
Submitted:
28 March 2024
Posted:
29 March 2024
You are already at the latest version
Abstract
Abstract: Congenital hyperinsulinism (CHI) is a rare disorder of glucose metabolism and is the most common cause of severe and persistent hypoglycemia (hyperinsulinemic hypoglycemia, HH) in the neonatal period and childhood. Most cases are caused by mutations in the ABCC8 and KCNJ11 genes that encode the ATP-sensitive potassium channel (KATP). We present the correlation between genetic heterogeneity and the variable phenotype in patients with early-onset hyperinsulinemic hypoglycemia caused by ABCC8 gene mutations. In the first patients, which presented persistent severe hypoglycemia since the first day of life, molecular genetic testing revealed the presence of a homozygous mutation in the ABCC8 gene [deletion in the ABCC8 gene c.(2390+1_2391-1)_(3329+1_3330- 1)del] correlated with a diffuse form of hyperinsulinism (the parents being healthy heterozygous carriers). In the second patient, the onset was on the third day of life with severe hypoglycemia, and genetic testing identified a heterozygous mutation in the ABCC8 gene c.1792C>T (p.Arg598*) inherited on the paternal line, which led to the diagnosis of focal form of hyperinsulinism. To locate the focal lesions, (18)F-DOPA (3,4-dihydroxy-6-[18F]fluoro-L-phenylalanine) PET/CT was recommended (an investigation that cannot be carried out in the country), but the parents refused to carry out the investigation abroad. In this case, early surgical treatment could have been curative. In addition, the second child also presented secondary adrenal insufficiency requiring replacement therapy. At the same time, she developed early recurrent seizures that required antiepileptic treatment. We emphasize the importance and of molecular genetic testing for diagnosis, management and genetic counseling in patients with HH.
Keywords:
hyperinsulinemic hypoglycemia
; ABCC8 gene
; mutation
; ATP- sensitive potassium channel
; genetic heterogeneity
; phenotypic variability
1. Introduction
Congenital hyperinsulinism (CHI) has an estimated incidence of 1/50,000 live births and is the most common cause of severe and persistent hypoglycaemia (Hyperinsulinaemic Hypoglycaemia, HH) in the neonatal, infancy and childhood period [1,2,3]. Early diagnosis and treatment positively influence the prognosis, preventing permanent brain damage. Most commonly, CHI is the consequence of loss-of-function (LOF) mutations in the ABCC8 (SUR1) and KCNJ11 (KIR6.2) genes, located on chromosome 11p15.1, which encode the adenosine triphosphate (ATP)-sensitive potassium channel (KATP) of pancreatic β-cells [4]. Mutations of other genes involved in the regulation of insulin secretion are rarely identified: GCK, GLUD1, HADH, SLC16A1, HNF4A, HNF1A, UCP2, HK1 and PGM1 (Figure 1) [5]
ABCC8: ATP-binding cassette, subfamily C, member 8 ; KCNJ11: Potassium channel, inwardly rectifying, subfamily J, member 11; GCK: Glucokinase gene; HK1: Hexokinase 1 gene; SLC16A1 / MCT1: Solute carrier family 16 (monocarboxylic acid transporter), member 1 gene; GLUD1: Glutamate dehydrogenase 1 gene; HADH: 3-hydroxyacyl-CoA dehydrogenase gene; UCP2: Uncoupling protein gene; CACNA1D: Calcium Channel, Voltage-Dependent, L Type, Alpha-1d Subunit gene; FOXA2: Forkhead box A2; HNF1A: HNF1 homeobox A; ETC: Electron transport chain [4,5].
KATP-CHI is associated with hyperplasia of Langherhans islets, which can be diffuse (all pancreatic β-cells are affected) or focal (localized islet dysfunction (30-40% of all CHI cases), with a correlation between the histological type (phenotype) and the genetic defect present (genotype) [5].
The two histopathological forms cannot be distinguished clinically. The best way to differentiate is by performing an (18)F-DOPA PET/CT (3,4-dihydroxy-6-[18F]fluoro-L-phenylalanine) positron emission tomography scan/ computed tomography [6]. Focal CHI begins at older ages and frequently associates with hypoglycemic seizures, compared to the diffuse form [5].
Patients with diffuse CHI frequently present homozygous recessive or a compound heterozygote mutation in ABCC8 or KCNJ11 genes (which encode the SUR/KIR6.2 components of the KATP channel in pancreatic β-cells) [5,7].
The molecular mechanism in focal CHI follows the “two-hit” model described by Knudson [8]. It involves the presence of a heterozygous paternally inherited mutation in ABCC8 or KCNJ11 genes and the appearance in some pancreatic cells of the second mutation in the chromosomal 11p15 region of maternal origin, with loss of heterozygosity (LOH). Another possible mechanism would be paternal uniparental isodisomy of chromosome 11p15.5 and the absence of the same region of maternal origin in focal lesions [5,9]. Mutations with loss of heterozygosity (LOH) in pancreatic somatic cells will determine the unbalanced expression of the imprinted genes (paternal IGF2, maternal H19 and CDKN1C) from the chromosomal 11p15.5 region, which regulates cell growth, with the appearance of focal adenomatous hyperplasia [10,11].The presence of the paternally inherited heterozygous KATP mutations has a predictive value for focal CHI in 94% of cases [10,12]. In the focal form of CHI, the lesions are unique. To date, few cases with multifocal lesions have been reported. The differential diagnosis between the two forms of CHI (focal and diffuse) is important for therapeutic approach. First-line drug treatment consists of oral diazoxide, glucagon, somatostatin analogues. In the focal CHI, the curative treatment involves the surgical excision of the focal lesion, while in the diffuse CHI, which does not respond to drug treatment, the symptoms can only be improved, through subtotal pancreatectomy, with the risk of complications, such as exocrine pancreatic insufficiency and diabetes mellitus [13,14,15].
We present the genotype-phenotype correlation in patients with early-onset hyperinsulinemic hypoglycemia (HH) caused by different mutations in the ABCC8 gene. First patient presents a homozygous autosomal recessive mutation in the ABCC8 gene associated with a diffuse CHI (both parents being healthy carriers of the same mutation); in the second patient, a paternally inherited heterozygous ABCC8 mutation was identified that led to a focal CHI.
2. Results
Genotype-Phenotype Correlation in Patients with Early-Onset Hyperinsulinemic Hypoglycemia (HH) Caused by Different Mutations in the ABCC8 Gene
2.1. Patient 1
We present the patient A.I.S, currently 3 months old, who was transferred from the neonatology department to the pediatric clinic at the age of 10 days due to persistent episodes of severe hypoglycemia. The baby comes from a noncomplicated pregnancy of a young and non-consanguineous couple. The mother was 28 years old, gesta II, para II, with no risk factors for diabetes neither before nor during gestation, no drug consumption, or other treatments until delivery. She has one previous child, healthy, 4 years old, born at term with 2950 g and an Apgar score of 9. The father was 31 years old, healthy with no history of chronic diseases.
