
Submitted:
28 March 2024
Posted:
29 March 2024
You are already at the latest version
Abstract
Maternal type 2 diabetes mellitus (T2DM) has been shown to result in fetal programming of the hypothalamus-pituitary-adrenal (HPA) axis, leading to adverse fetal outcomes. T2DM is preceded by prediabetes and shares similar pathophysiological complications. However, no studies have investigated the effects of maternal prediabetes on fetal HPA axis function and postnatal offspring development, hence this study. Pre-diabetic (PD) and non-pre-diabetic (NPD) female Sprague Dawley rats were mated with non-prediabetic males. After gestation, male pups born from the PD and NPD groups were collected. Markers of HPA axis function, adrenocorticotropin hormone (ACTH) and corticosterone were measured in all dams and pups. Glucose tolerance and expression of mineralocorticoid (MR) and glucocorticoid (GR) receptors were further measured in all pups at birth and their developmental milestones. The results demonstrated increased basal concentrations of ACTH and corticosterone in the dams from the PD group by comparison to NPD. Furthermore, the results show an increase in pups ACTH and corticosterone concentrations, impaired glucose tolerance and dysregulated MR and GR expression in the PD group at all developmental milestones. These observations reveal that pregestational prediabetes is associated with maternal dysregulation of the HPA axis, impacting offspring HPA axis development along with impaired glucose handling.
Keywords:
Maternal HPA axis
; fetal HPA axis
; pregestational prediabetes
; pups
1. Introduction
Fetal programming is a process whereby a stimulus or insult at a critical period in development results in permanent adaptation of the organism’s structure or physiology [1,2,3]. Evidence in fetal programming studies has shown that fetal overexposure to endogenous glucocorticoids may underpin the link between early life events and later disease [4,5,6,7]. It is proposed that dysregulation of the maternal hypothalamic-pituitary-adrenal (HPA) axis determines fetal exposure to stress hormones, influencing fetal development and programming the fetal HPA axis [8,9,10,11,12].
In humans, the physiologically active glucocorticoid (GC) is cortisol, whereas in rodents, it is corticosterone [13,14]. During pregnancy, the maternal HPA axis experiences significant changes, with the placenta secreting corticotropin-releasing hormone (CRH), which further elevates adrenocorticotrophin hormone (ACTH) and cortisol levels [15,16,17]. This creates a positive feedback loop where maternal cortisol stimulates placental CRH synthesis, ultimately resulting in higher glucocorticoid levels [4,18]. In addition, previous research has reported that despite the increasing circulating levels of glucocorticoid, the diurnal secretion of corticosterone is maintained throughout pregnancy [19,20,21].
In cases of maternal adversity, such as those seen in maternal stress, have been associated with prolonged activation and dysregulation of the maternal HPA axis, leading to elevated plasma ACTH and cortisol levels [9,22,23,24]. Additionally, research has indicated that type 2 diabetes mellitus (T2DM) shares similarities with maternal stress conditions during pregnancy, including the persistent activation and dysregulated function of the HPA axis with elevated glucocorticoid levels [25,26,27,28,29,30]. Given that T2DM is a complicated and multifaceted disease caused by a mix of genetic and environmental risk factors, it is considered a stressor to the human body [31,32,33,34,35]. Studies show that excessive levels of maternal can overwhelm the enzymatic barriers that effectively prevent excessive fetal exposure to maternal GCs, therefore exposing fetuses to excess glucocorticoids [5,29,30,36]. Studies have shown that pregnancies affected by T2DM in conjunction with uteroplacental vasculopathy and increased glucocorticoids exhibit intrauterine growth restriction (IUGR), often manifested as low birth weight [37,38,39]. Studies have also shown that excessive fetal exposure to GCs is associated with the downregulation of fetal glucocorticoid receptor (GR) and mineralocorticoid receptor (MR) and impairment of the feedback regulation of the HPA axis in both infancy and adulthood [5,13,40,41]. Cross-sectional research has indicated a connection between low birth weight and disrupted functioning of the HPA axis, leading to elevated levels of GC in adulthood [10,42,43,44]. In addition, the association between low birth weight and the development of T2DM was first reported in studies by Hales et al., who demonstrated a several-fold increase in the incidence of glucose intolerance and T2DM in adult men who were born with low birth weight compared with those born with normal birth weight [39,45]. Additionally, research suggests that individuals born with low birth weight often associated with catch-up growth with increased risk for various non-communicable diseases (NCDs) such as hypertension, cardiovascular diseases and mental disorders in adulthood, aligning with the developmental origins of health and disease (DOHaD) hypothesis [44,46,47,48,49,50,51].
Furthermore, studies have found that T2DM is often preceded by an early-onset condition known as prediabetes [52,53,54]. Prediabetes is a condition in which blood glucose concentrations are higher than the normal but do not meet the diagnostic criteria for T2DM [55,56,57]. Studies show that prediabetes is predicted to affect 453.8 million people by 2030 and usually 5-10% progress to T2DM each year due asymptomatic characteristics [58,59]. A diet-induced animal model for prediabetes was established in our laboratory and it was found to mimic the human condition [60,61,62]. In addition, this animal model showed similarities in pathophysiology with T2DM, including dysregulation in the HPA axis associated with increased basal corticosterone and impaired regulation of their GR and MR in male animals [63]. This raised the question of whether the difficulties associated with maternal stress and preexisting T2DM pregnancies and fetal programming of fetal HPA axis are also present during prediabetes and whether maternal basal corticosterone and ACTH levels in prediabetic dams may impact fetal HPA axis development. Therefore, using this animal model, the study investigated the effects of pregestational prediabetes on maternal HPA axis function and its effects on postnatal offspring development.
2. Materials and Methods
2.1. Animals and Housing
All animal experimentation was approved by the Animal Research Ethics Committee (AREC) of the University of Kwa-Zulu Natal (AREC/032/020D). Three-week old female Sprague-Dawley rats were bred and housed in the Biomedical Research Unit (BRU) of the University of Kwa-Zulu Natal were used in the study. The animals were maintained under standard laboratory conditions of constant temperature (22 ± 2 °C), carbon dioxide (CO2) content (<5000 p.m.), relative humidity (55 ± 5%) and illumination (12 h light/dark cycle, lights on at 07h00). The noise level was maintained at less than 65 decibels approved. The animals were allowed access to food and fluids ad libitum. The animals acclimatized to their new environment for 1 week while consuming standard rat chow and tap water before the induction of pre-diabetes by exposure to a well-established experimental diet (HFHC).
2.2. Induction of Prediabetes
Female Sprague–Dawley rats were randomly assigned to two diet groups, group A and B (n = 6 per group): Experimental pre-diabetes was induced in female Sprague–Dawley rats using a protocol previously described [60]. To summarize, group A labelled as the non-prediabetic group (NPD), was given a standard rat chow diet with normal water for 36-weeks period. The animals in group B were given a high-fat, high-carbohydrate (HFHC) diet with 15% fructose added for same duration of 36 weeks. After a duration of 36 weeks, the diagnostic criteria of the American Diabetes Association (ADA) was applied to identify pre-diabetes in all animals. This involved identifying animals exhibiting pre-diabetic indicators, such as fasting blood glucose levels ranging from 5.6 to 7.1 mmol/L, oral glucose tolerance test (OGTT) 2-hour glucose levels between 7.1 and 11.1 mmol/Land glycated haemoglobin (HbA1c) levels between 5.7% and 6.4% were used as an additional diagnostic criterion for prediabetes. Animals meeting these criteria were categorized as pre-diabetic, while those with measurements below the pre-diabetic thresholds were considered non-pre-diabetic.
2.3. Oral Glucose Tolerance Response
An oral glucose tolerance test was performed on all animals to assess their glucose tolerance response. This test was completed after carbohydrate loading, following a well-established laboratory technique [64]. After fasting for 16 hours, glucose levels were determined at time 0. Then, a monosaccharide syrup was administered orally using an 18-gauge gavage needle that is 38 mm long and curved, with a 21/4 mm ball end (Able Scientific, Canning Vale, Australia). The glucose concentration was determined by collecting blood using the tail-prick method [65] and measuring glucose concentrations using a OneTouch select glucometer (Lifescan, Mosta, Malta, United Kingdom). Glucose levels were subsequently assessed at 15-, 30-, 60- and 120-minutes following carbohydrate loading.
2.4. Mating
Prior to mating, all 12 female Sprague–Dawley rats underwent assessment for the proestrus stage through vaginal smear analysis under a microscope. Those in the proestrus stage were permitted to mate with non-prediabetic male Sprague–Dawley rats. Confirmation of pregnancy and assignment of gestational day (GND) 0 were determined the following morning by the presence of a vaginal plaque or vaginal smear containing spermatozoa observed under a microscope [66]. The male rats were then be removed from the cage and male rats were returned to their cage after successful mating [67]. Pregnant rats were then observed until the end of gestation (21 days).
2.5. Male Pups Were Collected for the Study
The animals were born naturally at gestational day 21 and kept with their dams. However, the animals at day 7 were immediately collected, weighed and were then carefully returned to the dams. According to studies, day 7 represents a neonatal/newborn development period which loosely correlate with similar developmental stages in humans (summarised in Table 1) [68,69,70]. The animals were kept with their mothers for a period of 21 days/3 weeks under standard laboratory conditions (for temperature and humidity) in a 12hr day and 12hr night cycle. This 21 day/3 weeks cycle allows rats to undergo crucial stages of development, including growth socialization and the establishing of basic behaviours [71,72,73]. Additionally, the regular day-night cycle helps regulate their circadian rhythms, which are important for overall health and wellbeing [74,75,76]. After 21 days/3 weeks the animals were weaned and 18 male Sprague Dawley pups born from the non-prediabetic (NPD) female group and 18 male pups born from prediabetic (PD) female group were collected. Afterwards, the dams were subjected to a period of fasting in order to evaluate the oral glucose tolerance (OGT) test and then euthanized for further investigations. This was considered phase one. In the second part of the investigation, the pups were divided into three independent experimental weeks, week 3, week 6 and week 16 with six pups in each group assigned to either the NPD or PD. These animals were given unlimited access to standard chow and normal drinking water throughout their experimental periods. With regards to the weeks chosen in our study, studies show that there are six recognized developmental time periods in rats [77,78]. These include the neonatal, infantile, juvenile, peripubertal, late pubertal and emerging adulthood which are recognized at week 3, 6 and 16 in our study respectively. These stages have been shown to roughly correspond to similar developmental stages in humans [79,80,81,82,83,84]. Table 1 provides a concise overview of the developmental phases in humans and rats, allowing for a comparison of their respective ages. In addition, at each of the different time points, 6 animals from each group were fasted for 16 hours to assess fasting glucose and OGT performance test. Subsequently, the animals were euthanized for terminal investigations at each subsequent week of investigation.
2.6. Blood Collection and Tissue Harvesting
For blood collection, all animals were sacrificed using the guillotine including the dams and blood was collected into the separated pre-cooled heparinised tubes and was centrifuged (Eppendorf centrifuge 5403, Germany) at 4°C 530g for 15 minutes. The separated plasma was stored at -80° C in a Bio Ultra freezer (Snijders Scientific, Holland) for Biochemical analysis. Following blood collection, the hippocampus was removed and placed in pre-cooled Eppendorf containers and snap-frozen in liquid nitrogen before storage in a Bio Ultra freezer (Snijers Scientific, Tilburg, Netherlands) at − 80 °C.
2.7. Biochemical Analysis
Plasma adrenocorticotropic hormone (ACTH) for dams and pups concentrations were measured using their respective rat competitive-ELISA kits (Elabscience Biotechnology Co., Ltd., Wuhan, China) according to the manufacturer’s instructions. The kits included a micro-ELISA plate that was coated with antibodies specific to each of the parameters measured. Standards and samples were pipetted into the appropriate wells of the micro-ELISA plate and incubated for 90 min at 37 °C. This was followed by adding the relevant biotinylated detection antibody (100 μl). After 60 min incubation at 37 °C, avidin–horseradish peroxidase conjugate (100 μl) was added to each microplate well. After a further 30 min incubation at 37 °C, the unbound components were washed away using the wash buffer provided. Substrate solution (100 μl) was added to each microplate well and after 15 min incubation at 37 °C, the stop solution (50 μl) was added. Optical density was measured using a nano spectrophotometer 47 (BMG Labtech, Ortenburg, Germany) at 450 nm. The concentrations of each parameter in the samples were extrapolated from a standard curve.
