
Submitted:
27 March 2024
Posted:
28 March 2024
You are already at the latest version
Abstract
In the last three decades the presence of phospholipids in the nucleus has been shown and thoroughly investigated. A great interest raised about nuclear inositol lipids mainly because of their role in signalling acting. Here we review the main issues of nuclear phospholipid localization and the role of nuclear inositol lipids and their related enzymes in cellular signaling both in physiological and pathological conditions.
Keywords:
nucleus
; phosphoinositides
; pholipases C
; myelodysplastic neoplasm (MDS)
; muscle
; brain
1. Introduction
Even though the presence of phospholipids in the nucleus has been reported since the 60s [1] it was difficult to assign a role to these molecules in the nuclear compartment.
In the sixties it was shown that the phospholipid content of active chromatin was 5-fold higher compared to repressed chromatin [2]. Few laboratories were interested in pursuing the biological significance of this observation with regards to cell cycle regulation, but the stumbling block was persisting controversy over the extent to which these phospholipids might arise by contamination from non-nuclear membranes. Manzoli and Cocco investigated this problem, and they concluded that non-contaminating phospholipids were genuinely associated with non-histone chromosomal proteins (NCHP), perhaps regulating DNA stability and replication. They also reported that NHCP fractions prepared from leukemic B-lymphocytes contained 40% less sphingomyelin and 4-fold higher levels of phosphatidylcholine, compared to normal B-cells [1]. The clear demonstration that lipids are really present at the chromatin level and that they do not derive from the nuclear membrane was given from Albi et al. [3] by using the radioiodination of the fatty acids technique. A good way to jump over the persisting controversy it seemed to investigate if there could be a role for nuclear phospholipids. Indeed in 1980 it has been described the analysis of Triton-treated nuclear material to remove nuclear membranes [4]. This short paper presented an important finding that put-to-bed the criticism that phospholipids in nuclear preparations must have arisen from contaminating membranes. Indeed, data had shown nuclear phospholipids need not even be associated with membranes. But still a bit of skepticism remained. That was the time of the emergence of inositol lipids as cellular signals [5]. They are present at very low levels and at that time were canonical plasma membrane signals. However, because of a hint by Smith and Wells [6], it was worthwhile to investigate the presence and possible role of these peculiar phospholipids in the nucleus. Smith and Wells had concluded inositol lipid synthesis occurred in the nuclear envelope, probably contaminated by the endoplasmic reticulum. But it was shown that membrane-free nuclei were able to synthesize phosphatidyl 4,5 bisphosphate (PtdIns(4,5)P2) in a cytoplasm-independent manner [7]. Several follow-up papers appeared that helped the nuclear inositol lipid idea become more generally accepted, and then others also began to publish studies into nuclear phosphoinositide signalling [8,9]. In 1992 it has been identified a nuclear-specific species of Phospholipase PLCβ that was activated within 2 minutes treatment of 3T3 mouse fibroblasts with insulin-like growth factor-1 [10].
2. Nuclear Phosphoinositides and Nuclear Functions
Polyphosphoinositides (PPIns) constitute a group of phospholipids originating from the primary compound Phosphatidylinositol (PtdIns). PtdIns consists of a myo-inositol hydrophilic head group linked to a hydrophobic diacylglycerol (DAG) tail backbone through a phosphodiester bond. The hydroxyl groups of the myoinositol head can be reversibly phosphorylated at the 3, 4 or 5 position yielding to seven distinct PPIns variants: PI3P, PI4P, PI5P, PI(3,4)P2, PI(3,5)P2, PI(4,5)P2 and PI(3,4,5)P3 [11].
As mentioned in the introductory paragraph, the nuclear presence of all phosphoinositides but one, PI(3,5)P2, has been reported in the 60s. Since then, biochemical studies ensured that the nuclear pool of PPIns was not a consequence of cytoplasmatic pool contamination and that it was regulated distinctly from the latter, while further studies defined nuclear targets potentially interacting with nuclear PPIns defining downstream signalling pathways.
Despite the fact that PPIs are amphiphilic, meaning they have a polar inositol head group that faces toward the cytoplasm/nucleoplasm and non-polar hydrophobic fatty acid tails embedded in the lipid bilayer [12], 40% of nuclear phosphoinositides can exist in a non-membrane state [13], mainly in nuclear speckles [14], membrane-less nuclear domains enriched in pre-mRNA splicing factors, located in the interchromatin regions of the nucleoplasm of mammalian cells [15]. Hydrophobic lipids probably exist within the aqueous nucleoplasm through the binding with nuclear proteins, but the exact mechanism and the specific proteins remain largely unclear. Steroidogenic factor-1 (SF-1; NR5A1 in the official nomenclature), a nuclear receptor transcription factor that plays a crucial role in the regulation of adrenal and gonadal development, function and maintenance [16], is one candidate nuclear protein binding PPIs. The research group guided by Blind showed that in a x-ray crystal structure of PI(4,5)P2 bound to the SF-1 ligand-binding domain the hydrophobic acyl chains of PI(4,5)P2 are hidden deep in the hydrophobic core of the SF-1 protein, while the hydrophilic phosphoinositide headgroup is solvent-exposed [17]. Additionally, they recently demonstrated the first evidence of association between SF-1 and nuclear PI(4,5)P2 in fixed human cells. They established two isogenic HEK cell line with a single, stably integrated tetracycline-inducible 3X-FLAG-tagged wild-type/mutated SF-1 and, after tetracycline treatment to induce SF-1 expression, they analyzed the nuclear accumulation of the immunofluorescence signal from antibodies directed against the headgroup of PI(4,5)P2. The results showed that the ectopic expression of wild-type SF-1 in HEK cells associates with the induction of a nuclear signal that cross-reacts with PI(4,5)P2 antibodies [18]. Further studies should analyze how SF-1 initially acquires the PI(4,5)P2 phospholipid and whether PIP2 antibody is directly recognizing PI(4,5)P2 bound to SF-1, or if the signal is an undiscovered, indirect byproduct of SF-1 expression.
Nuclear PPIs have several functions encompassing DNA synthesis, epigenetic signalling, transcription factor regulation and DNA damage signalling [19].
Concerning DNA synthesis, the research group of Cocco obtained two different clones of Swiss 3T3 cells, one unresponsive and one responsive to the mitogenic stimulus by insulin growth factor 1 (IGF1). They demonstrated that in highly purified nuclei of responsive cells treated with IGF1, incubated with [γ-32 P]adenosine triphosphate, the PIP and PIP2 labeling decreased compared to the controls and the Protein Kinase C (PKC) was activated. On the contrary, in unresponsive cells they did not notice nor the transient effect of PPIs neither the PKC activation. These results show a direct link between polyphosphoinositide metabolism, nuclear PKC and early events leading to cell division [20].
PPIs can participate in epigenetic and transcription signalling through histone modifications regulation and transcription complexes interaction. An example can be represented by PI5P binding to several nuclear proteins through plant homeodomains (PHD) domains that in turn interact with methylated and non-methylated lysine residues in histone tails and regulate gene expression [21]. The basal transcription complex TFIID has a component, TAF3, a critical factor that controls embryonic stem cell pluripotency and is critical for myogenic differentiation, with a PHD finger that interacts with H3K4me3 to regulate gene expression [22]. This enables changes in nuclear PI5P to impact on myogenic differentiation. Under non-differentiating conditions, PIP4K2β, a nuclear-localized lipid kinase, phosphorylates PI5P to PI(4,5)P2 [23], thereby attenuating the expression of genes required for myogenic differentiation induced by TAF3. Upon induction of differentiation, PIP4K2β is localized into the cytoplasm, leading to increased nuclear PI5P [24].
PPIs are finally involved in DNA damage repair as well. PIP5K is a P2(3,4)P2 synthetizing enzyme [25] which interacts with the p53 tumor suppressor protein, whose post-transcriptional expression is reduced by the knockdown or inhibited activity of PIP5K1A [26]. p53 also directly interacts with PIP2 inducing an interplay between p53 and the heat shock protein HSP27, leading to p53 stabilization [27]. Additionally, the PIP2-p53 complex is a substrate for Inositol Polyphosphate Multikinase (IPMK), which generates p53 bound to PI(3,4,5)P3, leading to the activation of nuclear PKB that in turn associates nucleophosmin (NPM/B23) to regulate the expression of genes involved in DNA repair and cell survival in response to a DNA damage signal [28]. PKB is one of the most well-studied targets of PI(3,4,5)P3, with the interaction occurring through the PH domain of PKB. Moreover, biochemical assays found that also nucleophosmin is a nuclear target of PI(3,4,5)P3, and that the interaction of B23 with PI(3,4,5)P3 is required for interaction with PKB in the nucleus [29].
What still remains to be investigated in this context is how the nuclear PPIns pool is established and how their associated modulating enzymes and downstream signalling pathways are controlled and maintained. Are PPIs formed in the cytoplasm and then translocated to the nucleus, or are they directly synthesized in the nucleus?
3. Nuclear PI-PLCs
Phosphoinositide-Specific Phospholipase C (PI-PLC) is a crucial family of enzymes encompassing 13 isoforms, further categorized into six families that exhibit varying expression patterns across different tissues: PI-PLCβ (1-4), PI-PLCγ (1-2), PI-PLCδ (1,3,4), PI-PLCε, PI-PLCζ e PI-PLCη (1-2) [30,31]. Structurally, these isoforms possess a catalytic domain consisting of X and Y domains, along with a regulatory domain comprising the C2 domain, EF-hand motif, and pleckstrin homology domain (PH domain), all of which are highly conserved [32]. However, each isoform displays a unique combination of these domains, resulting in distinct regulatory mechanisms, functions, and tissue distributions [32]. Broadly, these enzymes act as catalysts in the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) in response to various stimuli, including growth factors, hormones, and neurotransmitters [30,31,32,33,34]. IP3 and DAG function as second messengers, with IP3 facilitating intracellular calcium ion accumulation, and both IP3 and DAG collectively activating Protein Kinase C (PKC), thereby intricately regulating the cell cycle at multiple levels [30].
