
Submitted:
28 March 2024
Posted:
29 March 2024
You are already at the latest version
Abstract
Understanding mechanisms of ageing remains a complex challenge for biogerontologists, but recent adaptations of evolutionary ageing theories offer a compelling lens in which to view both age-related molecular and physiological deterioration. Ageing is commonly associated with a progressive loss of biochemical processes and degeneration of molecular material, however, the mechanisms of diminishing function are not simply a result of aberrant expression of these pathways. Natural selection pressures are at their highest in youthful periods to select genes advantageous to maximising reproductive capacity. After sexual maturation, selective pressure diminishes, subjecting individuals to maladaptive pleiotropic gene function once beneficial for developmental growth, but pathogenic later in life. Due to this selective ‘shadowing’ in ageing, mechanisms to counter hyperfunctional genes are unlikely to evolve within a given species. These genetic ‘run-on’ programmes are not dysfunctional per se and, in fact, remain highly robust. Thus, interventions capable of optimising gene function during the ageing process represent as attractive therapeutic modalities. Caenorhabditis elegans offers a unique model to explore high-throughput screening of interventions to combat hyperfunctional pathways involved in ageing. However, the majority of research to date employ whole-life vs late-life pathway manipulation of developmental/growth-related systems to claim ageing attenuation, despite significant impairments in early-life fitness and discordance with healthspan. Here, we discuss data supporting hyperfunctional changes at a global molecular and functional level in C. elegans, and how classical lifespan-extension mutants alter these dynamics. We later discuss the relevance of such mutant models for exploring mechanisms of ageing, highlighting that post-reproductive gene manipulation represents a far more translatable approach for C. elegans research not routed within pleiotropic constraints. More so, lifespan has become a routine readout for quantifying ‘ageing’, however, we make the argument that a compression of gerospan vs lifespan extension is a more meaningful goal for anti-ageing regimes with current knowledge. Similarly, mechanisms governing lifespan are distinct from those regulating health, and slowing senescent pathology accumulation appears more applicable for the abstraction to human ageing.
Keywords:
Hyperfunction
; C. elegans
; ageing
; gene optimisation
; healthspan
Highlights
- Widely-held views that ageing is caused by pathogenic gene function has limited our scope-of-view that an over-activation of wild-type gene function may explain proximate causes of ageing
- Genetic alleles that drive reproductive fitness are preferentially selected for, despite their (hyperfunctional) pleiotropic effects that contribute to ageing pathology later in life
- C. elegans represent a candidate model to explore therapeutic mechanisms to counter pleiotropic expression changes in ageing, but current experimental approaches are limited in their design and should be re-addressed
Introduction
Ageing remains the largest socio-economic burden in humans and the greatest risk factor for developing life-threating diseases [1]. Gerontological fields have proposed numerous potential explanations for ageing including the disposable soma [2], oxidative stress and damage accumulation [3] as well as developmental/evolutionary theories [4,5]. However, many of these lack the ability to explain key aspects of ageing deterioration as standalone models [6], and some have recently been largely discredited to explain causal pathophysiological events seen in ageing [7,8]. The current available explanations seem to fall exclusively into two categories; ageing caused by accumulative molecular damage, or programmatic events that define the ageing projection. Probabilistically, ageing is a combination of both processes, but defining the precise onset and nature of the proximate causes of ageing remains a significant challenge. The widely-held assumption that ageing is caused by the accumulation of damaged genetic material overlooks the possibility that unobserved (and seemingly robust) wild-type gene (hyper)function can drive ageing progression later in life. Two recent ageing reviews have explored key evolutionary frameworks in detail, namely, hyperfunction and developmental theories [6,9]. Importantly, these works challenge current understandings on proximate causes of ageing and provide frameworks for how evolutionary processes might explain physiological ageing deterioration. Therefore, there is pressing need to design pre-clinical research that is compatible for testing evolutionary theories of ageing, yet current approaches are somewhat limited in design.
Antagonistic pleiotropy (AP) was first proposed by George C. Williams in 1957, stating that genes which contribute to fitness and reproductive success in early life will persist throughout populations by natural selection, outweighing any detrimental affects they cause later in life [4]. Genomic surveillance is known to decline with age across species [10,11,12], reflecting the diminishing presence of selective forces after reproduction that allow these once beneficial genes to continue their unruly expression to drive tissue and organ failure [13]. Genes that are fitness-promoting early in life are unlikely to evolve mechanisms of repression, given that selective ‘shadowing’ becomes apparent after successfully passing on these genes to subsequent generations [6]. This blinding of selection windows with age imply that ageing, and its associated pathologies, are largely evolutionary diseases subject to an inappropriate continuation of developmental programmes concerned merely with maximising reproductive rates. In this view, one can appreciate how continued expression of growth-promoting pathways could drive aberrant hyperfunction within particular tissues and organs that depletes biochemical energy stores from excess biosynthesis, hypertrophic cellular growth and replicative senescence [14]. Optimisation of gene function to slow ageing rates after sexual maturation is, therefore, beyond evolutionary limits, and mitigation of molecular AP effects ultimately lies in the hands of genetic/pharmacological approaches.
Mikhail Blagosklonny was amongst the first to suggest a quasi-programmatic ‘hyperfunction’ view of ageing that incorporated AP concepts with experimental support (see [14] for original propositions and [6] for a detailed overview of career work to date). His work unifies programmatic and senescent theories of ageing, placing hyperfunction as an event that precedes, and is causal to the onset of senescence accumulation in ageing [15]. The key tenants of hyperfunction state that ageing is not a consequence of functional decline or damage accumulation per se, but in fact a persistent, overactivation of pathways once fundamental for developmental growth that drive pathological events later in life. Thus, senescence is a consequence of hyperfunctional processes that depletes/inhibits cellular plasticity. This view has gained attention in the ageing community as a candidate model for studying ageing both mechanistically and therapeutically [6]. Thus, a review of experimental evidence through the lens of molecular hyperfunction in ageing, and how these concepts should be experimentally addressed is needed. Here, we will review key literature in the established ageing model Caenorhabditis elegans to explore this concept.
C. elegans have contributed considerably to our understanding of ageing decline and tissue-specific genomic and proteomic deterioration across its short 3-week life-course [16,17,18]. With a fully mapped genome, ease of genetic manipulation techniques and a translucent outer cuticle allowing in vivo protein surveillance, C. elegans are an established invertebrate model for studying conserved mechanisms of ageing. Advancements in sequencing and mass-spectrometry techniques have improved our understanding of transcriptomic and proteomic alterations across the worm life-course, where elements of molecular hyperfunction (i.e., RNA and protein accumulation, late-life activation of developmental pathways) have been noted [19,20]. Reviews of transcriptional and proteomic alterations in ageing C. elegans are limited, and a hyperfunctional view of these molecular changes has not been considered to date. When ageing is viewed through this emerging paradigm, commonalities in transcriptional and proteomic aberrations on a global level (i.e., beyond gene to protein expression mapping) become clearer, where current dogma suggests a dissociation between the two [21].
Seminal findings have shown that insulin-like growth factor receptor mutants (IGF1/daf-2 in worms) live up to twice as long as wild-types in C. elegans [22], consolidation that reductions in growth-related/anabolic pathways attenuates ageing. Although these data are fundamental in highlighting potential pathways regulating ageing deterioration, the field continues to exploit loss-of-function mutants or life-long gene silencing of such pathways, challenging the applicability of these findings in the context of human ageing. Age-specific genetic or pharmacological manipulation (i.e., that begin during vs preceding the onset of healthspan decline) of pleiotropic pathways should be of priority when assessing the roles they play in ageing. Similarly, is the focus of lifespan extension (as opposed to gerospan suppression) in claims of reducing the ageing burden. It is not clear which strategy is optimal [23], where precise scaling of lifespan and healthspan needs to be more closely evaluated to make such claims. We will later discuss temporal considerations for future C. elegans ageing experiments in order to maximise the translation of these findings into people.
Ageing-induced genetic pleiotropy – an overlooked aspect of healthspan research?
Developmental success requires dynamic alterations in gene/protein expression to accommodate a period of fast growing physiological and molecular alterations [24]. These processes are tightly regulated in developing/young adult life across species when selective pressure is highest, however, begin to escape genetic surveillance after reproductive periods [2]. Importantly, data are emerging for the concept of AP-induced ageing in C. elegans. Namely, the GATA transcription circuit is an important regulator of germline, hypodermal and vulval development, however, expression of two member genes (namely, ELT-5 and ELT-6) continue progressively with ageing and are known to repress the function of their cytoprotective counterpart ELT-3 [19]. Post-reproductive RNAi of both ELT-5 and ELT-6 (i.e., starting from day 5 of adulthood) alleviates ELT-3 restraint and upregulates protective pathways to increase survival and stress resistance [19]. This represents a run-on programme effect (termed ‘quasi-programme’ by Blagosklonny [9] – a once beneficial developmental programme that continues its expression becoming, non-adaptive in late-life) that causes ageing deterioration via an impaired ability to respond to stress with age.
Such AP-induced quasi-programmes have been implicated in the development of teratoma-like tumors in C. elegans, where the continued activation of embryonic programmes drives unfertilized oocyte entry into the uterus, causing detrimental cellular hypertrophy via endoreduplicated chromatin masses [25]. Similarly, apoptosis of germ cells throughout embryogenesis contributes to the provision of developing oocytes and promotes fitness in early adulthood, however, continuation of this pathways causes gonad degeneration and senescence in ageing animals [20]. Interestingly, C. elegans conversion of gut biomass into yolk (regulated by autophagic processes) contributes to fitness in early life, but causes intestinal atrophy and senescence in late-life [26]. The IIS pathway has been shown to drive both early (adaptive) and late-life (pathogenic) biomass conversion processes, where inhibition of daf-2/IIS reduces yolk accumulation, intestinal atrophy and ultimately extends life [26], implicating the IIS system as a classical AP pathway.
Other examples more widely implicated in across-species ageing pathology include the mechanistic target of rapamycin (mTOR) protein complex, a key regulator of nutrient sensing, signalling and developmental growth across the animal kingdom, where absence of this pathway is lethal in embryogenesis [27,28]. However, continued late-life activation of the PI3K/AKT/mTOR axis plays fundamental roles in the development and progression of cancer cell survival, growth, motility and metabolism [29,30]. Functional pleiotropic effects of mTOR were first observed in C. elegans, where LET-363 mutants (the worm ortholog of mTOR) display marked developmental delay and arrest as L3 larvae, however, exhibit significant extension of lifespan thereafter (albeit, likely in an increasingly frail state) [28]. Importantly, these ageing AP effects also extend into higher mammals where, for example, increasing IGF-1 signalling in cardiac myocytes by overexpression of the IGF-1 receptor increased cardiac growth, function and exercise capacity in young mice. However, these same mice exhibit maladaptive cardiac remodeling, ventricular fibrosis, impaired contractile function and lung congestion compared to wild-type mice in later life, ultimately leading to increased late-life mortality and shorter lifespans [31].
