
Submitted:
30 March 2024
Posted:
01 April 2024
You are already at the latest version
Abstract
Pulmonary hypertension (PH) is a progressive and potentially fatal complication of sickle cell disease (SCD) affecting 6-10% of adult SCD patients. Various mechanisms and theories have been evaluated to explain the pathophysiology of the disease. However, questions remain, particularly regarding the clinical heterogeneity of the disease in terms of symptoms, complications, and survival. Beyond the classical mechanisms that have been thoroughly investigated and include hemolysis, nitric oxide availability, endothelial disorders, thrombosis, and left heart failure, attention is focused nowadays on the potential role of the genes involved in such processes. Potential candidate genes are investigated through next-generation sequencing, with the TGF-β pathway being the initial target. This field of research may also provide novel targets for pharmacologic agents in the future as is already the case with idiopathic PH. The collection and processing of data and samples from multiple centers can yield safe results that will allow a better understanding of SCD-related PH as a part of the disease’s clinical spectrum. The following review attempts to capture the most recent findings of studies on gene polymorphisms that have been associated with PH in SCD patients.
Keywords:
Sickle cell disease
; Pulmonary hypertension
; Gene polymorphisms
; Gene sequencing
1. Introduction
Sickle cell disease (SCD) refers to a spectrum of syndromes in which hemoglobin S (HbS) is produced as a result of a point mutation in the beta-globin chain where A is replaced by T at codon 6. The mutation is inherited in a homozygous state or coinherited with a thalassemic mutation at the other beta globin allele leading to reducedorabsent production of normal beta globin or coinherited with another abnormal beta-globin mutation.The most commongenotypes encountered include sickle cell anemia, sickle-beta thalassemia, hemoglobin SC disease, and others.
The point mutation in the beta-globin gene which results in SCD leads to the production of sickle hemoglobin which polymerizes in hypoxic conditions. The clinical manifestations of SCD cover a wide range of clinical entities. Nevertheless, the major features can be categorized on their connection to hemolytic anemia and vaso-occlusion mechanisms. Most common complications can lead to acute and chronic illness that may have a major impact on morbidity and mortality (Figure 1).
Among pulmonary complications of SCD, pulmonary hypertension (PH) is a relatively common and usually severe condition and an independent risk factor for mortality [1,2]. The reported incidence of PH in adult SCD patients ranges between 6% and 11% [3]. The presence of PH is often suspected when exertional dyspnea is observed. Initial evaluation requires Doppler echocardiography, measurement of N-terminal pro-brain natriuretic peptide (NT-proBNP) levels, and a six-minute walk test. Definitive diagnosis is based on right heart catheterization with demonstration of a high resting mean pulmonary arterial pressure (mPAP).Hemolytic anemia is a risk factor for the development of PH. Patients with PH have characteristic hemodynamic features, several comorbidities, and distinct phenotypes.
In an effort to focus on PH pathophysiology and novel findings regarding the potential influence of genetic modifying we searched articles indexed in PubMed using the keywords “Sickle cell disease” and “Pulmonary hypertension” and “Genetic polymorphisms”. Through a review of the articles identified we ended up sampling data for the genetic polymorphisms reported to be associated with pulmonary hypertension features in SCD patients.
2. Results
2.1. Classification of PH
According to the latest updates, patients with PH are potentially classified into one of five groups based on etiology as presented below [4,5]. The first group includes patients with pulmonary arterial hypertension, while the remaining four groups include patients who are considered to have PH. Regarding SCD, some patients with consistent hemodynamic features are classified in group 5, while others demonstrate features of PH related to left-sided heart disease or thromboembolic disease and are placed in groups 2 and 4, respectively [4].
- Group 1: Pulmonary arterial hypertension
- Group 2: PH due to left heart disease
- Group 3: PH due to chronic lung disease and/or hypoxemia
- Group 4: PH due to pulmonary artery obstructions (eg, chronic thromboembolic PH)
- Group 5: PH due to unclear multifactorial mechanisms (e.g., SCD, beta-thalassemia, myeloproliferative disorders, sarcoidosis, and metabolic disorders)
2.2. Clinical Presentation of PH
The diagnosis of PH in SCD patientsmay prove challenging in some cases. Prevalence of PH increases with increasing age and PH is a leading cause of early death as right heart failure may have been already established when patients become symptomatic. Typical symptoms include exertional dyspnea, fatigue, chest pain and signs of right heart failure such as lower extremity edema and palpitations [6]. On the other hand, other common accompanying conditions such as anemia, left ventricular dysfunction and hepatic cirrhosismay be responsible for the symptoms and signs above and further complicate diagnosis.
2.3. Diagnostic Assessment for PH
Diagnostic steps should include factors that may also contribute to PH pathogenesis, including iron overload, chronic liver disease, HIV infection, nocturnal hypoxemia and pulmonary thromboembolism. Initial laboratory studies usually demonstrate an elevated level of NT-proBNP [2]. Plasma NT-proBNP is a peptide released by the myocardium that can be useful in identifying patients with SCD at higher risk of PH, and increased mortality risk [7,8].
Doppler echocardiography is used to screen for PH and right heart failure presence. It is used to estimate pulmonary artery and right ventricular systolic pressures and assesses left and right ventricular size, thickness, and function. Tricuspid regurgitant velocity (TRV) measurement has been proposed as a useful tool to estimate the pulmonary artery systolic pressure (PASP) [9]. Several studies have reported that using a TRV of 2.5m/s as a cutoff point for elevated PASPpractically means that 20–30% of SCD patients are diagnosed with PH and it is suggested that even a mildly elevated TRV is associated with decreased survival [9,10]. However, every component of the echocardiogram should be evaluated by the clinician to estimate PH risk [11].
Asix-minute walking test with oximetry may be used to assess functional capacity and is inversely correlated with PH severity [12]. Measuring pulse oxygen saturation during ambulation on a flat surface and during stair climbing identifies patients who need a more complete evaluation for PH.
The use of pulmonary functional tests should be considered on a case-by-case basis as many SCD patients develop abnormal pulmonary function, with mild restriction patterns and abnormal diffusing capacity in the context of pulmonary fibrosis. A ventilation-perfusion scan is also necessary to exclude chronic pulmonary thromboembolic disease as it is superior to computed tomography pulmonary angiography [13].Lastly, cardiac magnetic resonance imaging is increasingly applied and may relate right and left ventricular cardiac output and function with prognostic features in SCD patients with PH [14,15].
Right heart catheterization remains the gold standard method for definitive diagnosis and hemodynamic aspects of PH [11]. Currently, PH diagnosis is established by the measuring of a resting mPAP>20 mmHg [16]. While 6 to 11 percent of SCD patients have PH based on an mPAP ≥25 mmHg, the hemodynamic features vary across patients with 40 percent of patients demonstrating a predominantly precapillary PH while in the rest,the hemodynamic pattern suggests postcapillary PH [4]. Some patients have mixed hemodynamic features. Mortality is significantly higher in SCD patients with PH defined by right heart catheterization [12].
As in many other cases, the clinician must be aware that each test must be evaluated both separately and in conjunction with the other available tests understanding its capabilities and limitations. For example, in a prospective study where echocardiography and catheterization were performed on SCD adult patients, an elevated TRV was reported in 27 percent, but only 6 percent had PH confirmed by right heart catheterization. When a TRV cutoff point of >2.9 m/sec was applied, the positive predictive value increased to 64 percent but also the false negative rate increased to 42 percent. This false negative rate was improved by using a six-minute walking test and an NT-proBNP level assessment [17].
