
Submitted:
28 March 2024
Posted:
28 March 2024
You are already at the latest version
Abstract
Prolylcarboxypeptidase (PRCP, PCP, Lysosomal Pro-X-carboxypeptidase, Angiotensinase C) controls angiotensin - and kinin- induced cell signaling. Elevation of PRCP appear to be activated in chronic inflammatory diseases (cardiovascular disease (CVD), diabetes) in proportion to severity. Vascular endothelial cell senescence and mitochondrial dysfunction have consistently been shown in models of CVD in aging. The cellular senescence, a driver of age-related dysfunction, can differentially alter the expression of lysosomal enzymes due to lysosomal membrane permeability. There is a lack of data demostrating the effect of age-related dysfunction on the expression and function of PRCP. To explore the changes in PRCP, PRCP-dependent prekallikrein (PK) pathway was charcterized in early – and late-passage human pulmonary artery endothelial cells (HPAECs). Detailed kinetic analysis of cells treated with high molecular weight kininogen (HK), a precursor of bradykinin (BK), and PK revealed a mechansim by which senescent HPAECs activate the generation of kallikrein upon the assembly of the HK-PK complex on HPAECs in parallel with an upregulation of PRCP and endothelial nitric oxide (NO) synthase (eNOS) and NO formation. The NO production and expression of both PRCP and eNOS increased in early-passage HPAECs while decreased in late-passage HPAECs. Low activity of PRCP in late passage HPAECs was associated with rapid decreased telomerase reverse transcriptase mRNA levels. We also found that, with an increase in the passage number of HPAECs, reduced PRCP altered respiration rate. These results indicated that aging dysregulates PRCP protein expression, and further studies will shed light into the complexity of PRCP-dependent signling pathway in aging.
Keywords:
cardiovascular dysfunction
; metabolic syndrome
; renin-angiotensin system
; Kallikrein-kinin system
; human telomerase reverse transcriptase
; complex I inhibitor
1. Introduction
As we age, all of our biological processes and function lose their efficiency. Age is not a direct cause of death, but the decreased function of our bodies, brought on by age, is. It has been long recognized that aging is as a major risk factor for numerous diseases including inflammation, metabolic syndrome, and CVD. Leonard Hayflick [1] observed that a culture of human tissue, in vitro, stopped dividing once the culture had reached a fixed threshold age via numerous cellular divisions. It was hypothesized that this phenomenon could be used to study human aging at the cellular level [2].
Endothelial metabolic changes associated with aging have been linked to vascular remodeling in blood vessels [3] and decline in blood flow and increased fluid shear stress, which become the driving force in activating platelets and inducing thrombosis. Aging-induced reduction in microcirculation plasticity and aging associated telomeric DNA damage contribute to the pathogenesis of range of age-related diseases, including brain [4], heart [5], kidney [6], and eye disease. The general consensus is that aging is driven largely by reactive oxygen species (ROS) produced in mitochondria, which can lead to oxidative damage to mitochondrial protein, membrane and DNA. ROS has been documented to damage telomeric DNA effectively [7,8]. ROS also causes lipid peroxidation [9] and promotes a prothrombotic state in vascular system. Hypertension, a limb of the metabolic syndrome, is associated with diabetes and impaired glucose tolerance. Notably, high glucose concentrations are associated with elevated ROS and reduced NO, highlighting the presence of a bidirectional response [10]. These studies brought the conclusions that aging endothelial cells undergo metabolic changes that these cells are simply unable to maintain antioxidant-ROS balance. Moreover, the aging of cells could be directly linked to genomic DNA [11] or environmental factors (such as salt intake and medications) that alter angiotensin II (Ang II, a potent vasoconstrictor) production [12].
It has been suggested that replicative senescent endothelial cell is a feature of type 2 diabetes mellitus (T2DM) and atherosclerosis. Although endothelial dysfunction is recognized as an initial step in atherosclerotic vascular disease, it is advanced in diabetes. PRCP is expressed in endothelial cells. All arteries, veins, and capillaries of the human circulatory system produce PRCP. Plasma PRCP was elevated in diabetic patients [13] while its substrate plasma kallikrein (the G allele of KLKB1 rs3733402) activity correlated with reduced history of CVD [14]. PRCP plays a role in regulating the function of activated cells in restoring and maintaining cellular homeostasis. For instance, PRCP metabolizes Ang II [15] to angiotensin (1-7) (Ang1-7, a vasodilator) and angiotensin III (Ang III, a vasoconstrictor) [16] to angiotensin (2-7) (Ang 2-7) at acidic pH [17]. A recent evidence indicates that PRCP is capable of protecting the heart from Ang II -induced hypertrophic remodeling via controlling myocardial Ang II levels [18]. Bradykinin (BK) protects endothelial cells. PRCP also activates the plasma PK to kallikrein in the presence of HK [19]. Formed kallikrein cleaves HK to liberate BK [20]. Activation of the BKB2 (B2) receptors [21] by BK and the Mas oncogene receptors [22] by Ang1-7 lead, among others, to vasorelaxation and improving cell metabolism via the generation of nitric oxide (NO), which was found to prevent endothelial senescence [23]. Strikingly, it is controversial as to whether Ang 1-7 mediates its effect via the proto-oncogene Mas receptors [24]. Regardless of the binding of the Ang 1-7 to Mas receptors, PRCP is capable of inactivating both Ang II and Ang III, both of which play crucial roles not only in the release of aldosterone from the adrenal glands, but also in the modulation of vascular tone. Thus, PRCP backs the endothelium -dependent relaxation.
PRCP expression has been found to be altered under pathologic conditions of inflammation, hyperlipidemia, diabetes, obesity, and hypertension. The central issue of PRCP involvement in the pathogenesis of experimental hypertension and heart transplant patients was defined as an essential identity for both renal and cardiac Ang1-7 formation [25]. Wu et al. [26] observed susceptibility for hypertension in Han Chinese without history of diabetes mellitus (DM) with the G allele of PRCP SNP rs7104980. Another interesting observation suggests that this SNP of PRCP may be a potential cardiovascular risk factor for percutaneous transluminal coronary angioplasty (PTCA) [14]. A decrease in PRCP level was reported following chronic ethanol regimen in spontaneously hypertensive rats (SHRs) [27]. Furthermore, their findings also indicated that the downregulation of PRCP in addition to enhanced RAS activity may provoke further deterioration of left ventricular (LV) systolic dysfunctions in SHRs. While PRCP gene variant affects the progression of hypertension [28], its depletion results in vascular dysfunction and faster arterial thrombosis in mice [29]. Endothelial dysfunction is induced in hypertensive patients [9]. Additional studies are required to confirm whether PRCP plays an essential role in endothelial cell regulation.
Here, we aimed to examine (1) whether senescent HPAECs are predisposed to enhanced PK activation due to enhanced HK binding, (2) whether senescent HPAECs express lower levels of PRCP mRNA, protein, and activity and produce less NO compared with early passage cells, and (3) whether the inhibition of PRCP is associated with the disruption of mitochondrial bioenergetics in HPAECs.
2. Results
2.1. Effect of Aging on Kallikrein Activity in HPAEC Line
Evidence indicates that the binding of PK to HK bound to HPAECs lead to its activation to kallikrein via PRCP [30]. Here, we examined whether PRCP-dependent PK activation is altered in HPAEC line at various passages. Since there was no defined working passage (subculture) number for HPAEC line, we investigated PK activation from the complex of HK and PK in working passage (P3-P20) and late passage (P21-P40). HPAECs were grown as previously described [31]. Briefly, monolayers of HPAECs (80-90% confluence) were washed 3 times with HEPES-NaHCO3 buffer and then blocked with 1% gelatin for 1 h at 37 °C. After washing, 20 nM HK was added to HPAECs and incubated for additional 1 h at 37 °C. At the end of the incubation, 20 nM PK was added and incubated for 1 h at 37 °C. Kallikrein activity was detected by the absorbance of freed para-nitoaniline (pNA) from chromogenic substrate S2302. Amidolytic activity was measured in 100 μl of assay buffer. Kallikrein activity was observed in all cell passages (Figure 1).
Figure 1.
Plasma kallikrein activity in various HPAE cell passages. Inset, endothelial (HPACs) PRCP activates PK (a zymogen) to kallikrein, which leads to the release of paranitroaniline (pNA) from S2302. All the values are expressed as mean standard error of the mean of at least three independent experiments.
Figure 1.
Plasma kallikrein activity in various HPAE cell passages. Inset, endothelial (HPACs) PRCP activates PK (a zymogen) to kallikrein, which leads to the release of paranitroaniline (pNA) from S2302. All the values are expressed as mean standard error of the mean of at least three independent experiments.
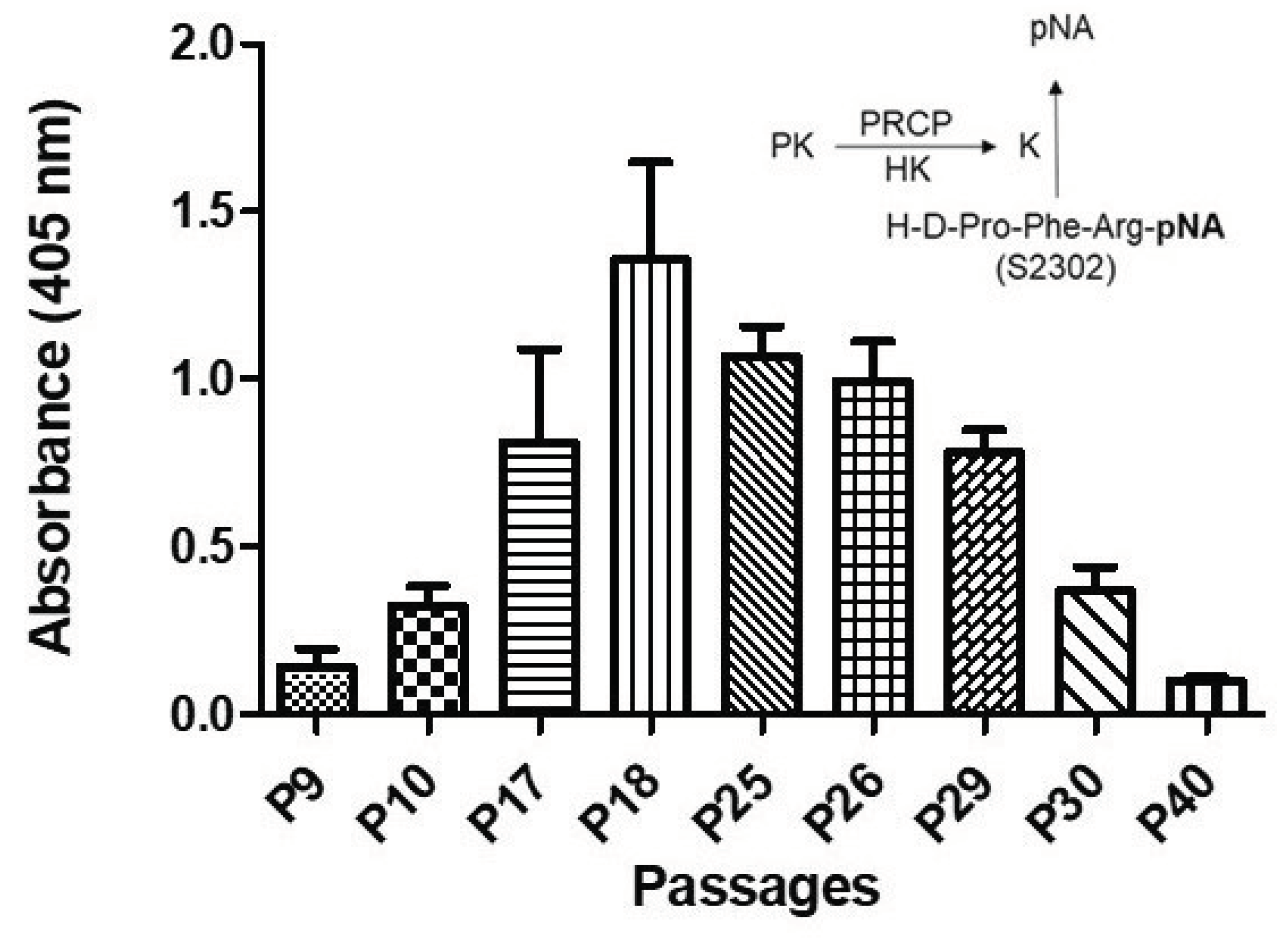
Kallikrein activity progressively increased from P7 to P19, peaking in working passage (P17-P19), and then a gradual decline in enzyme activity was observed in late passage (P21-P40) (Figure 1). Together these data demonstrated that PK activation is cell passage-dependent, and late culture cell passage experiences alteration in response to stimulus such as HK-PK complex.
