
You are currently viewing a beta version of our website. If you spot anything unusual, kindly let us know.
Preprint
Review
Treatment of Adamantinomatous Craniopharyngioma Using RNAi and CRISPR Targeting the WNT Pathway
Altmetrics
Downloads
107
Views
75
Comments
0

This version is not peer-reviewed
Abstract
Adamantinomatous Craniopharyngioma (ACP) is a rare epithelial tumor located in the craniopharyngeal duct, often associated with high morbidity and a high long-term recurrence rate. ACP is predominantly diagnosed in children below the age of 15 and accounts for a majority of Craniopharyngioma cases in the United States. It is most commonly caused by a mutation in exon 3 of the CTNNB1 gene, which promotes overaccumulation of β-Catenin in the WNT pathway. As β-Catenin accumulates, it over-activates the transcription factor, causing an over-transcription of WNT proteins and uncontrolled cell growth. Current methods of treatment for ACP are centered around surgical intervention and radiotherapy, including gross-total resection (GTR), subtotal resection (STR), and a combination of subtotal resection and adjuvant radiotherapy (STR+XTR). These methods of treatment are associated with high risk due to the proximity of ACP to critical structures near the craniopharyngeal duct. High recurrence rates and morbidity are closely associated with current methods of treatment and other methods have yet to be attempted in craniopharyngioma. This review will explore two possible methods of non-surgical and non-radiological therapeutic interventions. The use of RNAi therapy and CRISPR may provide insight and be a potential way to treat ACP with increased quality of life and decreased recurrence rates.
Keywords:
Subject: Biology and Life Sciences - Neuroscience and Neurology
Introduction
Craniopharyngioma is an aggressive benign tumor with a global recurrence rate of nearly 27% and may be associated with a highly decreased quality of life [1,2]. Although craniopharyngioma is a WHO class I benign tumor, growth can be unpredictable, affecting the sellar region and surrounding areas containing critical structures such as the optic chiasma, the pituitary gland, and the hypothalamus[3]*. Damage to these areas commonly manifests symptoms such as endocrine deficiencies, visual impairments, growth retardations, nausea, weight gain, and headaches [4]*. Age at diagnosis is represented by a bimodal peak, one peak at 5-15 years of age and another peak at 50-75 years of age [5,1,6].
Craniopharyngioma presents in two histopathological forms: adamantinomatous craniopharyngioma (ACP) and papillary craniopharyngioma (PCP). ACP is most commonly observed in children under the age of 15 but can also be observed in adults, whereas PCP is almost exclusively diagnosed in patients above the age of 30[7]. ACP differs histologically and pathologically from PCP. ACP is characterized by the nucleocytoplasmic accumulation of β-Catenin [8,9]. β-Catenin then over-activates the WNT pathway, causing uncontrolled proliferation [10,11,12]. On the other hand, PCP is associated with the mutation of BRAF V600e[13]. Despite the differences between ACP and PCP, outcomes following treatment for both tumors do not differ[13].
Two major problems with craniopharyngioma are morbidity and recurrence rates post-treatment. In a study analyzing the symptoms of 121 cases of craniopharyngioma in a follow-up study 10 years after initial surgery, patients had a 90% survival rate but a 48% probability of experiencing major visual field defects, 39% probability of experiencing hyperphagia or obesity, and a high probability of experiencing other major complications 14. Additionally, in a systematic review of 765 craniopharyngioma cases, the aggregate global rate of recurrence was 26.2%[2]. Currently, three methods of treatment for craniopharyngioma are prominently used; radiotherapy, surgery, and a combination of both methods.
The same meta-study reviewed the effects of the three main methods of treatment: GTR (gross total resection), STR (subtotal resection), and a combination of STR+XRT (adjuvant radiotherapy)[2,15]. In a three year follow-up, patients treated with GTR had a recurrence rate of 17%, patients treated with STR+XRT had a recurrence rate of 27%, and patients treated with STR had a recurrence rate of 45%, suggesting that GTR may be the optimal method to reduce the chances of recurrence [2]. Although GTR results in the lowest recurrence rates, there is a high risk of long-term complications such as panhypopituitarism, diabetes insipidus, hypothalamic obesity, and cognitive deficits, leading surgeons to predominantly treat the cases using STR and STR+XTR [16,17,18]. Because of the high risk of long-term complications and high recurrence rates presented by current methods of treatment, the need for a new treatment method is imperative. Therefore, this review will suggest RNA interference and CRISPR as alternative methods to target ACP, potentially without long-term complications.
β-Catenin, WNT Pathway, and the Cause of Adamaninomatous Craniopharyngioma
The WNT pathway is a critical pathway used in many vital processes such as cell proliferation and cell behavior [20]. When WNT ligands (secreted glycoproteins) are not present, β-Catenin attaches itself to a destruction complex consisting of Axin, APC, and GSK3β, and certain residues are produced by exon 3 in CTNNB1, the gene that codes for β-Catenin, and β-Catenin is phosphorylated. After phosphorylation, β-Catenin is rapidly degraded and destroyed by the destruction complex, thus, preventing unnecessary growth (Figure 1) [10,19]. When WNT ligands bind to frizzled, Dishevelled(dsh) is activated, which attracts GBP/Frat-1 and GSK3β is removed from the destruction compound [8,17,48]. The removal of GSK3β from the destruction compound prevents the ubiquitination of CTNNB1, and the stable β-Catenin penetrates the cell nucleus, which binds to the LEF/TCF transcription factors. Then, WNT target genes are transcribed, causing cell proliferation [10,19].
