
Submitted:
29 March 2024
Posted:
29 March 2024
You are already at the latest version
Abstract
Rett Syndrome (RTT) is a severe neurodevelopmental disorder predominately diagnosed in females and primarily caused by pathogenic variants in the X-linked gene Methyl-CpG Binding Protein 2 (MECP2). Most often, the disease causing MECP2 allele resides on the paternal X chromosome while a healthy copy is maintained on the maternal X chromosome with inactivation (XCI) resulting in mosaic expression of one allele in each cell. Preferential inactivation of the paternal X chromosome is theorized to result in reduced disease severity; however, establishing such a correlation is complicated by known MECP2 genotype effects and an age dependent increase in severity. To mitigate these confounding factors, we developed an age- and genotype-normalized measure of RTT severity by modeling longitudinal data collected in the US Rett Syndrome Natural History Study. This model accurately reflected individual increase in severity with age and preserved group-level genotype specific differences in severity allowing for the creation of a normalized clinical severity score. Applying this normalized score to a RTT XCI dataset revealed that XCI influence on disease severity depends on MECP2 genotype with a correlation between XCI and severity observed only in individuals with MECP2 variants associated with increased clinical severity. This normalized measure of RTT severity provides the opportunity for future discovery of additional factors contributing to disease severity that may be masked by age and genotype effects.
Keywords:
Rett Syndrome
; X Chromosome Inactivation
; longitudinal modeling
1. Introduction
Rett syndrome (RTT) is a severe neurodevelopmental disorder that predominantly, but not exclusively, affects females and is characterized by apparently normal early development followed by regression of purposeful hand use and acquired verbal communication, development of repetitive hand stereotypies, and impaired gait or the inability to walk [1,2]. Most cases of RTT are caused by de novo heterozygous pathogenic loss-of-function variants in the X-linked transcriptional regulator Methyl-CpG Binding Protein 2 (MECP2) [3]. Although all people with RTT share these characteristic clinical features, overall clinical severity is variable between affected individuals [4]. At the group level, MECP2 genotype-phenotype relationships have been shown to be a major driver of this clinical variability, with some MECP2 variants (e.g. R133C, R294X, and R306C) associated with milder clinical severity and other variants (e.g. R106W, T158M, R168X, R255X, R270X) associated with increased clinical severity [4,5]. Although this genotype-phenotype relationship is robust at the group level, some affected individuals display clinical severity discordant from expected severity predicted by this group-level genotype-phenotype relationship. For example, some people with “severe” MECP2 variants have mild clinical severity and vice-versa. This individual level variation suggests that additional factors such as differences in environment, treatment interventions, or biological/genetic features play a role in determining individual-level clinical severity in RTT.
Because most cases of RTT are caused by de novo heterozygous variants in the X-linked gene MECP2, differential X-chromosome inactivation (XCI) has been proposed as a source of variation in clinical severity. During female embryonic development, XCI randomly silences one X-chromosome in each cell. In RTT, this results in cellular mosaicism, with some cells expressing a wild-type copy of MECP2 and others expressing a disease-causing MECP2 allele. In the majority of RTT cases, de novo pathogenic MECP2 variants arise on the paternal X-chromosome during spermatogenesis, and increased silencing of the diseased paternal allele (pXCI) is expected to result in milder disease severity owing to more cells expressing functional MeCP2 [6]. In extreme cases, rare incidences of familial RTT have been reported where an unaffected mother has a disease-causing variant in MECP2 but is protected by preferential inactivation of the mutant allele [7].
The impact of pXCI on disease severity in the general RTT population is less clear. One study restricted analysis to affected individuals with either T158M or R168X variants and found that increased activation of the X chromosome harboring the disease allele correlates with increased severity [8]. However, a larger study including multiple MECP2 variants did not find a correlation between pXCI and clinical severity [9]. Additional studies reported conflicting results of the effect of non-random XCI on disease severity, though these studies were limited by the lack of information of the directionality of XCI skewing related to mutant MECP2 allele expression [10,11]. Furthermore, previous studies also had limitations related to the fact that measures of clinical severity in RTT are correlated with both age and specific MECP2 variants, with a clear age-dependent increase in clinical severity and well-established genotype-phenotype relationships that may outweigh contribution of pXCI [4,5]. Ideally, evaluation of the effect of XCI on clinical severity in RTT would use age- and genotype-matched cohorts; however, in a rare disorder such as RTT with a limited population this is not feasible. While previous work addressed the genotype-phenotype issue by restricting analysis to the recurrent T158M and R168X variants, variation in genotype frequency limits the feasibility of this approach for other variants [8,12].
To circumvent the problem of age and genotype dependent effects on severity, we utilized data from the US Rett Syndrome Natural History Study (RNHS) to develop age- and genotype-normalized clinical severity scores. Applying this normalized clinical severity score, we found that pXCI had a minor influence on severity in individuals with severe MECP2 variants but did not contribute to clinical severity in individuals with mild MECP2 variants. The ability to utilize this normalized clinical severity score provides the opportunity to compare severity across ages and MECP2 genotypes and has the potential to help identify other biological or environmental factors that contribute to overall clinical severity in RTT in future studies.
2. Materials and Methods
2.1. Participants
Participants in this study were enrolled in the Rett syndrome and RTT-related Disorders Natural History Study (RNHS, NCT02738281), a longitudinal investigation into caregiver provided historical and clinically observed information spanning from 2006 to 2021. 1,826 individuals participated in the RNHS with an average of 5 visits per individual (ranging from 1 to 17 visits). In addition to following individuals with RTT (Classic or Atypical), this study included people who did not meet RTT diagnostic criteria but had pathogenic variants in MECP2, and individuals with RTT-related disorders including MECP2 duplication syndrome, CDKL5 deficiency disorder, and FOXG1 syndrome.
The Clinical Severity Score (CSS) was employed as one metric to track severity progression in RNHS participants. The CSS is a summation of 13 individual clinical parameters assessed with Likert scores from 0-4 or 0-5. The total CSS score ranges from 0 to 58 with 0 corresponding to normal and 58 representing the most severe presentation of the disorder [4]. Clinicians rating the CSS were provided in-person training at site visits to ensure consistent scoring across study locations.
2.2. Data Used for this Manuscript
Longitudinal CSS and individual parameters used to calculate the CSS were retrieved from the RNHS. A systematic algorithmic method was used to identify and revise longitudinal data on individual CSS entries in the RNHS that were logically inconsistent to generate revised longitudinal CSS scores for 1,819 individuals (7 individuals from the total RNHS had missing data precluding further processing). A schematic of the data correction algorithm is presented in Table S1. Briefly, individuals who showed variation in fixed historical scores (e.g. Age of Onset of Regression) were isolated and values deviating from the earliest reported observation were replaced with this value. For metrics which can only increase with time or follow a plateau (e.g. Scoliosis), values which showed a decrease or deviation from the plateau were replaced with the nearest previous observation. Revised CSS scores were calculated from these cleaned parameters for use in downstream analysis.