Corticosterone concentrations for dams and pups were measured in plasma using different Linco-plex kits (Millipore, Billerica, MA, USA). Metabolic hormone kits (Merck-Millipore, MI, USA) were used to quantify plasma and Corticosterone concentrations. Assays were done according to the manufacturer’s instructions. Concentrations were read on Luminex® (50 µL, 50 beads per bead set) using a Luminex® 200™, HTS, FLEXMAP 3D®, MAGPIX® instrument with xPONENT® software. All samples, quality controls (QCs)and inter-plate control were duplicated. Concentrations of all the analytes in the QCs were within the expected ranges and the inter-plate variation was below 20%. The data generated were managed using Bio-Plex Manager Software, version 4.1.1.
2.8. Glucocorticoid Receptor and Mineralocorticoid Receptor Gene Expression via Real-Time- PCR
The hippocampus tissues collected at 3 weeks, 6 weeks and at 16 weeks were homogenised and total RNA was isolated using a ReliaPrep miRNA Cell and Tissue Miniprep System (Promega, USA) according to the manufacturer’s protocol. A Nanodrop 2000 (Thermo Scientific, Roche, South Africa) was used to determine the purity and concentration of RNA. A purity ratio (A260/A280) of 1.7–2.1 was considered acceptable for conversion of RNA to cDNA. Following the manufacturer’s instructions (GoTaq® 2-Step RT-Qpcr System as a cDNA synthesis kit, Promega, USA). Total RNA was reverse transcribed to cDNA. To perform the PCR amplification on ROCHE LightCycler96 (Roche, South Africa), the BIO-RAD iTaq Universal SYBR Green I master mix was used. Primer sequences (Metabion, Germany) used in this study are listed in Table 2 below. Primer sequences were adapted from [85,86]. After that, the Rattus primer sequences were blasted to verify the primer sequence and their accession number. PCR was performed using the following cycling conditions: an initial denaturation cycle at 95 °C for 10 min followed by PCR, which consisted of 45 cycles at 95 °C for 30 s, 65 °C for 30 s and a final elongation at 72 °C for 30 s with a single fluorescence measurement. Melting curve analysis was performed at 95 °C for 30 s, 65 °C for 20 s and 95 °C at a ramp rate of 0.05 °C/s and a continuous fluorescence measurement, followed by a final cooling step at 40 °C for 60 s. The RT-qPCR results were analysed using the 2−ΔΔCq comparative method relative to the control groups [87]. The housekeeping gene used in this study was Glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
2.9. Statistical Analysis
All data are expressed as means ± standard error of means (SEM). GraphPad Prism Instant Software (version 8.00, GraphPad Software, San Diego, California, (USA) was used for statistical analysis. All terminal data was analysed using the normality and lognormality test and a student t-test to assess differences between control and experimental groups. Values of p < 0.05 were considered statistically significantly different between the compared groups.
3. Results
3.1. Dams Oral Glucose Tolerance (OGT) Response and Glycated Hemoglobin (HbA1c) Concentrations
Figure 1 shows results of the OGT (Figure 1a) and HbA1c concentrations (Figure 1b) of non-prediabetic female group (NPD n=6) and prediabetic female group (PD n=6) at 36 weeks. In the OGTT (Figure 1a), the blood glucose concentration is significantly higher in the PD group at time 0 when compared to the NPD group. The blood glucose concentration in the PD group remained significantly higher when compared to the NPD group throughout the 2-hr test. The HbA1c concentration (Figure 1b) is significantly higher in the PD group when compared to the NPD group.
3.2. Dams Hypothalamic-Pituitary-Adrenal (HPA) Axis Components
Figure 2 shows results of the dams ACTH (Figure 2a) and corticosterone (Figure 2b) concentrations of non-prediabetic female group (NPD n=6) and prediabetic female group (PD n=6) at 21 days after birth. The ACTH concentration (Figure 2a) is significantly higher in the PD group when compared to the NPD group. Similarly, the corticosterone concentration (Figure 2b) is significantly higher in the PD group when compared to the NPD group.
3.3. Pups Hypothalamic-Pituitary-Adrenal (HPA) Axis Components
Figure 3 shows results of the ACTH (Figure 3a) and corticosterone (Figure 3b) concentrations of pups born from the non-prediabetic pregnant female group (NPD n=6) and pups born from the prediabetic pregnant female group (PD n=6) at week 3, 6 and 16 of the experimental periods. The ACTH concentration (Figure 3a) is significantly higher in the PD group when compared to the NPD group throughout the experimental weeks. Similarly, the corticosterone concentration (Figure 3b) is significantly higher in the PD groups when compared to the NPD groups throughout the experimental weeks.
3.4. Pups Hippocampal Glucocorticoid Receptors (GR) & Mineralocorticoid Receptor (MR)
Figure 4 shows results of the hippocampal GR (Figure 4a) and MR (Figure 4b) gene expressions measured in non-stressed pups born from non-prediabetic pregnant female group (NPD) and prediabetic pregnant female group (PD) at week 3, 6 and 16 of the experimental periods. The PD groups experienced a half-fold decrease in GR (Figure 4a) gene expression relative to the NPD groups in all experimental weeks. The PD groups experienced a two-fold increase in MR (Figure 4b) gene expression relative to the NPD groups in all experimental weeks.
3.5. Pups Adrenal Gland Weight
Figure 5 shows results of the adrenal gland weight of pups born from the non-prediabetic pregnant female group (NPD n=6) and pups born from the prediabetic pregnant female group (PD n=6) at week 3, 6 and 16 of the experimental periods. The adrenal weight in the PD group is significantly higher when compared to the NPD group throughout the experimental weeks.
3.6. Pups Bodyweights
Figure 5 shows results of the body weights of pups born from the non-prediabetic pregnant female group (NPD n=6) and pups born from the prediabetic pregnant female group (PD n=6) at day 7 (Figure 6a), week 3, 6 and 16 (Figure 6b) of the experimental periods. At day 7 (Figure 6a), the body weight in the PD group is significantly lower when compared to the NPD group. At week 3 (Figure 6b), bodyweight in the PD group shows no significant compared to the NPD group. At week 6 (Figure 6b), the bodyweight in the PD group is significantly higher when compared to the NPD group. Lastly, at week 16 (Figure 6b) the body weight in the PD group shows no significant when compared to the NPD group.
3.7. Pups Oral Glucose Tolerance (OGT) Response
Figure 7 shows results of the OGT of pups born from non-prediabetic pregnant female group (NPD) and prediabetic pregnant female group (PD) rats at week 3, 6 and 16 of the experimental periods. The OGTT blood glucose concentration is significantly higher in the PD groups at time 0 when compared to the NPD groups in all experimental weeks. The blood glucose concentration in the PD groups remained significantly higher throughout the 2-hr test when compared to the NPD groups in all experimental weeks.
4. Discussion
Fetal programming, a response to adverse fetal conditions, leads to lasting adaptations altering organ growth, physiology and metabolism thus increasing adult disease risk [88,89]. Excessive glucocorticoid (GC) exposure in utero, often due to maternal HPA axis dysregulation, has been shown to link early events with later diseases such as, hypertension, cardiovascular diseases, T2DM and mental disorders [5,90,91]. During normal pregnancy, the maternal HPA axis undergoes significant changes, yet diurnal GC secretion remains maintained [9,92]. Studies suggest that T2DM exhibits resemblances to maternal stress conditions during pregnancy, such as dysregulated HPA axis with increased levels of GC [25,26,27,28]. Fetal exposure to excess maternal GCs causes growth restriction, programme life-long changes in HPA axis activity which increases risk of developing T2DM and cardiometabolic diseases in adult life [41,92,93]. Several studies have suggested that the onset of complications associated with T2DM begin during the prediabetic state [52,53,54]. An experiment in our lab established a diet-induced prediabetic animal model and showed similarities with humans, including dysregulation in the function of HPA axis [63]. However, no studies have yet shown the influence of pre-existing prediabetes during pregnancy on the maternal-fetal HPA axis interaction. Therefore, this study aimed to investigate the effects of pregestational prediabetes on maternal HPA axis function and its effects on postnatal offspring development.
In non-diabetic individuals, glucose homeostasis is tightly regulated with fasting plasma glucose (FPG) maintained at 3·9–5·6 mmol/L and postprandial glucose level of less than 7.8 mmol/L [94]. In the postprandial state elevated blood glucose concentration stimulate pancreatic beta β cells to produce adequate insulin enough to clear glucose from the bloodstream through insulin signalling pathway [95,96]. Studies show that prediabetes can be diagnosed by at least two of these characteristics: impaired fasting glucose (IFG) (5.5-6.9 mmol/l), impaired glucose tolerance (IGT) (7.8-11.0 mmol/l) and elevated glycated haemoglobin A1c (HbA1c) (5.7-6.4%) [97,98]. In the present study, there was a significant increase in the fasting plasma glucose concentration before glucose loading and a failure of blood glucose concentration post-glucose load to return to baseline following a 2-hr OGT test in the PD group suggesting the presence of IGF and IGT in Figure 1a. This suggests that glucose utilization in insulin-dependent peripheral tissues such as skeletal muscles is decreased [99,100]. In addition, the PD group had significant increase in HbA1c concentration when compared to NPD group in our study in Figure 1b. The results align with previous research indicating that elevated plasma glucose concentrations, also seen in PD and T2DM, result in non-enzymatic glycation of hemoglobin [60,101,102]. This glycation process occurs throughout the entire 120-day lifespan of red blood cells through an Amadori reaction, forming a stable and irreversible ketoamine linkage [60]. These findings in our results indicate that the levels of glucose in the blood and the length of time that red blood cells are exposed to glucose are responsible for the production of HbAc1. The coexistence of IGF and IGT, together with elevated HbAc1 levels, in our PD group indicated the induction of prediabetes at 36 weeks. A prior investigation conducted in our lab revealed that male animals developed prediabetes after 20 weeks of being fed a diet high in fat and carbohydrates [60]. Our current study extends this timeframe by an additional 16 weeks. A previous study has attributed this to several physiological disparities, including genetic, hormonal differences such progesterone and estrogen that have been shown to exert protective effects that may have delayed the induction of prediabetes in females [103].
During pregnancy, the regulation of the maternal HPA axis undergoes dramatic changes such as regulating stress response and maintaining homeostasis for both mother and the developing fetus [22,24]. The HPA axis control the diurnal secretion of glucocorticoids which play a crucial role in fetal development [9,19]. Physiological active glucocorticoid is known as cortisol in humans and corticosterone in rats [13,14]. Previous studies have shown that maternal stress during pregnancy is associated with 3–4-time fold increase in GCs [9,104,105]. This is due to the constant activation and dysregulation of the HPA axis that is also observed in T2DM patients [25,26,27,28]. Studies show that pregnancy in women with T2DM worsen especially those who already have other complications such as uncontrolled hyperglycemia, hypertension or vascular diseases [106,107]. In the present study, we evaluated HPA axis activity by measuring two components of the HPA axis under basal non-stressful conditions and found both plasma ACTH levels and corticosterone concentrations were significantly increased in the PD pregnant female group when compared to the NPD pregnant female group in Figure 2. These results corroborated with the previous study that found a dysregulation of the HPA axis in male prediabetic animals evidenced by the elevated basal corticosterone concentration along with the unchanged ACTH concentration in non-stressed conditions [63]. In the rat, late pregnancy and in the postpartum period has been associated with reduced basal activity of the HPA axis with decreased ACTH and corticosterone [108,109,110]. Therefore, the basal increased concentrations of ACTH and corticosterone may be an indication that indeed the pre-existing prediabetic state in pregnancy maintained the impaired negative feedback and HPA axis dysregulation.
During development, fetuses need glucocorticoids for various aspects of brain development and late gestational lung maturation [111,112]. Exposure to adverse maternal cues, such as high glucocorticoids during critical developmental periods, have been shown to increase the risk of stress-related conditions such as depression, characterized by HPA-axis dysregulation [113,114]. Additionally, it could predispose individuals to cardiometabolic diseases and T2DM later in life [115,116]. The above is in line with the ‘fetal programming-hypothesis’. Exposure to glucocorticoid in utero is thought to compromise fetal brain development specifically, the prefrontal cortex, hippocampus and amygdala, brain areas associated with regulating the HPA axis [91,117,118]. Previous studies done in animals show that prenatal stress exposure alters hippocampal glucocorticoid receptor density, sensitivity which permanently alters the set-point and HPA axis regulation [119,120]. Studies show that these are observed from the very early neonatal, prepubertal and post-pubertal periods and appear to persist through to adulthood [121,122,123,124]. Research indicates that excessive cortisol exposure during early gestation triggers an early shift from tissue accretion to differentiation, thereby reducing fetal growth in various vital organs such as the brain, heart, liver, kidney and adrenal glands [125,126,127]. This process often leads to the clinical manifestation of intrauterine growth restriction (IUGR), characterized by the development of growth-retarded fetuses [128,129]. The diagnosis of IUGR is assigned to infants with a birth weight below the 10th percentile for gestational age [130,131]. In diabetic pregnancy, IUGR is observed most commonly in patients with vasculopathy (retinal, renal, or chronic hypertension) [37,38,39,132]. The association between low birth weight and elevated plasma cortisol concentrations, hypertension, cardiovascular diseases, T2DM and mental disorders has been documented by epidemiological studies [133,134]. However, the impact of maternal dysregulated HPA axis function in pregestational prediabetes and its influence on fetal HPA axis and postnatal offspring development has not yet been explored. Therefore, leading to the next phase of the study.
