cells activates integrins, which leads to cells adhering and sticking together (marked with anchors), which significantly impedes their migration and spread of the tumor [339]. B. Chronic inflammation in TME may activate proteinases, mainly tumor necrosis factor-alpha converting enzyme (TACE) also known as a disintegrin and metalloprotease 17 (ADAM17) TACE/ADAM17, transiently or in a tumor-specific manner [351,352]. As a result, mFKN on TC is cleaved and the mFKN/sFKN balance is shifted in favor of the soluble form. TC adhesion is significantly weakened (marked with a crossed out anchor) because sFKN, responsible for chemotaxis, has become the main CX3CR1 ligand. If this is accompanied by increased expression of CX3CR1 in TC, sFKN-induced cancer cell migration will occur [348]. Therefore, simultaneous increase in sFKN and CX3CR1 expression may cause pro-tumor effects of the FKN/CX3CR1 axis with increased risk of metastasis due to the presence of circulating tumor cells (CTCs), separated from the primary tumor and traveling through the blood stream [326]. Because TACE/ADAM17 activates epidermal growth factor receptor (EGFR), hypoxia-induced angiogenesis, uncontrolled proliferation and apoptosis resistance of cancer cells do not necessarily depend directly on CX3CR1, but on EFGR signaling [98]. Moreover, some FKN signaling pathways, including PI3K/Akt/IKK/Iκβ/NF-κβ, are also EGFR-mediated [176,185].
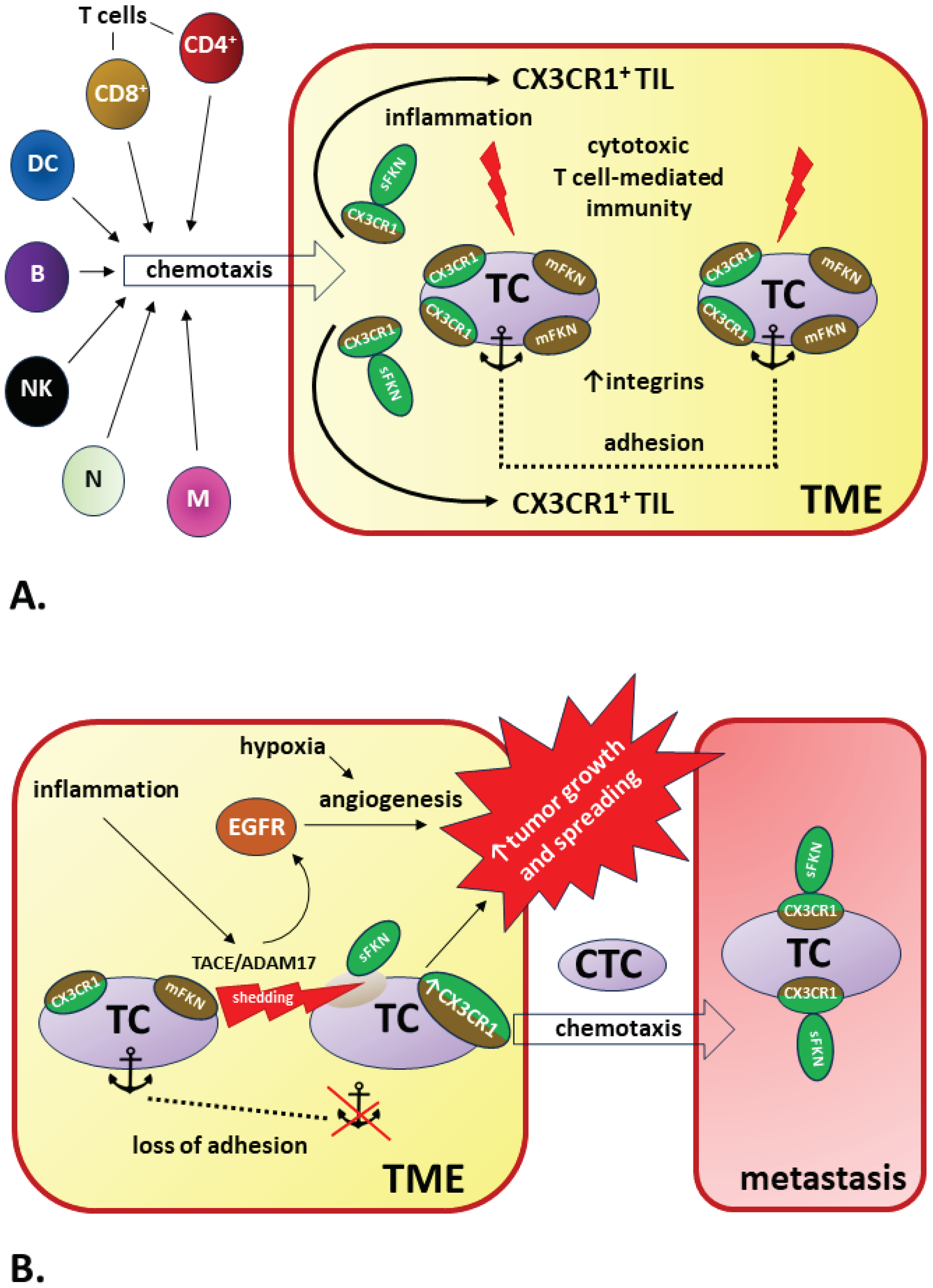

Table 1.