The cellular distribution of PLC enzymes varies based on the isoform and can change under diverse circumstances [35].
Observations on Swiss 3T3 mouse fibroblasts revealed that PI-PLC activity is activated within the nucleus independently of plasma membrane phosphoinositide metabolism. This activation, triggered by insulin-like growth factor-1 (IGF-1), resulted in changes in nuclear phosphatidylinositol levels and the translocation of protein kinase C (PKC) into the nucleus [8,20]. Subsequent studies identified PI-PLC β1 as the enzyme responsible for this nuclear activity. Additionally, PI-PLC β1, along with other enzymes such as diacylglycerol kinase ζ (DGK ζ) and phosphatidylinositol phosphate kinase α (PIPKα), was found to localize to nuclear speckles. These findings provide insights into the distinct roles of PI-PLC isoforms within the nucleus and their involvement in cellular signaling pathways [36].
The PI-PLCβ1 enzyme, encoded by a gene located on the short arm of chromosome 20 (20p12), is characterized by two splicing variants that differ at the C-terminal domain: PI-PLCβ1a (150 kDa) and PI-PLCβ1b (140 kDa) [37]. Both isoforms contain a nuclear localization sequence (NLS) at the 3′ end, but PI-PLCβ1a also features a nuclear export sequence (NES) downstream of the NLS sequence [38]. Consequently, PI-PLCβ1a localizes in both the nucleus and the cytoplasm, whereas PI-PLCβ1b is predominantly distributed in the nucleus, particularly in nuclear regions called speckles, where gene expression regulation occurs [39]. Researches have shown that phosphorylation of PLCβ1 by P42-MAPK enhances its nuclear activity, but subsequent phosphorylation by PKC deactivates PLCβ1 [40]. Following activation, nuclear PI-PLCB1 plays a role in regulating the cell cycle at both the G1/S transition and G2/M progression through various molecules. Furthermore, nuclear PI-PLCB1 has been linked to processes such as hematopoietic, osteogenic, myogenic, and adipogenic differentiation [41].
PI-PLCγ1 has been identified within the nucleus, with its SH3 domain reported to function as a guanine nucleotide exchange factor for specific nuclear GTPases, including phosphoinositide 3-kinase enhancer and dynamin-1. Notably, the assistance provided by PI-PLCγ1 to phosphoinositide 3-kinase enhancer is independent of its phospholipase activity, influencing various cellular functions such as proliferation and survival [42].
Also the PI-PLCδ family members exhibit bidirectional movement between the nucleus and cytoplasm, suggesting their potential involvement in regulating cell growth. Although PI-PLCδ1 is typically located in the cytoplasm of quiescent cells, it localizes inside nuclear structures at the G1/S boundary during the cell cycle. This isoform possesses both nuclear export and import sequences, facilitating its movement between the cytoplasm and nucleus [42].
Another variant, PI-PLCδ4, demonstrates nuclear localization in various cellular contexts, including regenerating rat liver, serum-stimulated Swiss 3T3 cells, AH794 rat ascites hepatoma cells, and src-transformed 3Y1 cells [43]. Upon stimulation by mitogens, PI-PLCδ4 expression is upregulated in the nucleus, where it plays a significant role in promoting cell growth and is among the early genes activated during the transition from G1 to S phase in the cell cycle [44]. Specifically, the nuclear δ4 isoform shows a substantial increase at the G1 to S phase transition and maintains high levels until the end of the M phase. While some studies suggest that this isoform is exclusive to the nucleus, others have not replicated this finding [43].
Given the emerging roles of these molecules within the nucleus, it is intriguing to explore the involvement of nuclear PLCs in various pathologies, potentially serving as promising new markers or therapeutic targets.
4. Nuclear PI-PLCs in Myelodysplastic Neoplasm (MDS)
PLCs signaling has emerged as a significant factor in both normal and malignant hematopoiesis, as evidenced by several studies [45,46,47]. The regulatory mechanisms governing PLCs signaling hold significance in hematological malignancies as well, with PLCβ1 enzyme being linked to the pathogenesis of myelodysplastic syndromes (MDS) [48].
Myelodysplastic Syndromes (MDS) are a heterogenous group of myeloid neoplasms depending on alterate function of hematopoietic stem cells (HSCs), leading to ineffective hematopoiesis, dysplasia and peripheral blood cytopenia and characterized by an increased risk of evolution to Acute Myeloid Leukemia (AML) in approximately one-third of cases [49]. The International Prognostic Scoring System (IPSS) and its revised version (IPSS-R) have served as pivotal tools for predicting outcomes and guiding treatment decisions in leukemia based on initial laboratory and cytogenetic assessments [50]. Recently, advancements in next generation sequencing (NGS) techniques have led to the creation of the Molecular International Prognostic Scoring System (IPSS-M), which classifies patients into six prognostic categories, ranked by their estimated survival and risk of leukemia progression: Very Low, Low, Moderate Low, Moderate High, High, and Very High [51]. Although allogeneic stem cell transplantation remains the only treatment opportunity for a limited fraction of MDS patients, epigenetic therapy, which include the use of hypomethylating agents (HMAs) such as Azacytidine, is the first-line approach for high-risk MDS patients [52].
Abnormalities in phosphoinositide-dependent signaling, epigenetic regulators, apoptosis, and cytokine interactions within the bone marrow microenvironment are defining features that contribute to the pathogenesis of diseases and neoplastic growth of MDS [53] (Figure 1). Among the enzymes of the nuclear PI cycle, nuclear PI-PLCβ1 plays a fundamental role in the regulation of hematopoietic differentiation, that seems to be related to the recruitment of Myeloid zinc finger-1 (MZF-1) [30], and suggesting that this enzyme has a significant impact on the initiation and progression from MDS to AML by affecting both genetic and epigenetic processes [54,55,56]. Through fluorescence in situ hybridization (FISH), an analysis of 80 MDS cases unveiled that 43.75% of patients displayed a monoallelic deletion in the PI-PLCβ1 gene situated on chromosome 20p12 [57,58]. In contrast, the PI-PLCB4 gene, encoding another signaling molecule and positioned on 20p12.3 within 1Mb of PI-PLCB1, remained unaffected, even in MDS patients exhibiting the deletion of the PLCB1 gene, suggesting a distinct and interstitial deletion pattern for PLCB1 [57,58]. Interestingly, it has been demonstrated that the monoallelic deletion of PI-PLCB1 gene correlates with adverse clinical outcomes and an increased risk of AML development both in high and low risk MDS patients [58].
At the epigenetic level, high-risk MDS often exhibits hypermethylation of the CpG islands within the promoter region of the PI-PLCβ1 gene, as indicated in a review by Cocco et al. [32]. Additionally, Follo et al. demonstrated that nuclear PI-PLCB1 serves as a direct target of Azacytidine, with PI-PLCB1 expression potentially predicting the efficacy of Azacytidine treatment. They observed that responsive MDS patients displayed higher levels of PI-PLCB1 during clinical improvement, as well increased expression levels of Cyclin D3, one of the molecular targets of PI-PLCβ1, a key regulator of G1/S cell cycle phase and hematopoietic differentiation [59]. Furthermore, their study revealed a correlation between PI-PLCB1 levels and activated AKT levels, suggesting opposite roles for PI-PLCB1 and AKT [60]. In a subsequent study [61], the analysis of PI-PLCB1 expression in 18 high-risk MDS patients undergoing Azacytidine treatment showed that changes in gene expression preceded clinical improvements or worsening by about two months.
Recent studies have highlighted the crucial role of the PI3K/Akt signaling pathway in various physiological processes and its persistent activation in high-risk MDS [45]. Elevated levels of phosphorylated Akt have been observed in MDS patients’ bone marrow and peripheral blood cells, suggesting its involvement in leukemia progression [62]. Additionally, there’s a notable increase in mTOR pathway activity in high-risk MDS patients, with rapamycin showing potential therapeutic effects, suggesting the therapeutic potential of targeting the PI3K/Akt/mTOR network in both acute myeloid leukemia (AML) and high-risk MDS cases [45].
These findings indicate a potential association between PI-PLCβ1 gene silencing and Akt activation in high-risk MDS, suggesting a role in disease progression towards AML. This connection may be influenced by PIP2 levels, crucial for both PI-PLCβ1 and PI3K/Akt axis activation [45]. Nevertheless, these findings are significant for high-risk MDS patients and the interplay between PI3K/Akt/mTOR and PI-PLCβ1 signaling pathways suggests potential combined therapeutic approaches to target disease progression.
4. Nuclear PLCs in Muscle Diseases
Nuclear PLCs play a crucial role also in muscle diseases by impairing the correct myogenic differentiation, in which PLCs have an important role.
For studying physiological myogenic differentiation, multiple studies have explored the PI-PLCβ1-dependent signaling pathways during myogenic differentiation employing C2C12 murine myoblasts. These cells serve as a reliable and widely used model for investigating myogenesis, as they demonstrate the capacity to differentiate into myotubes under low serum conditions, allowing for the study of numerous muscle-specific genes and proteins [63]. C2C12 differentiation is characterized by a marked increase in PI-PLC-β1 and cyclin D3 expression [64,65]. Indeed, at the onset of the differentiation process, myoblast determination protein 1 (MyoD) induces cyclin D3 expression, which in turn binds to the unphosphorylated retinoblastoma protein, leading to the withdrawal of myoblasts from the cell cycle [66]. In addition, it was shown that PI-PLCβ1 could activate cyclin D3 promoter during the differentiation of myoblasts to myotubes, only in the presence of the binding of the transcription factor c-jun to cyclin D3 promoter [67,68]. Another crucial player in myogenic differentiation is IPMK, that leads to the production of higher phosphorylate inositol species starting from the IP3 substrate, the PI-PLCβ1 second messenger. IPMK production of IP5 is able to induce the Wnt/β-catenin pathway, leading to the translocation of β-catenin to the nucleus where it induces downstream gene expression, among which c-jun which in turn activates cyclin D3 promoter, demonstrating IPMK acts on the same cyclin D3 promoter region targeted by PI-PLCβ1 [69]. In summary, the model proposed for myogenic differentiation suggests that PI-PLCβ1 induction triggers the hydrolysis of PIP2, resulting in the production of IP3. Subsequently, IP3 undergoes phosphorylation by IPMK, leading to the generation of increasingly phosphorylated inositol species such as IP4, IP5, and IP6. The accumulation of IP5 prompts the translocation of beta-catenin to the nucleus, where it activates c-jun, ultimately stimulating the cyclin D3 promoter.