2.3. XCI DATA Generation
Paternal X chromosome inactivation status was obtained as part of a previously published study [9]. Briefly, maternal and proband peripheral blood samples were obtained as part of the RNHS (NCT02738281) or Biobanking of Rett Syndrome and Related Disorders (NCT02705677). DNA samples isolated from peripheral blood samples were analyzed for repeat length and methylation status at the AR locus. The percentage of paternal X inactivation was extrapolated by comparing maternal and proband samples. From 320 individuals with available XCI records in the RNHS, 198 had an informative XCI result and met restrictions for the current study as outlined above.
2.4. CSS Model Development
Data processing and analysis were performed using R version 4.1.3.
2.4.1. Model Development
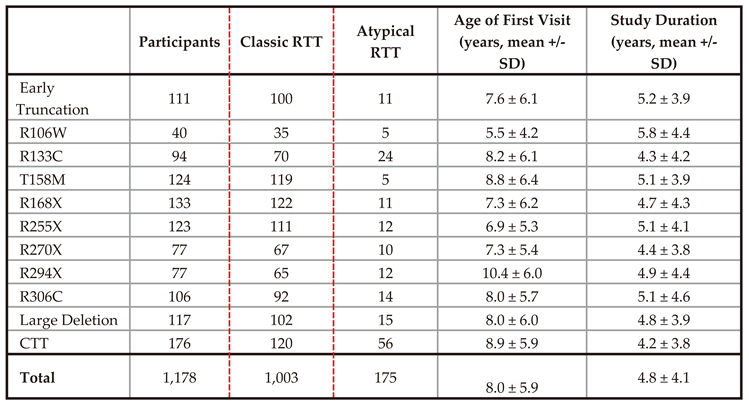
Training data for model development was restricted to female individuals with a confirmed pathogenic variant in MECP2 belonging to one of the eleven common variant groupings (R106W, R133C, T158M, R168X, R255X, R270X, R294X, R306C, large deletion, early truncation, or C-terminal truncating variant). Participants were also restricted to those with visits between 2 and 25 years of age to account for low numbers of observations beyond this window. 1,178 individuals met these requirements for downstream analysis (1,003 with a diagnosis of “Classic”, 175 with a diagnosis of “Atypical”, see Table 2 and Table S2 for demographic information). Following further restriction to individuals with a classic diagnosis for purposes of model training, 5,469 observations across 1,003 individuals were available for model development with an average of 5 observations per individual.
A mixed effects model was chosen for further analysis to accommodate the longitudinal nature of the data and between subject differences in observation number and interval of observation. Exploratory data analysis indicated CSS progression follows a logarithmic growth-like pattern (Figure S1), so age was log transformed to accommodate this observation. Transformed age values were further decomposed into between and within person components to parse out subject versus group effects. The between person component was calculated from the centered mean transformed age of observation within an individual (‘Mean_lnage_center’), while the within person component (‘Duration’) was defined as the distance from the between person component to the transformed age of observation within an individual. A conditional growth mixed effects model of MECP2 genotype and age impact on clinical severity (1) was fitted to the transformed decomposed data using the lme function within the nlme package. In this model, age was defined as the interaction of between- and within-subject time components with only the within-subject component considered for subject-specific random effects. Mean genotype specific clinical severity trajectories were calculated using the predict function from the nlme package to provide a top-level view of CSS progression with time for each common MECP2 genotype.

2.4.2. Model Validation
A pseudo leave-one-out cross validation approach was used to determine model accuracy. Model performance was assessed on any female with one of the above variants regardless of diagnosis (1,178 individuals total). The model was iteratively trained on individuals with a classic diagnosis leaving out the test subject if they had a classic diagnosis. For novel subjects with atypical diagnosis, no exclusion was warranted. A total of 1,178 test subject – model pairs were run. Individual level predictions were made for each test subject–model pair using the IndvPred_lme function in the JMbayes package to allow Bayesian prediction of the novel test subject. Root mean squared error was calculated by standard methods for predicted and observed values for each individual. For a subset of individuals with numerous observations, individual level predicted trajectories were plotted alongside observed CSS values for visual inspection (Figure S2).
2.4.3. CSS Trajectory Prediction
An overarching model was trained on individuals with a classic diagnosis as described in Model Development. Using the IndvPred_lme function in the JMbayes package, predicted clinical severity scores were calculated for ages 2 to 25 in 0.1 year bins for all 1,178 individuals in this study regardless of diagnosis. Mean predicted CSS were then calculated for each individual by averaging predicted severity scores across time. The mean predicted CSS is analogous to calculating the area under the curve of a predicted severity trajectory over time as equal numbers of predicted scores are calculated for each individual over the same intervals. The mean predicted CSS therefore provides an integrated view of severity progression without retaining age as a factor effectively normalizing the score across ages.
2.4.4. CSS Percentile Estimation and Normative CSS Calculation
Mean predicted CSS scores for individuals with a classic diagnosis were isolated and cumulative distributions of mean predicted CSS were calculated for each variant group using the ecdf function in the stats package. Using these genotype specific cumulative distributions of CSS, percentiles were assigned to the mean predicted CSS scores for all individuals in this study regardless of diagnosis. This mean percentile score was defined as normative CSS (nCSS), an age and genotype normalized clinical severity score, in downstream analysis.
2.5. Analysis of XCI Effect
Pearson’s correlation coefficients were calculated separately for the association of paternal XCI with CSS for all individuals, those with mild MECP2 variants, and those with severe variants. Potential interactions between MECP2 variant severity and XCI were assessed by regression analysis of a simplified model of normative CSS:
nCSS=SevGroup + SevGroup*pXCI + SevGroup
Here, nCSS is the age and genotype invariant normative severity score described above, SevGroup is the MECP2 variant severity grouping (mild or severe), and pXCI is the percentage of paternal X inactivation. Percentage variance explained by the simplified model was determined by dividing an individual parameter’s sum of squares by the model total sum of squares.
2.6. Data and Code Availability
Revised CSS scores, model predicted nCSS scores, and pXCI (if available) are provided in the supplementary materials. Code employed for model development, analysis, and figure generation is available at https://github.com/jkmerrit/2023_RTT_XCI
3. Results
3.1. pXCI Does Not Correlate with Raw CSS Scores
To evaluate the effect of skewed X-chromosome inactivation (XCI) on severity in RTT, we obtained XCI information on 320 people with RTT and maternal XCI data which allows the determination of paternal XCI (pXCI) in the affected individuals. For the 320 affected probands, informative pXCI results were obtained for 198 participants with RTT (Table 1, Classic RTT n=183, Atypical n=15).