2.2. Correlation between PRCP Expression and Kallikrein Activity in HPAECs at Various Passages
PRCP-dependent PK activation is evident in endothelial cells [32]. It appears to play a direct role in mediating vasodilation through the bradykinin B2 pathway, and opposing Ang II-mediated vascoconstriction via AT2R, helping maintain vascular endothelial integrity . Since PK activation peaked in working passage (P17-P19), PRCP mRNA and protein expression were measured and compared to those of lower and higher passages as shown in Figure 2.
Figure 2.
PRCP expression is age-dependent. A, Representative example of PRCP protein expression in working passage (P2-P19) assessed by Western blotting. B, Densitometric analysis of Western blotting. C, Densitometric analysis of RT-PCR of PRCP in cultured endothelial cells from HPAECs of both working and late passages of three independent total RNA preparations. Values are expressed as mean ± SEM of 3 experiments. .
Figure 2.
PRCP expression is age-dependent. A, Representative example of PRCP protein expression in working passage (P2-P19) assessed by Western blotting. B, Densitometric analysis of Western blotting. C, Densitometric analysis of RT-PCR of PRCP in cultured endothelial cells from HPAECs of both working and late passages of three independent total RNA preparations. Values are expressed as mean ± SEM of 3 experiments. .
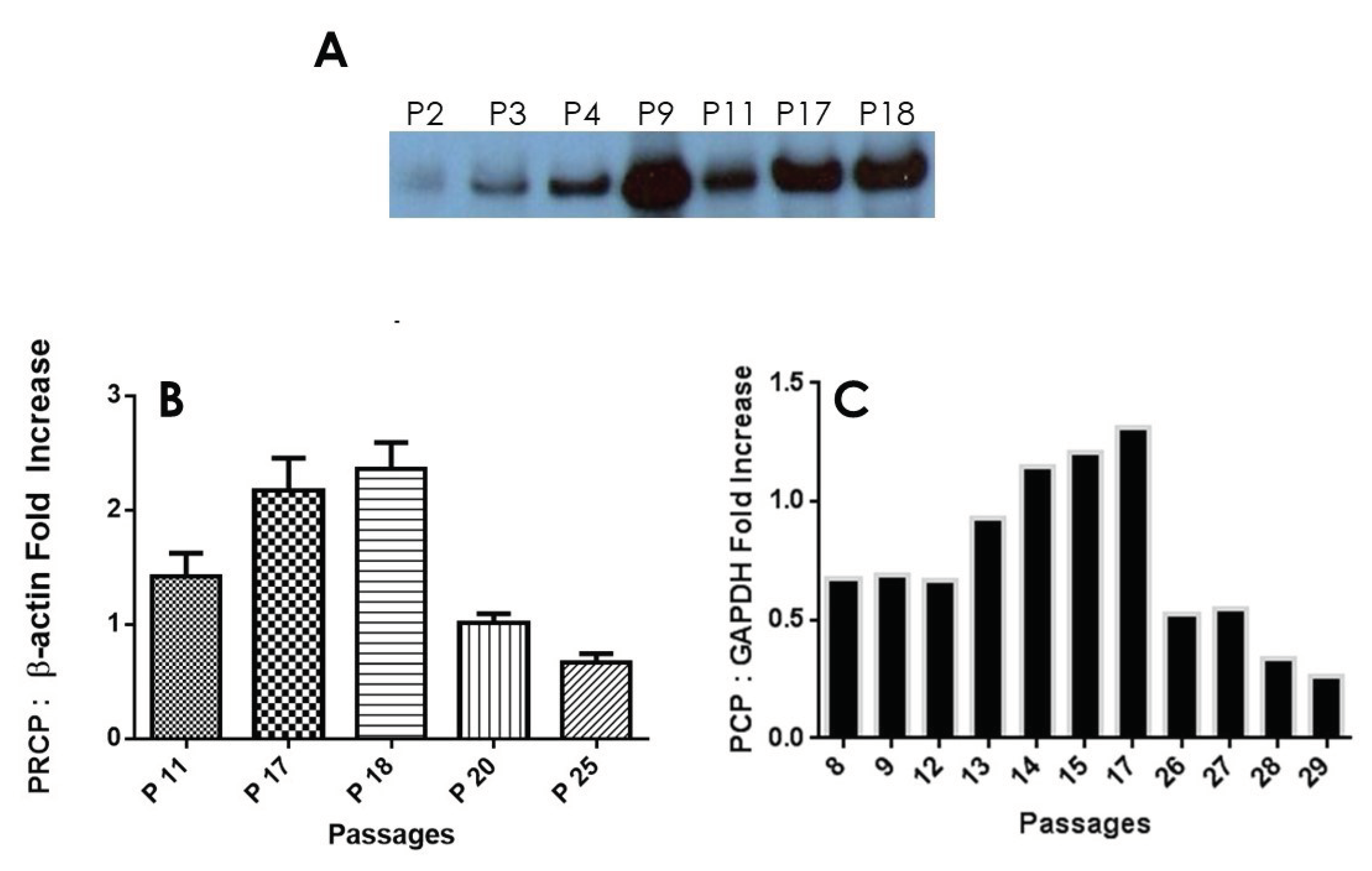
After performing multiple Western Blots to measure protein expression of PRCP, it was found that a trend similar to HK-PK activity can be found. PRCP protein levels increased from Passages 11 and 17 to peak at passages 18 and 19, followed by a significant decrease in passage 25 (Figure 2A). β-actine served as a loading control (data not shown). The unusal band (P9) in this data set that was significantly larger than the rest of the values in the working passage was considered to be an outlier. Densitometic analysis of bands showed a 40% to 65% reduction in PRCP expression in late passage (Figure 2B). Next, we investigated PRCP mRNA expression in resting cells at passages (P8 - P29). We observed an increase of PRCP mRNA from working passages 8 through 17 even though the endothelail cells of passages 8, 9, and 12 displayed unaltered expression of PRCP. However, a gradual decrease in PRCP mRNA expression was observed in late passage (P22 – P29). As a control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used. These changes mirrored PRCP protein levels (Figure 2C). A strong signal for PRCP mRNA was observed in working passages compared to late passages. PRCP production in late passages also was significantly lower than that of the working passages. These data demonstrated a role for PRCP in regulating PK in endothelial cells.
2.3. PRCP Delays Cellular Senescence through an NO-Dependent Mechansim
Prior studies have shown that the activation of PK to kallikrein by PRCP leads to the liberation of bradykinin (BK) [33]. Formed BK then binds to B2 receptors on the endothelial cell surface to release NO and prostaglandin I2 (PGI, prostacyclin) [34]. The assembly of the complex of HK and PK on HPAECs induced NO formation from in working passage (P5 -P19) (Figure 3A). However, there was a decline in NO production from late passage (P20). Understandably, as the cell passage increased, so did the amount of NO products. This supports the idea that the effects of the HK-PK complex on endothelium is becoming an important for understanding the pathophysiology of aging.
Figure 3.
eNOS plays a role in maintaining endothelial integrity during aging. A, NO production by HPAECs. PRCP-dependent PK activation induced NO elaboration from all cell in working passage (P5 – P20). B, Densitometric analysis of RT-PCR of eNOS mRNA in cultured endothelial cells from HPAECs of both working and late passages of three independent total RNA preparations. Values are expressed as mean ± SEM of three independent experiments.
Figure 3.
eNOS plays a role in maintaining endothelial integrity during aging. A, NO production by HPAECs. PRCP-dependent PK activation induced NO elaboration from all cell in working passage (P5 – P20). B, Densitometric analysis of RT-PCR of eNOS mRNA in cultured endothelial cells from HPAECs of both working and late passages of three independent total RNA preparations. Values are expressed as mean ± SEM of three independent experiments.
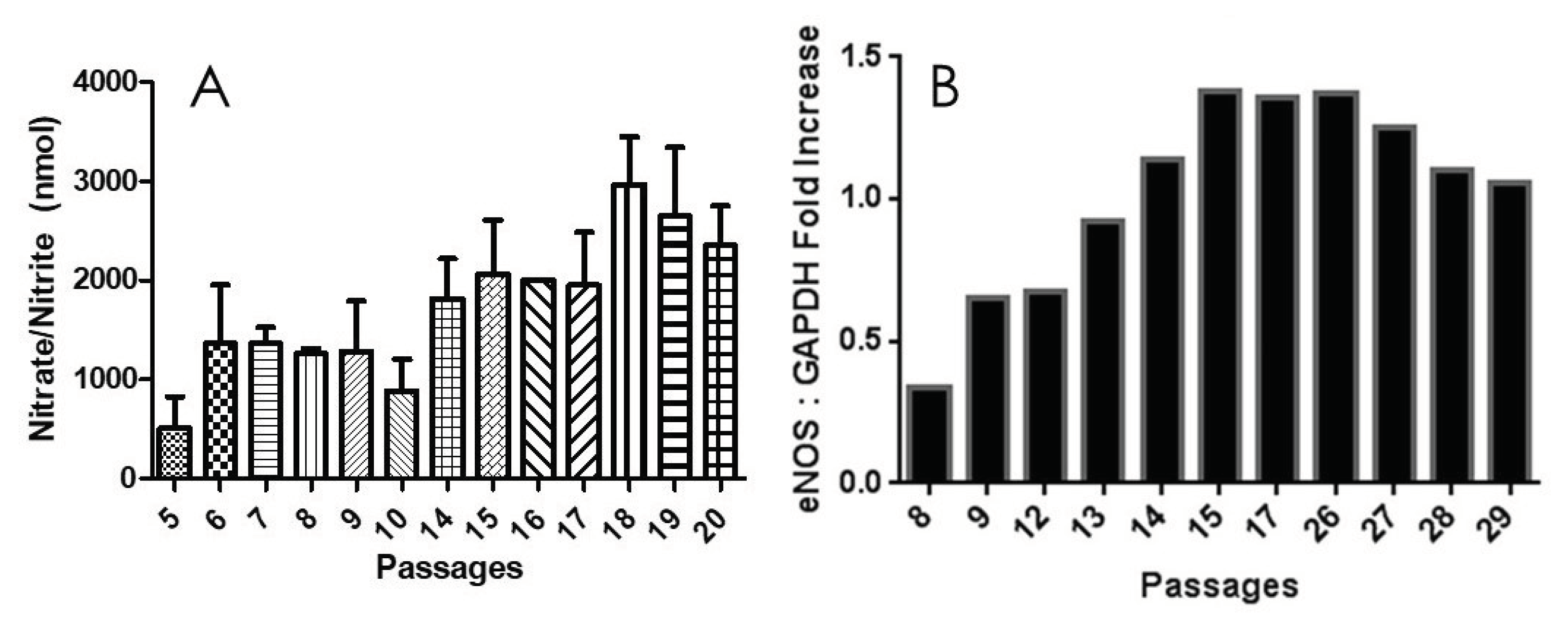
Next, we evaluated changes in the expression levels of endothelial nitric oxid synthase (eNOS). The relative levels of eNOS mRNA significantly changed during cell aging (Figure 3B). GAPDH served as a positive control. After cell passage 8, the expression of eNOS of passage (P9 – P26) was increased markedly upon binding of the HK-PK complex to cells, to a maximum of 3-times than that of the passage 8. After passage 26, eNOS expression levels gratually declined. This slow decrease is due to the rising need for NO to oxidize the increased quantity of reactive oxygen species (ROS) when enough substrate L-arginine and cofactor BH4 are present, or due to increased production of superoxide (O2•-) by eNOS (referred to as eNOS uncoupling) [35]. As NO is rapidly used up, eNOS remains steady for several late, senescent passages. Our results supported previous findings with NO in aging cells [36], showing that there is also a rapid increase in eNOS over time, followed by a slow decrease. These results indicated that the upregulation of eNOS mRNA was consistent with the increase in kallikrein formation (Figure 1) and elevation of PRCP enzyme activity (Figure 2), suggesting that PRCP might play a key role in protecting cell survival via scavenging ROS.