The birth occurred at term (gestational age of 38 weeks), birth weight of 3140 g, pelvic presentation, acute fetal distress, Apgar score 6 at 1 minute, and 8 at 5 minutes, prolapse of the left lower limb, necessitating hospitalization of the child in the neonatal intensive care unit. Transfontanelle ultrasonography identified a bilateral subependymal hemorrhage. From the first day of life, the baby presented severe hypoglycemia, with blood glucose values of 13 mg/dL at 11 hours postnatal and 30 mg/dL at 26 hours of life, requiring PEV with 12.5% glucose from the first days of life.
At blood glucose values below 50 mg/dL, plasma insulin showed a value of 41.35 uIU/ml (normal values 3-25 u IU/mL) and C peptide of 4.92 ng/mL (normal values 0.2-4.4 ng/ mL) - both increased, serum cortisol and thyroid hormones were within normal limits with growth hormone increased 133.67 uUI/ml (normal values 0-55 uUI/mlL) (Table 1).
A form of congenital hyperinsulinism was suspected and a genetic consultation was requested, which recommended molecular genetic testing (gene panel).
In pediatric unit the treatment with diazoxide was initiated in a progressively increasing dose, with frequent monitoring of blood glucose levels on a blood glucose meter, and at a dose of 10 mg/kg/day, hydrochlorothiazide was also added.
It did not require glucose IV infusions, the child being fed orally every 2.5-3 hours. At the maximum dose of diazoxide, hypoglycemic episodes became rarer, but severe hypoglycemia persisted, 1-2 episodes/day with a flat glycemic curve on a continuous glycemic monitoring system, reasons why we switched to the second-line medication, rapid somatostatin analogues (sandostatin). Depending on the glycemic profile, the dose of rapid somatostatin analogues was increased up to 25 mcg/kg/day in 3 subcutaneous doses under which remission of severe hypoglycemia was obtained with feeding at 2.5-3 hours intervals, without immediate adverse effects, with mild hypoglycemia 4% on continuous glycemic monitoring (CGM) / 7 days.
We tried to space out the meals, but severe hypoglycemia still occurred. In evolution, the initial total dose of rapid somastotatin analogues did not require adjustment, the dose related to the child’s weight decreasing over time to 16,8 mg/kg/day at the age of 3 months.
Abdominal MRI (Magnetic Resonance Imaging) did not identify pathological changes in the pancreas.
Molecular genetic testing (Hypoglycemia, Hyperinsulinism and Ketone Metabolism Panel, Bluprint Genetics Laboratory) identified a homozygous deletion in the ABCC8 gene c.(2390+1_2391-1)_(3329+1_3330-1)del, which encompasses exons 20-26, the result correlating with the diffuse CHI. Genetic testing of the parents revealed that both parents are heterozygous of the same mutation identified in the child (Figure 2).
Since under the current therapy the infant did not present apparent hypoglycemic episodes, the surgical team decided to postpone the surgery, taking into account the young age of the child and the intra- and postoperative possible risks, in this case a subtotal pancreatectomy will be probably recommended.
2.2. Patient 2
The second patient is also a girl, D.M.S, aged 1 year and 3 months, who was evaluated in the pediatric clinic at the age of 14 days for persistent severe hypoglycemia. The child comes from the first pregnancy of young and non-consanguineous couple (Figure 3).
The pregnancy progressed apparently normally, being monitored by ultrasound. There were no risk factors for maternal diabetes before or during pregnancy and no drugs were used during pregnancy. The birth occurred at term (gestational age of 38 weeks), by caesarean section, cranial presentation; birth weight of 2700 g, the Apgar score was 6 at 1 minute with good adaptation to extrauterine life, but with glycemic values of 60 mg/dL in the first days of life, the child being discharged at 48 hours of life.
At 3 days old, the parents initially contacted the territorial neonatology department because the child refusal to eat, on which occasion a severe hypoglycemia was detected. A neonatal sepsis was initially suspected, but the persistence of hypoglycemia up to a value of 17 mg/dL raised the suspicion of a possible congenital hyperinsulinism, the child being later admitted to the pediatric clinic.
Biochemical investigations revealed at a hypoglycemia of 41 mg/dL, respectively 46 mg/dL a plasma insulin levels of 15.28 uIU/mL, respectively 15.48 uIU/mL (normal values: 3-25 uIU/mL) and C-peptide 2.2 ng/mL (normal values: 0.9-7.1 ng/mL), respectively 2.01 ng/mL, in two consecutive days (Table 1).
Thyroid hormones and growth hormone were simultaneously dosed, with values within normal limits; on hypoglycemia the plasma cortisol level was low (2.27 mcg/mL) (normal values: 4.3-22.4 mcg/mL). Plasma ACTH dosage was recommended, low values below 5 pg/mL were detected (normal values: 5 - 46 pg/mL), which suggested a central adrenal insufficiency.
A genetic consultation was requested, which recommended molecular genetic testing (gene panel for hypoglycemia) later performed at the Invitae laboratory, which identified a heterozygous ABCC8 c.1792C>T (p.Arg598*) mutation present in the child and his father. This result raises the suspicion of a focal form of congenital hyperinsulinism (Figure 3).
The treatment with diazoxide and the attack dose of hydrocortisone was initiated, later in the substitution dose. The dose of diazoxide was progressively increased, associating hydrochlorothiazide at doses over 10 mg/kg/day of diazoxide, but severe hypoglycemia is maintained. Rapid somatostatin analogue (Santostatin) was then added up to a dose of 25 mcg/kg/body weight, in 4 subcutaneous doses, with the decrease of severe episodes of hypoglycemia under this treatment; after a month and a half of treatment with rapid somatostatin analogues, hypoglycemia was no longer evident during intermittent blood glucose monitoring, allowing even a 6-hour fasting period.
At the age of 1 month and 10 days, the child presented a tonico-clonic seizure in the left hemibody, of short duration, repeated, and antiepileptic therapy was instituted.
An urgent craniocerebral computed tomography was performed which identified the presence of symmetrical hypodense areas occipital and posterior parietal bilaterally - ischemic sequelae, and at the level of the pituitary gland, an inhomogeneous and hypocapturing nodule of 3/3 mm in the antero-inferior portion. Subsequently, brain MRI performed at 7 months of age revealed a normal appearance.
The result of the genetic test raised the suspicion of focal CHI, being recommended to perform a (18) F-DOPA PET/CT (3,4-dihydroxy-6-[18F]fluoro-L-phenylalanine) positron emission tomography scan/ computed tomography).
Taking into account that in Romania there was no possibility of carrying out this investigation, they were done steps to direct to a specialized clinic from abroad, but the parents have so far refused this investigation.
In the evolution, it was not necessary to increase the initial total dose of rapid somatostatin analogues, during intermittent blood glucose monitoring, hypoglycemia not being identified (declaratively).
At the age of 9 months, the endocrinological evaluation in a specialized clinic revealed the normalization of plasma levels of ACTH and cortisol, and Hb A1c = 4.9%. Glycemic monitoring during the respective hospitalization did not reveal hypoglycemia at the dose of rapid somatostin analogues initiated in infancy, at present 11 mcg/ kg body weight.