NT-proBNP can be proposed as a screening tool for PH when Doppler echocardiography is not available as plasma NT-proBNP levels ≥160 pg/mL can detect PH with a sensitivity and specificity of 57 and 91 percent, respectively. Of note, in SCD cases measurements may be confusing in patients with renal insufficiency or left heart failure [6,18].
In an observational study with SCD patients, patients with SCD and PH walked a shorter distance during thesix-minute walking test than SCD patients without PH and distance inversely correlated with the mPAP as measured by right heart catheterization [19]. In another study including children with SCD, during a walking test oxygen saturation declined in 68 percent of children with an elevated TRV compared with 32 percent of those with a normal TRV [20]. However, the walking test should be cautiously used as a screening tool for PH complicating SCD because chronic anemia and other conditions may affect result interpretation.
2.4. Pathophysiology
Various mechanisms have been proposed to explain the occurrence of PH in SCD patients. Apart from documented findings regarding hemolysis, nitric oxide (NO) depletion and cell-free hemoglobin, the research has now extended to the function of the activated endothelium, inflammatory response as well as tothe genetic polymorphisms of genes that are potentially involved in all the above mechanisms [21,22,23,24,25,26]. Traditionally, left heart disease and diastolic dysfunction may play a key role through vascular remodeling [27] (Figure 2).
It is known that hemolysis releases high amounts of cell-free hemoglobin that overcome the binding ability of haptoglobin and hemopexin. This was initially thought to lead to massive depletion of available circulating NO but conflicting reports on the impact of this phenomenon on NO availability exist [28]. Moreover, these effects on endothelial function through increased cell-free Hb may also provoke endothelial and vascular smooth muscle dysfunction [26].
In the case of SCD, NO bioavailability may additionally be affected by a number of other contributing factors. Increased oxidant-related metabolism of NO can reduce the vasodilating capacity of NO. NO acts mainly as a vasodilator and platelet aggregation inhibitor. Alteredredoxmechanisms occur in SCD, as increased levels of oxidative molecules including singlet oxygen, hydroxyl radical, hydrogen peroxide, and superoxide are formed within the red blood cells of the patients. These oxidative molecules can interact with NO to produce nitrate, nitrite, and peroxynitrite, which can be toxic [29].
Increasedplasma levels of arginase, released from red cells, also have been proposed as a potential factor of NO availability regulation. Arginase converts L-arginine to ornithine and therefore limits L-arginine availability, which is an important substrate for NO production [30]. Moreover, in SCD patients, plasma arginase activity has been positively associatedwith secondary PH and a lower ratio of arginine to ornithine has been associated with inflammation markers, increased soluble adhesion molecules, greater severity of PH, and an increased mortality risk [31].
The products of intravascular hemolysis, such as free heme, released upon hemoglobin oxidation and microparticles containing heme species, collectively mentioned as erythrocyte-danger associated molecules (e-DAMPS) are considered to mediate oxidative stress, neutrophil extracellular traps (NETS) formation, inflammasome activation, sterile inflammation and increased endothelial adhesion.
Plasma levels of another molecule, asymmetric dimethylarginine (ADMA), may also contribute independently to endothelial dysfunction. ADMA is a NO synthase inhibitor and in a study with SCD patients, ADMA levels correlated with hemolysis, low oxygen saturation, PH, and early death [32].
Limited NO bioavailability leads to enhanced platelet activation and endothelin-1 release. As a result, this leads to vasculopathy-related conditions that include endothelial dysfunction, vasoconstriction, inflammation, and a hypercoagulable state. These, in turn, may lead to vascular remodeling and increased pulmonary resistance with the imminent occurrence of PH features [33].
Many patients with SCD have functional asplenia or have undergone splenectomy. Splenectomy has been linked with the development of PH, particularly in patients withhemolysis. This can be partially explained by the loss of normal splenic function that also leads to platelet activation, promoting pulmonary vasculature microthrombosis and endothelium dysfunction. Furthermore, after splenectomy, the rate of intravascular hemolysis increases [24].
The role of coagulation mechanisms in the pathogenesis of SCD-related PH becomes prominent. Although it is known that vascular occlusions lead to thrombosis, coagulation may be activated in patients with SCD in the absence of vascular occlusions as suggested by increased tissue factor, increased thrombin generation, and platelet and endothelial activation. Increased circulating monocyte and endothelial cell microparticles along with RBC platelet and neutrophil and platelet are reported. On the other hand, anionic phospholipids, mainly phosphatidylserine, are transported to the surface of sickle RBCs enhancing coagulation activity. Soluble vascular cell adhesion molecule-1, a marker of endothelial dysfunction, is reported to correlate with the severity of hemolysis in patients with SCD-related PH [34].The increased prevalence of thromboembolic complications has led to acknowledging SCD as a prothrombotic, hypercoagulablecondition [35]. In our center (Laikon general hospital of Athens), the incidence of venous thromboembolism in SCD is as high as 23% and its presence was associated with cerebral ischemia. It is of note that D-Dimers are elevated in the vast majority of SCD patients, even in steady state [36].
Obstructive sleep apnea is a frequent complication of SCD in children that is regularly underdiagnosed and has been associated with stroke and PH [37]. Trials in adults include a small number of patients but indicate a high incidence of sleep-disordered breathing [38]. Several mechanisms have been proposed for nocturnal oxygen desaturation in SCD [39].
Despite long research on the above factors’ contribution to the pathophysiology of PH in SCD patients, there are still questions that cannot be fully answered. The exact reason for which some patients exhibit severe hemolytic phenotype, associated with vascular dysfunction while in other patients these phenomena are not so severe cannot be fully explained, nor can patients with a high risk for vascular complications be identified easily. Advances in genome-wide investigation methods and bioinformatics appear promising for identifying gene polymorphisms associated with PH pathogenesis and prognosis.
2.5. Role of Gene Polymorphisms in Pathogenesis and Prognosis of SCD-Related PH
The attempts to identify differentially expressed genes (DEGs)orsingle nuclear polymorphisms (SNPs)associated with specific complications of SCD, including PH have been amplified with the use of next-generation sequencing techniques and genome-wide association studies. The development of PH has not been studied as extensively as other SCD complications with genomic studies, but interesting data are gradually accumulating and indicate that some of the DEGs involved are associated with mechanisms such as extracellular exosomes, platelet degranulation, blood microparticles, immune response, and protein binding. Moreover, there are indications of involvement in crucial biological pathways such as hemopoiesis, cytokine-to-cytokine receptor interaction, TGF-β signaling pathway and extracellular interaction mechanisms.
TGF-β pathway was one of the first fields where interesting results were reported (Ashley-Koch et al., 2008). Analyses demonstrated a potential role of genetic variation in genes related tothe TGF-β pathway in PH risk. Several genes were initially implicated, including ACVRL1, ADRB1, ADCY6, BMP6, BMPR2, CR1,FY, LCAT, LTA4H, SELP SERPINC1, SLC12A6 and TGFBR3. After regression analysis, SNPs in ACVRL1, BMP6, and ADRB1 became significant while TGFBR3nearly demonstrated significance [40]. The associations with these genes were interesting becauseACVRL1 mutations have previously been reported in primary PH cases with hereditary hemorrhagic telangiectasia [41].ACVRL1and BMP6 belong to the TGF-β pathway. AlthoughBMPR2did not remain significant in the final analysis, it has also been elsewhere associated with primary PH in approximately 50% of familial PH cases [42]. In a complementary analysis in the same study, 2 SNPs in the ARG2 gene (rs12587111 and rs1885042) were reported to be nominally associated with PH. ARG2 encodes arginase which is implicated in NO dysfunction in SCD. As reported earlier,higher arginase levels have been reported in SCD patients with PH [31,40].