The fetal pituitary matures first, with fetal HPA activity beginning at midgestation [19,135]. The actions of the fetal HPA axis are essential in fetal development, maturation and homeostasis and eventually prepare for the survival of the neonate [135,136]. After birth, the HPA axis is able to regulate responses to adverse conditions, acting on the metabolism of carbohydrates, proteins and lipids and participating in anti-inflammatory effects and suppression of the immune response [137,138]. However, research has been shown that exposure to glucocorticoids in utero is associated with increased adrenocortical function in juvenile period with increased fasting cortisol concentrations in adults [139,140]. The upregulation of postnatal HPA function in other species may reflect changes in the HPA axis at the level of the hypothalamus, pituitary or adrenal gland itself [141,142]. Previous studies that have shown maternal and fetal/newborn cortisol levels are correlated and maternal cortisol levels are associated with reactivity of the newborn HPA axis [22,143]. Therefore, we evaluated ACTH and corticosterone concentrations and found a significant increase in all developmental stages in pups born from PD group when compared to pups born from NPD group in Figure 3. These results indeed correlate with maternal increased ACTH and corticosterone concentrations in this study. However, previous studies have shown that prolonged and continual increase in glucocorticoids travels to the brain where constant, elevated corticosterone in the highly regulated brain and cause constant activation of HPA axis thus HPA axis hyperactivity [144,145]. Therefore, the prolonged increase of ACTH and corticosterone in all developmental stages in our study may have resulted in HPA axis hyperactivity.
In addition, HPA axis activity is modulated by a feedback regulation of glucocorticoids exerted by two different types of receptors the MR and the GR [145,146]. The relationship between MR and GR is also critical to negative feedback as the two receptors act co-ordinately to reduce corticosterone secretion by inhibiting ACTH secretion following exposure to stress and maintain homeostatic balance at rest [147,148]. This balance is critical for the normal function of HPA axis [149]. However, excess glucocorticoid exposure in utero has been shown to reduce the number of both glucocorticoid and mineralocorticoid receptors in the hippocampus subsequently alters the set point of fetal HPA axis evident after birth [30,117,129]. Hence, the present study evaluated hippocampal GR and MR gene expressions. In this study, the pups born from PD group had significantly decreased in GR gene expression while MR gene expression had significant increased consistent in all developmental stages when compared to pups born from NPD group in Figure 4. The decreased GR expression correlates with previous study and supports that maternal glucocorticoids in our study may have overwhelmed the placenta barriers and crossed over the placenta and entered the pup brains and occupied GR leading to GR resistance and decreased it expression even in non-stressful conditions. However, the increase MR gene expression contrasted with other studies that show a decrease in it expression. Studies show that excess glucocorticoid exposure during utero can lead to changes in DNA methylation patterns, histone modification and microRNA expression that influence gene expression pattern [40,150]. These epigenetic changes may specifically up regulate MR gene expression and down regulate GR expression as a regulatory response to cope with the persistent elevated levels of glucocorticoid in utero and even after birth [42,151]. Therefore, the imbalances of GR and MR gene expression in our result may have been due to compensatory mechanism that occurred during utero and persistent after birth to emerging adulthood. Our study supports the hypothesis that prenatal exposure to excess glucocorticoid promotes persistent changes in the HPA axis evident by increased ACTH, corticosterone and imbalances in GR and MR gene expressions. Furthermore, studies show that the adrenal glands may undergo adrenocortical hypertrophy as a result of increased production of corticosterone seen in this study [152,153]. This excessive corticosterone secretion has been shown to lead to enlargement of the adrenal glands over time which correlates with the findings of the present study in Figure 5.
Moreover, other studies have provided further mechanistic insight into HPA axis programming [154,155,156,157]. Previous research that shown that fetal exposure to maternal high glucocorticoid can programme changes in the HPA axis while also reducing fetal growth evident as low birth weight [158,159]. In addition, previous studies shown that about 30% of all infants with lower birth weight show catch-up growth during the first 2 years of life and this is to compensate for their genetically determined growth trajectory [160,161,162]. The detrimental effects of catch-up growth in humans have been associated with development of glucose intolerance, insulin resistance, T2DM, hypertension and cardiovascular disease in adulthood [163,164,165]. In the present study, the pups born from PD group had significant lower body weight when compared to pups born from NPD group Figure 6a. The results corroborated with previous findings that have shown high maternal glucocorticoid also observed in the study disturbed fetus development and reduced fetal growth evident to low body weight. However, in this study, the pups born from PD group had no significant change in body weight in the juvenile and emerging adults periods while at prepubertal period there was a significant increase in body weight when compared to pups born from NPD group in Figure 6b. Therefore, we deduced that the body weight in the developmental stages showed absence of catch-up growth due to the body weight discrepancy. Studies show that the absence of catch-up growth may play important role in protecting the animals from adverse metabolic outcomes in the long term and to prevent catch up growth deterioration effect [166,167].
Moreover, studies show that elevated maternal GC levels during pregnancy influences both programming of the fetal HPA axis and metabolic pathways such as glucose metabolism [9,42]. This programming can lead to alterations in the development of tissues such as skeletal muscle involved in glucose homeostasis, as a result of fetal programming during the period of growth restriction [168,169]. Prenatal studies shown that GC exposure causes hyperglycemia following oral glucose in male offspring with alterations in the expression of genes that mediate GC [9,170]. Several studies show that low birth weight offspring have been associated with glucose intolerance, insulin resistance and T2DM in adulthood [50,171]. Hence, in our study we evaluated glucose tolerance in pups. In the present study, there was impaired fasting glucose as there was a significant increase in blood glucose concentration the PD group of all developmental stages in Figure 7. There was also evidence of impaired glucose tolerance in the PD group as the blood glucose concentrations remained higher and a failure of blood glucose concentration to return to base line after the 2-hr test in all developmental stages. Given that glucocorticoids have been shown to inhibit the pancreatic-β cells from secreting insulin directly, impair insulin-mediated glucose uptake and interfere in the insulin signalling cascade in peripheral tissues such as skeletal muscle while increasing energy availability by increasing glucose output from the liver [172,173]. Therefore, the observed HPA axis hyperactivity with increased basal glucocorticoid in our study may have caused a continuous increase in blood glucose concentration especially in a fasting state contributing to the impaired fasting glucose and glucose intolerance in our study. On the other hand, the absence of catch-up growth in our study did not diminish or prevent glucose intolerance.
5. Conclusion
In conclusion, the findings of this study showed that pregestational prediabetes may be associated with maternal dysregulated function of the HPA axis as evidenced by the elevated ACTH, corticosterone concentrations in the dams. The findings of the study further shows that maternal dysregulation in HPA axis alters the set-point and development of the offspring’s HPA axis as evidenced by the elevated ACTH, corticosterone concentrations, impaired GR and MR gene expression followed by increased adrenal gland weights in non-stressed conditions in the offspring. In addition, this study shows that maternal HPA axis dysregulation is also associated with reduced fetal growth manifested as lower body weight and lack of catch-up growth during development. However, the offspring still exhibit catch up growth detrimental effects such as impaired fasting glucose and glucose intolerance, resembling features seen in T2DM studies. Overall, maternal pregestational prediabetes leads to hyperactivity in the offspring’s HPA axis with impaired negative feedback mechanisms from childhood to early adulthood. These findings suggest parallels between pregestational prediabetes, T2DM and maternal stress in terms of their influence on HPA axis function and postnatal development, even under normal dietary and non-stressed conditions. Moreover, offspring from prediabetic pregnancies may be at risk of early-onset prediabetes and mental health disorders associated with HPA axis hyperactivity. Therefore, monitoring maternal periconceptional health status and conditions during pregnancy may be of great importance.
Author Contributions
Conceptualization, M.N., A.K., N.D.X. and P.S.N.; writing—original draft preparation, M.N.; writing—review and editing, A.K. and P.S.N.; visualization, M.N. and A.K.; supervision, A.K., N.D.X. and P.S.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The animal study protocol was reviewed and approved by the Animal Research Ethics Committee (AREC) of the University of KwaZulu-Natal, South Africa (AREC/032/020D).
Data Availability Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The authors would like to acknowledge the endocrine group for their support and the Biomedical Resource Units of KwaZulu-Natal for their technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| ADA | American Diabetes Association |
| AREC | Animal Research Ethics Committee |
| BBB | Blood brain barrier |
| BRU | Biomedical Research Unit |
| CRH | Corticotropic releasing hormone |
| DNA | Deoxyribonucleic acid |
| DOHaD | Developmental origins of health and disease |
| ELISA | Enzyme linked immunosorbent assay. |
| GC | Glucocorticoids |
| GND | Gestational day |
| GR | Glucocorticoid receptor |
| HbA1c | Glycated haemoglobin A1c |
| HFHC | High fat high carbohydrate |
| HPA | Hypothalamic–adrenal–pituitary |
| IFG | Impaired fasting glucose |
| IGT | Impaired glucose tolerance |
| IUGR | Intrauterine growth restriction |
| MR | Mineralocorticoid receptor |
| NPD | Non-prediabetes |
| OGTT | Oral glucose tolerance test |
| PD | Prediabetes |
| PCR | Polymerase chain reaction |
| RNA | Ribonucleic acid |
| T2DM | Type 2 diabetes mellitus |
| UKZN | University of KwaZulu-Natal |
References
- Lemley, C. , Fetal programming: maternal-fetal interactions and postnatal performance. Clinical Theriogenology 2020, 12, 252–267. [Google Scholar]
- Cerritelli, F.; Frasch, M.G.; Antonelli, M.C.; Viglione, C.; Vecchi, S.; Chiera, M.; Manzotti, A. A Review on the Vagus Nerve and Autonomic Nervous System During Fetal Development: Searching for Critical Windows. Front. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Gundacker, A.; Rico, L.C.; Stoehrmann, P.; Tillmann, K.E.; Weber-Stadlbauer, U.; Pollak, D.D. Interaction of the pre- and postnatal environment in the maternal immune activation model. Discov. Ment. Heal. 2023, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.J.; Tang, J.I.; Nyirenda, M.J. Mechanisms underlying the role of glucocorticoids in the early life programming of adult disease. Clin. Sci. 2007, 113, 219–232. [Google Scholar] [CrossRef]
- Braun, T.; Challis, J.R.; Newnham, J.P.; Sloboda, D.M. Early-Life Glucocorticoid Exposure: The Hypothalamic-Pituitary-Adrenal Axis, Placental Function, and Long-term Disease Risk. Endocr. Rev. 2013, 34, 885–916. [Google Scholar] [CrossRef] [PubMed]
- Solano, M.E.; Holmes, M.C.; Mittelstadt, P.R.; Chapman, K.E.; Tolosa, E. Antenatal endogenous and exogenous glucocorticoids and their impact on immune ontogeny and long-term immunity. Semin. Immunopathol. 2016, 38, 739–763. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.J.; De Oliveira, J.C.; Giachini, F.R.; Lima, V.V.; Tostes, R.C.; Bomfim, G.F. Programming of Vascular Dysfunction by Maternal Stress: Immune System Implications. Front. Physiol. 2022, 13, 787617. [Google Scholar] [CrossRef] [PubMed]
- Low, F.M., P. D. Gluckman, and M.A. Hanson, Epigenetic and developmental basis of risk of obesity and metabolic disease, in Cellular Endocrinology in Health and Disease. 2021, Elsevier. p. 289-313.