2.4. β-Galactosidase Activity Increases with Age
In order to better visualize the effect of aging on endothelial cell senescence, β-galactosidase (β-gal), a marker protein for senescence, staining was implemented on various HPAEC passages. Using light mircoscopy, the concentration of β-gal increased in HPAECs as cell aged (Figure 4A), confirming a previously published work [37], and secescence with maximimal effects was observed at late passage (P27). Relative cell count was measured and graphed (Figure 4B). β-gal concentration became apparent at passage P16 (at peaked increased transcription of PRCP, 20% dead cells) (Figure 4B) and was significant when cells reached passage 27. Suprisingly, senescent cells were functionally active at working passage (P16-P19), for instance changes in PRCP, eNOS and enhanced NO. The altered cell morphology was becoming increasingly evident following the passage 27 (Data are shown), highlighting a hallmark of aging.
Senescence is a cellular response characterized apparantly by numerous triggers including oxidative stress [38,39,40], telomere damage/shortening [41,42], mitochondrial dysfunction [39], and inflammation [40]. HPAECs well-correlated with a shortening of human telomerase reverse transcriptase (hTERT) length (Figure 4C), which decreased with increasing passages. As shown above, the buildup of PRCP mRNA reaches a high peak (passage 17) and begins to decline by cell passage 18 (FigURE 2C). Interestingly, the decrease in the transcription of telemerase preceded the decrease in mRNA transcription of PRCP. Fibroblast growth factor 2 (FGF-2) was found to be responsible for cell survival and the formation of new blood vessels [43,44]. FGF-2 mimicked PRCP, peaking slightly earlier, but followed the same trend as PRCP, reinforcing the belief that, it too, promotes cell survival and the delay of endothelial cell senescence (Figure 4D). Together these data demonstrated that the delayed loss of PRCP may be important in the delayed cell death, while its expression pattern may serve as an independent indicator for assessing cellular senescence.
2.5. Effect of UM8190-Induced PRCP Inhibition on Mitochondrial Functions
It is known that NO is a reversible inhibitor of mitochondrial respiratory chain. We have hypothesized that this effect is also mediated by the activation of PRCP-dependent pathway. To test this hypothesis, we determined the impact of UM8190 (Figure 5A) , a selective inhibitor of PRCP, on the mitochondrial respiratory function in both intact HPAECs and digitonin-permeabilized HPAECs, based on a previously published guidances [45] with some modifications.
To evaluate the integrity of cellular respiration, the respiratory rates of HPAECs were initially determined in 1 ml respiratory medium (bicarbonate free DMEM/F12 medium) with continuous stirring at 37°C according to a previously described report [46,47] (Figure 5). The rate of O2 consumption linearly decreased (Figure 5B), suggesting that mitochondria are functional in untreated HPAE cell line.
NO inhibits mitochondrial complex I [48], which is discussed in detail elsewhere [49]. Next, studies carried out to determine whether UM8190-induced PRCP inhibition could dose-dependently protect against NO-induced mitochondrial complex I inhibition. UM8190 is an inhibitor of PRCP (apparent Ki = 43 μM) [50]. Contrary to our hypothesis, treatment of HPAECs with UM8190, at pharmacological concentrations (10 – 150 μM), resulted in a reduction of mitochondrial O2 consumption rate in HPAE cell line (Figure 5C). The resulting UN8190 effects on OCR are plotted as a percentage of control (Figure 5D).
Figure 5.
UM8190 treatment decreases oxygen consumption. A, UM8190 structure. B, oxygen consumption of HPAECs as a result of mitochondrial activity was plotted in the absence of UM8190. Representative trace served as control. C, After a basal measurment of ORC, measurement of OCR for HPAECs (5x105 cells/ml) followed by the sequential addition of UM8190 (10-150 μM) with a measurment of OCR as indicated by arrows. OCRs were measured three times and plotted as a function of time. D, The resulting UM8190 effects on OCR are plotted as a percentage of control. Data shown are the mean ± SEM. N =3 of three independent experiments.
Figure 5.
UM8190 treatment decreases oxygen consumption. A, UM8190 structure. B, oxygen consumption of HPAECs as a result of mitochondrial activity was plotted in the absence of UM8190. Representative trace served as control. C, After a basal measurment of ORC, measurement of OCR for HPAECs (5x105 cells/ml) followed by the sequential addition of UM8190 (10-150 μM) with a measurment of OCR as indicated by arrows. OCRs were measured three times and plotted as a function of time. D, The resulting UM8190 effects on OCR are plotted as a percentage of control. Data shown are the mean ± SEM. N =3 of three independent experiments.
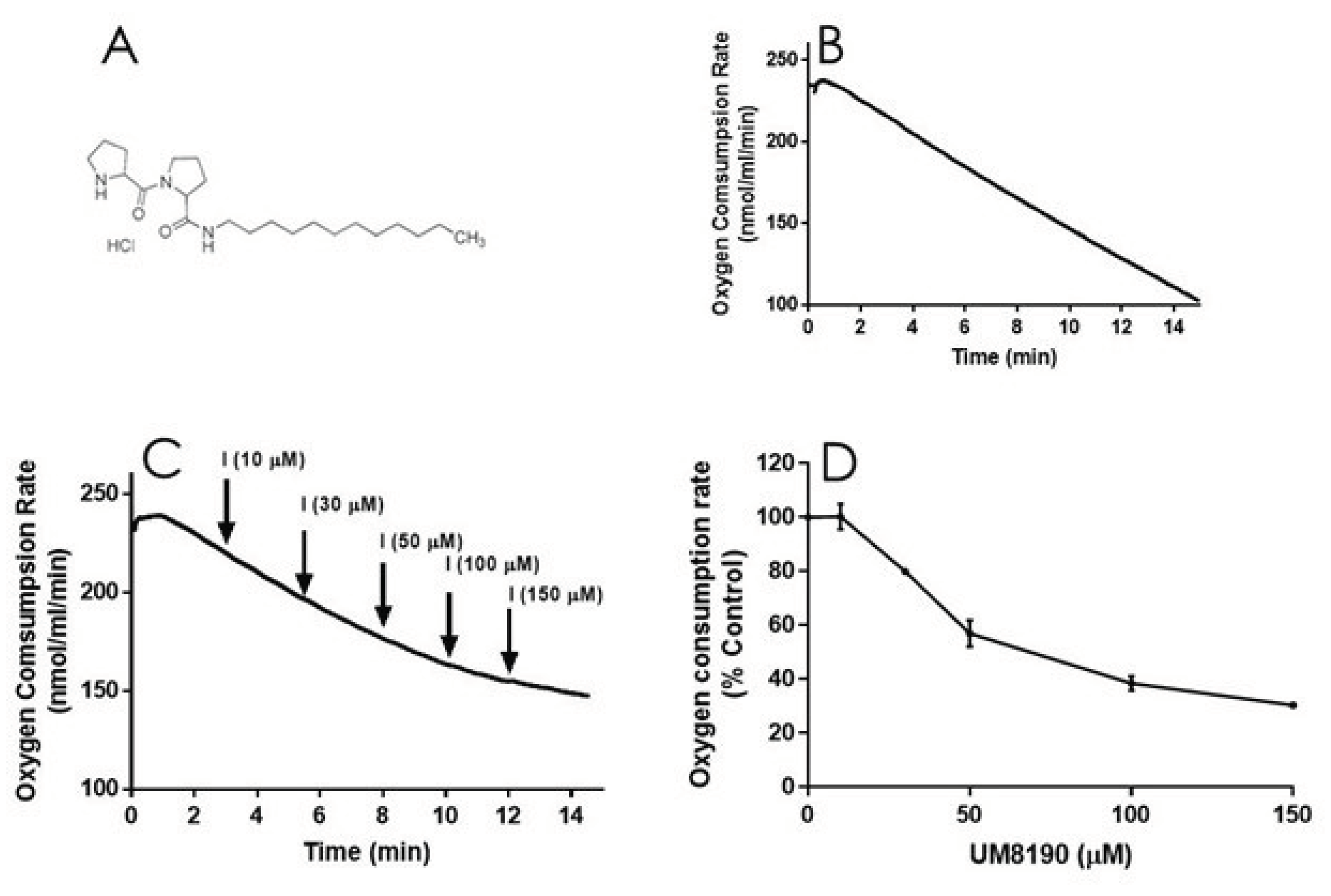
Low concentrations of digitonin has been used to selectively permeabilize the plasma membrane of various cell lines [51] with no apparent effect on the nucleas [52]. UM8190-induced PRCP inhibition blocked mitochondrial function at > 20 μM.