The most studied muscle diseases with impaired PLCs expression level, specifically PI-PLCβ1, are myotonic dystrophies (DM), the most common muscular dystrophies in adults [70]. They are autosomal dominant progressive multisystem disorders, which can indeed affect other non-skeletal muscle organs such as the heart, brain and gastrointestinal system. DM is divided in two subtypes: DM type 1 and type 2, both dominantly inherited with significant overlap in clinical manifestations such as myotonia, muscle weakness, insulin resistance, cardiac conduction defects, cataracts, cognitive dysfunction, and mental retardation. They are instead distinguished by the pathological origin: DM1 is triggered by the pathological expansion of a CTG triplet repeat in the gene coding for DMPK, the dystrophia myotonica-protein kinase, while DM2 by a CCTG tetranucleotide repeat expansion in the ZNF9 gene, which encodes a CCHC-type zinc-finger protein [71]. These expansions alterate the signalling pathway downstream the CUG triplet repeat RNA-binding protein 1 (CUGBP1), which plays a central role in alternative splicing of specific target genes and can interact with eukaryotic initiation factor 2 (eIF2) and cyclin D3 inducing normal myogenic differentiation. In normal myotubes, cyclin D3-cyclin dependent kinase 4/6 (cdk4/6) mediate indeed the phosphorylation of CUGBP1, increasing the interactions of CUGBP1 with eIF2 enabling translational function during normal myogenesis [72]. In DM-differentiating cells we assist to multiple alterations leading to ineffective myogenesis: CUGBP1-eIF2 interactions are reduced and both PI-PLCβ1 and cyclin D3 expression are decreased [73]. Figure 2 shows physiological and pathological myogenic differentiation.
Interestingly, the research group of Cocco demonstrated, by transfecting myoblasts from patients with DM1 and DM2, how forced PI-PLCβ1 expression can cause a cyclin D3 expression increase, determining a partially restored phenotype of the myotubes [74]. Additionally, Salisbury E. et al. showed that the ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1 [75].
To conclude this overview on DM, both PI-PLCβ1 and cyclin D3 could be investigated for future possible molecular therapies in order to induce a correct skeletal muscle differentiation in DM.
5. Nuclear PI-PLCs in Neurological Diseases
The healthy human brain comprises around 60% lipids, mainly found in myelin and white matter. Phospholipids (PLs) are the most abundant lipid species, crucial for neuronal membrane structure and function. Changes in phospholipid composition can disrupt membrane fluidity, impacting cellular signaling and leading to pathological conditions, including cancer. However, the mechanisms behind phospholipid alterations in tumors are not fully understood [76]. PI-PLCs are essential components in the intricate machinery of various brain regions, exerting influence over a spectrum of neural functions and implicated in the pathogenesis of numerous brain disorders [77].
Among the different isoforms, PI-PLCβs stand out as the prominent ones [78], with PI-PLCβ1 as the most extensively studied, at both cytoplasmatic and nuclear level, in brain contexts [79]. Its prevalence is notably observed in key regions like the hippocampus and cerebral cortex, as well as in specific neuronal populations such as cerebellar interneurons and telencephalic principal neurons [80]. Furthermore, PI-PLCδ isoforms, which possess both nuclear export and import sequences [81], including PI-PLCδ3 and PI-PLCδ4, exhibit distinct roles within neural architecture. PI-PLCδ3, primarily localized in the brain, contributes significantly to the radial migration of neurons during cerebral cortex development [32,82]. Meanwhile, PI-PLCδ4′s expression in both brain tissues and regenerating areas underscores its involvement not only in neural function but also in processes related to tissue repair [83].
PI-PLCβ1, in particular, has been identified as a crucial enzyme influencing various brain processes. For instance, nuclear PI-PLCB1 (i.e., PI-PLCB1b) is involved in the modulation of endocannabinoid neuronal excitability through DAG synthesis and can be linked to depolarization and receptor activation, maintaining the brain inhibitory pathways via 2-AG [77]. Moreover, PI-PLCB1 pathway, which is activated via metabotropic glutamate receptors (mGluRs), is also implicated in the development of normal cortical circuit and in activity-dependent development of the cerebral cortex [84,85]. These suggest that imbalances in PI-PLCβ1 could be associated with several brain disorders, including epilepsy, schizophrenia, and neuro-oncological pathologies [83,86,87].
In addition, research has revealed that PI-PLCβ1 exhibits co-localization and physical interaction with Lamin B1, a crucial component of the nuclear lamina [88]. These findings gain significance in the context of Autosomal Dominant Leukodystrophy (ADLD), a remarkably rare and fatal neurodegenerative disorder characterized by the overexpression of Lamin B1 due to genetic anomalies such as LMNB1 gene duplications or deletions. Recent investigations emphasize the pivotal role played by cellular signaling and morphological changes, particularly in astrocytic function, in the progression of ADLD [89]. Given the established role of PI-PLCβ1 in various cellular processes within the brain, further exploration is warranted to ascertain potential correlations between its interactions with Lamin B1 and the pathogenesis of this ADLD.
Notably, there is substantial evidence suggesting the involvement of PI metabolism in the development of Glioblastoma Multiforme (GBM), highlighting the intricate interplay between PLCs and other mediators regulating cell proliferation, differentiation, migration, and growth [90,91,92] (Figure 3). GBM represents a grade IV astrocytoma, distinguished by its diverse cellular composition, genetic instability, extensive infiltration, angiogenesis, and resilience against existing treatments [93]. GBM can arise either spontaneously or progress from a pre-existing astrocytoma, both scenarios carrying a bleak prognosis. Despite ongoing efforts, current therapies for GBM exhibit limited efficacy, with only a small fraction of patients—approximately 3-5%—surviving beyond three years [94].
Analyses of the Cancer Genome Atlas and Gene Expression Omnibus have shed light on the potential significance of PI-PLCβ1 as a biomarker in high-grade gliomas (HGG). Notably, an inverse relationship between PI-PLCβ1 expression and the pathological grade of gliomas has been elucidated, suggesting its potential as a prognostic indicator and a novel signature gene for proneural (PN) subtypes within the molecular classification of HGG. Additionally, insights from Kaplan-Meier survival analysis from the Repository for Molecular Brain Neoplasia Data (REMBRANDT) underscore a correlation between PI-PLCβ1 expression and prolonged patient survival [92].
More recently, Ratti et al. not only corroborated these data, but also demonstrated that in vitro downregulation of PI-PLCB1 is associated with increased expression of mesenchymal markers (e.g., Slug, N-Cadherin), matrix metalloproteinases MMP-2 and MMP-9, and the nuclear marker Ki-67. Furthermore, there was an overexpression of the active form of B-catenin and heightened activation of the ERK1/2 pathway. Taken together, these findings indicate that silencing of PI-PLCB1 correlates with increased invasion, migration, and cell proliferation [90]. Indeed, after silencing of PI-PLCB1, there was also observed increased activation of the Stat3 pathway, a well-established oncogenic transcription factor known to play a pivotal role in tumor resistance and aggressive cancer progression in glioblastoma [90], thereby strengthening the hypothesis that downregulation of PI-PLCB1 in glioblastoma promotes a more aggressive phenotype. The mechanisms behind PLCβ1 downregulation in high-grade tumors, particularly in glioblastoma, remain unclear. Understanding these processes and their specific roles in gliomas could lead to the discovery of diagnostic and prognostic biomarkers. Further research is necessary to identify epigenetic anomalies associated with this condition, which could inform the development of targeted therapies [90].