To account for the observed age-dependent increase in clinical severity in the entire cohort of people with RTT enrolled in the RNHS (Figure S1A), we assessed the association between pXCI and CSS score observed nearest to 9.7 years of age, the mean all-visit age for the cohort with informative pXCI information. For many participants with informative pXCI, the nearest CSS score was several years removed from the 9.7-year-old mean, and we observed the same overall increase in CSS with age in this group (Figure 1A). Using the CSS score closest to 9.7 years old for participants with informative pXCI data did not show any correlation between pXCI and CSS (Figure 1B). To control for known MECP2 genotype-phenotype relationships where some variants have a mild phenotype (R133C, R294X, R306C, C-terminal truncations, S. Figure 1B,C) and others present a more severe disorder (R106W, T158M, R168X, R255X, R270X, early truncations, and large deletions), we looked at pXCI/CSS relationships separately within these groups. While no correlation is present in individuals with mild MECP2 variants (Figure 1C), a trend towards a negative correlation exists in individuals with severe variants (Figure 1D). The presence of age and genotype effects in this analysis may mask less salient contributors to RTT severity and obscure true pXCI/CSS relationships highlighting the necessity of a severity metric independent of these factors.
3.2. Modeling Accurately Predicts Rett Syndrome Severity over Time
To develop an age- and genotype-normalized CSS, we constructed a mixed effects model incorporating the contribution of assessment age and MECP2 genotype on CSS score that enables the prediction of an individual’s CSS at any age from a limited set of observations. To generate this model, we focused on individuals in the RNHS with classic RTT and common MECP2 variants or variant groups to model the typical clinical severity progression (n = 1,003, Table 2 red dashed area; Table S2). Because the observed age-dependent increase in CSS follows a logarithmic growth pattern (Figure S1A), we incorporated a logarithmic transform for time. To accommodate variation in observed severity trajectories between individuals, we incorporated individual identifiers as a random effect, and decomposed time in the model into two components: the mean age of observation (between-subject effects) and the length of time separating an individual observation from the mean age (within-subject effects) [13]. Acknowledging known MECP2 genotype-phenotype relationships, we treated genotype as a fixed effect in the model such that each variant grouping would have a separate baseline intercept and trajectory consistent with clinical observations [4,5]. This accurately modeled the expected rapid increase in severity from 2 to 10 years of age with subsequent deceleration in the rate of CSS increase and approaching a plateau in later years (Figure 2A, black dashed line overall group). For individual variants, the model produced similar growth trajectories with a pattern of genotype specific severity consistent with previous reports (Figure 2A, colored lines) [4,5].

Table 2.
Model development and validation cohort. RNHS participants were restricted to female individuals with one of eleven common MECP2 variants and at least one visit between the ages of 2 and 25. These individuals were further restricted to those with a diagnosis of Classic RTT for model training (red box).
Table 2.
Model development and validation cohort. RNHS participants were restricted to female individuals with one of eleven common MECP2 variants and at least one visit between the ages of 2 and 25. These individuals were further restricted to those with a diagnosis of Classic RTT for model training (red box).
 |
To validate the model, we assessed the accuracy of individual participant model predicted scores compared to observed CSS scores. Visual inspection of predicted individual participant-level severity trajectories showed close relationship to observed CSS scores for all common variant groups (example images shown in Figure S2). We empirically assessed the accuracy of this model by using a leave-one-out cross validation approach to determine predictive error (Root Mean Square Error [RMSE]) of the model for all individuals with RTT (Classic and Atypical) in the RNHS (n = 1,178, Table 2, Table S1). The overall RMSE was ~2, indicating good fit and demonstrating that the model is able to predict an individual’s CSS within ± 2 points on the 58-point CSS (Figure 2A inset). Using individual mean predicted CSS as an age-normalized score, we found MECP2 genotype-phenotype relationships seen with the observed CSS score (S. Figure 1B,C) were preserved using the individual model predicated mean CSS score (Figure 2B-C), demonstrating that the model accurately represents observed RTT severity. However, this genotype-phenotype relationship highlights the need for a genotype normalized measure of RTT severity to enable comparison across genotypes as well as ages.
3.3. Age- and Genotype- Normalized CSS Scores Reveal a Genotype Dependent Correlation between pXCI and Severity
To control for MECP2 genotype effects on disease severity, we created genotype normalized CSS by calculating the within-genotype cumulative distribution of age-normalized mean predicted CSS scores from participants with Classic RTT and common MECP2 variants or variant groups (Figure 3A). This allows the conversion of the age-normalized mean predicted CSS score to a standardized MECP2 variant specific score based on the percentile within the variant-specific cumulative distribution (percentile/100, thus ranging from 0-1). We then calculated the MECP2 genotype normalized scores for all RTT participants (Classic and Atypical) from individual model-predicted mean CSS scores. Using these genotype-normalized scores as a normative CSS (nCSS) removes the genotype-phenotype impact on severity, as demonstrated by the cumulative distribution plot of nCSS (Figure 3B), which lacks the genotype-phenotype relationship observed in Figure 3A. This nCSS (which incorporates both age- and genotype effects) allows for standardized comparison of clinical severity in RTT between individuals at different ages and with different MECP2 genotypes.
We then used this age- and genotype-normalized nCSS to evaluate the contribution of pXCI to clinical severity in RTT. For the entire cohort with informative pXCI data (Table 1), we did not find a relationship between pXCI and nCSS (Figure 4A), consistent with the lack of observed relationship between raw CSS scores and pXCI (Figure 1A). Similarly, we found no relationship between pXCI and nCSS scores in individuals with mild MECP2 variants (Figure 4B), as observed for raw CSS and pXCI (Figure 1C). However, we found that pXCI was negatively correlated with nCSS scores for people with severe variants in MECP2 (Figure 4C, p = 0.013, r = -0.215), whereas there was only a trend towards the correlation between raw CSS and pXCI in this group (Figure 1C, p = 0.055, r = -0.17). The presence of a unique pXCI/nCSS relationship in the context of severe MECP2 variants (Figure 4C) suggests pXCI impacts disease severity through an interaction with MECP2 variant severity. To further explore this relationship, we assessed how pXCI and MECP2 variant severity contribute to the nCSS score (Figure 4D). As expected, MECP2 variant severity alone does not affect normative severity due to elimination of genotype-phenotype effects in the nCSS (see Figure 3B). pXCI alone was also not found to majorly explain variation in nCSS between individuals (Figure 4D) consistent with the lack of correlation between pXCI/nCSS in the entire cohort (Figure 4A). However, we found the interaction between MECP2 variant severity and pXCI notably contributes to nCSS and explains ~3% of the variation in severity between individuals (Figure 4D).
4. Discussion
Here we describe a model of RTT severity that allows for generation of an age and genotype normative CSS. We show that application of this method to analysis of an XCI dataset reveals a previously unidentified correlation between increased pXCI and decreased disease severity in individuals with severe MECP2 variants but not those with mild variants. From the current data, we estimate that three percent of the variation in severity between individuals with RTT is attributed to the interaction between pXCI and MECP2 variant severity driven by the specific interaction of pXCI and severe variants.