Pleiotropic ageing mechanisms continue to grow in experimental support, for example the role of trl-1 upregulation in C. elegans upon re-feeding from starvation to increase brood size at the cost of reduced lifespan later in life [32], as well as dual roles for aryl hydrocarbon receptors (AhR) promoting cell differentiation during embryogenesis but catalysing NAD+ degradation and stimulating oxidative stress in later life [33]. However, a common, persisting misconception with AP is that genes begin to elicit pleiotropic effects due to damage accumulation with ageing. This is a largely falsified claim, likely arising from the common presupposition that ageing is caused by macromolecular damage [3]. In fact, AP genes are in essence wild-type, but become contextually maladaptive depending on the physiological environment in which they continue to operate. The acknowledgement that ageing could, in fact, be caused by quasi-programmatic wild-type gene function is gaining consideration, and recent adaptions to Williams AP model have built on this.
Molecular hyperfunction as a candidate theory of ageing
An appreciated view is that key Hallmarks of ageing have a hierarchical sequence of events that drives ageing and senescence [34]. For example, Genomic instability, epigenetic alterations, loss of proteostasis and telomere attrition are proposed ‘primary’ drivers of ageing, with loss of mitochondrial function, nutrient sensing and chronic inflammation consequences of perturbations in primary features. Although these events undoubtedly play roles in ageing progression, this view is still somewhat comprised of individualistic components at its core and tells us little about the causal events that dictate primary hallmark collapse. Such approaches have struggled to explain causal mechanisms of ageing for decades [6], and a united theory that combines programmatic events with senescent phenotypes could progress experimental concepts for ageing studies.
Blagosklonny focuses heavily on IIS and mTOR pathways, and the anti-ageing properties of Rapamycin (inhibitor of mTOR) to build his key argument; developmental/growth-related pathways display quasi-programme AP effects later in life, and inhibition of these pathways reduces diseases of ageing through a suppression of molecular hyperfunction [15]. Simplistically, Blagosklonny analogises that ageing is akin to a car entering a low-speed zone without brakes and an inability to slow down, counter to a traditional view that ageing occurs from the accumulation of rusting with time [35]. This view eloquently ties together evolutionary theories of ageing (AP) that drive senescence and disease, and are not mutually dependent on theories of damage accumulation – of which experimental evidence continues to challenge its prospects in being causal to ageing [36,37]. In fact, there exists multiple genes that, when inhibited (via RNAi interference) from the first stage of larval development, C. elegans growth arrests [38]. Conversely, when these same genes are inhibited during adulthood, 42% extend adult lifespan, lending mechanistic support that ageing is driven by AP gene function that becomes hyperfunctional.
Conceptually, Blagosklonny has focused predominantly on biosynthetic pathways of growth and development, but these might not be the only key players. Recent across-species transcriptomic analysis identified translational fidelity and translational elongation as key terms associated with longer life, and genes associated with translational fidelity are under strong selective pressure across species [39]. If transcription/translation processes are a strongly selected feature, one could imagine aberrant hyperfunction of these apparatus in later life periods when AP onsets and selective shadowing come into effect. These data are discussed below, and potentially links hyperfunction of transcriptional machinery (either separately, or in combination) with protein complexes such as mTOR.
Transcriptomic remodeling towards a hyperfunctional state in ageing
The C. elegans transcriptome displays well-established alterations with ageing, with consistent declines in genes/pathways regulating mitochondrial function and metabolic processes, and increases in stress response, DNA damage repair and innate immunity-regulating pathways [18,40]. Up to 6,000 genes can be seen to significantly change in expression profiles from young adult to aged timepoints in the whole worm [11], highlighting both the transcriptional malleability to physiological changes with age as well as potential maladaptive expression changes. The assumption made using common pathway/gene set analysis pipelines is that these genetic alterations directly contribute to functional physiological changes. Whilst a proportion of gene expression changes inevitably contributes directly to healthspan deterioration (e.g., decreased gene expression and synthesis of mitochondrial proteins, a hallmark of ageing [34]), systems regulating transcriptional apparatus themselves are subject to age-related deterioration, suggesting a class of genetic changes that are by nature neither promoting nor deterring ageing progression but merely reflect a coordinative loss of internal regulatory systems.
Rangaraju and colleagues recently showed results to this effect, highlighting that transcriptional drift (where genes within a functional group change their expression in opposing directions vs youthful profiles) increases with ageing, and that mRNA stoichiometry between genes within the same functional group begin to conflict and disrupt homeostasis in C. elegans [11]. Specifically, individual genes that comprise a unit (i.e., a functional pathway) responsible for regulating a given biological process change their expression profile late in life compared to youthful ages. This opposing expressional ‘switch’ has been shown to dysregulate the communication within, and regulative capacity of functionally related signalling pathways [11]. Mechanistic explanations for transcriptional drift remain indefinable and, whilst being a difficult phenomenon to address experimentally, the role of diminishing selective pressure undoubtedly plays a role in allowing such a widespread transcriptional dysregulation.
Recent work revealed that RNA polymerase II transcriptional/elongation speeds increased progressively with chronological age in worms and higher mammals [41]. As a result, the time for splicing events is compromised, and increased amounts of circular RNA’s, rare protein isoforms and intron-retaining transcripts become prevalent. Genetic inhibition of subunits required for RNA polymerase II function (ama-1/POLR2A loss-of-function mutant) in C. elegans slowed elongation speeds, reduced circular RNA formation and extended lifespan and healthspan. This represents a similar phenomenon to hyperfunction of developmental/growth-related pathways in ageing at the level of transcriptional apparatus. Thus, it is interesting to consider the possible communication between hyperfunctional growth pathways such as mTOR and transcriptional/elongation apparatus. Specifically, is one driving the other, or are both responses a consequence of transcriptional drift/selection shadowing that occurs after sexual maturation? In the former, transcriptional drift could underlie increases in RNA polymerase II speeds that lead to greater translational flux and mTOR activity. Given that no mechanisms appear to have evolved to regulate either of these processes, the system would likely remain in a state of biosynthesis, where positive feedback between growth pathways and transcriptional machinery [42] could be driving the ageing-associated increase in both processes collectively. Alternatively, continuous increases in both are separate events, and result from AP-induced hyperfunction that compound together to affect ageing and healthspan (see Figure 1). Additionally, alterations in RNA/mRNA processing genes could be an underlying cause of aberrant transcriptional regulation with ageing. Ham and colleagues recently showed that age-associated increased usage of distal (vs proximal) alternative 3’ (A3) splice sites lead to an increase in non-coding RNA (ncRNA) formation that accumulate with ageing [43]. This increase in unprocessed RNA’s containing both exons and introns (i.e., mis-spliced transcripts) supports the notion that normal RNA processing is lost with ageing and contributes to significant increases in mis-spliced genes and RNA isoform accumulation. Although a crucial finding, proximate causes that lead to these impairments in RNA processing are currently poorly understood. Increased RNA polymerase II speeds are known to impair splicing events and increase intron-retaining transcripts [41], however the proximate cause underpinning RNA polymerase dysregulation is not known.
Progressive protein biosynthesis is a feature of the C. elegans proteome
Whilst numerous data exists exploring genomic remodeling with ageing in C. elegans, only a handful of papers have explored changes in the C. elegans ageing proteome. With significant improvements in mass-spectrometry resolution and fractionation techniques for reduced sample complexity and improved protein coverage, much has been learnt from the C. elegans ageing proteome. Additionally, targeted downstream interrogation of identified proteins using fluorescent reporter strains is beginning to couple global molecular changes to physiological function in vivo. First works from Hartl’s group profiled >5,000 proteins across days 1, 6, 12, 17 and 22 post-adulthood using the stable-isotope labelling with amino acids in cell culture (SILAC) approach, showing one-third of the worm proteome alters at least 2-fold with ageing, where 50% of these changes are accounted for by proteins increasing in abundancy progressively to day 22 [51]. Proteasomal proteins encompass a majority of those increasing with ageing and, interestingly, small heat-shock proteins were seen to increase up to 90-fold with age. This stark accumulation of protein degradative systems reflects a compensatory response for an imbalance of protein accumulation and effective clearance. Moreover, the authors compared the subset of proteins increasing in abundance between days 6 and 22 with transcript levels in previously published dicer mutants, defective in miRNA biosynthesis [52]. Of these, up to 40% of these accumulating proteins showed transcriptional upregulation in dicer mutants, suggesting that loss of miRNA derepression of transcriptional/translational processes contributes to ageing-induced protein accumulation. Moreover, whilst others have suggested mRNA levels are weakly correlated with protein abundances [53,54,55] this highlights the complex coupling of (post)transcriptional and proteomic dysregulation in driving aberrant protein accumulation in ageing. Subsequently, Kenyon’s group performed deep analysis of the ageing proteome using SILAC covering >9,300 proteins across days 1, 5 and 10 post-adulthood [17]. Of these, only 627 significantly changed in abundance across the three time points, with proteins linearly increasing with ageing enriched for terms such as nucleosome assembly, cellular macromolecular assembly tissue morphogenesis and response to stress. Conversely, protein groups declining in abundance with ageing were enriched for fatty acid metabolic processes, organic acid biosynthetic processes and oxidation reduction. These data began to highlight distinct biological differences in deteriorating vs accumulating pathways in C. elegans, with a loss in metabolic processes and continual accumulation in growth related processes.
Functional data went on to show that life-long inhibition of mRNA translation (thus, protein biosynthesis) via RNAi gene knock-down of ifg-1 (eIF4G homologue) and rsks-1 (ribosomal S6K, homologue of mammalian p70S6K) in wild-type C. elegans extended lifespan [56]. Additionally, the authors show that let-363 (mTOR) RNAi significantly extends lifespan, and combined let-363 (mTOR) and ifg-1 RNAi provided further lifespan extending effects, suggesting the additive effects of mTOR inhibition act through distinctive mechanisms to just translation-inhibition alone. Others have used pharmacological approaches to inhibit protein biosynthesis in worms throughout the life-course. Specifically, cycloheximide (used to block translation-elongation processes) exposure starting from the first day of adulthood in C. elegans significantly extended lifespan across all concentrations used (0.1, 1 and 10 µM) [57]. Using β-galactosidase staining as a readout of senescence, cycloheximide also reduced the intensity of senescence-associated β-galactosidase fluorescence in worms under protein synthesis-inhibiting conditions. In support, the authors also showed that mild protein synthesis restriction in normal and tumor-derived human cells reduced senescent features such as cell swelling and replicative senescence [57]. However, the authors did not assess reproductive rates in cycloheximide exposed worms, where the aforementioned study performing ifg-1, rsks-1 and let-363 RNAi (see [56]) showed that translation-inhibiting gene silencing significantly reduced reproductive rates in these animals, limiting the implications of these findings in the context of healthy ageing.