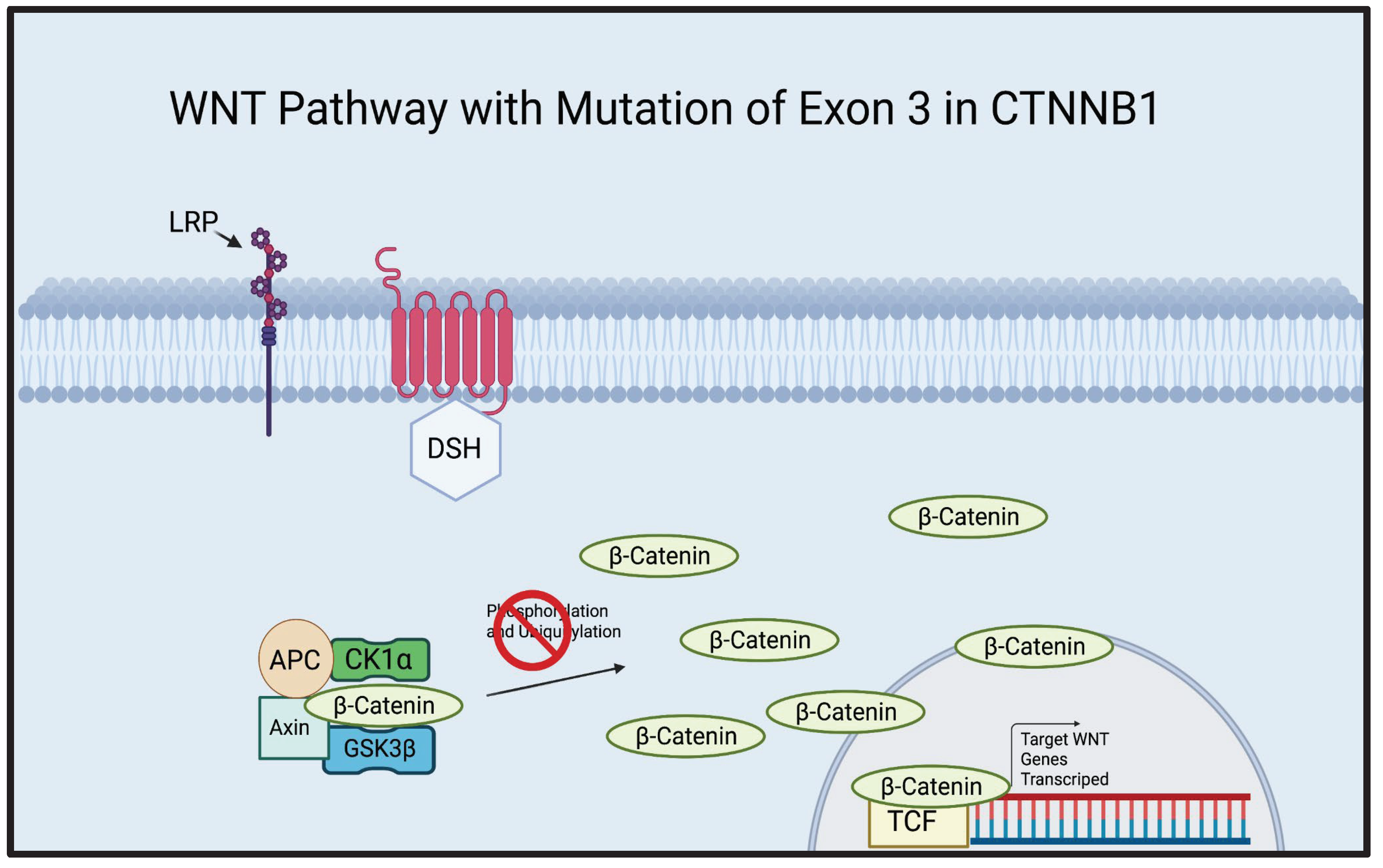
ACP is most commonly associated with the mutation of exon 3 in the CTNNB1 gene[8,9]. A whole genome sequencing study demonstrated that 68.75% of ACP cases presented a mutation of the exon 3 of the CTNNB1 gene and an exome sequencing study of ACP demonstrated that 91.7% of ACP cases were associated with the mutation of exon 3 in CTNNB1[8,9]. Furthermore, a mouse study demonstrated that the overactivation of the WNT pathway, created by excising exon 3 of CTNNB1 in pituitary/progenitor stem cells, resulted in either death or the development of tumors that closely resembled human ACP[12]. The amino acid sites critical for the phosphorylation of β-Catenin are encoded by exon 3 of CTNNB1 [21] (Figure 2). It is evident that exon 3 may play an important role in the development of ACP, therefore, targeting cells possessing exon 3 mutations may be significant in future therapeutic advancements.
RNAi Therapy in Treatments of ACP
sHRNA
Numerous studies have shown that shRNA is a viable method to treat various tumors and cancers [15,22,23]. In tumors such as Wilm’s Tumor, a form of kidney cancer that primarily manifests in children, apoptosis of cancerous cells has been acheived using shRNA to suppress critical exons of the WT1 gene in in vitro and in vivo studies[22,24]. In a study analyzing the effects of shRNA on other WT1 cell lines, a gene that has been found to code for many cancers such as leukemia, lung cancer, and ovarian cancer, plasmids containing shRNA were transfected into B16F10 cells, a cell line associated with melanoma, and incubated for 72 hours[15,22]. In this cell line, shRNA-WT1 was highly effective at decreasing cell viability compared to the control group suggesting that shRNA treatment may be effective for reducing tumor burden[22]. Additionally, when shRNA was used in combination with other therapeutics such as gemcitabine and cisplatin, apoptosis was induced at much higher rates when compared to just gemcitabine and cisplatin treatment[15]. In another study, shRNA was used to target critical exons 5 and 10, and ‘3UTR of the WT1 gene to induce apoptosis in WT1-expressing solid cancer cells of 4 different cell lines(CITE). In HT-1080, a cell line associated with human sarcoma, shRNA inhibiting exon 5 of WT1 induced apoptosis around five times the rate of the control shRNA and shRNA targeting exon 5 of WT1 induced apoptosis at twice the rate of the control shRNA in AZ-521, a cell line associated with human gastric carcinoma[22]. The same study displayed that in HT-1080 cell line, shRNA targeting exon 5 combined with radiotherapy-induced apoptosis at nearly three times the rate of treatment consisting of just radiotherapy[22]. Studies inducing apoptosis in cancerous cells by using shRNA to inhibit exons critical to the gene have been proven in WT1 induced cancers[15,22]. Similar methods of treatment may be used to inhibit critical exons of CTNNB1 in cells to induce apoptosis in cells with exon 3 mutations to treat ACP. Additionally, shRNA treatments could potentially be combined with current methods such as GTR, STR, and STR+XTR to amplify the results of these methods. shRNA is a promising method to treat ACP, and may be a crucial therapeutic intervention in increasing patients’ long-term quality of life.
siRNA
In addition to shRNA, siRNA may be an effective alternative method of combating ACP. Although siRNA is double-stranded and chemically created, and shRNA is vector-based, both siRNA and shRNA have different functions but similar outcomes [25,26,27]. Studies have used siRNA to combat mutations in other cell lines within the WNT pathway [28,29]. siRNA highly inhibited β-Catenin expression in the HCC cell line, a cell line associated with liver cancer[29]. siRNA treatment in MG-63 osteosarcoma cells also proved as a method to silence β-Catenin expression and suppress the invasion and motility of the osteosarcoma cells [28]. Treatments from similar diseases may provide insight into repurposing RNAi therapies to inhibit β-Catenin expression in ACP. No shRNA or siRNA treatment has previously been attempted in vitro or in vivo for the treatment of ACP, and RNAi may be pivotal in treating ACP with decreased patient burden and recurrence rates.
CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), in addition to RNAi therapies, could be an alternative method to treat ACP. CRISPR is a controversial gene therapy method in which DNA can be cut and replaced at will [30,31]. CRISPR has been used in a select number of cancer studies outside of humans and has been successful in reducing and inducing apoptosis in a large percentage of cancer cells [32,33]. To combat bladder cancer, CRISPR was engineered to detect hTERT, a protein that plays a large role in bladder cancer[32]. CRISPR induced apoptosis in specifically bladder cancer 5637 cells and T24 cells without inducing apoptosis in healthy HFF cells[32]. CRISPR has also proven to be effective against cervical cancer, caused by the E6 and E7 oncogenes[33]. A study analyzing the effect of using CRISPR to reduce the expression of the E6 and E7 oncogenes resulted in CRISPR reducing the expression of E6 and E7 by about eight times when compared to controls[33]. Due to the fact that most cases of craniopharyngioma are a mutation of the exon 3 in the CTNNB1 gene, CRISPR may be a viable solution to delete the abnormal mutation and insert a healthy exon 3 after GTR to prevent recurrence [8,9,34]. CRISPR may be the key to treating ACP effectively with minimal side effects.
