
Submitted:
28 March 2024
Posted:
29 March 2024
You are already at the latest version
Abstract
Programmed death ligand 1 (PD-L1) is a co-inhibitory molecule expressed on the surface of various
cell types, known for its suppressive effect on T cells through interaction with PD-1. Neutrophils also express
PD-L1, and its expression is elevated in specific situations; however, the immunobiological role of PD-L1+
neutrophil has not been fully characterized. Here, we report that PD-L1-expressing neutrophils increased in
methicillin-resistant Staphylococcus aureus (MRSA) infection are highly functional in bacterial elimination and
supporting inflammatory resolution. The frequency of PD-L1+ neutrophils was dramatically increased in
MRSA-infected mice, and this population exhibited enhanced activity in bacterial elimination compared to
PD-L1- neutrophils. An administration of PD-L1 monoclonal antibody did not impair PD-L1+ neutrophil
function suggesting that PD-L1 expression itself does not influence neutrophil activity. However, PD-1/PD-L1 blockade significantly delayed liver inflammation resolution in MRSA-infected mice, as indicated by increased plasma alanine transaminase (ALT) level and frequencies of inflammatory leukocytes in the liver implying that neutrophil PD-L1 suppresses the inflammatory response of these cells during the acute phase of MRSA infection. Our results reveal that elevated PD-L1 expression can be a marker for enhanced anti-bacterial function of neutrophils. Moreover, PD-L1+ neutrophils are an indispensable population attenuating inflammatory leukocyte activities assisting in a smooth transition into the resolution phase in MRSA infection.
Keywords:
PD-L1
; neutrophil
; MRSA
; PD-1
; immunosuppressive
1. Introduction
Neutrophils play a crucial role in innate defense against various pathogens [1]. They are abundantly present in the bloodstream and are continuously produced by the bone marrow (BM) allowing for rapid and abundant recruitment to sites of infection where they contribute to pathogen elimination [2]. Neutrophils employ a variety of mechanisms, including myeloperoxidase (MPO), neutrophil elastase (NE), neutrophil extracellular trap (NET), superoxide and cytokine productions as well as phagocytosis, to aggressively eliminate pathogens through those physical and chemical-mediated strategies [3]. While neutrophils were long understood as a homogeneous population, recent studies have revealed that they are a heterogeneous population particularly in abnormal conditions such as infectious diseases and cancers [4,5]. Therefore, characterizing each neutrophil population and their functions have been a necessary to understand the role of neutrophils in specific situations.
Recent studies have shown that neutrophils expressing programmed death-ligand 1 (PD-L1, also known as CD274) are immunosuppressors in cancers [5,6]. PD-L1 is a well-known suppressive molecule expressed on tumor cells and some myeloid cells in the tumor microenvironment (TME), and often found the upregulation in the peripheral immune cells [7,8,9]. In the context of tumor immunology, PD-L1-expressing cells suppress T cell function by interacting directly with PD-1 [10]. This interaction triggers the phosphorylation of the immunoreceptor tyrosine-based switch motif (ITSM) in the cytosolic tail of PD-1, recruiting SH2 domain-containing protein tyrosine phosphatase-2 (SHP-2), which results in the dephosphorylation of zeta-chain-associated protein kinase 70 (ZAP70) in T cells, thereby suppressing their functions [11,12]. This mechanism of functional suppression is widely conserved in PD-L1-expressing cells; therefore, PD-L1+ neutrophils are mostly considered to participate in an immunosuppressive role in cancer. However, other functional characterizations of PD-L1+ neutrophils, especially in non-cancer conditions, are still insufficient.
In this report, we demonstrate that PD-L1+ neutrophils exhibit higher immune activities than PD-L1- neutrophils which is characterized by their predominant role for bacterial elimination in MRSA-challenged mice. Moreover, PD-L1+ neutrophils play an important role in maintaining the inflammatory resolution mechanism during the acute phase of MRSA infection. Our finding provides a novel understanding of the immunological role of PD-L1+ neutrophils in bacterial infection and acute inflammation.
2. Materials and Methods
Reagents and antibodies. Anti-PD-L1 (10F.9G2), anti-CD45 (30-F11), anti-CD11b (M1/70), anti-Ly-6G (1A8), anti-Ly-6C (HK1.4), anti-F4/80 (BM8), anti-CD3 (17A2), anti-CD4 (GK1.5), anti-CD8 (53-6.7), anti-TCRγδ (UC7-13D5), anti-NK1.1 (S17016D), anti-IFN-γ (XMG1.2), anti-IL-17A (TC11-18H10.1), anti-IL-6 (MP5-20F3) and anti-TNF-α (MP6-XT22) were purchased from BioLegend (San Diego, CA, USA). Anti-IL-1β (EPR24895-116), anti-myeloperoxidase (MPO, EPR20257), anti-neutrophil erastase (NE, EPR7479) were purchased from Abcam (Cambridge, UK). Anti-Granzyme B (NGZB) and anti-perfolin (eBioOMAK-D), 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA), SYTOXTM Green and 7-amino-actinomycin D (7-AAD) were purchased from Thremo Fisher Scientific (Waltham, MA, USA). Anti-PD-L1 mAb (10F.9G2) and corresponding isotype antibody were purchased from BioXcell (Lebanon, NH, USA). phorbol-12-myristate-13-acetate (PMA), ionomycin, ALT activity assay kit were purchased from Sigma Aldrich (Laclede Avenue. St. Louis, MO, USA). Golgi StopTM was purchased from BD Bioscience (Franklin Lakes, NJ, USA). Percoll was purchased from GE health care (Chicago, IL, USA). Recombinant murine granulocyte-macrophage colony-stimulating factor (rmGM-CSF) was purchased from Peprotech (Cranbury, NJ, USA).
Methicillin-resistant Staphylococcus aureus culture and sample preparation. The frozen S. aureus (MRSA; USA300) stock was thawed on ice, then transferred to a tryptic soy broth (TSB; BD Bioscience, Franklin Lakes, NJ, USA) The bacterial suspension was cultured at 37 °C for 18 h with shaking, then the grown bacteria were further cultured in TSB at 1:100 dilution at 37°C for 6-8 hours. The colony forming unit (CFU) was calculated by standard curve method in each culture. For Heat-killed S. aureus (HK-SA) preparation, the bacterial suspension was heated at 95 °C for 15 min. The heated S. aureus suspension was centrifuged at 10,000 rpm for 1 min to harvest the bacteria cells and washed with phosphate buffered saline (PBS), then the cell pellet was resuspended in PBS. Cell wall extract (CWE), crude protein extract (CPE), Lipoprotein (LP) and nucleic acids were isolated from S. aureus by following a method described in previous reports with minor modifications [13,14]. Briefly, the cultured S. aureus (108-9 CFU/mL) was harvested by centrifuging at 5,000 g for 20 min, then the pellet was washed twice with Tris-HCl (20 mM, pH 8.0). The pellet was resuspended in Tris–HCl (20 mM, pH 8.0) and stored at -80℃ for overnight. The sample was centrifuged at 5,000 g for 20 min again, then bacterial cells were crushed with 0.3 mm stainless beads followed by sonication at 4℃ for 20 min. The treated sample was centrifuged at 5,000 g for 20 min, then the supernatant was harvested as containing proteins. The precipitated pellet was treated with protease (1 μg/mL) and lipoprotein lipase (1 μg/mL) at 37℃ for 1 h followed by heating at 95℃ for 15 min. The sample was washed with Tris–HCl (20 mM, pH 8.0) and centrifuged at 5,000 g for 20 min. The precipitated sample was used as CWE. The supernatant containing protein was mixed with equal volume of 100% of ethanol and kept at -80 °C for overnight. The sample was centrifuged at 12,000 g for 15 min, then the precipitated pellet was washed with 80% of ethanol and centrifuged again at 12,000 g for 15 min. The precipitated pellet was dissolved with 1 M urea/50 mM Tris-HCl, 50 mM ethylenediaminetetraacetic acid (EDTA) (pH 8.0) and treated with DNase (50 μg/mL) and RNase (50 μg/mL) at 37℃ for 1 h, then the sample was used as CPE. For LP isolation, Triton X-114 was added to the CPE (final 1%), then the suspension was incubated at 4 °C with gentle mixing. The sample was further incubated at 37 °C to form the micelle phase-containing LP. The micelle phase was collected, and LP was precipitated by following a method for CPE precipitation. The precipitated pellet was used as LP. DNA and RNA were isolated from bacteria using NucleoSpin® and TRIzol®, respectively.