A different study (Klings et al., 2009) reported that SNPs located in intron 1 of the NEDD4L gene may be associated with elevated serum NT-proBNP levels in SCD patients after genome-wide association studies [43]. This gene encodes a member of the Nedd4 family of HECT domain E3 ubiquitin ligases that target specific proteins for lysosomal degradation and thereby is involved in the regulation of various signaling pathways including TGF-β, autophagy, innate immunity or DNA repair [44]. On this basis, NEDD4L might be implicated directly in SCD-related PH pathogenesis and a target for further prognostic and therapeutic application [43].
Nouraie et al.,(2009), investigated CYBR5 T116S polymorphism and suggested a protective effect against PH. CYBR5 T116S polymorphism was associated with lower TRVs, possibly attributed to lower hemolysis levels in the carriers of this SNP [45].CYBR5 T116S is a frequently identified polymorphism in African children and may be associated with protection from severe malaria-related anemia. This polymorphism may be related to increased cytochrome b5 reductase activity which in turn may explain its protective effect on hemolysis through increased anti-oxidant [46].
Another preliminary study (Desai et al., 2012) investigated patients with an elevated versus normal TRV and catheterization-confirmed PH and reported a significant association with five SNPs of the GALNT13 gene and a single SNP ofthe PRELP gene. Moreover, a trait locus upstream of the adenosine-A2B receptor (ADORA2B) gene was identified [47,48]. The GALNT13 protein is a member of the GalNAcT protein family and is suggested that it can be a potential prognostic factor in lung cancer [49]. The protein encoded by the PRELP gene is a leucine-rich repeat protein present in connective tissue extracellular matrix. The protein functions as a molecule anchoring basement membranes to the underlying connective tissue. This protein has been investigated in the pathogenesis of Hutchinson-Gilford progeria, psoriatic and rheumatoid arthritis, bladder cancer and respiratory tract infections [50]. ADORA2B gene encodes an adenosine receptor that is a member of the G protein-coupled receptor superfamily. ADORA2B-related signaling has been reported to be involved in solid tumor migration mechanisms as well as tumor-derived exosome-induced angiogenesis [51].
MAPK8 encodes a protein member of the mitogen-activated protein (MAP) kinase family. MAP kinases are involved in many crucial cellular processes such as proliferation, differentiation, transcription regulation and development. This kinase is activated by various cell signals, including response to oxidative stress and targets specific transcription factors. The activation of this kinase by tumor necrosis factor alpha (TNF-alpha) is reported to be important for TNF-alpha-induced apoptosis. In a cohort of SCD patients, the A allele of a MAPK8 expression quantitative trait locus, rs10857560, was reported to be associated with precapillary PH. In a combined group, the homozygous state of AA genotype rs10857560 was characterized by lower MAPK8 expression and was present in all the precapillary PH cases [52].
The glutathione S-transferase (GST) gene family encodes genes involved in certain processes including detoxication and toxification mechanisms. The GST genes are known to be upregulated in response to oxidative stress. An Egyptian study measured the frequency distribution of the GSTM1, GSTT1 and GSTP1 gene polymorphisms in a group of adult SCD patients. It was demonstrated that the GSTM1 null genotype was associated with acute chest syndrome and veno-occlusive crisis while the GSTT1 null genotype was associated with increased blood transfusion requirements. Absence of both GSTM1 and GSTT1 genes was significantly associated with pulmonary hypertension [53].
Endothelial NOS (eNOS), also known as nitric oxide synthase 3, is an enzyme encoded by the NOS3 gene located in chromosome 7. It is responsible for NO generation in the vascular endothelium therefore it plays a crucial role in regulating vascular tone, cellular proliferation, leukocyte adhesion, and platelet aggregation. Yousry et al. (2016) genotyped a group of SCD patientsfor eNOS 4a/b and eNOS 786T>C polymorphisms and analyzed the results with the severity of SCD clinical manifestations. They demonstrated that the homozygous mutant eNOS-786T>T genotype was significantly associated with higher acute chest syndrome risk. On the other hand, the wild-type eNOS-4a/4b genotype seemed to be protective against vaso-occlusive crisis and PH. Lastly, the mutant homozygous haplotype (C-4a) was significantly associated with the risk of acute chest syndrome,veno-occlusive crisis, and PH [54].
Thrombospondin-1 (TSP1) regulates TGF-β pathway activation and endothelial and smooth muscle cell proliferation, processes known to be affected in PAH.In familial cases of PAH, mutations involving the TSP1 gene highlighted it as a potential modifier gene in these rare cases [55]. When TSP1 SNPs were studied in SCD-related PH, univariate regression analyses revealed that rs1478604 and rs1478605 SNPs were associated with differences in pulmonary artery systolic pressure as assessed by echocardiography. These SNPs are proximal to the transcription start site therefore they may have a potential transcription-regulation activity on the TSP1 gene with rs1478605 being the most possible to regulate TSP1 gene expression [56,57].
Endothelin-1 (ET-1) gene polymorphisms have been investigated extensively in the pathogenesis of various vascular diseases including SCD. The mutant T allele of the ET-1 G5665T polymorphism was reported to be associated with increased plasma ET-1 levels [58]. Khorshied et al.(2018), reported that in their study, statistical analysis comparing patients having the wild and the polymorphic genotypes demonstrated that pulmonary dysfunction in the form of pulmonary hypertension and acute chest syndrome, and severe vaso-occlusive crises were more frequent in patients with the polymorphic genotypes [59]. In the contrary, Thakur et al.(2014) reported that ET-1 G5665T SNP had no significance among his group. Differences were also recorded in the sex-related distribution of the polymorphic genotypes [60].
Interleukin-1 β (IL-1 β) is a cytokine necessary for host defense responses to infection, sepsis and injury. There are multiple studies on the role of cytokines in the pathophysiology of SCD showing increased levels of plasma IL-1β. It is believed that the release of cytokines in response to infection, endothelial cell activation and other harmful stimuli may play a key role in the pathophysiology of SCD complications. Afifi et al.,(2019),reported an increased prevalence of the mutant genotype of IL-1β +3954 SNP in a group of Egyptian SCD patients. The mutant genotype was more prevalent in cases with PH.The mean ESPAP was significantly higher among the TT genotype versus the combined genotypes CC + CT. This study confirms the findings of Vicari et al.,(2015)who reported similar findings among SCD Brazilian patients [61,62].
Methyltetrahydrofolate reductase (MTHFR) is responsible for the transformation of 5,10-methylenetetrahydrofolate into 5-methyltetrahydrofolate. The MTHFR gene has 677C>T and 1298A>C functional polymorphisms that produce a thermolabile form of MTHFR. Decreased MTHFR levels have been proposed as a risk factor for deep vein thrombosis in the past. Several studies have investigated the association between MTHFR 677C>T and vascular phenomena of SCD with conflicting results.Lakkakula et al., (2019), in a meta-analysis, reported that mutant genotypes (CT + TT vs. CC) of the MTHFR 677C>T polymorphism were associated with an increased risk of vascular events in SCD patients [63].
RASA3 is a GTPase-activating protein that is involved in R-Ras and Rap1 activity and related to vasculogenesis and endothelial mechanisms. When studied in SCD patients, RASA3 expression was lower in patients’ PH and was associated with higher mortality. The SNP rs9525228 correlated with PH risk, higher TRV values and higher pulmonary vascular resistance. Moreover, it was associated with precapillary PH values and decreased survival in a subgroup of the patients [64]. The above results are summarized in Table 1.