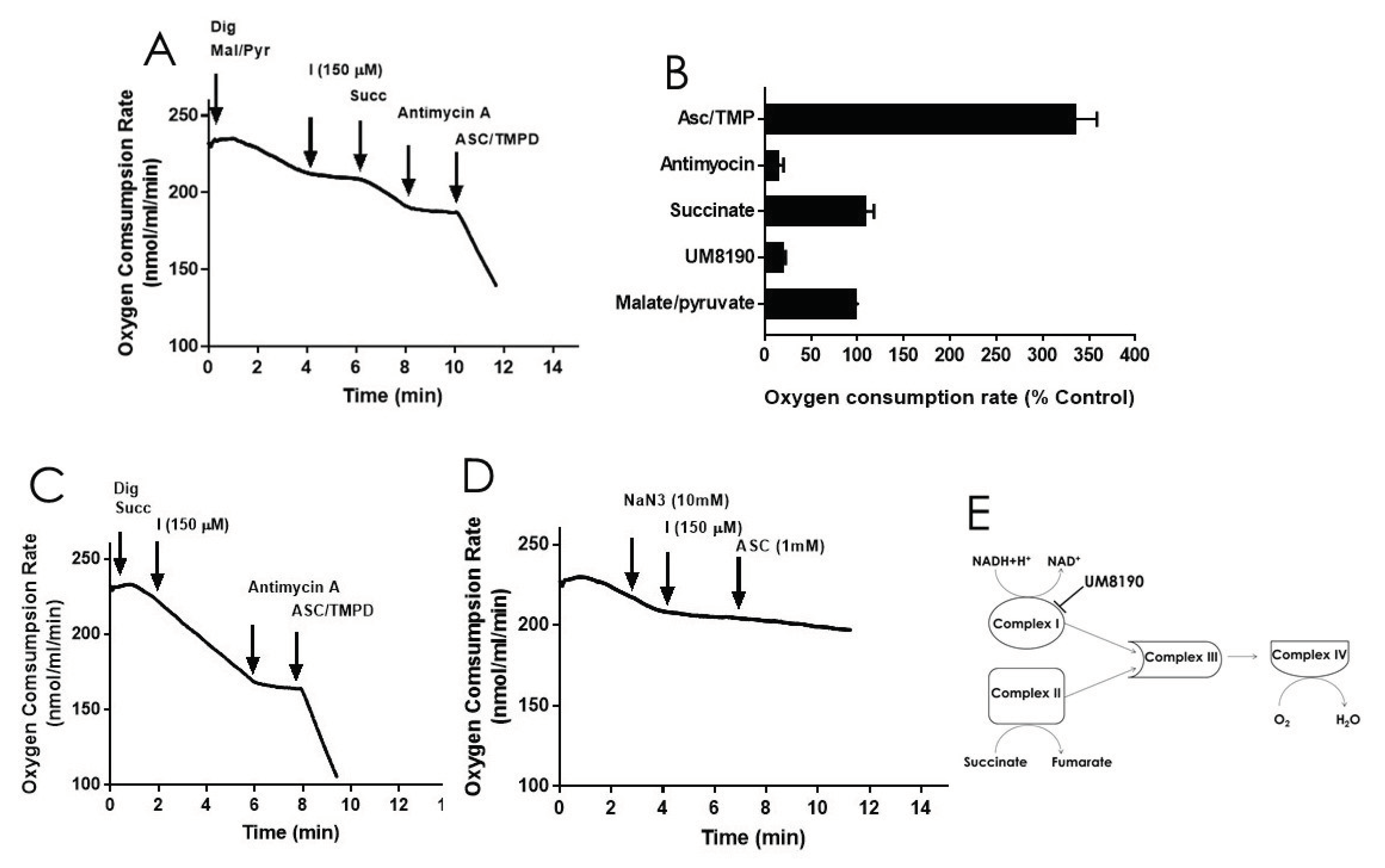
Addition of digitonin (12 μM) did not alter the rate of basal respiration of HPAE cell suspension, suggesting that digitonin permeablization maintains the mitochondrial function. Next, investigations were performed to detemine the effects of UM8190 on various complexes of mitochondrial in the present of digitonin-permeabilized HPAECs. Malate/pyruvate (5mM), a substrate of complex I, stimulated an increase in oxygen consumption. The subsequent addition of UM8190 (150 μM) inhibited NADH oxidation leading to reduced oxygen consumption, (Figure 6A). Succinate is a known substrate of complex II (succinate:quinone oxidoreductase) [53]. Succinate (5mM) addition restored oxygen consumption. However, respiration was inhibited by antimycin A (an inhibitor of complex III, 1 μM). Next, we examined the effect of N,N,N’,N’-tetramethyl-p-phenylenediamine (TMPD, 0.2 mM, an artificial electron donor) on complex IV in the presence of ascorbate (5 mM), which maintains TMPD in a reduced state. Addition of them to the respiratory medium rescued antimycin A effect and the integrity of mitochondria (Figure 6A). The data is summarized by OCR (Figure 6B). Because succinate bypassed UM8190-induced PRCP inhibition effect, electrons entered through QH2 resuming oxidation.
Like complex I, complex II generates ROS [54]. To assess whether UM8190 (150 μM) -induced PRCP inhibition would affect succinate oxidation, digitonin-treated HPAE cells were incubated with succinate (5 mM) (Figure 6C). While UM8190-induced PRCP inhibition (150 μM) was ineffective and antimycin A stopped oxidation, it showes that UM8190 is neither an inhibitor of complex II or complex III of mitochodria. Addition of TMPD and ascorbate rescued the respiration of mitochodria, indicating the mitochondrial integrity is intact. Taken together, UM8190 was ineffective in inhibiting complex II (Figure 6C).
Sodium azide (NaN3) is an inhibitor of cytochrome C oxidase (complex IV). The addition of sodium azide (NaN3, 10 mM) to the non-permeabilized HPAE cell suspension rapidly inhibited mitochodrial respiration rate and addition of UM8190 did not rescue NaN3 effect, which blocks cytochrome C oxidase (Figure 6D). While ROS production sites in mitochondria are complex I, complex II, and complex III, the scheme shown in Figure 6E, summarizes that UM8190 inhibits complex I. These data indicated that UM8190-induced PRCP inhibition may inhibit the accumulation of potentially damaging levels of ROS.
2.6. Effect of UM8190 on Mitochondrial Generation of Reactive Oxygen Species (ROS)
Complex I, as a major ROS production site, plays an important role during the normoxic condition.[16]. UM8190-induced PRCP inhibition blocked the mitochondrial complex I. CM-H2DCFDA, an indicator for ROS in cells, has been used to detect level of ROS in endothelial cells [55]. Briefly, it detects intracellular superoxide radical anion levels in live cells at Ex/Em: 485/530 nm [56]. Investigations were performed to determine the effect of UM8190-induced PRCP inhibition on CM-H2DCFDA in working passage (P10, 10% of growing cells scored as β-Gal positive) HPAECs by mitochondria. To avoid non-specific product, we used the lowest CM-H2DCFDA concentration [57]. Hydrogen peroxide (H2O2) was used as positive control. The cells were treated with different concentrations of UM8190 ( 10 - 150 μM) and H2O2 (10 μM) for 19 hr at 37°C incubator with 5% CO2. At the end of the incubation, the cells were washed 2 times with 1x PBS followed by treatment of CM-H2DCFDA (5 μM) for 1hr at 37°C incubator with 5% CO2. Following the incubation, CM-H2DCFDA solution was removed and the cells were washed with PBS to remove free dye, then 100 μl of HBSS was added to each well and read at 485 nm in Synergy 2 plate reader (Bio-TEK) (Figure 7). Compared to H2O2 treated cells, UM8190-induced PRCP inhibition dose-dependently showed the generation of ROS at two concentrations (10 μM, 30 μM), but protecting the cells at higher concentrations, suggesting that inhibition of PRCP results in an accumulation of ROS at low concentration (Figure 7).
Figure 7.
UM8190 compound inhibits mitochondrial Complex I ROS production. All the values are expressed as mean standard error of the mean of at least three independent experiments. Inset, a schematic diagram showing the proposed mechansim by which UM8190 exerts its effects on the HPAECs including the kallikrein-kinin forming pathway and mitochondrial complex I. .
Figure 7.
UM8190 compound inhibits mitochondrial Complex I ROS production. All the values are expressed as mean standard error of the mean of at least three independent experiments. Inset, a schematic diagram showing the proposed mechansim by which UM8190 exerts its effects on the HPAECs including the kallikrein-kinin forming pathway and mitochondrial complex I. .
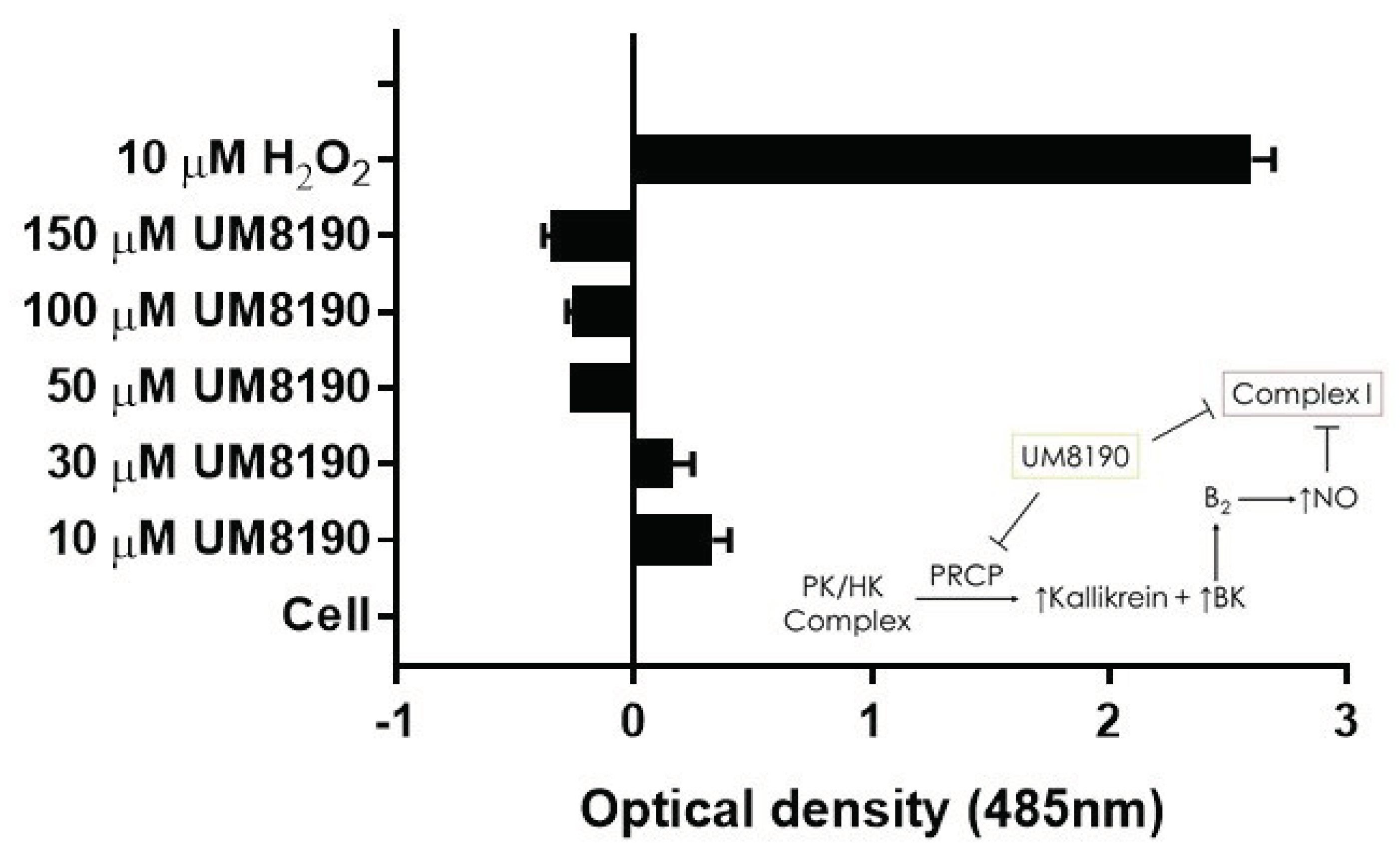
However, this ratio was significantly inhibited by concentrations above 30 μM UM8190. Currently, we are unable to provide a readily available explanation for this finding. One possible explanation for the decrease in ROS formation is that UM8190 directly blocks complex I, which could be potentially clinically useful. The schematic diagram to the right shows activation of B2 receptor by PRCP-dependent PK activation leads to bradykinin mediated NO formation and and inhibtion of complex I. It appears that UM8190 inhibits both PRCP-dependent PK activation and complex I. This new finding will provide a new lead compound and target for further in vitro and in vivo studies in mitochondria.
3. Discussion
PRCP has emerged as a cardioprotective protease with imperative implications for cardiovascular health. PRCP may have crucial physiological functions in the kidney, heart, brain, and in particular, in endothelial cells, which regulate in coordination with vascular smooth muscle cells to help the blood flow to tissues. Recent research studies provided compelling evidence that PRCP protects against the impairment of both heart [18] and kidney [58] and help to dampen elevated blood pressure [17]. PRCP is associated with a significantly increase risk of preeclampsia [28], metabolic condition [13], including IgA nephropathy (IgAN), an autoimmune disease, pathogenesis [59]; however, several of the underlying mechanisms have yet to be fully elucidated.
The aim of the present study was to examine the effects of aging on PRCP in cultured HPAE cells. Due to its ability to regulate various vasoactive peptides that help to control water and electrolytes, cause smooth muscles in the heart and the blood vessels to relax, and delay thrombus formation through the activation of the plasma kallikrein-kinin system (KKS) and regulation of angiotensin molecules. PRCP-dependent pathways appear to represent a remarkable role in vascular physiology and in the development of cardiovascular disease. The major findings of this study are as follows: (1) this study addressed significant gap in PRCP data related to endothelial injury during the aging process, (2) as the cell ages the expression of PRCP and the activity of PK, a PRCP substrate, are increased in the early stages of senescent cells, (3) the possible molecular mechanisms underlying the PRCP-induced vascular protection is found to be via a progressive elevation of the production of NO during the replicative cell senescence, (4) Increased PRCP expression is associated with progressively reduced hTERT expression in age-related HPAEC dysfunction, suggesting PRCP could be an early predictor of endothelial dysfunction, and (5) UM8190 may also act as an inhibitor of complex I of mitochondrial respiratory chain.