Animal experiment. C57BL/6 mice were purchased from CLEA Japan (Tokyo, Japan) and The Jackson Laboratory (Bar Harbor, ME, USA). TLR2-KO mice (B6.129-Tlr2tm1Kir/J), TLR4-KO mice (B6(Cg)-Tlr4tm1.2Karp/J) and MyD88-KO mice (B6.129P2(SJL)-Myd88tm1.1Defr/J) were purchased from The Jackson Laboratory. All mice were bred in-house and maintained in a specific pathogen-free (SPF) facility with 12-hour light/dark cycles and allowed free access to food and water. Adult mice of both genders aged 8-20 weeks were used for each experiment. All animal experimental protocols were approved by the animal care and use committee of Jichi Medical University (20038-01), University of South China (202005053) and Shibata Gakuen University (2107).
MRSA infection. MRSA infection was performed by following a method described in previous report with minor modification [14]. Briefly, the MRSA was washed and resuspended in PBS at 1.0x109 CFU/mL concentration. The mice received an intravenous (i.v.) injection of MRSA (100 µl of suspension) through the retro orbital sinus. For in vivo PD-1/PD-L1 blockade, the MRSA-challenged mice were i.v. injected isotype antibody or anti-PD-L1 mAb (200 µg in 100 µl of saline). The blood samples and tissue samples (spleen, liver and lung) were collected at 48 h of post infection. Before organ extraction, the mice were perfused with PBS. The tissues were washed with PBS, then were chopped and homogenized in PBS. The processed tissue samples were filtered through a 70 μm strainer and centrifuged at 300 g for 5 min. The supernatants were collected and serially diluted with PBS prior to seeding on TSB plates. The plates were incubated at 37°C for overnight, then MRAS CFUs were determined in the samples.
Primary cell isolation. Peripheral blood was collected using ethylenediaminetetraacetic acid (EDTA)-2Na coated tube and treated with red blood cell (RBC) lysis buffer at room temperature (RT) for 10 min. The sample was washed with PBS and the leukocytes were collected by centrifugation at 300 g for 5 min. Spleen and Lung were mechanically crushed on a 70 µm cell strainer in RPMI complete medium, then the cells were collected by centrifugation at 300 g for 5 min. The cells were treated with RBC lysis buffer at RT for 10 min, then were washed with RPMI complete medium. After centrifugation at 300 g for 5 min, the precipitated cells were used as splenocytes and lung-isolated cells, respectively. Bone marrow (BM) cells were isolated from femurs and tibias. The cells were flushed from the bones using 10 mL syringe with 27G needle in RPMI complete medium. The cells were collected by centrifugation at 300 g for 5 min, then treated with RBC lysis buffer at RT for 10 min. After washing with PBS, the cells were collected by centrifugation at 300 g for 5 min. The precipitated cells were used as BM-isolated cells. Neutrophils were enriched from BM-isolated cells using Neutrophil Isolation kit, mouse (Miltenyi Biotec, Bergisch Gladbach, North Rhine-Westphalia, Germany). Hepatic leukocytes were isolated from whole liver. The liver was excised from PBS-perfused mouse and chopped and mechanically crashed on a 70 µm cell strainer in RPMI complete medium followed by digesting with collagenase (1 mg/mL) at 37℃ for 30 min. The samples were re-filtrated by passing through a 70 µm cell strainer and the cells were collected by centrifugation at 300 g for 5 min and the precipitated cells were re-suspended in 35% Percoll (diluted with PBS) for density gradient separation by centrifugation at 600 g for 20 min without acceleration and brake. After centrifugation, the precipitated cells were further treated with RBC lysis buffer at RT for 10 min. The samples were washed with RPMI complete medium and centrifuged at 300 g for 5 min. The precipitated cells were used as hepatic leukocytes. Splenic CD4+ and CD8+ T cells were isolated from splenocytes using CD4+ T cell isolation kit, mouse (Miltenyi Biotec, Bergisch Gladbach, North Rhine-Westphalia, Germany) and CD8a+ T cell isolation kit, mouse (Miltenyi Biotec), respectively. The isolation procedures were performed by following the product manuals. The purity of neutrophils, CD4+ or CD8+ T cells was assessed by flow cytometry and the sample with more than 90% of CD11b+Ly-6G+, CD4+ or CD8+ T cells was used for subsequent experiments.
Flow cytometry and cell sorting. Flow cytometry analysis was performed by using LSRII (BD Bioscience, Franklin Lakes, NJ, USA) and cell sorting was performed by using FACS Aria II (BD Bioscience). For extracellular marker staining, the samples were stained with fluorochrome-conjugated mAb or tetramer in the presence of anti-CD16/CD32 mAb for blocking of Fc gamma Receptor (FcγR) II/III at 4°C for 30 min. Intracellular staining was performed by using the BD Cytofix/Cytoperm™ Fixation/Permeabilization Kit (BD Bioscience) kit For intracellular staining, the extracellular-stained cells were fixed at 4℃ for 20 min followed by staining of intracellular targets at 4°C for 30 min. The samples studied for IL-1β production in netrophils were treated with GolgiStopTM (1 μg/mL) during the in vitro reaction. The samples studied for cytokine production in T cells were stimulated with PMA (100 ng/mL) and ionomycin (250 ng/mL) in the presence of GolgiStopTM (1 μg/mL) at 37°C for 5 h. ROS production was measured by staininig with H2DCFDA (5 μM) at 37°C for 30 min. The data was analyzed by FlowJo 10 (BD Bioscience).
Real-Time Polymerase Chain Reaction (Real-time PCR). The total RNA was isolated from neutrophils using TRizol (Thermo Fisher Scientific). The concentration and purity or RNA were measured by NanoDrop 2000c (Thermo Fisher Scientific). Total RNA (250-500 ng) was used for reverse transcription for making complementary DNA (cDNA) using a PrimeScript™ RT-PCR Kit (Takara, Tokyo, Japan). The cDNA was used for quantitative PCR performed using a Thermal Cycler Dice® Real Time System III (Takara, Tokyou, Japan). The mRNA expressions were quantified by ΔCt method. The primer sequences used in the assay were represented in Supplemental Table S1.
Culture for generating PD-L1+ neutrophils. BM-isolated cells (3.0-5.0x106/mL) were culture with RPMI complete medium supplemented with GM-CSF (100 ng/mL) at 37°C for 24 h. The PD-L1- and PD-L1+ cells were isolated from CD11b+Ly-6G+ population in the pre-cultured BM cells by cell sorter.
Phagocytosis assay. BM-isolated neutrophils (1.0x107/mL) were incubated FITC labeled S. aureus (SA-FITC; 25 µg/mL) in RPMI complete medium at 37°C for 2 h. The cells were washed with PBS and were analyzed by flow cytometry. The fluorescence signal (mean fluorescence intensity; MFI) originated from intracellular incorporated bacteria was used for assessing phagocytosis activity in the neutrophils.
Neutrophil stimulation. BM-isolated neutrophils (1.0x107/mL) were seeded in 96 well plats with RPMI complete medium. For PD-L1 upregulation studies, the cells were treated with vehicle (PBS), HK-SA, CWE, CPE, LP, DNA or RNA at indicated concentrations at 37°C for 6 h. After incubation, the cells were subjected to flow cytometry. Alternatively, the neutrophils were cultured at 37℃ for 3 h in total RNA isolation. For functional studies, the neutrophils were treated with PBS or HK-SA (1.0x108 CFU/mL, MOI=1:10) at 37°C for 6 h, then the cells were subjected to flow cytometry assay. For IL-1β production assay, the neutrophils were cultured with PBS or HK-SA (same MOI to functional studies) at 37°C for 18 h, then the plate was immediately frozen at -80℃. The plate was centrifuged at 300 g for 5 min, and supernatant was subjected to IL-1β ELISA.
In vitro MRSA killing. Neutrophils (1.0x107/mL) were mixed with MRSA (1.0x108 CFU/mL, MOI=1:10) in RPMI complete medium. The samples were incubated at 37°C for 5 h. The samples were centrifuged at 300 g for 5 min, then the supernatants were discarded, and the precipitated cells were washed with PBS. The cells were again collected by centrifugation at 300 g for 5 min, then treated with PBS/0.1% Triton X-100 at RT for 15 min for lysing the cells. The supernatant samples were used for determination of MRSA CFUs by seeding on a TSB agar plate. The plates were incubated at 37°C for overnight, then MRAS CFUs were determined in each sample.