- Sheng, J.A.; Bales, N.J.; Myers, S.A.; Bautista, A.I.; Roueinfar, M.; Hale, T.M.; Handa, R.J. The Hypothalamic-Pituitary-Adrenal Axis: Development, Programming Actions of Hormones, and Maternal-Fetal Interactions. Front. Behav. Neurosci. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Zhang, L. Role of the hypothalamic–pituitary–adrenal axis in developmental programming of health and disease. Front. Neuroendocr. 2012, 34, 27–46. [Google Scholar] [CrossRef]
- Seckl, J.R. , Glucocorticoids, developmental ‘programming’and the risk of affective dysfunction. Progress in brain research 2007, 167, 17–34. [Google Scholar]
- Lesage, J.; Sebaai, N.; Leonhardt, M.; Dutriez-Casteloot, I.; Breton, C.; Deloof, S.; Vieau, D. Perinatal maternal undernutrition programs the offspring hypothalamo–pituitary–adrenal (HPA) axis. Stress 2006, 9, 183–198. [Google Scholar] [CrossRef]
- Kapoor, A.; Petropoulos, S.; Matthews, S.G. Fetal programming of hypothalamic–pituitary–adrenal (HPA) axis function and behavior by synthetic glucocorticoids. Brain Res. Rev. 2008, 57, 586–595. [Google Scholar] [CrossRef]
- Spencer, R.L.; Deak, T. A users guide to HPA axis research. Physiol. Behav. 2016, 178, 43–65. [Google Scholar] [CrossRef]
- Kalantaridou, S.; Zoumakis, E.; Makrigiannakis, A.; Lavasidis, L.; Vrekoussis, T.; Chrousos, G. Corticotropin-releasing hormone, stress and human reproduction: an update. J. Reprod. Immunol. 2010, 85, 33–39. [Google Scholar] [CrossRef]
- Sirianni, R.; Rehman, K.S.; Carr, B.R.; Parker, C.R.; Rainey, W.E. Corticotropin-Releasing Hormone Directly Stimulates Cortisol and the Cortisol Biosynthetic Pathway in Human Fetal Adrenal Cells. J. Clin. Endocrinol. Metab. 2005, 90, 279–285. [Google Scholar] [CrossRef]
- Iliodromiti, Z.; Antonakopoulos, N.; Sifakis, S.; Tsikouras, P.; Daniilidis, A.; Dafopoulos, K.; Botsis, D.; Vrachnis, N. Endocrine, paracrine, and autocrine placental mediators in labor. Hormones 2012, 11, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Stocker, C.J.; Arch, J.R.S.; Cawthorne, M.A. Fetal origins of insulin resistance and obesity. Proc. Nutr. Soc. 2005, 64, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Mastorakos, G.; Ilias, I. Maternal and Fetal Hypothalamic-Pituitary-Adrenal Axes During Pregnancy and Postpartum. Ann. New York Acad. Sci. 2003, 997, 136–149. [Google Scholar] [CrossRef] [PubMed]
- St-Jean, M., I. Bourdeau, and A. Lacroix, Adrenal cortex and medulla physiology during pregnancy, labor, and puerperium, in Maternal-Fetal and Neonatal Endocrinology. 2020, Elsevier. p. 101-116.
- Lee, J.H. and D.J. Torpy, Adrenal insufficiency in pregnancy: Physiology, diagnosis, management and areas for future research. Reviews in Endocrine and Metabolic Disorders. 2023, 24, 57–69.
- Duthie, L.; Reynolds, R.M. Changes in the Maternal Hypothalamic-Pituitary-Adrenal Axis in Pregnancy and Postpartum: Influences on Maternal and Fetal Outcomes. Neuroendocrinology 2013, 98, 106–115. [Google Scholar] [CrossRef]
- Ruffaner-Hanson, C.; Noor, S.; Sun, M.S.; Solomon, E.; Marquez, L.E.; Rodriguez, D.E.; Allan, A.M.; Caldwell, K.K.; Bakhireva, L.N.; Milligan, E.D. The maternal-placental-fetal interface: Adaptations of the HPA axis and immune mediators following maternal stress and prenatal alcohol exposure. Exp. Neurol. 2022, 355, 114121–114121. [Google Scholar] [CrossRef]
- Valsamakis, G.; Chrousos, G.; Mastorakos, G. Stress, female reproduction and pregnancy. Psychoneuroendocrinology 2018, 100, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M. The potential influence of maternal stress hormones on development and mental health of the offspring. Brain, Behav. Immun. 2005, 19, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Sze, Y. and P.J. Brunton, How is prenatal stress transmitted from the mother to the fetus? Journal of Experimental Biology 2024, 227(Suppl_1).
- Joseph, J.J.; Golden, S.H. Cortisol dysregulation: the bidirectional link between stress, depression, and type 2 diabetes mellitus. Ann. New York Acad. Sci. 2016, 1391, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Kyrou, I., H. Randeva, and C. Tsigos, Stress, insulin resistance, and type 2 diabetes. Stress: Neuroendocrinology and neurobiology, handbook of stress 2017, 2, 351–358. [Google Scholar]
- Nugent, J.L. , Effects of Glucocorticoids on Placental Development and Function: Implications for Fetal Growth Restriction. 2012: The University of Manchester (United Kingdom).
- Krontira, A.C.; Cruceanu, C.; Binder, E.B. Glucocorticoids as Mediators of Adverse Outcomes of Prenatal Stress. Trends Neurosci. 2020, 43, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Verma, A.; Garg, R.; Singh, J.; Verma, H. Cardiometabolic Risk Factors Associated With Type 2 Diabetes Mellitus: A Mechanistic Insight. Clin. Med. Insights: Endocrinol. Diabetes 2023, 16. [Google Scholar] [CrossRef]
- Lima, J.E.; Moreira, N.C.; Sakamoto-Hojo, E.T. Mechanisms underlying the pathophysiology of type 2 diabetes: From risk factors to oxidative stress, metabolic dysfunction, and hyperglycemia. Mutat. Res. Toxicol. Environ. Mutagen. 2021, 874-875, 503437. [Google Scholar] [CrossRef] [PubMed]
- Boekelheide, K.; Blumberg, B.; Chapin, R.E.; Cote, I.; Graziano, J.H.; Janesick, A.; Lane, R.; Lillycrop, K.; Myatt, L.; States, J.C.; et al. Predicting Later-Life Outcomes of Early-Life Exposures. Environ. Heal. Perspect. 2012, 120, 1353–1361. [Google Scholar] [CrossRef]
- Agarwal, P.; Morriseau, T.S.; Kereliuk, S.M.; Doucette, C.A.; Wicklow, B.A.; Dolinsky, V.W. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit. Rev. Clin. Lab. Sci. 2018, 55, 71–101. [Google Scholar] [CrossRef]
- Langley-Evans, S.C. Early life programming of health and disease: The long-term consequences of obesity in pregnancy. J. Hum. Nutr. Diet. 2022, 35, 816–832. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wang, W.; Zuo, R.; Sun, K. Mechanisms for establishment of the placental glucocorticoid barrier, a guard for life. Cell. Mol. Life Sci. 2018, 76, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.R. , Fetal growth in diabetic pregnancy. Clinical obstetrics and gynecology 1997, 40, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Weiss, U.; Cervar, M.; Puerstner, P.; Schmut, O.; Haas, J.; Mauschitz, R.; Arikan, G.; Desoye, G. Hyperglycaemia in vitro alters the proliferation and mitochondrial activity of the choriocarcinoma cell lines BeWo, JAR and JEG-3 as models for human first-trimester trophoblast. Diabetologia 2001, 44, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Weiss, U.; Cervar, M.; Puerstner, P.; Schmut, O.; Haas, J.; Mauschitz, R.; Arikan, G.; Desoye, G. Hyperglycaemia in vitro alters the proliferation and mitochondrial activity of the choriocarcinoma cell lines BeWo, JAR and JEG-3 as models for human first-trimester trophoblast. Diabetologia 2001, 44, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Moisiadis, V.G.; Matthews, S.G. Glucocorticoids and fetal programming part 2: mechanisms. Nat. Rev. Endocrinol. 2014, 10, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Howland, M.A.; Sandman, C.A.; Glynn, L.M. Developmental origins of the human hypothalamic-pituitary-adrenal axis. Expert Rev. Endocrinol. Metab. 2017, 12, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Grace, C.E.; Kim, S.-J.; Rogers, J.M. Maternal influences on epigenetic programming of the developing hypothalamic-pituitary-adrenal axis. Birth Defects Res. Part A: Clin. Mol. Teratol. 2011, 91, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Selvam, N.; K, J.; Mithra, P. Mediation effect of cord blood cortisol levels between maternal prepregnancy body mass index and birth weight: a hospital-based cross-sectional study. Clin. Exp. Pediatr. 2022, 65, 500–506. [Google Scholar] [CrossRef]
- de Mendonça, E.L.S.S., et al., Premature birth, low birth weight, small for gestational age and chronic non-communicable diseases in adult life: A systematic review with meta-analysis. Early human development. 2020, 149, 105154.
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Starikov, R.; Has, P.; Wu, R.; Nelson, D.M.; He, M. Small-for-gestational age placentas associate with an increased risk of adverse outcomes in pregnancies complicated by either type I or type II pre-gestational diabetes mellitus. J. Matern. Neonatal Med. 2020, 35, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.; Jacob, C.; Barker, M.; Fall, C.H.D.; Hanson, M.; Harvey, N.C.; Inskip, H.M.; Kumaran, K.; Cooper, C. Developmental Origins of Health and Disease: A Lifecourse Approach to the Prevention of Non-Communicable Diseases. Healthcare 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Chacko, A.; Carpenter, D.O.; Callaway, L.; Sly, P.D. Early-life risk factors for chronic nonrespiratory diseases. Eur. Respir. J. 2014, 45, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D. BIRTH WEIGHT AND ADULTHOOD DISEASE AND THE CONTROVERSIES. Fetal Matern. Med. Rev. 2006, 17, 205–227. [Google Scholar] [CrossRef]
- Tian, M.; Reichetzeder, C.; Li, J.; Hocher, B. Low birth weight, a risk factor for diseases in later life, is a surrogate of insulin resistance at birth. J. Hypertens. 2019, 37, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Grella, P. , Low birth weight and early life origins of adult disease: insulin resistance and type 2 diabetes. Clinical and Experimental Obstetrics & Gynecology 2007, 34, 9–13. [Google Scholar]
- Buysschaert, M.; Medina, J.L.; Bergman, M.; Shah, A.; Lonier, J. Prediabetes and associated disorders. Endocrine 2014, 48, 371–393. [Google Scholar] [CrossRef]
- Khan, R.M.M.; Chua, Z.J.Y.; Tan, J.C.; Yang, Y.; Liao, Z.; Zhao, Y. From Pre-Diabetes to Diabetes: Diagnosis, Treatments and Translational Research. Medicina 2019, 55, 546. [Google Scholar] [CrossRef]
- Magliano, D.J.; Sacre, J.W.; Harding, J.L.; Gregg, E.W.; Zimmet, P.Z.; Shaw, J.E. Young-onset type 2 diabetes mellitus — implications for morbidity and mortality. Nat. Rev. Endocrinol. 2020, 16, 321–331. [Google Scholar] [CrossRef]
- Kaur, G.; Lakshmi, P.V.M.; Rastogi, A.; Bhansali, A.; Jain, S.; Teerawattananon, Y.; Bano, H.; Prinja, S. Diagnostic accuracy of tests for type 2 diabetes and prediabetes: A systematic review and meta-analysis. PLOS ONE 2020, 15, e0242415. [Google Scholar] [CrossRef]
- Colagiuri, S. Definition and Classification of Diabetes and Prediabetes and Emerging Data on Phenotypes. Endocrinol. Metab. Clin. North Am. 2021, 50, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Hostalek, U. Global epidemiology of prediabetes - present and future perspectives. Clin. Diabetes Endocrinol. 2019, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Li, J.; Yan, N.; Li, N.; Liu, X.; Li, X.; Zhang, J.; Wang, Q.; Yang, C.; Qiu, J.; et al. Increased TG Levels and HOMA-IR Score Are Associated With a High Risk of Prediabetes: A Prospective Study. Asia Pac. J. Public Heal. 2023, 35, 413–419. [Google Scholar] [CrossRef]
- Tabák, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimäki, M. Prediabetes: a high-risk state for diabetes development. Lancet 2012, 379, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Khathi, A.; Luvuno, M.; Mabandla, M. VOLUNTARY INGESTION OF A HIGH-FAT HIGH-CARBOHYDRATE DIET: A MODEL FOR PREDIABETES. 2018, 74. 74. [CrossRef]
- Mzimela, N.C.; Ngubane, P.S.; Khathi, A. The changes in immune cell concentration during the progression of pre-diabetes to type 2 diabetes in a high-fat high-carbohydrate diet-induced pre-diabetic rat model. Autoimmunity 2019, 52, 27–36. [Google Scholar] [CrossRef]
- Naidoo, K.; Ngubane, P.S.; Khathi, A. Investigating the Effects of Diet-Induced Pre-Diabetes on the Functioning of Calcium-Regulating Organs in Male Sprague Dawley Rats: Effects on Selected Markers. Front. Endocrinol. 2022, 13, 914189. [Google Scholar] [CrossRef]
- Mosili, P.; Mkhize, B.C.; Ngubane, P.; Sibiya, N.; Khathi, A. The dysregulation of the hypothalamic–pituitary–adrenal axis in diet-induced prediabetic male Sprague Dawley rats. Nutr. Metab. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Mapanga, R.F.; Tufts, M.A.; Shode, F.O.; Musabayane, C.T. Renal Effects of Plant-Derived Oleanolic Acid in Streptozotocin-Induced Diabetic Rats. Ren. Fail. 2009, 31, 481–491. [Google Scholar] [CrossRef]
- S, P.; R, R.; R, K. Blood sample collection in small laboratory animals. J. Pharmacol. Pharmacother. 2010, 1, 87–93. [Google Scholar] [CrossRef]
- Qulu, L.; Daniels, W.; Mabandla, M.V. Exposure to prenatal stress has deleterious effects on hippocampal function in a febrile seizure rat model. Brain Res. 2015, 1624, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Mkhize, N.V.P.; Qulu, L.; Mabandla, M.V. The Effect of Quercetin on Pro- and Anti-Inflammatory Cytokines in a Prenatally Stressed Rat Model of Febrile Seizures. J. Exp. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.S., et al., Development and maturation of the male reproductive system. Birth Defects Research Part B Developmental and Reproductive Toxicology. 2003, 68, 125–136.