6. Conclusions
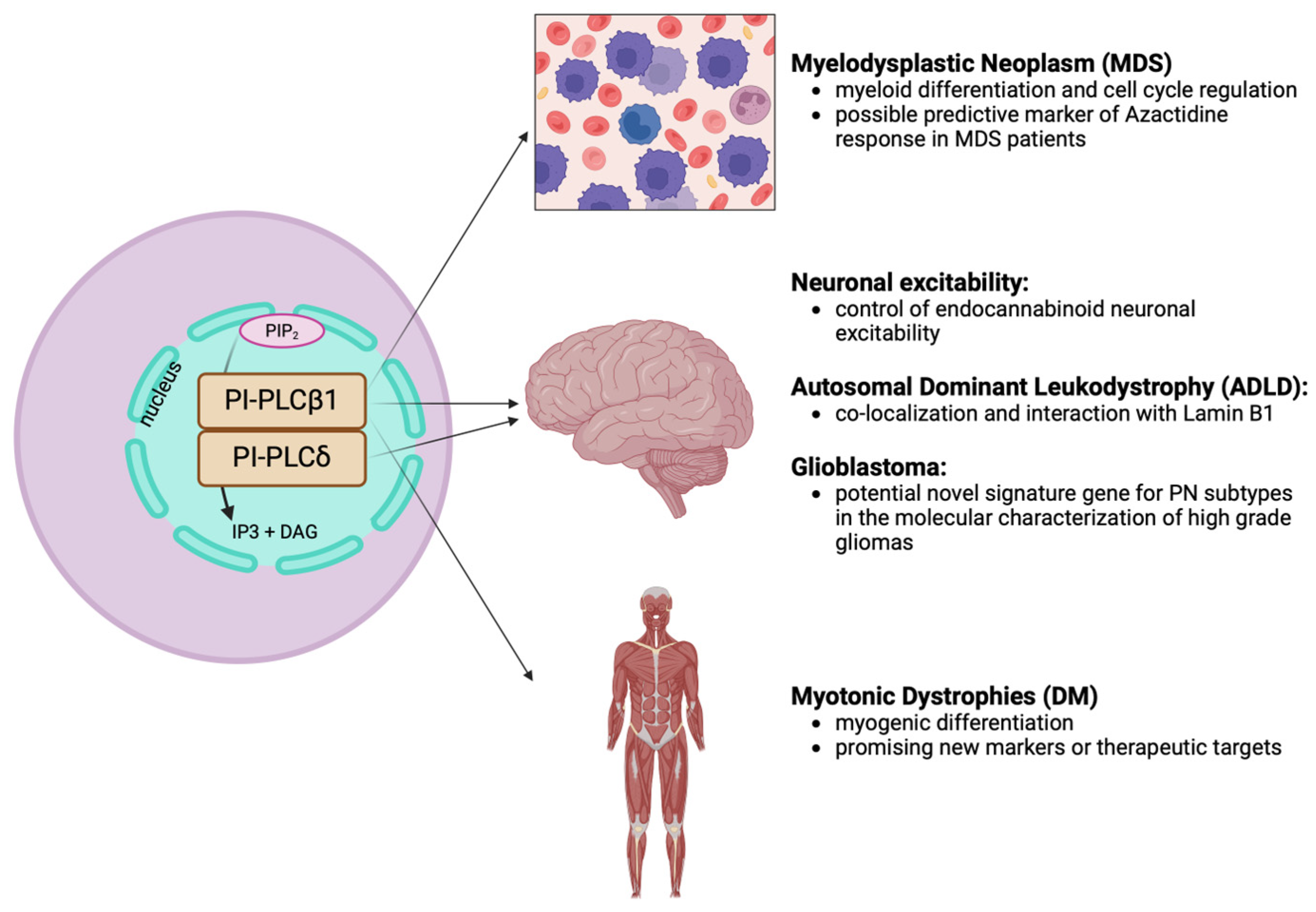
In conclusion, this review has shed light on the significant role of nuclear PIs and nuclear PI-PLCs, particularly in hematological neoplasms, muscular diseases, and neurological disorders, including brain neoplasms. The intricate interplay of these signaling molecules in regulating cellular functions underscores their relevance in disease pathogenesis and their potential role as new markers or therapeutic targets, aiming at mitigating diseases burden and improving clinical outcome. Figure 4 provides a comprehensive overview, highlighting the involvement of nuclear PLCs in different human diseases.
Author Contributions
Conceptualization, L.C. and M.Y.F.; writing—original draft preparation, I.C. and E.C.; writing—review and editing, L.M. and S.Ratti.
Funding
The publication was supported by the International Visegrad Fund through Visegrad Grants within the project “Inclusion of Young Researchers from the V4 Region in Top Research on the Cell Nucleus” (project ID 22230207).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Manzoli, F.A.; Maraldi, N.M.; Cocco, L.; Capitani, S.; Facchini, A. Chromatin phospholipids in normal and chronic lymphocytic leukemia lymphocytes. Cancer Res. 1977, 37, 843–849. [Google Scholar] [PubMed]
- Rose, H.G.; Frenster, J.H. Composition and metabolism of lipids within repressed and active chromatin of interphase lymphocytes. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1965, 106, 577–591. [Google Scholar] [CrossRef]
- Albi, E.; Mersel, M.; Leray, C.; Tomassoni, M.L.; Viola-Magni, M.P. Rat liver chromatin phospholipids. Lipids 1994, 29, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Cocco, L.; Maraldi, N.M.; Manzoli, F.A.; Gilmour, R.S.; Lang, A. Phospholipid interactions in rat liver nuclear matrix. Biochem. Biophys. Res. Commun. 1980, 96, 890–898. [Google Scholar] [CrossRef]
- Michell, R.H.; Kirk, C.K.; Jones, L.M.; Downes, C.P.; Creba, J.A. The stimulation of inositol lipid metabolism that accompanies calcium mobilization in stimulated cells: Defined characteristics and unanswered questions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1981, 296, 123–138. [Google Scholar] [CrossRef]
- Smith, C.D.; Wells, W.W. Phosphorylation of rat liver nuclear envelopes. II. Characterization of in vitro lipid phosphorylation. J. Biol. Chem. 1983, 258, 9368–9373. [Google Scholar] [CrossRef]
- Cocco, L.; Martelli, A.M.; Gilmour, R.; Ognibene, A.; Manzoli, F.A.; Irvine, R.F. Rapid changes in phospholipid metabolism in the nuclei of Swiss 3T3 cells induced by treatment of the cells with insulin-like growth factor I. Biochem. Biophys. Res. Commun. 1988, 154, 1266–1272. [Google Scholar] [CrossRef]
- Divecha, N.; Banfić, H.; Irvine, R. The polyphosphoinositide cycle exists in the nuclei of Swiss 3T3 cells under the control of a receptor (for IGF-I) in the plasma membrane, and stimulation of the cycle increases nuclear diacylglycerol and apparently induces translocation of protein kinase. EMBO J. 1991, 10, 3207–3214. [Google Scholar] [CrossRef]
- Payrastre, B.; Nievers, M.; Boonstra, J.; Breton, M.; Verkleij, A.; Henegouwen, P.V.B.E. A differential location of phosphoinositide kinases, diacylglycerol kinase, and phospholipase C in the nuclear matrix. J. Biol. Chem. 1992, 267, 5078–5084. [Google Scholar] [CrossRef]
- Martelli, A.M.; Gilmour, R.S.; Bertagnolo, V.; Neri, L.M.; Manzoli, L.; Cocco, L. Nuclear localization and signalling activity of phosphoinositidase Cβ in Swiss 3T3 cells. Nature 1992, 358, 242–245. [Google Scholar] [CrossRef]
- Fiume, R.; Faenza, I.; Sheth, B.; Poli, A.; Vidalle, M.; Mazzetti, C.; Abdul, S.; Campagnoli, F.; Fabbrini, M.; Kimber, S.; et al. Nuclear Phosphoinositides: Their Regulation and Roles in Nuclear Functions. Int. J. Mol. Sci. 2019, 20, 2991. [Google Scholar] [CrossRef]
- Dickson, E.J.; Hille, B. Understanding phosphoinositides: Rare, dynamic, and essential membrane phospholipids. Biochem. J. 2019, 476, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.M.; Blind, R.D. Signaling through non-membrane nuclear phosphoinositide binding proteins in human health and disease. J. Lipid Res. 2019, 60, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Boronenkov, I.V.; Loijens, J.C.; Umeda, M.; Anderson, R.A. Phosphoinositide Signaling Pathways in Nuclei Are Associated with Nuclear Speckles Containing Pre-mRNA Processing Factors. Mol. Biol. Cell 1998, 9, 3547–3560. [Google Scholar] [CrossRef] [PubMed]
- Spector, D.L.; Lamond, A.I. Nuclear Speckles. Cold Spring Harb. Perspect. Biol. 2010, 3, a000646. [Google Scholar] [CrossRef] [PubMed]
- Relav, L.; Doghman-Bouguerra, M.; Ruggiero, C.; Muzzi, J.C.D.; Figueiredo, B.C.; Lalli, E. Steroidogenic Factor 1, a Goldilocks Transcription Factor from Adrenocortical Organogenesis to Malignancy. Int. J. Mol. Sci. 2023, 24, 3585. [Google Scholar] [CrossRef] [PubMed]
- Blind, R.D.; Sablin, E.P.; Kuchenbecker, K.M.; Chiu, H.-J.; Deacon, A.M.; Das, D.; Fletterick, R.J.; Ingraham, H.A. The signaling phospholipid PIP 3 creates a new interaction surface on the nuclear receptor SF-1. Proc. Natl. Acad. Sci. USA 2014, 111, 15054–15059. [Google Scholar] [CrossRef]
- Chi, E.S.; Stivison, E.A.; Blind, R.D. SF-1 Induces Nuclear PIP2. Biomolecules 2023, 13, 1509. [Google Scholar] [CrossRef] [PubMed]
- Vidalle, M.C.; Sheth, B.; Fazio, A.; Marvi, M.V.; Leto, S.; Koufi, F.-D.; Neri, I.; Casalin, I.; Ramazzotti, G.; Follo, M.Y.; et al. Nuclear Phosphoinositides as Key Determinants of Nuclear Functions. Biomolecules 2023, 13, 1049. [Google Scholar] [CrossRef]
- Martelli, A.; Gilmour, R.; Neri, L.; Manzoli, L.; Corps, A.; Cocco, L. Mitogen-stimulated events in nuclei of Swiss 3T3 cells. Evidence for a direct link between changes of inositol lipids, protein kinase C requirement and the onset of DNA synthesis. FEBS Lett. 1991, 283, 243–246. [Google Scholar] [CrossRef]
- Poli, A.; Zaurito, A.E.; Abdul-Hamid, S.; Fiume, R.; Faenza, I.; Divecha, N. Phosphatidylinositol 5 Phosphate (PI5P): From Behind the Scenes to the Front (Nuclear) Stage. Int. J. Mol. Sci. 2019, 20, 2080. [Google Scholar] [CrossRef] [PubMed]
- Lauberth, S.M.; Nakayama, T.; Wu, X.; Ferris, A.L.; Tang, Z.; Hughes, S.H.; Roeder, R.G. H3K4me3 Interactions with TAF3 Regulate Preinitiation Complex Assembly and Selective Gene Activation. Cell 2013, 152, 1021–1036. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, M.R.; Goncalves, M.D.; Loughran, R.M.; Possik, E.; Vijayaraghavan, T.; Yang, A.; Pauli, C.; Ravi, A.; Verma, A.; Yang, Z.; et al. Phosphatidylinositol-5-Phosphate 4-Kinases Regulate Cellular Lipid Metabolism By Facilitating Autophagy. Mol. Cell 2018, 70, 531–544.e9. [Google Scholar] [CrossRef] [PubMed]