While previous analysis of the same XCI dataset did not identify a correlation between XCI and disease severity, our ability to uncover a relationship in this study is aided by the use of an age- and genotype- normalized severity score in addition to restricting our analysis to common MECP2 variants [9]. Our findings are consistent with previous reports of correlation between XCI and severity in individuals with R168X and T158M, variants that are classified as severe in the current study [8]. Though a higher degree of correlation between XCI and disease severity was previously reported for these variants, the current study encompasses variants beyond R168X and T158M and employs a different severity scale, so only qualitative comparisons between these two works can be made.
A key limitation in this and previous investigations into the contribution of XCI to RTT severity has been the use of peripheral blood as the source of genomic DNA for determining XCI status. Concerns have been raised that XCI in peripheral blood may not accurately represent states of mosaicism in the central nervous system relevant to RTT [10]. However, recent analysis by the GTEx consortium suggest XCI trends in peripheral blood are maintained in the brain [14]. Having evidence that peripheral blood samples accurately reflect CNS mosaicism supports continued use of whole blood as a surrogate for XCI in the brain, as obtaining suitable numbers of CNS samples for a well powered investigation of the impact of pXCI on RTT severity is not feasible.
The interaction between MECP2 variant severity and XCI contributing to nCSS is an unexpected finding. Genotype-phenotype relationships are collapsed in the nCSS (Figure 3.B), so the presence of a genotypic interaction with XCI suggests a more complex association. Mild variants invariant to the effects of XCI might represent a base level of MECP2 dysfunction sufficient to cause RTT. The presence of even relatively small numbers of mild mutant allele expressing cells elicits hallmarks of the disorder; however, disease severity is largely unaffected by the relative ratio of cells expressing healthy versus mild mutant alleles. Conversely, for more severe variants, a greater ratio of dysfunctional cells could have an additive effect on worsening disease severity. Preferential skewing of XCI and silencing of the disease allele in this case provides a measurable reduction in clinical severity. This is a key consideration as we enter the era of MECP2 targeted therapeutics, as this may indicate variant specific differences in the percentage of functional MECP2 expressing cells needed to elicit observable clinical improvement. For people with mild variants, expression of functional MECP2 in a large percentage of cells may be required for observable clinical improvement, whereas individuals with severe variants may see clinically meaningful differences with a smaller percentage of functional MECP2 expressing cells.
Beyond exploring impacts of XCI on disease severity, the age- and genotype-normalized nCSS score provides a means to assess other factors that contribute to RTT severity. Because the nCSS score allows for direct comparisons between individuals regardless of their age and MECP2 variant, we are able to conduct well-powered studies without restriction to specific ages and/or genotypes. This feature also allows for analysis of historical datasets where sample collection may not have been age matched nor controlled for genotype. Moreover, the nCSS allows for interrogation of factors contributing to disease severity that may have a relatively small impact on clinical presentation compared to the well-established MECP2 genotype-phenotype relationship. This approach could contribute to the identification of other biological factors (genetic, metabolomic), environmental factors, or therapies (physical, occupational, speech) that influence clinical severity, but may be masked by the strong MECP2 genotype-phenotype relationship. Finally, our strategy of modeling longitudinal data to obtain age- and genotype-normalized scores could see application to other measures of clinical outcomes in RTT including the Revised Motor Behavior Assessment (RMBA), the Rett Syndrome Behavior Questionnaire (RSBQ), and the Rett Syndrome Caregiver Assessment of Symptom Severity (RCASS) [15,16,17].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Rett syndrome severity increases with age and depends on MECP2 genotype; Figure S2: Example model predicted clinical severity trajectories for individuals with repeated measures; Table S1: Summary of data corrections used in this study; Table S2: Study Participant Demographics.
Author Contributions
JKM and JLN conceptualized this article, with input from SS, MJF, and AKP. XF and RCC generated the X chromosome inactivation data used in this article. JKM and JLN compiled and organized the data, and JKM conducted the analysis of the data. JKM prepared the draft of this manuscript, and all authors provided writing review and edits. The authors read and approved the final manuscript.
Funding
This work was supported by funding from the National Institutes of Health grants U54 HD061222 (AKP), P50HD103537 (JLN), the International Rett Syndrome Foundation (JLN), and the Vanderbilt Postdoctoral Training Program in Functional Neurogenomics 5T32MH065215 (JKM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Eunice Kennedy Shriver Child Health and Human Development Institute (NICHD).
Institutional Review Board Statement
This study was approved by Institutional Review Board (IRB) by a single-IRB agreement provided through the University of Alabama at Birmingham and IRB approval at the Greenwood Genetic Center. Written informed consent was obtained for each participant according to the Declaration of Helsinki. A Certificate of Confidentiality was provided by the National Institute of Child Health and Development (NICHD). This non-interventional clinical trial protocol was registered with Clinical Trials.Gov (NCT02738281).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Revised CSS scores, model predicted nCSS scores, and pXCI (if available) are pro-vided in Supplementary File 1. Code employed for model development, analysis, and figure generation is available at https://github.com/jkmerrit/2023_RTT_XCI.