Table 1.Summary of the findings concerning gene polymorphisms that presented an association with pulmonary hypertension. SCD: sickle cell disease, SNP: single-nucleotide polymorphism, PH: pulmonary hypertension, N/A: not available, TRV: tricuspid regurgitation velocity, ACS: acute chest syndrome, VOC: veno-occlusive crisis, ESPAP: estimated peak systolic pulmonary arterial pressure, ACVRL1: activin receptor-like kinase 1, BMP6: bone morphogenetic protein 6, ADRB1: beta-1 adrenergic receptor, TGFBR3: transforming growth factor beta receptor 3, TSP1: thrombospondin 1, NEDD4L: neural precursor cell expressed developmentally downregulated gene 4-like, MTHFR: methylenetetrahydrofolate reductase, ET-1: endothelin-1, PRELP: proline/arginine-rich end leucine-rich repeat protein, RASA3: RAS P21 protein activator 3, MAPK8: mitogen-activated protein kinase 8, GST: glutathione S-transferase, ADORA2B: adenosine A2B receptor, GALNT13: polypeptide N-acetylgalactosaminyltransferase 13, eNOS: endothelial nitric oxide synthase, CYBR5: cytochrome b5 reductase.
3. Discussion
PH is one of the most severe complications of SCD. Mechanisms involved in pathogenesis are complex and etiology is multifactorial. Established pathogenetic mechanisms include intravascular hemolysis, chronic lung involvement, hypercoagulable status, asplenia and thromboembolic disease, but questions on identifying patients predisposed to develop PH remain. In recent years genome-wide association studies (GWASs) have become a new standard in genomic studies in several medical areas. The search for genetic risk factors for common diseases in an effort to explain clinical diversity led to remarkable findings and applications on cardiovascular diseases, autoimmune diseases, neoplasms and many others. The utilization of GWASs has been followed by deep DNA sequencing methods that play a crucial role in investigating genetic variants in a large number of samples from different populations and should further extend our understanding of the differential expression of genes in normal and pathological samples.
Of course, these methods have certain limitations and problems in their application. GWASs require specifically organized and large quantities of data to avoid false-positive results. Usually, larger sample numbers are needed compared to gene-specific studies to validate data and false negative findings may result if SNPs are analyzed separately. Lastly, as it has been demonstrated in SCD, replication and validationof the findings in groups of different ancestry and across all genotypic subgroups is important. On the other hand, for the time being, the cost of deep DNA sequencing remains high and access to the techniques and computational systems is not universal [65]. Other issues that may arise in GWAS include linkage disequilibrium with causative polymorphisms, interpretation of associations with SNPs without a known function, gene-gene interaction, gene-environment interference, and precise phenotype definition [66].
It is a reasonable approach to focus on pathways and mediators involved in the pathogenesis of complications for identifying genetic modifiers. As expected, findings of genomic studies in SCD patients confirm the importance of pathways related to hemolysis, NO bioavailability, vascular remodeling, endothelial integrity, inflammation, and oxidant injury for PH pathogenesis. Most studies so far have investigated targeted candidate genes and more comprehensive GWAS are expected to follow. To date, much emphasis has been placed on polymorphisms in genes related to the TGF-β pathway.
Another reasonable approach for future studies is to examine SCD patients for polymorphisms identified in patients with idiopathic pulmonary arterial hypertension, unrelated to SCD.For example, Tang et al., (2022), suggested several genes as possible markers for PH pathogenesis and potential targets for the prevention of pulmonary vascular restructure including SLC4A1, AHSP, ALAS2, CA1, HBD, SNCA, HBM, SELENBP1, SERPINE1, ITGA2B, TEAD4, TGIF2LY, GATA5, GATA1, GATA2, and FOS [67].It would be interesting to investigate whether these data can be confirmed in SCD, with special attention to the ancestral composition of the patient population. The recent identification of the RASA3 gene as a candidate gene involved in the pathogenesis and prognosis of PH in SCD as well as pulmonary arterial hypertension in non-SCD patients is also an important finding [64]. Validation of each study in larger groups as well as wider ancestral groups is necessary to confirm causative associations.
4. Conclusions
Ultimately, the aim of identifying genetic markers predisposing to PH might help in selecting high-risk patients for more frequent screening for PH, early introduction of disease-modifying treatments such as hydroxyurea or blood transfusions, development of new targeted therapies and monitoring response to treatment.
5. Future Directions
The future of research on SCD-related complications, including PH, must utilize every available data sourcefrom multiple centers. A typical example of an initiative in this direction was the creation of the International Hemoglobinopathy Research Network (INHERENT) in 2020, as a formally coordinated network aiming to thorough investigation of genetic modifiers in hemoglobinopathies.Through a large-scale multi-center genome-wide association study, researchers may overcome the obstacles met in previous studies related to limited sample sizes and decreased statistical power. The main targets of this effort include the identification of potential genetic modifiers, the reproduction of previous results,the adoption of universally accepted genetic, phenotypic, and clinical definitions based on validated standards and the availability of high-quality data [68]. This kind of oriented approach to gathering, analyzing, and validating data will be of major importance. The complexity of molecular events during time, in addition to established pathogenetic mechanisms must be investigated to explain the clinical diversity in SCD. Only then can research progress be focused to identify patients at risk for specific complications and potentially change the disease course.
Author Contributions
MD contributed to the conception and design of the work, SC and PF acquired the data and drafted the work, IT and GA revised the work. All authors have approved the submitted version and agree to be personally accountable for the author’s own contributions and for ensuring that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and documented in the literature.
Funding
This research received no external funding.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Acknowledgments
The authors have nothing to acknowledge.
Conflicts of Interest
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no funding for this work that could have influenced its outcome.