At the clinical examination, the child showed inappropriate weight gain and moderate neuromotor retardation, as well as the recurrence of epileptic seizures, necessitating the adjustment of the antiepileptic treatment.
3. Discussion
In both children, the onset of hypoglycemia was early, from the first days of birth, being severe. First-line treatment with diazoxide was initiated in the first two weeks of life, but both children were unresponsive, switching to second-line medication (somatostatin).
To control hypoglycemia, a higher dose of somatostatin than recommended by international guidelines was initially required, the dose which, however, did not have to be adjusted to the child’s weight, maintaining the initial dose.
The second child also had a central adrenal insufficiency that required substitution treatment. At the same time, he developed early recurrent seizures that required the initiation of anticonvulsant therapy.
We mention that in Romania it is not possible to perform (18)F-DOPA PET/CT, which is necessary in this case for the localization of focal lesions. This form of hyperinsulinism would have required surgical treatment, which could have been curative, if it had been instituted early.
Both children presented an inappropriate weight curve with an early weight deficit in case 1 with a weight index of 0.72 at 3.5 months despite an appropriate food intake. The anthropometric data of the second patient were within normal limits initially, later a progressive stature-weight growth delays became constant, with stature value of -2.16 SD and weight value of -2.16 SD at the age of 1 year. The second child had also a moderate psychomotor retardation.
The two cases presented reflect genetic heterogeneity (different mutations in the ABCC8 gene) associated with phenotypic variability. Thus, in the first case, a homozygous mutation of the ABCC8 gene was correlated with the diffuse CHI, while in the second case, the patient had a paternally inherited heterozygous ABCC8 mutation, which was correlated with a posible focal CHI. The presented data are consistent with those from the specialized literature.
The Arg598Ter variant has been reported in over 10 individuals with congenital hyperinsulinism [16,17]. About 0.006% of African Americans are healthy heterozygous carriers of this mutation, according to The Genome Aggregation Database (gnomAD; dbSNP rs139328569) (http://gnomad.broadinstitute.org, accessed on 7 March 2024) [18].
This variant (Arg598Ter) was also reported as pathogenic in ClinVar (VariationID: 434056). Of the 11 affected individuals, at least 4 were compound heterozygotes carrying a reported pathogenic variant in trans, increasing the likelihood that the Arg598Ter variant is pathogenic (Variation ID: 434053) (https://www.ncbi.nlm.nih.gov/clinvar/variation/434053/, accessed on 7 March 2024) (Damaj, 2008; De Vroede, 2004; Bellanné-Chantelot, 2010; [19,20,21,22]
In vitro functional studies provided evidence that the Arg598Ter variant may slightly affect protein function [21,23].This variant causes a premature stop codon at position 598, leading to a truncated or absent protein. Paternally-inherited loss-of-function (LOF) ABCC8 mutations represent a known mechanism in autosomal recessive hyperinsulinemic hypoglycemia [24,25].
Carriers of a single heterozygous pathogenic FHI (familial hyperinsulinemic hypoglycemia, type 1) -associated variant inherited from the father may be at risk for focal FHI. Focal FHI occurs when a single pathogenic FHI-associated variant is inherited from a carrier father and a second change occurs in only some of the pancreatic cells, causing the loss of the normal maternal gene. In the area of the pancreas in which only the paternal FHI gene is represented, insulin is overproduced and may cause hyperinsulinism of variable severity [24,25].
In the second case, the patient’s father presented the same ABCC8 (Arg598Ter) heterozygous mutation as the child, but did not present symptoms of hypoglycemia. The allelic expression imbalance (AEI) could explain the variable phenotypic expressivity in this case, the father and the child presenting different phenotypes, although the same ABCC8 mutation was present. AEI refers to the different gene expression in intensity of the two alleles of the genes that encode the same protein. Initially, it was thought that the expression of maternal and paternal alleles is balanced, and this balance could reduce the effect of recessive mutations. However, several mechanisms are involved in the regulation of gene expression, including epigenetic ones. Subsequent research demonstrated that AEI occurred when the expression of one of the alleles was inhibited or exacerbated or in the case of post-transcriptional degradation of mature mRNA [25].The existence of AEI in the case of the ABCC8 gene will be elucidated through future studies.
ABCC8 (OMIM 600509) encodes ATP-binding cassette transporter subfamily C member 8 member 8 which is expressed in pancreatic β-cells and in the nervous system [4]. Together with the proteins encoded by the KCNJ11, KCNJ8 and ABCC9 genes, ABCC8 forms the ATP-sensitive potassium channel (KATP) that detects metabolic changes in pancreatic β-cells and regulates insulin secretion. LOF mutations in the ABCC8 or KCNJ11 genes lead to KATP channel dysfunction and hyperinsulinism. Depolarization of the cell membrane occurs even in the absence of an increased intracellular ATP/ADP ratio, initiating the insulin secretion cascade, even in the absence of glucose [26].
In the case of the ABCC8 gene, a genetic heterogeneity correlated with the phenotypic variability is described, being reported over 890 variants in ABCC8 annotated as disease-causing mutation (DCM) in the HGMD Professional variant database, including both missense and truncating variants (nonsense, frameshift, variants affecting splicing, gross deletions). Of these, over 400 are associated with hyperinsulinemic hypoglycemia and at least 14 mutations have been associated with permanent neonatal diabetes mellitus (PNDM) [16,27].
Pathogenic mutations in the ABCC8 gene cause autosomal dominant and recessive familial hyperinsulinemic hypoglycemia, type 1 (FHI) (OMIM 256450) and dominant leucine-sensitive hypoglycemia of infancy (OMIM 240800) [4]. Other pathogenic ABCC8 variants are associated with autosomal dominant noninsulin-dependent (OMIM 125853), permanent neonatal (PNDM) (OMIM 606176) and transient neonatal type 2 diabetes mellitus (OMIM 610374). Although pathogenic variants in ABCC8 are more frequently associated with permanent and transient neonatal diabetes, late-onset cases are described. It is proven that autosomal dominant hyperinsulinism caused by LOF ABCC8 mutations develops reduced glucose tolerance and, in some cases, diabetes mellitus [12,27,28].
Gain-of function (GOF) missense mutations in the ABCC8 gene are detected in cases of PNDM. KATP channels carrying these mutations lose regulatory inhibition by ATP [29].
Thus, in PNDM patients, persistent hyperglycemia is caused by loss of pancreatic β-cell membrane excitability to glucose and loss of pancreatic insulin. Glucose normally increases β-cell excitability by inhibiting KATP channels, opening voltage-dependent calcium channels, increasing intracellular calcium [Ca2+]i, which triggers insulin secretion [30,31].
ABCC8 mutations are detected in more than 45% of FHI cases [32,33]. Recessive LOF ABCC8 mutations are detected in patients with FHI/congenital hyperinsulinism, in which heterozygous individuals are healthy carriers [34].