Discussion
This review discussed two potential therapeutic methods to treat ACP. With unreasonable recurrence rates and significant long-term decrease in quality of life, it is clear the current methods are not effective enough to reduce the long-term burden for patients [2,14].
In an in vivo study analyzing the recurrence rates of colon cancer after surgical treatments in mice, the recurrence rate was 71%, whereas, mice treated with shRNA after surgery displayed a recurrence rate of 16% [23]. With results showing that shRNA could be effective in reducing recurrence rates in other cancers, may be just as effective at reducing recurrence rates of ACP. shRNA can also be used after current methods of treatment such as GTR or STR+XRT to prevent the recurrence of ACP. In a study analyzing the effect of shRNA combined with other treatments on Wilms tumor, shRNA treatment combined with cisplatin and gemcitabine reduced the colonies formed by 97% compared to control colonies and another study displayed that shRNA in combination with radiotherapy inhibited the growth of cervical cancer in vivo [15,35]. However, shRNA and siRNA have been known to result in off-target effects, which can induce apoptosis in non-targeted cells [36,37]. Though off-target effects of RNAi may be concerning, the recurrence of ACP can potentially be life-threatening, affect the endocrine system, and severely reduce the quality of life of patients38. As of today, no testing has been conducted to find the effects of shRNA on recurrence in CTNNB1 cell lines or in ACP models but it has been shown in other diseases such as colon cancer[23]. RNAi and CRISPRi treatments could be used along with current methods to prevent recurrence and could be a pivotal step in improving the long-term quality of life and reducing recurrence rates of ACP.
However, a challenge arises in testing the proposed methods due to a lack of establishment of a solid cell line for ACP. An in vivo mouse study in which exon 3 in the Rathke’s pouch progenitors cells were excised, mice in which β-Catenin was not degraded were produced and contained a tumor very similar to the human ACP (HesxlCre; CTNNB1+flax(ex3))[12]. Cell lines for ACP need to be established soon, but during the absence of advancements in the research of ACP cell lines, RNAi therapies and CRISPR methods can be initially tested with the HesxlCre; CTNNB1+flax(ex3) mice and further tested in vitro when a solid cell line is established.
Using targeted genetic approaches to ACP may not apply to all patients because while a majority of cases are associated with a mutation in exon 3 of CTNNB1, not all cases have a defect in β-catenin. A whole genome sequencing demonstrated that only 11 out of 16 cases of ACP were associated with the mutation in the exon 3 of CTNNB1, and another study found that only 76.1% of 117 cases contained a mutation in exon 3, leading to the conclusion that a mutation of exon 3 in CTNNB1 is not the sole cause of ACP [9,34]. CRISPR and RNAi treatment methods proposed in this review are specifically geared towards ACP caused by a mutation of exon 3 in CTNNB1 and patients diagnosed with ACP without a mutation of exon 3 of CTNNB1 must be treated with other methods.
In recent years, scientists have strived to find cost efficient and simple methods to manipulate DNA. The use of previous methods such as transcription activator-like effector nuclease (TALEN) and zinc-finger nuclease (ZFN) posed many difficulties[39,40]. The costs associated with TALEN and ZFN were unrealistic in therapeutic use and both methods were not nearly as effective as CRISPR [39,40]. CRISPR is a very effectively gene editing method in which DNA can be excised and replaced at will[41].
Although CRISPR can be used to cure many diseases with ease, CRISPR is highly regulated due to moral and legal implications. The main concern lies in CRISPR’s ability to edit embryonic DNA. This can lead to advancements in eugenics, an ideology in which medically uncommon or abnormal genes are eliminated [42,43,44]. On the counterpart, CRISPR could also be used to cure many potent diseases that cannot be treated effectively using methods currently developed. Rules and regulations for gene-editing are already in effect, and in the future, further rules and modifications of rules can be implemented to effectively use CRISPR. Studies outside of humans have shown that CRISPR is very effective in many diseases that cannot be effectively treated today such as bladder cancer [32], cervical cancer [45,46], HIV[45], and even the craniopharyngioma counterpart; PCP [47]. In the future, changes and leniency in gene editing could allow the use of CRISPR to be used against certain diseases. With low associated costs, high convenience, and studies displaying positive results in other diseases, CRISPR could be the primary method in treating ACP.
Conclusion
Adamantinomatous Craniopharyngioma (ACP) is an aggressive tumor primarily found in children and is associated with high recurrence rates and low long-term quality of life. This review proposed two new therapeutic methods in which ACP could be treated to address these issues - RNAi therapy and CRISPR. These methods could benefit patients’ quality of life and end the long term effects associated with current treatments of ACP.
References
- Karavitaki, N., Cudlip, S., Adams, C. B. T. & Wass, J. A. H. Craniopharyngiomas. Endocr. Rev. 27, 371–397 (2006). [CrossRef]
- Dandurand, C., Sepehry, A. A., Asadi Lari, M. H., Akagami, R. & Gooderham, P. Adult Craniopharyngioma: Case Series, Systematic Review, and Meta-Analysis. Neurosurgery 83, 631 (2018). [CrossRef]
- Muller, H. L. Childhood craniopharyngioma. Recent advances in diagnosis, treatment and follow-up. Horm. Res. 69 (2008). [CrossRef]
- Müller, H. L. The Diagnosis and Treatment of Craniopharyngioma. Jinko Mondai Kenkyusho. Nenpo 110, 753–766 (2020). [CrossRef]
- Bunin, G. R. et al. The descriptive epidemiology of craniopharyngioma. J. Neurosurg. 89, 547–551 (1998). [CrossRef]
- Olsson, D. S., Andersson, E., Bryngelsson, I.-L., Nilsson, A. G. & Johannsson, G. Excess Mortality and Morbidity in Patients with Craniopharyngioma, Especially in Patients with Childhood Onset: A Population-Based Study in Sweden. J. Clin. Endocrinol. Metab. 100, 467–474 (2015). [CrossRef]
- Momin, A. A. et al. Descriptive epidemiology of craniopharyngiomas in the United States. Pituitary 24, (2021) . [CrossRef]
- Brastianos, P. et al. GE-05 * EXOME SEQUENCING REVEALS BRAF MUTATIONS IN PAPILLARY CRANIOPHARYNGIOMAS. Neuro-Oncology vol. 16 v97–v97 (2014). [CrossRef]
- He, J. et al. Characterization of novel CTNNB1 mutation in Craniopharyngioma by whole-genome sequencing. Mol. Cancer 20 (2021). [CrossRef]
- Martinez-Barbera, J. P. Molecular and cellular pathogenesis of adamantinomatous craniopharyngioma. Neuropathology and Applied Neurobiology vol. 41 721–732 (2015). [CrossRef]
- Sekine, S. et al. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am. J. Pathol. 161 (2002). [CrossRef]
- Gaston-Massuet, C. et al. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc. Natl. Acad. Sci. U. S. A. 108, 11482–11487 (2011) . [CrossRef]
- Duff, J. et al. Long-term outcomes for surgically resected craniopharyngiomas. Neurosurgery 46, (2000). [CrossRef]
- Karavitaki, N. et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clinical Endocrinology vol. 62 397–409 (2005) . [CrossRef]
- Zapata-Benavides, P. et al. shRNA-WT1 Potentiates Anticancer Effects of Gemcitabine and Cisplatin Against B16F10 Lung Metastases In Vitro and In Vivo. In Vivo 33, (2019). [CrossRef]
- Stripp, D. C. H. et al. Surgery with or without radiation therapy in the management of craniopharyngiomas in children and young adults. International Journal of Radiation Oncology*Biology*Physics vol. 58 714–720 . [CrossRef]
- Fahlbusch, R., Honegger, J., Paulus, W., Huk, W. & Buchfelder, M. Surgical treatment of craniopharyngiomas: experience with 168 patients. Journal of Neurosurgery vol. 90 237–250 (1999). [CrossRef]
- Kalapurakal, J. A., Goldman, S., Hsieh, Y. C., Tomita, T. & Marymont, M. H. Clinical outcome in children with craniopharyngioma treated with primary surgery and radiotherapy deferred until relapse. Med. Pediatr. Oncol. 40, (2003). [CrossRef]
- Huelsken, J. & Behrens, J. The Wnt signalling pathway. Journal of Cell Science vol. 115 3977–3978 (2002). [CrossRef]
- Logan, C. Y. & Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, (2004). [CrossRef]
- Gao, C. et al. Exon 3 mutations of CTNNB1 drive tumorigenesis: a review. Oncotarget 9, 5492 (2018). [CrossRef]
- Tatsumi, N. et al. Wilms’ tumor gene WT1-shRNA as a potent apoptosis-inducing agent for solid tumors. International Journal of Oncology (2008). [CrossRef]
- Hwang, J.-E. et al. Intravenous KITENIN shRNA Injection Suppresses Hepatic Metastasis and Recurrence of Colon Cancer in an Orthotopic Mouse Model. J. Korean Med. Sci. 26, 1439–1445 (2011). [CrossRef]
- Kijima, N. et al. Wilms’ Tumor 1 Is Involved in Tumorigenicity of Glioblastoma by Regulating Cell Proliferation and Apoptosis. Anticancer Res. 34, 61–67 (2014).