References
- Gladwin, M.T.; Vichinsky, E. Pulmonary Complications of Sickle Cell Disease. N. Engl. J. Med. 2008, 359, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Castro, O.L.; Machado, R.F. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood 2016, 127, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Savale, L.; Habibi, A.; Lionnet, F.; Maitre, B.; Cottin, V.; Jais, X.; Chaouat, A.; Artaud-Macari, E.; Canuet, M.; Prevot, G.; et al. Clinical phenotypes and outcomes of precapillary pulmonary hypertension of sickle cell disease. Eur. Respir. J. 2019, 54, 1900585. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.; Brida, M.; Carlsen, J.; Coats, A.J.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2022, 61, 2200879. [Google Scholar] [CrossRef] [PubMed]
- Klings, E.S.; Machado, R.F.; Barst, R.J.; Morris, C.R.; Mubarak, K.K.; Gordeuk, V.R.; Kato, G.J.; Ataga, K.I.; Gibbs, J.S.; Castro, O.; et al. An Official American Thoracic Society Clinical Practice Guideline: Diagnosis, Risk Stratification, and Management of Pulmonary Hypertension of Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2014, 189, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Barst, R.J.; Gibbs, J.S.R.; Hildesheim, M.; Sachdev, V.; Nouraie, M.; Hassell, K.L.; Little, J.A.; Schraufnagel, D.E.; Krishnamurti, L.; et al. Risk Factors for Death in 632 Patients with Sickle Cell Disease in the United States and United Kingdom. PLOS ONE 2014, 9, e99489. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.F.; Hildesheim, M.; Mendelsohn, L.; Remaley, A.T.; Kato, G.J.; Gladwin, M.T. NT-pro brain natriuretic peptide levels and the risk of death in the cooperative study of sickle cell disease. Br. J. Haematol. 2011, 154, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Sachdev, V.; Jison, M.L.; Shizukuda, Y.; Plehn, J.F.; Minter, K.; Brown, B.; Coles, W.A.; Nichols, J.S.; Ernst, I.; et al. Pulmonary Hypertension as a Risk Factor for Death in Patients with Sickle Cell Disease. N. Engl. J. Med. 2004, 350, 886–895. [Google Scholar] [CrossRef]
- Mehari, A.; Gladwin, M.T.; Tian, X.; Machado, R.F.; Kato, G.J. Mortality in Adults With Sickle Cell Disease and Pulmonary Hypertension. JAMA 2012, 307, 1254–1256. [Google Scholar] [CrossRef]
- Liem, R.I.; Lanzkron, S.; Coates, T.D.; DeCastro, L.; Desai, A.A.; Ataga, K.I.; Cohen, R.T.; Haynes, J.J.; Osunkwo, I.; Lebensburger, J.D.; et al. American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease. Blood Adv. 2019, 3, 3867–3897. [Google Scholar] [CrossRef] [PubMed]
- Mehari, A.; Alam, S.; Tian, X.; Cuttica, M.J.; Barnett, C.F.; Miles, G.; Xu, D.; Seamon, C.; Adams-Graves, P.; Castro, O.L.; et al. Hemodynamic Predictors of Mortality in Adults with Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2013, 187, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Mehari, A.; Igbineweka, N.; Allen, D.; Nichols, J.; Thein, S.L.; Weir, N.A. Abnormal Ventilation–Perfusion Scan Is Associated with Pulmonary Hypertension in Sickle Cell Adults. J. Nucl. Med. 2018, 60, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Niss, O.; Fleck, R.; Makue, F.; Alsaied, T.; Desai, P.; Towbin, J.A.; Malik, P.; Taylor, M.D.; Quinn, C.T. Association between diffuse myocardial fibrosis and diastolic dysfunction in sickle cell anemia. Blood 2017, 130, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.-L.; Tian, X.; Alam, S.; Mehari, A.; Leung, S.W.; Seamon, C.; Allen, D.; Minniti, C.P.; Sachdev, V.; Arai, A.E.; et al. Elevated transpulmonary gradient and cardiac magnetic resonance-derived right ventricular remodeling predict poor outcomes in sickle cell disease. Haematologica 2015, 101, e40–e43. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Parent, F.; Bachir, D.; Inamo, J.; Lionnet, F.; Driss, F.; Loko, G.; Habibi, A.; Bennani, S.; Savale, L.; Adnot, S.; et al. A Hemodynamic Study of Pulmonary Hypertension in Sickle Cell Disease. N. Engl. J. Med. 2011, 365, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.F.; Anthi, A.; Steinberg, M.H.; Bonds, D.; Sachdev, V.; Kato, G.J.; Taveira-DaSilva, A.M.; Ballas, S.K.; Blackwelder, W.; Xu, X.; et al. N-Terminal Pro-Brain Natriuretic Peptide Levels and Risk of Death in Sickle Cell Disease. JAMA 2006, 296, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Anthi, A.; Machado, R.F.; Jison, M.L.; Taveira-DaSilva, A.M.; Rubin, L.J.; Hunter, L.; Hunter, C.J.; Coles, W.; Nichols, J.; Avila, N.A.; et al. Hemodynamic and Functional Assessment of Patients with Sickle Cell Disease and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2007, 175, 1272–1279. [Google Scholar] [CrossRef]
- Minniti, C.P.; Sable, C.; Campbell, A.; Rana, S.; Ensing, G.; Dham, N.; Onyekwere, O.; Nouraie, M.; Kato, G.; Gladwin, M.T.; et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: association with hemolysis and hemoglobin oxygen desaturation. Haematologica 2009, 94, 340–347. [Google Scholar] [CrossRef]
- Bunn, H.F.; Nathan, D.G.; Dover, G.J.; Hebbel, R.P.; Platt, O.S.; Rosse, W.F.; Ware, R.E. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood 2010, 116, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; McGowan, V.; Machado, R.F.; Little, J.A.; Taylor, J.; Morris, C.R.; Nichols, J.S.; Wang, X.; Poljakovic, M.; Morris, S.M.; et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood 2006, 107, 2279–2285. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Gladwin, M.T.; Steinberg, M.H. Deconstructing sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007, 21, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, C.C.; Machado, R.F. The different facets of sickle cell disease-related pulmonary hypertension. Curr. Opin. Pulm. Med. 2021, 27, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Liu, Y.; Liu, B. Integrated bioinformatics analysis reveals marker genes and immune infiltration for pulmonary arterial hypertension. Sci. Rep. 2022, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The Clinical Sequelae of Intravascular Hemolysis and Extracellular Plasma Hemoglobin. JAMA 2005, 293, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Niss, O.; Quinn, C.T.; Lane, A.; Daily, J.; Khoury, P.R.; Bakeer, N.; Kimball, T.R.; Towbin, J.A.; Malik, P.; Taylor, M.D. Cardiomyopathy With Restrictive Physiology in Sickle Cell Disease. JACC: Cardiovasc. Imaging 2016, 9, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P. Reconstructing sickle cell disease: A data-based analysis of the “hyperhemolysis paradigm” for pulmonary hypertension from the perspective of evidence-based medicine. Am. J. Hematol. 2011, 86, 123–154. [Google Scholar] [CrossRef]
- Akinsheye, I.; Klings, E.S. Sickle cell anemia and vascular dysfunction: The nitric oxide connection. J. Cell. Physiol. 2010, 224, 620–625. [Google Scholar] [CrossRef]
- Jison, M.L.; Gladwin, M.T. Hemolytic Anemia–associated Pulmonary Hypertension of Sickle Cell Disease and the Nitric Oxide/Arginine Pathway. Am. J. Respir. Crit. Care Med. 2003, 168, 3–4. [Google Scholar] [CrossRef]
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated Arginine Metabolism, Hemolysis-Associated Pulmonary Hypertension, and Mortality in Sickle Cell Disease. JAMA 2005, 294, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Wang, Z.; Machado, R.F.; Blackwelder, W.C.; Taylor, J.G.; Hazen, S.L. Endogenous nitric oxide synthase inhibitors in sickle cell disease: abnormal levels and correlations with pulmonary hypertension, desaturation, haemolysis, organ dysfunction and death. Br. J. Haematol. 2009, 145, 506–513. [Google Scholar] [CrossRef]
- Farmakis, D.; Aessopos, A. Pulmonary Hypertension Associated With Hemoglobinopathies. Circ. 2011, 123, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Nasimuzzaman, M. D.; Malik, P. Role of the coagulation system in the pathogenesis of sickle cell disease. Blood Adv. 2019, 3, 3170–3180. [Google Scholar] [CrossRef]
- Shet, A.S.; Lizarralde-Iragorri, M.A.; Naik, R.P. The molecular basis for the prothrombotic state in sickle cell disease. Haematologica 2020, 105, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulou, M.; Politou, M.; Stavroula, K.; Koutsouri, D.; Tsioutsias, P.; Flevari, P.; Voskaridou, E. SILENT CEREBRAL ISCHEMIA AND THROMBOEMBOLIC EVENTS IN SICKLE CELL DISEASE: ANALYSIS OF COAGULATION PARAMETERS AND THROMBOELASTOGRAPHY. Abstract release date: 05/18/17. EHA Library. Dimopoulou M. 05/18/2017; 181261; E1485.