Both plasma KKS and the renin-angiotensin system (RAS) serve a central role in the regulation of renal, cardiac, and vascular physiology. The activation of these two intertwined pathways have significant role in numerous common pathology conditions including inflammation, heart failure, renal disease, and diabetes. While both KKS and RAS are tightly regulated system and recognize different receptors that are expressed on endothelial cells, they keep their effect at just the right level by sharing enzymes (angiotensin converting enzyme, PRCP) that perform more than one function. While kinins (BK, des-Arg9-bradykinin), metabolites of both plasma KKS and tissue KKS, can increase vascular permeability and vasodilation, angiotensin II and angiotensin III molecules, metabolites of RAS, can cause vasoconstriction in vivo. The endothelium maintains the delicate balance between these two physiological functions [60] to give instantaneous power to generate the forward-moving pushing blood wave in order to prevent coagulation, fibrinolysis, and inflammation by producing NO and other regulatory factors [61], while accepting nutrients from the blood.
The process of vascular disease is complex and is co-regulated by multiple interwoven signaling pathways leading to endothelial dysfunction or damage, whereas increasing evidence suggests that chronic low-grade inflammation in the pathology of numerous age-related chronic conditions (such as insulin resistance, hypertension, vascular aging) is one of the initiating events [62,63,64]. Endothelial senescence is considered to be a hallmarks of aging [65]. Most importantly, the impairment of endothelium contributes to compromised tissue perfusion and induces functional decline in older individuals [66].
By investigating the effects of HK and PK complex on senescent endothelium cells, we found a great starting point to investigate whether the PK activation in the presence of cell- bound HK may help to delay cell dysfunction. Evidence has revealed a strong connection between PRCP certain facets of autophagy [67], and a causal relationship was found among kallikrein activity, angiotensin II and PRCP [18,68]. This study showed that the PRCP-mediated kallikrein-kinin pathway delayed endothelial cell senescence by promoting cellular survival through generation of NO, a vasodilative molecule. Taken together, this finding may be explained by the idea that PRCP helps to prevent free radical induced cell injury via not only the production of NO, but it also through a previously described process of autophagy [67], a complex and diverse homeostatic phenomenon.
Cellular replicative senescence is a slow, biological process of aging that involves the accumulation of various changes to the internal environment of a cell, most notably the buildup of acidic β-gal. These changes, both structural and molecular in nature, disable metabolism of many cellular processes and eventually lead to the induction of apoptosis. As cells divide, the ends of chromosomes, and telomeres, slowly shorten. Once telomere shortening reaches critical levels, those chromosomes may no longer replicate properly, leading to cellular process complication and apoptosis. Telomerase is a reverse transcriptase that is responsible to counteract telomere shortening and prolong cellular life span. To explore whether PRCP possesses a novel function in pro-survival pathway, we compared its expression against the expression pattern of hTERT. PRCP serves as a focal point in response to a variety of extracellular stimuli including Ang II, Ang III, kinin, and the plasma HK-PK complex. One of the crucial findings of this study demonstrated that the expression and function of PRCP was age-dependent. We also found that in comparison with low hTERT expression, PRCP expression was elevated in HPAECs. Since our study found age-dependent increase in PRCP expression that was proportional to decrease in telomerase expression, monitoring PRCP activity may be predictive of biological age.
Prior studies have shown that the endothelial senescence is triggered by numerous senescence stressors including oxidative stress and mitochondrial dysfunction [36,69]. In a previous work, we showed that overexpression of PRCP enhanced certain markers of mitochondrial autophagy [67], the process by which damaged mitochondria are removed under mitochondrial toxicity conditions [70] or may help to delay cell death. The chronic defects in mitochondrial proton-pumping NADH:ubiquinone oxidoreductase (complex I) [71] or metformin-induced inhibition of complex I [72] with an IC50 value of 20 mM [73] suppressed basal autophagy [71] and prevented ROS generation by complex I.
The kallikrein proteomics analysis of the diabetic macular edema [74] and antibody-array interaction mapping method to detect the activation of the plasma PK [75], and hydrolysis of PK to kallikrein when bound to HK on cells to liberate BK [76], have demonstrated their crucial role involved in disease-related signaling networks. Multiple lines of evidence indicate that damage occurs to the vascular endothelium is an early event in cardiovascular disease, switching from anti-thrombotic properties to prothrombotic state, and may therefore play a key pathogenic role in the disease. Ang II display numerous physiological actions, related mainly to the regulation of electrolytes and fluid osmolarity. While chronic elevation of Ang II induces hypertension that is accompanied by enhanced thrombosis in arterioles through the angiotensin type 2 receptor-dependent pathway [77], treatment with angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists appear to affect the balance between the RAS and KKS axis, regulating not only blood pressure but also thrombosis [78]. In contrast, prekallikrein null mice (Klkb1-/-) exhibited a delayed artery occlusion times on the rose Bengal and ferric chloride thrombosis models [79]. Interestingly, ablation of Klkb1 dampened the progression of atherosclerosis in mice on Apoe-deficient background [80]. We also showed that B2 receptor knockout mice were protected from thrombosis by increased NO and prostacyclin [81]. In brain, a compelling evidence demonstrated that the inhibition of plasma kallikrein reduced matrix metalloproteinase-9 activity [82] following stroke and tissue plasminogen activator (tPA) therapy. Thus, PK activation is tissue specific. Remarkably, BK has been recognized as an inducer of tPA [83,84].
UM8190 inhibits PRCP [50]. It also appears to inhibit complex I of mitochondrial respiratory chain. Evidence indicates that complex I inhibitors are capable of improving glucose homeostasis [85]. Thus, while the clinical safety of UM8190 compound is largely unproven, its effect on complex I offers a promising and exciting strategy to control hyperglycemia and help prevent diabetes complications. Together, these data indicated that PRCP-dependent PK activation may serve as cell signaling molecule for a normal biologic process. Moreover, PRCP expression is elevated when endothelial cells are activated and may provide a novel strategy for the prediction of the risk and severity of vascular disease.
4. Materials and Methods
4.1. Materials
The HPAECs, EGM, FBS growth supplement, HEPES, 0.25% Trypsin-EDTA, and trypsin neutralizing solution were used as supplied by Clonetics (San Diego, CA). The HK and PK were purchased from Enzyme Research Laboratory (South Bend, IN). S2302 was purchased from DiaPharma (Franklin, OH). The primary antibody, Goat Anti-Human PRCP and the secondary Anti-Goat IgG: Whole Molecule, Peroxidase Conjugate were purchased from Bioscience (Long Beach, CA). Mouse Anti-Human β-Actin (Santa Cruz, CA). The “Nitrate/Nitrite Fluorometric Assay Kit” was purchased from Cayman Chemicals. The Cellular Senescence Assay Kit was purchased from Chemicon International (Temecula, CA). TRIzol, Super Signal West Femto Maximum Sensitivity Substrate, RiPA Buffer, Super Signal West Femto Maximum Sensitivity Substrate were supplied by ThermoFisher Scientific (Rockford, IL). the Precision Plus Protein standard (Dual Color) was purchased from Bio-Rad (Rockford, IL). Ethidium bromide was from Sigma-Aldrich (St. Louis, MO).
4.2. Endothelial Cell Culture
Human pulmonary artery endothelial cells (HPAECs) were grown and subcultured over time, from Passage 7 to Passage 40 in endothelial growth medium (EGM) according to manufacture recommendations. Cells were allowed three days to proliferate before each subculture procedure using 0.25% Trypsin-EDTA, DPSB, and the trypsin neutralizing solution (TNS). Right before detaching the cells from the flask to be subcultured, condition medium was removed and aliquoted and saved for further study. The cells were grown in fetal bovine serum (FBS) supplemented with EGM at a constant temperature of 37°C, in an environment with a constant carbon dioxide level of 5%.
4.3. HK/PK Activity
After obtaining a cell pellet from the trypsinized flask, 100 µL of the HPAECs were seeded at 30,000 cells / well of the 96 wells plate and incubated at 37°C incubator with 5% CO2. The cells were washed with HEPES-NaHCO3 buffer (137 mM NaCl; 3 mM KCl; 14.7 mM HEPES; 1 mM MgCl2; 2 mM CaCl2; 5.5 mM glucose and 0.1% gelatin, pH 7.1) and blocked with 1% HEPES-gelatin mixture for an additional 1 hr to reduce any non specific binding. At the end of incubation, the cells was washed twice with HEPES and treated with 20 nM HK for one hour. At the end of the incubation, the cells were washed and treated with 20 nM PK and incubated for additional hour. Finally, the cells were treated with 0.5 mM a HD-Pro-Phe-Arg-paranitroaniline substrate (S2302) and incubated for an hour. All incubations took place at a constant temperature of 37°C incubator with 5% CO2. The activity of formed plasma kallikrein was quantified by detecting the amount of free paranitroanilide in vitro, at 405 nm using the ELx800 Plate Reader (Bio-Tek).
4.4. Western Blot
Adherent cell monolayer were washed with DPSB. The protein cell lysates were extracted using the 1x RIPA Buffer (ThermoFisher Scientific) containg protease inhibitors and separated by electrophoresis in a 10% SDS-PAGE. After electrophoresis, the gel was transferred to a nitrocellulose membrane using electro-blot buffer ( 20% methanol, in 1x Tris/glycine) at 4 °C for one hour. Once transferred, the membrane was blocked with 5% non-fat, dry milk in PBS containing 0.1% Tween-20 (PBST) for an hour at room tempeture. Then, the membrane is treated overnight at 4 °C with 1:20 dilution of the primary antibody (Goat Anti-Human PRCP from Bioscience). The next day, the membrane was washed thrice with PBST and then treated with 1:1000 dilution of secondary antibody (Anti-Goat IgG: Whole Molecule, Peroxidase Conjugate from Bioscience). The membrane was washed thrice again with PBST before being treated with the “Super Signal West Femto Maximum Sensitivity Substrate,” courtesy of Thermo Scientific (Rockford, IL), a chemiluminescent substrate to be used for imaging with the ChemiDoc Imager (Bio-Rad).
Following the first set of imaging, the membrane was washed with 1x PBS and stripped with Thermo Scientific’s “Restore Western Blot Stripping Buffer” at room temperature for 15 minutes before being blocked again and re-probed with 1:100 dilution of the primary antibody (Mouse Anti-Human B-Actin from (Santa Cruz, CA), followed by 1:1000 dilution of secondary antibody (Anti-Goat IgG: Whole Molecule, Peroxidase Conjugate from Bioscience). After blocking, antibody treatments, and washing, the membrane was imaged again and a ratio between the two densities was calculated.
4.5. Nitrite/Nitrate Assay
The condition medium which was collected and stored over the course of growing HPAECs passages was used to measure nitric oxide. The “Nitrate/Nitrite Fluorometric Assay Kit” (Cayman Chemicals) was used to quantify the metabolites of endothelial nitric oxide. Samples of condition medium were adjusted to 80 µL with a 50/50 mix of fresh growth medium and assay Buffer. Enzyme Cofactors and Nitrate Reductase was added to each well and the plate was incubated for an hour at room temperature. Afterwards, DAN (2,3-diaminoaphthalene) was added, followed by sodium hydroxide. The plate was read (Ex 360-365 nm, Em 430 nm). By using the standard curve, the concentration of Nitrate + Nitrite of the sample was calculated.
4.6. Cellular Senescence Assay Kit
The “Chemicon International” Cellular Senescence Assay Kit was used on various passages of growing HPAE cells to qualify their percentage of senescent cells. Senescent-associated β-galactosidase (SA-β-gal) is only present in senescent cells, and Chemicon’s kit provides all reagents needed to detect SA-β-gal activity at pH 6.0 in cell cultures. SA-β-gal catalyzes the hydrolysis of X-gal, causing the accumulation of the blue dye in senescent cells.