The phenotypic severity in FHI cases varies, from severe hypoglycemia with neonatal onset, difficult to treat, to milder manifestations of the disease, with reduced symptoms, which begin in childhood, in their case, there are difficulties related to the diagnosis of hypoglycemia [35].
KATP-channel inactivating mutations in ABCC8 associated with mutations in the KCNJ11 gene cause 97% of cases of diazoxide-unresponsive hyperinsulinism [35,36]. An autosomal recessive and, more rarely, an autosomal dominant transmission are detected most frequently, but de novo mutations have also been reported [35].
In approximately 97% of FHI cases detected in the Ashkenazi Jewish population, two ABCC8 founder mutations (c.3989-9g>a and p.F1387del) were detected [37]. The pathogenic variant c.3989-9G>A has been identified in several different ethnic groups suggesting that this is a hotspot mutation. Homozygous recessive KATP channel mutations associated with impaired insulin secretion are associated with diffuse pancreatic islet damage. These allelic variants can also cause focal adenomatosis of β-cells when a paternally derived KATP variant becomes expressed through embryonic loss of heterozygosity for the maternal allele in a clone of β-cells [37].
More than 40% of cases with FHI-KATP have pancreatic adenomatous hyperplasia involving a limited region of the pancreas (focal CHI). In this case, the transmission is autosomal dominant, but only manifests when the pathogenic variant occurs on the paternally derived allele and a somatic event result in the loss of the maternal allele in a β-cell precursor [37]. The clinical manifestations are similar to those of autosomal recessive FHI-KATP, but the genetic and therapeutic aspects are clearly different. In the form of autosomal dominant FHI, the onset is after the age of 6-9 months, and the clinical manifestations are less severe than in the recessive form and usually respond to treatment with diazoxide [34].
The recessive forms of diffuse or focal FHI are associated postnatally with severe hypoglycemia that does not respond to treatment with diazoxide or octreotide, requiring surgical treatment (subtotal pancreatectomy) [38].
Sporadic forms of HH are associated with moderate/severe episodes of hypoglycemia and hyperinsulinism evident from the first days of life and usually have a poor response to treatment, but the prognosis improves after partial pancreatectomy [38].
4. Genetic Counseling
In the case of the first patient, genetic testing of the parents was indicated, which revealed that both are healthy carriers of the mutation present in the child [ABCC8 c.(2390+1_2391-1) (3329+1_3330-1)del]. Their risk of having a new affected pregnancy is 25%, taking into account the autosomal recessive transmission of the disease. In the second case, the risk of the couple in which the father is heterozygous of the ABCC8 c.1792C>T (p.Arg598*) mutation of having a new pregnancy that inherits the paternal mutation is 50%, correlated with autosomal dominant pattern of inheritance. The probability that a sibling of a child with focal CHI will inherit the paternal ABCC8 mutation is 50%, but the probability that he will also have somatic paternal UPD for chromosome 11p15.5 is low [38].
5. Material and Method
We studied the correlations between different mutations in the ABCC8 gene (genetic heterogeneity) and the variable phenotype in the case of two patients with hyperinsulinemic hypoglycemia diagnosed with different forms of the disease (focal form, respectively diffuse form of CHI). Molecular genetic testing (gene panel for hypoglycemia) of the patients and their parents was performed at laboratories abroad, two different ABCC8 variants were identified, correlated with the histological type of the disease. The obtained results were compared with those present in the specialized literature (Clinvar and The Genome Aggregation Database - gnomAD) being consistent with the data presented in similar studies, and revealed the importance of genetic testing in achieving early diagnosis and management and prognosis in the case of CHI patients.
6. Conclusions
CHI is a major cause of hypoglycemia in the neonatal and childhood period. Early diagnosis and appropriate management of HH are important to avoid long-term neurological complications.
The use of (18)F-DOPA PET/CT to differentially diagnose diffuse from focal CHI has completely changed the approach to diagnosis and management in these patients in recent years. For the future, the management of the diffuse CHI that does not respond to drug treatment remains a challenge, and the identification of genetic mechanisms will provide new insights into the physiology of pancreatic β-cells. Also, genetic testing must be included in the management of patients with HH, as there is a correlation between the genetic mutation and the clinical manifestations.
The two presented cases illustrate variable phenotypes (diffuse / focal) in patients with hyperinsulinemic hypoglycemia caused by different mutations in the gene. They demonstrate the importance and clinical utility of genetic analysis for diagnosis and treatment guidance.
In both cases, the hypoglycemia started in the first days of life, and treatment with diaxoxide was initiated. Both patients were unresponsive to this treatment, requiring the change of diazoxide to somatostatin. Patients with heterozygous mutations in ABCC8 and their family members require long-term monitoring as they are at increased risk of developing diabetes mellitus.
Author Contributions
Conceptualization, L.I.B., D.A.B., G.P., S.P., G. G, D.A.B., L.P. and L.M.T.; methodology, L.I.B., L.P., G.P., G.G., N.G., A.L., I.V., M.C.B. and L.M.T.; investigation, data curation, L.I.B., D.A.B., G.P., L.P., S.P., Ș.M.M. and L.M.T.; writing—original draft preparation, L.I.B., G.P., Ș.M.M., A.L., S.P., I.V., and L.M.T.; writing—review and editing, L.I.B., N.G., D.A.B, S.P. and L.M.T.; visualization, D.A.B., A.L., M.C.B. and L.M.T.; supervision, L.I.B., D.A.B., L.P., and L.M.T. The authors D.A.B, G.P., L.P., G.G., N.G., S.P., Ș.M.M, A.L., M.C.B., I.V. and L.M.T., contributed equally with L.I.B to this article. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the St. Mary’s Emergency Children Hospital, Iasi, approved number 7485 / 5 March 2024.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank Dr. Delia Bizim and Dr. Camen Oltean for the effort made in the diagnosis and management of patients, as well as for the support given to the writing of the paper, data collection and their intellectual advice.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
CHI: Congenital hyperinsulinism; HH: Hyperinsulinemic hypoglycemia; (18)F-DOPA PET/CT: (18)F-DOPA PET/CT: (3,4-dihydroxy-6-[18F]fluoro-L-phenylalanine) positron emission tomography scan/ computed tomography; KATP: adenosine triphosphate (ATP)-sensitive potassium channel; ABCC8: ATP-binding cassette, subfamily C, member 8 ; KCNJ11: Potassium channel, inwardly rectifying, subfamily J, member 11; GCK: Glucokinase gene; HK1: Hexokinase 1 gene; SLC16A1 / MCT1: Solute carrier family 16 (monocarboxylic acid transporter), member 1 gene; GLUD1: Glutamate dehydrogenase 1 gene; HADH: 3-hydroxyacyl-CoA dehydrogenase gene; UCP2: Uncoupling protein gene; CACNA1D: Calcium Channel, Voltage-Dependent, L Type, Alpha-1d Subunit gene; FOXA2: Forkhead box A2; HNF1A: HNF1 homeobox A; ETC: Electron transport chain; FHI: Autosomal dominant and recessive familial hyperinsulinemic hypoglycemia, type 1; OMIM: Online Inheritance of Man; AEI: Allelic expression imbalance; PNDM: Permanent neonatal type 2 diabetes mellitus; LOF: Loss-of function; GOF: Gain-of function; DCM: Disease-causing mutation (DCM).