- Tantawy, A.; El-Sherif, N.; Makkeyah, S.; Eldeen, N.S.; Farghal, N.B.E.-D.; Soliman, N.; Ebeid, F.S.E. Sleep disordered breathing and its relation to stroke and pulmonary hypertension in children with sickle cell disease: a single-center cross-sectional study. Ann. Hematol. 2023, 102, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Efird, J.T.; Knupp, C.; Kadali, R.; Liles, D.; Shiue, K.; Boettger, P.; Quan, S.F. Sleep Disorders in Adult Sickle Cell Patients. Sleep Med. 2015, 11, 219–223. [Google Scholar] [CrossRef]
- Mehari, A.; Klings, E.S. Chronic Pulmonary Complications of Sickle Cell Disease. Chest 2016, 149, 1313–1324. [Google Scholar] [CrossRef]
- Ashley-Koch, A.E.; Elliott, L.; Kail, M.E.; De Castro, L.M.; Jonassaint, J.; Jackson, T.L.; Price, J.; Ataga, K.I.; Levesque, M.C.; Weinberg, J.B.; et al. Identification of genetic polymorphisms associated with risk for pulmonary hypertension in sickle cell disease. Blood 2008, 111, 5721–5726. [Google Scholar] [CrossRef]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M.; et al. Clinical and Molecular Genetic Features of Pulmonary Hypertension in Patients with Hereditary Hemorrhagic Telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef]
- Roberts, K. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur. Respir. J. 2004, 24, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Klings, E.S.; A Dworkis, D.; Sedgewick, A.; Hartley, S.W.; Allison, A.-K.; Telen, M.J.; Kato, G.J.; Gladwin, M.; Sebastiani, P.; Baldwin, C.T.; et al. Genetic Polymorphisms in NEDD4L Are Associated with Pulmonary Hypertension of Sickle Cell Anemia. Blood 2009, 114, 2562–2562. [Google Scholar] [CrossRef]
- Rotin, D.; Prag, G. Physiological Functions of the Ubiquitin Ligases Nedd4-1 and Nedd4-2. Physiology 2024, 39, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Nouraie, M.; Reading, N.S.; Campbell, A.; Minniti, C.; Rana, S.R.; Luchtman-Jones, L.; Sable, C.; Dham, N.; Ensing, G.; Kato, G.J.; et al. Cytochrome b5 Reductase T116S Mutation and Hemolysis in Sickle Cell Disease. Blood 2009, 114, 903–903. [Google Scholar] [CrossRef]
- Gordeuk, V.R.; Nouraie, M.; Niu, X.; DelBove, J.N.; Prchal, J.T. Cytochrome B5 Reductase T116S Polymorphism Is Associated with Decreased Risk of Severe Anemia Among Zambian Children with Malaria. Blood 2010, 116, 4233–4233. [Google Scholar] [CrossRef]
- Desai, A.A.; Zhou, T.; Ahmad, H.; Zhang, W.; Mu, W.; Trevino, S.; Wade, M.S.; Raghavachari, N.; Kato, G.J.; Peters-Lawrence, M.H.; et al. A Novel Molecular Signature for Elevated Tricuspid Regurgitation Velocity in Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2012, 186, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Geard, A.; Pule, G.D.; Chelo, D.; Bitoungui, V.J.N.; Wonkam, A. Genetics of Sickle Cell-Associated Cardiovascular Disease: An Expert Review with Lessons Learned in Africa. OMICS: A J. Integr. Biol. 2016, 20, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Nogimori, K.; Hori, T.; Kawaguchi, K.; Fukui, T.; Mii, S.; Nakada, H.; Matsumoto, Y.; Yamauchi, Y.; Takahashi, M.; Furukawa, K.; et al. Increased expression levels of ppGalNAc-T13 in lung cancers: Significance in the prognostic diagnosis. Int. J. Oncol. 2016, 50, 746–746. [Google Scholar] [CrossRef]
- Shozu, K.; Kaneko, S.; Shinkai, N.; Dozen, A.; Kosuge, H.; Nakakido, M.; Machino, H.; Takasawa, K.; Asada, K.; Komatsu, M.; et al. Repression of the PRELP gene is relieved by histone deacetylase inhibitors through acetylation of histone H2B lysine 5 in bladder cancer. Clin. Epigenetics 2022, 14, 1–13. [Google Scholar] [CrossRef]
- Ludwig, N.; Yerneni, S.S.; Azambuja, J.H.; Gillespie, D.G.; Menshikova, E.V.; Jackson, E.K.; Whiteside, T.L. Tumor-derived exosomes promote angiogenesis via adenosine A2B receptor signaling. Angiogenesis 2020, 23, 599–610. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, W.; Ma, S.-F.; Desai, A.A.; Saraf, S.; Miasniakova, G.; Sergueeva, A.; Ammosova, T.; Xu, M.; Nekhai, S.; et al. Hypoxic Response Contributes to Altered Gene Expression and Precapillary Pulmonary Hypertension in Patients With Sickle Cell Disease. Circ. 2014, 129, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Ellithy, H.N.; Yousri, S.; Shahin, G.H. Relation between glutathione S-transferase genes (GSTM1, GSTT1, and GSTP1) polymorphisms and clinical manifestations of sickle cell disease in Egyptian patients. Hematology 2015, 20, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Yousry, S.M.; Ellithy, H.N.; Shahin, G.H. Endothelial nitric oxide synthase gene polymorphisms and the risk of vasculopathy in sickle cell disease. Hematology 2016, 21, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Maloney, J.P.; Stearman, R.S.; Bull, T.M.; Calabrese, D.W.; Tripp-Addison, M.L.; Wick, M.J.; Broeckel, U.; Robbins, I.M.; Wheeler, L.A.; Cogan, J.D.; et al. Loss-of-function thrombospondin-1 mutations in familial pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2012, 302, L541–L554. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.A.; Novelli, E.M.; Isenberg, J.S.; Garrett, M.E.; Chu, Y.; Soldano, K.; Ataga, K.I.; Telen, M.J.; Ashley-Koch, A.; Gladwin, M.T.; et al. Thrombospondin-1 gene polymorphism is associated with estimated pulmonary artery pressure in patients with sickle cell anemia. Am. J. Hematol. 2017, 92, E31–E34. [Google Scholar] [CrossRef]