Acknowledgments
We thank Cristan A. Farmer, PhD (NIMH) for her advice and expertise regarding the longitudinal models employed in this study. We also extend our gratitude to the people and caregivers who took part in the Rett Syndrome Natural History Study. We acknowledge the many contributors to the Rett Syndrome Natural History Study including Eric Marsh, MD, PhD (Children’s Hospital of Philadelphia/University of Pennsylvania); Tim Benke, MD, PhD (University of Colorado-Denver); Shannon Standridge, DO (Cincinnati Children’s Hospital); Sarika Peters, PhD and Cary Fu, MD (Vanderbilt Medical Center); Peter Heydemann, MD (Rush Memorial Medical Center); Robin Ryther, MD (Washington University); Mary Jones, MD (Children’s Hospital-Oakland/UCSF); Richard Haas MD (University of California San Diego); Daniel Glaze, MD and Bernhard Suter MD (Baylor College of Medicine).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett Syndrome: Revised Diagnostic Criteria and Nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Benke, T.A.; Marsh, E.D.; Skinner, S.A.; Merritt, J.; Lieberman, D.N.; Standridge, S.; Feyma, T.; Heydemann, P.; Peters, S.; et al. The Array of Clinical Phenotypes of Males with Variants in Methyl-CpG Binding Protein 2. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2019, 180, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett Syndrome Is Caused by Variants in X-Linked MECP2, Encoding Methyl-CpG-Binding Protein 2. Nat Genet 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific Variants in Methyl-CpG-Binding Protein 2 Confer Different Severity in Rett Syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Pillai, R.B.; Shekar, K.V.; Lane, J.B.; Motil, K.J.; Skinner, S.A.; Tarquinio, D.C.; Glaze, D.G.; McGwin, G.; Kaufmann, W.E.; et al. Methyl-CpG-Binding Protein 2 (MECP2) Variant Type Is Associated with Disease Severity in Rett Syndrome. J. Med. Genet. 2014, 51, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Trappe, R.; Laccone, F.; Cobilanschi, J.; Meins, M.; Huppke, P.; Hanefeld, F.; Engel, W. MECP2 Variants in Sporadic Cases of Rett Syndrome Are Almost Exclusively of Paternal Origin. The American Journal of Human Genetics 2001, 68, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Lee, S.S.; Zhang, X.; Houwink-Manville, I.; Song, H.R.; Amir, R.E.; Budden, S.; Naidu, S.; Pereira, J.L.; Lo, I.F.; et al. Rett Syndrome and beyond: Recurrent Spontaneous and Familial MECP2 Variants at CpG Hotspots. Am. J. Hum. Genet. 1999, 65, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Archer, H.; Evans, J.; Leonard, H.; Colvin, L.; Ravine, D.; Christodoulou, J.; Williamson, S.; Charman, T.; Bailey, M.E.S.; Sampson, J.; et al. Correlation between Clinical Severity in Patients with Rett Syndrome with a p.R168X or p.T158M MECP2 Variant, and the Direction and Degree of Skewing of X-Chromosome Inactivation. Journal of Medical Genetics 2007, 44, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Butler, K.M.; Abidi, F.; Gass, J.; Beisang, A.; Feyma, T.; Ryther, R.C.; Standridge, S.; Heydemann, P.; Jones, M.; et al. Analysis of X-inactivation Status in a Rett Syndrome Natural History Study Cohort. Mol Genet Genomic Med 2022, 10, e1917. [Google Scholar] [CrossRef] [PubMed]
- Xiol, C.; Vidal, S.; Pascual-Alonso, A.; Blasco, L.; Brandi, N.; Pacheco, P.; Gerotina, E.; O’Callaghan, M.; Pineda, M.; Armstrong, J.; et al. X Chromosome Inactivation Does Not Necessarily Determine the Severity of the Phenotype in Rett Syndrome Patients. Sci Rep 2019, 9, 11983. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van Den Veyver, I.B.; Schultz, R.; Malicki, D.M.; Tran, C.Q.; Dahle, E.J.; Philippi, A.; Timar, L.; Percy, A.K.; Motil, K.J.; et al. Influence of Variant Type and X Chromosome Inactivation on Rett Syndrome Phenotypes. Annals of Neurology 2000, 47, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, F.; Jacobsen, A.; Rigau, M.; Bosio, M.; Kaliyaperumal, R.; Laros, J.F.J.; Willighagen, E.L.; Valencia, A.; Roos, M.; Capella-Gutierrez, S.; et al. A Catalogue of 863 Rett-Syndrome-Causing MECP2 Variants and Lessons Learned from Data Integration. Sci Data 2021, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Curran, P.J.; Bauer, D.J. The Disaggregation of Within-Person and Between-Person Effects in Longitudinal Models of Change. Annual Review of Psychology 2011, 62, 583–619. [Google Scholar] [CrossRef] [PubMed]
- Tukiainen, T.; Villani, A.-C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X Chromosome Inactivation across Human Tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Raspa, M.; Bann, C.M.; Gwaltney, A.; Benke, T.A.; Fu, C.; Glaze, D.G.; Haas, R.; Heydemann, P.; Jones, M.; Kaufmann, W.E.; et al. A Psychometric Evaluation of the Motor-Behavioral Assessment Scale for Use as an Outcome Measure in Rett Syndrome Clinical Trials. American Journal on Intellectual and Developmental Disabilities 2020, 125, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.K.; Neul, J.L.; Benke, T.A.; Marsh, E.D.; Glaze, D.G. A Review of the Rett Syndrome Behaviour Questionnaire and Its Utilization in the Assessment of Symptoms Associated with Rett Syndrome. Frontiers in Pediatrics 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Raspa, M.; Gwaltney, A.; Bann, C.; von Hehn, J.; Benke, T.A.; Marsh, E.D.; Peters, S.U.; Ananth, A.; Percy, A.K.; Neul, J.L. Psychometric Assessment of the Rett Syndrome Caregiver Assessment of Symptom Severity (RCASS). J Autism Dev Disord 2024. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
X chromosome inactivation does not correlate with raw clinical severity scores. A) Raw clinical severity scores (CSS) at visit closest to 9.7 years old, the mean age of all visits for the XCI cohort. Linear regression (red line) shows an age dependent increase in severity. B) Raw CSS scores plotted against percentage of pXCI for all participants (n=198, Red line: linear regression of plotted values, Shaded area: standard error) showing no correlation (Pearson’s r = 0.074; p = 0.298; 95% CI: -0.066 to 0.212). C) No correlation between pXCI and raw CSS for individuals with mild MECP2 variants (R133C, R294X, R306C, or CTT; n = 66; Pearson’s r = 0.20; p = 0.103; 95% CI: -0.042 to 0.424). D) A trend towards a weak negative correlation between pXCI and raw CSS is seen for individuals with severe MECP2 variants (early truncation, R106W, T158M, R168X, R255X, R270X, or large deletions; n = 132; Pearson’s r = -0.17; p = 0.055; 95% CI: -0.329 to 3.33e-03).
Figure 1.
X chromosome inactivation does not correlate with raw clinical severity scores. A) Raw clinical severity scores (CSS) at visit closest to 9.7 years old, the mean age of all visits for the XCI cohort. Linear regression (red line) shows an age dependent increase in severity. B) Raw CSS scores plotted against percentage of pXCI for all participants (n=198, Red line: linear regression of plotted values, Shaded area: standard error) showing no correlation (Pearson’s r = 0.074; p = 0.298; 95% CI: -0.066 to 0.212). C) No correlation between pXCI and raw CSS for individuals with mild MECP2 variants (R133C, R294X, R306C, or CTT; n = 66; Pearson’s r = 0.20; p = 0.103; 95% CI: -0.042 to 0.424). D) A trend towards a weak negative correlation between pXCI and raw CSS is seen for individuals with severe MECP2 variants (early truncation, R106W, T158M, R168X, R255X, R270X, or large deletions; n = 132; Pearson’s r = -0.17; p = 0.055; 95% CI: -0.329 to 3.33e-03).
