
Submitted:
28 March 2024
Posted:
29 March 2024
You are already at the latest version
Abstract
Atopic dermatitis (AD) is a chronic inflammatory disorder presenting a dysregulated immune response and derangement of the epidermal barrier lipids. The Th2-type cytokines interleukin IL-4 and IL-13 play a prominent role in AD by activating the Janus Kinase/Signal Transduction and Activator of Transcription (JAK/STAT) intracellular signal axis. This study aimed to investigate the role of JAK/STAT in the lipid perturbations induced by Th2 signaling in 3D-epidermal equivalents. We used tofacitinib, a low-molecular-mass JAK inhibitor, to screen for the JAK/STAT-mediated deregulation of lipid metabolism. Th2 cytokines decreased the expression of elongases 1, 3, and 4, and serine-palmitoyl-transferase and increased that of sphingolipid delta(4)-desaturase, and carbonic anhydrase 2. Th2 cytokines inhibited the synthesis of palmitate and caused a depletion of triglycerides. This depletion was associated with altered PC profiles and fatty acid (FA) metabolism. Overall, the ceramide profiles were minimally affected. Except for most sphingolipids and very long chain FAs, the effects of Th2 on lipid pathways were reversed by co-treatment with tofacitinib. An increase in the mRNA levels of CPT1A and ACAT1, reduced by tofacitinib, suggests that Th2 cytokines promote FA beta-oxidation. In conclusion, pharmacological inhibition of JAK/STAT activation prevents the lipid disruption caused by halted homeostasis of FA metabolism.
Keywords:
3D-Skin model
; Th2 cytokines
; skin lipidomics
; JAK/STAT
; Atopic Dermatitis
1. Introduction
Atopic dermatitis (AD) is an immune disorder characterized by a Th2-predominant inflammation. Although AD is a multifactorial disorder, a major feature of the disease is a dysfunction of the epidermal permeability barrier (EPB) in the stratum corneum (SC). In AD, EPB impairment is caused by a disruption in the epidermal proteins and lipids produced by keratinocytes, which can lead to critical features of AD skin [1]. For quite some time, experts have debated whether the impairment of the skin barrier is a direct consequence of the TH2 cytokines environment [2], genetic predisposition, altered microbiome, or environmental insults [3]. It is widely recognized that the proteins filaggrin (FLG), loricrin (LOR), and involucrin (IVL), which are essential for proper skin differentiation, are suppressed by the cytokines IL-4 and IL-13. These proteins are notably decreased in acute AD skin lesions [4,5,6,7,8]. However, the mechanisms responsible for the lipid changes observed in AD still need to be fully understood.
We hypothesized that Th2 cytokines exacerbate barrier abnormalities by disrupting the process underlying barrier lipid formation. This new study using 3D human epidermal equivalents sheds light on the potential role of the JAK/STAT axis in this process, specifically on lipid metabolism of the skin barrier.
Th2 cytokines bind to the IL-4Rα/IL-13Rα1 receptor complex on keratinocytes, activating the JAK/STAT signaling pathway. The STAT family consists of seven transcription factors, each phosphorylated and activated by different members of the JAK family [9,10]. STATs dimerize and translocate from the cytosol into the nucleus, where they regulate gene transcription. The JAK/STAT pathway is essential in AD’s exaggerated Th2 cell response [10]. JAK1- and JAK3-associated receptors modulate the IL-4 signaling pathway via phosphorylation of STAT3, STAT5, and STAT6 [9]. In turn, IL-4 potentiates Th2 cell differentiation, stimulating the release of additional cytokines [10]. Several studies have demonstrated the benefits of inhibiting the JAK/STAT pathway in AD [11,12,13]. Most JAK inhibitors screened for AD treatment affect the IL-4 pathway [14].
Tofacitinib is a small molecule that modulates critical cytokine signals in the progression of immune and inflammatory processes and affects innate and adaptive immune responses. Tofacitinib is an inhibitor of JAK1 and JAK3 and, to a lesser extent, of JAK2 [15]. Improvement in Eczema Area and Severity Index (EASI) scores has been observed after four weeks of topical treatment with tofacitinib [16]. Studies in vitro and in vivo show improved keratinocyte differentiation and EPB function [17]. Tofacitinib has been proven to modulate the activity of the JAK/STAT pathway in keratinocytes [18].
Emerging evidence suggests the involvement of JAK/STAT pathway in Th2 cytokine-mediated changes in the epidermal lipid profile of EPB [19]. Our study aimed to investigate the role of the JAK/STAT axis in the deregulation of the lipid homeostasis induced by Th2 cytokines.
2. Materials and Methods
2.1. Materials
The immortalized human keratinocyte cell line Ker-CT (ATCC® CRL-4048TM) was purchased from ATCC (Manassas, VA, USA). M154, calcium chloride (0.2 M), human keratinocyte growth supplements (HKGS), L-glutamine (2 mM), penicillin (100 u/mL), streptomycin (100 µg/mL), fetal bovine serum (FBS), trypsin/EDTA and D-PBS were purchased from Invitrogen Technologies (Monza, Italy). AurumTM Total RNA Mini kit, SYBR Green PCR Master Mix and Bradford reagent were from Bio-Rad (Milan, Italy). RevertAidTM First Strand cDNA synthesis kit was from Thermo Fisher Scientific (Monza, Italy). IL-4 and IL-13 were from Peprotech (Cranbury, NJ, USA). GAPDH antibody (G9545) was from Sigma-Aldrich (Milan, Italy). The antibodies for STAT1 (#14994), phospho-STAT1 (Tyr701) (#9167), STAT3 (#9139), phospho-STAT3 (Tyr105) (#9145), STAT6 (#9362), phospho-STAT6 (Tyr641) (#9361), secondary anti-mouse IgG HRP-conjugated antibody and anti-rabbit IgG HRP-conjugated antibody were purchased from Cell Signaling (Danvers, MA, USA). The anti-IVL (ab53112), anti-FLG (ab24584), anti-cytokeratin 10 (ab76318) and anti-LOR (ab85679) antibodies were purchased from Abcam (Cambridge, UK). The anti-ELOVL1 (NBP312302) was purchased from Novus Biologicals™ (Centennial, CO, USA). Amersharm ECL Western Blotting Detection Reagent was from GE Healthcare (Buckinghamshine, UK). Protease inhibitor cocktail was from Roche (Mannheim, Germany). RIPA lysis buffer, broad spectrum protease inhibitor cocktail, and broad-spectrum phosphatase inhibitor cocktail were from Boster Biological Technology Co (Pleasanton, CA, USA).
2.2. Chemicals
Deuterated Ceramide LIPIDOMIX® Mass Spec Standard Solution, EquiSPLASH™ LIPIDOMIX® Mass Spec Standard Solution and N-palmitoyl-d31-D-erythro-sphingosine (d31-Cer16:0, MW 569.1) were purchased from Avanti Polar Lipids (Alabaster, Alabama, US). Deuterated cholesterol-2,2,3,4,4,6-d6 (d6-CH MW 392), deuterated cholesterol sulfate sodium salt (d7-CHS, MW 495) and hexadecanoic-9,9,10,10,11,11,12,12,13,13,14,14,15,15,16,16,16-d17 acid (d17-PA, MW 273), glyceryl trihexadecanoate-d98 (d98TG 48:0, MW 906) and n-hexadecyl-1,1,2,2-d4 hexadecanoate-16,16,16-d3 (d7WE, MW 488) were purchased from CDN Isotopes Inc., (Pointe-Claire, Quebec, Canada). Details on the internal standards used are reported in Table S1. HPLCMS-grade ethyl acetate, acetone and chloroform were purchased from Carlo Erba (Milan, Italy). HPLCMS-grade acetonitrile, isopropanol and methanol were purchased from Biosolve (Chimie SARL, Dieuze, France; BV, Valkenswaard, Netherlands). Ultra-HPLCMS-grade water was purchased from LiChrosolv by Merck (Darmstadt, Germany). The butylated hydroxytoluene (BHT) and the mobile phase modifiers ammonium formate (NH4COOH) and ammonium fluoride (NH4F), were purchased from Sigma Aldrich (Milan, Italy). The mass calibration solution was prepared in acetonitrile from Agilent Technologies Tuning mix (HP0321 solution, Agilent Technologies, Santa Clara, CA, USA).
2.3. Culture of 3D Epidermal Equivalents and Stimulation with Cytokines and the JAK1/3 Inhibitor
The immortalized human keratinocyte Ker-CT cell line was maintained at 37°C under 5% CO2 in the defined medium M154 with HKGS, 2 mM L-glutamine, 100 u/mL penicillin, 100 µg/mL streptomycin, and 100 µM CaCl2. For routine cell cultivation cells were passaged when the 60-70% confluence was reached. Ker-CT cell line was used to generate 3D human epidermal equivalents (HEEs). Briefly, Ker-CT were seeded on cell culture inserts (Thermo Scientific, Roskilde, Denmark; 0.4 µm pore size; 2x105 cells per insert) and maintained submerged for 3 days in CnT-Prime Epithelial Culture Medium (CnT-PR) (CellnTEC, Bern, Switzerland) and switched in CnT-Prime 3D Barrier Medium (CnT-PR-3D) in an air-liquid condition for 12 days. Fresh medium was replaced every alternate day. To reproduce in vitro the effects of Th2 cytokines observed in AD skin, IL-4 (10 ng/mL) and IL-13 (10 ng/mL) were added during the last 5 days of air-liquid culture. HEEs were treated with Th2 cytokines in the absence or presence of 2 µM tofacitinib diluted in the culture medium 1 hour before the addition of cytokines. HEEs samples were processed for gene and protein expression analyses, lipidomic profile and immunohistochemistry. For routine histological procedures samples were formalin-fixed and paraffin-embedded for hematoxylin and eosin (H&E) staining, morphometry and immunofluorescence analyses.
2.4. Isolation of RNA and Analysis of mRNA by Real-Time RT-PCR
Total RNA was isolated from 3D HEEs samples using the AurumTM Total RNA Mini kit according to the manufacturer’s procedures. Total RNA samples were stored at -80°C until use. Following DNAse I treatment, cDNA was synthesized using a mix of oligo-dT and random primers and RevertAidTM First Strand cDNA synthesis kit according to the manufacturer’s instructions. Real-time RT-PCR was performed in a total volume of 10 μL with SYBR Green PCR Master Mix and 200 nM concentration of each primer. Sequences of all primers used are shown in Table S2. Reactions were carried out in triplicates using a CFX96 Real Time System (Bio-Rad Laboratories Srl). Melting curve analysis was performed to confirm the specificity of the amplified products. The relative expression of mRNA was normalized to the expression of GAPDH mRNA by the change in the Δ cycle threshold (ΔCt) method and calculated based on 2-ΔCt. The cycle time (Ct) read of GAPDH mRNA confirmed that the expression level of the gene was stable in all treatment groups.
Table S3 reports mean values and standard deviation (SD) of fold change (FC) of mRNA expression of inflammatory and lipid genes in treated HEEs compared to vehicle. Results were expressed as the FC between treatment and vehicle (taken as 1-fold). Data represented the mean ± SD of three independent experiments.
2.5. Western Blot Analysis
HEEs were lysed in RIPA buffer supplemented with a protease/phosphatase inhibitor cocktail and then sonicated. Total lysates were centrifuged at 12.000 rpm for 10 minutes at 4°C and then stored at -80°C until analysis. Following spectrophotometric protein measurement, equal amounts of protein were resolved on acrylamide SDS-PAGE and transferred onto a nitrocellulose membrane (Amersham Biosciences, Milan, Italy). Protein transfer efficiency was checked with Ponceau S staining (Sigma-Aldrich). Membranes were first washed with water, blocked with EveryBlot Blocking Buffer (Bio-Rad Laboratories Srl, Milan, Italy) for 10 min at room temperature and then treated overnight with primary antibodies at 4°C, according to instructions. Secondary anti-mouse or anti-rabbit IgG HRP-conjugated antibodies were used. Antibody complexes were visualized using enhanced chemiluminescence (ECL). A subsequent hybridization with anti-GAPDH was used as the loading control. Protein levels were quantified by measuring the optical densities of specific bands using UVITEC Imaging System (Cambridge, UK). Results were expressed as the FC relative to vehicle (taken as 1-fold). Data represented the mean ± SD of three independent experiments.