- Ghasemi, A., S. Jeddi, and K. Kashfi, The laboratory rat: Age and body weight matter. EXCLI journal. 2021, 20, 1431.
- Arellano, J.I., A. Duque, and P. Rakic, A coming-of-age story: adult neurogenesis or adolescent neurogenesis in rodents? Frontiers in Neuroscience. 2024, 18, 1383728.
- Alberts, J.R. , Olfactory contributions to behavioral development in rodents. Mammalian olfaction, reproductive processes, and behavior, 1976, 67-94.
- Peijs, G.L.A.M. , Development of social behaviour in the rat. 1977, [Sl: sn].
- Makowska, I.J. , Understanding the welfare of rats living in standard versus semi-naturalistic laboratory environments. 2016, University of British Columbia.
- Foster, R.G. Sleep, circadian rhythms and health. Interface Focus 2020, 10, 20190098. [Google Scholar] [CrossRef] [PubMed]
- González, M.M.C. Dim Light at Night and Constant Darkness: Two Frequently Used Lighting Conditions That Jeopardize the Health and Well-being of Laboratory Rodents. Front. Neurol. 2018, 9, 609. [Google Scholar] [CrossRef] [PubMed]
- Schuler, C.B. and K.M. Hope, Circadian rhythm: Light-dark cycles. Integrative and Functional Medical Nutrition Therapy: Principles and Practices, 2020, 577-594.
- Sengupta, P. The Laboratory Rat: Relating Its Age With Human's. 2013, 4, 624–630.
- Sheard, P.; McCaig, C.; Harris, A. Critical periods in rat motoneuron development. Dev. Biol. 1984, 102, 21–31. [Google Scholar] [CrossRef]
- Picut, C.A.; Ziejewski, M.K.; Stanislaus, D. Comparative Aspects of Pre- and Postnatal Development of the Male Reproductive System. Birth Defects Res. 2017, 110, 190–227. [Google Scholar] [CrossRef]
- Beckman, D.A. and M. Feuston, Landmarks in the development of the female reproductive system. Birth Defects Research Part B: Developmental and Reproductive Toxicology. 2003, 68, 137–143.
- Bell, M.R. Comparing Postnatal Development of Gonadal Hormones and Associated Social Behaviors in Rats, Mice, and Humans. Endocrinology 2018, 159, 2596–2613. [Google Scholar] [CrossRef] [PubMed]
- Stanley, D.P.; Shetty, A.K. Aging in the rat hippocampus is associated with widespread reductions in the number of glutamate decarboxylase-67 positive interneurons but not interneuron degeneration. J. Neurochem. 2004, 89, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R. , Comparing rat’s to human’s age: how old is my rat in people years? Nutrition 2005, 21, 775. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S. and P. Sengupta, Rabbits and men: relating their ages. Journal of basic and clinical physiology and pharmacology. 2018, 29, 427–435.
- Wang, C.; Liu, Y.; Wang, H.; Gao, F.; Guan, X.; Shi, B. Maternal Exposure to Oxidized Soybean Oil Impairs Placental Development by Modulating Nutrient Transporters in a Rat Model. Mol. Nutr. Food Res. 2021, 65, 2100301. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, X.; Fang, M.; Chen, G.; Cao, J.; Qu, H.; Wang, H. Maternally derived low glucocorticoid mediates adrenal developmental programming alteration in offspring induced by dexamethasone. Sci. Total. Environ. 2021, 797, 149084. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.J.; Kim, Y.J. What is fetal programming?: a lifetime health is under the control of in utero health. Obstet. Gynecol. Sci. 2017, 60, 506–519. [Google Scholar] [CrossRef] [PubMed]
- ztürk, H.N.O. and P.F. Türker, Fetal programming: could intrauterin life affect health status in adulthood? Obstetrics & Gynecology Science. 2021, 64, 473.
- Moisiadis, V.G.; Matthews, S.G. Glucocorticoids and fetal programming part 1: outcomes. Nat. Rev. Endocrinol. 2014, 10, 391–402. [Google Scholar] [CrossRef]
- van Bodegom, M.; Homberg, J.R.; Henckens, M.J.A.G. Modulation of the Hypothalamic-Pituitary-Adrenal Axis by Early Life Stress Exposure. Front. Cell. Neurosci. 2017, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Pofi, R.; Tomlinson, J.W. Glucocorticoids in pregnancy. Obstet. Med. 2019, 13, 62–69. [Google Scholar] [CrossRef]
- Grilo, L.F.; Tocantins, C.; Diniz, M.S.; Gomes, R.M.; Oliveira, P.J.; Matafome, P.; Pereira, S.P. Metabolic Disease Programming: From Mitochondria to Epigenetics, Glucocorticoid Signalling and Beyond. Eur. J. Clin. Investig. 2021, 51, e13625. [Google Scholar] [CrossRef] [PubMed]
- Nakanga, W.P. , Accuracy and utility of fasting and stimulated glucose for diagnosis of diabetes in Sub-Saharan Africa. 2022: University of Exeter (United Kingdom).
- Newsholme, P.; Cruzat, V.; Arfuso, F.; Keane, K. Nutrient regulation of insulin secretion and action. J. Endocrinol. 2014, 221, R105–R120. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.D.; Maratou, E.; Kountouri, A.; Board, M.; Lambadiari, V. Regulation of Postabsorptive and Postprandial Glucose Metabolism by Insulin-Dependent and Insulin-Independent Mechanisms: An Integrative Approach. Nutrients 2021, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Hollander, and C. Spellman, Controversies in prediabetes: do we have a diagnosis? Postgraduate medicine. 2012, 124, 109–118.
- Kashyap, S.R.; Desouza, C.; Aroda, V.R.; Kim, S.H.; Neff, L.M.; Wu, S.S.; Raskin, P.; Pratley, R. Glycemic and metabolic sub-classification of prediabetes and risk factors for cardiovascular disease in the D2d cohort. Am. J. Prev. Cardiol. 2023, 15, 100525. [Google Scholar] [CrossRef] [PubMed]
- Solis-Herrera, C. , et al., Pathogenesis of type 2 diabetes mellitus. 2021. [Google Scholar]
- Chia, C.W.; Egan, J.M.; Ferrucci, L.; Lleva, R.R.; Inzucchi, S.E.; Egan, B.M.; Hennes, M.M.; Stepniakowski, K.T.; O’shaughnessy, I.M.; Kissebah, A.H.; et al. Age-Related Changes in Glucose Metabolism, Hyperglycemia, and Cardiovascular Risk. Circ. Res. 2018, 123, 886–904. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, A.; Mavropulo-Stolyarenko, G.; Karonova, T.; Thieme, D.; Hoehenwarter, W.; Ihling, C.; Stefanov, V.; Grishina, T.; Frolov, A. Multiple Glycation Sites in Blood Plasma Proteins as an Integrated Biomarker of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2019, 20, 2329. [Google Scholar] [CrossRef]
- Awasthi, S.; Saraswathi, N.T. Non-enzymatic glycation mediated structure–function changes in proteins: case of serum albumin. RSC Adv. 2016, 6, 90739–90753. [Google Scholar] [CrossRef]
- Díaz, A.; López-Grueso, R.; Gambini, J.; Monleón, D.; Mas-Bargues, C.; Abdelaziz, K.M.; Viña, J.; Borrás, C. Sex Differences in Age-Associated Type 2 Diabetes in Rats—Role of Estrogens and Oxidative Stress. Oxidative Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sandman, C.A.; Glynn, L.; Schetter, C.D.; Wadhwa, P.; Garite, T.; Chicz-DeMet, A.; Hobel, C. Elevated maternal cortisol early in pregnancy predicts third trimester levels of placental corticotropin releasing hormone (CRH): Priming the placental clock. Peptides 2006, 27, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Bleker, L.S.; van Dammen, L.; Leeflang, M.M.; Limpens, J.; Roseboom, T.J.; de Rooij, S.R. Hypothalamic-pituitary-adrenal axis and autonomic nervous system reactivity in children prenatally exposed to maternal depression: A systematic review of prospective studies. Neurosci. Biobehav. Rev. 2018, 117, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Kokkinopoulou, I., A. Diakoumi, and P. Moutsatsou, Glucocorticoid receptor signaling in diabetes. International journal of molecular sciences. 2021, 22, 11173.
- Bruehl, H.; Rueger, M.; Dziobek, I.; Sweat, V.; Tirsi, A.; Javier, E.; Arentoft, A.; Wolf, O.T.; Convit, A. Hypothalamic-Pituitary-Adrenal Axis Dysregulation and Memory Impairments in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2007, 92, 2439–2445. [Google Scholar] [CrossRef]
- Johnstone, H.A.; Wigger, A.; Douglas, A.J.; Neumann, I.D.; Landgraf, R.; Seckl, J.R.; Russell, J.A. Attenuation of Hypothalamic-Pituitary-Adrenal Axis Stress Responses in Late Pregnancy: Changes in Feedforward and Feedback Mechanisms. J. Neuroendocr. 2000, 12, 811–822. [Google Scholar] [CrossRef]
- Neumann, I.D. INCREASED BASAL ACTIVITY OF THE HYPOTHALAMO–PITUITARY–ADRENAL AXIS DURING PREGNANCY IN RATS BRED FOR HIGH ANXIETY-RELATED BEHAVIOUR. Psychoneuroendocrinology 1998, 23, 449–463. [Google Scholar] [CrossRef]
- Brunton, P.J. Resetting the Dynamic Range of Hypothalamic-Pituitary-Adrenal Axis Stress Responses Through Pregnancy. J. Neuroendocr. 2010, 22, 1198–1213. [Google Scholar] [CrossRef]
- Morsi, A.; DeFranco, D.; Witchel, S.F. The Hypothalamic-Pituitary-Adrenal Axis and the Fetus. Horm. Res. Paediatr. 2018, 89, 380–387. [Google Scholar] [CrossRef]
- Berens, A.E. and C.A. Nelson, Neurobiology of fetal and infant development. Handbook of infant mental health, 2019, 41-62.
- Bale, T.L.; Baram, T.Z.; Brown, A.S.; Goldstein, J.M.; Insel, T.R.; McCarthy, M.M.; Nemeroff, C.B.; Reyes, T.M.; Simerly, R.B.; Susser, E.S.; et al. Early Life Programming and Neurodevelopmental Disorders. Biol. Psychiatry 2010, 68, 314–319. [Google Scholar] [CrossRef]
- O’Leary, N. , An investigation of the role of antenatal depression, maternal cortisol and postnatal interactive behaviour on infant neurodevelopment in the first year of life. 2019, Trinity College.
- Rajendram, R., V. R. Preedy, and V.B. Patel, Diet, nutrition, and fetal programming. 2017, Springer.