References
- Vora, S.; Chandran, S.; Rajadurai, V.S.; Hussain, K. Hyperinsulinemic hypoglycemia in infancy: Current concepts in diagnosis and management. Indian Pediatr. 2015, 52, 1051–1059. [Google Scholar] [CrossRef]
- Kapoor, R.R.; Flanagan, S.E.; James, C.; Shield, J.; Ellard, S.; Hussain, K. Hyperinsulinaemic Hypoglycaemia. Arch Dis Child. 2009, 94, 450–457. [Google Scholar] [CrossRef]
- Sousa-Santos, F.; Simões, H.; Castro-Feijóo, L.; Rodríguez, P.C.; Fernández-Marmiesse, A.; Fiaño, R.S.; Rego, T.; Carracedo. ; Conde, J.B. Congenital hyperinsulinism in two siblings with ABCC8 mutation: same genotype, different phenotypes. Arq. Bras. de Endocrinol. Metabol. 2018, 62, 560–565. [Google Scholar] [CrossRef] [PubMed]
- OMIM—Online Mendelian Inheritance in Man. Available online: https://www.omim.org/, (accessed on 9 March 2024).
- Gϋemes, M.; Rahman, S.A.; Kapoor, R.R.; Flanagan, S.; Houghton, J.A.L.; Misra, S.; Oliver, N.; Dattani, M.T.; Shah, P. Hyperinsulinemic hypoglycemia in children and adolescents: Recent advances in understanding of pathophysiology and management. Rev. Endocr. Metab. Disord. 2020, 21, 577–597. [Google Scholar] [CrossRef]
- States, L.J.; Davis, J.C.; Hamel, S.M.; Becker, S.A.; Zhuang, H. 18F-6-Fluoro-l-Dopa PET/CT Imaging of Congenital Hyperinsulinism. J. Nucl. Med. 2021, 62, 51S–56S. [Google Scholar] [CrossRef]
- Rasmussen, A.G.; Melikian, M.; Globa, E.; Detlefsen, S.; Rasmussen, L.; Petersen, H.; Brusgaard, K.; Rasmussen, A.H.; Mortensen, M.B.; Christesen, H.T. The difficult management of persistent, non-focal congenital hyperinsulinism: A retrospective review from a single, tertiary center. Pediatr Diabetes. 2020, 21, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Sims, K. Congenital Hyperinsulinism. Neoreviews. 2021, 22, e230–e240. [Google Scholar] [CrossRef] [PubMed]
- Fournet, J.-C.; Mayaud, C.; de Lonlay, P.; Gross-Morand, M.-S.; Verkarre, V.; Castanet, M.; Devillers, M.; Rahier, J.; Brunelle, F.; Robert, J.-J.; et al. Unbalanced Expression of 11p15 Imprinted Genes in Focal Forms of Congenital Hyperinsulinism: Association with a Reduction to Homozygosity of a Mutation in ABCC8 or KCNJ11. Am. J. Pathol. 2001, 158, 2177–2184. [Google Scholar] [CrossRef]
- Rosenfeld, E.; Mitteer, L.; Boodhansingh, K.; Becker, S.A.; McKnight, H.; Boyajian, L.; Ackermann, A.M.; Kalish, J.M.; Bhatti, T.R.; States, L.J.; et al. Case Report: Two Distinct Focal Congenital Hyperinsulinism Lesions Resulting From Separate Genetic Events. Front. Pediatr. 2021, 9, 699129. [Google Scholar] [CrossRef]
- Snider, K.E.; Becker, S.; Boyajian, L.; Shyng, S.-L.; MacMullen, C.; Hughes, N.; Ganapathy, K.; Bhatti, T.; Stanley, C.A.; Ganguly, A. Genotype and Phenotype Correlations in 417 Children With Congenital Hyperinsulinism. J. Clin. Endocrinol. Metab. 2013, 98, E355–E363. [Google Scholar] [CrossRef]
- ElSheikh, A.; Shyng, S.-L. KATP channel mutations in congenital hyperinsulinism: Progress and challenges towards mechanism-based therapies. Front. Endocrinol. 2023, 14, 1161117. [Google Scholar] [CrossRef]
- Krawczyk, S.; Urbanska, K.; Biel, N.; Bielak, M.J.; Tarkowska, A.; Piekarski, R.; Prokurat, A.I.; Pacholska, M.; Ben-Skowronek, I. Congenital Hyperinsulinaemic Hypoglycaemia—A Review and Case Presentation. J. Clin. Med. 2022, 11, 6020. [Google Scholar] [CrossRef] [PubMed]
- Hewat, T.I.; Johnson, M.B.; Flanagan, S.E. Congenital Hyperinsulinism: Current Laboratory-Based Approaches to the Genetic Diagnosis of a Heterogeneous Disease. Front. Endocrinol. 2022, 13, 873254. [Google Scholar] [CrossRef]
- Roženková, K.; Güemes, M.; Shah, P.; Hussain, K. The Diagnosis and Management of Hyperinsulinaemic Hypoglycaemia. J. Clin. Res. Pediatr. Endocrinol. 2015, 7, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Salomon-Estebanez, M.; Shah, P.; Nicholson, J.; Cosgrove, K.E.; Dunne, M.J. Therapies and outcomes of congenital hyperinsulinism-induced hypoglycaemia. Diabet. Med. 2018, 36, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Ge, J.; Zhang, M.; Hussain, K.; Guan, Y.; Cheng, R.; Xi, L.; Zheng, Z.; Ren, S.; Luo, F. Genotype and phenotype analysis of a cohort of patients with congenital hyperinsulinism based on DOPA-PET CT scanning. Eur. J. Pediatr. 2019, 178, 1161–1169. [Google Scholar] [CrossRef]
- http://gnomad.broadinstitute.org, (accesed on 7 March 2024).
- https://clinvarminer.genetics.utah.edu/submissions-by-variant/NM_000352.6%28ABCC8%29%3Ac.1792C%3ET%20%28p.Arg598Ter%29, (accesed on 7 March 2024).