2.6. Histology, Morphometry and Immunofluorescence Analysis of HEEs
For histological and morphometric analyses, de-paraffinized HEEs sections were stained with hematoxylin and eosin (H&E). Serial sections were analyzed by recording stained images using a cooled CCD color digital camera (Zeiss, Oberkochen, Germany). The blue edition of the Zen 2.6 software (Zeiss) was used for the evaluation of epidermal and SC thickness. At least 100 measurements were taken on the images acquired under the different experimental conditions for either the epidermis or the SC. Sample sections were analyzed along their entire length. The results were expressed as the average thickness value ± SD obtained from three different experiments. For the immunofluorescence analysis, sections were dewaxed in xylene and rehydrated through graded ethanol in PBS. The antigen retrieval was obtained by heating the sections at 95.7°C in slightly acidic conditions (pH 6). Then the sections were blocked for 15 min with 5% normal goat serum in PBS and incubated overnight at 4°C with the following primary antibodies: anti-IVL (1:200 in PBS), anti-LOR (1:300 in PBS), anti-FLG (1:200 in PBS). The primary antibodies were visualized by incubating the sections for 2 h at room temperature with the following secondary antibodies: anti-rabbit IgG-Alexa Fluor 555 conjugated antibody (1:800 in PBS) and anti-mouse IgG-Alexa Fluor 488 conjugated antibody (1:800 in PBS) (Cell Signaling). Sections were mounted using ProLong mounting with DAPI (TermoFisher). For the immunohistochemical staining of ELOVL1, the tissue sections were dewaxed, processed for antigen-retrieval by heating at 95.7°C in alkaline solution (pH 9), and then incubated with the primary antibody (1:200 in PBS). The staining was visualized by the Thermo Ultravision Quanto Detection System HRP, using 3,3′-diaminobenzidine as substrate chromogen. All the sections were counterstained with hematoxylin. Images of stained sections were recorded using a CCD camera on a Zeiss microscope (Axioskop 2 Plus) and the signal intensity was quantified using Zeiss Zen 2.6 (blue edition) software for image analysis.
2.7. Lipid Extraction
The epidermal sheet was isolated from the insert following incubation overnight with dispase. HEEs were dried on absorbent paper, then, the epidermal sheet was extracted with water/methanol/chloroform (1/3.32/1.66 v/v/v) in presence of a mixture of deuterated internal standards, which included deuterated cholesterol, fatty acids (FAs), ceramides, triglycerides (TGs) and phospholipids (the latter ones from the EquiSPLASH-LIPIDOMIX) in 20 µL of a methanol solution containing BHT 1,2 mM to prevent autoxidation. The lipid extract was dried under nitrogen flow and suspended in 200 µL chloroform/methanol 2/1 v/v prior to analysis.
2.8. GCMS Analysis
GCMS served the quantitative measurement of free FAs (FFAs), fatty alcohols (FOHs), and cholesterol, following derivatization. Specifically, the analyzed saturated FFAs (SFAs) were 12-26 carbon atoms long. Four branched SFAs (iso and anteisobranched FAs) with carbon number between 15 and 17 were also detected. Ten monounsaturated FAs (MUFAs) with chain length between C14 and C24 and the polyunsaturated FA (PUFA) linoleic acid (FA 18:2) were detected. Twenty µL of the lipid extract obtained from the 3D epidermis models were dried under nitrogen and then derivatized with 40 µL BSTFA added of 1% trimethylchlorosilane in pyridine. The reaction was carried out at 60 °C for 60 minutes to produce the trimethylsilyl (TMS) derivatives of most lipids. The GCMS analysis was performed with the 8890 GC system combined with the 5977B Series MSD single quadrupole (Agilent Technologies, CA, USA). Helium was used as the carrier gas at the flow rate 1.2 mL/min. The analysis was conducted on the HP-5MS UI fused silica column (30 m x 0.250 mm internal diameter x 0.25 µm film thickness, chemically bound with a 5%-phenyl-methylpolysiloxane phase (Agilent Technologies, CA, USA). The GC oven program was as follows: initial temperature 80°C, held for 2 min, 280°C at 33 min, 310°C to final run time of 49 minutes. Samples were acquired in scan mode following EI ionization [20].
2.9. LC Separations
Reversed Phase-High Performance LC (RP-HPLC) was applied to the separation of lipids with a relatively broad range of hydrophobicity [21] (Camera et al., 2010). TGs and diglycerides (DGs) were detected under positive electrospray ionization (+ESI) conditions; ceramides, cholesterol sulfate, and long chain SFAs (27-30 carbon atoms) were analyzed under negative ESI (-ESI) conditions. Hydrophilic Interaction Liquid Chromatography (HILIC) was used to separate polar and hydrophilic lipids. Quantification of phosphatidylcholines (PCs), lysophosphatidylcholines (LPCs) and sphingomyelins (SMs) was performed in +ESI mode. Phosphatidylethanolamines (PEs), ether-linked phosphatidyl-ethanolamine (PE O-), phosphatidylinositols (PIs) and phosphatidylglycerols (PGs) were detected in -ESI mode. RPLC separation was conducted on the Infinity II 1260 series HPLC system equipped with a degasser, a quaternary pump, an autosampler and a column compartment (Agilent Technologies, CA, USA). The RP-HPLC separation was performed using the Zorbax Eclipse Plus C18 column (2.1 x 50 mm, 1.8 µm particle size) (Agilent Technologies, CA, USA). The maximum operating pressure was 600 bar/9000 psi. Cell extracts were eluted with a binary gradient of (A) 0.2 mM NH4F in water (18.2 Ω) and (B) 0.2 mM NH4F in methanol/isopropanol 80/20 [22]. Following a hold time of 2 min in 40% B, the gradient 40-99% occurred between 2.0 and 36.0 min; 99% B was held from 36.0 to 46.0 min. The mobile phase returned to 40% B between 46.0 and 48.0 min. A 10 min post-run time of 40% B was included. The column was thermostated at 60°C, the flow rate was 0.3 mL/min, and the injection volume was 0.6 µL and 1 µL, in +ESI and -ESI modes, respectively. HILIC separation was performed with a HALO HILIC column, 2.1 x 50 mm, 2.7 µm particle size, with maximum operating pressure at 600 bar/9000 psi (Advanced Materials Technology, AZ, USA). The column temperature was set at 40°C. The mobile phase consisted of (A) aqueous solutions of 5 mM NH4COOH in water (18.2 Ω) and (C) acetonitrile. The elution program was 98% C, 0-1.0 min; 98-80% C, 1.0-18.0 min; 80% C, 18.0-20.0 min; 80-98% C, 20.0-21.0 min, 98% C, 21.0-22.0 min. A 10 min post run of the initial condition was added. The mobile phase flow rate was 0.4 mL/min, the injection volume was 0.4 µL and 1 µL in +ESI and -ESI modes, respectively.
2.10. HRMS
The HPLC instrument was connected by the ESI Dual Agilent Jet Stream (AJS) interface with the 6545 Quadrupole Time of Flight (QTOF) as the mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). Gaseous nitrogen was used for both nebulization and desolvation processes. The ion source gas temperature was set at 200°C and the flow rate at 12 L/min; the nebulizer pressure was 40 psi. Sheath gas temperature was set at 350 °C; sheath gas flow rate was 12 L/min. The capillary voltage parameter was 4000. The fragmentor voltage was 120 V and the skimmer 40 V. Data independent acquisition (DIA) was performed for a preliminary investigation in all-ions MS/MS mode, at three collision energy (CE) values, i.e., 0, 20, and 40 eV. Data dependent acquisition (DDA) was accomplished with targeted MS/MS for structural characterization of lipids. The m/z range for MS and MS/MS was 59-1700 at a mass resolving power of 40.000. Internal mass calibration for accurate mass measurement used m/z 121.0509 and m/z 922.0098 in +ESI; reference ions in –ESI were m/z 112.9856 and m/z 966.0007 using NH4COOH; m/z 119.0363 and m/z 940.0015 using NH4F.
2.11. Data Processing and Statistical Analysis
Data derived from Western blot analysis, Real-Time RT-PCR, histology, morphometry and immunofluorescence analysis were represented as mean ± SD of three independent experiments. The values were expressed as relative to the vehicle (taken as 1). Statistical significance was assessed using paired Student’s t-test or ANOVA followed by Tukey’s multiple comparisons test using GraphPad Prism (GraphPad Software). The minimal level of significance was p<0.05. GCMS data were acquired using MassHunter GC/MSD 5977B acquisition software and processed with MassHunter Workstation Software Quantitative Analysis (version 10.1). LC-HRMS data were acquired and deconvoluted using the MassHunter Data Acquisition Software (B.09.00, Agilent Technologies). The data acquired by LCMS were processed with Agilent MassHunter Workstation Profinder (version 10.0). All data were derived by normalizing the response of the individual lipid by the response of the same class labelled internal standard (e.g., FFAs vs d17-PA, cholesterol vs d6-cholesterol, etc.) and multiplied by the deuterated internal standard pmole. In turn, mole amounts were normalized by the protein content and reported as pmol/mg protein.
The multivariate statistical analysis was performed by Agilent MassHunter Mass Profiler Professional (MPP) (version 15.1). The mole amounts calculated for the lipid species were transferred into a template (.csv) and imported into the software to determine relevant differences of compounds across groups. The data in linear scale were log2 transformed and a baseline to the same-experiment controls was applied. To create interpretations, samples were divided into groups according to the specific treatment, i.e., vehichle, tofacitinib, Th2-cytokines, and their combination. One-way analysis of variance (one-way ANOVA) was performed. FC ratio was calculated in selected conditions. Tukey HSD was used as the post hoc test to explore differences between treatments. For the multiple testing correction Storey’s approach was chosen using the bootstrap method with a q-value cut-off ≤ 0.05. Data were further analyzed using XLSTAT 2020.1.2 (Addinsoft, New York, USA). Continuous variables were represented as mean ± SD. Significant differences between and within multiple groups were examined using Kruskal-Wallis. The Dunn method was used for multiple pairwise comparisons and Bonferroni’s correction of the significance level was applied. P-values were calculated using the approximation of the distribution of K by a chi-square distribution with (k-1) degree of freedom. Differences and correlations were considered statistically significant with p ≤ 0.05.
3. Results
3.1. Effects of JAK/STAT Inhibition by Tofacitinib on Th2 Cytokine-Mediated Changes in Epidermal Morphology and Barrier Protein Expression
To reproduce the effects of Th2 cytokines in vitro, human epidermal equivalents (HEEs) were treated with IL-4 and IL-13, each at a concentration of 10 ng/mL, for five days. Conventional H&E staining of the 3D HEEs treated with the citokines revealed histological features resembling AD epidermis in vivo. Spongiosis was observed due to the expanded space between adjacent keratinocytes (Figure 1A). Morphometric analysis and quantitative evaluation of the thickness of both viable epidermis and SC showed a significant increase in the thickness of epidermal portion in Th2-treated HEEs compared to control HEEs. Tofacitinib partially counteracted the increased epidermal thickness induced by Th2 cytokines. Compared to Th2 alone, increased SC thickness was observed when the Th2 cytokines were combined with tofacitinib (Figure 1B). No significant modulation of the mRNA expression of the early differentiation marker K10 and the late differentiation markers LOR and IVL was detected after stimulation with Th2 cytokines (Figure 1C). According to previous evidence [23,24], Th2 cytokines caused a significant decrease in the mRNA levels of FLG and CASP14, which is the enzyme crucial for the FLG degradation process [25]. Tofacitinib treatment abolished the reduction of FLG and CASP14 mRNA observed in Th2-treated HEEs (Figure 1C). Consistent with the mRNA levels, protein quantification by both Western blot (Figure 1D) and immunofluorescence analyses (Figure 1E,F) showed a significant decrease in FLG expression in Th2-treated HEEs, which was prevented by tofacitinib. A similar result was obtained when profilaggrin (pro-FLG) and FLG expression were analyzed separately in the stratum granulosum (SG) and the stratum corneum (SC) (Figure 1G).