4.7. Reverse-Transcriptase Polymerase Chain reaction and Agarose Gel
Cell pellets were resuspended using TRIzol (Life Technologies). The mRNA was extracted from subcultured HPAEC using QIAGEN’s “RNase-Free DNase Set.” RNA was put through reverse-transcription via Invitrogen’s “SuperScript III One-Step RT-PCR (with Platinum Taq) Kit.” We bought multiple human DNA primers from Invitrogen, including hTERT (SENSE – 5’ ATG GGG ACA TGG AGA ACA AG 3’ and ANTISENSE – 5’ GTG AAC CTG CGG AAG ACG GT 3’), B-Actin (SENSE – 5’ TGA ATG GAC AGC CAT CAT GGA C 3’ and ANTISENSE – 5’ TCT CAA GTC AGT GTA CAG GAA AGC 3’), FGF-2 (SENSE – 5’ TCA GCT CTT AGC AGA CAT TGG AAG AAA AAG 3’ and ANTISENSE – 5’ GGA GTG TGT GCT AAC CGT TAC CTG GCT ATG 3’), PRCP (SENSE – 5’ GTG GCT GAG GAA CTG AAA GC 3’ and ANTISENSE – 5’ TGT CAC CAA AGG GGA GAG AC 3’) , and eNOS (SENSE – 5’ ATG TTT GTC TGC GGC GAT GTT AC 3’ and ANTISENSE – 5’ ATG CGG CTT GTC ACT TCC TG 3’). The PCR products were visualized using 1-2% agarose gel containg ethidium bromide (agarose powder and ethidium bromide were purchased from Invitrogen and Sigma respectively) depending on the size of the expected PCR product.
4.8. Oxygen Consumption Rate Measurements on HPAE Cells
Mitochondrial respiration is measured using Oxytherm Clarke-type electrode System (Hansatech, Germany, distributed by PP System, MA) to monitor oxygen concentration in non-permeabilized or digitonin permebilized human pulmonary artery endothelial (HPAE) cells.
The confluent HPAE cells were dissociated using trypsin-EDTA solution. Trypsinized cells were centrifuged at 180 x g, for 5 mins at RT and the HPAE cell pellet was washed twice with DPBS and re-centrifuged. The final HPAE cell pellet was re-suspended in 1x PBS to a final cell density of 50 x 104 cells/ mL. 100 μl of cell suspention was used in 900 μl of Sodium Carbonate free DMEM/F/12 medium (Sigma, MO) for each non-permebilize assay ( 50 x 103 cells per run).
HPAECs mitochondrial respiration rate was measured in 1.0 mL of total respiration solution (Sodium Carbonate free DMEM/F/12 medium) at 37°C under continuous stirring. 100 μl of HPAE cells (50x104 cells/mL) was added to euilibrated media in Oxytherm incubation chamber with constant stirring. The mitochodrial respiration was performed in the absence or presence of UM8190.
To check the mechanistic mitochodrial function for each complex, respiration rate was measured either through complex I or complex II in mitochondria buffer (20 mm HEPES, 120 mm KCl, 2 mm KH2PO4, 2 mm MgCl2, 1 mm EGTA, pH = 7.3). We examine complex I function by using digitonin (12 μm) permeabilize HPAE cells in mitochondria buffer and start the respiration through complex I by adding malate (5 mM) and pyruvate (5 mM). The respiration rate of malate/pyruvate was inhibited with 1 μM rotinone ( a complex I inhibitor) and then the respiration was restored by adding succinate (5 mM) to stimulate mitochodrial respiration through complex II and generate ATP via OXPHOS (succinate dehydrogenase). The mitochondrial respiration, after addition of succinate was inhibited by antimycin A (an inhibitor of coenzyme Q–cytochrome C reductase and complex III or cytochrome bc1). Finally, the integrity of mitochondria was checked by adding ascorbate (5 mM) and N,N,N,N-tetramethyl-p-phenylenediamine (TMPD; 0.2 mM; Sigma, MO) to rescue respiration by stimulation of cytochrome C oxidase (complex IV). Respiration rates were measured for at least 15 min in each step or a steady state was reached. Baseline was recorded for normalization, then the test compounds were added alone or in combination. To examine the mitochodrial respiration rate through complex II, we used digitonin (12 μm) permeabilize HPAE cells in mitochondria buffer and add succinate (5mM) to start respiration through complex II. The respiration was inhibited by antimycin A (an inhibitor of coenzyme Q–cytochrome C reductase and complex III or cytochrome bc1). Finally, the integrity of mitochondria was checked by adding ascorbate (5 mM) and N,N,N,N-tetramethyl-p-phenylenediamine (TMPD; 0.2 mM) to rescue respiration by stimulation of cytochrome C oxidase (complex IV).
5. Conclusions
In summary, the evidence presented herein indicates an actively protective role for PRCP-dependent pathway against oxidative stress in aging endothelial senescence. This study provided a link between dysfunctional (excessively short telomeres) telomeres and the generation of PRCP. Making PRCP as a highly promising diagnostic marker for visualization of CVD.
The role of senescent cells has been confirmed in numerous age-related diseases such as insulin resistance [86] and macular degeneration [87]. The dysregulated inflammation induces ROS production [88], which is implicated in aging. Our data may provide a novel strategy for the prediction of vascular disease. Mitochondria play numerous roles in cancer cell metabolism [89]. Our studies gave new insights into the mechanism of action of UM8190 that may inhibit complex I. The findings of this study facilitate the future studies attempting to characterize the functional consequences of UM8190 on mitochondria. For instance, UM8190 along with other UM8190 analogs may be used to target oxidative phosphorylation and mitochondria-related metabolism to prevent metastasis and progression of cancers. Testing these drug-like UM8190 may shed light on the clinical application of UM8190 as a therapeutic agent.
Author Contributions
Conceptualization, Z.S. and N.B.; methodology, N.B. and F.M.; validation, N.B., F.M., and Z.S.; formal analysis, N.B. and Z.S.; investigation, N.B.; resources, Z.S.; writing—original draft preparation, Z.S.; writing—review and editing, N.B., J.P., Z.S., and F.M.; visualization, N.B.; supervision, Z.S.; project administration, Z.S.; funding acquisition, Z.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions in the study are included in the article, further inquiries can be directed to the corresponding author.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| Ang III | Angiotensin III |
| Ang 2-7 | Angiotensin 2-7 |
| ATP | Adenosine triphosphate |
| β-Gal | β-galactosidase |
| BKB2 | Bradykinin B2 receptor |
| BK | Bradykinin |
| CVD | Cardiovascular diseases |
| EGFR | Epidermal Growth Factor Receptor |
| eNOS | Endothelial nitric oxide synthase |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| H2O2 | Hydrogen Peroxide |
| HK | High molecular weight |
| HPAEC | Human pulmonary artery endothelial cells |
| hTERT | Human telomerase reverse transcriptase |
| IgAN | IgA nephropathy |
| KKS | Kallikrein-kinin system |
| mDNA | Mitochondrial DNA |
| Mal/Pyr | Malate/ pyruvate |
| NAD | Nicotinamide Adenine Dinucleotide |
| NADH | Nicotinamide Adenine Dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NaN3 | Sodium azide |
| NO | Nitric oxide |
| PAR | Protease-activated receptor |
| PK | Prekallikrein |
| PRCP | Prolylcarboxypeptidase |
| PTCA | percutaneous transluminal coronary angioplasty |
| RAS | Renin-angiotensin system |
| ROS | Reactive oxygen species |
| SHRs | Spontaneously hypertensive rats |
| T2DM | Type 2 diabetes mellitus |
| TMPD | N,N,N’,N’-tetramethyl-p-phenylenediamine |
References
- Hayflick, L., The Cell Biology of Aging. Journal of Investigative Dermatology 1979, 73, (1), 8-14. [CrossRef]
- Shay, J. W.; Wright, W. E., Hayflick, his limit, and cellular ageing. Nature reviews. Molecular cell biology 2000, 1, (1), 72-6. [CrossRef]
- Ungvari, Z.; Tarantini, S.; Donato, A. J.; Galvan, V.; Csiszar, A., Mechanisms of Vascular Aging. Circulation Research 2018, 123, (7), 849-867. [CrossRef]
- Saretzki, G.; Wan, T. Telomerase in Brain: The New Kid on the Block and Its Role in Neurodegenerative Diseases. Biomedicines 2021, 9, 490. [CrossRef]
- Brandt, M.; Dörschmann, H.; Khraisat, S.; Knopp, T.; Ringen, J.; Kalinovic, S.; Garlapati, V.; Siemer, S.; Molitor, M.; Göbel, S.; et al. Telomere Shortening in Hypertensive Heart Disease Depends on Oxidative DNA Damage and Predicts Impaired Recovery of Cardiac Function in Heart Failure. Hypertension 2022, 79, 2173–2184. [CrossRef]
- Levstek, T.; Podkrajšek, K.T. Telomere Attrition in Chronic Kidney Diseases. Antioxidants 2023, 12, 579. [CrossRef]
- Ahmed, W.; Lingner, J. PRDX1 Counteracts Catastrophic Telomeric Cleavage Events That Are Triggered by DNA Repair Activities Post Oxidative Damage. Cell Rep. 2020, 33, 108347. [CrossRef]
- Hayashi, T.; Kotani, H.; Yamaguchi, T.; Taguchi, K.; Iida, M.; Ina, K.; Maeda, M.; Kuzuya, M.; Hattori, Y.; Ignarro, L.J. Endothelial cellular senescence is inhibited by liver X receptor activation with an additional mechanism for its atheroprotection in diabetes. Proc. Natl. Acad. Sci. 2014, 111, 1168–1173. [CrossRef]
- Ferroni, P.; Basili, S.; Paoletti, V.; Davì, G. Endothelial dysfunction and oxidative stress in arterial hypertension. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 222–233. [CrossRef]
- Arabshomali, A.; Bazzazzadehgan, S.; Mahdi, F.; Shariat-Madar, Z. Potential Benefits of Antioxidant Phytochemicals in Type 2 Diabetes. Molecules 2023, 28, 7209. [CrossRef]
- Aubert, G.; Lansdorp, P. M., Telomeres and Aging. Physiological reviews 2008, 88, (2), 557-579.
- Brown, N.J.; E Vaughan, D. Prothrombotic effects of angiotensin.. 2000, 45, 419–29.