- Stijf-Bultsma, Y.; Sommer, L.; Tauber, M.; Baalbaki, M.; Giardoglou, P.; Jones, D.R.; Gelato, K.A.; van Pelt, J.; Shah, Z.; Rahnamoun, H.; et al. The Basal Transcription Complex Component TAF3 Transduces Changes in Nuclear Phosphoinositides into Transcriptional Output. Mol. Cell 2015, 58, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.D.; Lee, A.A.; Duewell, B.R.; Groves, J.T. Membrane-mediated dimerization potentiates PIP5K lipid kinase activity. eLife 2022, 11, e73747. [Google Scholar] [CrossRef]
- Choi, S.; Chen, M.; Cryns, V.L.; Anderson, R.A. A nuclear phosphoinositide kinase complex regulates p53. Nat. Cell Biol. 2019, 21, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Sheetz, M.P. When PIP2 Meets p53: Nuclear Phosphoinositide Signaling in the DNA Damage Response. Front. Cell Dev. Biol. 2022, 10, 903994. [Google Scholar] [CrossRef]
- Chen, M.; Choi, S.; Wen, T.; Chen, C.; Thapa, N.; Lee, J.H.; Cryns, V.L.; Anderson, R.A. A p53–phosphoinositide signalosome regulates nuclear AKT activation. Nat. Cell Biol. 2022, 24, 1099–1113. [Google Scholar] [CrossRef]
- Kwon, I.-S.; Lee, K.-H.; Choi, J.W.; Ahn, J.-Y. PI(3,4,5)P3 regulates the interaction between Akt and B23 in the nucleus. BMB Rep. 2010, 43, 127–132. [Google Scholar] [CrossRef]
- Ratti, S.; Ramazzotti, G.; Faenza, I.; Fiume, R.; Mongiorgi, S.; Billi, A.M.; McCubrey, J.A.; Suh, P.-G.; Manzoli, L.; Cocco, L.; et al. Nuclear inositide signaling and cell cycle. Adv. Biol. Regul. 2018, 67, 1–6. [Google Scholar] [CrossRef]
- Yang, Y.R.; Follo, M.Y.; Cocco, L.; Suh, P.-G. The physiological roles of primary phospholipase C. Adv. Biol. Regul. 2013, 53, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Cocco, L.; Follo, M.Y.; Manzoli, L.; Suh, P.-G. Phosphoinositide-specific phospholipase C in health and disease. J. Lipid Res. 2015, 56, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Follo, M.Y.; Faenza, I.; Piazzi, M.; Blalock, W.L.; Manzoli, L.; McCubrey, J.A.; Cocco, L. Nuclear PI-PLCβ1: An appraisal on targets and pathology. Adv. Biol. Regul. 2014, 54, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, D.; Aluigi, M.; Manzoli, L.; Billi, A.M.; Di Giorgio, F.P.; Morleo, M.; Martelli, A.M.; Cocco, L. Molecular characterization of the human PLC β1 gene. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2002, 1584, 46–54. [Google Scholar] [CrossRef]
- Di Raimo, T.; Leopizzi, M.; Mangino, G.; Della Rocca, C.; Businaro, R.; Longo, L.; Vasco, V.R.L. Different expression and subcellular localization of Phosphoinositide-specific Phospholipase C enzymes in differently polarized macrophages. J. Cell Commun. Signal. 2016, 10, 283–293. [Google Scholar] [CrossRef]
- Cocco, L.; Faenza, I.; Follo, M.Y.; Billi, A.M.; Ramazzotti, G.; Papa, V.; Martelli, A.M.; Manzoli, L. Nuclear inositides: PI-PLC signaling in cell growth, differentiation and pathology. Adv. Enzym. Regul. 2009, 49, 2–10. [Google Scholar] [CrossRef]
- Bahk, Y.; Lee, Y.; Lee, T.; Seo, J.; Ryu, S.; Suh, P. Two forms of phospholipase C-beta 1 generated by alternative splicing. J. Biol. Chem. 1994, 269, 8240–8245. [Google Scholar] [CrossRef] [PubMed]
- Gil Kim, C.; Park, D.; Rhee, S.G. The Role of Carboxyl-terminal Basic Amino Acids in Gqα-dependent Activation, Particulate Association, and Nuclear Localization of Phospholipase C-β1. J. Biol. Chem. 1996, 271, 21187–21192. [Google Scholar] [CrossRef]
- Gałgański, Ł.; Urbanek-Trzeciak, M.O.; Krzyzosiak, W.J. Nuclear speckles: Molecular organization, biological function and role in disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef]
- Vidalle, M.C.; Sheth, B.; Fazio, A.; Marvi, M.V.; Leto, S.; Koufi, F.-D.; Neri, I.; Casalin, I.; Ramazzotti, G.; Follo, M.Y.; et al. Nuclear Phosphoinositides as Key Determinants of Nuclear Functions. Biomolecules 2023, 13, 1049. [Google Scholar] [CrossRef]
- Ramazzotti, G.; Faenza, I.; Fiume, R.; Billi, A.M.; Manzoli, L.; Mongiorgi, S.; Ratti, S.; McCubrey, J.A.; Suh, P.-G.; Cocco, L.; et al. PLC-β1 and cell differentiation: An insight into myogenesis and osteogenesis. Adv. Biol. Regul. 2017, 63, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Faenza, I.; Fiume, R.; Piazzi, M.; Colantoni, A.; Cocco, L. Nuclear inositide specific phospholipase C signalling—Interactions and activity. FEBS J. 2013, 280, 6311–6321. [Google Scholar] [CrossRef] [PubMed]
- Manzoli, L.; Martelli, A.M.; Billi, A.M.; Faenza, I.; Fiume, R.; Cocco, L. Nuclear phospholipase C: Involvement in signal transduction. Prog. Lipid Res. 2005, 44, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Fiume, R.; Ramazzotti, G.; Faenza, I.; Piazzi, M.; Bavelloni, A.; Billi, A.M.; Cocco, L. Nuclear PLCs affect insulin secretion by targeting PPARγ in pancreatic β cells. FASEB J. 2011, 26, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Follo, M.Y.; Mongiorgi, S.; Finelli, C.; Clissa, C.; Ramazzotti, G.; Fiume, R.; Faenza, I.; Manzoli, L.; Martelli, A.M.; Cocco, L. Nuclear inositide signaling in myelodysplastic syndromes. J. Cell. Biochem. 2010, 109, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Faenza, I.; Billi, A.M.; Follo, M.Y.; Fiume, R.; Martelli, A.M.; Cocco, L.; Manzoli, L. Nuclear phospholipase C signaling through type 1 IGF receptor and its involvement in cell growth and differentiation. . 2005, 25, 2039–2041. [Google Scholar]
- Ramazzotti, G.; Faenza, I.; Fiume, R.; Matteucci, A.; Piazzi, M.; Follo, M.Y.; Cocco, L. The physiology and pathology of inositide signaling in the nucleus. J. Cell. Physiol. 2010, 226, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Follo, M.Y.; Manzoli, L.; Poli, A.; McCubrey, J.A.; Cocco, L. PLC and PI3K/Akt/mTOR signalling in disease and cancer. Adv. Biol. Regul. 2014, 57, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G. Myelodysplastic syndromes: 2023 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2023, 98, 1307–1325. [Google Scholar] [CrossRef]
- Aguirre, L.E.; Al Ali, N.; Sallman, D.A.; Ball, S.; Jain, A.G.; Chan, O.; Tinsley-Vance, S.M.; Kuykendall, A.; Sweet, K.; Lancet, J.E.; et al. Assessment and validation of the molecular international prognostic scoring system for myelodysplastic syndromes. Leukemia 2023, 37, 1530–1539. [Google Scholar] [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Ossa, J.E.A.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Zeidan, A. Trends in Clinical Investigation for Myelodysplastic Syndromes. Clin. Lymphoma Myeloma Leuk. 2016, 16 (Suppl. S1), S57–S63. [Google Scholar] [CrossRef] [PubMed]
- Casalin, I.; De Stefano, A.; Ceneri, E.; Cappellini, A.; Finelli, C.; Curti, A.; Paolini, S.; Parisi, S.; Zannoni, L.; Boultwood, J.; et al. Deciphering signaling pathways in hematopoietic stem cells: The molecular complexity of Myelodysplastic Syndromes (MDS) and leukemic progression. Adv. Biol. Regul. 2024, 91, 101014. [Google Scholar] [CrossRef] [PubMed]