Examples of proven actions within the FKN/CX3CR1 axis in physiological states and pathophysiology of diseases in selected tissues, organs and systems.
Table 1.
Examples of proven actions within the FKN/CX3CR1 axis in physiological states and pathophysiology of diseases in selected tissues, organs and systems.
| Location in the body | Regulation of biological processes via FKN/CX3CR1 axis in normal and pathological conditions | References |
|---|---|---|
| Central nervous system (CNS) | - In brain tissues, FKN is mostly expressed in neurons while microglia express CX3CR1. - FKN/CX3CR1 signaling enables precise interactions between neurons, microglia, and immune cells. - Through its key role in microglia-neuron communication, the FKN/CX3CR1 axis regulates a broad spectrum of microglial properties including microglial cell migration and dynamic surveillance of the brain parenchyma, neuronal survival, synaptic plasticity, and a variety of synaptic functions, as well as neuronal excitability via cytokine release modulation, chemotaxis, and phagocytosis. - FKN suppresses lipopolysaccharide (LPS)-induced microglia activation by reducing the production of nitric oxide (NO), interleukin-6 (IL-6) and transforming growth factor alpha (TNF-α), and therefore inhibits neuronal cell death in response to LPS cytotoxicity in the brain tissue. - FKN/CX3CR1 signal disruption is one of the most important elements in the pathogenesis of CNS-related disorders, especially neurodegenerative diseases and brain traumatic injuries. However, the results of studies on the modulation of inflammation in the CNS by FKN/CX3CR1 are often ambiguous or contradictory. For example, disruption of FKN signaling is beneficial in limiting the effects of CNS ischemia, but detrimental in other neurodegenerative diseases, including Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS). Furthermore, deletion of CX3CR1 in Alzheimer’s disease may, possibly depending on the stage of disease progression, lead to both neuroprotective and detrimental effects. There is also no complete agreement on the importance of the involvement of FKN isoforms in the development of neuropathological processes. - The specific response, neurotoxic or neuroprotective, most likely depends on the type of destructive factor, the CNS area influencing the regional heterogeneity of microglial cells, and the local concentrations of FKN and CX3CR1. |
Sheridan and Murphy 2013 [188] Pawelec et al. 2020 [189] Paolicelli et al. 2014 [190]; Camacho-Hernández and Peña-Ortega 2023 [191] Mizuno et al. 2003 [192]; Lyons et al. 2009 [161]; Mecca et al. 2018 [193] Subbarayan et al. 2022 [194]; Cipriani et al. 2011 [195]; Poniatowski et al. 2017 [57]; Luo et al. 2019 [196]; Bivona et al. 2023 [197]; Nash et al. 2015 [198]; Juliani et al. 2021 [199]; Lee et al. 2010 [200], Cho et al. 2011 [201]; Fuhrmann et al. 2010 [202]; Pawelec et al. 2020 [189]; Eugenín et al. 2023 [203] Sheridan and Murphy 2013 [188]; Stratoulias et al. [204] |
| Bone marrow and immune tissue | - The expression of CX3XR1 increases with the maturation of myeloid cells and shows an inverse correlation with the Ly6C marker and the C-C chemokine receptor 2 (CCR2) in the blood. This may indicate that CX3CR1 reduces the motility of Ly6C(high) monocytes in the bone marrow and thereby controls their release into the bloodstream. - FKN-CX3CL1 axis plays a significant role in an early stage of osteoblast differentiation, possibly through their trans and cis interactions. - FKN regulates mouse osteoclast precursors (OCPs) survival and primes OCPs for subsequent osteoclast differentiation. - Autoimmune and inflammatory response in rheumatoid arthritis (RA) is positively correlated with concentration of FKN in the serum and synovial fluid. The associated chemotaxis primarily involves the recruitment of CD16+ monocytes into synovial tissues, as CX3CR1 expression in CD16+ monocytes is markedly higher compared to other populations (e.g., CD14+ and CD16-). - Bone marrow (BM) FKN levels are significantly increased in the multiple myeloma (MM) patients and positively correlated with BM microvessel