Attenuation of protein accumulation, transcriptional drift and elongation speeds from life-long mutations in the Insulin signalling pathway
In attempts to provide a comparative proteomic model for ‘healthy’ ageing, researchers have analysed proteomic changes in long-lived (daf-2/IGF-1 receptor mutants) and short-lived (daf-16/FOXO and hif-1/hypoxia-inducible factor) mutants. Crucially, the age-related increase in protein abundance seen in wild-types was significantly decreased in daf-2 mutants, and further elevated in short-lived daf-16 mutants, highlighting the functional benefit of lowering protein abundance with ageing [51]. Others have previously shown that translational flux and mRNA levels are lower in daf-2 mutants compared to wild-type, controlled by daf-16 expression [58]. Together, one would expect lower protein aggregation in these long-lived mutants, however, daf-2 accumulate more insoluble proteins compared to wild-type, particularly enriched for small heat-shock species and proteasomal complexes [51]. Interestingly, aggregate-forming proteins were less hydrophobic, more charged and displayed higher disorder compared to wild-type aggregating proteins, suggesting that the accumulation of potentially toxic protein species could augment global proteostasis, demonstrated by daf-2’s improved ability to maintain the expression of a metastable FlucDM-GFP protein. Importantly, decreasing protein abundancy is suggested to aid in the solubility of proteins, where excessive protein formation can drive insoluble aggregate formation by overcrowding [59], highlighting the importance of late-life repression of protein level increase with ageing (see Figure 2).
More recently, Braeckman’s group studied protein turnover rates and protein half-lives in control vs daf-2 mutants, with the assumption that protein accumulation and subsequent their ‘dwell’ time are likely detrimental to protein homeostasis. Interestingly, 54% of analysed peptides showed slower turnover rates in daf-2 mutants (particularly in translation-related machinery), with a near doubling of average protein half-life time from 103 to 173-hours in daf-2 vs controls [60]. Given that age-related increases in protein abundance are lower in daf-2 mutants, increased protein dwell time represents an interesting paradox in the mechanisms underlying lifespan/healthspan improvements in daf-2. However, gene ontology enrichment showed those with reduced turnover and increased half-lives were enriched for mitochondrial, stress-response and cytoskeletal/muscle-related proteins. Protein translation is one of the cells most expensive energetic tasks [61], thus, reductions in translation and protein accumulation together with suppressed turnover rates of cytoprotective proteins represents a potential energy-saving phenomenon whilst still permitting the presence of protective species within cellular environments [60].
Interestingly, daf-2 mutants display attenuation of both drift and RNA processing dysfunction with ageing, prominent features in wild-type ageing (see above). RNAi (starting throughout and beyond development) against daf-2 dramatically suppressed transcriptome-wide drift, where daf-2 day 6 adults showed profiles similar to those of day 1 adults in wild-type [11]. When combined daf-2 and daf-16 RNAi was tested, this attenuation of transcriptional drift was abolished and in fact greater than in wild-type animals [11]. These data implicate the FOXO transcription-factor as a crucial regulator of the ageing transcriptome, in line with others who show that daf-16 expression acts to stabilize transcriptomic environments [62]. Additionally, daf-2 mutants show lower usage of distal A3 splice-sites, decreased A3-induced isoform accumulation with ageing and attenuation of RNA processing-related gene expression [43], suggesting that the slower rates of transcriptional flux and reduced age-related gene co-expression drift can improve the function of RNA surveillance mechanisms and limit aberrant protein formation. These data, combined with markedly reduced translation rates and total mRNA levels compared to wild-type [63], suggest the late-life expression of growth pathways such as daf-2/IGF-1 contribute to ageing, and limiting its expression positively effects lifespan/healthspan via an attenuation of age-related hyperfunction across multiple systems (see Figure 2).
Lifespan-extending mutants for healthspan research – a complex story
Gene inhibition experiments mentioned to this point (daf-2, ifg-1, rsks-1, let-363 etc.) were performed with either life-long RNAi inhibition (i.e., starting during and beyond developmental periods) or substitution (e.g. loss-of-function) mutants. Claims of ageing attenuation from a life-long loss of pathways that are a prerequisite to healthy development in higher mammals have minimal translational impact. It is well known that animals under translation-inhibiting conditions show reduced fecundity and slower developmental timings, where observed lifespan extension might have resulted from AP-induced (i.e., soma vs reproductive investment) effects. Genetic alterations that induce impairments in reproductive fitness in early life, but lifespan extension in later life are, in affect, fitness preventing and would not be selected for in the wild. Therefore, whilst studies of this nature provide valuable insight into the functional effects of modulating organismal growth, the results are confounded by AP effects and are limited in their physiological relevancy for higher mammals.
The consensus that these mutants also display extensions in healthspan in accordance with their longer life has been subject to much scrutiny over the years, with adequate reports of a dissociation between the two and longer periods of frailty in these mutants [64,65]. However, others have shown that daf-2 mutants exhibit elevated expression levels of an odorant receptor, promoting these mutants to favor food over exploration that is common in wild-types, masking the true movement effect of these animals [66]. When daf-2 mutants are assayed in the absence of food, life-course movement rates do in fact scale with longer lifespans. Nevertheless, this feature appears exclusive to daf-2 mutants, where many other lifespan extending mutants display significant fitness costs when accounted for by lifespan extension and early life fitness impairments [23]. Also, daf-2 inhibition-mediated healthspan extension under permitting experimental conditions does not circumvent the early fitness defects in these mutants such as reduced fecundity, egg retention, growth defects and germline shrinkage [67]. In fact, these very effects could (partly) underpin the observed lifespan extension in these animals (and others such as clk-1 [68]), in line with the disposable soma and AP theories of ageing (see Figure 3). Such mechanisms are largely irrelevant for physiologically meaningful, and implementable regimes to combat human ageing: i.e., interventions that induce lifespan/healthspan benefits later in human life, at the cost of reduced fitness/reproductive capacity in early life, are evolutionarily unsustainable approaches to combat ageing. Experimental optimisation of gene function would need to be efficacious when implemented after developmental/reproductive periods to hold translational potential, intervening solely outside of selective windows where the dysregulation of growth/synthesis-promoting pathways begin to impair healthspan.
Similarly, earlier mentioned studies (see above) employing cycloheximide for translation-inhibition did so under 5-fluro-2ʹ-deoxyuridine (FUdR) conditions, a chemical used to inhibit the enzyme thymidylate synthetase, interfering with DNA synthesis and blocking reproductive capacity [69]. Thus, whilst cycloheximide-induced translation inhibition might extend lifespan when reproduction is abolished, similar effects might not be seen if these adult worms require the diversion of energetic resources (ultimately under control of protein synthesis) towards reproduction. Similar arguments can be made for the aforementioned findings that ama-1 mutant worms (thus, life-long inhibition of RNA polymerase II-directed transcription) have slower elongation rates that positively affects ageing rates [41]. Again, reproductive capacity was not assessed in these mutants, thus, significantly reduced rates of transcription could be contextually beneficial given that allocation of resources towards significantly costly processes such as reproduction was not a requirement in these animals. If the proximate-ultimate causes of ageing are a result of continued developmental/growth-related pathway activation [5], then ablating the normal function of this system (thus, the expression of genes required during sexual maturation) could have profound effects on the late-life health of these animals that does not reflect physiological ageing attenuation (see Figure 3 and Figure 4). If the roles of AP and hyperfunction in ageing are to be addressed experimentally, it is crucial that conditions permit normal reproductive capacity.
Re-thinking genetic and therapeutic screening in C. elegans for ageing research
In humans, peak physical strength and aerobic capacity typically occurs early in young adult life [70]. Similarly, C. elegans exhibit a period of physiological fitness within the first few days of adult life [71]. Age-related muscle loss, termed sarcopenia, starts to manifest in the fourth decade of life in people, with an estimated 3-8% loss of muscle mass per decade after the age of 30 [72]. Additionally, human aerobic capacity [73] and metabolic processes [74,75] start to deteriorate at similar time frames, contributing to the progressive increase in frailty. Despite this, the majority of C. elegans translational research exploit intervention-onset from the first day of life after hatching, with continuous exposure throughout all developmental stages and adulthood until cessation of life. This represents a puzzling approach for C. elegans ageing research, commencing genetic or pharmacological interventions that intervene with healthy periods of the life-course in attempts to delay ageing. Many essential biochemical processes are established during C. elegans development; developmental starvation imprints adult foraging behavior [76], mitochondrial dynamics can determine adult respiration levels and lifespan [77] and individual variances in levels of ROS through development can predict adult longevity [78]. Thus, developmental genetic/pharmacological onset likely interacts with, and remodels communicative signalling mechanisms of normal (i.e., non-pathogenic) molecular ageing events, confounding the applicability of reports for ageing attenuation. Whilst preventative therapeutic care is an attractive approach to counter ageing, pharmacological approaches to reduce the age-associated increase in molecular hyperfunction (e.g., Metformin, Rapamycin) are likely to be exclusive to older ages in humans. Therefore, pre-clinical research should imitate the timing of these therapeutic windows, and post-reproductive intervention onset (i.e., at time periods of sub-cellular and physiological decline) are likely to help us understand physiologically relevant signalling mechanisms that contribute to ageing hyperfunction.
Such optimisation of late-life gene function was assessed by Kenyon’s group, using adult-only RNAi against daf-2 in wild-type C. elegans showing a dramatic increase in lifespan that bypassed any interference with reproduction [79]. These results, however, were not corroborated with simultaneous measures of healthspan, where it is not inconceivable that these animals spent longer periods in frail conditions [23,64]. Additionally, the effects on lifespan began to narrow when treatments began on day 5 of adulthood, and were no longer visible from day 8 treatments, suggesting a potential limitation of double stranded RNA feeding approaches in aged post-mitotic animals. Further influential work by Makalov’s group explored life extension-induced fitness costs in C. elegans using life-long, adulthood or post-reproductive genetic inhibition (via RNAi) of well-known longevity genes converging nutrient-sensing signalling (age-1/PI3K), mTOR (raga-1/Ras-related GTPase RagA), global protein synthesis (ifg-1/eIF4G), somatic-cell protein synthesis (ife-2/eIF4E) and mitochondrial respiration (nuo-6/ NDUFB4) pathways [80]. Inhibition of age-1 extended life under all RNAi conditions, remarkably, with no fitness costs on reproductive success, egg size or individual fitness. Interestingly, only adulthood and post-reproductive RNAi induced life extension under raga-1 inhibition with no effects on fitness, and ife-2 inhibition extended life across all ages with no fitness costs. Conversely, knock-down of nuo-6 displayed a strong negative correlation between reproduction and lifespan extension across RNAi timings, and ablation of reproduction was seen in life-long and adulthood ifg-1 RNAi despite no changes in lifespan. Interestingly, post-reproductive ifg-1 inhibition showed dramatic lifespan extension beyond periods that could influence early-life fitness, supporting the notion that hyperfunction of growth-pathways/ protein synthesis negatively impacts C. elegans ageing trajectory.
Therapeutically, previous work has shown worms treated with metformin (to induce mTOR inhibition) on the first day of adulthood exhibited lifespan and healthspan extension (Chen et al, 2017), circumventing the developmental arrest previously reported with mTOR inhibition during development [28] (however, again, performed under FUdR-inhibiting conditions). Rapamycin treatment starting from the 600th day of life in male and female mice significantly extended late-life survivability compared to untreated controls [81]. Although healthspan was not assessed by the authors, these lifespan effects were more prominent when treatment began from 600 days (~60y in humans) vs 270 days. Others have shown that delivery of monoclonal antibodies for the IGF-1 receptor in 78-week-old mice was sufficient to extend lifespan and healthspan [82]. More recently, others also showed that transient rapamycin treatments (8mg/kg injection for 3-months) in middle- aged male mice significantly extended lifespan and healthspan [83]. This highlights the contextual importance of mTOR in early life, where late-life inhibition can ameliorate the negative consequences of this pleiotropic system in mammalian ageing progression, also providing experimental support for treatments commencing at periods of physiological deterioration.