Neutrophil and T cell coculture. The neutrophils (2.5x106/mL) and CD4+ or CD8+ T cells (2.5x106/mL) (at a ratio=1:1) were co-cultured in the presence of anti-CD3 mAb (10 µg/mL) and anti-CD28 mAb (2 µg/mL) at 37°C for 24 h. For PD-1/PD-L1 blockade, the co-cultures were further treated with anti-PD-L1 mAb (10 µg/mL) or isotype antibody (10 µg/mL). At the last 5 h, the cells were stimulated with PMA (100 ng/mL) and ionomycin (250 ng/mL) in the presence of GolgiStopTM (1 µg/mL). The IFN-γ production in CD4+ and CD8+ T cells were analyzed by flow cytometry.
Enzyme-linked immunosorbent assay (ELISA). Mouse IL-1 beta/IL-1F2 DuoSet ELISA kit was purchased from R&D Systems (Minneapolis, MN, USA). All procedures were followed by manufacture’s instruction.
Statistics. Student t-test and one-way analysis of variance (ANOVA) were used to analyze the data for significant differences. Values of p < 0.05, p < 0.01, and p < 0.001 were regarded as significant.
3. Results
3.1. MRSA Infection Increases PD-L1 Expression in Neutrophil
We first investigated how PD-L1 expression changes in response to pathogenic invasion by establishing a murine MRSA infection model. Mice were intravenously infected with MRSA, and the cell surface expressions of PD-L1 in neutrophils from peripheral blood (PB), liver, lung, and spleen were analyzed by flow cytometry at 48 hours post-infection. In naïve mice, PD-L1 was already expressed in some neutrophils, while the frequency of PD-L1+ neutrophil was less than 20% in each sample (PB: 7.312±1.178, liver: 16.25±1.937, lung: 14.58±0.999, spleen: 14.98±1.915) (Figure 1A; top, B; black dots). The PD-L1 expression was significantly increased in neutrophil from MRSA-challenged mice. Neutrophils in all samples consistently exhibited around 2.5 to 3-fold increases in the frequencies of PD-L1+ cells compared to the naïve status (PB: 20.57±2.189, liver: 48.47±3.640, lung: 43.25±4.071, spleen: 41.48±2.736) (Figure 1A; bottom, B; red dots). Interestingly, the increase in PD-L1+ neutrophils was more pronounced in the liver, lung, and spleen compared to the PB. PD-L1 mRNA expression was also analyzed in PB neutrophils from naïve or MRSA-challenged mice. Consistent with the protein expression, PD-L1 mRNA expression was significantly elevated in the PB neutrophils isolated from MRSA-challenged mice compared to that of naïve mice (Figure S1). Thus, MRSA infection upregulates PD-L1 expression in neutrophils.
3.2. PD-L1-Expressing Neutrophil Possesses a Predominant Activity Contributing to Effective MRSA Elimination
Next, we investigated the function of PD-L1+ neutrophils in MRSA infection. Neutrophil activities were assessed in the livers of MRSA-infected mice at 48 hours post-bacterial challenge following PD-L1 expression status. Intriguingly, all measured activities of neutrophils, including reactive oxygen species (ROS), interleukin (IL)-1β, MPO, NE productions and NETs formation, were significantly increased in PD-L1+ neutrophils compared to PD-L1- cells (Figure 2A–E). These findings were further investigated by in vitro experiments using neutrophils with artificially induced PD-L1 expression as model cells. To do so, whole BM-isolated cells were cultured with GM-CSF at 37°C for 24 h that enabled us to obtain a sufficient number of PD-L1+ neutrophils. Subsequently, PD-L1- and PD-L1+ neutrophils in the CD11b+Ly-6G+ population were respectively sorted by cell sorter and subjected to each experiment (Figure 2F). In a phagocytosis assay using FITC-labeled S. aureus (SA), PD-L1+ neutrophils captured more bacteria than PD-L1- cells (Figure 2G). Additionally, ROS production, IL-1β production, and NETs formation under heat-killed SA (HK-SA) stimulation were all significantly increased in PD-L1+ neutrophils compared to PD-L1- neutrophils (Figure 2H–J). Moreover, PD-L1+ neutrophils exhibited greater efficacy in eliminating live-MRSA than PD-L1- neutrophils in an in vitro bacterial killing assay (Figure 2K). Thus, PD-L1+ neutrophils have an increased anti-bacterial activity which may serve as a major effector cell for bacterial elimination in MRSA infection.
3.3. MRSA Recognition via TLRs Increases PD-L1 Expression in Neutrophil
We investigated the crucial mechanism involved in PD-L1 upregulation in neutrophils during MRSA infection. For this purpose, we prepared HK-SA and isolated structural components, such as cell wall extracts (CWE), cell protein extracts (CPE), lipoproteins (LP), and nucleic acids (DNA, RNA), from MRSA culture following the represented procedures based on previous publications (Figure 3A) [13,14,15]. In vitro HK-SA stimulation showed that PD-L1 expressions were significantly increased in neutrophils treated with HK-SA at concentrations greater than 5.0x107 CFU/mL (MOI=1:5), and the expressions were dose dependently increased resulted in the largest PD-L1+ cells in the culture stimulated with 5.0x108 CFU/mL (MOI=1:50) of HK-SA (Figure 3A). Next, we investigated the bioactivities of MRSA structural components in neutrophil PD-L1 upregulation. We found that CWE dramatically increased PD-L1 expression in neutrophils with the frequency of PD-L1+ neutrophils exceeding 50% at the highest dose (10 µg/mL). CPE also increased PD-L1 expression in neutrophils, while it was lesser extent than CWE. Interestingly, LP isolated from CPE increased the frequencies of PD-L1+ neutrophils even at concentrations one-tenth that of CPE. In contrast, nucleic acids did not increase PD-L1 expression in neutrophils (Figure 3C).
Previous publications have described the crucial roles of TLRs in S. aureus recognition [13,14,15,16,17,18]. Therefore, we decided to investigate whether TLRs and their downstream factor, myeloid differentiation factor 88 (Myd88), contribute to PD-L1 upregulation in neutrophils. To determine the responsible TLRs in neutrophil PD-L1 upregulation, we first analyzed the mRNA expressions of TLRs in the HK-SA stimulated neutrophils. The mRNA expression of TLR2 and TLR4, which express cell surface, were predominantly increased in the HK-SA exposed neutrophils (Figure 3D,E). Other cell surface receptors, TLR1 and TLR6 mRNA levels were equivalent between control and HK-SA stimulation. Additionally, intracellular TLRs, such as TLR3, TLR7, TLR8 and TLR9, were also similar expressions between control and HK-SA exposed neutrophils (Figure S2). The mRNA expression of MyD88, a universal adaptor molecule predominantly involved in TLR signal transduction except for TLR3, also increased in HK-SA stimulated neutrophils (Figure 3F).
Following the RNA expression profiles, we performed stimulation assays using neutrophils isolated from WT, TLR2-KO, TLR4-KO, or MyD88-KO mice with HK-SA, CWE, or LP stimulation. In HK-SA stimulation, the upregulation of PD-L1 expression in WT neutrophils was completely abolished in TLR2 or TLR4-deficient cells. Additionally, the deficiency of MyD88 also impaired PD-L1 upregulation under HK-SA stimulation (Figure 3G). Either TLR or MyD88 deficiency also suppressed PD-L1 upregulation in CWE-stimulated neutrophils (Figure 3H). However, only TLR2 or MyD88 deficiency, but not TLR4 deficiency, abolished PD-L1 upregulation in LP-stimulated neutrophils (Figure 3I). Thus, S. aureus directly triggers PD-L1 upregulation through TLR signaling in neutrophils.
3.4. PD-L1+ Neutrophil Contributes Not Only for Enhancing Bacterial Clearance but also for Inflammatory Resolution
We investigated the immunological role of elevated PD-L1 expression in neutrophils in MRSA infection. Given that PD-L1 has an immunosuppressive role through interaction with PD-1 expressed on other immune cells, we performed in vivo PD-1/PD-L1 blockade to investigate the influence of abolishing the suppressive effect in MRSA-infected mice [16]. Mice were infected with MRSA via i.v. injection, and either isotype or anti-PD-L1 mAb was concurrently administered by i.v. injection. After 48 hours of MRSA infection, surviving mice were sacrificed, and MRSA colonization was measured in peripheral blood (PB), liver, lung, and spleen. Additionally, liver neutrophil function was characterized in each mouse (Figure 4A). PD-1/PD-L1 blockade did not alter MRSA colonization in the mice. All samples showed similar MRSA colony-forming units (CFUs) between the mice injected with isotype antibody and anti-PD-L1 mAb (Figure 4B). Application of anti-PD-L1 mAb did not change MPO and NE productions in neutrophils (Figure 4C,D). Additionally, the frequencies of NETotic cells were equivalent between the mice treated with isotype antibody and those treated with anti-PD-L1 mAb (Figure 4E).