- Damaj, L.; le Lorch, M.; Verkarre, V.; Werl, C.; Hubert, L.; Nihoul-Fékété, C.; Aigrain, Y.; de Keyzer, Y.; Romana, S.P.; Bellanne-Chantelot, C.; et al. Chromosome 11p15 Paternal Isodisomy in Focal Forms of Neonatal Hyperinsulinism. J. Clin. Endocrinol. Metab. 2008, 93, 4941–4947. [Google Scholar] [CrossRef]
- De Vroede, M.; Bax, N.; Brusgaard, K.; Dunne, M.J.; Groenendaal, F. Laparoscopic Diagnosis and Cure of Hyperinsulinism in Two Cases of Focal Adenomatous Hyperplasia in Infancy. PEDIATRICS 2004, 114, e520–e522. [Google Scholar] [CrossRef]
- Bellanne-Chantelot, C.; Saint-Martin, C.; Ribeiro, M.-J.; Vaury, C.; Verkarre, V.; Arnoux, J.-B.; Valayannopoulos, V.; Gobrecht, S.; Sempoux, C.; Rahier, J.; et al. ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism. J. Med Genet. 2010, 47, 752–759. [Google Scholar] [CrossRef]
- Wieland, I.; Schanze, I.; Felgendreher, I.M.; Barthlen, W.; Vogelgesang, S.; Mohnike, K.; Zenker, M. Integration of genomic analysis and transcript expression of ABCC8 and KCNJ11 in focal form of congenital hyperinsulinism. Front. Endocrinol. 2022, 13, 1015244. [Google Scholar] [CrossRef]
- Arya, V.B.; Guemes, M.; Nessa, A.; Alam, S.; Shah, P.; Gilbert, C.; Senniappan, S.; Flanagan, S.E.; Ellard, S.; Hussain, K. Clinical and histological heterogeneity of congenital hyperinsulinism due to paternally inherited heterozygous ABCC8/KCNJ11 mutations. Eur J Endocrinol. 2014, 171, 685–695. [Google Scholar] [CrossRef]
- Chang, G.; Ying, L.; Zhang, Q.; Feng, B.; Yao, R.; Ding, Y.; Li, J.; Huang, X.; Shen, Y.; Yu, T.; et al. Genetic variants of ABCC8 and clinical manifestations in eight Chinese children with hyperinsulinemic hypoglycemia. BMC Endocr. Disord. 2024, 24, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.; Ahamed, A.; Unnikrishnan, A.G.; Kumar, H.; Ellard, S. Permanent neonatal diabetes mellitus due to an ABCC8 mutation: a case report. . 2014, 15, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Xu, A; Cheng, J.; Sheng, H.; Wen, Z.; Lin, Y.; Zhou, Z.; Zeng, C., Shao, Y.; Li, C., Liu, L.; Li, X. Clinical Management and Gene Mutation Analysis of Children with Congenital Hyperinsulinism in South China. J Clin Res Pediatr Endocrinol. 2019, 11, 400–409.
- Hashemian, S.; Esfehani, R.J.; Karimdadi, S.; Ghaemi, N.; Eshraghi, P.; Gonabadi, N.M.; Sahebkar, A.; Vakili, R.; Abbaszadegan, M.R. Genotyping of ABCC8, KCNJ11, and HADH in Iranian Infants with Congenital Hyperinsulinism. Case Rep. Endocrinol. 2021, 2021, 1–6. [Google Scholar] [CrossRef]
- Ortiz, D.; Bryan, J. Neonatal Diabetes and Congenital Hyperinsulinism Caused by Mutations in ABCC8/SUR1 are Associated with Altered and Opposite Affinities for ATP and ADP. Front Endocrinol (Lausanne). 2015, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Beltrand, J.; Busiah, K.; Vaivre-Douret, L.; Fauret, A.L.; Berdugo, M.; Cavé, H.; Polak, M. Neonatal Diabetes Mellitus. Front Pediatr. 2020, 8, 540718. [Google Scholar] [CrossRef]
- Denton, J.S.; Jacobson, D.A. Channeling dysglycemia: ion-channel variations perturbing glucose homeostasis. Trends Endocrinol. Metab. 2012, 23, 41–48. [Google Scholar] [CrossRef]
- Gillis, D.; Familial Hyperinsulinism. 2003 Aug 19 [updated 2019 Mar 21]. In: Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1375/, (accessed on 10 March 2024).
- Nestorowicz, A.; Glaser, B.; Wilson, B.A.; Shyng, S.-L.; Nichols, C.G.; Stanley, C.A.; Thornton, P.S.; Permutt, M.A. Genetic Heterogeneity in Familial Hyperinsulinism. Hum. Mol. Genet. 1998, 7, 1119–1128. [Google Scholar] [CrossRef]
- Giri, D.; Hawton, K.; Senniappan, S. Congenital hyperinsulinism: recent updates on molecular mechanisms, diagnosis and management. J. Pediatr. Endocrinol. Metab. 2021, 35, 279–296. [Google Scholar] [CrossRef]
- Stanley, C.A. Perspective on the Genetics and Diagnosis of Congenital Hyperinsulinism Disorders. J. Clin. Endocrinol. Metab. 2016, 101, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ackermann, A.M.; Boodhansingh, K.E.; Bhatti, T.R.; Liu, C.; Schug, J.; Doliba, N.; Han, B.; Cosgrove, K.E.; Banerjee, I.; et al. Functional and Metabolomic Consequences of KATP Channel Inactivation in Human Islets. Diabetes 2017, 66, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Glaser, B.; Blech, I.; Krakinovsky, Y.; Ekstein, J.; Gillis, D.; Mazor-Aronovitch, K.; Landau, H.; Abeliovich, D. ABCC8 mutation allele frequency in the Ashkenazi Jewish population and risk of focal hyperinsulinemic hypoglycemia. Anesthesia Analg. 2011, 13, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Salomon-Estebanez, M.; Shah, P.; Nicholson, J.; Cosgrove, K.E.; Dunne, M.J. Therapies and outcomes of congenital hyperinsulinism-induced hypoglycaemia. Diabet. Med. 2018, 36, 9–21. [Google Scholar] [CrossRef]
- Li, M.; Han, X.; Ji, L. Clinical and Genetic Characteristics of ABCC8 Nonneonatal Diabetes Mellitus: A Systematic Review. J. Diabetes Res. 2021, 2021, 1–14. [Google Scholar] [CrossRef]
- Taylor-Miller, T.; Houghton, J.; Munyard, P.; Kumar, Y.; Puvirajasinghe, C.; Giri, D. Congenital hyperinsulinism due to compound heterozygous mutations in ABCC8 responsive to diazoxide therapy. J. Pediatr. Endocrinol. Metab. 2020, 33, 671–674. [Google Scholar] [CrossRef]
Figure 1.
Genetic Heterogeneity and Molecular Pathways in Hyperinsulinism [5].
Figure 1.
Genetic Heterogeneity and Molecular Pathways in Hyperinsulinism [5].