3.2. JAK/STAT Inhibition Counteracts the Induction of Inflammatory Genes in Th2-Treated Epidermal Equivalents
To evaluate the effect of tofacitinib on STAT signalling, Western blot analysis of native STAT1, STAT3, and STAT6 and their phosphorylated forms was performed (Figure S1). As expected, the stimulation with Th2 cytokines upregulated the phosphorylation of STAT3 and STAT6; tofacitinib efficiently counteracted their activation. No significant modulation of STAT1 phosphorylation was observed after Th2 exposure (Figure S1). The expression of genes involved in the inflammatory response was evaluated in HEEs and Th2-treated HEEs in the presence of tofacitinib. The compound hindered the Th2-mediated increase in the mRNA expression of IL-1α, IL-1β, IL-6, IL-8 cytokines, CCL26 chemokine, and podoplanin (PDPN), a keratinocyte glycoprotein upregulated via JAK/STAT signaling (Figure 2A) [26].
3.3. Modulation of Lipid Genes by Th2 Cytokines and Counteracting Effects of Tofacitinib
Next, we investigated the activity of IL-4/IL13 in the synthesis of sphingolipids. Th2 cytokines modulated several genes involved in lipid metabolism (e.g., elongation of FFAs and synthesis of sphingolipids), as determined by Real-Time RT-PCR. A significant reduction in the mRNA levels of FA elongases (ELOVLs) 1, 3, and 4 was observed in Th2-treated HEEs (Figure 2B). Co-treatment with tofacitinib prevented the reduction of ELOVL3 and ELOVL4 mRNA induced by Th2 cytokines. The decrease of ELOVL1 expression following Th2 cytokines and the recovery in the presence of tofacitinib were also observed at the protein level (Figure 2C–E). The expression of genes involved in the biosynthesis of sphingolipids, such as serine palmitoyltransferase (SPT), and sphingolipid delta(4)-desaturase (DEGS2), showed an opposite response to Th2 treatment. Th2 cytokines decreased and increased the mRNA expression of SPT and DEGS2, respectively. Co-treatment with tofacitinib was able to counteract the Th2-mediated effects (Figure 2F). The mRNA levels of carbonic anhydrase 2 (CA2) and the transcription factor Peroxisome Proliferator-Activated Receptor-Gamma (PPARγ), genes involved in the lipid metabolism [27], were up- and down-regulated, respectively, after Th2 exposure. Tofacitinib counteracted the effects induced by the Th2 cytokines (Figure 2F).
3.4. Alteration of Lipid Profiles Induced by Th2 Cytokines and Modulating Effects of Tofacitinib
To understand the impact of Th2 cytokine-mediated signaling and the significance of JAK/STAT pathway inhibition in the 3D epidermal models, lipid extracts were analyzed using a combination of analytical approaches. The abundance profiles of FFAs, FOHs, and cholesterol in the HEEs were determined by GCMS. Polar and neutral lipid profiles were determined by LCMS. One-way ANOVA was performed on the lipidomics data to compare effects between groups. The test retrieved 51 lipid species that showed statistically significant differences among the 300 target species. Table S4 shows the results of one-way ANOVA with Tukey HSD post-hoc multiple comparisons. Normalized abundance (pmol/mg protein) was compared to vehicle and expressed as the average of six samples per group. To explore relationships and correlations between lipids, their expression profiles were organized in a hierarchical clustering (Figure 3A), showing trends in lipid modulation characteristics of each treatment. The dendrogram indicates linkage among regulated species. The clustering of individual lipids according to their similarity in changes of expression supported the identification of three principal groups.
The first cluster is characterized by an overall lowering effect of Th2 cytokines, which was unmodified upon JAK/STAT inhibition. Lower abundance of hexosylceramides (HexCers), ultra-long FFAs, and saturated DGs was observed in all treatments. Interestingly, the reduction in HexCers abundance occurred to a statistically significant extent with tofacitinib alone (Table S5). This finding may indicate an effect of tofacitinib on the recycling pathways of ceramides [28]. The Th2 cytokines significantly downregulated long-chain SFAs (Table S6). The effects of tofacitinib were not remarkable (Figure 3B). The abundance of the detected DGs was significantly lower than the control regardless of the treatment (Table S7).
The second cluster of the heatmap is defined by an induction stimulated by Th2 cytokines. It consists of LPC 20:4, sterol-like 458, some DGs and SMs, and PCs. Its overall abundance is lower in HEEs treated with tofacitinib compared to vehicle. Sterol-like species, whose identification awaits characterization, were observed together with their precursor cholesterol, whose concentration was only slightly affected, although tending to be lower in Th2 HEEs (Figure S2A). Phospholipids account for the major components of the plasma membrane. Phosphocholine-containing lipids depicted in the heat map, i.e., PCs, LPCs, and SMs, are important components of the plasma membrane and precursors in lipid synthesis [29] (Tables S8 and S9). Several members of the PC family were significantly modified in HEEs treated with Th2 cytokines (Figure 3C). Tofacitinib per se exerted no effect on the PC profile of HEEs. Irrespective of the direction of the modulation by Th2 cytokines, these effects were abrogated by tofacitinib. Interestingly, the PCs members that were decreased and increased by Th2-type cytokines were dominated by the MUFAs FA 16:1 and FA 18:1 side chain (Table S9). Other phospholipids, i.e., PEs, ether PEs, PGs and PIs were minimally affected by the treatments (Table S10).
The third cluster of the heatmap represents all the species that were significantly downregulated by Th2 cytokines and that the inhibition of the JAK/STAT pathway by tofacitinib was able to revert the lipids-lowering effects of Th2 cytokines. Palmitoleic acid (FA 16:1n-7), a MUFA with 16 carbon atoms and a double bond (DB) at the delta-9 position, and FOH 17:1 were significantly reduced in the Th2-treated HEEs (Table S6). The panel of metabolites determined by GCMS included several species that showed decreased concentration (pmol/mg protein) in all the treatments, although significance was not reached (Table S6). As shown in Figure 3D, tofacitinib abrogated the Th2 effects on FA 16:1n-7. The observation of a lower abundance of FA 16:1-containing PCs could be related to the significantly reduced concentration of FA 16:1n-7 in its free form.
The amounts of two other sterol metabolites, tentatively assigned as lanosterol (sterol-like 498) and dihydrolanosterol (sterol-like 500) based on their EI-MS fragmentation spectrum, showed a similar trend (Figure S2B,C).
The most significant effects on TGs are reported in Figure 3D and Table S11. Interpretation of the MS/MS fragmentation spectra supported the prevalence of FA 16:1 as the bound FA in one or two sn positions of the TGs depleted by Th2 cytokines contained FA 16:1 (Table S11) [29].
The observation of significantly depleted TGs suggested a disruption of lipid storage. Lipid droplets (LDs) are ubiquitous subcellular structures that serve the sequestration of TGs. We determined the mRNA levels of PLIN1 and PLIN2, proteins involved in the formation and regulation of intracellular LDs [30,31]. A significant reduction in the expression of both PLIN1 and PLIN2 was observed in Th2-treated HEEs, supporting the hypothesis of a Th2-driven depletion of the TG storage. Co-treatment with tofacitinib reversed the reduction in PLIN1 and PLIN2 mRNA expression caused by Th2 cytokines alone (Figure 3E). Since data in the literature show that FA oxidation is promoted in AD models, we preliminarily explored the hypothesis that in our model, FAs arising from TGs could undergo β-oxidation in mitochondria and peroxisomes [32,33]. To this end, we analyzed the mRNA expression of CPT1A, ACAT1, ACADS, genes encoding proteins involved in mitochondrial FA oxidation, and ACOX1 gene associated with peroxisomal FA oxidation [9,33,34,35,36]. Th2 cytokines significantly induced CPT1A and ACAT1 mRNA levels; co-treatment with tofacitinib abolished this effect (Figure 3F). ACADS and ACOX1 expression showed the same trend without reaching statistical significance. These results support the hypothesis of a link between the increased mitochondrial function observed in AD epidermis [33,37] and the increased utilization of FAs as energetic substrates under Th2 signaling.
Changes in concentrations of cholesterol sulfate and ceramides, although apparent, did not reach significance (Table S12). The interpretation of the profiles of ceramide abundance in HEEs treated with tofacitinib and Th2 cytokines and their combination was not obvious. As expected, there was a trend towards decreased levels of ceramides in HEEs treated with Th2 cytokines (Table S12). Nevertheless, CerNP species with 38 to 44 carbon atoms increased after Th2 cytokine treatment, as expected due to the significant upregulation of DEGS2. Ceramides bearing linoleic acid ester bound to omega hydroxylated long-chain FAs have been characterized in the epidermis (EOS, EODS, EOP, EOH) [38]. Several acylceramides showed a decrease in Th2-HEEs, which was recovered with tofacitinib, but significance was not reached. The changes in the concentration of two Cer[EOH] species, which decreased upon Th2 and recovered in presence of tofacitinib approached significance (Table S12).
Since we expected to observe more pronounced modulation of ceramide profiles, also supported by the downmodulation of SPT mRNA, we undertook the analysis of ceramides according to their sphingoid base (SB). First, we checked the abundance of ceramides containing the sphingoid bases (SBs) sphingosine (S) and phytosphingosine (P) with chain length between 16 and 24, and between 16 and 20 carbon atoms, respectively, bound to a non-hydroxylated fatty acid (N). The results of the abundance of the main SBs were consistent with the overall profiles of the ceramides (Table S12).
4. Discussion
Skin barrier defects in AD result from the dysregulation of multiple pathways. The incretion of Th2 cytokines in AD is at the interface between immune activation and barrier disruption. Lipids play a pivotal role in the water-holding capacity of the epidermis, and they are key components of the skin barrier, a complex system in which different compartments can be distinguished: physical, chemical, immunological and microbiological barrier [39]. In AD, the hydrophobic barrier is not properly formed, due to mechanisms that have been only partly eviscerated.
In this study, we report the effects of Th2 cytokines on lipid distribution using 3D human epidermal equivalents. We first assessed whether our 3D epidermal model mimicked the changes associated with stimulation by the cytokines IL-4 and IL-13. To this end, we performed morphometric analysis and evaluated specific differentiation markers relevant to the disease phenotype. As expected, FLG expression was reduced in HEEs treated with Th2 cytokines. In addition, we demonstrated a decrease in both pro-FLG and FLG localized in the SG and SC, respectively. However, we did not observe the downregulation of IVL and LOR, contrary to what has been shown in other studies [8]. These discrepancies may be due either to differences in both the experimental models and the doses of cytokines used. The most innovative aspect of this study is the in-depth analysis of the lipid profile after stimulation with the cytokines IL-4 and IL-13 using lipidomic strategies. Despite some limitations due to differences in lipid composition and organization when compared to native skin, e.g., overall shorter chain length of FFAs, the 3D epidermal equivalent is a suitable model to study barrier properties in pathophysiological conditions reproduced in vitro [39,40,41]. Activation of the JAK/STAT axis by phosphorylation drives alterations in keratinocyte differentiation and abnormalities in skin lipids.