Although PD-L1 blockade did not alter the antibacterial activity of PD-L1+ neutrophil, we found that inflammation resolution was delayed by PD-L1 blockade in MRSA-challenged mice. ALT levels, which is inflammatory parameter in the liver [17], was dramatically increased in mice at 48 h (day 2) of post-MRSA challenge, and the levels were comparable between two groups. ALT levels gradually decreased in both groups of mice; however, anti-PD-L1 mAb treatment showed significantly higher levels of ALT at day 5 and day 7 of post-MRSA challenge compared to isotype antibody treatment (Figure 4F). To understand the cause of this delayed inflammation resolution by PD-L1 blockade, we investigated the activity of other immune cells in the liver involved in the inflammatory response. Inflammatory cytokine productions in macrophages were at equivalent levels with or without PD-1/PD-L1 blockade (Figure 4 G-I). While inflammatory responses in some types of leukocytes were significant upregulated by PD-L1 blockade. Pro-inflammatory cytokine and cytotoxic mediator-producing effector CD4+ and CD8+ T cells were significantly increased in MRSA-challenged mice treated with anti-PD-L1 mAb compared to those treated with isotype antibody (Figure 4J, red and blue plots). Pro-inflammatory cytokine-producing innate lymphocytes, such as γδT cells and NKT cells, and granzyme B or perforin-producing NK cells were also increased in the mice subjected to PD-L1 blockade (Figure 4J, green, orange, and pink plots). To demonstrate the suppressive function of PD-L1+ neutrophil, we performed in vitro co-culture of T cells and neutrophils. PD-L1- and PD-L1+ neutrophils were obtained as described in Figure 2F, and the cells were co-cultured with splenic CD4+ T cells or CD8+ T cells with T cell receptor (TCR) stimulation. The cultures were also treated with isotype antibody or anti-PD-L1 mAb. In the presence of PD-L1+ neutrophils, IFN-γ producing CD4+ and CD8+ T cells were significantly decreased in the cultures compared to those without neutrophils. PD-L1- neutrophils did not suppress cytokine production in both CD4+ and CD8+ T cells. While PD-1/PD-L1 blockade rescued IFN-γ production in CD4+ and CD8+ T cells in the presence of PD-L1+ neutrophils (Figure 4K,L). Thus, PD-L1 mAb administration does not regulate the antibacterial activity of neutrophils; however, it interferes inflammatory resolution by sustaining inflammatory response of leukocytes in MRSA-challenged mice.
4. Discussion
The functional suppression based on PD-1/PD-L1 interaction was originally found in anti-tumor immunity, particularly between PD-L1 expressing tumor cells and PD-1 upregulated CD8+ T cells, which impairs the function of CD8+ T cells [16,18]. Recent studies have revealed that PD-L1 expression is widely conserved in myeloid lineage cells, and the protein also interacts with PD-1 expressed on T cells to suppress their activity. This finding results in targeting dendritic cells (DCs) and macrophages for PD-L1 blockade to recover anti-tumor immunity [19,20]. While the study of PD-L1+ neutrophils is still a minor field and has far less accumulated evidence than other myeloid cells. Additionally, there are some possibilities that PD-L1 may has unknown roles distinct from general immunosuppressive role in cancer. In fact, a previous report documented another function of PD-L1 as stabilizing neutrophil extracellular traps (NETs) formation [21]. Given that neutrophils are abundantly circulating in our bloodstream and frequently involved in diverse immune responses, we decided to investigate the role of PD-L1+ neutrophils in infectious diseases.
An interesting finding in our results is the functional difference between PD-L1- and PD-L1+ neutrophil in MRSA infection. It was clear that PD-L1+ neutrophil possesses significantly enhanced anti-bacterial activity compared to PD-L1- neutrophil. The increased population of PD-L1+ neutrophil in MRSA-infected mice suggests that this population may predominantly combat bacteria during infection. This evidence supports the concept that neutrophil population is composed by heterogeneous manner, and each population has specific role in various conditions including infection [22]. However, we have not elucidated the mechanism by which neutrophil control PD-L1 expression in their population. We found that MRSA cell wall extract (CWE) and lipoproteins (LP) trigger PD-L1 expression through Toll-like receptors (TLRs); however, this response spontaneously occurs in the neutrophils of MRSA-challenged mice. Therefore, this finding cannot explain the selective upregulation of PD-L1 expression in neutrophils. Indeed, it is challenging to elucidate the PD-L1 upregulation mechanism under physiological conditions, as various factors may influence neutrophil characteristics. Additionally, we have not explored mechanistic aspects of how PD-L1 expression enhances neutrophil function against MRSA invasion. A previous report revealed that PD-L1 expression prevents apoptosis and extends their survival in human neutrophils [23]. If this functional modification is also occurred in MRSA infection, it might be a possible mechanistic change that prolongs the survival of neutrophils and maintains their effective function against pathogens. Recent finding suggests that PD-L1 can act as a transcription factor [24], raising the possibility that neutrophils also possess a PD-L1-mediated transcriptional regulation mechanism in immune activity. Further investigation is required to elucidate whether increased PD-L1 expression is merely a marker or serves a specific function in neutrophil.
As another new insight into the role of PD-L1+ neutrophils in MRSA infection, we propose a supportive role in inflammatory resolution by suppressing inflammatory activity of leukocytes via PD-1/PD-L1 interaction. Our data suggest that inflammatory leukocytes were increased in the liver of MRSA-challenged mice upon administration of anti-PD-L1 mAb. PD-L1 expression is predominantly elevated in neutrophils in the liver of MRSA-infected mice compared to the naïve condition; therefore, we suspect that these cells are playing a suppressive role against inflammatory leukocytes. We also provide strong evidence of PD-L1+ neutrophil-mediated suppressive effects through in vitro neutrophil and T cell co-culture. In summary, blocking PD-L1 on neutrophils impairs indispensable negative regulation of leukocyte inflammatory responses in MRSA-infected livers that consequently delays inflammatory resolution. This represents a new understanding of the role of PD-L1+ neutrophils in regulating excessive inflammation during infectious diseases. Although our results strongly support the suppressive role of PD-L1+ neutrophils against leukocytes, our study has some limitations. We have not performed neutrophil depletion to prove the importance of their suppressive role against leukocytes. Since neutrophil depletion simply accelerates MRSA colonization during the acute phase, we were unable to take this approach. One possibility is to use neutrophil-specific PD-L1 knockout (KO) mice, which can be created by crossing Pdl1-floxed mice and S100 calcium-binding protein A8 (calgranulin A)-Cre mice [25,26]. Additionally, PD-L1 expression in other cells may need to be characterized more deeply under MRSA infection. For instance, other myeloid cells, such as DCs and macrophages, and hepatocytes might express PD-L1 in MRSA-infected mice by excessive inflammation. Since neutrophils frequently circulate in the bloodstream and reside in tissues, PD-L1+ neutrophils may have a higher frequency of contact with leukocytes than other PD-L1+ cells. However, the background effect of elevated PD-L1 expressions in other cells must be considered.
Recent studies have revealed novel roles and functions of neutrophils that far exceed traditional understanding. This report also provides new evidence in neutrophil biology. Since the evidence is still accumulating and some findings are contradictory, immunotherapy targeting neutrophils has not yet been applied clinically. However, summarizing solid findings may support the creation of new strategies for neutrophil-based therapy, similar to targeting T cells, in the future.
5. Conclusions
The result in this study is summarized as a diagram represented in Figure 5. Under MRSA infection, PD-L1 expression is upregulated in neutrophil by recognition of bacterial structural components through extra cellular TLRs. The PD-L1+ neutrophil is highly reactive than PD-L1- neutrophil, and the population effectively eliminate MRSA by enhanced phagocytic activity as well as increased productions of cytokine, ROS, MPO, NE and NETs. On the other hand, PD-L1+ neutrophil ceases excessive tissue inflammation associated with accumulating inflammatory leukocytes through PD-1/PD-L1 interaction results in supporting inflammatory resolution.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
A.T. and S.S. established the original study concept. A.T, F.A., A.O., Z.P. and S.S. performed experiments. A.T., A.O. and S.S. performed data analysis. A.O., Z.P., D.Y.C. and S.S. had responsibility for resources. A.T., D.Y.C. and S.S. finalized data. A.T. and S.S. wrote the original manuscript. A.T. and S.S. finalized manuscript. S.S. supervised this study.
Funding
This study was supported by the Japan Society for the Promotion of Science (16H06814 (SS), 21K15958 (SS)), 21K20573 (AO), 22K05543 (AO)), Mishima Kaiun Memorial Fund (SS), the National Science Foundation of Hunan Province (2022JJ40382 (ZP)) and American Heart Association’s Career Development award 23CDA1052548 (DYC).