- Rogers, N.M.; Yao, M.; Sembrat, J.; George, M.P.; Knupp, H.; Ross, M.; Sharifi-Sanjani, M.; Milosevic, J.; Croix, C.S.; Rajkumar, R.; et al. Cellular, Pharmacological, and Biophysical Evaluation of Explanted Lungs from a Patient with Sickle Cell Disease and Severe Pulmonary Arterial Hypertension. Pulm. Circ. 2013, 3, 936–951. [Google Scholar] [CrossRef] [PubMed]
- Navarro, K.G.; Agyingi, S.E.; Nwabuobi, C.K.; Thomas, B.N. Polymorphism of the endothelin-1 gene (rs5370) is a potential contributor to sickle cell disease pathophysiology. Genes Dis. 2016, 3, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Khorshied, M.M.; Mohamed, N.S.; Hamza, R.S.; Ali, R.M.; El-Ghamrawy, M.K. Protein Z and Endothelin-1 genetic polymorphisms in pediatric Egyptian sickle cell disease patients. J. Clin. Lab. Anal. 2017, 32, e22264. [Google Scholar] [CrossRef] [PubMed]
- Thakur, T.J.; Guindo, A.; Cullifer, L.R.; Li, Y.; Imumorin, I.G.; Diallo, D.A.; Thomas, B.N. Endothelin-1 but not Endothelial Nitric Oxide Synthase Gene Polymorphism is Associated with Sickle Cell Disease in Africa. Gene Regul. Syst. Biol. 2014, 8, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Afifi, R.A.-R.A.-A.; Sedky, Y.M.; Abd-Elkareem, H.; Botros, S.K.A. IL-Iβ +3954 C / T Polymorphism and Its Clinical Associations in Egyptian Sickle Cell Disease Patients. Int. J. Hematol. Stem Cell Res. 2019, 13, 35–41. [Google Scholar] [CrossRef]
- Vicari, P.; Adegoke, S.A.; Mazzotti, D.R.; Cançado, R.D.; Noguti, M.A.E.; Figueiredo, M.S. Corrigendum to “Interleukin-1β and interleukin-6 gene polymorphisms are associated with manifestations of sickle cell anemia” [Blood Cells Mol. Dis. 54/3(2014), 244–249]. Blood Cells, Mol. Dis. 2015, 54, 328. [Google Scholar] [CrossRef]
- Lakkakula, B. Association between MTHFR 677C>T polymorphism and vascular complications in sickle cell disease: A meta-analysis. Transfus. Clin. et Biol. 2019, 26, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, C.C.; Zhang, X.; Schwantes-An, T.L.; Stearman, R.S.; Hooker, S.; Kittles, R.A.; Aldred, M.A.; Lutz, K.A.; Pauciulo, M.W.; Nichols, W.C.; et al. RASA3 is a candidate gene in sickle cell disease-associated pulmonary hypertension and pulmonary arterial hypertension. Pulm. Circ. 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Fertrin, K.Y.; Costa, F.F. Genomic polymorphisms in sickle cell disease: implications for clinical diversity and treatment. Expert Rev. Hematol. 2010, 3, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H. Genetic Etiologies for Phenotypic Diversity in Sickle Cell Anemia. Sci. World J. 2009, 9, 46–67. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Liu, Y.; Liu, B. Integrated bioinformatics analysis reveals marker genes and immune infiltration for pulmonary arterial hypertension. Sci. Rep. 2022, 12, 1–14. [Google Scholar] [CrossRef]
- Kountouris, P.; Stephanou, C.; Archer, N.; Bonifazi, F.; Giannuzzi, V.; Kuo, K.H.M.; Maggio, A.; Makani, J.; Mañú-Pereira, M.d.M.; Michailidou, K.; et al. The International Hemoglobinopathy Research Network (INHERENT): An international initiative to study the role of genetic modifiers in hemoglobinopathies. Am. J. Hematol. 2021, 96, E416–E420. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of sickle cell disease complications. SCD: sickle cell disease, CNS: central nervous system. Created with GitMind.
Figure 1.
Schematic representation of sickle cell disease complications. SCD: sickle cell disease, CNS: central nervous system. Created with GitMind.
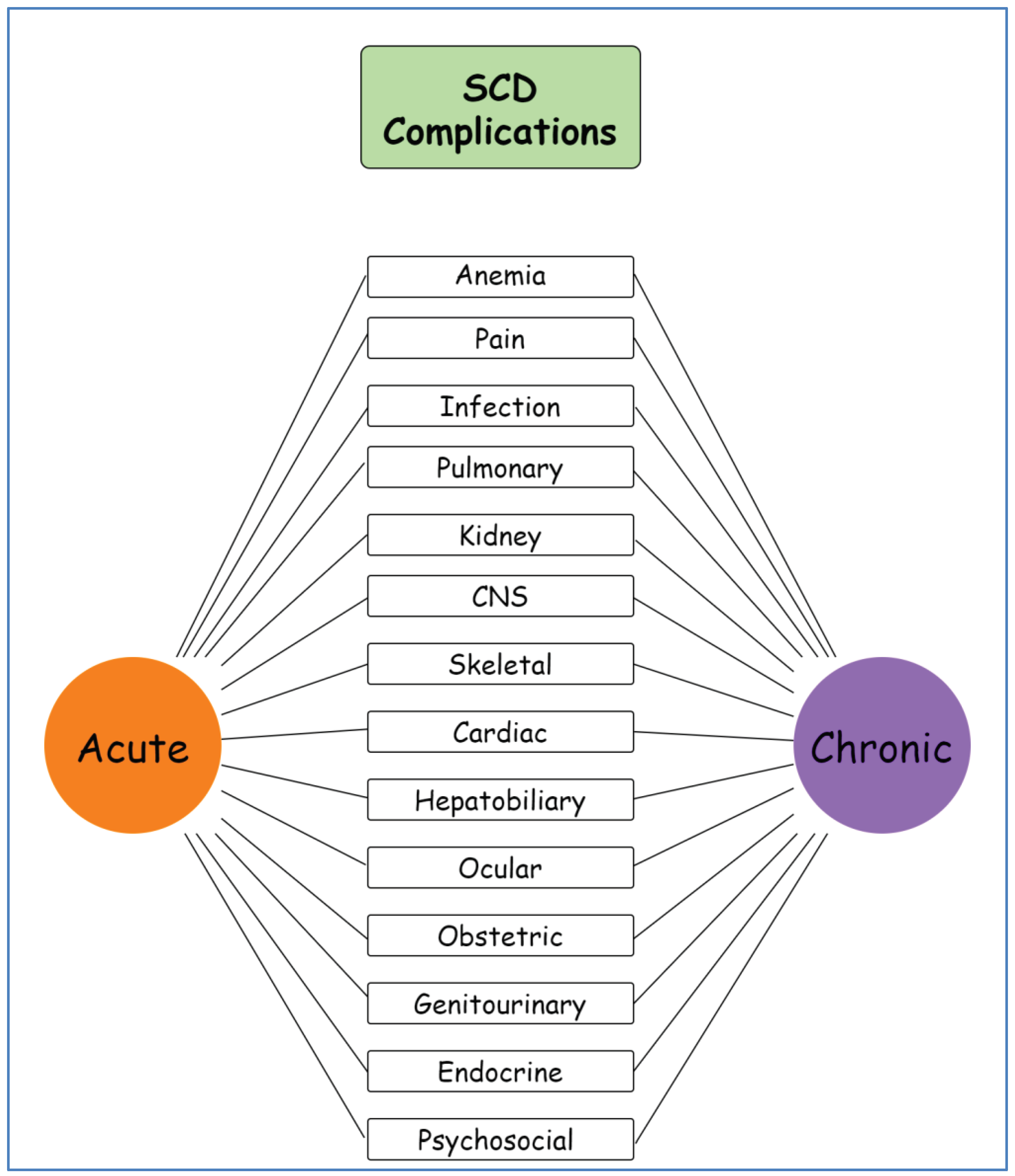
Figure 2.
Pathophysiology of sickle cell-related pulmonary hypertension and related gene polymorphisms. TGF-β:transforming growth factor-β, ACVRL1: activin receptor-like kinase 1, BMP6: bone morphogenetic protein 6, ADRB1: beta-1 adrenergic receptor, TGFBR3: transforming growth factor beta receptor 3, TSP1: thrombospondin 1, NEDD4L: neural precursor cell expressed developmentally downregulated gene 4-like, MTHFR: methylenetetrahydrofolate reductase, ET-1: endothelin-1, PRELP: proline/arginine-rich end leucine-rich repeat protein, RASA3: RAS P21 protein activator 3, MAPK8: mitogen-activated protein kinase 8, GST: glutathione S-transferase, ADORA2B: adenosine A2B receptor, GALNT13: polypeptide N-acetylgalactosaminyltransferase 13, eNOS: endothelial nitric oxide synthase, CYBR5: cytochrome b5 reductase, NO: nitric oxide.
Figure 2.