- Cocco, L.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Russo, D.; Bosi, C.; Quaranta, M.; Malagola, M.; Parisi, S.; Stanzani, M.; et al. An increased expression of PI-PLCβ1 is associated with myeloid differentiation and a longer response to azacitidine in myelodysplastic syndromes. J. Leukoc. Biol. 2015, 98, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, G.M.; Alves, J.; Reckziegel, L.; Lazzaretti, G.; Abujamra, A.L. DNA Methylation Events as Markers for Diagnosis and Management of Acute Myeloid Leukemia and Myelodysplastic Syndrome. Dis. Markers 2017, 2017, 5472893. [Google Scholar] [CrossRef] [PubMed]
- Follo, M.Y.; Marmiroli, S.; Faenza, I.; Fiume, R.; Ramazzotti, G.; Martelli, A.M.; Gobbi, P.; McCubrey, J.A.; Finelli, C.; Manzoli, F.A.; et al. Nuclear phospholipase C β1 signaling, epigenetics and treatments in MDS. Adv. Biol. Regul. 2013, 53, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Vasco, V.R.L.; Calabrese, G.; Manzoli, L.; Palka, G.; Spadano, A.; Morizio, E.; Guanciali-Franchi, P.; Fantasia, D.; Cocco, L. Inositide-specific phospholipase c β1 gene deletion in the progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia 2004, 18, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Follo, M.Y.; Finelli, C.; Clissa, C.; Mongiorgi, S.; Bosi, C.; Martinelli, G.; Baccarani, M.; Manzoli, L.; Martelli, A.M.; Cocco, L. Phosphoinositide-Phospholipase C β1 Mono-Allelic Deletion Is Associated With Myelodysplastic Syndromes Evolution Into Acute Myeloid Leukemia. J. Clin. Oncol. 2009, 27, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Ramazzotti, G.; Billi, A.M.; Manzoli, L.; Mazzetti, C.; Ruggeri, A.; Erneux, C.; Kim, S.; Suh, P.-G.; Cocco, L.; Faenza, I. IPMK and β-catenin mediate PLC-β1-dependent signaling in myogenic differentiation. Oncotarget 2016, 7, 84118–84127. [Google Scholar] [CrossRef]
- Follo, M.Y.; Finelli, C.; Bosi, C.; Martinelli, G.; Mongiorgi, S.; Baccarani, M.; Manzoli, L.; Blalock, W.L.; Martelli, A.M.; Cocco, L. PI-PLCβ-1 and activated Akt levels are linked to azacitidine responsiveness in high-risk myelodysplastic syndromes. Leukemia 2007, 22, 198–200. [Google Scholar] [CrossRef]
- Follo, M.Y.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Bosi, C.; Testoni, N.; Chiarini, F.; Ramazzotti, G.; Baccarani, M.; Martelli, A.M.; et al. Reduction of phosphoinositide-phospholipase C beta1 methylation predicts the responsiveness to azacitidine in high-risk MDS. Proc. Natl. Acad. Sci. USA 2009, 106, 16811–16816. [Google Scholar] [CrossRef]
- Nyåkern, M.; Tazzari, P.L.; Finelli, C.; Bosi, C.; Follo, M.Y.; Grafone, T.; Piccaluga, P.P.; Martinelli, G.; Cocco, L.; Martelli, A.M. Frequent elevation of Akt kinase phosphorylation in blood marrow and peripheral blood mononuclear cells from high-risk myelodysplastic syndrome patients. Leukemia 2005, 20, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Scheffold, J.; Røst, L.M.; Cheon, H.; Bruheim, P. Serum-free cultures of C2C12 cells show different muscle phenotypes which can be estimated by metabolic profiling. Sci. Rep. 2022, 12, 827. [Google Scholar] [CrossRef]
- Faenza, I.; Bavelloni, A.; Fiume, R.; Lattanzi, G.; Maraldi, N.M.; Gilmour, R.S.; Martelli, A.M.; Suh, P.; Billi, A.M.; Cocco, L. Up-regulation of nuclear PLCβ1 in myogenic differentiation. J. Cell. Physiol. 2003, 195, 446–452. [Google Scholar] [CrossRef]
- Luca, G.; Ferretti, R.; Bruschi, M.; Mezzaroma, E.; Caruso, M. Cyclin D3 critically regulates the balance between self-renewal and differentiation in skeletal muscle stem cells. STEM CELLS 2013, 31, 2478–2491. [Google Scholar] [CrossRef] [PubMed]
- Cenciarelli, C.; De Santa, F.; Puri, P.L.; Mattei, E.; Ricci, L.; Bucci, F.; Felsani, A.; Caruso, M. Critical Role Played by Cyclin D3 in the MyoD-Mediated Arrest of Cell Cycle during Myoblast Differentiation. Mol. Cell. Biol. 1999, 19, 5203–5217. [Google Scholar] [CrossRef]
- Faenza, I.; Ramazzotti, G.; Bavelloni, A.; Fiume, R.; Gaboardi, G.C.; Follo, M.Y.; Gilmour, R.S.; Martelli, A.M.; Ravid, K.; Cocco, L. Inositide-Dependent Phospholipase C Signaling Mimics Insulin in Skeletal Muscle Differentiation by Affecting Specific Regions of the Cyclin D3 Promoter. Endocrinology 2007, 148, 1108–1117. [Google Scholar] [CrossRef]
- Ramazzotti, G.; Faenza, I.; Gaboardi, G.C.; Piazzi, M.; Bavelloni, A.; Fiume, R.; Manzoli, L.; Martelli, A.M.; Cocco, L. Catalytic activity of nuclear PLC-β1 is required for its signalling function during C2C12 differentiation. Cell. Signal. 2008, 20, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Ramazzotti, G.; Billi, A.M.; Manzoli, L.; Mazzetti, C.; Ruggeri, A.; Erneux, C.; Kim, S.; Suh, P.-G.; Cocco, L.; Faenza, I. IPMK and β-catenin mediate PLC-β1-dependent signaling in myogenic differentiation. Oncotarget 2016, 7, 84118–84127. [Google Scholar] [CrossRef]
- Ratti, S.; Follo, M.Y.; Ramazzotti, G.; Faenza, I.; Fiume, R.; Suh, P.-G.; McCubrey, J.A.; Manzoli, L.; Cocco, L. Nuclear phospholipase C isoenzyme imbalance leads to pathologies in brain, hematologic, neuromuscular, and fertility disorders. J. Lipid Res. 2019, 60, 312–317. [Google Scholar] [CrossRef]
- Soltanzadeh, P. Myotonic Dystrophies: A Genetic Overview. Genes 2022, 13, 367. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, L. Correction of RNA-Binding Protein CUGBP1 and GSK3β Signaling as Therapeutic Approach for Congenital and Adult Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2019, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Faenza, I.; Blalock, W.; Bavelloni, A.; Schoser, B.; Fiume, R.; Pacella, S.; Piazzi, M.; D’Angelo, A.; Cocco, L. A role for PLCβ1 in myotonic dystrophies type 1 and 2. FASEB J. 2012, 26, 3042–3048. [Google Scholar] [CrossRef] [PubMed]
- Faenza, I.; Blalock, W.; Bavelloni, A.; Schoser, B.; Fiume, R.; Pacella, S.; Piazzi, M.; D’Angelo, A.; Cocco, L. A role for PLCβ1 in myotonic dystrophies type 1 and 2. FASEB J. 2012, 26, 3042–3048. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, E.; Sakai, K.; Schoser, B.; Huichalaf, C.; Schneider-Gold, C.; Nguyen, H.; Wang, G.-L.; Albrecht, J.H.; Timchenko, L.T. Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp. Cell Res. 2008, 314, 2266–2278. [Google Scholar] [CrossRef]
- Marvi, M.V.; Neri, I.; Evangelisti, C.; Ramazzotti, G.; Asioli, S.; Zoli, M.; Mazzatenta, D.; Neri, N.; Morandi, L.; Tonon, C.; et al. Phospholipases in Gliomas: Current Knowledge and Future Perspectives from Bench to Bedside. Biomolecules 2023, 13, 798. [Google Scholar] [CrossRef]