Institutional Review Board Statement
All animal experimental protocols were approved by the animal care and use committee of Jichi Medical University (20038-01), University of South China (202005053) and Shibata Gakuen University (2107).
Acknowledgments
We thank xxx.
Conflicts of Interest
Authors declare no conflict of interest.
References
- Witter, A.R.; Okunnu, B.M.; Berg, R.E. The Essential Role of Neutrophils during Infection with the Intracellular Bacterial Pathogen Listeria monocytogenes. J Immunol. 2016, 197, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kubes, P. NETosis: how vital is it? Blood. 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, C.C.; Malanchi, I. Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol. 2022, 22, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Pylaeva, E.; Korschunow, G.; Spyra, I.; Bordbari, S.; Siakaeva, E.; Ozel, I.; Domnich, M.; Squire, A.; Hasenberg, A.; Thangavelu, K.; Hussain, T.; Goetz, M.; Lang, K.S.; Gunzer, M.; Hansen, W.; Buer, J.; Bankfalvi, A.; Lang, S.; Jablonska, J. During early stages of cancer, neutrophils initiate anti-tumor immune responses in tumor-draining lymph nodes. Cell Rep. 2022, 40, 111171. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, S.; Xue, J.; Qi, M.; Liu, X.; Huang, Y.; Hu, J.; Dong, H.; Ling, K. PD-L1 tumor-intrinsic signaling and its therapeutic implication in triple-negative breast cancer. JCI Insight. 2021, 6, e131458. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. 2016, 5, e1247135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Yan, Z.; Yang, H.; Sun, W.; Yao, Y.; Chen, Y.; Jiang, R. IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J Immunother Cancer. 2020, 8, e000285. [Google Scholar] [CrossRef]
- Cha,J. H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol Cell. 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Marasco, M.; Berteotti, A.; Weyershaeuser, J.; Thorausch, N.; Sikorska, J.; Krausze, J.; Brandt, H.J.; Kirkpatrick, J.; Rios, P.; Schamel, W.W.; Köhn, M.; Carlomagno, T. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci Adv. 2020, 6, eaay4458. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jie, H.B.; Lei, Y.; Gildener-Leapman, N.; Trivedi, S.; Green, T.; Kane, L.P.; Ferris, R.L. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. 2015, 75, 508–518. [Google Scholar] [CrossRef]
- Saito, S.; Quadery, A.F. Staphylococcus aureus Lipoprotein Induces Skin Inflammation, Accompanied with IFN-γ-Producing T Cell Accumulation through Dermal Dendritic Cells. Pathogens. 2018, 7, 64. [Google Scholar] [CrossRef]
- Peng, Z.; Cao, D.Y.; Wu, H.Y.; Saito, S. Immunization with a Bacterial Lipoprotein Establishes an Immuno-Protective Response with Upregulation of Effector CD4+ T Cells and Neutrophils Against Methicillin-Resistant Staphylococcus aureus Infection. Pathogens. 2020, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Okuno, A.; Cao, D.Y.; Peng, Z.; Wu, H.Y.; Lin, S.H. Bacterial Lipoteichoic Acid Attenuates Toll-Like Receptor Dependent Dendritic Cells Activation and Inflammatory Response. Pathogens. 2020, 9, 825. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Shahbaz, S.; Sligl, W.; Osman, M.; Tyrrell, D.L.; Elahi, S. Differential Impact of SARS-CoV-2 Isolates, Namely, the Wuhan Strain, Delta, and Omicron Variants on Erythropoiesis. Microbiol Spectr. 2022, 10, e0173022. [Google Scholar] [CrossRef]
- Saito, S.; Kawamura, T.; Higuchi, M.; Kobayashi, T.; Yoshita-Takahashi, M.; Yamazaki, M.; Abe, M.; Sakimura, K.; Kanda, Y.; Kawamura, H.; Jiang, S.; Naito, M.; Yoshizaki, T.; Takahashi, M.; Fujii, M. RASAL3, a novel hematopoietic RasGAP protein, regulates the number and functions of NKT cells. Eur J Immunol. 2015, 45, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Li, K,; Li, T. ; Feng, Z.; Huang, M.; Wei, L.; Yan, Z.; Long, M.; Hu, Q.; Wang, J.; Liu, S.; Sgroi, D.C.; Demehri, S. CD8+ T cell immunity blocks the metastasis of carcinogen-exposed breast cancer. Sci Adv. 2021, 7, eabd8936. [CrossRef]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.X.; Tang, H. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun. 2020, 11, 4835. [Google Scholar] [CrossRef]
- Xia, Q.; Jia, J.; Hu, C.; Lu, J.; Li, J.; Xu, H.; Fang, J.; Feng, D.; Wang, L.; Chen, Y. Tumor-associated macrophages promote PD-L1 expression in tumor cells by regulating PKM2 nuclear translocation in pancreatic ductal adenocarcinoma. Oncogene. 2022, 41, 865–877. [Google Scholar] [CrossRef]
- Zhu, C.L.; Xie, J.; Zhao, Z.Z.; Li, P.; Liu, Q.; Guo, Y.; Meng, Y.; Wan, X.J.; Bian, J.J.; Deng, X.M.; Wang, J.F. PD-L1 maintains neutrophil extracellular traps release by inhibiting neutrophil autophagy in endotoxin-induced lung injury. Front Immunol. 2022, 13, 949217. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood. 2016, 127, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Wang, Y.P.; Xie, J.; Zhao, Z.Z.; Gupta, S.; Guo, Y.; Jia, S.H.; Parodo, J.; Marshall, J.C.; Deng, X.M. Upregulated PD-L1 delays human neutrophil apoptosis and promotes lung injury in an experimental mouse model of sepsis. Blood. 2021, 138, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Nihira, N.T.; Bu, X.; Chu, C.; Zhang, J.; Kolodziejczyk, A.; Fan, Y.; Chan, N.T.; Ma, L.; Liu, J.; Wang, D.; Dai, X.; Liu, H.; Ono, M.; Nakanishi, A.; Inuzuka, H.; North, B.J.; Huang, Y.H.; Sharma, S.; Geng, Y.; Xu, W.; Liu, X.S.; Li, L.; Miki, Y.; Sicinski, P.; Freeman, G.J.; Wei, W. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol. 2020, 22, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Petzold, T.; Zhang, Z.; Ballesteros, I.; Saleh, I.; Polzin, A.; Thienel, M.; Liu, L.; Ul Ain, Q.; Ehreiser, V.; Weber, C.; Kilani, B.; Mertsch, P.; Götschke, J.; Cremer, S.; Fu, W.; Lorenz, M.; Ishikawa-Ankerhold, H.; Raatz, E.; El-Nemr, S.; Görlach, A.; Marhuenda, E.; Stark, K.; Pircher, J.; Stegner, D.; Gieger, C.; Schmidt-Supprian, M.; Gaertner, F.; Almendros, I.; Kelm, M.; Schulz, C.; Hidalgo, A.; Massberg, S. Neutrophil "plucking" on megakaryocytes drives platelet production and boosts cardiovascular disease. Immunity. 2022, 55, 2285–2299. [Google Scholar] [CrossRef]
- Németh, T.; Futosi, K.; Sitaru, C.; Ruland, J.; Mócsai, A. Neutrophil-specific deletion of the CARD9 gene expression regulator suppresses autoantibody-induced inflammation in vivo. Nat Commun. 2016, 7, 11004. [Google Scholar] [CrossRef]
Figure 1.
PD-L1 expression is increased in neutrophils of MRSA-infected mice. The mice were infected to MRSA by i.v. injection and the PD-L1 expression in neutrophils were analyzed in PB, liver, lung, and spleen at 48 h of post infection. Representative plots (A) and cumulative percentages of PD-L1+ neutrophils (B) were shown. The cumulative data were shown as mean ± standard error (SD) of six samples. Student t-test was used to analyze data for significant differences. Values of *p < 0.001 was regarded as significant.
Figure 1.
PD-L1 expression is increased in neutrophils of MRSA-infected mice. The mice were infected to MRSA by i.v. injection and the PD-L1 expression in neutrophils were analyzed in PB, liver, lung, and spleen at 48 h of post infection. Representative plots (A) and cumulative percentages of PD-L1+ neutrophils (B) were shown. The cumulative data were shown as mean ± standard error (SD) of six samples. Student t-test was used to analyze data for significant differences. Values of *p < 0.001 was regarded as significant.