Ceramides, FFAs and cholesterol and its conjugates, i.e., cholesterol sulfate and cholesterol esters are key components of the permeability barrier in the uppermost epidermal layers. These three major classes are present in equimolar abundance. Alterations in their molar ratio have been implicated in several skin diseases, particularly AD [42]. The hallmarks of lipid abnormalities in the SC of AD [43,44,45,46] include shortening of the chain length of FAs bound in ceramides and a decrease in the absolute and relative abundance of acylceramides [47]. IL-4 markedly reduces the levels of long-chained ceramides in the epidermis by downregulating the expression of serine-palmitoyl transferase-2 (SPT2), acid sphingomyelinase (aSMase), and β-glucocerebrosidase (GCase) [48]. Ceramides play a crucial role in maintaining the homeostasis of the epidermal barrier and are also involved in cell signaling, proliferation, differentiation and apoptosis in human epidermis [42]. Ceramides are generated de novo in the endoplasmic reticulum by the condensation of serine and palmitoyl-CoA catalyzed by SPT [49,50].
Our results showed that Th2 cytokines decreased the mRNA expression of SPT. Although there was a trend towards the reduction of SPT end products, the effects were not straightforward. The experimental conditions used in this study, in particular the air-liquid phase, favored the detection of lipid changes that preceded the apparent change in ceramide abundance. Nevertheless, inhibition of the JAK/STAT pathway by tofacitinib tended to restore cytokine modulation of both SPT gene and ceramides, suggesting a potential benefit of JAK/STAT blockade in reversing lipid perturbations induced by Th2 cytokines.
During keratinocyte differentiation, the total amount of ceramides substantially increases [42,51]. To prevent intracellular ceramides from reaching cytotoxic levels, they are further processed into glucosylceramides and SMs and then transported into extracellular space [42]. The decrease we observed in ceramides induced by Th2 cytokines is even more pronounced in HexCers. HexCers include glucosylceramides, which are important components of the lamellar bodies, whose deficiency may contribute to a dysfunction of the epidermal barrier homeostasis. It is likely that the JAK/STAT3 pathway is implicated in the biotransformation of ceramides via ceramide-glucosyltransferase, a key enzyme in glycosphingolipid synthesis [52]. SMs were minimally altered. Sphingomyelin synthase 2 (SGMS2) produces DGs and SMs by transferring phosphocholine from PC to the ceramide terminal OH group [53]. We found an increase and a decrease in the intermediate products PCs and DGs, respectively. This may be an indication of a disturbance in the SGMS2 pathway, which deserves further investigation in future studies.
FAs play a crucial role in the formation and dynamics of biological membranes, and they are essential for cell metabolism and energy balance [54]. In our study, the absolute amounts of FFAs, including MUFAs, were profiled. Conflicting results have been reported in the literature regarding the abundance of FFAs in AD. Both increased abundance of MUFAs and decreased levels of FA 16:1 and FA 18:1 have been described, together with increased susceptibility to S. aureus infection and disruption of the epidermal barrier [55,56]. Furthermore, MUFA depletion correlates with skin dryness in AD. Both natural moisturizing factors (NMFs) and skin surface lipids contribute to skin hydration. Recently, we described a significant depletion of MUFAs in the skin surface lipids derived from both sebum and SC [57]. Although palmitoleic acid (FA 16:1n-7) decreased significantly following Th2 cytokines, the involvement of the SCD1 desaturase pathway in the observed depletion of FA 16:1n-7 was unclear due to unchanged SCD1 mRNA levels. However, it cannot be excluded that FA 16:1n-7 is also degraded by beta-oxidation [58]. Due to the importance of SCD1 in skin integrity, further investigation is needed to clarify the mechanisms causing FA 16:1n-7 depletion upon Th2 signaling [59]. The simultaneous addition of tofacitinib abrogated the delipidizing effects of Th2-cytokines. Indeed, we observed a significant recovery of FA 16:1n-7 when Th2 cytokines were co-administered with tofacitinib.
ELOVLs are responsible for elongating the FA chain by adding 2-carbon units. The ELOVL1 enzyme is involved in the elongation of FA 18:0 to FA 26:0 and FA 18:1 to FA 22:1 when activated by coenzyme A (CoA) binding. The ELOVL3 and ELOVL4 enzymes are involved in the elongation of saturated FA 16:0 to FA 22:0-CoA and ultra-long FAs (C26-36), respectively [60]. The observation of decreased expression of ELOVLs in 3D-HEEs treated with IL-4 and IL-13 is consistent with findings of their reduced expression in human SC from AD lesional areas [61,62]. Recent studies have identified significant metabolic changes in the SC and plasma of AD patients following treatment with dupilumab, a monoclonal antibody targeting the receptors for IL-4 and IL-13 [63,64]. Halting the IL-4/IL-13 signaling proved to revert abundance of short chain NS-ceramides to normal and to restore the relative and absolute abundance of EOS-ceramides. The observed improvement following dupilumab treatment supports the involvement of ELOVLs pathways in the downstream effects of IL-4/IL-13 signaling [64]. Inhibition of STATs phosphorylation by tofacitinib treatment restored the expression of ELOVL1, 3 and 4. The relationship between the levels of STAT6 and ELOVL3 and ELOVL6 has been reported [61]. Consistent with previous evidence, IL-4 and IL-13 promote the phosphorylation of STAT6 in human keratinocytes [24,65]. Inhibition of the JAK/STAT pathway by tofacitinib re-equilibrated the unbalanced lipid composition in human keratinocytes. However, the reported data are conflicting and the mechanisms underlying the lipid chain shortening process remain unclear. The conflicting data on the expression and regulation of ELOVLs is likely due to the different experimental approaches used (e.g., in vitro models, cytokine doses, treatment duration), each presenting limitations in mimicking the complexity of the AD skin microenvironment [62].
TG depletion is an initiating event and aggravating condition in AD [61,66]. The observation of decreased TGs is associated with the suppression of key genes involved in lipid synthesis, such as DGAT1, DGAT2, FADS1 and ELOVL1 [67]. In our study, we observed that the TGs species depleted upon exposure to Th2 cytokines through a JAK/STAT-dependent mechanism presented specific features, as demonstrated by the MS/MS data. A large body of evidence supports the role of JAK/STAT signaling in the regulation of metabolic processes underlying energy expenditure and the turnover of lipid stores [68]. Cultured human keratinocytes vary their lipid composition in a density-dependent manner. Lipid neosynthesis is active before keratinocytes reach confluence, resulting in the accumulation of TGs in post-confluent cultures [69]. Normally, TGs are stored in LDs [70]. However, the presence of LDs in keratinocytes has only been studied in the context of epidermal dysfunction. PLINs bind to the surface of LDs and have both structural and regulatory functions [71]. The role of PLINs in the epidermis has been little investigated. Although the modulation of PLIN1 and PLIN2 transcripts in the Th2-treated HEEs awaits clarification, it has been observed to follow the changes in the TGs levels in this study. The reservoir of TGs is plastic and provides a pool of fatty acyl residues for phospholipid biosynthesis [69,72]. Defects in the catabolism of TGs have been described in humans with ichthyosis-bearing mutations in the ABHD5/CGI-58 gene. Mice with a deficiency of genes involved in TG metabolism develop permeability barrier dysfunction. Defective TG metabolism results in severe disruption in the formation of acylceramides, essential for the build-up of the cornified lipid envelope in the epidermis [73]. Linoleic acid is mainly derived from TGs and is specifically incorporated into acylceramides [42].
There is evidence for metabolic relationships between glycerolipids, i.e., TGs and DGs, and PCs in mammalian cells. Specifically, the Kennedy pathway implicates the activation of phosphocholine with cytidine triphosphate (CTP), which is then transferred to DGs to produce PCs. Two enzymes of the Lands cycle, LPCAT1 and LPCAT2, synthesize PCs directly at the surface of the LDs where TGs are stored [74]. Our model showed a significant increase in PCs species and a significant decrease in TGs members when HEEs were treated with Th2 cytokines, suggesting a potential interaction between these lipid domains. Our findings support previous studies demonstrating a significantly high percentage of MUFAs bound in PCs, in AD [75]. The significant increase of PCs is in line with the evidence of PCs and phospholipid accumulation, which is an indicator of atopic pathogenic mechanism [66]. Indeed, it has been reported that although phospholipids are entirely degraded physiologically, they persist in AD epidermis [75]. This highlights the significance of phospholipid metabolism at the SG-SC interface for the potential release of FFAs in facilitating proper barrier formation. It also indicates how the inadequate metabolism of phospholipids and their resultant accumulation represent a hallmark of AD [66]. Therefore, reversing PCs accumulation represents a benefit of tofacitinib.
Cholesterol is fundamental to the vital function of mammalian cells. In cutaneous tissues, cholesterol is involved in the maintenance of barrier architecture and function and acts as a precursor for steroid synthesis. Cholesterol levels are finely regulated as even slight changes can have dramatic effects. While the HEEs treated with Th2 cytokines compensated for the Th2 effects on cholesterol, upstream precursors were affected, suggesting that the cholesterol biosynthetic pathway is a target of the Th2 signaling that might be controlled by JAK/STAT inhibition. It is important that future studies address the role of the PPARγ transcription factor in the lipid response to Th2 cytokines. Indeed, effects of perturbed PPARγ transcriptional activity may contribute, in different directions, to the overall mechanisms of action of tofacitinib.
In conclusion, our results highlight that inhibition of the JAK/STAT pathway effectively abrogates lipid perturbations induced by Th2 cytokines. These findings suggest that JAK/STAT inhibitors, especially if applied topically, can correct the epidermal barrier lipid abnormalities induced by Th2 cytokines and be of great benefit to AD management.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Effects of tofacitinib on STAT signalling (STAT1, STAT3, and STAT6) and their phosphorylated forms; Figure S2: Box plots of cholesterol and sterol like metabolites; Table S1: List of the deuterated internal standards used for extraction protocol and semi-quantitative analyses; Table S2: Forward (F) and reverse (R) primers used for the Real time RT-PCR analysis; Table S3: mRNA expression of inflammatory and lipid genes in treated HEEs compared to vehicle; Table S4: Abundance profiles of FAs, Sterol-like, HexCers, TGs, DGs, PCs, SMs, Pes in HEEs treated with tofacitinib and Th2 cytokines and their combination. Table S5: Abundance profile of HexCers in HEEs treated with tofacitinib and Th2 cytokines and their combination; Table S6: The panel of abundance of FFAs FOHs, cholesterol, and related metabolites; Table S7: The abundance of the detected DGs in HEEs treated with tofacitinib and Th2 cytokines and their combination; Table S8: LPCs and SMs abundance in HEEs treated with tofacitinib and Th2 cytokines and their combination; Table S9: Abundance of PCs members and molecular species characterized for candidate PCs in HEEs treated with tofacitinib and Th2 cytokines and their combination; Table S10: Abundance of PEs, PGs and PIs in HEEs treated with tofacitinib and Th2 cytokines and their combination; Table S11: TGs amounts and molecular species characterized for candidates TGs in HEEs treated with tofacitinib and Th2 cytokines and their combination; Table S12: Cholesterol sulfate, ceramides and of the selected SBs amounts in HEEs treated with tofacitinib and Th2 cytokines and their combination.
Author Contributions
E.C., G.C., and E.F. conceptualization; E.F., A.C., S.M., A.D.N., D.K., M.Z., M.M., G.B., E.C., and G.C. formal analysis; E.F., A.C., S.M., D.K., M.Z., G.B., M.M., and G.C. investigation; E.F., A.C., S.M., D.K., G.B., M.M., E.C., and G.C. writing–original draft; E.F., A.C., S.M., D.K., C.C., A.D.N., G.B., M.M., E.C., and G.C. writing–review & editing; E.C. funding acquisition.