density. - CX3CR1 expression is an additional marker of natural killer (NK) cells differentiation and is closely related to their ability to migrate to the central nervous system (CNS) from the periphery. - CX3CR1 is prevalently expressed on killer cell lectin-like receptor subfamily G member 1 (KLRG1)+ NK cells, a subset considered terminally differentiated. Therefore, CX3CR1 may represent a marker of a KLRG1(+) NK-cell population with unique properties that can irreversibly differentiate from the KLRG1(+)/CX3CR1(-) NK cells during steady state conditions in the bone marrow. - FKN activates Jak2-Stat5α-ERK1/2 signaling through CX3CR1, thereby triggering integrin-dependent machinery of the cytoskeleton reorganization to allow chemotactic migration of bone marrow-derived mesenchymal stem cells (BMSCs) towards an ischemic cerebral lesion. |
Jacquelin et al. 2013 [205] Hoshino et al. 2013 [206] Kuboi et al. 2022 [207] Yano et al. 2007 [208] Marchica et al. 2019 [209] Hamman et al. 2011 [90] Sciumè et al. 2011 [210] Zhang et al. 2015 [211] |
| Cardiovascular system | - FKN and CX3CR1 are expressed in atherosclerotic lesions and FKN is involved in the initiation step of atherosclerotic plaque formation. Monocyte-endothelial cell interactions are partly mediated by expression of monocyte CX3CR1 and endothelial cell FKN. Activation of these lymphocytes upon ligand/receptor binding leads to release of lysis granules that destroy vascular endothelium. - After endothelial damage, the release of FKN from apoptotic cells results in subsequent recruitment of macrophages, which promotes the removal of apoptotic debris; However, in more advanced stages of atherosclerosis, signaling through the FKN/CX3CR1 axis enhances foam cell formation, promoting the development of atherosclerotic plaques. - CX3CR1 expression on vascular smooth muscle cells (VSMCs) within atherosclerotic plaque causes the functional state of the FKN/CX3CR1 axis to play an important role in plaque stability. Events responsible for cardiovascular mortality and morbidity are predominantly caused by rupture of “vulnerable” atherosclerotic lesions. - FKN promotes the development of atherosclerotic lesions by activating platelets and causing their adhesion to the endothelium. - Early activation of the cardiac FKN/CX3CR1 axis delays β-adrenergic-induced heart failure. - FKN levels are markedly elevated during acute myocardial infarction (AMI), compared to patients with stable angina pectoris (AP), although they do not correlate with infarct size. The inverse pattern in gene expression of CX3CR1 might be here a compensatory mechanism. - In addition to demonstrating a positive correlation of FKN concentration with an increased risk of developing poorer cardiac function after AMI, levels of FKN had also been proven to be prognostic for the likelihood of developing major adverse cardiovascular events (MACEs) in an acute ST-elevation myocardial infarction (STEMI) patients. - Inhibition of the FKN/CX3CR1 interaction has a beneficial effect on the final infarct size after reperfusion, as it reduces the severity of an important complication - ischemia/reperfusion injury. This complication is directly related to the action of CX3CR1+ lymphocytes towards microvascular obstruction (MVO). |
Teupser et al. 2004 [212]; Ma et al. 2022 [213]; Riopel et al. 2019 [214]; Liu and Jiang 2011 [215] Elliott et al. 2017 [216]; White et al. 2014[186]; Landsman et al. 2009 [167] Lucas et al. 2003 [217]; Harman and Jørgensen 2019 [218]; Apostolakis and Spandidos 2013 [219]; Skoda et al. 2018 [220] Noels et al. 2019 [221]; Flierl et al. 2015 [76] Flamant et al. 2021 [222] Njerve et al. 2014 [223]; Yao et al. 2015 [224] Yao et al. 2015 [224];Xu et al. 2019 [225] Loh et al. 2023 [44]; Boag et al. 2015 [226] |