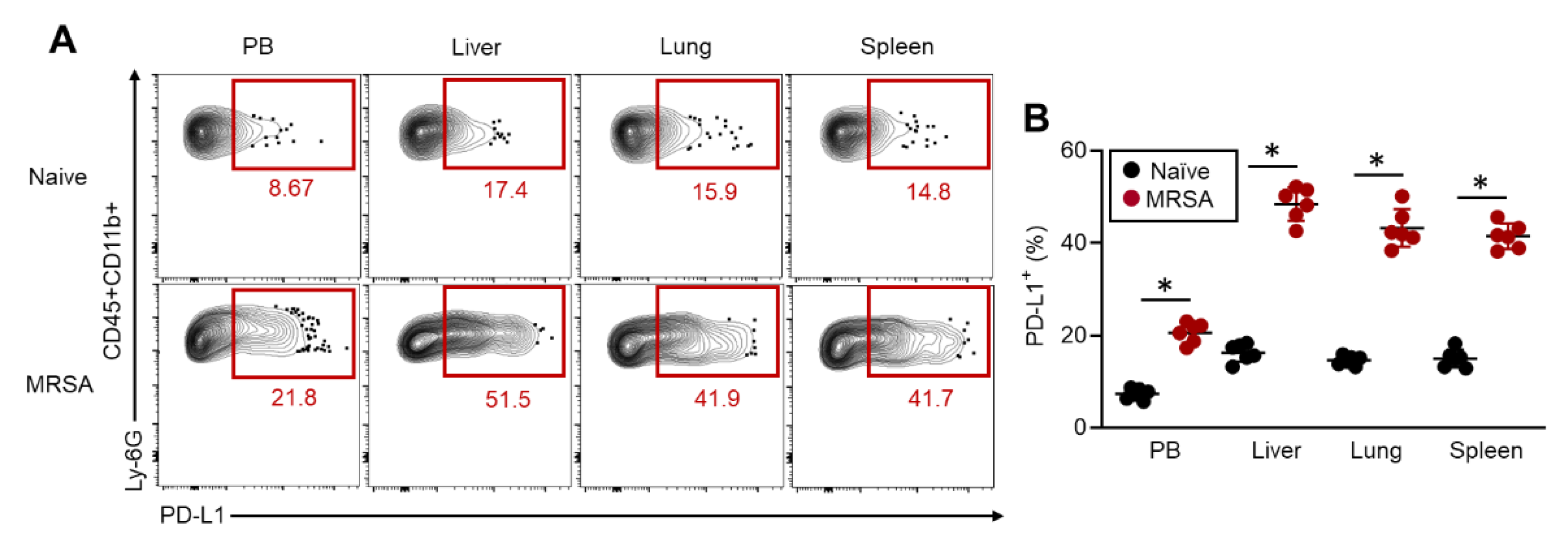
Figure 2.
PD-L1+ neutrophil has enhanced anti-bacterial activity. A-E) In vivo functional assay of neutrophils following PD-L1 expression. The activities were measured in the liver neutrophils after 48 h of MRSA challenge. The productions of ROS (A), IL-1β (B), MPO (C), neutrophils elastase (D) and NETs formation (E) were measured by flow cytometry. F-K) In vitro functional assay of neutrophils. F) The diagram of in vitro PD-L1 induction in neutrophils and sorting for PD-L1+ and PD-L1- neutrophils. G) Phagocytic activity against S. aureus (FITC-labeled). ROS (H) and IL-1β production (I) in HK-SA (MOI=1:50) stimulated neutrophils. J) Cell-free-DNA (CFD) level measured in the cultured medium of HK-SA (MOI-1:50)-stimulated neutrophils. K) In vitro MRSA killing of neutrophils. Survival intracellular MRSA was quantified by measuring CFU. The cumulative data were shown as mean ± standard error (SD) of six samples. Student t-test was used to analyze data for significant differences. Values of *p < 0.05, **p < 0.01 and ***p < 0.001 were regarded as significant.
Figure 2.
PD-L1+ neutrophil has enhanced anti-bacterial activity. A-E) In vivo functional assay of neutrophils following PD-L1 expression. The activities were measured in the liver neutrophils after 48 h of MRSA challenge. The productions of ROS (A), IL-1β (B), MPO (C), neutrophils elastase (D) and NETs formation (E) were measured by flow cytometry. F-K) In vitro functional assay of neutrophils. F) The diagram of in vitro PD-L1 induction in neutrophils and sorting for PD-L1+ and PD-L1- neutrophils. G) Phagocytic activity against S. aureus (FITC-labeled). ROS (H) and IL-1β production (I) in HK-SA (MOI=1:50) stimulated neutrophils. J) Cell-free-DNA (CFD) level measured in the cultured medium of HK-SA (MOI-1:50)-stimulated neutrophils. K) In vitro MRSA killing of neutrophils. Survival intracellular MRSA was quantified by measuring CFU. The cumulative data were shown as mean ± standard error (SD) of six samples. Student t-test was used to analyze data for significant differences. Values of *p < 0.05, **p < 0.01 and ***p < 0.001 were regarded as significant.
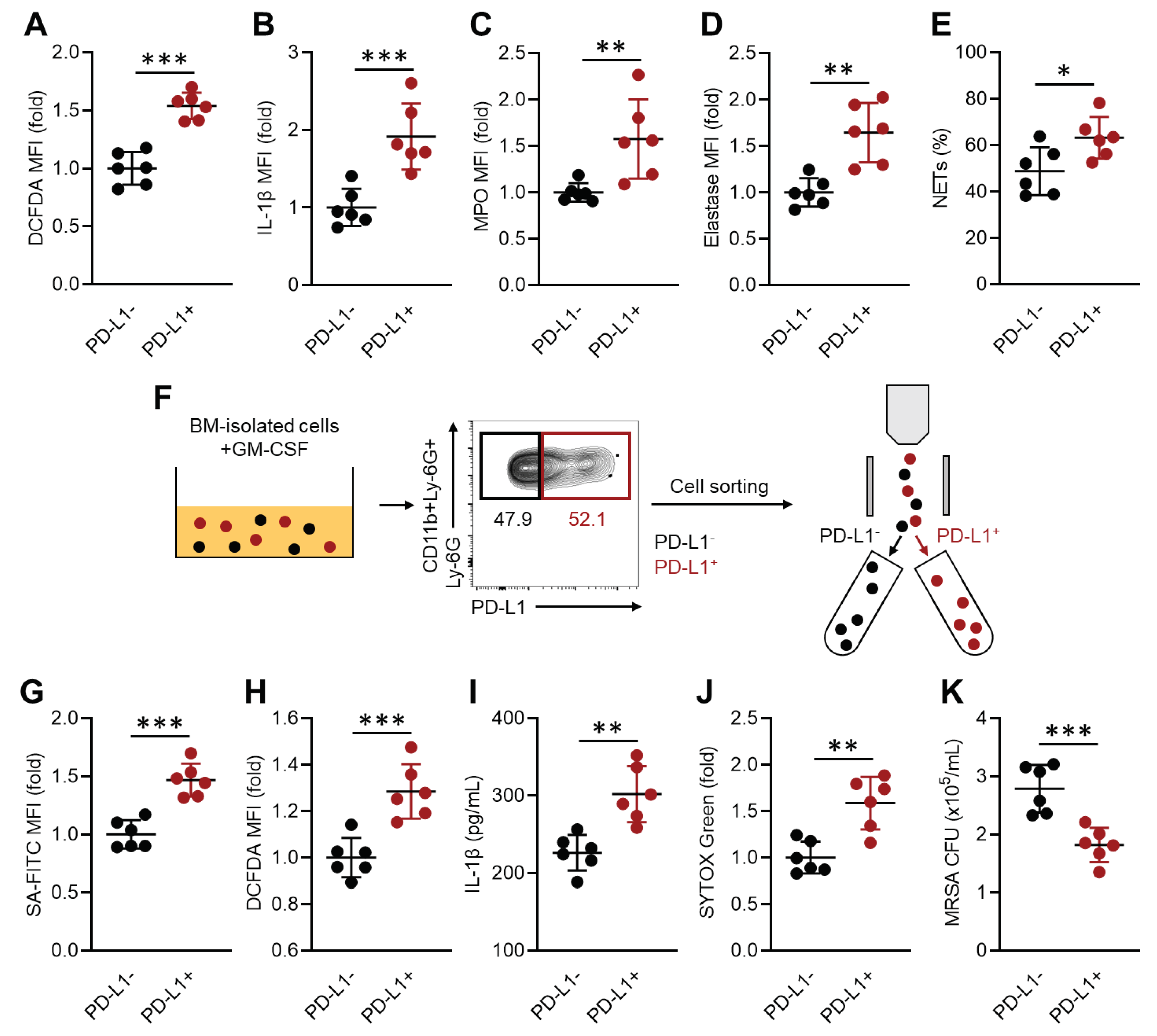
Figure 3.