Funding
This research was supported by the Global Medical Grants program sponsored by Pfizer. This work was supported through funding from the institutional ‘Ricerca Corrente 2024′ granted by the Italian Ministry of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the supporting agencies.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data described are contained within the manuscript.
Acknowledgments
We acknowledge the Global Medical Grants program provided by Pfizer and the National Institute of Health for the support received. The authors would like to thank Dr. Claudia Messina (Scientific Direction, San Gallicano Dermatological Institute-IRCCS) for her valuable support in the manuscript preparation.
Conflicts of Interest
E.C. and G.C. were, respectively, the principal investigator (PI) and the Co-PI of the research project funded by Pfizer. A.C. received the research fellowship granted by Pfizer.
Abbreviations
ACADS (Acyl-CoA Dehydrogenase Short Chain), ACOX1 (acyl-coenzyme A oxidase 1), ACAT1 (Acetyl-CoA Acetyltransferase 1), AD (atopic dermatitis), CA2 (carbonic anhydrase 2), CASP14 (caspase 14), CCL26 (C-C Motif Chemokine Ligand 26), CPT1A (carnitine palmitoyltransferase 1A), DEGS2 (sphingolipid delta(4)-desaturase), DGs (diglycerides), EPB (epidermal permeability barrier), EASI (Eczema Area and Severity Index), ELOVLs (fatty acids elongases), FFAs (free fatty acids), FLG (filaggrin), FOHs (fatty alcohols), GAPDH (Glyceraldehyde-3-Phosphate Dehydrogenase), GCMS (Gas chromatography-mass spectrometry), H&E (hematoxylin and eosin), HEEs (human epidermal equivalents), HexCers (hexosylceramides), HPLC (High Performance Liquid Chromatography), Hydrophilic Interaction Liquid Chromatography (HILIC), IL (interleukin), IVL (involucrin), JAK (Janus kinase), K10 (keratin 10), LOR (loricrin), LDs (lipid droplets), LPCs (lysophosphatidylcholines), MUFAs (monounsaturated fatty acids), PCs (phosphatidylcholines), PEs (phosphatidylethanolamines), PGs (phosphatidylglycerols), PIs (phosphatidylinositols), PDPN (Podoplanin), PLIN2 (Perilipin-2), PPARγ (Peroxisome Proliferator Activated Receptor gamma), RP-HPLC (Reversed Phase-HPLC), SB (sphingoid base), SC (stratum corneum), SFAs (saturated fatty acids), SG (stratum granulosum), SMs (sphingomyelins), SPT (serine palmitoyltransferase), STAT (signal transducer and activator of transcription), TGs (triglycerides), Th2 (T-helper2).
References
- Wolf, R.; Wolf, D. Abnormal epidermal barrier in the pathogenesis of atopic dermatitis. Clin Dermatol 2012, 30, 329-334.
- Gandhi, N.A.; Pirozzi, G.; Graham, N.M.H. Commonality of the IL-4/IL-13 pathway in atopic diseases. Expert Rev Clin Immunol 2017, 13, 425-437.
- Yang, G.; Seok, J.K.; Kang, H.C.; Cho, Y.; Lee, H.S.; Lee, J.Y. Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis. Int J Mol Sci 2020, 21, 2867. (accessed on Mar 25, 2024). [CrossRef]
- Karim, N.; Phinney, B.S.; Salemi, M.; Wu, P.; Naeem, M.; Rice, R.H. Human stratum corneum proteomics reveals cross-linking of a broad spectrum of proteins in cornified envelopes. Exp Dermatol 2019, 28, 618-622. (accessed on Jul 24, 2023). [CrossRef]
- Ishitsuka, Y.; Roop, D.R. Loricrin: Past, Present, and Future. International Journal of Molecular Sciences 2020, 21, 2271. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32218335. [CrossRef]
- Furue, M. Regulation of Filaggrin, Loricrin, and Involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic Implications in Atopic Dermatitis. Int J Mol Sci 2020, 21, 5382. (accessed on Jul 24, 2023). [CrossRef]
- Drislane, C.; Irvine, A.D. The role of filaggrin in atopic dermatitis and allergic disease. Annals of allergy, asthma, & immunology 2020, 124, 36-43. Available online:. https://doi.org/10.1016/j.anai.2019.10.008. [CrossRef]
- Kim, B.E.; Leung, D.Y.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol 2008, 126, 332-337.
- He, A.; Chen, X.; Tan, M.; Chen, Y.; Lu, D.; Zhang, X.; Dean, J.M.; Razani, B.; Lodhi, I.J. Acetyl-CoA Derived from Hepatic Peroxisomal β-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Molecular cell 2020, 79, 30-42.e4. Available online:. https://doi.org/10.1016/j.molcel.2020.05.007. [CrossRef]
- Bao, L.; Zhang, H.; Chan, L.S. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT 2013, 2, e24137.
- Gunduz, O. JAK/STAT pathway modulation: Does it work in dermatology? Dermatol Ther 2019, 32, e12903.
- Rodrigues, M.A.; Torres, T. JAK/STAT inhibitors for the treatment of atopic dermatitis. J Dermatolog Treat 2020, 31, 33-40. (accessed on Aug 3, 2023). [CrossRef]
- Chovatiya, R.; Paller, A.S. JAK inhibitors in the treatment of atopic dermatitis. J Allergy Clin Immunol 2021, 148, 927-940. (accessed on Aug 3, 2023). [CrossRef]
- Solimani, F.; Meier, K.; Ghoreschi, K. Emerging Topical and Systemic JAK Inhibitors in Dermatology. Front Immunol 2019, 10, 2847.
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542-550.
- Bissonnette, R.; Papp, K.A.; Poulin, Y.; Gooderham, M.; Raman, M.; Mallbris, L.; Wang, C.; Purohit, V.; Mamolo, C.; Papacharalambous, J.; Ports, W.C. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. British journal of dermatology (1951) 2016, 175, 902-911. Available online: https://api.istex.fr/ark:/67375/WNG-2B42NBR1-3/fulltext.pdf. [CrossRef]
- Nakashima, C.; Yanagihara, S.; Otsuka, A. Innovation in the treatment of atopic dermatitis: Emerging topical and oral Janus kinase inhibitors. Allergology International 2022, 71, 40-46. Available online:. https://doi.org/10.1016/j.alit.2021.10.004. [CrossRef]
- Srivastava, A.; Stahle, M.; Pivarcsi, A.; Sonkoly, E. Tofacitinib Represses the Janus Kinase-Signal Transducer and Activators of Transcription Signalling Pathway in Keratinocytes. Acta Derm Venereol 2018, 98, 772-775.
- Hatano, Y.; Adachi, Y.; Elias, P.M.; Crumrine, D.; Sakai, T.; Kurahashi, R.; Katagiri, K.; Fujiwara, S. The Th2 cytokine, interleukin-4, abrogates the cohesion of normal stratum corneum in mice: implications for pathogenesis of atopic dermatitis. Exp Dermatol 2013, 22, 30-35.
- Ludovici, M.; Kozul, N.; Materazzi, S.; Risoluti, R.; Picardo, M.; Camera, E. Influence of the sebaceous gland density on the stratum corneum lipidome. Sci Rep 2018, 8, 11500-7.
- Camera, E.; Ludovici, M.; Galante, M.; Sinagra, J.L.; Picardo, M. Comprehensive analysis of the major lipid classes in sebum by rapid resolution high-performance liquid chromatography and electrospray mass spectrometry. J Lipid Res 2010, 51, 3377-3388.
- Maiellaro, M.; Bottillo, G.; Cavallo, A.; Camera, E. Comparison between ammonium formate and ammonium fluoride in the analysis of stratum corneum lipids by reversed phase chromatography coupled with high resolution mass spectrometry. Sci Rep 2024, 14, 40. (accessed on Jan 8, 2024). [CrossRef]
- Amano, W.; Nakajima, S.; Kunugi, H.; Numata, Y.; Kitoh, A.; Egawa, G.; Dainichi, T.; Honda, T.; Otsuka, A.; Kimoto, Y.; Yamamoto, Y.; Tanimoto, A.; Matsushita, M.; Miyachi, Y.; Kabashima, K. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol 2015, 136, 667-677.e7.
- Clarysse, K.; Pfaff, C.M.; Marquardt, Y.; Huth, L.; Kortekaas Krohn, I.; Kluwig, D.; Lüscher, B.; Gutermuth, J.; Baron, J. JAK1/3 inhibition preserves epidermal morphology in full-thickness 3D skin models of atopic dermatitis and psoriasis. J Eur Acad Dermatol Venereol 2019, 33, 367. [CrossRef]
- Hoste, E.; Kemperman, P.; Devos, M.; Denecker, G.; Kezic, S.; Yau, N.; Gilbert, B.; Lippens, S.; De Groote, P.; Roelandt, R.; Van Damme, P.; Gevaert, K.; Presland, R.B.; Takahara, H.; Puppels, G.; Caspers, P.; Vandenabeele, P.; Declercq, W. Caspase-14 Is Required for Filaggrin Degradation to Natural Moisturizing Factors in the Skin. J Invest Dermatol 2011, 131, 2233-2241.
- Honma, M.; Minami-Hori, M.; Takahashi, H.; Iizuka, H. Podoplanin expression in wound and hyperproliferative psoriatic epidermis: regulation by TGF-beta and STAT-3 activating cytokines, IFN-gamma, IL-6, and IL-22. J Dermatol Sci 2012, 65, 134-140.
- Jiang, Y.J.; Lu, B.; Kim, P.; Paragh, G.; Schmitz, G.; Elias, P.M.; Feingold, K.R. PPAR and LXR Activators Regulate ABCA12 Expression in Human Keratinocytes. Journal of Investigative Dermatology 2008, 128, 104-109. Available online:. https://doi.org/10.1038/sj.jid.5700944. [CrossRef]
- Iwabuchi, K.; Nakayama, H.; Oizumi, A.; Suga, Y.; Ogawa, H.; Takamori, K. Role of Ceramide from Glycosphingolipids and Its Metabolites in Immunological and Inflammatory Responses in Humans. Mediators Inflamm 2015, 2015, 120748.
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Köfeler, H.; Merrill, A.H.; Murphy, R.C.; O’Donnell, V.B.; Oskolkova, O.; Subramaniam, S.; Wakelam, M.J.O.; Spener, F. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J Lipid Res 2020, 61, 1539-1555. (accessed on Jul 25, 2023). [CrossRef]
- Dahlhoff, M.; Camera, E.; Picardo, M.; Zouboulis, C.C.; Chan, L.; Chang, B.H.; Schneider, M.R. PLIN2, the major perilipin regulated during sebocyte differentiation, controls sebaceous lipid accumulation in vitro and sebaceous gland size in vivo. Biochim Biophys Acta 2013, 1830, 4642-4649.
- Zhang, C.; Liu, P. The New Face of the Lipid Droplet: Lipid Droplet Proteins. Proteomics 2019, 19, e1700223.