- Xu, S.; Lind, L.; Zhao, L.; Lindahl, B.; Venge, P. Plasma Prolylcarboxypeptidase (Angiotensinase C) Is Increased in Obesity and Diabetes Mellitus and Related to Cardiovascular Dysfunction. Clin. Chem. 2012, 58, 1110–1115. [CrossRef]
- Gittleman, H.R.; Merkulova, A.; Alhalabi, O.; Stavrou, E.X.; Veigl, M.L.; Barnholtz-Sloan, J.S.; Schmaier, A.H. A Cross-sectional Study of KLKB1 and PRCP Polymorphisms in Patient Samples with Cardiovascular Disease. Front. Med. 2016, 3, 17–17. [CrossRef]
- Skidgel, R.A.; Erdös, E.G. Cellular carboxypeptidases. Immunol. Rev. 1998, 161, 129–141. [CrossRef]
- Mallela, J.; Perkins, R.; Yang, J.; Pedigo, S.; Rimoldi, J.; Shariat-Madar, Z. The functional importance of the N-terminal region of human prolylcarboxypeptidase. Biochem. Biophys. Res. Commun. 2008, 374, 635–640. [CrossRef]
- Maier, C.; Schadock, I.; Haber, P.K.; Wysocki, J.; Ye, M.; Kanwar, Y.; Flask, C.A.; Yu, X.; Hoit, B.D.; Adams, G.N.; et al. Prolylcarboxypeptidase deficiency is associated with increased blood pressure, glomerular lesions, and cardiac dysfunction independent of altered circulating and cardiac angiotensin II. J. Mol. Med. 2017, 95, 473–486. [CrossRef]
- Nguyen, B.Y.; Zhou, F.; Binder, P.; Liu, W.; Hille, S.S.; Luo, X.; Zi, M.; Zhang, H.; Adamson, A.; Ahmed, F.Z.; et al. Prolylcarboxypeptidase Alleviates Hypertensive Cardiac Remodeling by Regulating Myocardial Tissue Angiotensin II. J. Am. Hear. Assoc. 2023, 12, e028298. [CrossRef]
- Merkulova, A.A.; Abdalian, S.; Silbak, S.; Pinheiro, A.; Schmaier, A.H. C1 inhibitor and prolylcarboxypeptidase modulate prekallikrein activation on endothelial cells. J. Allergy Clin. Immunol. 2023, 152, 961–971.e7. [CrossRef]
- Wang, J.; Matafonov, A.; Madkhali, H.; Mahdi, F.; Watson, D.; Schmaier, A.; Gailani, D.; Shariat-Madar, Z. Prolylcarboxypeptidase Independently Activates Plasma Prekallikrein (Fletcher Factor). Curr. Mol. Med. 2014, 14, 1173–1185. [CrossRef]
- Zhang, M.; Lin, D.; Luo, C.; Wei, P.; Cui, K.; Chen, Z. Tissue Kallikrein Protects Rat Prostate against the Inflammatory Damage in a Chronic Autoimmune Prostatitis Model via Restoring Endothelial Function in a Bradykinin Receptor B2-Dependent Way. Oxidative Med. Cell. Longev. 2022, 2022, 1–16. [CrossRef]
- Molaei, A.; Molaei, E.; Hayes, A.W.; Karimi, G. Mas receptor: a potential strategy in the management of ischemic cardiovascular diseases. Cell Cycle 2023, 22, 1654–1674. [CrossRef]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.-F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [CrossRef]
- Gaidarov, I.; Adams, J.; Frazer, J.; Anthony, T.; Chen, X.; Gatlin, J.; Semple, G.; Unett, D.J. Angiotensin (1–7) does not interact directly with MAS1, but can potently antagonize signaling from the AT1 receptor. Cell. Signal. 2018, 50, 9–24. [CrossRef]
- Kovarik, J.J.; Kopecky, C.; Antlanger, M.; Domenig, O.; Kaltenecker, C.C.; Werzowa, J.; Hecking, M.; Mahr, S.; Grömmer, M.; Wallner, C.; et al. Effects of angiotensin-converting-enzyme inhibitor therapy on the regulation of the plasma and cardiac tissue renin-angiotensin system in heart transplant patients. J. Hear. Lung Transplant. 2017, 36, 355–365. [CrossRef]
- Wu, Y.; Yang, H.; Yang, B.; Yang, K.; Xiao, C. Association of polymorphisms in prolylcarboxypeptidase and chymase genes with essential hypertension in the Chinese Han population. J. Renin-Angiotensin-Aldosterone Syst. 2012, 14, 263–270. [CrossRef]
- Liu, J.; Hakucho, A.; Fujimiya, T. Angiotensinase C mRNA and Protein Downregulations Are Involved in Ethanol-Deteriorated Left Ventricular Systolic Dysfunction in Spontaneously Hypertensive Rats. BioMed Res. Int. 2015, 2015, 1–10. [CrossRef]
- Wang, L.; Feng, Y.; Zhang, Y.; Zhou, H.; Jiang, S.; Niu, T.; Wei, L.-J.; Xu, X.; Xu, X.; Wang, X. Prolylcarboxypeptidase gene, chronic hypertension, and risk of preeclampsia. Am. J. Obstet. Gynecol. 2006, 195, 162–171. [CrossRef]
- Adams, G.N.; Stavrou, E.X.; Fang, C.; Merkulova, A.; Alaiti, M.A.; Nakajima, K.; Morooka, T.; Merkulov, S.; LaRusch, G.A.; I Simon, D.; et al. Prolylcarboxypeptidase promotes angiogenesis and vascular repair. Blood 2013, 122, 1522–1531. [CrossRef]
- Kolte, D.; Bryant, J.; Holsworth, D.; Wang, J.; Akbari, P.; Gibson, G.; Shariat-Madar, Z. Biochemical characterization of a novel high-affinity and specific plasma kallikrein inhibitor. Br. J. Pharmacol. 2010, 162, 1639–1649. [CrossRef]
- Zhao, Y.; Qiu, Q.; Mahdi, F.; Shariat-Madar, Z.; Røjkjær, R.; Schmaier, A.H. Assembly and activation of HK-PK complex on endothelial cells results in bradykinin liberation and NO formation. Am. J. Physiol. Circ. Physiol. 2001, 280, H1821–H1829. [CrossRef]
- Schmaier, A.H. Physiologic activities of the Contact Activation System. Thromb. Res. 2014, 133, S41–S44. [CrossRef]
- Ngo, M.-L.; Mahdi, F.; Kolte, D.; Shariat-Madar, Z. Upregulation of prolylcarboxypeptidase (PRCP) in lipopolysaccharide (LPS) treated endothelium promotes inflammation. J. Inflamm. 2009, 6, 3–3. [CrossRef]
- Kichuk, M.R.; Seyedi, N.; Zhang, X.; Marboe, C.C.; Michler, R.E.; Addonizio, L.J.; Kaley, G.; Nasjletti, A.; Hintze, T.H.; D, G.; et al. Regulation of Nitric Oxide Production in Human Coronary Microvessels and the Contribution of Local Kinin Formation. Circ. 1996, 94, 44–51. [CrossRef]
- Förstermann, U.; Münzel, T., Endothelial Nitric Oxide Synthase in Vascular Disease. Circulation 2006, 113, (13), 1708-1714.
- Wada, Y.; Umeno, R.; Nagasu, H.; Kondo, M.; Tokuyama, A.; Kadoya, H.; Kidokoro, K.; Taniguchi, S.; Takahashi, M.; Sasaki, T.; et al. Endothelial Dysfunction Accelerates Impairment of Mitochondrial Function in Ageing Kidneys via Inflammasome Activation. Int. J. Mol. Sci. 2021, 22, 9269. [CrossRef]
- Vasa, M.; Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Nitric Oxide Activates Telomerase and Delays Endothelial Cell Senescence. Circ. Res. 2000, 87, 540–542. [CrossRef]
- Pyo, I.S.; Yun, S.; Yoon, Y.E.; Choi, J.-W.; Lee, S.-J. Mechanisms of Aging and the Preventive Effects of Resveratrol on Age-Related Diseases. Molecules 2020, 25, 4649. [CrossRef]
- Sas, K.; Szabó, E.; Vécsei, L. Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23, 191. [CrossRef]
- Jun, J.-I.; Lau, L.F. Cellular senescence controls fibrosis in wound healing. Aging 2010, 2, 627–631. [CrossRef]
- Aon-Im, P.; Monthakantirat, O.; Daodee, S.; Chulikhit, Y.; Sriya, N.; Boonyarat, C.; Chumwangwapee, T.; Khamphukdee, C.; Kijjoa, A., Evaluation of the Impact of Alternanthera philoxeroides (Mart.) Griseb. Extract on Memory Impairment in D-Galactose-Induced Brain Aging in Mice through Its Effects on Antioxidant Enzymes, Neuroinflammation, and Telomere Shortening. Molecules 2024, 29, (2).
- Quiles, J.; Cabrera, M.; Jones, J.; Tsapekos, M.; Caturla, N. In Vitro Determination of the Skin Anti-Aging Potential of Four-Component Plant-Based Ingredient. Molecules 2022, 27, 8101. [CrossRef]
- Benington, L.; Rajan, G.; Locher, C.; Lim, L.Y. Fibroblast Growth Factor 2—A Review of Stabilisation Approaches for Clinical Applications. Pharmaceutics 2020, 12, 508. [CrossRef]
- Chen, P. Y.; Qin, L.; Li, G.; Tellides, G.; Simons, M., Fibroblast growth factor (FGF) signaling regulates transforming growth factor beta (TGFbeta)-dependent smooth muscle cell phenotype modulation. Sci Rep 2016, 6, 33407.
- Dranka, B.P.; Hill, B.G.; Darley-Usmar, V.M. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic. Biol. Med. 2010, 48, 905–914. [CrossRef]
- Pandya, J. D.; Sullivan, P. G.; Leung, L. Y.; Tortella, F. C.; Shear, D. A.; Deng-Bryant, Y., Advanced and High-Throughput Method for Mitochondrial Bioenergetics Evaluation in Neurotrauma. Methods Mol Biol 2016, 1462, 597-610.
- Finlin, B.S.; Memetimin, H.; Confides, A.L.; Kasza, I.; Zhu, B.; Vekaria, H.J.; Harfmann, B.; Jones, K.A.; Johnson, Z.R.; Westgate, P.M.; et al. Human adipose beiging in response to cold and mirabegron. J. Clin. Investig. 2018, 3. [CrossRef]
- Stuehr, D.J.; Nathan, C.F. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells.. J. Exp. Med. 1989, 169, 1543–1555. [CrossRef]
- Brown, G.C.; Borutaite, V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2004, 1658, 44–49. [CrossRef]
- Rabey, F.M.; Gadepalli, R.S.; Diano, S.; Cheng, Q.; Tabrizian, T.; Gailani, D.; Rimoldi, J.M.; Shariat-Madar, Z. Influence of a novel inhibitor (UM8190) of prolylcarboxypeptidase (PRCP) on appetite and thrombosis.. Curr. Med. Chem. 2012, 19, 4194–206. [CrossRef]
- Reversible Membrane Permeabilization of Mammalian Cells Treated with Digitonin and Its Use for Inducing Nuclear Reprogramming by Xenopus Egg Extracts. Cloning and Stem Cells 2008, 10, (4), 535-542.
- Liu, J.; Xiao, N.; DeFranco, D. B., Use of digitonin-permeabilized cells in studies of steroid receptor subnuclear trafficking. Methods 1999, 19, (3), 403-9.
- Iverson, T. M.; Singh, P. K.; Cecchini, G., An evolving view of complex II-noncanonical complexes, megacomplexes, respiration, signaling, and beyond. J Biol Chem 2023, 299, (6), 104761.
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [CrossRef]
- Fink, B.; Laude, K.; McCann, L.; Doughan, A.; Harrison, D.G.; Dikalov, S. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am. J. Physiol. Physiol. 2004, 287, C895–C902. [CrossRef]
- Zielonka, J.; Kalyanaraman, B., Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol Med 2010, 48, (8), 983-1001.
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [CrossRef]
- Qin, Y.-F.; Tian, H.-H.; Sun, F.; Qin, X.-P. [Effect of losartan on the protection of the kidney and PRCP-kallikrein axis of the two-kidney, one-clipped renovascular hypertensive rats].. 2013, 48, 59–65.
- Krochmal, M.; Cisek, K.; Filip, S.; Markoska, K.; Orange, C.; Zoidakis, J.; Gakiopoulou, C.; Spasovski, G.; Mischak, H.; Delles, C.; et al. Identification of novel molecular signatures of IgA nephropathy through an integrative -omics analysis. Sci. Rep. 2017, 7, 1–13. [CrossRef]
- Herrera, M.D.; Mingorance, C.; Rodriguez-Rodriguez, R.; Alvarez de Sotomayor, M. Endothelial dysfunction and aging: An update. Ageing Res. Rev. 2010, 9, 142–152. [CrossRef]
- Vita, J. A., Endothelial Function. Circulation 2011, 124, (25), e906-e912.