More recently, our group compared the effects of mitochondria-targeted hydrogen sulfide treatments in treating C. elegans ageing progression across the life-course [40], with drug exposures starting from the first developmental stage, or days 0, 2 and 4 post-adulthood. Interestingly, developmentally treated animals displayed lifespan and healthspan extension across the life-course, whereas all adult-onset treatments extended healthspan up to the limit of, but did not extend, normal ageing lifespans. Importantly, adult-onset, but not developmental drug treatments significantly restored ageing transcriptomic profiles in late-life via the downregulation of ELT-6 gene/protein expression and upregulation of genes controlled by ELT-3. Additionally, mitochondrial health and muscle-structural integrity were maintained for longer periods in adult vs developmentally treated animals. These greater transcriptomic and sub-cellular improvements of putative ageing-related targets in adult vs developmental treatments raise the question: is lifespan extension a fundamental requirement for interventional success in C. elegans studies? Could healthspan extension up to the limit of existing animal lifespans (i.e., gero-compression) be a more biologically meaningful finding for translational research?
Lifespan extension or gero-compression?
Living longer without simultaneous improvements in physiological health is a major societal burden, increasing time periods spent in frail conditions [84]. Recent mathematical projections suggest that a compression of morbidity, rather than an extension of lifespan, would provide greater socio-economic gain [85]. Despite this, an overwhelming majority of lower organism research focuses on genetic and/or therapeutic strategies to extend animal lifespan, infrequently corroborating these findings with life-course measures of healthspan or offset in senescent phenotypes, with the assumption these readouts are inextricably linked. Importantly, we now know there are distinct mechanisms controlling lifespan vs healthspan in C. elegans [23]. Thus, the absence of lifespan extension in response to genetic/pharmacological intervention in isolation does not necessarily indicate ineffective healthspan effects. The coordinative scaling of healthspan with lifespan extension is a non-trivial requirement to make the claim of ageing attenuation. Consequently, could interventions that extend healthspan up to the limits of natural lifespan be preferential over those that extend both lifespan and healthspan?
For example, if i) the onset of physical deterioration was suppressed until later in life vs normal ageing, albeit with identical lifespan proportions, healthspan is sufficiently extended via a compression of gerospan (Figure 5B). Similarly, if ii) such intervention significantly extended lifespan and the onset of deterioration was further delayed proportionally to this increased lifespan, the time spent in gerospan is further compressed relative to its lifespan (Figure 5C). Crucially, iii) lifespan and healthspan extension where the onset of deterioration does not scale with longer life would reduce time in gerospan compared to normal ageing projections, however, would be less effective compared to both previous examples (Figure 5D). Lastly, iv) were lifespan extension to occur, but with no delayed onset of physiological deterioration, the relative time sent in gerospan is exacerbated beyond normal ageing levels (Figure 5E). Thus, small but meaningful differences in relative lifespan/healthspan ratios induced by genetic or pharmacological interventions are an important, but somewhat overlooked, aspect for translational research.
Recent individual ageing experiments in C. elegans lend support for this concept [86]. Specifically, isogenic populations of C. elegans show uniform levels of physiological fitness in early adulthood, but lifespan and healthspan begin to diverge across individuals of the same age throughout ageing, an event previously believed to be stochastic in nature [16], but might in fact involve genetic [87] and programmatic [88] control. Importantly, some individuals that live longer exhibit lower rates of physiological decline, however, undergo a disproportionately extended period of frailty [86]. In humans, medical advancements have contributed significantly to increased life expectancies, however, improvements in sanitation, vaccinations and nutrition encompassed the vast majority of longer life [1]. Therefore, extending the human lifespan to similar degrees is an unlikely near-term possibility, where it has recently been suggested that humans are approaching their maximal lifespan, and further modulation would require interventions beyond improvements in healthspan [89]. Thus, we argue interventions that can extend healthspan up to species-specific lifespan limits (e.g., a gerospan compression model) could be most favourable translational candidates to explore in higher models.
Protein-degron systems represent a new candidate approach for antagonistic pleiotropy and hyperfunction research
The Auxin-inducible degron (AID) systems was first identified in plants [90], but has been adapted for use in mammalian cell lines and animal models such as C. elegans, Drosophila, zebrafish and mice (reviewed previously [91]). Briefly, proteins are engineered to include small AID peptide tags for recognition by auxin upon its introduction into the system. These proteins are then subject to ubiquitin-induced degradation and removal of the protein from the system, commonly with ~90% efficiency, a stark improvement on the inconsistent knock-down of genes using RNAi approaches [91]. This allows for temporal control of protein levels either within the entire system, or within tissue-specific regions (by expressing the ubiquitin ligase substrate recognition subunit, TIR1, in the tissue of interest [92]) using promoters for tissue-targeted degradation of AID tagged proteins. AID can also be coupled to GFP motifs, and use in C. elegans, for example, allows in vivo conformation of protein degradation using fluorescence microscopy that provides many benefits to ex vivo qt-PCR against RNAi inhibited genes.
Venz and colleagues recently made the first steps to exploring IIS inhibition from post-developmental stages in C. elegans via AID degradation. The authors employed both global and tissue-specific (muscle, neurons and intestine) AID of daf-2 for temporal and spatial reductions in daf-2 protein levels [67]. Interestingly, systemic daf-2 degradation from young-adulthood resulted in a 70-135% lifespan extension, surpassing that of both daf-2(e1368) and daf-2(e1370) mutants, suggesting degradation of wild-type daf-2 protein elicits greater effects than allelic manipulation of daf-2 at a genetic level. Both neuronal and intestinal (but not muscle) AID were sufficient to induce lifespan extension, however to lesser extends than daf-2(e1370). Although not addressed by the authors, this greater magnitude of life extension in daf-2(e1370) could highlight AP-related soma tradeoffs, where this mutant (but not tissue AID’s) has a marked reduction in reproductive investment. A remarkable finding in this work was the extent of lifespan extension when systemic AID of daf-2 was induced from days 10 (48-72% increase) and 12 (49-57% increase), confirming earlier suppositions that diminished life extension from daf-2 RNAi after day 6 merely reflects methodological limitations of double-stranded RNA interference [79]. Most strikingly, AID starting from days 21 and 25 post-adulthood (when up to 75% of the population had died) allowed these animals to live for a further 22 and 36 days, respectively – a period of time that represents a full wild-type life-cycle. A caveat to this finding, however, is the omittance of concordant healthspan measures in these animals, where such a drastic addition of living days in frail conditions would represent an extension of animal gerospan.
Subsequent work employed similar daf-2 AID in C. elegans, recapitulating findings from Venz [67] that neuronal and intestinal, but not muscle AID, was sufficient to extend lifespan [92]. The authors added to this, finding that epidermal and germline depletion was also insufficient to extend life. Crucially, despite their ability to extend life, intestinal and neuronal daf-2 AID did not improve neuromuscular performance in later life, reflecting a (negative) gerospan extension model (see above section and Figure 5E as an example). Conversely, daf-2 depletion in muscle did not affect lifespan, but significantly improved late-life neuromuscular performance, representing compressive changes in gerospan (see Figure 5B as an example). This data highlights the fundamental importance of assessing combined lifespan and healthspan measures for implications of healthy ageing.
The AID system was more recently exploited for both global somatic inhibition of raga-1 and let-363, as well as neuron-specific depletion of both in C. elegans [93]. Severe impairments in early-life fitness are well known for both of these loss-of-function mTOR mutants [28,93]. Similarly, somatic AID of raga-1 from day 1 of adulthood extends lifespan but impairs reproduction and developmental growth, whereas neuronal-only AID extends life and circumvents perturbations in developmental fitness. Surprisingly, day 1-onset somatic degradation of let-363 impairs survival and induces a severe reproductive defect leading to internal hatching, but neuron-only AID significantly extends life and avoids impairments in developmental and reproductive fitness [93]. Unfortunately, neuromuscular health was not assessed in any of these cohorts, making it difficult to accurately decipher true alterations in healthy ageing projections. These data do, however, re-affirm that hyperfunction of growth-related complexes (IIS and mTOR here) negatively affects lifespan (and where data permits, ageing [67]), potentially partly reflected by an increased insoluble protein load as recently shown [94]. Crucially, AID systems represent a robust method for the temporal and spatial optimisation of gene function in vivo, effective in geriatric animals where AP is known to onset and senescent pathology to accumulate from the hyperfunction of wild-type genes.
Next steps: using C. elegans to map the ageing pleiotropome on a systems and functional level
Whilst experimental data for AP and hyperfunction are developing within the field, the identification of AP genes and their late-life effects are largely limited to individual pathway effects [19,20,25,26,40]. Attempts to characterise putative pleiotropic gene candidates on a systems level (i.e., combined transcriptomic and proteomic) would greatly advance our understandings into the proximate-ultimate roles of AP/hyperfunction in ageing. However, genetic and proteomic environments exhibit stark expression-direction changes with ageing across species, and a vast portion of upregulated genetic responses are adaptive vs hyperfunctional in a pathogenic sense. For example, multiple stress-response pathways increase their expression in late-life, and physiological health is exacerbated when these pathways are inhibited. These precise changes make mapping an ageing ‘pleiotropome’ (i.e., global AP/hyperfunctional gene changes) a difficult quest.
C. elegans offers a unique opportunity to manipulate gene function in vivo in order to map genetic alterations to physiological health. The continual development of high-throughput screening platforms (and automated downstream quantitation pipelines) for functional genomic screens allows the controlled study of population ageing rates with minimal laboursome input. With >5,000 differentially expressed gene changes across the worm life-course [11], putative pleiotropic gene targets could be in their thousands, rendering targeted genetic-inhibition screens challenging at best. Thus, incorporation of transcription-factor binding analysis of putative pleiotropic gene clusters would drastically minimise the number of gene targets for functional gene inhibition screens (demonstrated recently [95]) to map potential pleiotropic/hyperfunctional genes to improvements in healthspan. Additionally, the incorporation of novel AID systems, both within the whole animal and tissue specific regions [67], represents a more favourable approach for such genetic screening that can circumvent problems with classical RNAi approaches and allow gene inhibition during geriatric periods of worm ageing. This represents an exciting area for furthering our understanding into causal ageing mechanisms.
Conclusions and future directions
AP and hyperfunction theories of ageing have gained substantial attention in recent years as potential causal players in ageing deterioration. The view that ageing is driven not by passive deterioration of molecular processes, but in fact a hyperfunction of wild-type genes that have not evolved mechanisms of suppression later in life appears seemingly underappreciated within ageing fields. Mechanisms (new genes/alleles) to suppress late-life hyperfunctional pathways that are beneficial to fitness early in life are unlikely to evolve within populations due to impossibility constraints [102], and restricting their pleiotropic effects is inevitably in the hands of targeted therapeutics. As a candidate model for ageing, we provide a focused overview of transcriptomic and proteomic data in ageing C. elegans that experimentally supports AP and hyperfunction mechanisms of ageing at the molecular and functional level. Additionally, we discuss otherwise unmentioned limitations for research of this nature with current experimental approaches in C. elegans, and suggest required modifications needed to improve the translational impact of future work. We discuss how selective processes are concerned merely with the propagation of genes that foster reproductive fitness, and it is these processes in which pleiotropic aberrations are commonly seen later in life. Thus, it is important to note that the use of chemicals which block reproduction (such as FUdR) interfere with the very pathways that might be fundamental in driving natural ageing progression. We stress the importance of maintaining normal reproductive capacity in C. elegans when attempting to uncover pathways that contribute to ageing (particularly, when assessing pleiotropic alterations later in life). Overall, C. elegans offer a unique model to explore generational and temporal aspects of AP and hyperfunction in ageing, with the benefit of high-throughout screening of genetic and pharmacological regimes that could mitigate these late-life effects.