- Ratti, S.; Follo, M.Y.; Ramazzotti, G.; Faenza, I.; Fiume, R.; Suh, P.-G.; McCubrey, J.A.; Manzoli, L.; Cocco, L. Nuclear phospholipase C isoenzyme imbalance leads to pathologies in brain, hematologic, neuromuscular, and fertility disorders. J. Lipid Res. 2019, 60, 312–317. [Google Scholar] [CrossRef]
- Suh, P.-G.; Park, J.-I.; Manzoli, L.; Cocco, L.; Peak, J.C.; Katan, M.; Fukami, K.; Kataoka, T.; Yun, S.-U.; Ryu, S.-H. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 2008, 41, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Bohm, D. Disruption of PLC-$beta;1-Mediated Signal Transduction in Mutant Mice Causes Age-Dependent Hippocampal Mossy Fiber Sprouting and Neurodegeneration. Mol. Cell. Neurosci. 2002, 21, 584–601. [Google Scholar] [CrossRef]
- Fukaya, M.; Uchigashima, M.; Nomura, S.; Hasegawa, Y.; Kikuchi, H.; Watanabe, M. Predominant expression of phospholipase Cβ1 in telencephalic principal neurons and cerebellar interneurons, and its close association with related signaling molecules in somatodendritic neuronal elements. Eur. J. Neurosci. 2008, 28, 1744–1759. [Google Scholar] [CrossRef]
- Stallings, J.D.; Tall, E.G.; Pentyala, S.; Rebecchi, M.J. Nuclear Translocation of Phospholipase C-δ1 Is Linked to the Cell Cycle and Nuclear Phosphatidylinositol 4,5-Bisphosphate. J. Biol. Chem. 2005, 280, 22060–22069. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, X.; Wang, L.; Zhu, B.; Zhang, C.; Jia, W.; Zhu, H.; Liu, X.; Zhong, M.; Xie, D.; et al. PLCD3, a flotillin2-interacting protein, is involved in proliferation, migration and invasion of nasopharyngeal carcinoma cells. Oncol. Rep. 2017, 39, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Rusciano, I.; Marvi, M.V.; Obeng, E.O.; Mongiorgi, S.; Ramazzotti, G.; Follo, M.Y.; Zoli, M.; Morandi, L.; Asioli, S.; Fabbri, V.P.; et al. Location-dependent role of phospholipase C signaling in the brain: Physiology and pathology. Adv. Biol. Regul. 2020, 79, 100771. [Google Scholar] [CrossRef] [PubMed]
- Hannan, A.J.; Blakemore, C.; Katsnelson, A.; Vitalis, T.; Huber, K.M.; Bear, M.; Roder, J.; Kim, D.; Shin, H.-S.; Kind, P.C. PLC-β1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat. Neurosci. 2001, 4, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Vasco, V.R.L. Phosphoinositide pathway and the signal transduction network in neural development. Neurosci. Bull. 2012, 28, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.R.; Kang, D.-S.; Lee, C.; Seok, H.; Follo, M.Y.; Cocco, L.; Suh, P.-G. Primary phospholipase C and brain disorders. Adv. Biol. Regul. 2016, 61, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Kurian, M.A.; Meyer, E.; Vassallo, G.; Morgan, N.V.; Prakash, N.; Pasha, S.; Hai, N.A.; Shuib, S.; Rahman, F.; Wassmer, E.; et al. Phospholipase C beta 1 deficiency is associated with early-onset epileptic encephalopathy. Brain 2010, 133, 2964–2970. [Google Scholar] [CrossRef] [PubMed]
- Fiume, R.; Ramazzotti, G.; Teti, G.; Chiarini, F.; Faenza, I.; Mazzotti, G.; Billi, A.M.; Cocco, L. Involvement of nuclear PLCβl in lamin B1 phosphorylation and G2/M cell cycle progression. FASEB J. 2008, 23, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Ratti, S.; Rusciano, I.; Mongiorgi, S.; Obeng, E.O.; Cappellini, A.; Teti, G.; Falconi, M.; Talozzi, L.; Capellari, S.; Bartoletti-Stella, A.; et al. Cell signaling pathways in autosomal-dominant leukodystrophy (ADLD): The intriguing role of the astrocytes. Cell. Mol. Life Sci. 2020, 78, 2781–2795. [Google Scholar] [CrossRef]
- Ratti, S.; Marvi, M.V.; Mongiorgi, S.; Obeng, E.O.; Rusciano, I.; Ramazzotti, G.; Morandi, L.; Asioli, S.; Zoli, M.; Mazzatenta, D.; et al. Impact of phospholipase C β1 in glioblastoma: A study on the main mechanisms of tumor aggressiveness. Cell. Mol. Life Sci. 2022, 79, 195. [Google Scholar] [CrossRef]
- Ramos, A.R.; Edimo, W.E.; Erneux, C. Phosphoinositide 5-phosphatase activities control cell motility in glioblastoma: Two phosphoinositides PI(4,5)P2 and PI(3,4)P2 are involved. Adv. Biol. Regul. 2018, 67, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Chang, J.T.; Liu, Z.; Chen, Y.; Li, M.; Zhu, J.-J. Phospholipase C Beta 1: A Candidate Signature Gene for Proneural Subtype High-Grade Glioma. Mol. Neurobiol. 2015, 53, 6511–6525. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, Y.P.; Weatherbee, J.L.; Wheelhouse, R.T.; Ross, A.H. Glioblastoma Multiforme Therapy and Mechanisms of Resistance. Pharmaceuticals 2013, 6, 1475–1506. [Google Scholar] [CrossRef] [PubMed]
- Batash, R.; Asna, N.; Schaffer, P.; Francis, N.; Schaffer, M. Glioblastoma Multiforme, Diagnosis and Treatment; Recent Literature Review. Curr. Med. Chem. 2017, 24, 3002–3009. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Role of nuclear PI-PLCβ1 in Myelodysplastic Neoplasm (MDS). (A) Correlation between increased risk of AML progression in MDS patients with the mono-allelic deletion of the PI-PLCB1 gene (del20p). (B) Increased expression of nuclear PI-PLCβ1, induced by Azacytidine sensitivity due to hypermethylation of the PI-PLCB1 promoter, promotes normal myeloid differentiation in MDS cells. MDS: Myelodysplastic Neoplasm; AML: Acute Myeloid Leukemia; PI-PLCB1: phosphoinositide-specific phospholipase C β1. [Created by Biorender.com].
Figure 1.
Role of nuclear PI-PLCβ1 in Myelodysplastic Neoplasm (MDS). (A) Correlation between increased risk of AML progression in MDS patients with the mono-allelic deletion of the PI-PLCB1 gene (del20p). (B) Increased expression of nuclear PI-PLCβ1, induced by Azacytidine sensitivity due to hypermethylation of the PI-PLCB1 promoter, promotes normal myeloid differentiation in MDS cells. MDS: Myelodysplastic Neoplasm; AML: Acute Myeloid Leukemia; PI-PLCB1: phosphoinositide-specific phospholipase C β1. [Created by Biorender.com].
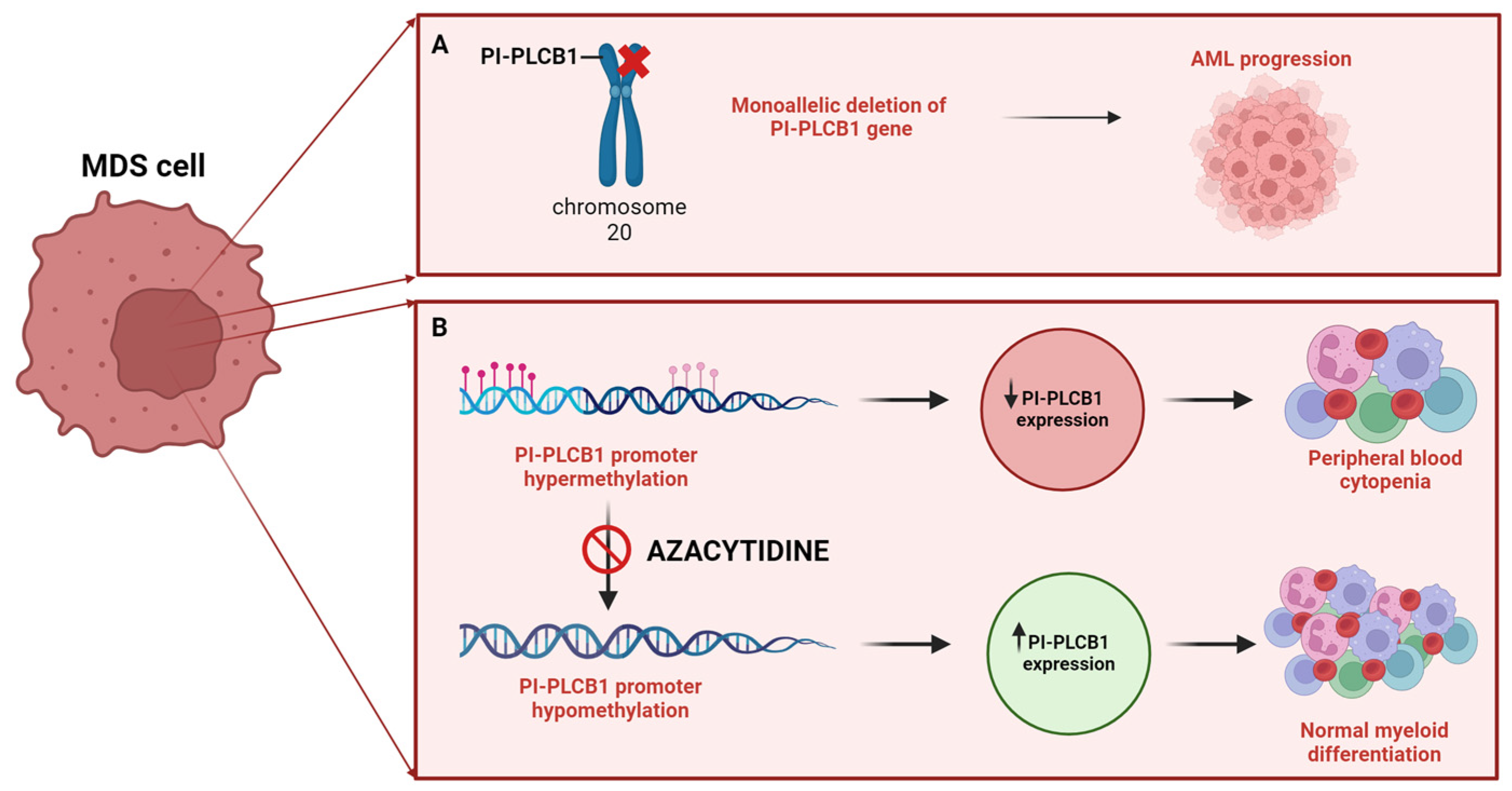
Figure 2.
Role of nuclear PI-PLCβ1 in physiological myogenic differentiation and in Myotonic dystrophies (DM). In physiological myogenic differentiation, PI-PLCβ1 metabolizes PIP2 leading to IP3 formation, that once phosphorylated by IPMK activates the Wnt/β catenin pathway. β catenin activates c-jun which in turn activates the cyclin D3 promoter. Cyclin D3-Cdk4/6 regulates the phosphorylation of CUGBP1 enabling its interaction with eIF2 activating translational activity. The downregulation of PI-PLCβ1 and consequent decrease in cyclin D3 expression leads to decreased CUGBP1-eIF2 interaction culminating in dysregulated myogenic differentiation. PIP2: phosphatidylinositol 4,5-bisphosphate; PI-PLCB1: phosphoinositide-specific phospholipase C β1; DAG: diacylglycerol; IP3: inositol 1,4,5-trisphosphate; IP5: inositol pentakisphosphate; IPMK: inositol polyphosphate multikinase; CUGBP1: CUG-binding protein 1; eIF2: eukaryotic initiation factor 2. [Created by Biorender.com].
Figure 2.