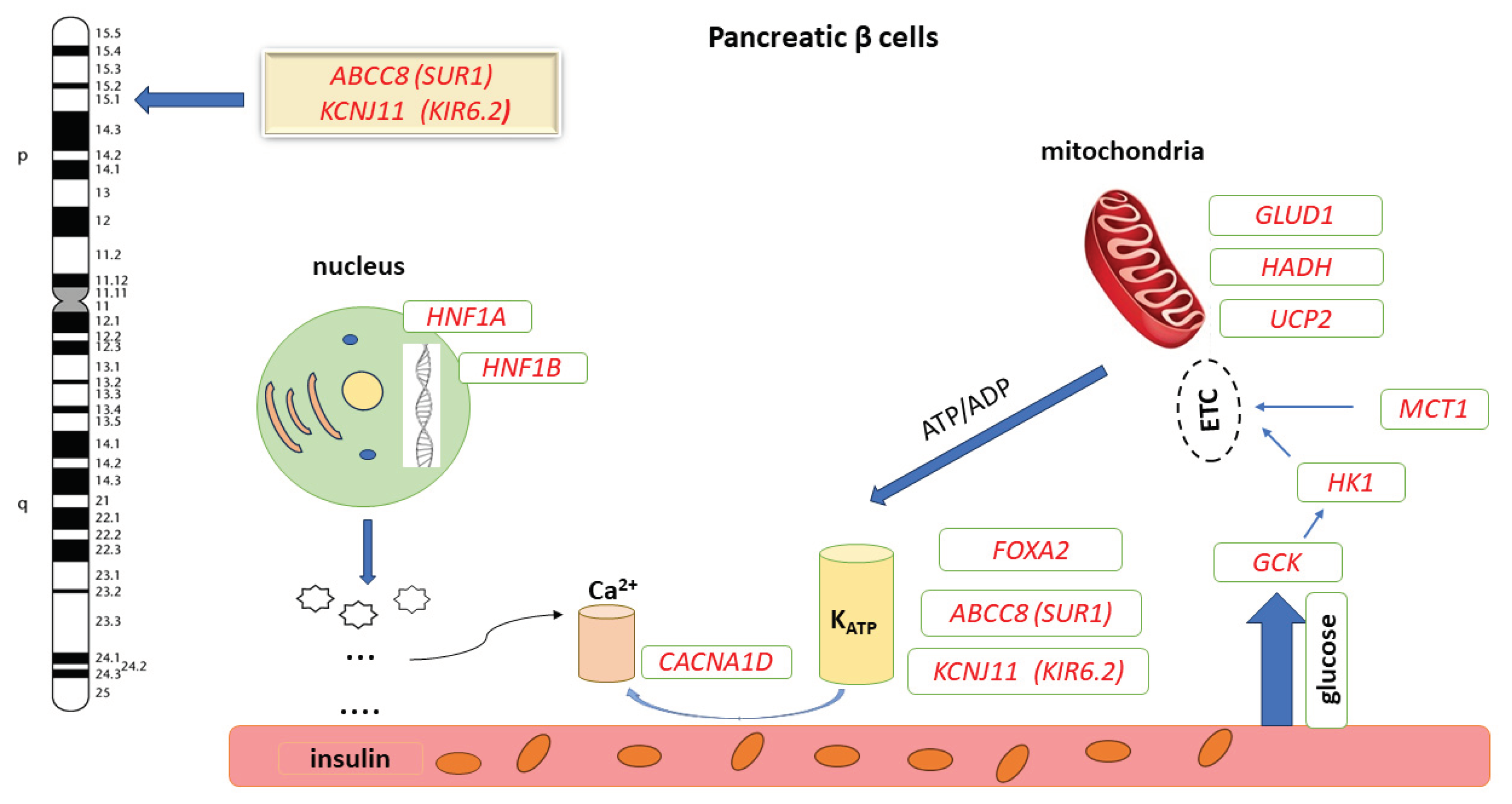
Figure 2.
Family Tree of Patient 1 (A.I.S.). Na: Heterozygous genotype (healthy carrier); aa: Homozygous genotype (affected individual).
Figure 2.
Family Tree of Patient 1 (A.I.S.). Na: Heterozygous genotype (healthy carrier); aa: Homozygous genotype (affected individual).
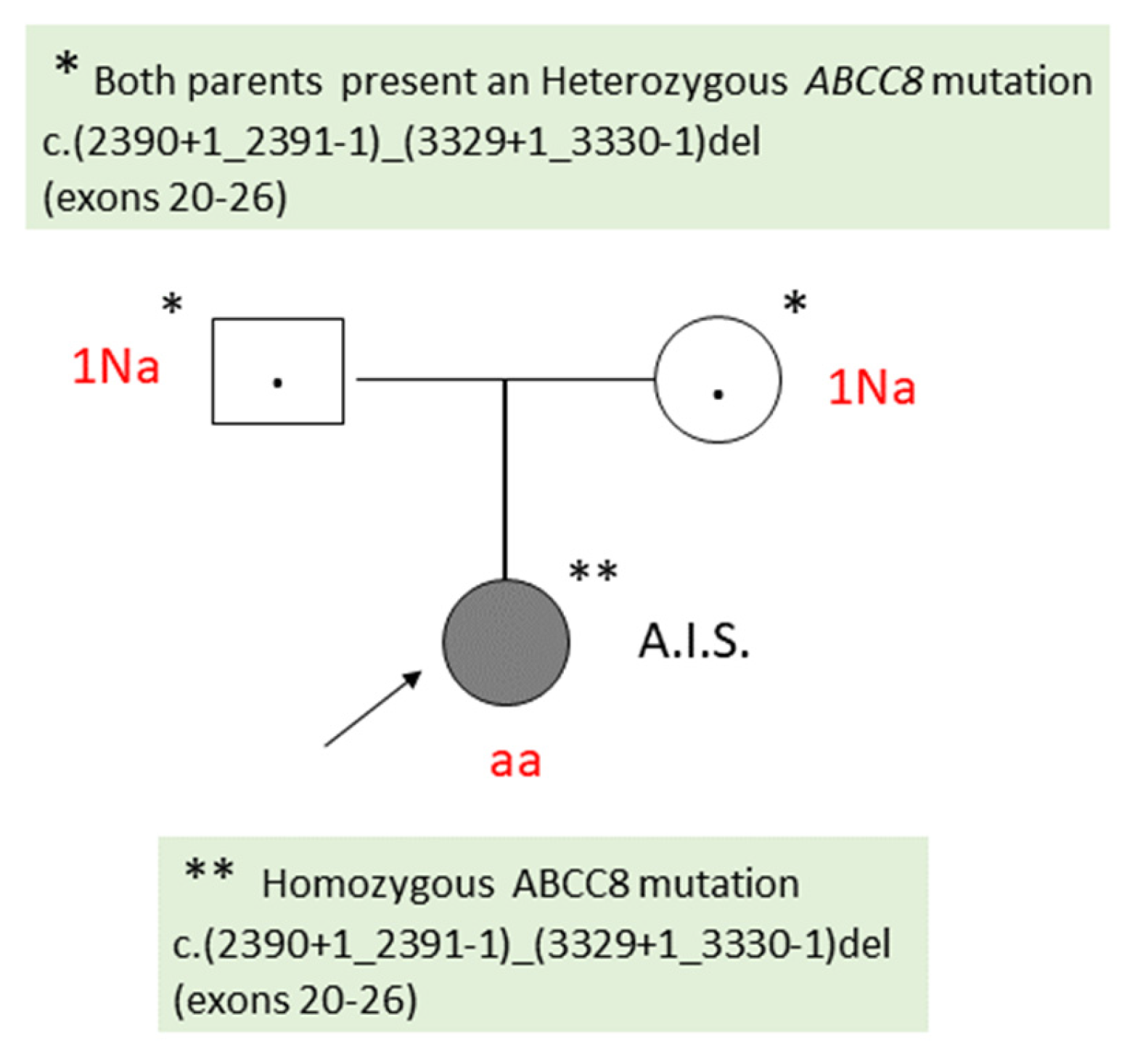
Figure 3.