- Hu, B. et al. Therapeutic siRNA: state of the art. Signal Transduction and Targeted Therapy 5, 1–25 (2020). [CrossRef]
- Rao, D. D., Vorhies, J. S., Senzer, N. & Nemunaitis, J. siRNA vs. shRNA: similarities and differences. Adv. Drug Deliv. Rev. 61, (2009). [CrossRef]
- Moore, C. B., Guthrie, E. H., Huang, M. T.-H. & Taxman, D. J. Short Hairpin RNA (shRNA): Design, Delivery, and Assessment of Gene Knockdown. Methods Mol. Biol. 629, 141 (2010). [CrossRef]
- Zhang, F., Chen, A., Chen, J., Yu, T. & Guo, F. SiRNA-mediated silencing of beta-catenin suppresses invasion and chemosensitivity to doxorubicin in MG-63 osteosarcoma cells. Asian Pac. J. Cancer Prev. 12, 239–245 (2011).
- siRNA-Mediated β-Catenin Knockdown in Human Hepatoma Cells Results in Decreased Growth and Survival. Neoplasia 9, 951–959 (2007). [CrossRef]
- Ledford, H. CRISPR, the disruptor. Nature Publishing Group UK (2015). [CrossRef]
- Katti, A., Diaz, B. J., Caragine, C. M., Sanjana, N. E. & Dow, L. E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 22, 259–279 (2022). [CrossRef]
- Zhuang, C. et al. Engineered CRISPR/Cas13d Sensing hTERT Selectively Inhibits the Progression of Bladder Cancer. Front Mol Biosci 8, 646412 (2021). [CrossRef]
- Transl. [CrossRef]
- Goschzik, T. et al. Genomic Alterations of Adamantinomatous and Papillary Craniopharyngioma. J. Neuropathol. Exp. Neurol. 76, 126–134 (2017). [CrossRef]
- Qi, L., Xing, L. N., Wei, X. & Song, S. G. Effects of VEGF suppression by small hairpin RNA interference combined with radiotherapy on the growth of cervical cancer. Genet. Mol. Res. 13, (2014). [CrossRef]
- Rao, D. D., Senzer, N., Cleary, M. A. & Nemunaitis, J. Comparative assessment of siRNA and shRNA off target effects: what is slowing clinical development. Cancer Gene Ther. 16, 807–809 (2009). [CrossRef]
- Baek, S. T. et al. Off-target effect of doublecortin family shRNA on neuronal migration associated with endogenous microRNA dysregulation. Neuron 82, 1255–1262 (2014). [CrossRef]
- Jazbinšek, S. et al. Prevalence of Endocrine and Metabolic Comorbidities in a National Cohort of Patients with Craniopharyngioma. HRP 93, 46–57 (2020). [CrossRef]
- Mahfouz, M. M., Piatek, A. & Stewart, C. N., Jr. Genome engineering via TALENs and CRISPR/Cas9 systems: challenges and perspectives. Plant Biotechnol. J. 12, 1006–1014 (2014). [CrossRef]
- Gaj, T., Gersbach, C. A. & Barbas, C. F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, (2013). [CrossRef]
- Doudna, J. A. & Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, (2014). [CrossRef]
- Kevles, D. J. Eugenics and human rights. BMJ 319, 435–438 (1999). [CrossRef]
- Brokowski, C. & Adli, M. CRISPR Ethics: Moral Considerations for Applications of a Powerful Tool. J. Mol. Biol. 431, (2019). [CrossRef]
- Zhang, D. et al. Genome editing with the CRISPR-Cas system: an art, ethics and global regulatory perspective. Plant Biotechnol. J. 18, (2020). [CrossRef]
- Ebina, H., Misawa, N., Kanemura, Y. & Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 3, 1–7 (2013). [CrossRef]
- Zhen, S. et al. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by CRISPR/Cas9. Biochem. Biophys. Res. Commun. 450, 1422–1426 (2014). [CrossRef]
- Targeted Disruption of V600E-Mutant BRAF Gene by CRISPR-Cpf1. Molecular Therapy - Nucleic Acids 8, 450–458 (2017). [CrossRef]
- Zeng, G. et al. ; Sirna-mediated beta-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia (New York, N.Y.). [CrossRef]
- Moon, Randall T. “Wnt/β-Catenin Pathway | Science's STKE.” Wnt/β-Catenin Pathway. Science Signaling. https://www.science.org/doi/10.1126/stke.2712005cm1 (2005).
Figure 1.
Illustration of the canonical WNT Pathway. A representation of the pathway in which WNT ligands are not present (left) where the destruction complex consisting of APC, CK1, Axin, and GSK3β surrounds β-Catenin and guides it through the degradation pathway. A representation of the pathway in which WNT ligands are present (right). When WNT ligands are present, the destruction complex detaches from and releases β-Catenin, allowing nuclear accumulation. β-Catenin then infiltrates the nucleus of the cell, replacing Groucho and activating the TCF. The activation of TCF transcribes the WNT target genes .[19] Created using Biorender.com. .
Figure 1.
Illustration of the canonical WNT Pathway. A representation of the pathway in which WNT ligands are not present (left) where the destruction complex consisting of APC, CK1, Axin, and GSK3β surrounds β-Catenin and guides it through the degradation pathway. A representation of the pathway in which WNT ligands are present (right). When WNT ligands are present, the destruction complex detaches from and releases β-Catenin, allowing nuclear accumulation. β-Catenin then infiltrates the nucleus of the cell, replacing Groucho and activating the TCF. The activation of TCF transcribes the WNT target genes .[19] Created using Biorender.com. .