Acknowledgements
L.S and T.E received no external funding for this work. N.J.S acknowledges the support of the Osteopathic Heritage Foundation through funding for the Osteopathic Heritage Foundation Ralph S. Licklider, D.O., Research Endowment in the Heritage College of Osteopathic Medicine.
Contributions
L.S, T.E and N.J.S conceived ideas. L.S wrote the manuscript and curated figures. L.S, T.E and N.J.S reviewed the manuscript.
Competing interests
The authors declare no competing interests.
References
- Garmany, A., S. Yamada, and A. Terzic, Longevity leap: mind the healthspan gap. NPJ Regen Med 2021, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B. , Evolution of ageing. Nature 1977, 270, 301–4. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. , Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.C. , Pleiotropy, Natural-Selection, and the Evolution of Senescence. Evolution 1957, 11, 398–411. [Google Scholar] [CrossRef]
- de Magalhaes, J.P. and G.M. Church, Genomes optimize reproduction: aging as a consequence of the developmental program. Physiology (Bethesda) 2005, 20, 252–9. [Google Scholar] [PubMed]
- Gems, D. , The hyperfunction theory: An emerging paradigm for the biology of aging. Ageing Res Rev 2022, 74, 101557. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. , Why the disposable soma theory cannot explain why women live longer and why we age. Aging-Us 2010, 2, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N. , The Free Radical Theory of Aging Is Dead. Long Live the Damage Theory! Antioxidants & Redox Signaling 2014, 20, 727–731. [Google Scholar]
- Lemaitre, J.F.; et al. A unified framework for evolutionary genetic and physiological theories of aging. PLoS Biol 2024, 22, e3002513. [Google Scholar] [CrossRef] [PubMed]
- Min, B.; et al. Age-associated bimodal transcriptional drift reduces intergenic disparities in transcription. Aging (Albany NY) 2018, 10, 789–807. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; et al. Suppression of transcriptional drift extends C. elegans lifespan by postponing the onset of mortality. Elife 2015, 4, e08833. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gomez, A., J. N. Buxbaum, and M. Petrascheck, The aging transcriptome: read between the lines. Current Opinion in Neurobiology 2020, 63, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. , The hyperfunction theory of aging: three common misconceptions. Oncoscience 2021, 8, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. , Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle 2006, 5, 2087–102. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. , Cell senescence, rapamycin and hyperfunction theory of aging. Cell Cycle 2022, 21, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Herndon, L.A.; et al. Stochastic and genetic factors influence tissue-specific decline in ageing. Nature 2002, 419, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; et al. Deep Proteome Analysis Identifies Age-Related Processes in C. elegans. Cell Syst 2016, 3, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; et al. Ageing induces tissue-specific transcriptomic changes in Caenorhabditis elegans. EMBO J 2022, 41, e109633. [Google Scholar] [CrossRef] [PubMed]
- Budovskaya, Y.V.; et al. An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell 2008, 134, 291–303. [Google Scholar] [CrossRef] [PubMed]
- de la Guardia, Y.; et al. Run-on of germline apoptosis promotes gonad senescence in C. elegans. Oncotarget 2016, 7, 39082–39096. [Google Scholar] [CrossRef] [PubMed]
- Takemon, Y.; et al. Proteomic and transcriptomic profiling reveal different aspects of aging in the kidney. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; et al. A C-Elegans Mutant That Lives Twice as Long as Wild-Type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; et al. Uncoupling lifespan and healthspan in Caenorhabditis elegans longevity mutants. Proc Natl Acad Sci U S A 2015, 112, E277–86. [Google Scholar] [CrossRef] [PubMed]
- Boeck, M.E.; et al. The time-resolved transcriptome of C. elegans. Genome Res 2016, 26, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; et al. A parthenogenetic quasi-program causes teratoma-like tumors during aging in wild-type C. elegans. NPJ Aging Mech Dis 2018, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Ezcurra, M.; et al. C. elegans Eats Its Own Intestine to Make Yolk Leading to Multiple Senescent Pathologies. Curr Biol 2018, 28, 3352. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, Y.G.; et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Molecular and Cellular Biology 2004, 24, 9508–9516. [Google Scholar] [CrossRef] [PubMed]
- Vellai, T.; et al. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; et al. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Frontiers in Oncology 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; et al. TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr Biol 2002, 12, 1448–61. [Google Scholar] [CrossRef]
- Abdellatif, M.; et al. Fine-Tuning Cardiac Insulin-Like Growth Factor 1 Receptor Signaling to Promote Health and Longevity. Circulation 2022, 145, 1853–1866. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; et al. An antagonistic pleiotropic gene regulates the reproduction and longevity tradeoff. Proc Natl Acad Sci U S A 2022, 119, e2120311119. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A. , Aryl hydrocarbon receptor (AhR) reveals evidence of antagonistic pleiotropy in the regulation of the aging process. Cellular and Molecular Life Sciences 2022, 79. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; et al. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. , TOR-driven aging Speeding car without brakes. Cell Cycle 2009, 8, 4055–4059. [Google Scholar] [CrossRef] [PubMed]
- de Magalhaes, J.P. , Ageing as a software design flaw. Genome Biology 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Gems, D. and J.P. de Magalha, The hoverfly and the wasp: A critique of the hallmarks of aging as a paradigm. Ageing Research Reviews 2021, 70. [Google Scholar]
- Chen, D.; et al. Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging Cell 2007, 6, 525–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Q.; et al. Large-scale across species transcriptomic analysis identifies genetic selection signatures associated with longevity in mammals. Embo Journal 2023, 42. [Google Scholar] [CrossRef] [PubMed]
- Vintila, A.R.; et al. Mitochondrial sulfide promotes life span and health span through distinct mechanisms in developing versus adult treated Caenorhabditis elegans. Proc Natl Acad Sci U S A 2023, 120, e2216141120. [Google Scholar] [CrossRef] [PubMed]
- Debes, C.; et al. Ageing-associated changes in transcriptional elongation influence longevity. Nature 2023, 616, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.M.; et al. The Translational Regulation in mTOR Pathway. Biomolecules 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.; et al. Systematic transcriptome analysis associated with physiological and chronological aging in Caenorhabditis elegans. Genome Res 2022, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, J.; et al. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; et al. Age-associated changes in expression of small, noncoding RNAs, including microRNAs, in C. elegans. RNA 2011, 17, 1804–20. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.Y.; et al. An mTORC1-Mdm2-Drosha axis for miRNA biogenesis in response to glucose- and amino acid-deprivation. Molecular Cancer Research 2016, 14. [Google Scholar] [CrossRef]
- Johnson, S.C.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–8. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A. and S.L. Schreiber, Direct control of mitochondrial function by mTOR. Proceedings of the National Academy of Sciences of the United States of America 2009, 106, 22229–22232. [Google Scholar] [CrossRef] [PubMed]
- Migliavacca, E.; et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat Commun 2019, 10, 5808. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, S.; et al. Reducing the metabolic burden of rRNA synthesis promotes healthy longevity in Caenorhabditis elegans. Nat Commun 2024, 15, 1702. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.M.; et al. Widespread Proteome Remodeling and Aggregation in Aging. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Welker, N.C., J. W. Habig, and B.L. Bass, Genes misregulated in C. elegans deficient in Dicer, RDE-4, or RDE-1 are enriched for innate immunity genes. RNA 2007, 13, 1090–102. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; et al. Correlation between protein and mRNA abundance in yeast. Molecular and Cellular Biology 1999, 19, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Washburn, M.P.; et al. Protein pathway and complex clustering of correlated mRNA and protein expression analyses in. Proceedings of the National Academy of Sciences of the United States of America 2003, 100, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Ideker, T.; et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 2001, 292, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.Z.; et al. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell 2007, 6, 111–9. [Google Scholar] [CrossRef] [PubMed]
- Takauji, Y.; et al. Restriction of protein synthesis abolishes senescence features at cellular and organismal levels. Sci Rep 2016, 6, 18722. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, G.; et al. Increased Protein Stability and Decreased Protein Turnover in the Caenorhabditis elegans Ins/IGF-1 daf-2 Mutant. J Gerontol A Biol Sci Med Sci 2016, 71, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. and A.P. Minton, Protein aggregation in crowded environments. Biological Chemistry 2006, 387, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Dhondt, I.; et al. FOXO/DAF-16 Activation Slows Down Turnover of the Majority of Proteins in C. elegans. Cell Rep 2016, 16, 3028–3040. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F. and M.D. Brand, A Hierarchy of Atp-Consuming Processes in Mammalian-Cells. Biochemical Journal 1995, 312, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Li, S.T.; et al. DAF-16 stabilizes the aging transcriptome and is activated in mid-aged Caenorhabditis elegans to cope with internal stress. Aging Cell 2019, 18, e12896. [Google Scholar] [CrossRef] [PubMed]
- Stout, G.J.; et al. Insulin/IGF-1-mediated longevity is marked by reduced protein metabolism. Mol Syst Biol 2013, 9, 679. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; et al. A demographic analysis of the fitness cost of extended longevity in. Journals of Gerontology Series a-Biological Sciences and Medical Sciences 2007, 62, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Austad, S.N. and J.M. Hoffman, Is antagonistic pleiotropy ubiquitous in aging biology? Evolution Medicine and Public Health 2018, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Hahm, J.H.; et al. C. elegans maximum velocity correlates with healthspan and is maintained in worms with an insulin receptor mutation. Nat Commun 2015, 6, 8919. [Google Scholar] [CrossRef] [PubMed]