| Respiratory system | - CX3CR1+ leukocyte attachment to and migration through the lung vascular endothelium lead to mononuclear cell accumulation in the lung vessel walls and parenchyma. Infiltrated CX3CR1+ immune cells can release mediators to induce injury, stimulate proliferation, and/or chemoattract inflammatory cells. This contributes to structural destruction and remodeling in the development of inflammatory lung diseases. - FKN/CX3CR1 signaling may be involved in the pathophysiology of hypoxia-induced pulmonary arterial hypertension (PAH) developing due to chronic inflammation. Both increased FKN concentrations and upregulated CX3CR1 expression cause PAH progression with vascular remodeling and proliferation of pulmonary artery smooth muscle cells. - Soluble FKN chemoattracts and activates CX3CR1+ leukocytes such as CD8+, CD4+, and γδ T lymphocytes, natural killer cells, dendritic cells, and monocytes/macrophages leading to mononuclear cell accumulation in the lung vessel walls and parenchyma. During the resolution phase of acute lung injury, apoptotic cell-derived CX3CL1 attracts alveolar macrophages transmigration toward apoptotic cells for phagocytosis. - In allergic asthma CX3CR1 is required for airway inflammation by promoting T helper cell survival and maintenance in inflamed lung together with chemotaxis recruited mast cells into bronchial mucosa. - CX3CL1 (fractalkine) is elevated in both bronchoalveolar lavage fluid and sputum from human asthmatics sensitized to fungi, implicating an association with CX3CL1 in fungal asthma severity. However, CX3CL1/CX3CR1 axis preserves lung function during fungal-associated allergic airway inflammation through a nonclassical immunoregulatory mechanism. Hence, absence of CX3CR1 signaling resulted in a profound impairment in lung function during fungal-associated allergic airway inflammation. - In pulmonary infections, the role of FKN/CX3CR1 axis remains unclear. For example, FKN may be involved in both immunopathological and anti-viral immune responses to rhinovirus infection. |
Zhang and Patel 2010 [227] Balabanian et al. 2002 [228]; Amsellem et al. 2017 [229] Tsai et al. 2021 [230] Mionnet et al. 2010 [91]; El-Shazly et al. 2006 [231] Godwin et al. 2021 [232] Upton et al. 2017 [233] |
| Liver | - FKN/CX3CR1 is upregulated during liver damage including chronic inflammatory liver diseases such as chronic hepatitis C, nonalcoholic steatohepatitis (NASH)/nonalcoholic fatty liver disease (NAFLD) and cirrhosis. - Assessment of the impact of increased FKN/CX3CR1 activity on the severity of steatosis, inflammation and liver fibrosis is still ambiguous. In addition to reports indicating that FKN-CX3CR1 interaction inhibits inflammatory properties in Kupffer cells/macrophages, resulting in decreased liver inflammation and fibrosis, there are also contradictory research data. - FKN/CX3CR1 upregulation was reported in injured bile ducts of primary cirrhosis with its involvement in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Moreover, the correlation between primary biliary cirrhosis and FKN expression is significantly proportional. |
Efsen et al. 2002 [234]; Bourd-Boittin et al. 2009 [59]; Sutti et al. 2015 [235]; Nagata et al. 2022 [236] Aoyama et al. 2010 [237]; Zhang et al. 2020 [238]; Ni et al. 2022 [71]; Sutti et al. 2015 [235]; Karlmark et al. 2010 [239]; Wasmuth et al. 2008 [240]; Hassan et al. 2023 [241] Isse et al. 2005 [242]; Shimoda et al. 2010 [243] |
| Gut | - Most macrophages and some dendritic cell (DC) subsets express CX3CR1 in the gut. In resting intestinal mucosa, the role of lamina propria CX3CR1+ macrophage is to pass captured antigen via trans-epithelial dendrites or phagocytosis onto DC for transport to mesenteric lymph node (MLN) to prime immune responses like lamina propria DC. - Deletion of FKN or CX3CR1 leads to a specific and significant reduction in lamina propria macrophages with decreased translocation of bacteria to MLNs and their ability to take up pathogens. Therefore, CX3CR1 may be treated as a specific marker for lamina propria macrophages and a critical component in maintaining lamina propria macrophage homeostasis. Contradictory, it has been reported that CX3CR1 deficient mice shows normal number of macrophages. - Intestinal microbiota can influence local accumulation of CX3CR1+ phagocytes, and the number of CX3CR1+ cells is reduced in germ-free mouse. - Enhanced recruitment of CX3CR1+ T cells by mucosal human intestinal microvascular endothelial cell (HIMECs)-derived FKN has been demonstrated in inflammatory bowel disease (IBD). |