Family Tree of Patient 2 (D.M.S.).
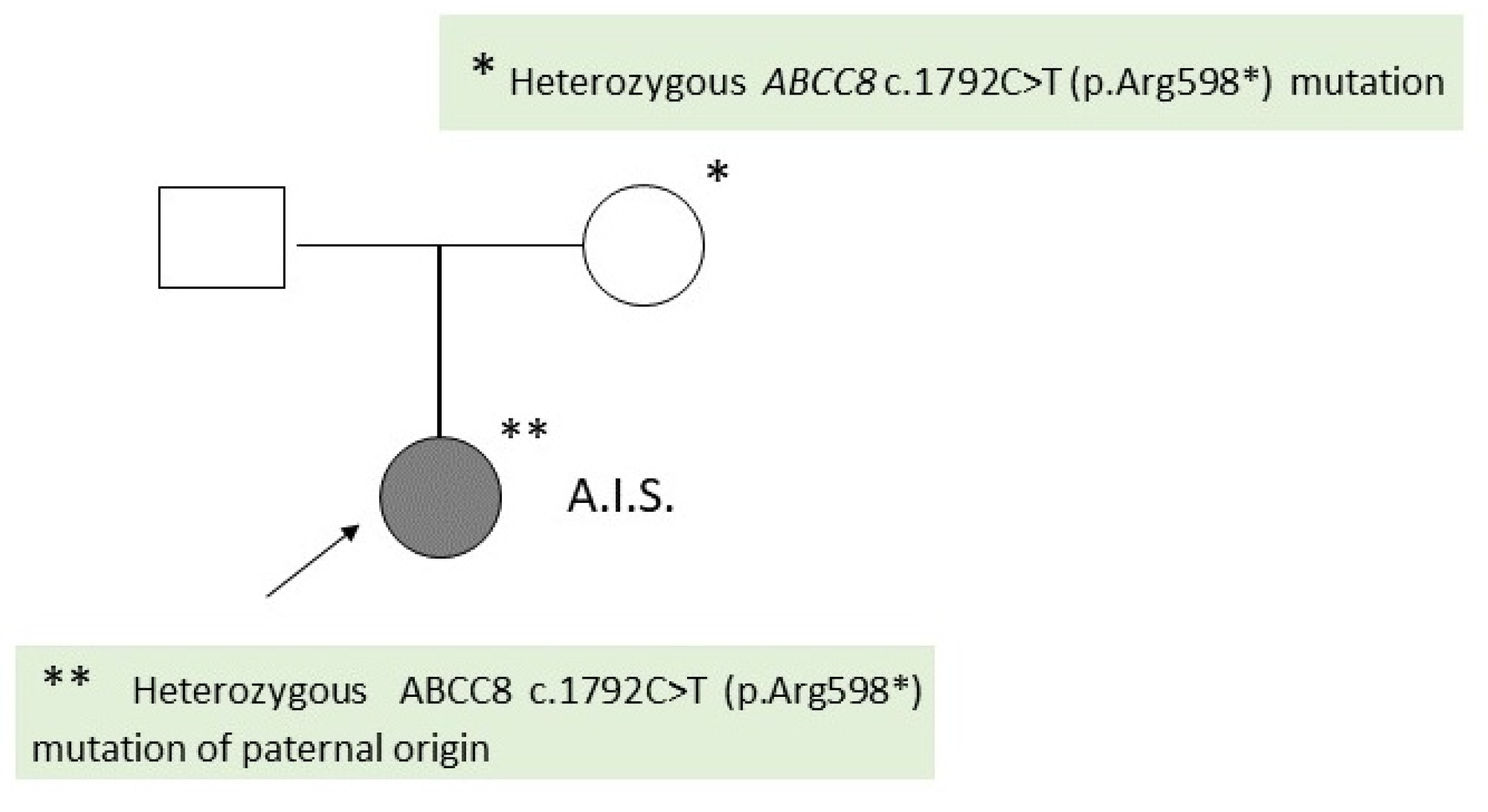

Table 1.
Clinical and Paraclinical Data of Patients with Congenital Hyperinsulinemic hypoglycemia.
| Criteria | Patient A.I.S. | Patient D.M.S. |
|---|---|---|
| Histologic type | Diffuse CHI | Focal CHI |
| The result of the patient’s genetic testing | Homozygous ABCC8 c.(2390+1_2391-1)_(3329+1_3330-1)del (exons 20-26) | Heterozygous ABCC8 c.1792C>T (p.Arg598*) |
| The result of genetic testing of the patient’s parents |
Both parents: Heterozygous ABCC8 c.(2390+1_2391-1)_(3329+1_3330-1)del (exons 20-26) |
Father: Heterozygous ABCC8 mutation c.1792C>T (p.Arg598*) Mother: normal result |
| Gender | F | F |
| Family history | no | no |
| Gestation | Term (38 weeks) | Term (38 weeks) |
| Parents’ consanguinity | No | No |
| Birth weight | 3140 g | 2700g |
| Onset of symptoms | 1st day | 3rd day |
| Persistent hypoglycaemia | 13-32mg/dL | 17-45 mg/dL |
| Insulin plasma level | ↑ (41,45 uUI/ mL; normal value: 3-25 uUI/ mL) | 15.28 uUI/ ml (normal value: 3-25 uUI/ml) |
| C- peptid plasma level | ↑ (4,92 ng/ mL; normal value: 0,2-4.4 ng/ mL) | 2,2 ng/ ml (normal value: 0,9-7,1 ng/mL |
| hGH | ↑ (133,67 uUI/mL ; normal value: 0-5 uUI/ mL) | Normal value |
| Thyroid hormones | Normal value | Normal value |
| Cortisol plasma level | Normal value | ↓ (2,27 µg/ dL(normal value: 4,3-22,4 µg/dL) |
| ACTH | Not performed | ACTH < 5 pg/mL (normal value: 5 -46 pg/ mL) |
| Macrosomia | No | No |
| Neurological manifestations | No | Tonic-clonic seizures Generalized hypotonia |
| Perinatal asphyxia | Yes | No |
| Transfontanelle ultrasonography |
Bilateral subependymal hemorrhage | Not performed |
| Brain CT | Not performed | Patologic |
| Abdominal IRM | normal | Normal |
| EEG | normal | Hypsarrhythmia |
| Diazoxide responsiveness | No | No |
| (18)F-DOPA PET/CT | Not indicated | Not performed yet |
* C-peptide, insulin, cortisol collected at hypoglycemia values below 50 mg/dL; (18)F-DOPA PET/CT: (3,4-dihydroxy-6-[18F]fluoro-L-phenylalanine) positron emission tomography scan/ computed tomography.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