Bacterial structural components trigger PD-L1 expression in neutrophils through extracellular TLRs. A) The diagram of purification procedure of MRSA structural components. B-C) In vitro neutrophil stimulation. Neutrophils were isolated from WT mice BM and subjected to stimulation assay for measuring PD-L1 upregulation. B) The percentages of PD-L1+ cells in neutrophils cultured with vehicle (PBS) or HK-SA. C) The percentages of PD-L1+ cells in neutrophils cultured with vehicle (PBS), CWE, CPE, LP, DNA or RNA isolated from MRSA. The mRNA expressions of TLR2 (D), TLR4 (E) and MyD88 (F) in vehicle (PBS) or HK-C60 (MOI=1:50) cultured neutrophils. The percentages of PD-L1+ neutrophils cultured with HK-SA (MOI=1:50) (G), CWE (10 μg/mL) (H) or LP (1 μg/mL) (I). The neutrophils were isolated from WT, TLR2-KO, TLR4-KO or MyD88-KO mice. The cumulative data were shown as mean ± standard error (SD) of six samples. One-Way ANOVA was used to analyze data for significant differences. Values of *p < 0.05, **p < 0.01 and ***p < 0.001 were regarded as significant.
Figure 3.
Bacterial structural components trigger PD-L1 expression in neutrophils through extracellular TLRs. A) The diagram of purification procedure of MRSA structural components. B-C) In vitro neutrophil stimulation. Neutrophils were isolated from WT mice BM and subjected to stimulation assay for measuring PD-L1 upregulation. B) The percentages of PD-L1+ cells in neutrophils cultured with vehicle (PBS) or HK-SA. C) The percentages of PD-L1+ cells in neutrophils cultured with vehicle (PBS), CWE, CPE, LP, DNA or RNA isolated from MRSA. The mRNA expressions of TLR2 (D), TLR4 (E) and MyD88 (F) in vehicle (PBS) or HK-C60 (MOI=1:50) cultured neutrophils. The percentages of PD-L1+ neutrophils cultured with HK-SA (MOI=1:50) (G), CWE (10 μg/mL) (H) or LP (1 μg/mL) (I). The neutrophils were isolated from WT, TLR2-KO, TLR4-KO or MyD88-KO mice. The cumulative data were shown as mean ± standard error (SD) of six samples. One-Way ANOVA was used to analyze data for significant differences. Values of *p < 0.05, **p < 0.01 and ***p < 0.001 were regarded as significant.
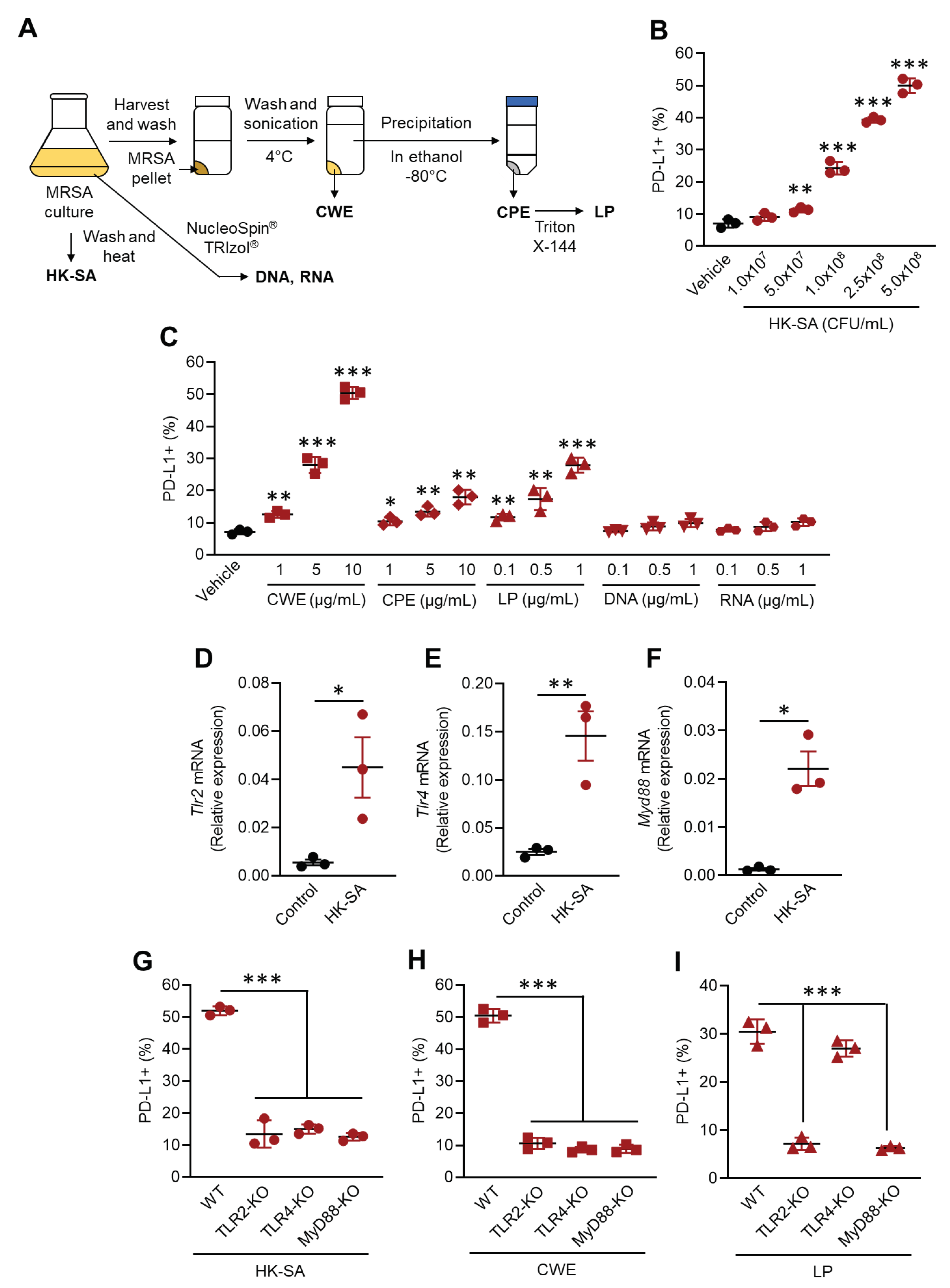
Figure 4.
PD-L1 blockade does not compromise neutrophil anti-bacterial immunity; however, it delay the resolution of inflammation in MRSA-infected mice. A) Experimental design of MRSA challenge and anti-PD-L1 mAb administration. Mice were infected to MRSA and received isotype or anti-PD-L1 mAb administration. The bacterial colonization and immune functions of neutrophils were analyzed in the mice at 48 h of post MRSA challenge. B) MRSA colonization in PB, liver, lung and spleen in MRSA-challenged mice. The CFUs were indicated as per mL for PB and per 100 mg for organs. C-E) In vivo neutrophil functional assay. MPO (C), neutrophil elastase (D) productions and percentage of NETosis cell (E) were shown. F) Plasma ALT concentration in MRSA-challenged mice. The IL-6 (G), TNF-α (H) or IL-1β (I) production of liver macrophages in MRSA-challenged mice in day 7. J) Liver leukocyte functional profile of MRSA-challenged mice. K-L) In vitro T cell suppression assay. T cells and neutrophils (PD-L1- or PD-L1+) were co-cultured with or without isotype Ab or anti-PD-L1 mAb, the IFN-γ producing populations of CD4+ (K) and CD8+ (L) T cells were analyzed, respectively. The cumulative data were shown as mean ± standard error (SD) of six samples. Student t-test was used to analyze data for significant differences. Values of *p < 0.05 and **p < 0.01 were regarded as significant. ns=not significant.
Figure 4.