- Venz, R.; et al. End-of-life targeted degradation of DAF-2 insulin/IGF-1 receptor promotes longevity free from growth-related pathologies. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; et al. A demographic analysis of the fitness cost of extended longevity in Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci 2007, 62, 126–35. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.H.; et al. Synchronous growth and aging of Caenorhabditis elegans in the presence of fluorodeoxyuridine. J Gerontol 1979, 34, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Lexell, J., C. C. Taylor, and M. Sjostrom, What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J Neurol Sci 1988, 84, 275–94. [Google Scholar] [CrossRef] [PubMed]
- Hosono, R.; et al. Age-Dependent Changes in Mobility and Separation of the Nematode Caenorhabditis-Elegans. Experimental Gerontology 1980, 15, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.K.; et al. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front Physiol 2012, 3, 260. [Google Scholar] [CrossRef] [PubMed]
- Fleg, J.L.; et al. Accelerated longitudinal decline of aerobic capacity in healthy older adults. Circulation 2005, 112, 674–82. [Google Scholar] [CrossRef] [PubMed]
- Shur, N.F.; et al. Age-related changes in muscle architecture and metabolism in humans: The likely contribution of physical inactivity to age-related functional decline. Ageing Res Rev 2021, 68, 101344. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.; et al. Weight stability masks sarcopenia in elderly men and women. Am J Physiol Endocrinol Metab 2000, 279, E366–75. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; et al. Environmental Programming of Adult Foraging Behavior in C. elegans. Curr Biol 2019, 29, 2867–2879. [Google Scholar] [CrossRef] [PubMed]
- Dillin, A.; et al. Rates of behavior and aging specified by mitochondrial function during development. Science 2002, 298, 2398–401. [Google Scholar] [CrossRef]
- Bazopoulou, D.; et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 2019, 576, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Dillin, A., D. K. Crawford, and C. Kenyon, Timing requirements for insulin/IGF-1 signaling in. Science 2002, 298, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.I.; et al. Cost-free lifespan extension via optimization of gene expression in adulthood aligns with the developmental theory of ageing. Proc Biol Sci 2021, 288, 20201728. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–U108. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; et al. Late-life targeting of the IGF-1 receptor improves healthspan and lifespan in female mice. Nat Commun 2018, 9, 2394. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R., S. C. Ding, and J. Wang, Digital health for aging populations. Nature Medicine 2023, 29, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.J., M. Ellison, and D.A. Sinclair, The economic value of targeting aging. Nat Aging 2021, 1, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.B.; et al. Extended Twilight among Isogenic C. elegans Causes a Disproportionate Scaling between Lifespan and Health. Cell Syst 2016, 3, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.Y.; et al. Single-worm quantitative proteomics reveals aging heterogeneity in isogenic Caenorhabditis elegans. Aging Cell 2023, e14055. [Google Scholar] [CrossRef] [PubMed]
- Rando, T.A. and T. Wyss-Coray, Asynchronous, contagious and digital aging. Nat Aging 2021, 1, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Dong, X., B. Milholland, and J. Vijg, Evidence for a limit to human lifespan. Nature 2016, 538, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; et al. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 2009, 6, 917–22. [Google Scholar] [CrossRef] [PubMed]
- Phanindhar, K. and R.K. Mishra, Auxin-inducible degron system: an efficient protein degradation tool to study protein function. Biotechniques 2023, 74, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.; et al. DAF-2/insulin IGF-1 receptor regulates motility during aging by integrating opposite signaling from muscle and neuronal tissues. Aging Cell 2022, 21, e13660. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; et al. Neuronal mTORC1 inhibition promotes longevity without suppressing anabolic growth and reproduction in C. elegans. PLoS Genet 2023, 19, e1010938. [Google Scholar] [CrossRef] [PubMed]
- Yee, Z.; et al. Inhibition of mTOR decreases insoluble proteins burden by reducing translation in C. elegans. Biogerontology 2021, 22, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.E.; et al. Individual cell types in C. elegans age differently and activate distinct cell-protective responses. Cell Rep 2023, 42, 112902. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.R.; et al. Association between muscular strength and mortality in men: prospective cohort study. BMJ 2008, 337, a439. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, J.M.; et al. Mammalian target of rapamycin complex 1 activation is required for the stimulation of human skeletal muscle protein synthesis by essential amino acids. J Nutr 2011, 141, 856–62. [Google Scholar] [CrossRef] [PubMed]
- Terzis, G.; et al. Resistance exercise-induced increase in muscle mass correlates with p70S6 kinase phosphorylation in human subjects. Eur J Appl Physiol 2008, 102, 145–52. [Google Scholar] [CrossRef] [PubMed]
- Phillips, B.E.; et al. Molecular networks of human muscle adaptation to exercise and age. PLoS Genet 2013, 9, e1003389. [Google Scholar] [CrossRef]
- Plaza-Diaz, J.; et al. Impact of Physical Activity and Exercise on the Epigenome in Skeletal Muscle and Effects on Systemic Metabolism. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- AgeUK. Later life in the United Kingdom. 2019; Available from: https://www.ageuk.org.uk/globalassets/age-uk/documents/reports-and-publications/later_life_uk_factsheet.pdf.
- Kern, D.G.a.C. Biological Constraint as a Cause of Aging. PrePrints 2022. [Google Scholar]
Figure 1.
Potential interactions between transcriptional apparatus and growth-related pathways that drive ageing progression. Ageing is associated with a continuous increase in both transcriptional machinery and the activation of developmental/growth-related pathways. The interaction between these two processes has not been explored however, where we present two potential hypothesis for these ageing events. (1) Transcriptional drift allows for the hyperactivation of RNA polymerase II and elongation speeds. In turn, mTOR senses the accumulation of mRNA transcripts as a state of anabolism and synthesis, maintaining/increasing its expression in accordance. This could establish a feedback loop between the two processes, where both systems signal states of growth and synthesis that feed into each other. (2) The hyperactivation of these systems are separate events that result from pleiotropic events. Specifically, genetic drift allows the dysregulation of transcriptional apparatus that causes increased elongation speeds and higher rates of mis-splicing and aberrant mRNA biosynthesis. Separately (but simultaneously), developmental/growth-related pathways continue their expression and activity beyond reproductive periods due to selective shadowing. In this view, the effects of transcriptional dysregulation and molecular hyperfunction compound (separately) together to drive ageing. This figure was created with BioRender.
Figure 1.
Potential interactions between transcriptional apparatus and growth-related pathways that drive ageing progression. Ageing is associated with a continuous increase in both transcriptional machinery and the activation of developmental/growth-related pathways. The interaction between these two processes has not been explored however, where we present two potential hypothesis for these ageing events. (1) Transcriptional drift allows for the hyperactivation of RNA polymerase II and elongation speeds. In turn, mTOR senses the accumulation of mRNA transcripts as a state of anabolism and synthesis, maintaining/increasing its expression in accordance. This could establish a feedback loop between the two processes, where both systems signal states of growth and synthesis that feed into each other. (2) The hyperactivation of these systems are separate events that result from pleiotropic events. Specifically, genetic drift allows the dysregulation of transcriptional apparatus that causes increased elongation speeds and higher rates of mis-splicing and aberrant mRNA biosynthesis. Separately (but simultaneously), developmental/growth-related pathways continue their expression and activity beyond reproductive periods due to selective shadowing. In this view, the effects of transcriptional dysregulation and molecular hyperfunction compound (separately) together to drive ageing. This figure was created with BioRender.
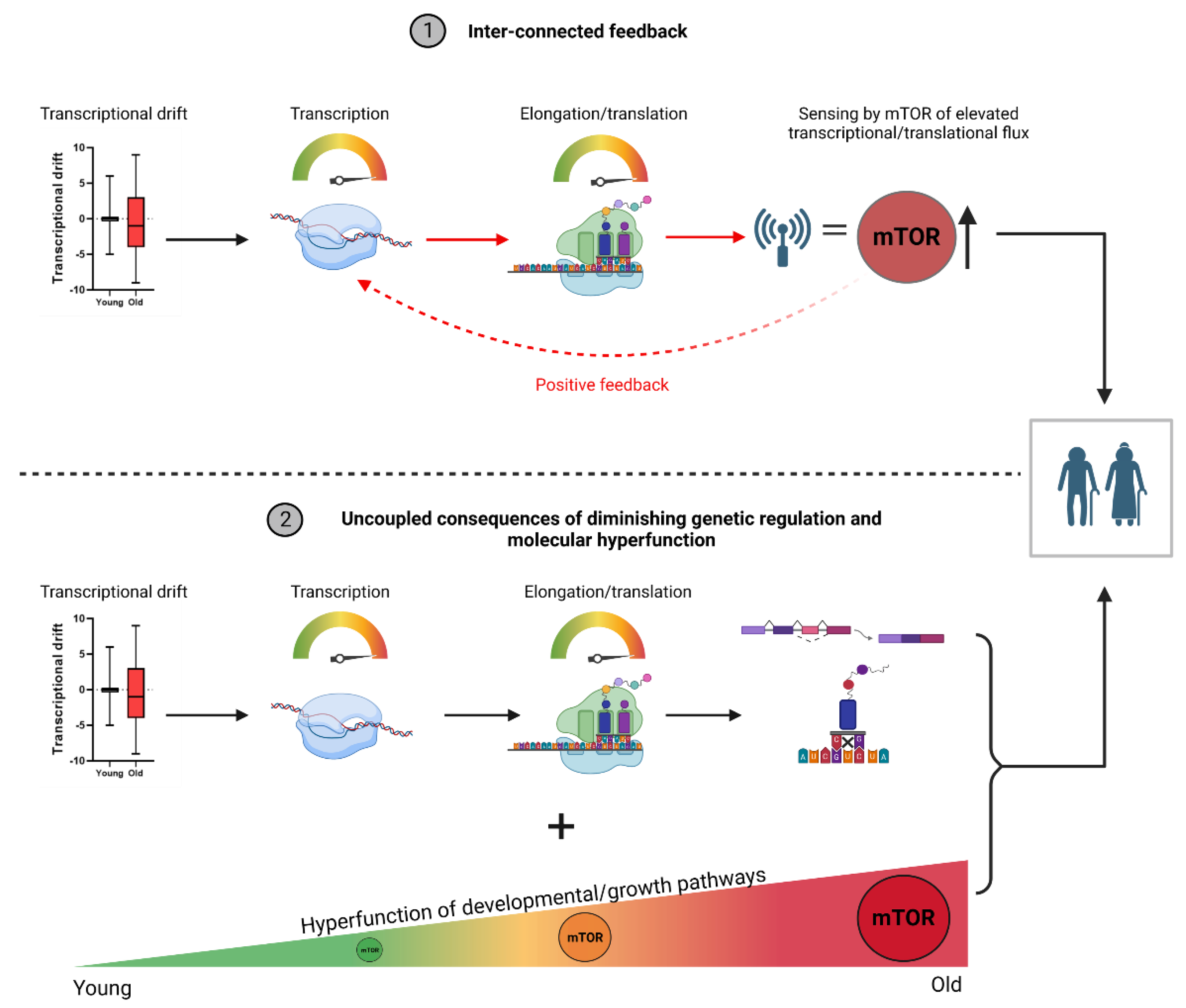
Figure 2.
Aberrant transcriptional changes in C. elegans ageing compromise mRNA stoichiometry and translational fidelity, events that are reversed in healthspan-extending ageing models. Ageing induces a loss of transcriptional regulation that impairs mRNA stoichiometry between functionally related genes and leads to opposing ‘drift’ like effects from youthful profiles. The loss of genetic regulation associates with linear increases in elongation speeds, subsequently reducing the time for accurate splice-site recognition and formation of mis-spliced sequences. Moreover, the ageing-induced loss of miRNA-mediated derepression of (maladaptive) mRNA species play roles in excess formation of mis-translated protein molecules, and align with putative protein targets that accumulate in late life. Subsequently, removal of these aberrant protein molecules is impaired by age-related declines in both the expression and functional capacity of proteasomal subunits. Aggregate-forming protein molecules remain within cells and contribute to molecular overcrowding. This accumulation of soluble and insoluble protein molecules reflects both pleiotropic molecular hyperfunction and a systems decline in removal pathways, respectively, simultaneously culminating in cellular environments that hinder homeostatic processes. Contextually, increased protein turnover would seem a likely protective countermeasure for such accumulation, but healthspan extending conditions show reduced turnover rates and increased half-lives of cytoprotective peptides within cells. Thus, maintenance of transcriptional/translational stoichiometry and reduced costs of required protein expression (via longer dwell times of cytoprotective peptides) highlights the importance of late-life reductions in anabolic pathways for slowing ageing deterioration.