- Jakobs, B.S.; Wanders, R.J. Fatty acid beta-oxidation in peroxisomes and mitochondria: the first, unequivocal evidence for the involvement of carnitine in shuttling propionyl-CoA from peroxisomes to mitochondria. Biochem Biophys Res Commun 1995, 213, 1035-1041. (accessed on Jul 24, 2023). [CrossRef]
- Pavel, P.; Leman, G.; Hermann, M.; Ploner, C.; Eichmann, T.O.; Minzaghi, D.; Radner, F.P.W.; Del Frari, B.; Gruber, R.; Dubrac, S. Peroxisomal Fatty Acid Oxidation and Glycolysis Are Triggered in Mouse Models of Lesional Atopic Dermatitis. JID Innov 2021, 1, 100033. (accessed on Sep 15, 2023). [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annual review of physiology 2016, 78, 23-44. Available online:. https://doi.org/10.1146/annurev-physiol-021115-105045. [CrossRef]
- Moreno-Fernández, S.; Garcés-Rimón, M.; Vera, G.; Astier, J.; Landrier, J.F.; Miguel, M. High Fat/High Glucose Diet Induces Metabolic Syndrome in an Experimental Rat Model. Nutrients 2018, 10, 1502. (accessed on Aug 3, 2023). [CrossRef]
- Vamecq, J.; Andreoletti, P.; El Kebbaj, R.; Saih, F.; Latruffe, N.; El Kebbaj, M.H.S.; Lizard, G.; Nasser, B.; Cherkaoui-Malki, M. Peroxisomal Acyl-CoA Oxidase Type 1: Anti-Inflammatory and Anti-Aging Properties with a Special Emphasis on Studies with LPS and Argan Oil as a Model Transposable to Aging. Oxidative Medicine and Cellular Longevity 2018, 2018, 6986984-13. Available online:. https://doi.org/10.1155/2018/6986984. [CrossRef]
- Leman, G.; Pavel, P.; Hermann, M.; Crumrine, D.; Elias, P.M.; Minzaghi, D.; Goudounèche, D.; Roshardt Prieto, N.M.; Cavinato,M., Wanner,A.; Blunder, S.; Gruber, R.; Jansen-Dürr, P.; Dubrac, S. Mitochondrial Activity Is Upregulated in Nonlesional Atopic Dermatitis and Amenable to Therapeutic Intervention. J Invest Dermatol 2022, 142, 2623-2634.e12.
- Ge, F.; Sun, K.; Hu, Z.; Dong, X. Role of Omega-Hydroxy Ceramides in Epidermis: Biosynthesis, Barrier Integrity and Analyzing Method. Int J Mol Sci 2023, 24, 5035. [CrossRef]
- Niehues, H.; Bouwstra, J.A.; El Ghalbzouri, A.; Brandner, J.M.; Zeeuwen, P.L.J.M.; van den Bogaard, E.H. 3D skin models for 3R research: The potential of 3D reconstructed skin models to study skin barrier function. Exp Dermatol 2018, 27, 501-511.
- Smits, J.P.H.; Niehues, H.; Rikken, G.; van Vlijmen-Willems, I.M.J.J.; van de Zande, G. W. H. J. F.; Zeeuwen, P.L.J.M.; Schalkwijk, J.; van den Bogaard, E.H. Immortalized N/TERT keratinocytes as an alternative cell source in 3D human epidermal models. Sci Rep 2017, 7, 11838-y.
- Rikken, G.; Niehues, H.; van den Bogaard, E.H. Organotypic 3D Skin Models: Human Epidermal Equivalent Cultures from Primary Keratinocytes and Immortalized Keratinocyte Cell Lines. Methods Mol Biol 2020, 2154, 45-61.
- Rabionet, M.; Gorgas, K.; Sandhoff, R. Ceramide synthesis in the epidermis. Biochim Biophys Acta 2014, 1841, 422-434. (accessed on Mar 21, 2024). [CrossRef]
- Janssens, M.; van Smeden, J.; Gooris, G.S.; Bras, W.; Portale, G.; Caspers, P.J.; Vreeken, R.J.; Hankemeier, T.; Kezic, S.; Wolterbeek, R.; Lavrijsen, A.P.; Bouwstra, J.A. Increase in short-chain ceramides correlates with an altered lipid organization and decreased barrier function in atopic eczema patients. J Lipid Res 2012, 53, 2755-2766. (accessed on Dec 1, 2023). [CrossRef]
- Goleva, E.; Berdyshev, E.; Leung, D.Y. Epithelial barrier repair and prevention of allergy. J Clin Invest 2019, 129, 1463-1474. (accessed on Dec 1, 2023). [CrossRef]
- Leung, D.Y.M.; Berdyshev, E.; Goleva, E. Cutaneous barrier dysfunction in allergic diseases. J Allergy Clin Immunol 2020, 145, 1485-1497. (accessed on Dec 1, 2023). [CrossRef]
- Langan, S.M.; Irvine, A.D.; Weidinger, S. Atopic dermatitis. Lancet 2020, 396, 345-360. (accessed on Dec 1, 2023). [CrossRef]
- Boer, D.E.C.; van Smeden, J.; Al-Khakany, H.; Melnik, E.; van Dijk, R.; Absalah, S.; Vreeken, R.J.; Haenen, C.C.P.; Lavrijsen, A.P.M.; Overkleeft, H.S.; Aerts, J.M.F.G.; Bouwstra, J.A. Skin of atopic dermatitis patients shows disturbed beta-glucocerebrosidase and acid sphingomyelinase activity that relates to changes in stratum corneum lipid composition. Biochim Biophys Acta Mol Cell Biol Lipids 2020, 1865, 158673.
- Sawada, E.; Yoshida, N.; Sugiura, A.; Imokawa, G. Th1 cytokines accentuate but Th2 cytokines attenuate ceramide production in the stratum corneum of human epidermal equivalents: an implication for the disrupted barrier mechanism in atopic dermatitis. J Dermatol Sci 2012, 68, 25-35.
- Cha, H.J.; He, C.; Zhao, H.; Dong, Y.; An, I.; An, S. Intercellular and intracellular functions of ceramides and their metabolites in skin (Review). Int J Mol Med 2016, 38, 16-22. (accessed on Mar 21, 2024). [CrossRef]
- Turpin-Nolan, S.M.; Brüning, J.C. The role of ceramides in metabolic disorders: when size and localization matters. Nat Rev Endocrinol 2020, 16, 224-233. (accessed on Mar 21, 2024). [CrossRef]
- Blaess, M.; Deigner, H. Derailed Ceramide Metabolism in Atopic Dermatitis (AD): A Causal Starting Point for a Personalized (Basic) Therapy. Int J Mol Sci 2019, 20, 3967. (accessed on Mar 21, 2024). [CrossRef]
- Tsurumaki, H.; Katano, H.; Sato, K.; Imai, R.; Niino, S.; Hirabayashi, Y.; Ichikawa, S. WP1066, a small molecule inhibitor of the JAK/STAT3 pathway, inhibits ceramide glucosyltransferase activity. Biochem Biophys Res Commun 2017, 491, 265-270.
- Murakami, C.; Sakane, F. Sphingomyelin synthase-related protein generates diacylglycerol via the hydrolysis of glycerophospholipids in the absence of ceramide. J Biol Chem 2021, 296, 100454. (accessed on Mar 21, 2024). [CrossRef]
- Günenc, A.N.; Graf, B.; Stark, H.; Chari, A. Fatty Acid Synthase: Structure, Function, and Regulation. Subcell Biochem 2022, 99, 1-33. (accessed on Dec 1, 2023). [CrossRef]
- van Smeden, J.; Janssens, M.; Gooris, G.S.; Bouwstra, J.A. The important role of stratum corneum lipids for the cutaneous barrier function. Biochim Biophys Acta 2014, 1841, 295-313. (accessed on Dec 1, 2023). [CrossRef]
- Li, S.; Villarreal, M.; Stewart, S.; Choi, J.; Ganguli-Indra, G.; Babineau, D.C.; Philpot, C.; David, G.; Yoshida, T.; Boguniewicz, M.; Hanifin, J.M.; Beck, L.A.; Leung, D.Y.; Simpson, E.L.; Indra, A.K. Altered composition of epidermal lipids correlates with Staphylococcus aureus colonization status in atopic dermatitis. Br J Dermatol 2017, 177, e125-e127.
- Alessia Cavallo, Emanuela Camera, Grazia Bottillo, Miriam Maiellaro, Mauro Truglio, Federico Marini, Marlène Chavagnac-Bonneville, Aurélie Fauger, Eric Perrier, Flavia Pigliacelli, Mauro Picardo, Antonio Cristaudo, Maria Mariano Biosignatures of defective sebaceous gland activity in sebum-rich and sebum-poor skin areas in adult atopic dermatitis. Experimental Dermatology. [CrossRef]
- Kawabata, K.; Karahashi, M.; Sakamoto, T.; Tsuji, Y.; Yamazaki, T.; Okazaki, M.; Mitsumoto, A.; Kudo, N.; Kawashima, Y. Fatty Acid β-Oxidation Plays a Key Role in Regulating cis-Palmitoleic Acid Levels in the Liver. Biol Pharm Bull 2016, 39, 1995-2008. (accessed on Dec 4, 2023). [CrossRef]
- Sampath, H.; Flowers, M.T.; Liu, X.; Paton, C.M.; Sullivan, R.; Chu, K.; Zhao, M.; Ntambi, J.M. Skin-specific deletion of stearoyl-CoA desaturase-1 alters skin lipid composition and protects mice from high fat diet-induced obesity. J Biol Chem 2009, 284, 19961-19973.
- Zwara, A.; Wertheim-Tysarowska, K.; Mika, A. Alterations of Ultra Long-Chain Fatty Acids in Hereditary Skin Diseases-Review Article. Front Med (Lausanne) 2021, 8, 730855.
- Berdyshev, E.; Goleva, E.; Bronova, I.; Dyjack, N.; Rios, C.; Jung, J.; Taylor, P.; Jeong, M.; Hall, C.F.; Richers, B.N.; Norquest, K.A.; Zheng, T.; Seibold, M.A.; Leung, D.Y. Lipid abnormalities in atopic skin are driven by type 2 cytokines. JCI Insight 2018, 3, 10.1172/jci.insight.98006. eCollection 2018 Feb 22.
- Pavel, P.; Blunder, S.; Moosbrugger-Martinz, V.; Elias, P.M.; Dubrac, S. Atopic Dermatitis: The Fate of the Fat. Int J Mol Sci 2022, 23, 2121. (accessed on Sep 15, 2023). [CrossRef]
- Zhang, L.; Wen, X.; Hou, Y.; Yang, Y.; Song, W.; Zeng, Y.; Sun, J. Integrated metabolomics and lipidomics study of patients with atopic dermatitis in response to dupilumab. Front Immunol 2022, 13, 1002536. (accessed on Sep 15, 2023). [CrossRef]
- Berdyshev, E.; Goleva, E.; Bissonnette, R.; Bronova, I.; Bronoff, A.S.; Richers, B.N.; Garcia, S.; Ramirez-Gama, M.; Taylor, P.; Praestgaard, A.; Agueusop, I.; Jurvilliers, P.; Boguniewicz, M.; Levit, N.A.; Rossi, A.B.; Zhang, A.; Leung, D.Y.M. Dupilumab significantly improves skin barrier function in patients with moderate-to-severe atopic dermatitis. Allergy 2022, 77, 3388-3397. (accessed on Sep 15, 2023). [CrossRef]
- Khanna, D.; Padilla, C.; Tsoi, L.C.; Nagaraja, V.; Khanna, P.P.; Tabib, T.; Kahlenberg, J.M.; Young, A.; Huang, S.; Gudjonsson, J.E.; Fox, D.A.; Lafyatis, R. Tofacitinib blocks IFN-regulated biomarker genes in skin fibroblasts and keratinocytes in a systemic sclerosis trial. JCI Insight 2022, 7, e159566. [CrossRef]
- Bhattacharya, N.; Sato, W.J.; Kelly, A.; Ganguli-Indra, G.; Indra, A.K. Epidermal Lipids: Key Mediators of Atopic Dermatitis Pathogenesis. Trends Mol Med 2019, 25, 551-562.
- Zhang, C.; Chinnappan, M.; Prestwood, C.A.; Edwards, M.; Artami, M.; Thompson, B.M.; Eckert, K.M.; Vale, G.; Zouboulis, C.C.; McDonald, J.G.; Harris-Tryon, T.A. Interleukins 4 and 13 drive lipid abnormalities in skin cells through regulation of sex steroid hormone synthesis. Proc Natl Acad Sci U S A 2021, 118, 10.1073/pnas.2100749118.