Pathophysiology of sickle cell-related pulmonary hypertension and related gene polymorphisms. TGF-β:transforming growth factor-β, ACVRL1: activin receptor-like kinase 1, BMP6: bone morphogenetic protein 6, ADRB1: beta-1 adrenergic receptor, TGFBR3: transforming growth factor beta receptor 3, TSP1: thrombospondin 1, NEDD4L: neural precursor cell expressed developmentally downregulated gene 4-like, MTHFR: methylenetetrahydrofolate reductase, ET-1: endothelin-1, PRELP: proline/arginine-rich end leucine-rich repeat protein, RASA3: RAS P21 protein activator 3, MAPK8: mitogen-activated protein kinase 8, GST: glutathione S-transferase, ADORA2B: adenosine A2B receptor, GALNT13: polypeptide N-acetylgalactosaminyltransferase 13, eNOS: endothelial nitric oxide synthase, CYBR5: cytochrome b5 reductase, NO: nitric oxide.
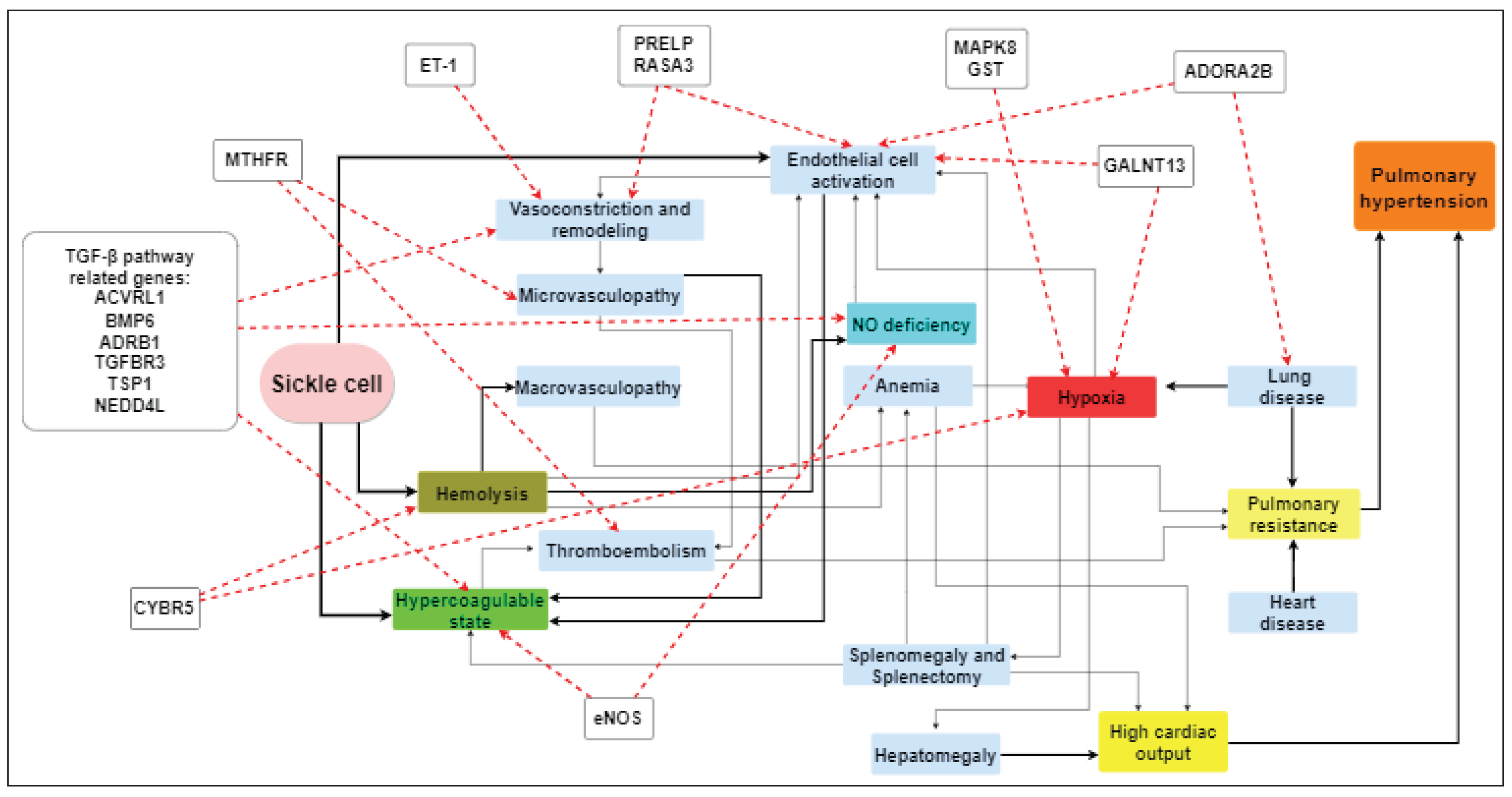

Table 1.
Gene polymorphisms associated with SCD-related pulmonary hypertension.
| Gene name | SNP/mutation | Study, Year | Number of patients | Ancestry | PH-related findings |
|---|---|---|---|---|---|
| ACVRL1 | rs3847859, rs706814 | Ashley-Koch et al., 2008 | 518 | N/A | Associated with the occurrence of PH |
| BMP6 | rs267192 | ||||
| ADRB1 | rs1801253, rs7921133 | ||||
| TGFBR3 | rs10874940 | ||||
| ARG2 | rs12587111, rs1885042 | Nominally associated with PH | |||
| NEDD4L | rs559046, rs1624292 | Klings et al., 2009 | 59 | N/A | Associated with a TRV ≥ 2.5 m/sec |
| 4 SNPs in intron 1 | 139 | Associated with elevated NT-pro-BNP levels | |||
| CYBR5 | T116S | Nouraie et al.,2009 | 261 | N/A | Heterozygosity and homozygosity for the CYBR5 T116S associated with lower TRV |
| GALNT13 | rs799813, rs10497120, rs13407922, rs16833378, rs9808145 |
Desai et al., 2012 | 27 | African American | Associated with elevated TRV |
| PRELP | rs2794452 | ||||
| ADORA2B | rs7208480 | ||||
| MAPK8 | rs10857560 | Zhang et al., 2014 | 61 | African American | Associated with precapillary PH |
| GST |
GSTM1, GSTT1, GSTP1 |
Ellithy et al., 2015 | 100 | Egyptian | Absence of both GSTM1 and GSTT1 genes significantly associated with developing PH |
| eNOS | eNOS 4a/b, eNOS 786T>C, C-4a |
Yousry et al., 2016 | 100 | Egyptian | Wild-type eNOS-4a/4b genotype seemed protective against VOC and PH. Mutant homozygous haplotype (C-4a) associated with the risk of ACS, VOC, and PH |
| TSP1 | rs1478604, rs1478605 | Jacob et al., 2017 | 406 | N/A | Associated with elevated TRV |
| ET-1 | G5665T | Khorshied et al., 2018 | 100 | Egyptian | Pulmonary dysfunction (PH and ACS) more frequent in patients with the polymorphic genotypes |
| IL-1 β | +3954 | Afifi et al.,2019 | 50 | Egyptian | Mutant genotype more prevalent in cases with PH. Mean ESPAP significantly higher among mutant genotypes. |
| Vicari et al., 2015 | 107 | Brazilian | |||
| MTHFR | 677C>T | Lakkakula et al.,2019 | 614 | N/A | Mutant genotype associated with increased risk of vascular events |
| RASA3 | rs9525228 | Prohaska et al.,2023 | 171 | African American | SNP correlated with PH risk, higher TRV and pulmonary vascular resistance and associated with precapillary PH values and decreased survival in a subgroup of the patients |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