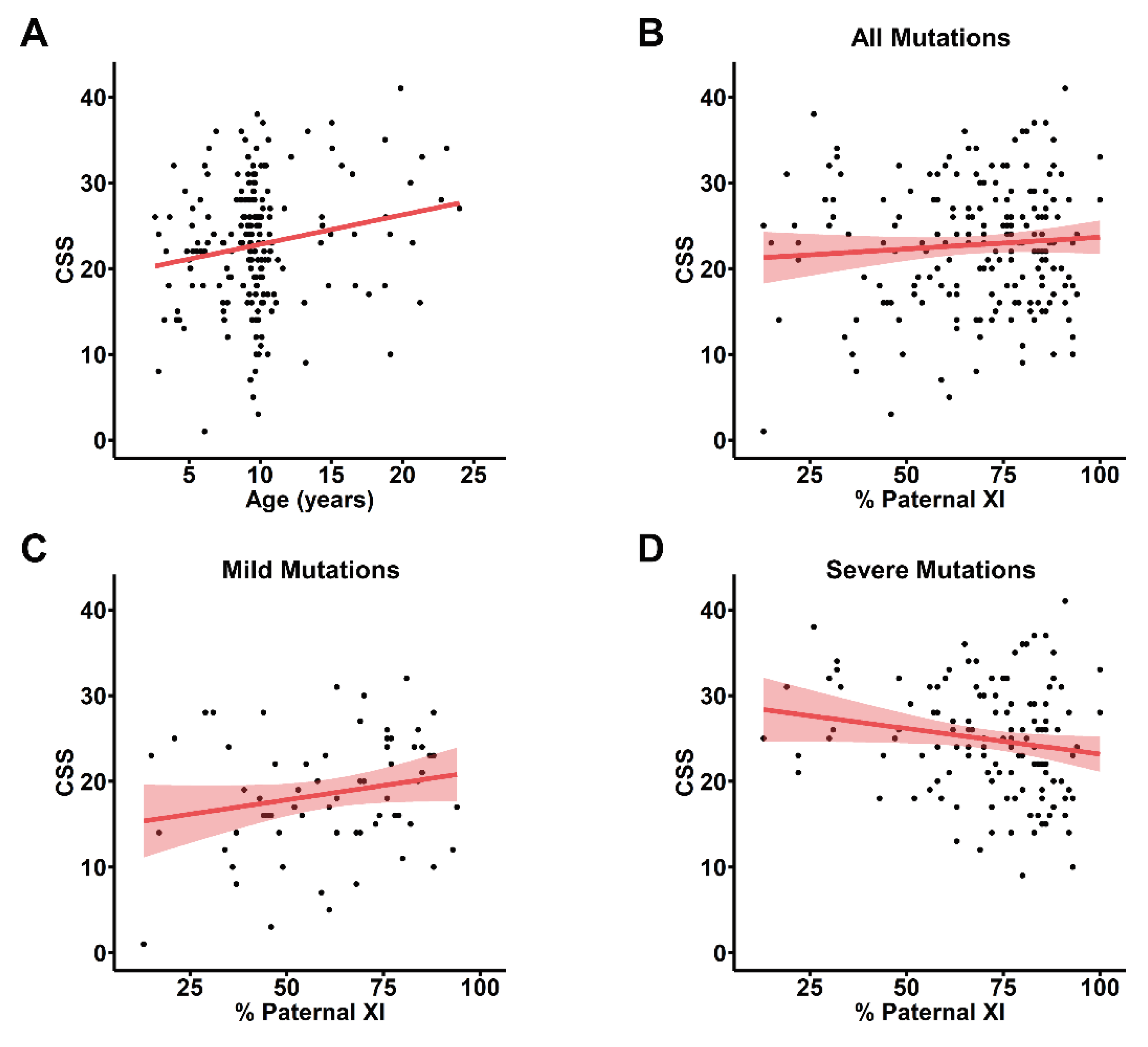
Figure 2.
Mixed effects model of Rett Syndrome clinical severity. A) Best fit model of age-related CSS trajectory for all participants (black dotted line) and individuals with common variants (colored lines for variant groups as shown in legend). Inset: Root mean squared error (RMSE) for mixed effects model predicted clinical severity scores determined by leave-one-out cross validation. Bars: median, minimum, and maximum RMSE, Boxes: interquartile range, Dots: outliers B) Within-subject mean predicted CSS (+/- SEM) for variant groups (X-axis) recapitulates previously established differences in severity between MECP2 variant groups. C) Heatmap showing pairwise comparisons of mean predicted CSS by variant (ANOVA with post-hoc pairwise comparisons using Tukey’s HSD) with color indicating p-value cutoffs as indicated in panel legend.
Figure 2.
Mixed effects model of Rett Syndrome clinical severity. A) Best fit model of age-related CSS trajectory for all participants (black dotted line) and individuals with common variants (colored lines for variant groups as shown in legend). Inset: Root mean squared error (RMSE) for mixed effects model predicted clinical severity scores determined by leave-one-out cross validation. Bars: median, minimum, and maximum RMSE, Boxes: interquartile range, Dots: outliers B) Within-subject mean predicted CSS (+/- SEM) for variant groups (X-axis) recapitulates previously established differences in severity between MECP2 variant groups. C) Heatmap showing pairwise comparisons of mean predicted CSS by variant (ANOVA with post-hoc pairwise comparisons using Tukey’s HSD) with color indicating p-value cutoffs as indicated in panel legend.
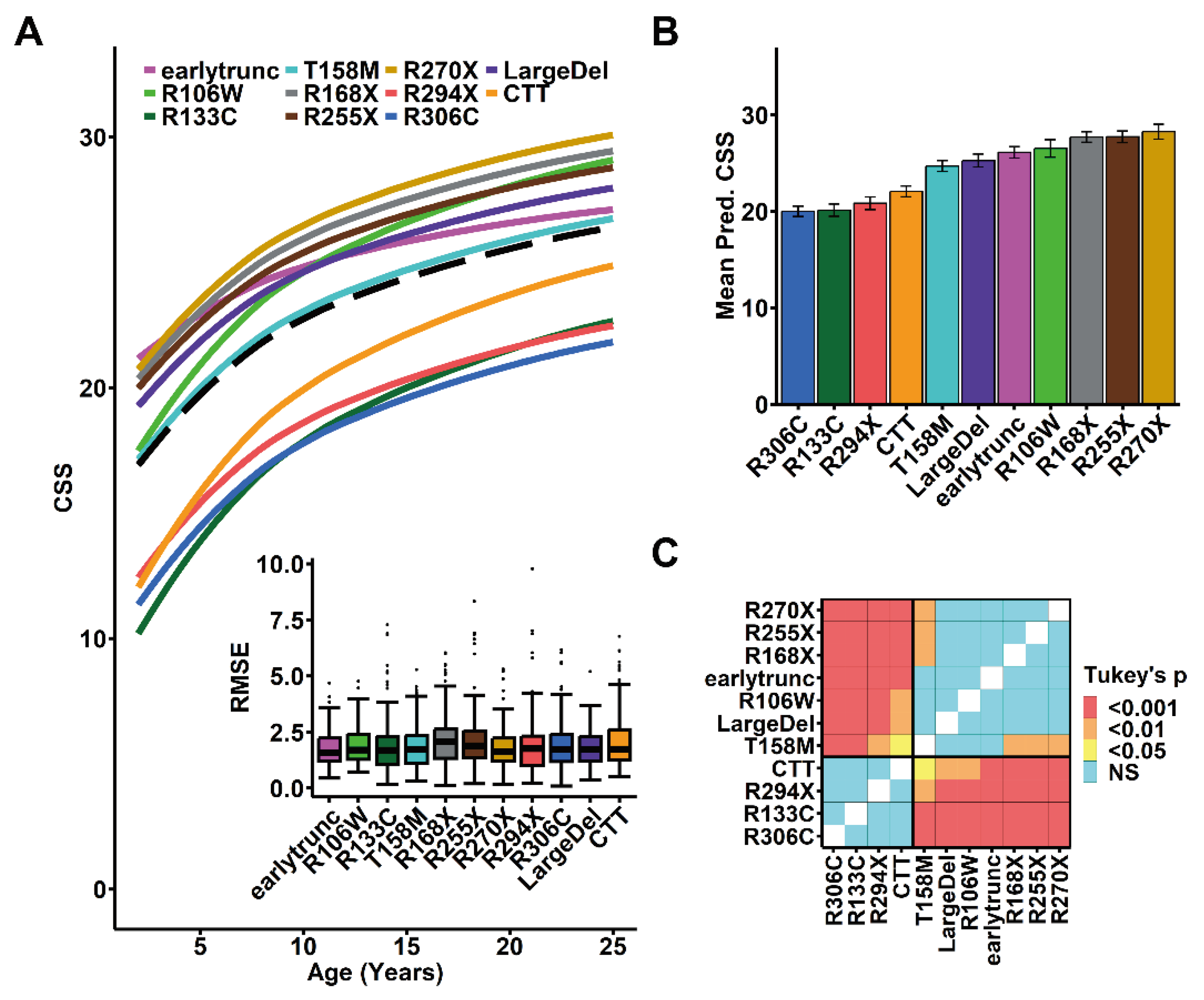
Figure 3.