- Castellano, J.M. and M. Tena-Sempere, Animal modeling of early programming and disruption of pubertal maturation. Puberty from Bench to Clinic. 2016, 29, 87–121.
- Miranda, A.; Sousa, N. Maternal hormonal milieu influence on fetal brain development. Brain Behav. 2018, 8, e00920. [Google Scholar] [CrossRef] [PubMed]
- Rufaka, A. , Does maternal stress, depression and anxiety affect fetal neurobehavioral development? a review. 2021, Brac University.
- Welberg, L.A. and J.R. Seckl, Prenatal stress, glucocorticoids and the programming of the brain. Journal of neuroendocrinology. 2001, 13, 113–128.
- Galeeva, A.; Ordyan, N.; Pivina, S.; Pelto-Huikko, M. Expression of glucocorticoid receptors in the hippocampal region of the rat brain during postnatal development. J. Chem. Neuroanat. 2006, 31, 216–225. [Google Scholar] [CrossRef]
- Pryce, C.R.; Aubert, Y.; Maier, C.; Pearce, P.C.; Fuchs, E. The developmental impact of prenatal stress, prenatal dexamethasone and postnatal social stress on physiology, behaviour and neuroanatomy of primate offspring: studies in rhesus macaque and common marmoset. Psychopharmacol. 2010, 214, 33–53. [Google Scholar] [CrossRef]
- Coe, C.L.; Kramer, M.; Czéh, B.; Gould, E.; Reeves, A.J.; Kirschbaum, C.; Fuchs, E. Prenatal stress diminishes neurogenesis in the dentate gyrus of juvenile Rhesus monkeys. Biol. Psychiatry 2003, 54, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Juruena, M.F. Early-life stress and HPA axis trigger recurrent adulthood depression. Epilepsy Behav. 2014, 38, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.G.; Owen, D.; Kalabis, G.; Banjanin, S.; Setiawan, E.B.; Dunn, E.A.; Andrews, M.H. Fetal Glucocorticoid Exposure and Hypothalamo-Pituitary-Adrenal (HPA) Function After Birth. Endocr. Res. 2004, 30, 827–836. [Google Scholar] [CrossRef]
- FowdenA. L.; ForheadA.J. Endocrine mechanisms of intrauterine programming. Reproduction 2004, 127, 515–526. [Google Scholar] [CrossRef]
- Fowden, A.L.; Forhead, A.J. Glucocorticoids as regulatory signals during intrauterine development. Exp. Physiol. 2015, 100, 1477–1487. [Google Scholar] [CrossRef]
- Katugampola, H., E. F. Gevers, and M.T. Dattani, Fetal Endocrinology. Brook’s Clinical Pediatric Endocrinology, 2019, 47-104.
- Fisher, D.A. , Endocrinology of fetal development. Williams textbook of endocrinology, 1998, 811-841.
- Fowden, A.; Valenzuela, O.; Vaughan, O.; Jellyman, J.; Forhead, A. Glucocorticoid programming of intrauterine development. Domest. Anim. Endocrinol. 2016, 56, S121–S132. [Google Scholar] [CrossRef] [PubMed]
- Suhag, A.; Berghella, V. Intrauterine Growth Restriction (IUGR): Etiology and Diagnosis. Curr. Obstet. Gynecol. Rep. 2013, 2, 102–111. [Google Scholar] [CrossRef]
- Dinu, M.; Stancioi-Cismaru, A.F.; Gheonea, M.; Luciu, E.D.; Aron, R.M.; Pana, R.C.; Marinas, C.M.; Degeratu, S.; Sorop-Florea, M.; Carp-Veliscu, A.; et al. Intrauterine Growth Restriction—Prediction and Peripartum Data on Hospital Care. Medicina 2023, 59, 773. [Google Scholar] [CrossRef] [PubMed]
- AlKhalifa, M.A.; Hsu, S.; Raza, G.; Ismail, M.S. Diabetes in pregnancy: A comparison of guidelines. 2021, 7. 7. [CrossRef]
- Phillips, D.I.W.; Walker, B.R.; Reynolds, R.M.; Flanagan, D.E.H.; Wood, P.J.; Osmond, C.; Barker, D.J.P.; Whorwood, C.B. Low Birth Weight Predicts Elevated Plasma Cortisol Concentrations in Adults From 3 Populations. Hypertension 2000, 35, 1301–1306. [Google Scholar] [CrossRef]
- Levitt, N.S., et al., Impaired glucose tolerance and elevated blood pressure in low birth weight, nonobese, young South African adults: early programming of cortisol axis. The Journal of Clinical Endocrinology & Metabolism 2000, 85, 4611–4618. 2000, 85, 4611–4618.
- Katugampola, H., M. Cerbone, and M.T. Dattani, Normal hypothalamic and Pituitary Development and Physiology in the Fetus and Neonate, in Maternal-Fetal and Neonatal Endocrinology. 2020, Elsevier. p. 527-545.
- Antonini, S.R., M. F. Stecchini, and F.S. Ramalho, Development and function of the adrenal cortex and medulla in the fetus and neonate, in Maternal-Fetal and Neonatal Endocrinology. 2020, Elsevier. p. 611-623.
- Androulakis, I.P. Circadian rhythms and the HPA axis: A systems view. Wiley Interdiscip. Rev. Syst. Biol. Med. 2021, 13, e1518–e1518. [Google Scholar] [CrossRef]
- Kamgang, V.W., M. Murkwe, and M. Wankeu-Nya, Biological Effects of Cortisol. 2023.
- Poore, K. and A. Fowden, The effect of birth weight on hypothalamo-pituitary-adrenal axis function in juvenile and adult pigs. The Journal of physiology. 2003, 547, 107–116.
- Phillips, D., et al., Elevated plasma cortisol concentrations: a link between low birth weight and the insulin resistance syndrome? The Journal of Clinical Endocrinology & Metabolism. 1998, 83, 757–760.
- Campana, G.; Loizzo, S.; Fortuna, A.; Rimondini, R.; Maroccia, Z.; Scillitani, A.; Falchetti, A.; Spampinato, S.M.; Persani, L.; Chiodini, I. Early post-natal life stress induces permanent adrenocorticotropin-dependent hypercortisolism in male mice. Endocrine 2021, 73, 186–195. [Google Scholar] [CrossRef]
- Fowden, A.L.; Vaughan, O.R.; Murray, A.J.; Forhead, A.J. Metabolic Consequences of Glucocorticoid Exposure before Birth. Nutrients 2022, 14, 2304. [Google Scholar] [CrossRef]
- Reynolds, R.M. Glucocorticoid excess and the developmental origins of disease: Two decades of testing the hypothesis – 2012 Curt Richter Award Winner. Psychoneuroendocrinology 2013, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.R.; Spencer-Segal, J.L. Glucocorticoids and the Brain after Critical Illness. Endocrinology 2021, 162. [Google Scholar] [CrossRef] [PubMed]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef]
- Harris, A.; Holmes, M.; de Kloet, E.; Chapman, K.; Seckl, J. Mineralocorticoid and glucocorticoid receptor balance in control of HPA axis and behaviour. Psychoneuroendocrinology 2012, 38, 648–658. [Google Scholar] [CrossRef]
- Ladd, C.; Huot, R.L.; Thrivikraman, K.; Nemeroff, C.B.; Plotsky, P.M. Long-term adaptations in glucocorticoid receptor and mineralocorticoid receptor mrna and negative feedback on the hypothalamo-pituitary-adrenal axis following neonatal maternal separation. Biol. Psychiatry 2004, 55, 367–375. [Google Scholar] [CrossRef]
- Castro, M. , et al., Physiology and pathophysiology of the HPA axis. Cushing’s Syndrome: Pathophysiology, Diagnosis and Treatment, 2011, 1-20.
- de Kloet, E.R.; van Acker, S.A.; Sibug, R.M.; Oitzl, M.S.; Meijer, O.C.; Rahmouni, K.; de Jong, W. Brain mineralocorticoid receptors and centrally regulated functions. Kidney Int. 2000, 57, 1329–1336. [Google Scholar] [CrossRef]
- Gheorghe, C.P.; Goyal, R.; Mittal, A.; Longo, L.D. Gene expression in the placenta: maternal stress and epigenetic responses. Int. J. Dev. Biol. 2010, 54, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R. Prenatal glucocorticoids and long-term programming. Eur. J. Endocrinol. 2004, 151, U49–U62. [Google Scholar] [CrossRef]
- Harvey, P.W.; Sutcliffe, C. Adrenocortical hypertrophy: establishing cause and toxicological significance. J. Appl. Toxicol. 2010, 30, 617–626. [Google Scholar] [CrossRef]
- Ulrich-Lai, Y.M.; Figueiredo, H.F.; Ostrander, M.M.; Choi, D.C.; Engeland, W.C.; Herman, J.P. Chronic stress induces adrenal hyperplasia and hypertrophy in a subregion-specific manner. Am. J. Physiol. Metab. 2006, 291, E965–E973. [Google Scholar] [CrossRef]
- A Coffman, J. Chronic stress, physiological adaptation and developmental programming of the neuroendocrine stress system. Futur. Neurol. 2020, 15, FNL39. [Google Scholar] [CrossRef]
- Bourque, S.L.; Davidge, S.T. Developmental programming of cardiovascular function: a translational perspective. Clin. Sci. 2020, 134, 3023–3046. [Google Scholar] [CrossRef]
- Matthews, S.G. , Early programming of the hypothalamo–pituitary–adrenal axis. Trends in Endocrinology & Metabolism 2002, 13, 373–380. [Google Scholar]
- Acevedo-Rodriguez, A.; Kauffman, A.S.; Cherrington, B.D.; Borges, C.S.; Roepke, T.A.; Laconi, M. Emerging insights into hypothalamic-pituitary-gonadal axis regulation and interaction with stress signalling. J. Neuroendocr. 2018, 30, e12590. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.M.V.; Syddall, H.E.; Wood, P.J.; Chrousos, G.P.; Phillips, D.I.W. Fetal Programming of the Hypothalamic-Pituitary-Adrenal (HPA) Axis: Low Birth Weight and Central HPA Regulation. J. Clin. Endocrinol. Metab. 2004, 89, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Tegethoff, M.; Pryce, C.; Meinlschmidt, G. Effects of Intrauterine Exposure to Synthetic Glucocorticoids on Fetal, Newborn, and Infant Hypothalamic-Pituitary-Adrenal Axis Function in Humans: A Systematic Review. Endocr. Rev. 2009, 30, 753–789. [Google Scholar] [CrossRef] [PubMed]
- Möllers, L.S.; Yousuf, E.I.; Hamatschek, C.; Morrison, K.M.; Hermanussen, M.; Fusch, C.; Rochow, N. Metabolic-endocrine disruption due to preterm birth impacts growth, body composition, and neonatal outcome. Pediatr. Res. 2021, 91, 1350–1360. [Google Scholar] [CrossRef]
- Singhal, A. , Should we promote catch-up growth or growth acceleration in low-birthweight infants? Low-birthweight baby: born too soon or too small 2015, 81, 51–60. [Google Scholar]
- Kosinska, M., B. Stoinska, and J. Gadzinowski, Catch-up growth among low birth weight infants: Estimation of the time of occurrence of compensatory events. Anthropological Review. 2004, 67, 87–95.