Joeris et al. 2017 [244]; Niess et al. 2005 [114]; Bain and Mowat 2011 [245]; Lee et al. 2018 [70] Ferretti et al. 2014 [246]; Bain et al. 2013 [247] Bain et al. 2014 [248] Sans et al. 2007 [249] |
| Placenta | - Human placenta is a source of FKN, which is expressed in the syncytiotrophoblast and can be released into the maternal circulation by constitutive MMP-dependent shedding. - FKN content within the apical microvillous plasma membrane increases significantly in the placenta of full-term pregnancy compared to the first trimester. - FKN/CX3CR1 axis mediates adhesion of monocytes to the villous trophoblast. - Increased expression and release of placental FKN may contribute to low grade systemic inflammatory responses in third trimester of normal pregnancy. - Placental FKN is upregulated in severe early-onset preeclampsia (PE). Significant underdevelopment of placental vascular network with significantly lowered the vascular/extravascular tissue index (V/EVTI) in PE is associated with dysregulation of the FKN/CX3CR1 system, especially in fetal growth restriction (FGR)-complicated pregnancies. - Increased average FKN content in the diabetic placenta is accompanied by an increase in the density of placental microvessels and a higher expression of CX3CR1, compared to the placenta from a normal pregnancy. Therefore, FKN/CX3CR1 signaling pathway is involved in the pathomechanism of placental microvasculature remodeling during diabetes class C (after White). - Placental hypoxia increases FKN production and upregulates CX3CR1 expression in the placental endothelial cells. Under these conditions, tumor necrosis factor alpha (TNFα) induces FKN, influencing a mechanism of FKN autoregulation by CX3CR1 expression. - Increased FKN concentration, accompanied by a higher mean FKN gene expression level in the tissues of pregnant women with missed abortion may be responsible for abnormal placental invasion. |
Siwetz et al. 2014 [250] Siwetz et al. 2015 [251] Siwetz et al. 2015 [252] Vishnyakova et al. 2021 [253] Szewczyk et al. 2021 [254]; Ullah et al. 2023 [255] Szukiewicz et al. 2013 [256]; Ullah et al. 2023 [255]; Szukiewicz et al. 2014 [257] Gokce et al. 2022 [258] |
| Joint and bone tissue | - The number of circulating CX3CR1high T cell is elevated in the circulation of rheumatoid arthritis (RA) patients. Joint-infiltrated CX3CR1high T cells strongly adhere to fibroblast-like synoviocytes (FLSs) in the synovium in an FKN-dependent manner. - FKN/CX3CR1 axis promotes inflammation-free osteoclastogenesis by enhancing precursor cell survival and differentiation. - Apoptosis of chondrocytes during joint osteoarthritis upregulates of the FKN-CX3CR1-p38 axis, which results in enhanced chemotaxis of osteoclast precursors (OCPs) and promotes bone resorption. - Development of osteoarthritis (OA) is largely driven by low-grade local background inflammation based on FKN-mediated chemotaxis. - FKN/CX3CR1 signaling in hemophilia is involved in the pathomechanism of irreversible joint degeneration (hemophilic arthropathy). - High concentrations of FKN in human blood serum are accompanied by increased concentrations of bone turnover and inflammatory factors in the serum, such as tartrate-resistant acid phosphatase 5b(TRACP-5b), cross-linked N-telopeptides of type I collagen (NTx) and interleukins (IL-1β, IL-6). - FKN knockdown ameliorates inflammation and apoptosis after exposure to LPS, and accelerates osteogenic differentiation. These effects related to FKN deficiency can be reversed by increased expression of CX3CR1. - FKN axis signaling alleviates intervertebral disc degeneration (IDD) by reducing inflammation and apoptosis of human nucleus pulposus cells (HNPCs) via macrophages. |
Tanaka et al. 2020 [259] Kuboi et al. 2022 [207]; Koizumi et al. 2009 [260] Koizumi et al. 2009 [260]; Guo et al. 2022 [261] Wojdasiewicz et al. 2014 [43,262] Wojdasiewicz et al. 2020 [184] Wojdasiewicz et al. 2019 [160] Lu et al. 2023 [263] Gao et al. 2023 [264] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