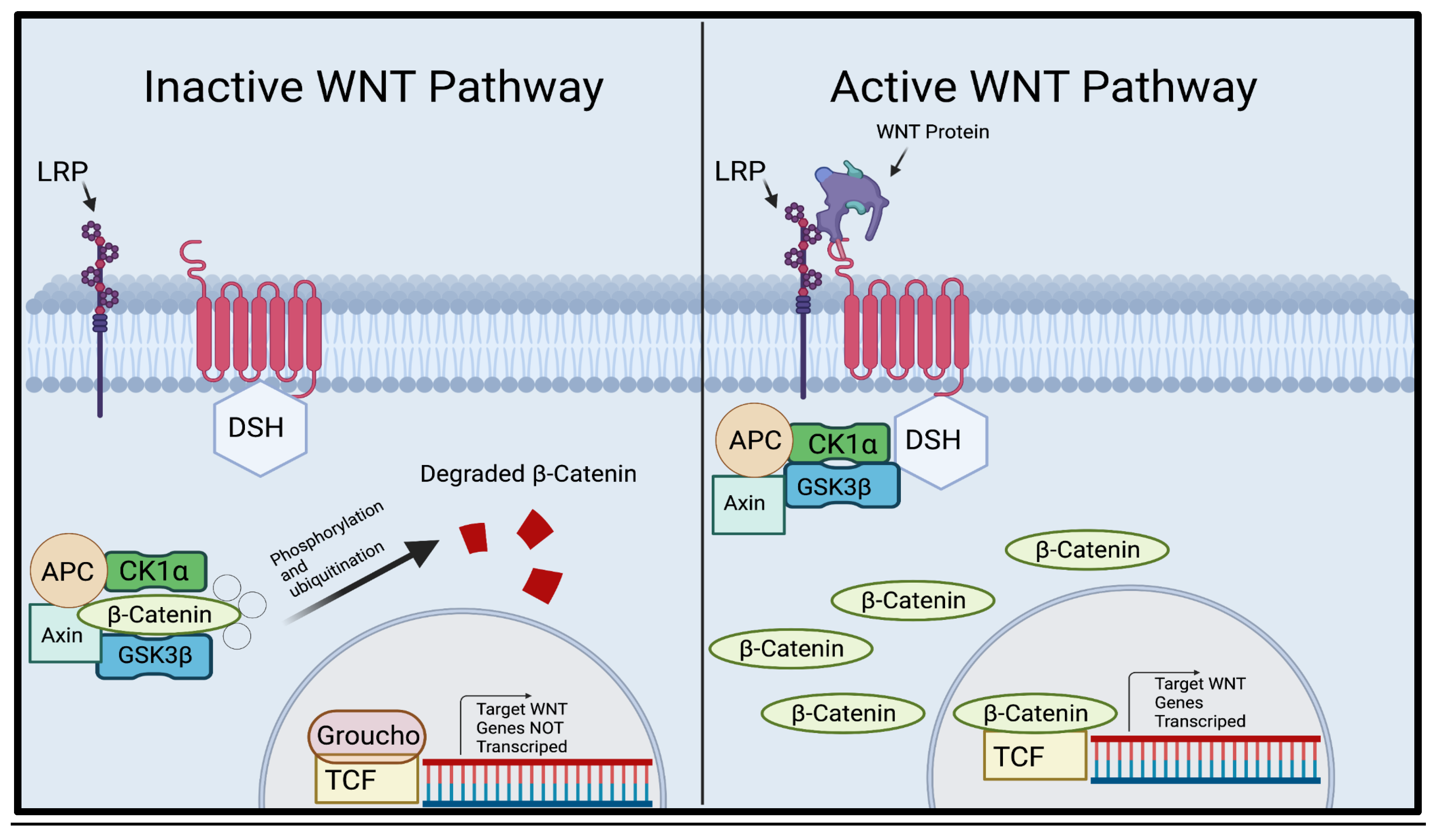
Figure 2.
Illustration of the canonical WNT Pathway with a mutation in exon 3 of CTNNB1. CTNNB1, a gene that codes for β-Catenin, contains an exon known as exon 3, which codes for the residues vital for the phosphorylation of β-Catenin. Without these residues, β-Catenin cannot be properly degraded. As seen in the illustration above, β-Catenin is attached to the destruction complex but cannot be phosphorylated and ubiquitinated due to the mutation of exon 3 in CTNNB1, leading to an overaccumulation of β-Catenin. β-Catenin continues to infiltrate the nucleus and activate the TCF, transcribing WNT genes and leading to uncontrolled cell proliferation.[21] Created using BioRender.com.
Figure 2.
Illustration of the canonical WNT Pathway with a mutation in exon 3 of CTNNB1. CTNNB1, a gene that codes for β-Catenin, contains an exon known as exon 3, which codes for the residues vital for the phosphorylation of β-Catenin. Without these residues, β-Catenin cannot be properly degraded. As seen in the illustration above, β-Catenin is attached to the destruction complex but cannot be phosphorylated and ubiquitinated due to the mutation of exon 3 in CTNNB1, leading to an overaccumulation of β-Catenin. β-Catenin continues to infiltrate the nucleus and activate the TCF, transcribing WNT genes and leading to uncontrolled cell proliferation.[21] Created using BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Submitted:
27 March 2024
Posted:
01 April 2024
You are already at the latest version
Alerts
This version is not peer-reviewed
Submitted:
27 March 2024
Posted:
01 April 2024
You are already at the latest version
Alerts
Abstract
Adamantinomatous Craniopharyngioma (ACP) is a rare epithelial tumor located in the craniopharyngeal duct, often associated with high morbidity and a high long-term recurrence rate. ACP is predominantly diagnosed in children below the age of 15 and accounts for a majority of Craniopharyngioma cases in the United States. It is most commonly caused by a mutation in exon 3 of the CTNNB1 gene, which promotes overaccumulation of β-Catenin in the WNT pathway. As β-Catenin accumulates, it over-activates the transcription factor, causing an over-transcription of WNT proteins and uncontrolled cell growth. Current methods of treatment for ACP are centered around surgical intervention and radiotherapy, including gross-total resection (GTR), subtotal resection (STR), and a combination of subtotal resection and adjuvant radiotherapy (STR+XTR). These methods of treatment are associated with high risk due to the proximity of ACP to critical structures near the craniopharyngeal duct. High recurrence rates and morbidity are closely associated with current methods of treatment and other methods have yet to be attempted in craniopharyngioma. This review will explore two possible methods of non-surgical and non-radiological therapeutic interventions. The use of RNAi therapy and CRISPR may provide insight and be a potential way to treat ACP with increased quality of life and decreased recurrence rates.
Keywords:
Subject: Biology and Life Sciences - Neuroscience and Neurology
Introduction
Craniopharyngioma is an aggressive benign tumor with a global recurrence rate of nearly 27% and may be associated with a highly decreased quality of life [1,2]. Although craniopharyngioma is a WHO class I benign tumor, growth can be unpredictable, affecting the sellar region and surrounding areas containing critical structures such as the optic chiasma, the pituitary gland, and the hypothalamus[3]*. Damage to these areas commonly manifests symptoms such as endocrine deficiencies, visual impairments, growth retardations, nausea, weight gain, and headaches [4]*. Age at diagnosis is represented by a bimodal peak, one peak at 5-15 years of age and another peak at 50-75 years of age [5,1,6].