- Dulloo, A.G.; Jacquet, J.; Seydoux, J.; Montani, J.-P. The thrifty ‘catch-up fat’ phenotype: its impact on insulin sensitivity during growth trajectories to obesity and metabolic syndrome. Int. J. Obes. 2006, 30, S23–S35. [Google Scholar] [CrossRef]
- Dulloo, A.G. Thrifty energy metabolism in catch-up growth trajectories to insulin and leptin resistance. Best Pr. Res. Clin. Endocrinol. Metab. 2008, 22, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Kelishadi, R.; Haghdoost, A.A.; Jamshidi, F.; Aliramezany, M.; Moosazadeh, M. Low birthweight or rapid catch-up growth: which is more associated with cardiovascular disease and its risk factors in later life? A systematic review and cryptanalysis. Ann. Trop. Paediatr. 2014, 35, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.N.; Fu, Q.; Callaway, C.W.; McKnight, R.A.; McMillen, I.C.; Ross, M.G.; Lane, R.H.; Desai, M. Epigenetics of programmed obesity: alteration in IUGR rat hepatic IGF1 mRNA expression and histone structure in rapid vs. delayed postnatal catch-up growth. Am. J. Physiol. Liver Physiol. 2010, 299, G1023–G1029. [Google Scholar] [CrossRef] [PubMed]
- Guilloteau, P.; Zabielski, R.; Hammon, H.M.; Metges, C.C. Adverse effects of nutritional programming during prenatal and early postnatal life, some aspects of regulation and potential prevention and treatments. 2009, 17–35. [Google Scholar]
- Isganaitis, E. Developmental Programming of Body Composition: Update on Evidence and Mechanisms. Curr. Diabetes Rep. 2019, 19, 60. [Google Scholar] [CrossRef]
- Warner, M.J.; Ozanne, S.E. Mechanisms involved in the developmental programming of adulthood disease. Biochem. J. 2010, 427, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Correia-Branco, A.; Keating, E.; Martel, F. Maternal Undernutrition and Fetal Developmental Programming of Obesity: The Glucocorticoid Connection. Reprod. Sci. 2014, 22, 138–145. [Google Scholar] [CrossRef]
- Lindsay, R.S.; Dabelea, D.; Roumain, J.; Hanson, R.L.; Bennett, P.H.; Knowler, W.C. Type 2 diabetes and low birth weight: the role of paternal inheritance in the association of low birth weight and diabetes. Diabetes 2000, 49, 445–449. [Google Scholar] [CrossRef]
- Rose, A.J.; Vegiopoulos, A.; Herzig, S. Role of glucocorticoids and the glucocorticoid receptor in metabolism: Insights from genetic manipulations. J. Steroid Biochem. Mol. Biol. 2010, 122, 10–20. [Google Scholar] [CrossRef]
- Beaupere, C.; Liboz, A.; Fève, B.; Blondeau, B.; Guillemain, G. Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 623. [Google Scholar] [CrossRef]
Figure 1.
Shows dams OGTT (a) and HbA1c concentrations (b) between NPD and PD groups at 36 weeks. Values are expressed as mean ± SEM. *p < 0.011; ***p < 0.001 denotes comparison with NPD.
Figure 1.
Shows dams OGTT (a) and HbA1c concentrations (b) between NPD and PD groups at 36 weeks. Values are expressed as mean ± SEM. *p < 0.011; ***p < 0.001 denotes comparison with NPD.
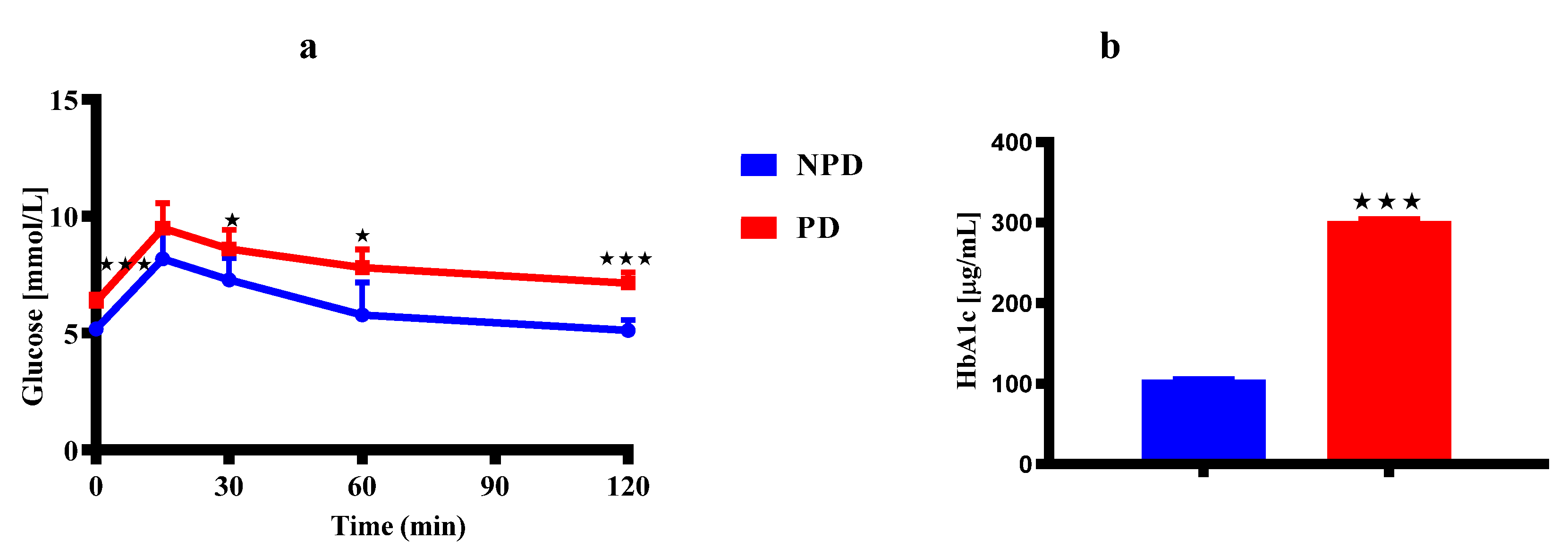
Figure 2.
Shows dams ACTH (a) and corticosterone (b) concentrations between NPD and PD groups at 21 days after birth. Values are expressed as mean ± SEM. ***p < 0.001 denotes comparison with NPD.
Figure 2.
Shows dams ACTH (a) and corticosterone (b) concentrations between NPD and PD groups at 21 days after birth. Values are expressed as mean ± SEM. ***p < 0.001 denotes comparison with NPD.
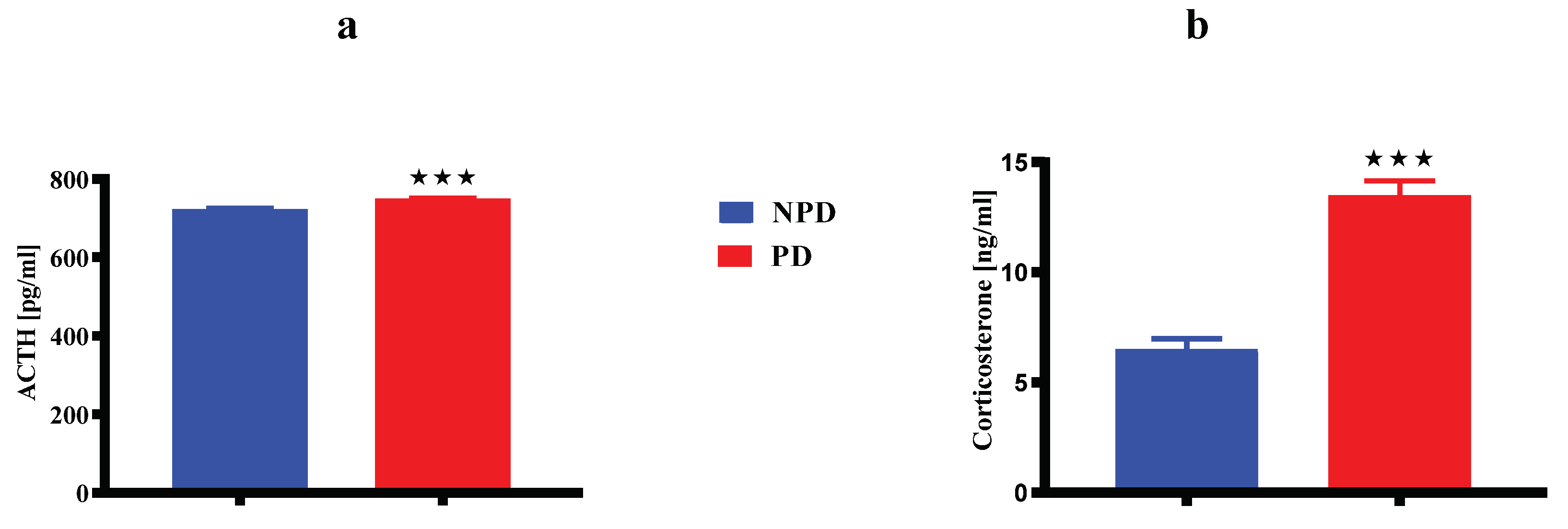
Figure 3.
Shows pups ACTH (a) and corticosterone (b) concentrations born between NPD and PD groups at week 3, 6 and 16 of the experimental periods. Values are expressed as mean ± SEM. *p < 0.0144; **p < 0.0022; ***p < 0.001 denotes comparison with NPD.
Figure 3.
Shows pups ACTH (a) and corticosterone (b) concentrations born between NPD and PD groups at week 3, 6 and 16 of the experimental periods. Values are expressed as mean ± SEM. *p < 0.0144; **p < 0.0022; ***p < 0.001 denotes comparison with NPD.

Figure 4.
Shows pups GR (a) and MR (b) gene expressions born between NPD and PD groups rats at week 3, 6 and 16 of the experimental periods. Values are expressed as mean ± SEM. **p < 0.0022; ***p < 0.001 denotes comparison with NPD.
Figure 4.
Shows pups GR (a) and MR (b) gene expressions born between NPD and PD groups rats at week 3, 6 and 16 of the experimental periods. Values are expressed as mean ± SEM. **p < 0.0022; ***p < 0.001 denotes comparison with NPD.
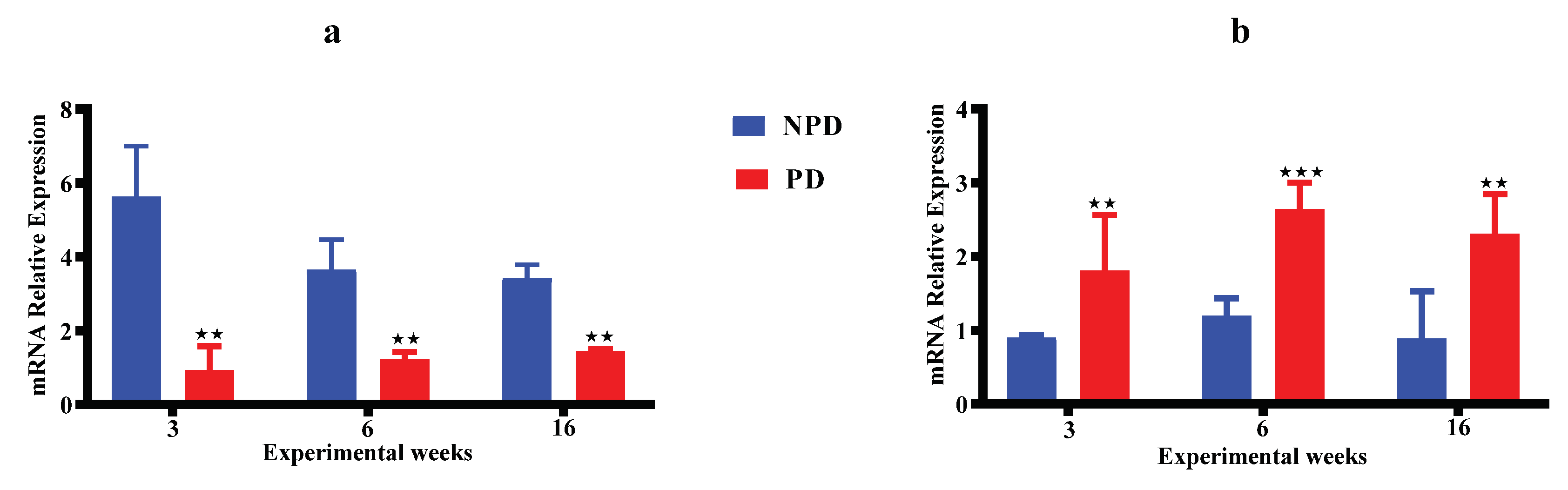
Figure 5.
Shows pups adrenal gland weight born between NPD and PD groups at week 3, 6 and 16 of the experimental periods. Values are expressed as mean ± SEM. **p < 0.0022; ***p < 0.001 denotes comparison with NPD.
Figure 5.
Shows pups adrenal gland weight born between NPD and PD groups at week 3, 6 and 16 of the experimental periods. Values are expressed as mean ± SEM. **p < 0.0022; ***p < 0.001 denotes comparison with NPD.