Role of nuclear PI-PLCβ1 in physiological myogenic differentiation and in Myotonic dystrophies (DM). In physiological myogenic differentiation, PI-PLCβ1 metabolizes PIP2 leading to IP3 formation, that once phosphorylated by IPMK activates the Wnt/β catenin pathway. β catenin activates c-jun which in turn activates the cyclin D3 promoter. Cyclin D3-Cdk4/6 regulates the phosphorylation of CUGBP1 enabling its interaction with eIF2 activating translational activity. The downregulation of PI-PLCβ1 and consequent decrease in cyclin D3 expression leads to decreased CUGBP1-eIF2 interaction culminating in dysregulated myogenic differentiation. PIP2: phosphatidylinositol 4,5-bisphosphate; PI-PLCB1: phosphoinositide-specific phospholipase C β1; DAG: diacylglycerol; IP3: inositol 1,4,5-trisphosphate; IP5: inositol pentakisphosphate; IPMK: inositol polyphosphate multikinase; CUGBP1: CUG-binding protein 1; eIF2: eukaryotic initiation factor 2. [Created by Biorender.com].
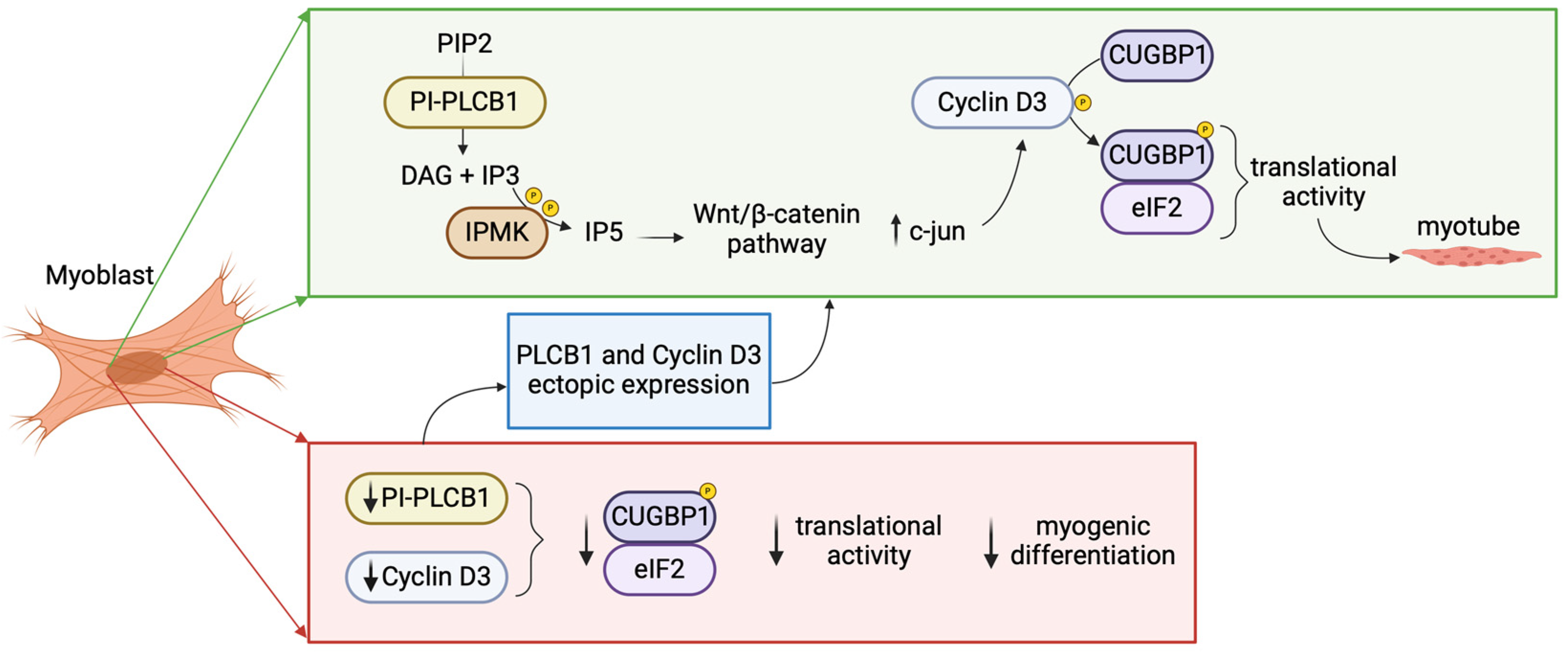
Figure 3.
Role of nuclear PI-PLCβ1 in Glioblastoma (GBM). The downregulation of PLCβ1 dictates various significant physio-pathological alterations, culminating in enhanced cellular migratory, invasive capacities, proliferation, and survival, which lead to a more aggressive phenotype. GBM: Glioblastoma Multiforme; PI-PLCB1: phosphoinositide-specific phospholipase C β1; MMP: matrix metalloproteinase; STAT3: Signal Transducer and Activator of Transcription 3 Signal Transducer and Activator of Transcription 3; ERK: Extracellular Signal-Regulated Kinase [Created by Biorender.com].
Figure 3.
Role of nuclear PI-PLCβ1 in Glioblastoma (GBM). The downregulation of PLCβ1 dictates various significant physio-pathological alterations, culminating in enhanced cellular migratory, invasive capacities, proliferation, and survival, which lead to a more aggressive phenotype. GBM: Glioblastoma Multiforme; PI-PLCB1: phosphoinositide-specific phospholipase C β1; MMP: matrix metalloproteinase; STAT3: Signal Transducer and Activator of Transcription 3 Signal Transducer and Activator of Transcription 3; ERK: Extracellular Signal-Regulated Kinase [Created by Biorender.com].
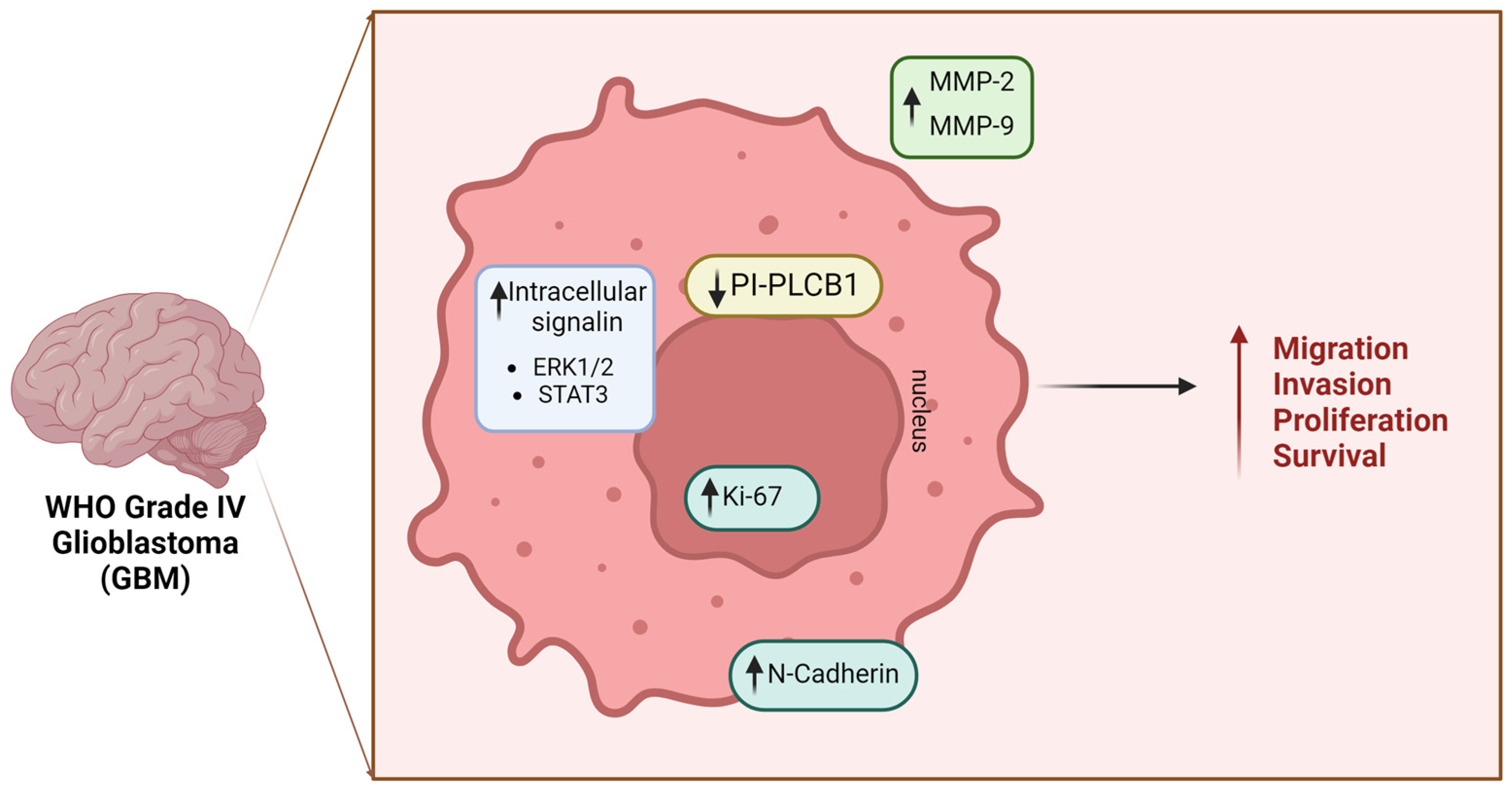
Figure 4.
Schematic representation of the role of nuclear PI-PLCβ1 and PI-PLCδ in different diseases. PI-PLCB1: phosphoinositide-specific phospholipase C β1; PI-PLCδ: phosphoinositide-specific phospholipase C δ; IP3: inositol triphosphate; DAG: diacylglycerol. [Created by Biorender.com].
Figure 4.
Schematic representation of the role of nuclear PI-PLCβ1 and PI-PLCδ in different diseases. PI-PLCB1: phosphoinositide-specific phospholipase C β1; PI-PLCδ: phosphoinositide-specific phospholipase C δ; IP3: inositol triphosphate; DAG: diacylglycerol. [Created by Biorender.com].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