- Dodington, D.W.; Desai, H.R.; Woo, M. JAK/STAT - Emerging Players in Metabolism. Trends Endocrinol Metab 2018, 29, 55-65.
- Williams, M.L.; Rutherford, S.L.; Feingold, K.R. Effects of cholesterol sulfate on lipid metabolism in cultured human keratinocytes and fibroblasts. J Lipid Res 1987, 28, 955-967.
- Ponec, M.; Kempenaar, J.; Weerheim, A.; de Lannoy, L.; Kalkman, I.; Jansen, H. Triglyceride metabolism in human keratinocytes cultured at the air-liquid interface. Arch Dermatol Res 1995, 287, 723-730. (accessed on Jul 24, 2023). [CrossRef]
- Schneider, M.R.; Zhang, S.; Li, P. Lipid droplets and associated proteins in the skin: basic research and clinical perspectives. Arch Dermatol Res 2016, 308, 1-6. Available online: https://link.springer.com/article/10.1007/s00403-015-1599-2. [CrossRef]
- Schurer, N.Y.; Monger, D.J.; Hincenbergs, M.; Williams, M.L. Fatty acid metabolism in human keratinocytes cultivated at an air-medium interface. J Invest Dermatol 1989, 92, 196-202.
- Radner, F.P.W.; Fischer, J. The important role of epidermal triacylglycerol metabolism for maintenance of the skin permeability barrier function. Biochim Biophys Acta 2014, 1841, 409-415.
- Moessinger, C.; Klizaite, K.; Steinhagen, A.; Philippou-Massier, J.; Shevchenko, A.; Hoch, M.; Ejsing, C.S.; Thiele, C. Two different pathways of phosphatidylcholine synthesis, the Kennedy Pathway and the Lands Cycle, differentially regulate cellular triacylglycerol storage. BMC Cell Biol 2014, 15, 43. (accessed on Dec 4, 2023). [CrossRef]
- Schäfer, L.; Kragballe, K. Abnormalities in Epidermal Lipid Metabolism in Patients with Atopic Dermatitis. Journal of Investigative Dermatology 1991, 96, 10-15. Available online:. https://doi.org/10.1111/1523-1747.ep12514648. [CrossRef]
Figure 1.
Effects of JAK/STAT inhibition by tofacitinib on the Th2-mediated changes in epidermal morphology and barrier proteins/enzymes expression. (A) Hematoxylin and eosin staining (H&E) of paraffin-embedded 3D HEEs treated with vehicle Th2 (IL-4 and IL-13), and tofacitinib. Histological analysis of Th2-treated HEEs showed characteristic AD morphologic features such as epidermal thickening and increased spaces between adjacent keratinocytes (arrows). (B) Quantitative analysis of epidermal and SC thickness. Scale bars: 50 µm, 20 µm. ***p˂0.001 vs vehicle; $p˂0.05 vs Th2; $$p˂0.01 vs Th2. (C) Quantitative RT-PCR analysis of K10, CASP14, FLG, LOR, and IVL, performed on 3D HEEs and HEEs treated with Th2 cytokines and tofacitinib. All values of mRNA expression were normalized against the expression of GAPDH and were expressed relative to vehicle (taken as 1). Data represented the mean ± SD of three independent experiments; *p˂0.05 vs vehicle; $p˂0.05 vs Th2. (D) Western blot analysis of K10, FLG, LOR and IVL protein expression performed on 3D HEEs and Th2-HEEs treated with tofacitinib. Representative blots are shown. GAPDH was used as endogenous loading control. Densitometric scanning of band intensities was performed to quantify the change in protein expression. Results were expressed as the fold change respect to vehicle (taken as 1-fold). Data represented the mean ± SD of three independent experiments; *p˂0.05 vs vehicle; $$p˂0.01 vs Th2. (E) Immunofluorescence and (F) quantitative analyses of FLG (green), LOR (red) and IVL (red) and of (G) pro-FLG and FLG on stratum granulosum and stratum corneum, respectively on serial sections of 3D HEEs and HEEs treated with tofacitinib and Th2 cytokines. Nuclei were counterstained with DAPI (blue). Scale bar: 50 µm.
Figure 1.
Effects of JAK/STAT inhibition by tofacitinib on the Th2-mediated changes in epidermal morphology and barrier proteins/enzymes expression. (A) Hematoxylin and eosin staining (H&E) of paraffin-embedded 3D HEEs treated with vehicle Th2 (IL-4 and IL-13), and tofacitinib. Histological analysis of Th2-treated HEEs showed characteristic AD morphologic features such as epidermal thickening and increased spaces between adjacent keratinocytes (arrows). (B) Quantitative analysis of epidermal and SC thickness. Scale bars: 50 µm, 20 µm. ***p˂0.001 vs vehicle; $p˂0.05 vs Th2; $$p˂0.01 vs Th2. (C) Quantitative RT-PCR analysis of K10, CASP14, FLG, LOR, and IVL, performed on 3D HEEs and HEEs treated with Th2 cytokines and tofacitinib. All values of mRNA expression were normalized against the expression of GAPDH and were expressed relative to vehicle (taken as 1). Data represented the mean ± SD of three independent experiments; *p˂0.05 vs vehicle; $p˂0.05 vs Th2. (D) Western blot analysis of K10, FLG, LOR and IVL protein expression performed on 3D HEEs and Th2-HEEs treated with tofacitinib. Representative blots are shown. GAPDH was used as endogenous loading control. Densitometric scanning of band intensities was performed to quantify the change in protein expression. Results were expressed as the fold change respect to vehicle (taken as 1-fold). Data represented the mean ± SD of three independent experiments; *p˂0.05 vs vehicle; $$p˂0.01 vs Th2. (E) Immunofluorescence and (F) quantitative analyses of FLG (green), LOR (red) and IVL (red) and of (G) pro-FLG and FLG on stratum granulosum and stratum corneum, respectively on serial sections of 3D HEEs and HEEs treated with tofacitinib and Th2 cytokines. Nuclei were counterstained with DAPI (blue). Scale bar: 50 µm.
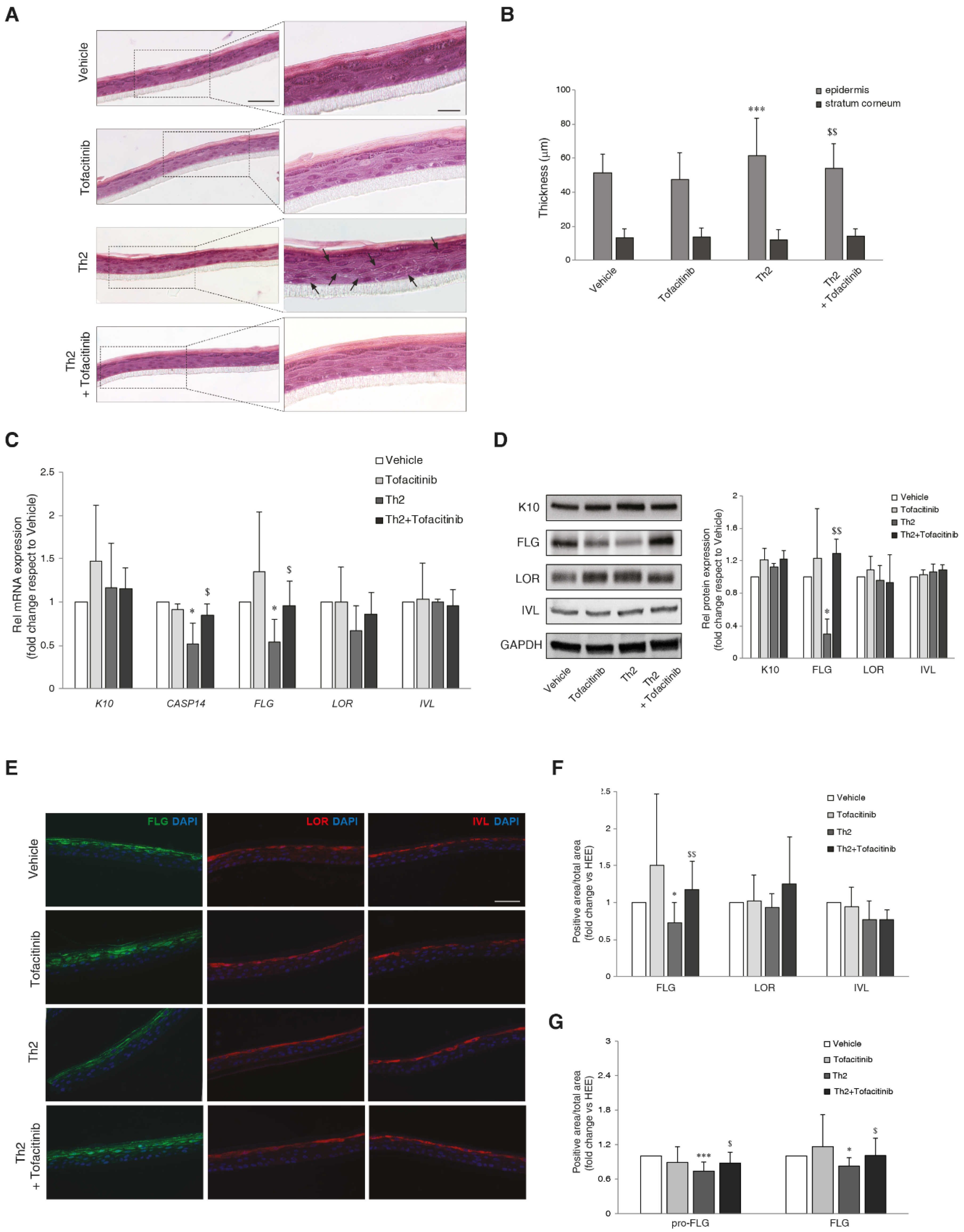
Figure 2.
Effects of JAK/STAT inhibition by tofacitinib on the Th2-mediated changes in the expression of inflammatory and lipid genes. Quantitative RT-PCR analysis of (A) IL-1α, IL-1β, IL-6, IL-8, CCL26, and PDPN (B) ELOVL1, ELOVL3 and ELOVL4, performed on 3D HEEs and Th2-HEEs treated with tofacitinib. All values of mRNA expression were normalized against the expression of GAPDH and were expressed relative to vehicle (taken as 1). Data represented the mean ± SD of three independent experiments; *p˂0.05, **p˂0.01 vs vehicle; $p˂0.05, $$p˂0.01 vs Th2. (C) Western blot analysis of ELOV1 protein expression performed on 3D HEEs and Th2-HEEs treated with tofacitinib. Representative blots are shown. GAPDH was used as endogenous loading control. Densitometric scanning of band intensities was performed to quantify the change in protein expression. Results were expressed as the fold change respect to vehicle (taken as 1-fold). Data represented the mean ± SD of three independent experiments; *p˂0.05 vs vehicle; $p˂0.05 vs Th2; $$p˂0.01 vs Th2 (D) Immunohistochemical and (E) quantitative analyses of ELOV1 on 3D HEEs and Th2-HEEs treated with tofacitinib. Nuclei were counterstained with hematoxylin. Scale bars: 20 µm, 10 µm. **p˂0.01 vs vehicle; $$p˂0.01 vs Th2. (F) Quantitative RT-PCR analysis of SPT, DEGS2, CA2, and PPARγ, performed on 3D HEEs and Th2-HEEs treated with tofacitinib. All values of mRNA expression were normalized against the expression of GAPDH and were expressed relative to vehicle (taken as 1). Data represented the mean ± SD of three independent experiments; *p˂0.05, **p˂0.01 vs vehicle; $p˂0.05, $$p˂0.01 vs Th2.
Figure 2.