Craniopharyngioma presents in two histopathological forms: adamantinomatous craniopharyngioma (ACP) and papillary craniopharyngioma (PCP). ACP is most commonly observed in children under the age of 15 but can also be observed in adults, whereas PCP is almost exclusively diagnosed in patients above the age of 30[7]. ACP differs histologically and pathologically from PCP. ACP is characterized by the nucleocytoplasmic accumulation of β-Catenin [8,9]. β-Catenin then over-activates the WNT pathway, causing uncontrolled proliferation [10,11,12]. On the other hand, PCP is associated with the mutation of BRAF V600e[13]. Despite the differences between ACP and PCP, outcomes following treatment for both tumors do not differ[13].
Two major problems with craniopharyngioma are morbidity and recurrence rates post-treatment. In a study analyzing the symptoms of 121 cases of craniopharyngioma in a follow-up study 10 years after initial surgery, patients had a 90% survival rate but a 48% probability of experiencing major visual field defects, 39% probability of experiencing hyperphagia or obesity, and a high probability of experiencing other major complications 14. Additionally, in a systematic review of 765 craniopharyngioma cases, the aggregate global rate of recurrence was 26.2%[2]. Currently, three methods of treatment for craniopharyngioma are prominently used; radiotherapy, surgery, and a combination of both methods.
The same meta-study reviewed the effects of the three main methods of treatment: GTR (gross total resection), STR (subtotal resection), and a combination of STR+XRT (adjuvant radiotherapy)[2,15]. In a three year follow-up, patients treated with GTR had a recurrence rate of 17%, patients treated with STR+XRT had a recurrence rate of 27%, and patients treated with STR had a recurrence rate of 45%, suggesting that GTR may be the optimal method to reduce the chances of recurrence [2]. Although GTR results in the lowest recurrence rates, there is a high risk of long-term complications such as panhypopituitarism, diabetes insipidus, hypothalamic obesity, and cognitive deficits, leading surgeons to predominantly treat the cases using STR and STR+XTR [16,17,18]. Because of the high risk of long-term complications and high recurrence rates presented by current methods of treatment, the need for a new treatment method is imperative. Therefore, this review will suggest RNA interference and CRISPR as alternative methods to target ACP, potentially without long-term complications.
β-Catenin, WNT Pathway, and the Cause of Adamaninomatous Craniopharyngioma
The WNT pathway is a critical pathway used in many vital processes such as cell proliferation and cell behavior [20]. When WNT ligands (secreted glycoproteins) are not present, β-Catenin attaches itself to a destruction complex consisting of Axin, APC, and GSK3β, and certain residues are produced by exon 3 in CTNNB1, the gene that codes for β-Catenin, and β-Catenin is phosphorylated. After phosphorylation, β-Catenin is rapidly degraded and destroyed by the destruction complex, thus, preventing unnecessary growth (Figure 1) [10,19]. When WNT ligands bind to frizzled, Dishevelled(dsh) is activated, which attracts GBP/Frat-1 and GSK3β is removed from the destruction compound [8,17,48]. The removal of GSK3β from the destruction compound prevents the ubiquitination of CTNNB1, and the stable β-Catenin penetrates the cell nucleus, which binds to the LEF/TCF transcription factors. Then, WNT target genes are transcribed, causing cell proliferation [10,19].
ACP is most commonly associated with the mutation of exon 3 in the CTNNB1 gene[8,9]. A whole genome sequencing study demonstrated that 68.75% of ACP cases presented a mutation of the exon 3 of the CTNNB1 gene and an exome sequencing study of ACP demonstrated that 91.7% of ACP cases were associated with the mutation of exon 3 in CTNNB1[8,9]. Furthermore, a mouse study demonstrated that the overactivation of the WNT pathway, created by excising exon 3 of CTNNB1 in pituitary/progenitor stem cells, resulted in either death or the development of tumors that closely resembled human ACP[12]. The amino acid sites critical for the phosphorylation of β-Catenin are encoded by exon 3 of CTNNB1 [21] (Figure 2). It is evident that exon 3 may play an important role in the development of ACP, therefore, targeting cells possessing exon 3 mutations may be significant in future therapeutic advancements.
RNAi Therapy in Treatments of ACP
sHRNA
Numerous studies have shown that shRNA is a viable method to treat various tumors and cancers [15,22,23]. In tumors such as Wilm’s Tumor, a form of kidney cancer that primarily manifests in children, apoptosis of cancerous cells has been acheived using shRNA to suppress critical exons of the WT1 gene in in vitro and in vivo studies[22,24]. In a study analyzing the effects of shRNA on other WT1 cell lines, a gene that has been found to code for many cancers such as leukemia, lung cancer, and ovarian cancer, plasmids containing shRNA were transfected into B16F10 cells, a cell line associated with melanoma, and incubated for 72 hours[15,22]. In this cell line, shRNA-WT1 was highly effective at decreasing cell viability compared to the control group suggesting that shRNA treatment may be effective for reducing tumor burden[22]. Additionally, when shRNA was used in combination with other therapeutics such as gemcitabine and cisplatin, apoptosis was induced at much higher rates when compared to just gemcitabine and cisplatin treatment[15]. In another study, shRNA was used to target critical exons 5 and 10, and ‘3UTR of the WT1 gene to induce apoptosis in WT1-expressing solid cancer cells of 4 different cell lines(CITE). In HT-1080, a cell line associated with human sarcoma, shRNA inhibiting exon 5 of WT1 induced apoptosis around five times the rate of the control shRNA and shRNA targeting exon 5 of WT1 induced apoptosis at twice the rate of the control shRNA in AZ-521, a cell line associated with human gastric carcinoma[22]. The same study displayed that in HT-1080 cell line, shRNA targeting exon 5 combined with radiotherapy-induced apoptosis at nearly three times the rate of treatment consisting of just radiotherapy[22]. Studies inducing apoptosis in cancerous cells by using shRNA to inhibit exons critical to the gene have been proven in WT1 induced cancers[15,22]. Similar methods of treatment may be used to inhibit critical exons of CTNNB1 in cells to induce apoptosis in cells with exon 3 mutations to treat ACP. Additionally, shRNA treatments could potentially be combined with current methods such as GTR, STR, and STR+XTR to amplify the results of these methods. shRNA is a promising method to treat ACP, and may be a crucial therapeutic intervention in increasing patients’ long-term quality of life.
siRNA
In addition to shRNA, siRNA may be an effective alternative method of combating ACP. Although siRNA is double-stranded and chemically created, and shRNA is vector-based, both siRNA and shRNA have different functions but similar outcomes [25,26,27]. Studies have used siRNA to combat mutations in other cell lines within the WNT pathway [28,29]. siRNA highly inhibited β-Catenin expression in the HCC cell line, a cell line associated with liver cancer[29]. siRNA treatment in MG-63 osteosarcoma cells also proved as a method to silence β-Catenin expression and suppress the invasion and motility of the osteosarcoma cells [28]. Treatments from similar diseases may provide insight into repurposing RNAi therapies to inhibit β-Catenin expression in ACP. No shRNA or siRNA treatment has previously been attempted in vitro or in vivo for the treatment of ACP, and RNAi may be pivotal in treating ACP with decreased patient burden and recurrence rates.
CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), in addition to RNAi therapies, could be an alternative method to treat ACP. CRISPR is a controversial gene therapy method in which DNA can be cut and replaced at will [30,31]. CRISPR has been used in a select number of cancer studies outside of humans and has been successful in reducing and inducing apoptosis in a large percentage of cancer cells [32,33]. To combat bladder cancer, CRISPR was engineered to detect hTERT, a protein that plays a large role in bladder cancer[32]. CRISPR induced apoptosis in specifically bladder cancer 5637 cells and T24 cells without inducing apoptosis in healthy HFF cells[32]. CRISPR has also proven to be effective against cervical cancer, caused by the E6 and E7 oncogenes[33]. A study analyzing the effect of using CRISPR to reduce the expression of the E6 and E7 oncogenes resulted in CRISPR reducing the expression of E6 and E7 by about eight times when compared to controls[33]. Due to the fact that most cases of craniopharyngioma are a mutation of the exon 3 in the CTNNB1 gene, CRISPR may be a viable solution to delete the abnormal mutation and insert a healthy exon 3 after GTR to prevent recurrence [8,9,34]. CRISPR may be the key to treating ACP effectively with minimal side effects.
Discussion
This review discussed two potential therapeutic methods to treat ACP. With unreasonable recurrence rates and significant long-term decrease in quality of life, it is clear the current methods are not effective enough to reduce the long-term burden for patients [2,14].
In an in vivo study analyzing the recurrence rates of colon cancer after surgical treatments in mice, the recurrence rate was 71%, whereas, mice treated with shRNA after surgery displayed a recurrence rate of 16% [23]. With results showing that shRNA could be effective in reducing recurrence rates in other cancers, may be just as effective at reducing recurrence rates of ACP. shRNA can also be used after current methods of treatment such as GTR or STR+XRT to prevent the recurrence of ACP. In a study analyzing the effect of shRNA combined with other treatments on Wilms tumor, shRNA treatment combined with cisplatin and gemcitabine reduced the colonies formed by 97% compared to control colonies and another study displayed that shRNA in combination with radiotherapy inhibited the growth of cervical cancer in vivo [15,35]. However, shRNA and siRNA have been known to result in off-target effects, which can induce apoptosis in non-targeted cells [36,37]. Though off-target effects of RNAi may be concerning, the recurrence of ACP can potentially be life-threatening, affect the endocrine system, and severely reduce the quality of life of patients38. As of today, no testing has been conducted to find the effects of shRNA on recurrence in CTNNB1 cell lines or in ACP models but it has been shown in other diseases such as colon cancer[23]. RNAi and CRISPRi treatments could be used along with current methods to prevent recurrence and could be a pivotal step in improving the long-term quality of life and reducing recurrence rates of ACP.
However, a challenge arises in testing the proposed methods due to a lack of establishment of a solid cell line for ACP. An in vivo mouse study in which exon 3 in the Rathke’s pouch progenitors cells were excised, mice in which β-Catenin was not degraded were produced and contained a tumor very similar to the human ACP (HesxlCre; CTNNB1+flax(ex3))[12]. Cell lines for ACP need to be established soon, but during the absence of advancements in the research of ACP cell lines, RNAi therapies and CRISPR methods can be initially tested with the HesxlCre; CTNNB1+flax(ex3) mice and further tested in vitro when a solid cell line is established.
Using targeted genetic approaches to ACP may not apply to all patients because while a majority of cases are associated with a mutation in exon 3 of CTNNB1, not all cases have a defect in β-catenin. A whole genome sequencing demonstrated that only 11 out of 16 cases of ACP were associated with the mutation in the exon 3 of CTNNB1, and another study found that only 76.1% of 117 cases contained a mutation in exon 3, leading to the conclusion that a mutation of exon 3 in CTNNB1 is not the sole cause of ACP [9,34]. CRISPR and RNAi treatment methods proposed in this review are specifically geared towards ACP caused by a mutation of exon 3 in CTNNB1 and patients diagnosed with ACP without a mutation of exon 3 of CTNNB1 must be treated with other methods.
In recent years, scientists have strived to find cost efficient and simple methods to manipulate DNA. The use of previous methods such as transcription activator-like effector nuclease (TALEN) and zinc-finger nuclease (ZFN) posed many difficulties[39,40]. The costs associated with TALEN and ZFN were unrealistic in therapeutic use and both methods were not nearly as effective as CRISPR [39,40]. CRISPR is a very effectively gene editing method in which DNA can be excised and replaced at will[41].
Although CRISPR can be used to cure many diseases with ease, CRISPR is highly regulated due to moral and legal implications. The main concern lies in CRISPR’s ability to edit embryonic DNA. This can lead to advancements in eugenics, an ideology in which medically uncommon or abnormal genes are eliminated [42,43,44]. On the counterpart, CRISPR could also be used to cure many potent diseases that cannot be treated effectively using methods currently developed. Rules and regulations for gene-editing are already in effect, and in the future, further rules and modifications of rules can be implemented to effectively use CRISPR. Studies outside of humans have shown that CRISPR is very effective in many diseases that cannot be effectively treated today such as bladder cancer [32], cervical cancer [45,46], HIV[45], and even the craniopharyngioma counterpart; PCP [47]. In the future, changes and leniency in gene editing could allow the use of CRISPR to be used against certain diseases. With low associated costs, high convenience, and studies displaying positive results in other diseases, CRISPR could be the primary method in treating ACP.
Conclusion
Adamantinomatous Craniopharyngioma (ACP) is an aggressive tumor primarily found in children and is associated with high recurrence rates and low long-term quality of life. This review proposed two new therapeutic methods in which ACP could be treated to address these issues - RNAi therapy and CRISPR. These methods could benefit patients’ quality of life and end the long term effects associated with current treatments of ACP.