- Püschel, G.P.; Klauder, J.; Henkel, J. Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases. J. Clin. Med. 2022, 11, 4358. [CrossRef]
- Drummond, G.R.; Vinh, A.; Guzik, T.J.; Sobey, C.G. Immune mechanisms of hypertension. Nat. Rev. Immunol. 2019, 19, 517–532. [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [CrossRef]
- Han, Y.; Kim, S.Y. Endothelial senescence in vascular diseases: current understanding and future opportunities in senotherapeutics. Exp. Mol. Med. 2023, 55, 1–12. [CrossRef]
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.D.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565. [CrossRef]
- Duan, L.; Motchoulski, N.; Danzer, B.; Davidovich, I.; Shariat-Madar, Z.; Levenson, V.V. Prolylcarboxypeptidase Regulates Proliferation, Autophagy, and Resistance to 4-Hydroxytamoxifen-induced Cytotoxicity in Estrogen Receptor-positive Breast Cancer Cells. J. Biol. Chem. 2011, 286, 2864–2876. [CrossRef]
- Guevara-Lora, I.; Sordyl, M.; Niewiarowska-Sendo, A.; Bras, G.; Korbut, E.; Goralska, J.; Malczewska-Malec, M.; Solnica, B.; Kozik, A. The effect of bradykinin on the pro-inflammatory response of human adipocytes. Acta Biochim. Pol. 2022, 69, 495–505. [CrossRef]
- Dai, D.-F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and Cardiovascular Aging. Circ. Res. 2012, 110, 1109–1124. [CrossRef]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 1–15. [CrossRef]
- Owen, M. R.; Doran, E.; Halestrap, A. P., Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000, 348 Pt 3, (Pt 3), 607-14.
- Abrosimov, R.; Baeken, M. W.; Hauf, S.; Wittig, I.; Hajieva, P.; Perrone, C. E.; Moosmann, B., Mitochondrial complex I inhibition triggers NAD(+)-independent glucose oxidation via successive NADPH formation, "futile" fatty acid cycling, and FADH(2) oxidation. Geroscience 2024.
- Degli Esposti, M., Inhibitors of NADH–ubiquinone reductase: an overview. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1998, 1364, (2), 222-235.
- Kita, T.; Clermont, A.C.; Murugesan, N.; Zhou, Q.; Fujisawa, K.; Ishibashi, T.; Aiello, L.P.; Feener, E.P. Plasma Kallikrein-Kinin System as a VEGF-Independent Mediator of Diabetic Macular Edema. Diabetes 2015, 64, 3588–3599. [CrossRef]
- Bergsma, D.; Chen, S.; Buchweitz, J.; Gerszten, R.; Haab, B.B. Antibody-Array Interaction Mapping, a New Method to Detect Protein Complexes Applied to the Discovery and Study of Serum Amyloid P Interactions with Kininogen in Human Plasma. Mol. Cell. Proteom. 2010, 9, 446–456. [CrossRef]
- Pinheiro, A.S.; Silbak, S.; Schmaier, A.H. Bradykinin – An elusive peptide in measuring and understanding. Res. Pr. Thromb. Haemost. 2022, 6, e12673. [CrossRef]
- Senchenkova, E. Y.; Russell, J.; Almeida-Paula, L. D.; Harding, J. W.; Granger, D. N., Angiotensin II-mediated microvascular thrombosis. Hypertension 2010, 56, (6), 1089-95.
- Dielis, A.W.; Smid, M.; Spronk, H.M.; Hamulyak, K.; Kroon, A.A.; Cate, H.T.; de Leeuw, P.W. The Prothrombotic Paradox of Hypertension. Hypertension 2005, 46, 1236–1242. [CrossRef]
- Bird, J. E.; Smith, P. L.; Wang, X.; Schumacher, W. A.; Barbera, F.; Revelli, J. P.; Seiffert, D., Effects of plasma kallikrein deficiency on haemostasis and thrombosis in mice: murine ortholog of the Fletcher trait. Thromb Haemost 2012, 107, (6), 1141-50.
- Wang, J.-K.; Li, Y.; Zhao, X.-L.; Liu, Y.-B.; Tan, J.; Xing, Y.-Y.; Adi, D.; Wang, Y.-T.; Fu, Z.-Y.; Ma, Y.-T.; et al. Ablation of Plasma Prekallikrein Decreases Low-Density Lipoprotein Cholesterol by Stabilizing Low-Density Lipoprotein Receptor and Protects Against Atherosclerosis. Circ. 2022, 145, 675–687. [CrossRef]
- Shariat-Madar, Z.; Mahdi, F.; Warnock, M.; Homeister, J.W.; Srikanth, S.; Krijanovski, Y.; Murphey, L.J.; Jaffa, A.A.; Schmaier, A.H. Bradykinin B2 receptor knockout mice are protected from thrombosis by increased nitric oxide and prostacyclin. Blood 2006, 108, 192–199. [CrossRef]
- Simão, F.; Ustunkaya, T.; Clermont, A.C.; Feener, E.P. Plasma kallikrein mediates brain hemorrhage and edema caused by tissue plasminogen activator therapy in mice after stroke. Blood 2017, 129, 2280–2290. [CrossRef]
- Czokało-Plichta, M.; Skibinska, E.; Kosiorek, P.; Musiał, W.J. Kallikrein-kinin system activation and its interactions with other plasma haemostatic components in the coronary artery disease.. 2001, 46, 209–24.
- Aspelin, T.; Eriksen, M.; Ilebekk, A.; Björkman, J.-A.; Lyberg, T. Different cardiac tissue plasminogen activator release patterns by local stimulation of the endothelium and sympathetic nerves in pigs. Blood Coagul. Fibrinolysis 2012, 23, 714–722. [CrossRef]
- Hou, W.; Yin, J.; Alimujiang, M.; Yu, X.; Ai, L.; Bao, Y.; Liu, F.; Jia, W. Inhibition of mitochondrial complex I improves glucose metabolism independently of AMPK activation. J. Cell. Mol. Med. 2017, 22, 1316–1328. [CrossRef]
- Chang, A. M.; Halter, J. B., Aging and insulin secretion. American Journal of Physiology-Endocrinology and Metabolism 2003, 284, (1), E7-E12.
- Harris, A.; Wirostko; Ehrlich, R.; Kheradiya, N.S.; Winston, D.M.; A Ciulla, T.; Wirostko, B. Age-related macular degeneration and the aging eye. Clin. Interv. Aging 2008, ume 3, 473–482. [CrossRef]
- Cucu, I. Signaling Pathways in Inflammation and Cardiovascular Diseases: An Update of Therapeutic Strategies. Immuno 2022, 2, 630–650. [CrossRef]
- Liu, Y.; Sun, Y.; Guo, Y.; Shi, X.; Chen, X.; Feng, W.; Wu, L.-L.; Zhang, J.; Yu, S.; Wang, Y.; et al. An Overview: The Diversified Role of Mitochondria in Cancer Metabolism. Int. J. Biol. Sci. 2023, 19, 897–915. [CrossRef]
Figure 4.
Senescence and telomere length. A, β-gal staining determined in various HPAEC passage (n=3), representative figure. B, Quantification of the number of β-gal positive cells. C, Reduced transcription of telomerase (hTERT) mRNA. The decrease in telomerase was accompanied by an increase in PRCP at working passage (P5-P20).
Figure 4.
Senescence and telomere length. A, β-gal staining determined in various HPAEC passage (n=3), representative figure. B, Quantification of the number of β-gal positive cells. C, Reduced transcription of telomerase (hTERT) mRNA. The decrease in telomerase was accompanied by an increase in PRCP at working passage (P5-P20).
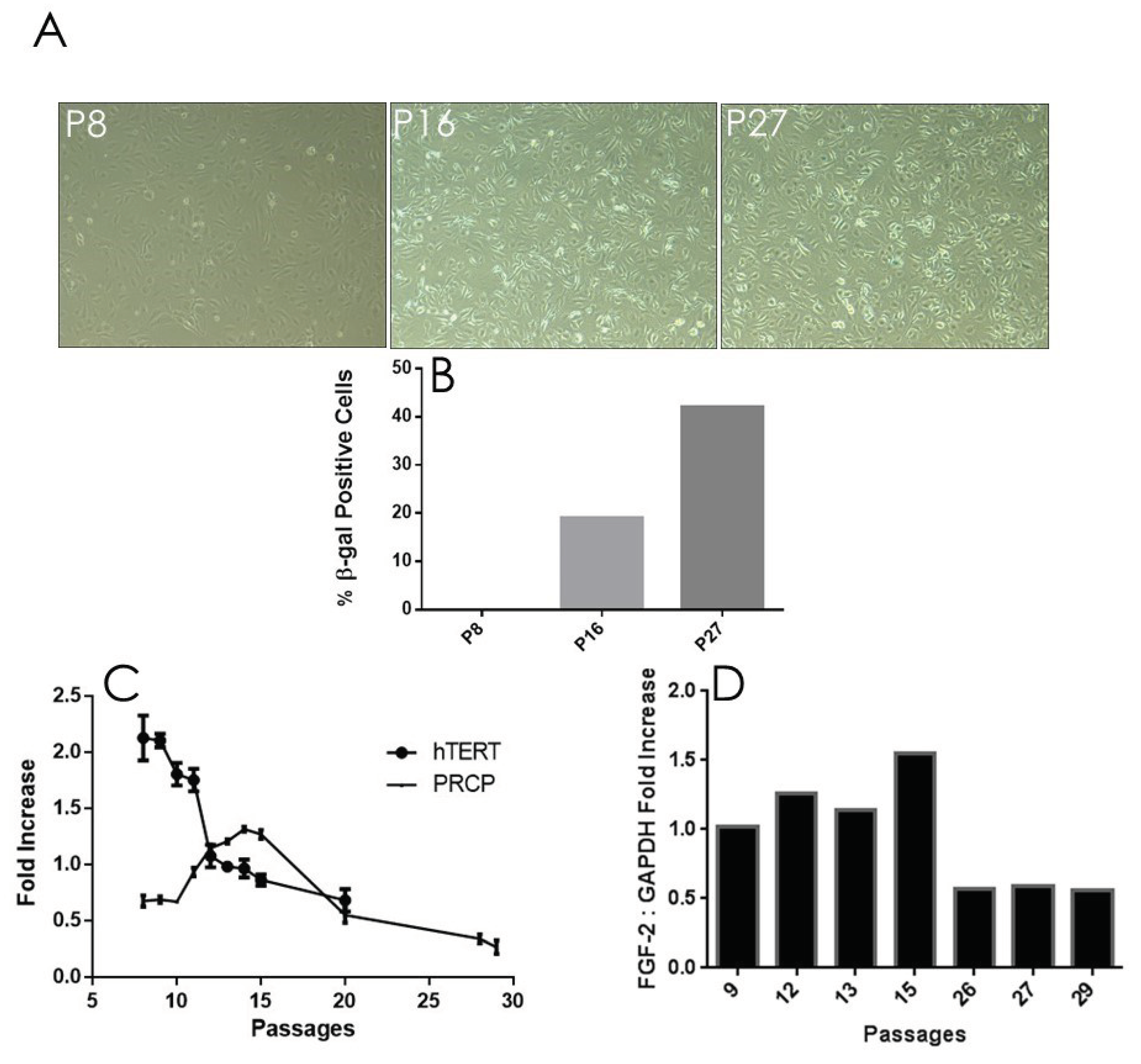
Figure 6.
Effect of UM8190 on the respiration of digitonin permeabilized HPAE cells. A, Effect of UM8190 on Complex I. B, Oxygen consumption rate as a persentage of control. C, The inhibitory effect of UM8190 on Complex II. D, Effect of UM8190 on cytochrome C oxidase. E, A schematic diagram summarizes the effects of UM8190 on complexes of mitochondrial respiratory chain. Data are representative traces from three independent experiments.
Figure 6.
Effect of UM8190 on the respiration of digitonin permeabilized HPAE cells. A, Effect of UM8190 on Complex I. B, Oxygen consumption rate as a persentage of control. C, The inhibitory effect of UM8190 on Complex II. D, Effect of UM8190 on cytochrome C oxidase. E, A schematic diagram summarizes the effects of UM8190 on complexes of mitochondrial respiratory chain. Data are representative traces from three independent experiments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