Figure 2.
Aberrant transcriptional changes in C. elegans ageing compromise mRNA stoichiometry and translational fidelity, events that are reversed in healthspan-extending ageing models. Ageing induces a loss of transcriptional regulation that impairs mRNA stoichiometry between functionally related genes and leads to opposing ‘drift’ like effects from youthful profiles. The loss of genetic regulation associates with linear increases in elongation speeds, subsequently reducing the time for accurate splice-site recognition and formation of mis-spliced sequences. Moreover, the ageing-induced loss of miRNA-mediated derepression of (maladaptive) mRNA species play roles in excess formation of mis-translated protein molecules, and align with putative protein targets that accumulate in late life. Subsequently, removal of these aberrant protein molecules is impaired by age-related declines in both the expression and functional capacity of proteasomal subunits. Aggregate-forming protein molecules remain within cells and contribute to molecular overcrowding. This accumulation of soluble and insoluble protein molecules reflects both pleiotropic molecular hyperfunction and a systems decline in removal pathways, respectively, simultaneously culminating in cellular environments that hinder homeostatic processes. Contextually, increased protein turnover would seem a likely protective countermeasure for such accumulation, but healthspan extending conditions show reduced turnover rates and increased half-lives of cytoprotective peptides within cells. Thus, maintenance of transcriptional/translational stoichiometry and reduced costs of required protein expression (via longer dwell times of cytoprotective peptides) highlights the importance of late-life reductions in anabolic pathways for slowing ageing deterioration.
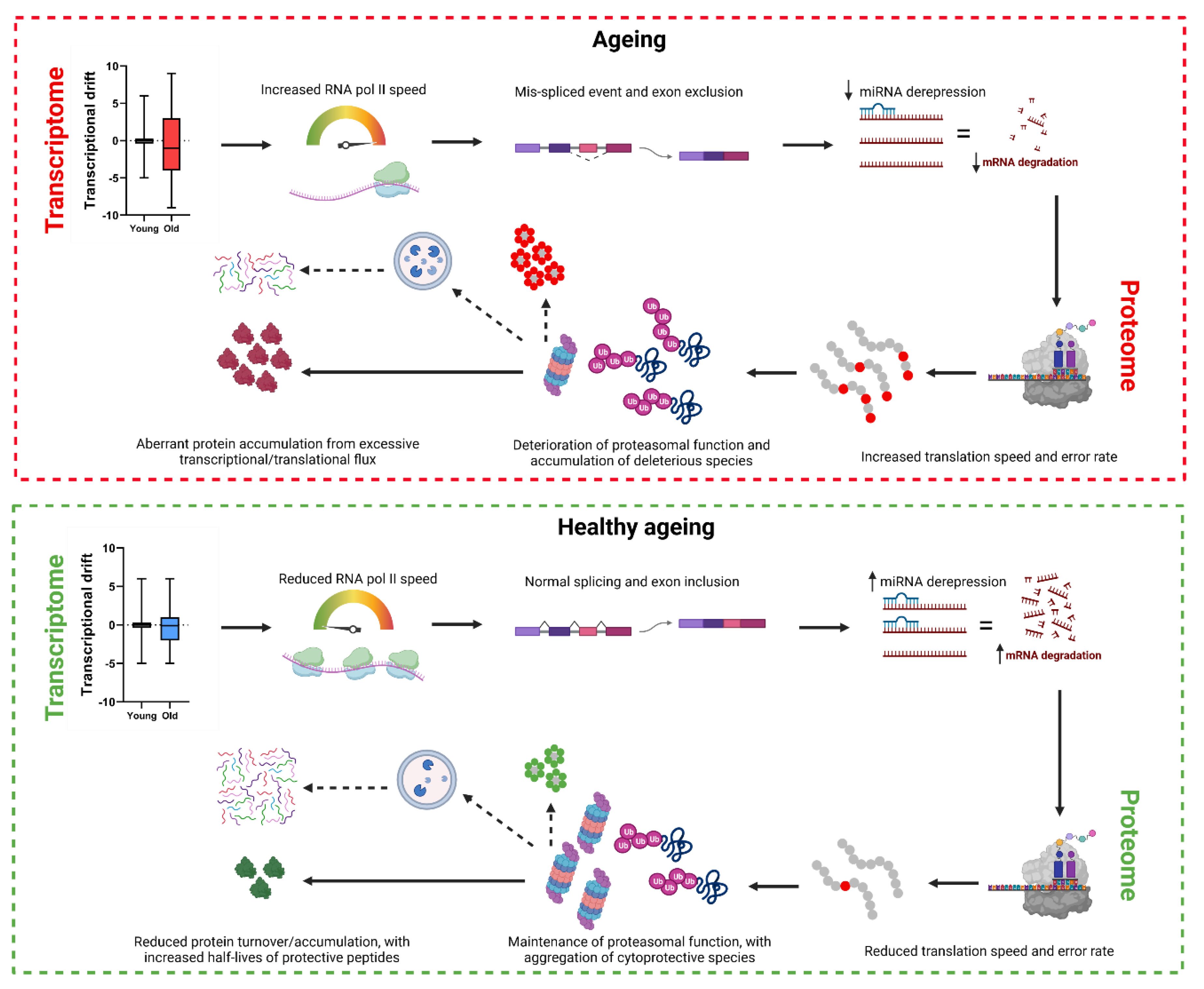
Figure 3.
Healthy ageing regimes should exclusively target aberrant late-life molecular drift/hyperfunction and not impede early life fitness. In humans, healthy reproductive periods are paramount, and (mutant)models that exhibit reduced growth and reproduction that foster longer life likely have minimal translational impact. For example, mutations in daf-2, eat-2 and clk-2 can dramatically extend lifespan, but these mutants exhibit reduced reproductive rates in early adulthood. Whilst resource allocation is still a debated phenomenon (i.e., Disposable Soma theory), reduced investment into reproduction could free up biochemical energy resources for somatic maintenance and regulation of energetically demanding ageing tissue (i.e., muscle, neurons). In addition, these IGF/mTOR ablation models would reduce the negative consequences of age-related hyperfunction of developmental pathways that are causal in ageing progression, also potentially indirectly restoring resources via a reduction in energetically expensive growth pathways. However, extended lifespan/healthspan in response to compromised reproduction is an unuseful approach to think about healthy ageing. In normal ageing, animals exhibit high reproductive rates that increase biphasically over the first few days of adulthood, where the majority of selective pressure is had. Physiological deterioration ensues shortly after, where hyperfunction of growth/developmental pathways that are no longer required continue their expression and cost on resources, ultimately leading to senescence. In these contexts, an ideal ageing intervention would not impair reproductive health, but instead begin its effects from post-reproductive periods. For example, later-life treatments using compounds to reduce the expression of growth pathways (i.e., mTOR, IGF) could stop aberrant hyperfunction, freeing resources for somatic maintenance and limiting the progression of senescence to positively affect animal healthspan.
Figure 3.
Healthy ageing regimes should exclusively target aberrant late-life molecular drift/hyperfunction and not impede early life fitness. In humans, healthy reproductive periods are paramount, and (mutant)models that exhibit reduced growth and reproduction that foster longer life likely have minimal translational impact. For example, mutations in daf-2, eat-2 and clk-2 can dramatically extend lifespan, but these mutants exhibit reduced reproductive rates in early adulthood. Whilst resource allocation is still a debated phenomenon (i.e., Disposable Soma theory), reduced investment into reproduction could free up biochemical energy resources for somatic maintenance and regulation of energetically demanding ageing tissue (i.e., muscle, neurons). In addition, these IGF/mTOR ablation models would reduce the negative consequences of age-related hyperfunction of developmental pathways that are causal in ageing progression, also potentially indirectly restoring resources via a reduction in energetically expensive growth pathways. However, extended lifespan/healthspan in response to compromised reproduction is an unuseful approach to think about healthy ageing. In normal ageing, animals exhibit high reproductive rates that increase biphasically over the first few days of adulthood, where the majority of selective pressure is had. Physiological deterioration ensues shortly after, where hyperfunction of growth/developmental pathways that are no longer required continue their expression and cost on resources, ultimately leading to senescence. In these contexts, an ideal ageing intervention would not impair reproductive health, but instead begin its effects from post-reproductive periods. For example, later-life treatments using compounds to reduce the expression of growth pathways (i.e., mTOR, IGF) could stop aberrant hyperfunction, freeing resources for somatic maintenance and limiting the progression of senescence to positively affect animal healthspan.
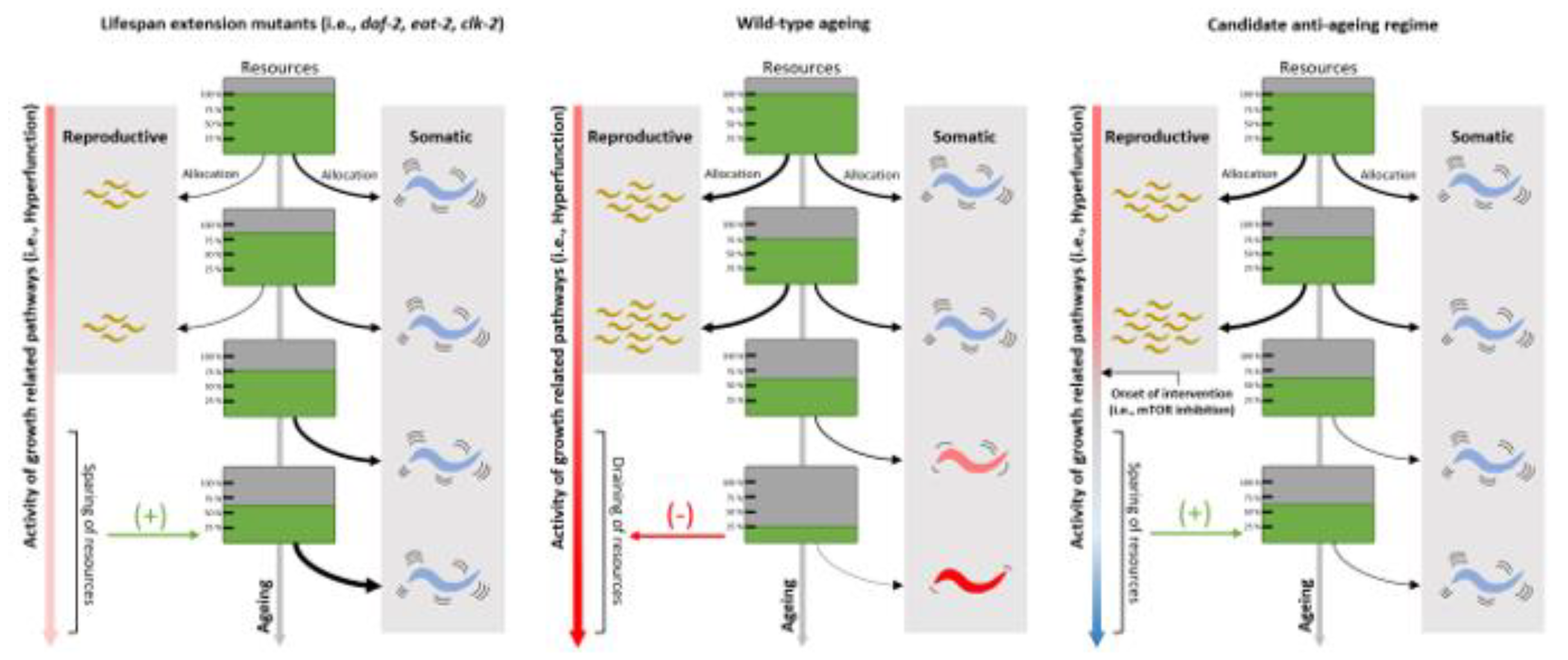
Figure 4.