PD-L1 blockade does not compromise neutrophil anti-bacterial immunity; however, it delay the resolution of inflammation in MRSA-infected mice. A) Experimental design of MRSA challenge and anti-PD-L1 mAb administration. Mice were infected to MRSA and received isotype or anti-PD-L1 mAb administration. The bacterial colonization and immune functions of neutrophils were analyzed in the mice at 48 h of post MRSA challenge. B) MRSA colonization in PB, liver, lung and spleen in MRSA-challenged mice. The CFUs were indicated as per mL for PB and per 100 mg for organs. C-E) In vivo neutrophil functional assay. MPO (C), neutrophil elastase (D) productions and percentage of NETosis cell (E) were shown. F) Plasma ALT concentration in MRSA-challenged mice. The IL-6 (G), TNF-α (H) or IL-1β (I) production of liver macrophages in MRSA-challenged mice in day 7. J) Liver leukocyte functional profile of MRSA-challenged mice. K-L) In vitro T cell suppression assay. T cells and neutrophils (PD-L1- or PD-L1+) were co-cultured with or without isotype Ab or anti-PD-L1 mAb, the IFN-γ producing populations of CD4+ (K) and CD8+ (L) T cells were analyzed, respectively. The cumulative data were shown as mean ± standard error (SD) of six samples. Student t-test was used to analyze data for significant differences. Values of *p < 0.05 and **p < 0.01 were regarded as significant. ns=not significant.
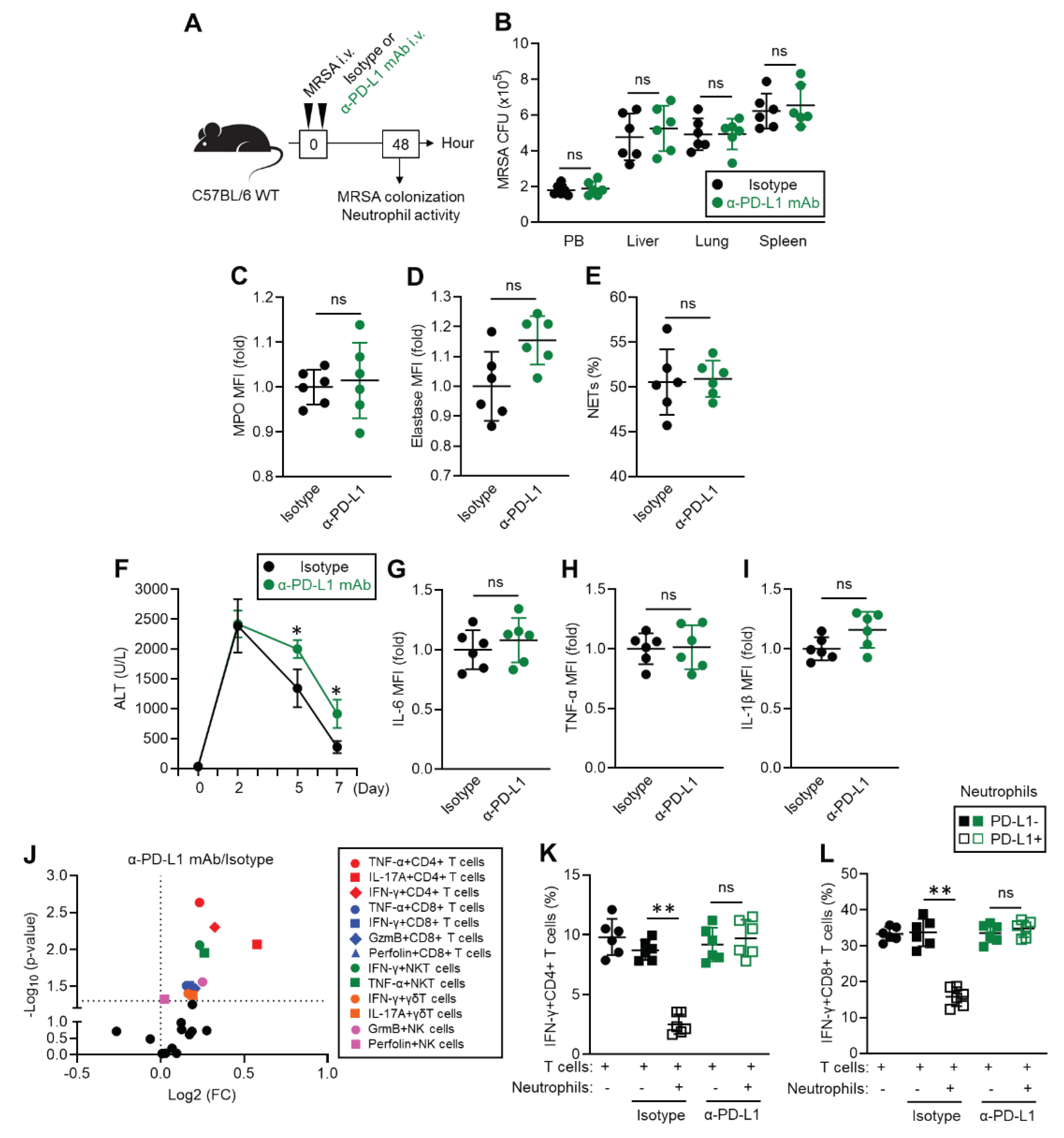
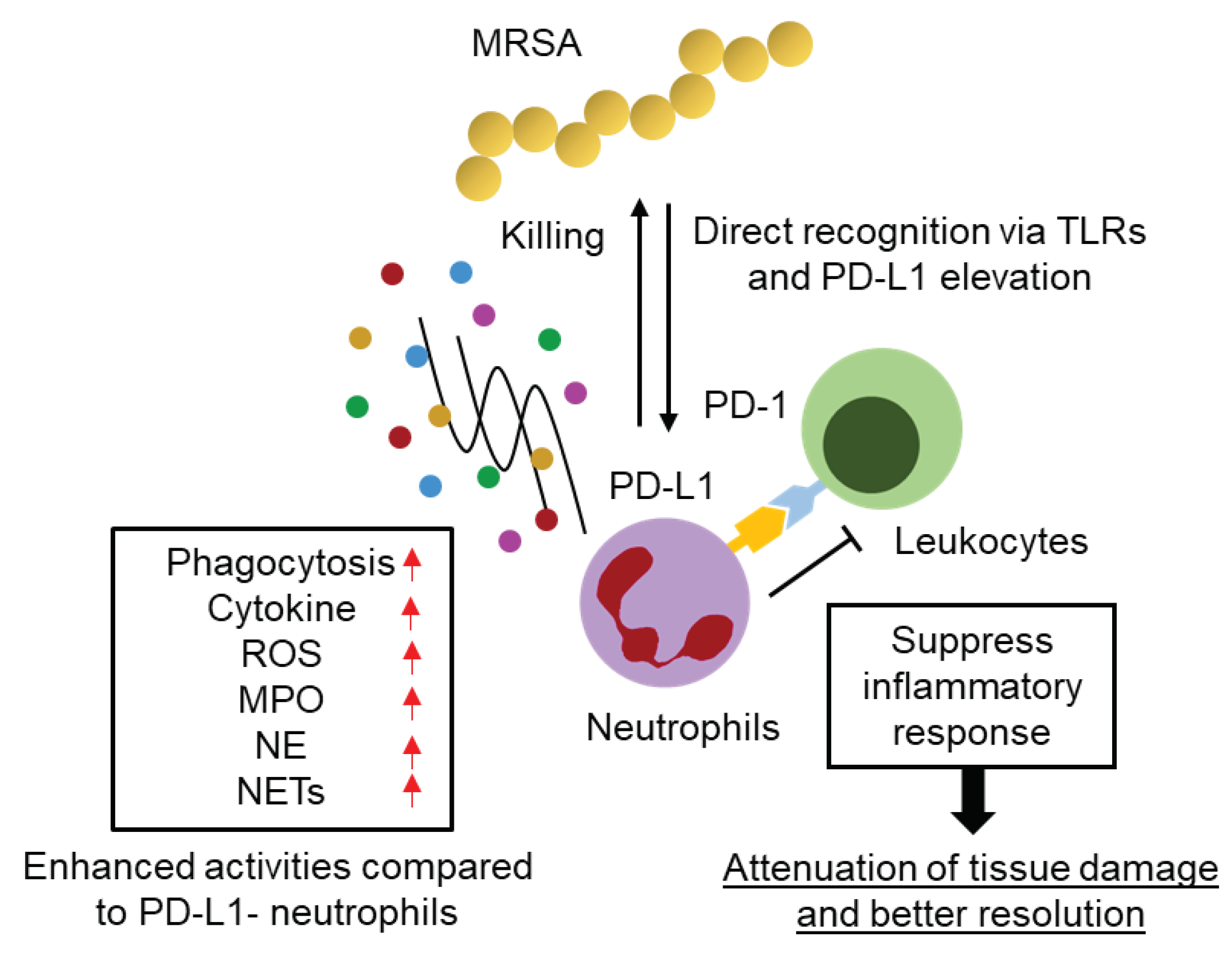
Figure 5.
Hypothetical diagram of PD-L1+ neutrophil function in the acute phase of MRSA infection.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