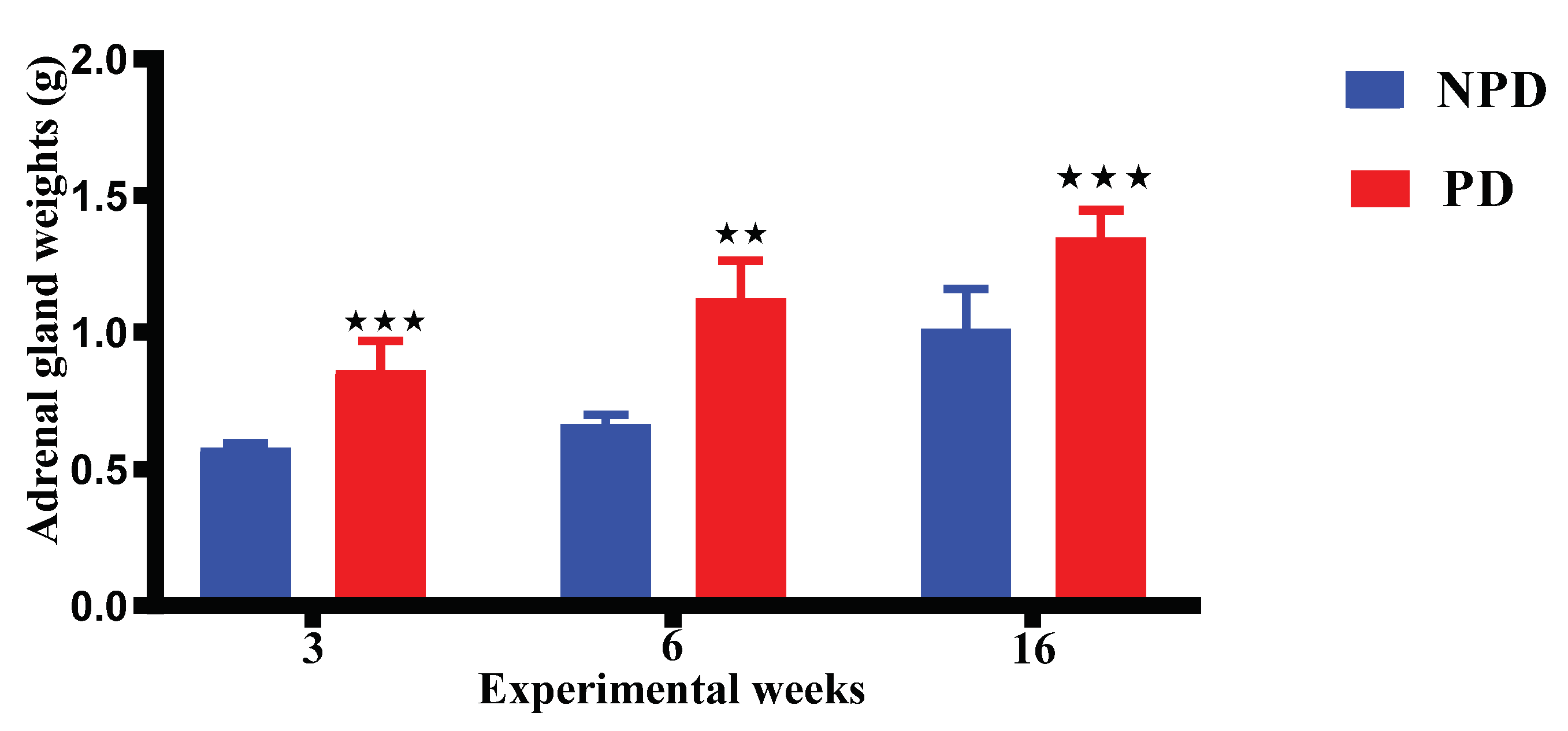
Figure 6.
Shows pups body weights born between NPD and PD groups at day7 (a), week 3, 6 and 16 (b). Values are expressed as mean ± SEM. **p < 0.0022; ***p < 0.001 denotes comparison with NPD.
Figure 6.
Shows pups body weights born between NPD and PD groups at day7 (a), week 3, 6 and 16 (b). Values are expressed as mean ± SEM. **p < 0.0022; ***p < 0.001 denotes comparison with NPD.
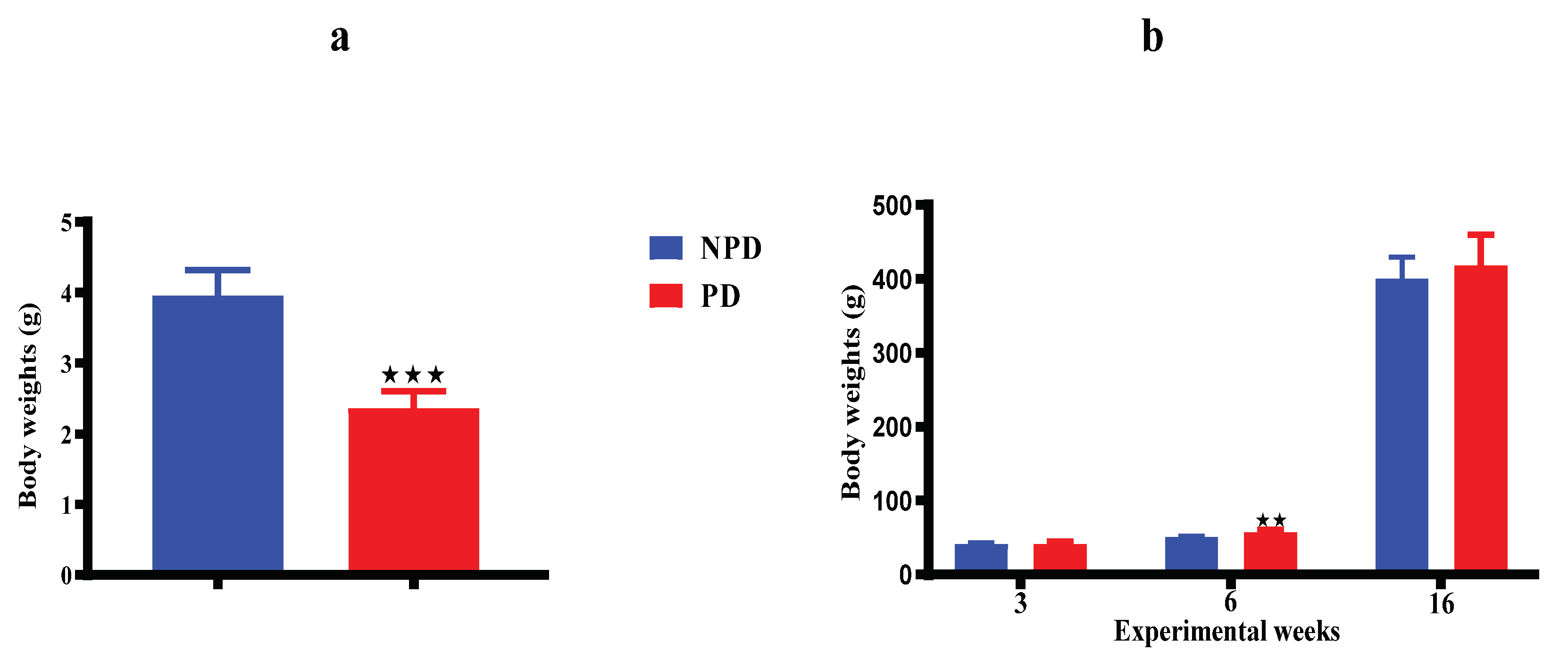
Figure 7.
Shows pups OGTT born between NPD and PD groups at week 3, 6 and 16 of the experimental periods. Values are expressed as mean ± SEM. *p < 0.0144; **p < 0.0022; ***p < 0.001 denotes comparison with NPD.
Figure 7.
Shows pups OGTT born between NPD and PD groups at week 3, 6 and 16 of the experimental periods. Values are expressed as mean ± SEM. *p < 0.0144; **p < 0.0022; ***p < 0.001 denotes comparison with NPD.
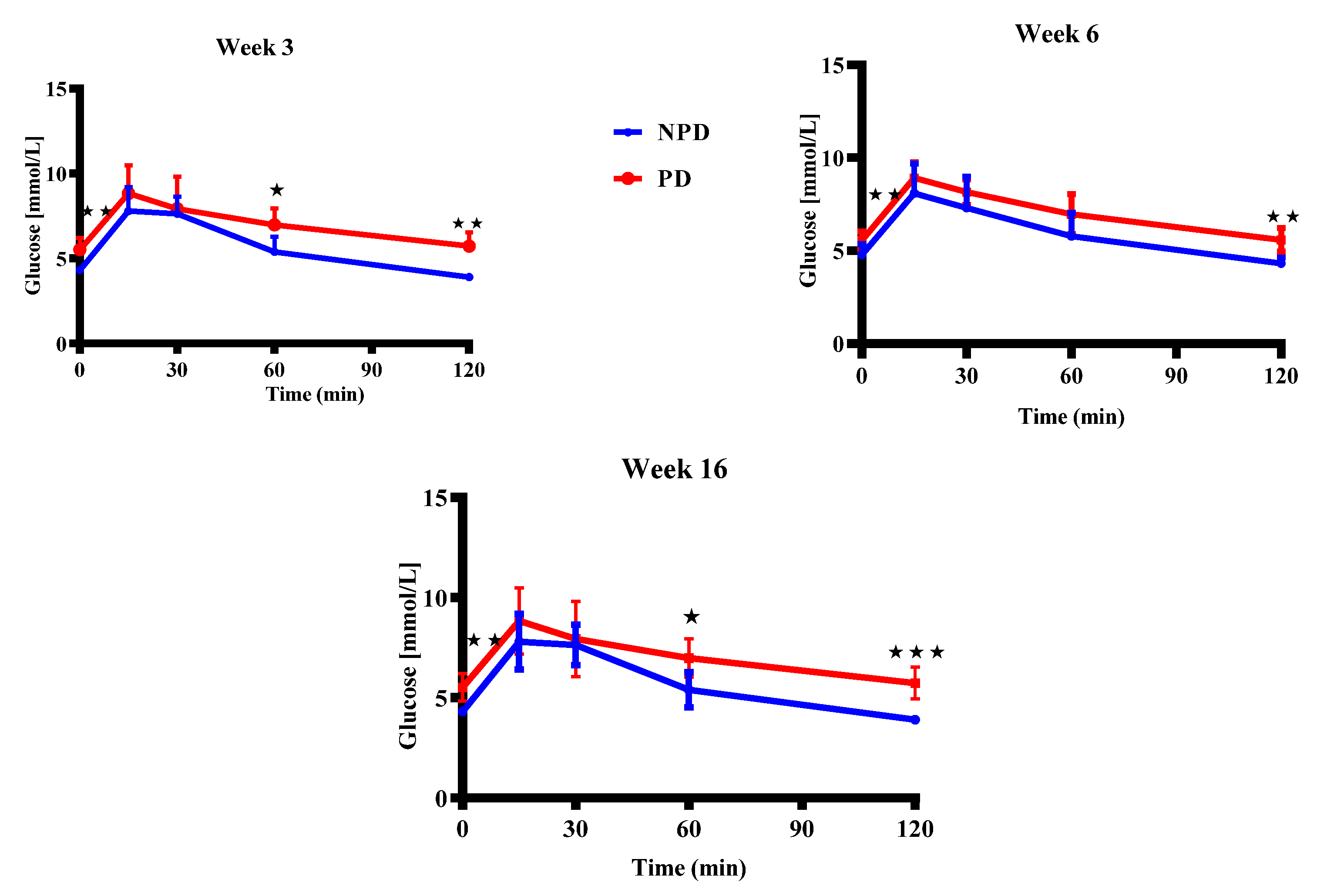

Table 1.
Postnatal developmental stages in Rats and Human.
| Developmental Stages in Rat/Human | Rat (Male) | Human |
|---|---|---|
| Neonatal/newborn | 0-7 days | 0-28 days |
| Infantile/infant | 8-20 days (1-2 weeks) | 1-23 months |
| Juvenile/child | 21-32 days (3-5 weeks) | 2-12 years |
| Peripubertal/adolescent | 33-55 days (5-8 weeks) | 12-16 years |
| Late puberty/adolescent | 56-70 days (8-10 weeks) | |
| Emerging adulthood | 70-150 days (10-21 weeks) | 18-25 years |
Adapted from Barrow (2011), Ojeda and skinner (2006), Parker and Picut (2016).
Table 2.
List of primers used in the study.
| Sequence Name | Sequence |
|---|---|
| Glucocorticoid receptor (Nr3c1 gene) | Forward: 5′-ACCTCGATGACCAAATGACC-3′ Reverse: 5′-AGCAAAGCAGAGCAGGTTTC-3′ |
| Mineralocorticoid receptor (Nr3c2 gene) | Forward: 5′-AAAGGGTAGTGTGTGCAGGG-3′ Reverse 5′-GTTCTCCTAGTTCCCGGAGG-3′ |
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Forward: 5′-AGTGCCAGCCTCGTCTCATA-3′ Reverse 5′-GATGGTGATGGGTTTCCCGT-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