Generation of variant-specific normative CSS scores. A) Cumulative distributions of within-subject mean predicted CSS for MECP2 variant groups showing expected genotype-phenotype relationship as presented in Figure 2. Normative CSS for each participant is generated by determining the variant group specific percentile for the participant’s mean predicted CSS. B) Cumulative distributions of normative CSS scores by MECP2 variant group demonstrates expected loss of genotype-phenotype relationship for variant specific normative CSS scores.
Figure 3.
Generation of variant-specific normative CSS scores. A) Cumulative distributions of within-subject mean predicted CSS for MECP2 variant groups showing expected genotype-phenotype relationship as presented in Figure 2. Normative CSS for each participant is generated by determining the variant group specific percentile for the participant’s mean predicted CSS. B) Cumulative distributions of normative CSS scores by MECP2 variant group demonstrates expected loss of genotype-phenotype relationship for variant specific normative CSS scores.
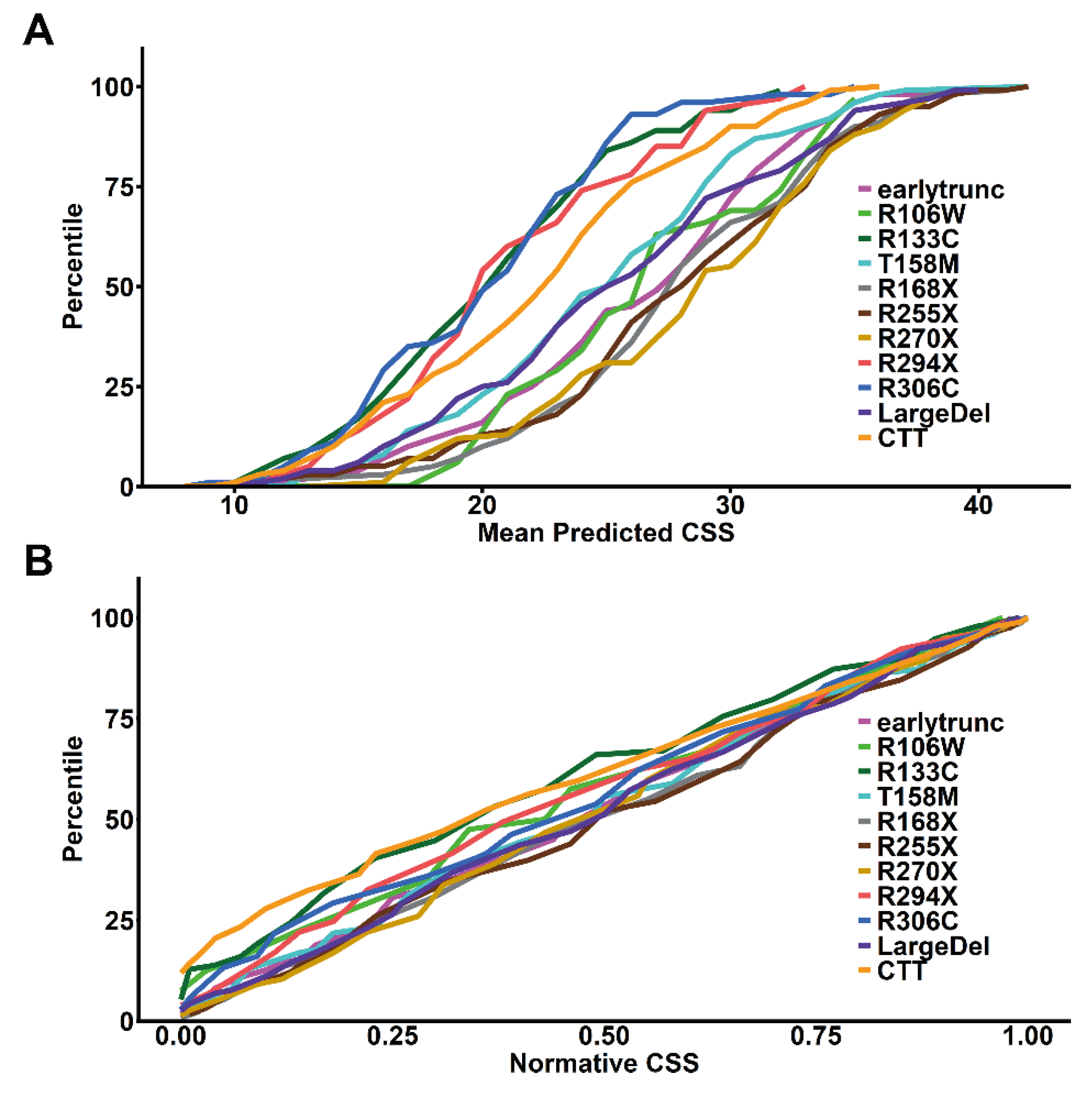
Figure 4.