Reproduction-inhibiting chemicals routinely employed in ageing experiments mask physiological processes that form the basis of late-life hyperfunctional pathways. C. elegans ageing experiments almost exclusively exploit the reproduction inhibitor 5-fluro-2ʹ-deoxyuridine (FUdR) to limit potential progeny contamination within populations. However, if AP and hyperfunction theories are correct, genes required during sexual maturation are bound for late-life overactivation due to selection shadowing, with run-on expression detrimental to organismal health. Thus, experimentally addressing proximate-ultimate ageing effects resulting from genetic hyperfunction must be done under normal physiological conditions for such findings to be translationally relevant.
Figure 4.
Reproduction-inhibiting chemicals routinely employed in ageing experiments mask physiological processes that form the basis of late-life hyperfunctional pathways. C. elegans ageing experiments almost exclusively exploit the reproduction inhibitor 5-fluro-2ʹ-deoxyuridine (FUdR) to limit potential progeny contamination within populations. However, if AP and hyperfunction theories are correct, genes required during sexual maturation are bound for late-life overactivation due to selection shadowing, with run-on expression detrimental to organismal health. Thus, experimentally addressing proximate-ultimate ageing effects resulting from genetic hyperfunction must be done under normal physiological conditions for such findings to be translationally relevant.
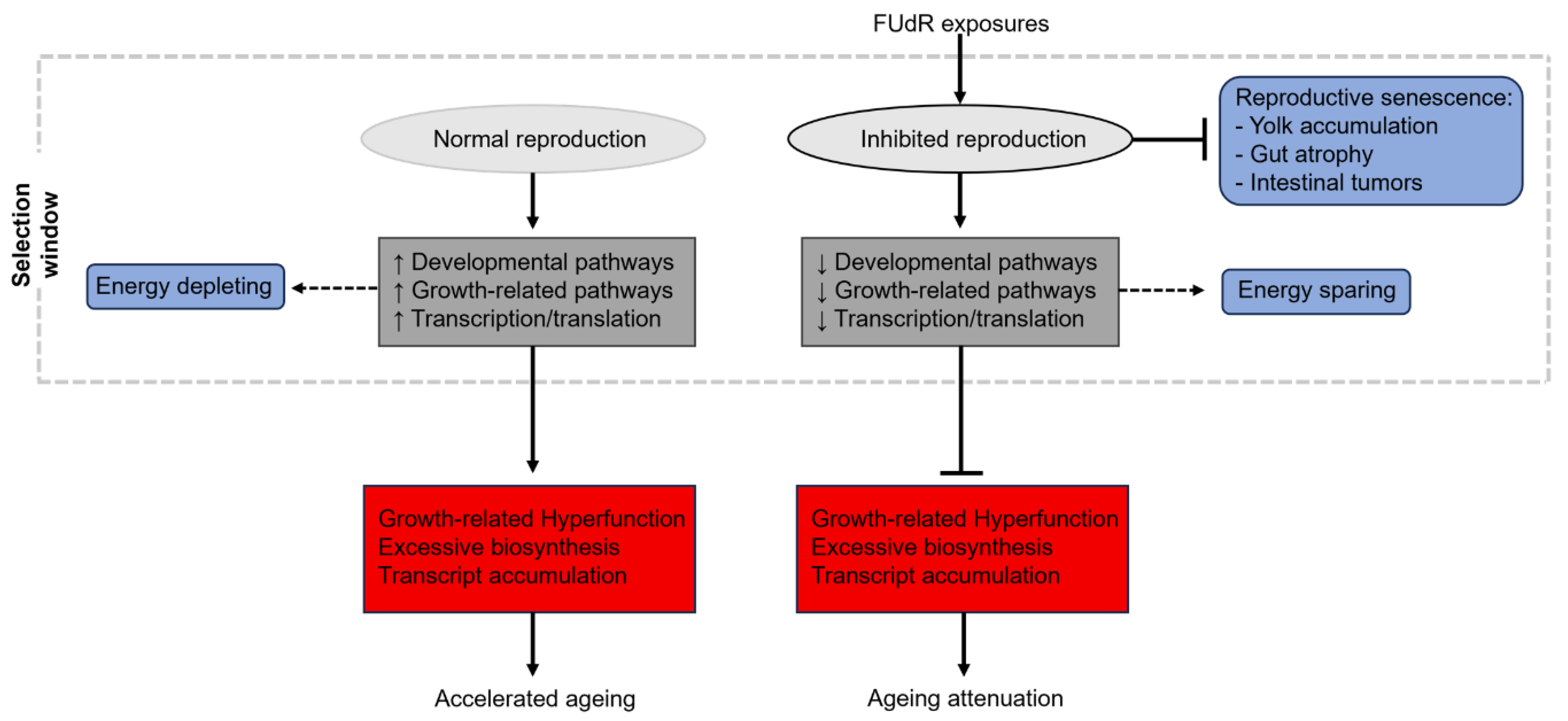
Figure 5.
Modulation of lifespan with varying changes in healthspan can have distinct and meaningful effects on relative time spent in gerospan. A) Normal ageing projections, where the grey line depicts the onset of physiological deterioration. B) Example of an intervention where lifespan remains unaltered, but healthspan improvements delay the onset of deterioration, compressing the relative time spent in gerospan. C) Dual lifespan and healthspan extension, where functional decline is proportionally delayed accordingly with longer life. D) Example of lifespan and healthspan extension where deteriorative onset is not scaled to longer life, thus, the proportion of time in gerospan is higher than situations (B) and (C). E) Interventions where there is no delay in the onset of physical deterioration, coupled with an extended lifespan, increases the period of time spent in gerospan. Bars graphs on the right depict area under the curve summations from the lifespan/healthspan-normalised time spent in gerospan.
Figure 5.
Modulation of lifespan with varying changes in healthspan can have distinct and meaningful effects on relative time spent in gerospan. A) Normal ageing projections, where the grey line depicts the onset of physiological deterioration. B) Example of an intervention where lifespan remains unaltered, but healthspan improvements delay the onset of deterioration, compressing the relative time spent in gerospan. C) Dual lifespan and healthspan extension, where functional decline is proportionally delayed accordingly with longer life. D) Example of lifespan and healthspan extension where deteriorative onset is not scaled to longer life, thus, the proportion of time in gerospan is higher than situations (B) and (C). E) Interventions where there is no delay in the onset of physical deterioration, coupled with an extended lifespan, increases the period of time spent in gerospan. Bars graphs on the right depict area under the curve summations from the lifespan/healthspan-normalised time spent in gerospan.
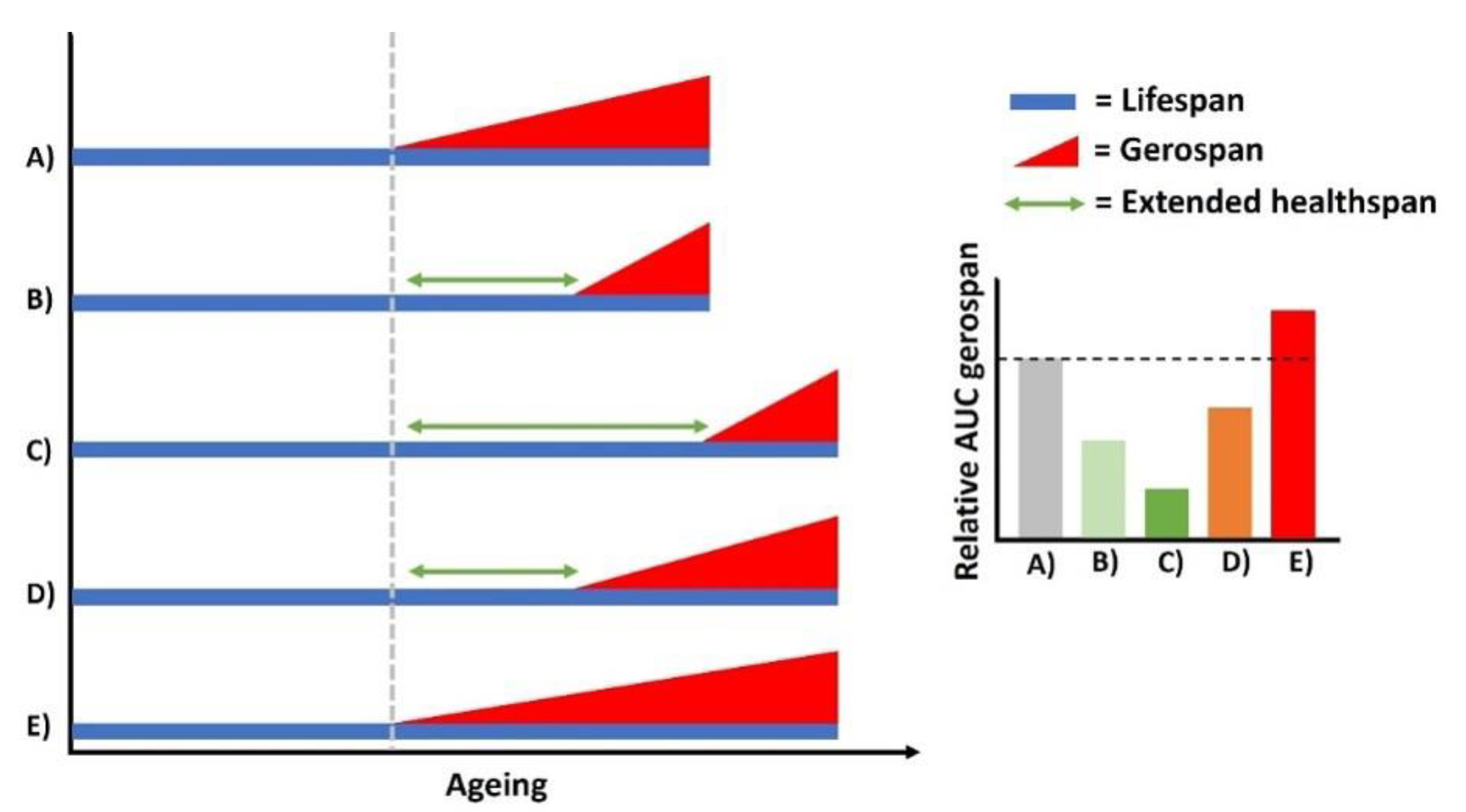
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