Effects of JAK/STAT inhibition by tofacitinib on the Th2-mediated changes in the expression of inflammatory and lipid genes. Quantitative RT-PCR analysis of (A) IL-1α, IL-1β, IL-6, IL-8, CCL26, and PDPN (B) ELOVL1, ELOVL3 and ELOVL4, performed on 3D HEEs and Th2-HEEs treated with tofacitinib. All values of mRNA expression were normalized against the expression of GAPDH and were expressed relative to vehicle (taken as 1). Data represented the mean ± SD of three independent experiments; *p˂0.05, **p˂0.01 vs vehicle; $p˂0.05, $$p˂0.01 vs Th2. (C) Western blot analysis of ELOV1 protein expression performed on 3D HEEs and Th2-HEEs treated with tofacitinib. Representative blots are shown. GAPDH was used as endogenous loading control. Densitometric scanning of band intensities was performed to quantify the change in protein expression. Results were expressed as the fold change respect to vehicle (taken as 1-fold). Data represented the mean ± SD of three independent experiments; *p˂0.05 vs vehicle; $p˂0.05 vs Th2; $$p˂0.01 vs Th2 (D) Immunohistochemical and (E) quantitative analyses of ELOV1 on 3D HEEs and Th2-HEEs treated with tofacitinib. Nuclei were counterstained with hematoxylin. Scale bars: 20 µm, 10 µm. **p˂0.01 vs vehicle; $$p˂0.01 vs Th2. (F) Quantitative RT-PCR analysis of SPT, DEGS2, CA2, and PPARγ, performed on 3D HEEs and Th2-HEEs treated with tofacitinib. All values of mRNA expression were normalized against the expression of GAPDH and were expressed relative to vehicle (taken as 1). Data represented the mean ± SD of three independent experiments; *p˂0.05, **p˂0.01 vs vehicle; $p˂0.05, $$p˂0.01 vs Th2.
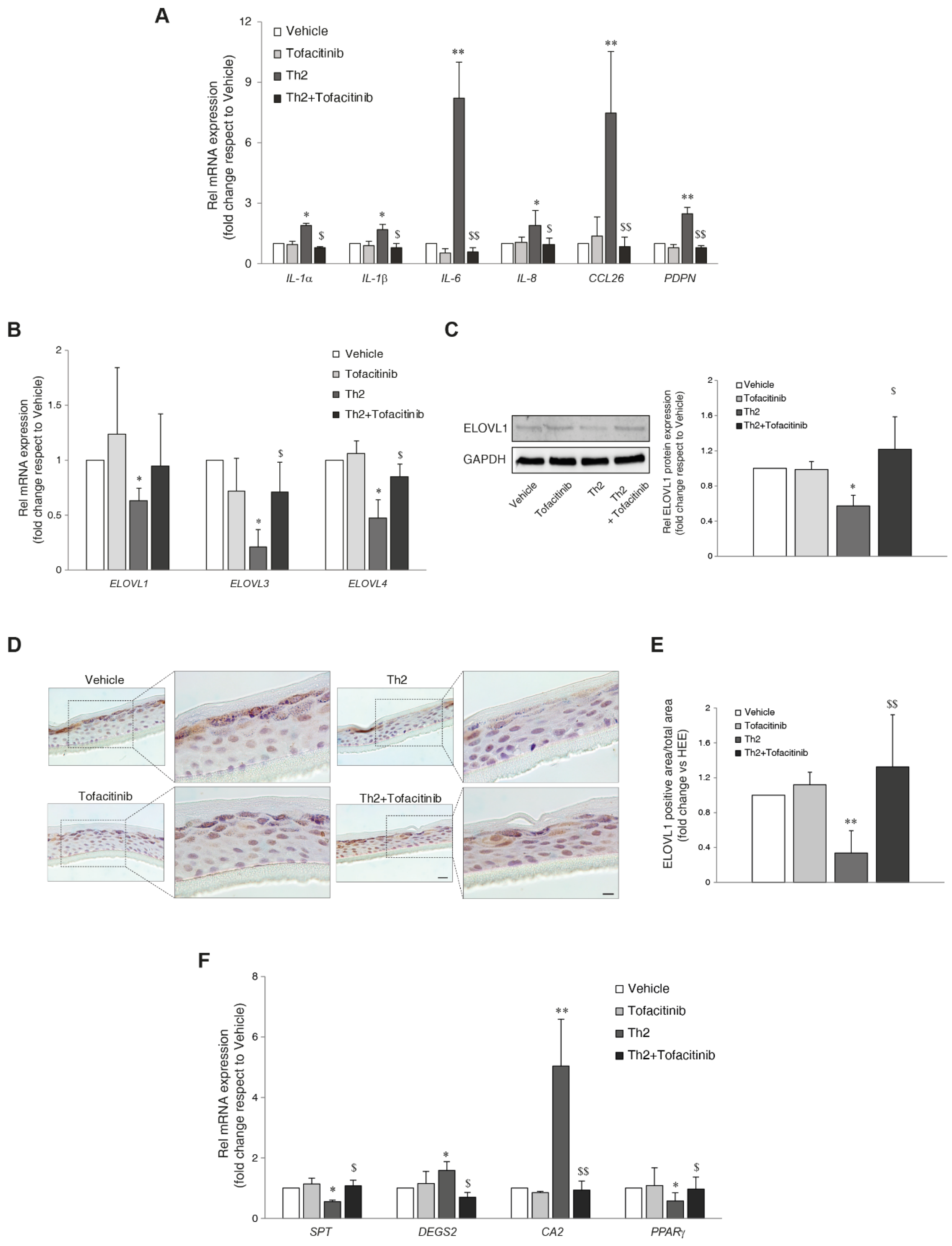
Figure 3.
Th2 cytokines IL-4 and IL-13 induce lipid changes through the activation of JAK/STAT pathway. (A) Hierarchical clustering of 51 lipid species resulted significantly modulated in the one-way ANOVA applied to the amounts of the 300 characterized lipids. (B) Ultra-long chain SFAs determined by LCMS. Profiles of abundance of (C) PCs determined by HILIC-MS and of (D) Palmitoleic acid (FA 16:1n-7) determined by GCMS and TGs determined by RP-LCMS in lipid extracts of 3D HEEs treated with vehicle, tofacitinib, Th2 cytokines, and combined tofacitinib and Th2 cytokines. Molar amounts of individual lipids were calculated against same-class deuterated internal standards and were normalized by the protein concentration and expressed as pmol/mg protein. Real Time RT-PCR analysis of (E) PLIN1 and PLIN2, (F) CPT1α, ACAT1, ACADS and ACOX1, performed on 3D HEEs and HEEs treated with Th2 cytokines and tofacitinib. All values of mRNA expression were normalized against the expression of GAPDH and were expressed as relative to the vehicle (taken as 1). Data represented the mean ± SD of three independent experiments; *p˂0.05, **p˂0.01 vs vehicle; $p˂0.05, $$p˂0.01 vs Th2.
Figure 3.
Th2 cytokines IL-4 and IL-13 induce lipid changes through the activation of JAK/STAT pathway. (A) Hierarchical clustering of 51 lipid species resulted significantly modulated in the one-way ANOVA applied to the amounts of the 300 characterized lipids. (B) Ultra-long chain SFAs determined by LCMS. Profiles of abundance of (C) PCs determined by HILIC-MS and of (D) Palmitoleic acid (FA 16:1n-7) determined by GCMS and TGs determined by RP-LCMS in lipid extracts of 3D HEEs treated with vehicle, tofacitinib, Th2 cytokines, and combined tofacitinib and Th2 cytokines. Molar amounts of individual lipids were calculated against same-class deuterated internal standards and were normalized by the protein concentration and expressed as pmol/mg protein. Real Time RT-PCR analysis of (E) PLIN1 and PLIN2, (F) CPT1α, ACAT1, ACADS and ACOX1, performed on 3D HEEs and HEEs treated with Th2 cytokines and tofacitinib. All values of mRNA expression were normalized against the expression of GAPDH and were expressed as relative to the vehicle (taken as 1). Data represented the mean ± SD of three independent experiments; *p˂0.05, **p˂0.01 vs vehicle; $p˂0.05, $$p˂0.01 vs Th2.
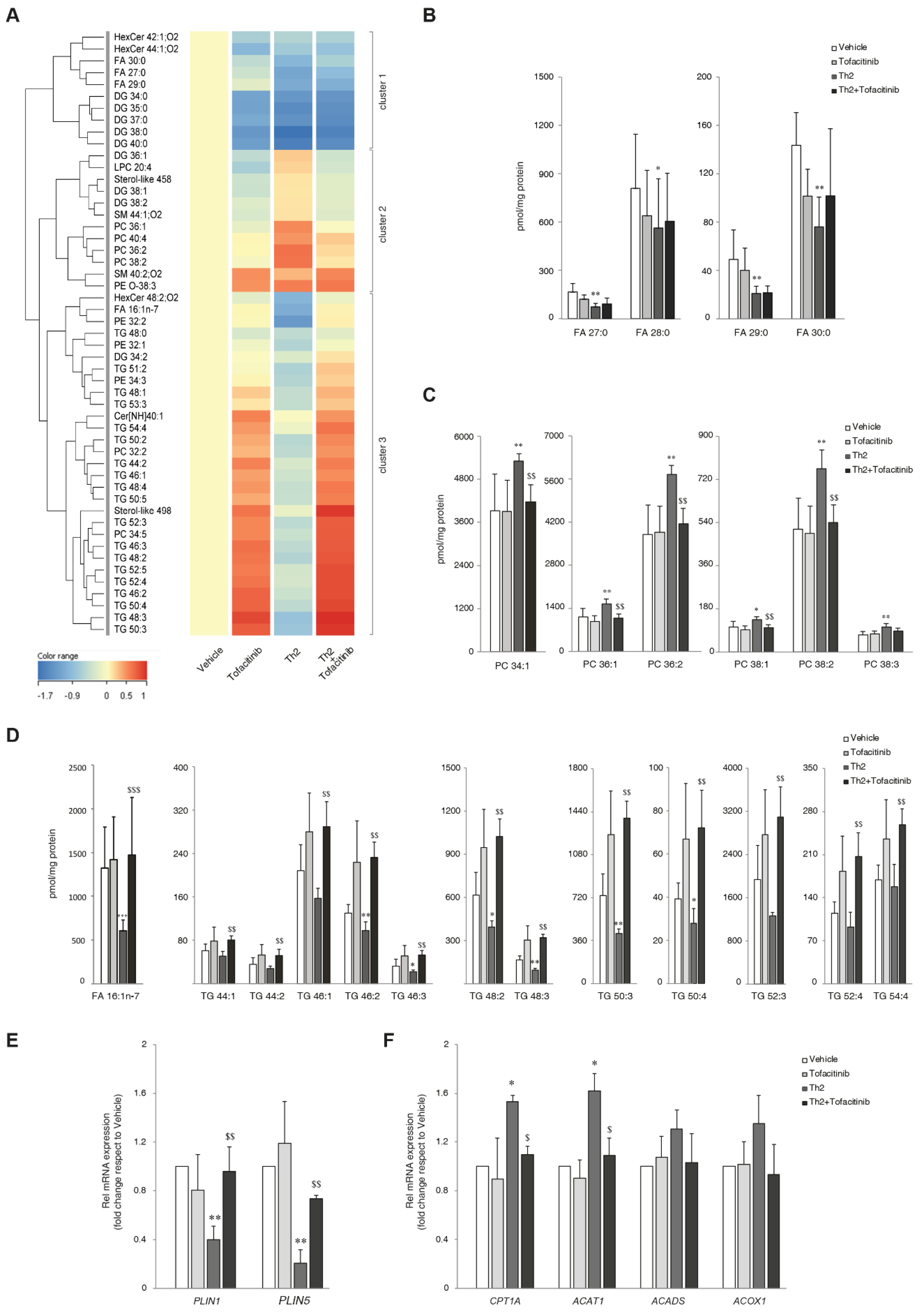
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