Paternal X inactivation correlates with reduced normative CSS for severe MECP2 variants. A) Normative CSS plotted against pXCI for all individuals shows no correlation (n = 198; Pearson’s r = -0.073; p = 0.310; 95% CI: -0.210 to 0.068). Regression line and standard error shading as in Figure 1. B) pXCI shows no correlation with normative CSS scores in individuals with mild MECP2 variants (n = 66; Pearson’s r = 0.139; p = 0.265; 95% CI: -0.106 to 0.369). C) Increased pXCI shows a minor correlation with reduced clinical severity as measured by normative CSS in individuals with severe MECP2 variants (n = 132; Pearson’s r = -0.215; p = 0.013; 95% CI: -0.372 to -0.0456). D) Summary of a simplified model where normative CSS is described by the interaction between pXCI and MECP2 variant severity.
Figure 4.
Paternal X inactivation correlates with reduced normative CSS for severe MECP2 variants. A) Normative CSS plotted against pXCI for all individuals shows no correlation (n = 198; Pearson’s r = -0.073; p = 0.310; 95% CI: -0.210 to 0.068). Regression line and standard error shading as in Figure 1. B) pXCI shows no correlation with normative CSS scores in individuals with mild MECP2 variants (n = 66; Pearson’s r = 0.139; p = 0.265; 95% CI: -0.106 to 0.369). C) Increased pXCI shows a minor correlation with reduced clinical severity as measured by normative CSS in individuals with severe MECP2 variants (n = 132; Pearson’s r = -0.215; p = 0.013; 95% CI: -0.372 to -0.0456). D) Summary of a simplified model where normative CSS is described by the interaction between pXCI and MECP2 variant severity.
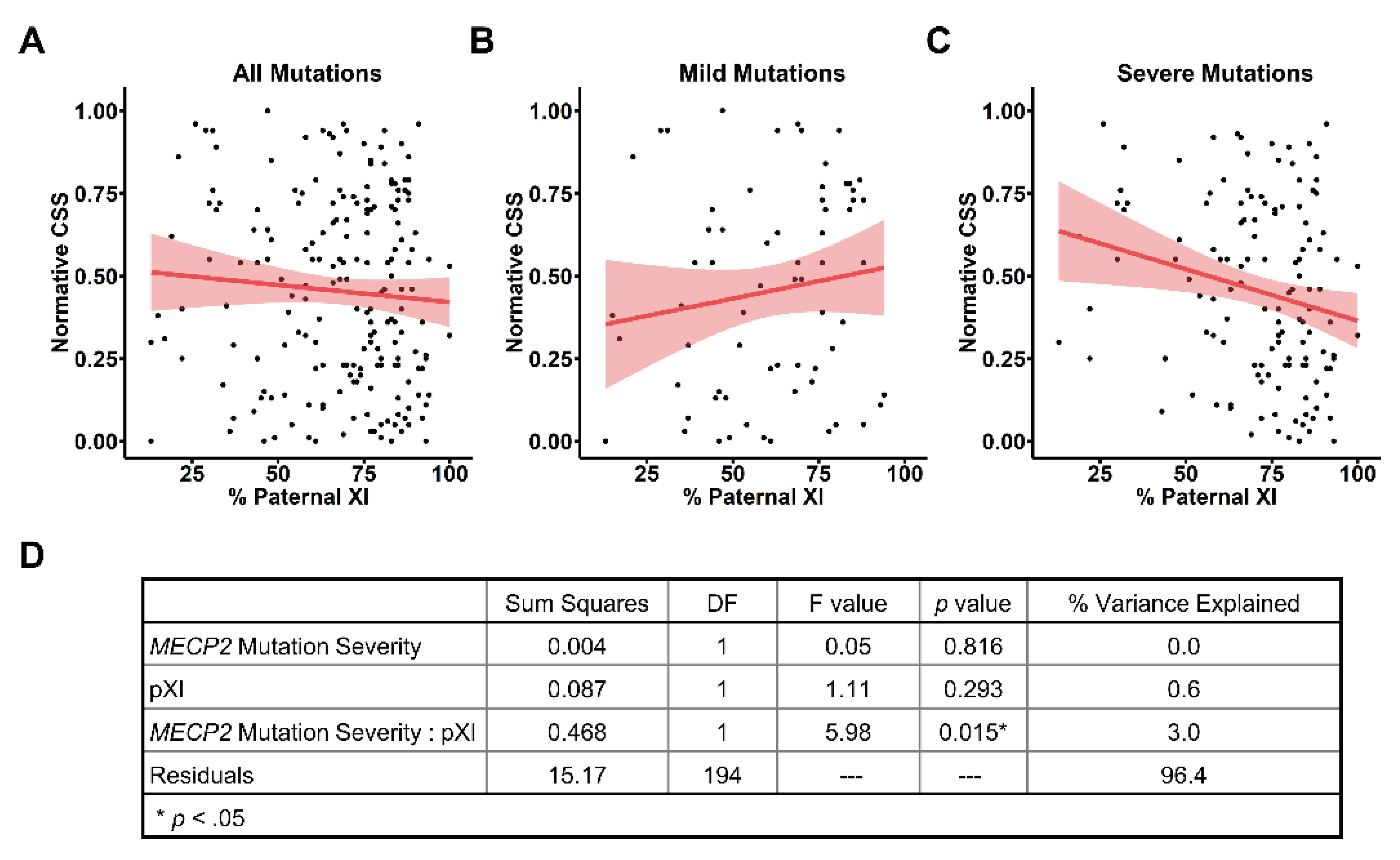
Table 1.
X chromosome inactivation study cohort. Phased XCI records were available for 320 individuals with RTT and related disorders. 198 individuals met the restriction of having one of eleven common RTT-causing MECP2 variants. CTT: C-Terminal Truncation.
Table 1.
X chromosome inactivation study cohort. Phased XCI records were available for 320 individuals with RTT and related disorders. 198 individuals met the restriction of having one of eleven common RTT-causing MECP2 variants. CTT: C-Terminal Truncation.
| Participants | Classic RTT | Atypical RTT | Age of First Visit (years, mean +/- SD) |
|
|---|---|---|---|---|
| Early Truncation | 17 | 16 | 1 | 5.7 ± 6.2 |
| R106W | 8 | 7 | 1 | 6.7 ± 4.5 |
| R133C | 10 | 9 | 1 | 6.1 ± 5.6 |
| T158M | 26 | 26 | 0 | 7.8 ± 6.5 |
| R168X | 25 | 25 | 0 | 5.9 ± 4.3 |
| R255X | 24 | 23 | 1 | 5.7 ± 5.3 |
| R270X | 14 | 13 | 1 | 6.0 ± 4.3 |
| R294X | 16 | 14 | 2 | 8.2 ± 4.2 |
| R306C | 19 | 17 | 2 | 7.2 ± 4.8 |
| Large Deletion | 18 | 17 | 1 | 6.9 ± 6.4 |
| CTT | 21 | 16 | 5 | 6.6 ± 5.4 |
| Total | 198 | 183 | 15 | 6.6 ± 5.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