References
- Karavitaki, N., Cudlip, S., Adams, C. B. T. & Wass, J. A. H. Craniopharyngiomas. Endocr. Rev. 27, 371–397 (2006). [CrossRef]
- Dandurand, C., Sepehry, A. A., Asadi Lari, M. H., Akagami, R. & Gooderham, P. Adult Craniopharyngioma: Case Series, Systematic Review, and Meta-Analysis. Neurosurgery 83, 631 (2018). [CrossRef]
- Muller, H. L. Childhood craniopharyngioma. Recent advances in diagnosis, treatment and follow-up. Horm. Res. 69 (2008). [CrossRef]
- Müller, H. L. The Diagnosis and Treatment of Craniopharyngioma. Jinko Mondai Kenkyusho. Nenpo 110, 753–766 (2020). [CrossRef]
- Bunin, G. R. et al. The descriptive epidemiology of craniopharyngioma. J. Neurosurg. 89, 547–551 (1998). [CrossRef]
- Olsson, D. S., Andersson, E., Bryngelsson, I.-L., Nilsson, A. G. & Johannsson, G. Excess Mortality and Morbidity in Patients with Craniopharyngioma, Especially in Patients with Childhood Onset: A Population-Based Study in Sweden. J. Clin. Endocrinol. Metab. 100, 467–474 (2015). [CrossRef]
- Momin, A. A. et al. Descriptive epidemiology of craniopharyngiomas in the United States. Pituitary 24, (2021) . [CrossRef]
- Brastianos, P. et al. GE-05 * EXOME SEQUENCING REVEALS BRAF MUTATIONS IN PAPILLARY CRANIOPHARYNGIOMAS. Neuro-Oncology vol. 16 v97–v97 (2014). [CrossRef]
- He, J. et al. Characterization of novel CTNNB1 mutation in Craniopharyngioma by whole-genome sequencing. Mol. Cancer 20 (2021). [CrossRef]
- Martinez-Barbera, J. P. Molecular and cellular pathogenesis of adamantinomatous craniopharyngioma. Neuropathology and Applied Neurobiology vol. 41 721–732 (2015). [CrossRef]
- Sekine, S. et al. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am. J. Pathol. 161 (2002). [CrossRef]
- Gaston-Massuet, C. et al. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc. Natl. Acad. Sci. U. S. A. 108, 11482–11487 (2011) . [CrossRef]
- Duff, J. et al. Long-term outcomes for surgically resected craniopharyngiomas. Neurosurgery 46, (2000). [CrossRef]
- Karavitaki, N. et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clinical Endocrinology vol. 62 397–409 (2005) . [CrossRef]
- Zapata-Benavides, P. et al. shRNA-WT1 Potentiates Anticancer Effects of Gemcitabine and Cisplatin Against B16F10 Lung Metastases In Vitro and In Vivo. In Vivo 33, (2019). [CrossRef]
- Stripp, D. C. H. et al. Surgery with or without radiation therapy in the management of craniopharyngiomas in children and young adults. International Journal of Radiation Oncology*Biology*Physics vol. 58 714–720 . [CrossRef]
- Fahlbusch, R., Honegger, J., Paulus, W., Huk, W. & Buchfelder, M. Surgical treatment of craniopharyngiomas: experience with 168 patients. Journal of Neurosurgery vol. 90 237–250 (1999). [CrossRef]
- Kalapurakal, J. A., Goldman, S., Hsieh, Y. C., Tomita, T. & Marymont, M. H. Clinical outcome in children with craniopharyngioma treated with primary surgery and radiotherapy deferred until relapse. Med. Pediatr. Oncol. 40, (2003). [CrossRef]
- Huelsken, J. & Behrens, J. The Wnt signalling pathway. Journal of Cell Science vol. 115 3977–3978 (2002). [CrossRef]
- Logan, C. Y. & Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, (2004). [CrossRef]
- Gao, C. et al. Exon 3 mutations of CTNNB1 drive tumorigenesis: a review. Oncotarget 9, 5492 (2018). [CrossRef]
- Tatsumi, N. et al. Wilms’ tumor gene WT1-shRNA as a potent apoptosis-inducing agent for solid tumors. International Journal of Oncology (2008). [CrossRef]
- Hwang, J.-E. et al. Intravenous KITENIN shRNA Injection Suppresses Hepatic Metastasis and Recurrence of Colon Cancer in an Orthotopic Mouse Model. J. Korean Med. Sci. 26, 1439–1445 (2011). [CrossRef]
- Kijima, N. et al. Wilms’ Tumor 1 Is Involved in Tumorigenicity of Glioblastoma by Regulating Cell Proliferation and Apoptosis. Anticancer Res. 34, 61–67 (2014).
- Hu, B. et al. Therapeutic siRNA: state of the art. Signal Transduction and Targeted Therapy 5, 1–25 (2020). [CrossRef]
- Rao, D. D., Vorhies, J. S., Senzer, N. & Nemunaitis, J. siRNA vs. shRNA: similarities and differences. Adv. Drug Deliv. Rev. 61, (2009). [CrossRef]
- Moore, C. B., Guthrie, E. H., Huang, M. T.-H. & Taxman, D. J. Short Hairpin RNA (shRNA): Design, Delivery, and Assessment of Gene Knockdown. Methods Mol. Biol. 629, 141 (2010). [CrossRef]
- Zhang, F., Chen, A., Chen, J., Yu, T. & Guo, F. SiRNA-mediated silencing of beta-catenin suppresses invasion and chemosensitivity to doxorubicin in MG-63 osteosarcoma cells. Asian Pac. J. Cancer Prev. 12, 239–245 (2011).
- siRNA-Mediated β-Catenin Knockdown in Human Hepatoma Cells Results in Decreased Growth and Survival. Neoplasia 9, 951–959 (2007). [CrossRef]
- Ledford, H. CRISPR, the disruptor. Nature Publishing Group UK (2015). [CrossRef]
- Katti, A., Diaz, B. J., Caragine, C. M., Sanjana, N. E. & Dow, L. E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 22, 259–279 (2022). [CrossRef]
- Zhuang, C. et al. Engineered CRISPR/Cas13d Sensing hTERT Selectively Inhibits the Progression of Bladder Cancer. Front Mol Biosci 8, 646412 (2021). [CrossRef]
- Transl. [CrossRef]
- Goschzik, T. et al. Genomic Alterations of Adamantinomatous and Papillary Craniopharyngioma. J. Neuropathol. Exp. Neurol. 76, 126–134 (2017). [CrossRef]
- Qi, L., Xing, L. N., Wei, X. & Song, S. G. Effects of VEGF suppression by small hairpin RNA interference combined with radiotherapy on the growth of cervical cancer. Genet. Mol. Res. 13, (2014). [CrossRef]
- Rao, D. D., Senzer, N., Cleary, M. A. & Nemunaitis, J. Comparative assessment of siRNA and shRNA off target effects: what is slowing clinical development. Cancer Gene Ther. 16, 807–809 (2009). [CrossRef]
- Baek, S. T. et al. Off-target effect of doublecortin family shRNA on neuronal migration associated with endogenous microRNA dysregulation. Neuron 82, 1255–1262 (2014). [CrossRef]
- Jazbinšek, S. et al. Prevalence of Endocrine and Metabolic Comorbidities in a National Cohort of Patients with Craniopharyngioma. HRP 93, 46–57 (2020). [CrossRef]
- Mahfouz, M. M., Piatek, A. & Stewart, C. N., Jr. Genome engineering via TALENs and CRISPR/Cas9 systems: challenges and perspectives. Plant Biotechnol. J. 12, 1006–1014 (2014). [CrossRef]
- Gaj, T., Gersbach, C. A. & Barbas, C. F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, (2013). [CrossRef]
- Doudna, J. A. & Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, (2014). [CrossRef]
- Kevles, D. J. Eugenics and human rights. BMJ 319, 435–438 (1999). [CrossRef]
- Brokowski, C. & Adli, M. CRISPR Ethics: Moral Considerations for Applications of a Powerful Tool. J. Mol. Biol. 431, (2019). [CrossRef]
- Zhang, D. et al. Genome editing with the CRISPR-Cas system: an art, ethics and global regulatory perspective. Plant Biotechnol. J. 18, (2020). [CrossRef]
- Ebina, H., Misawa, N., Kanemura, Y. & Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 3, 1–7 (2013). [CrossRef]
- Zhen, S. et al. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by CRISPR/Cas9. Biochem. Biophys. Res. Commun. 450, 1422–1426 (2014). [CrossRef]
- Targeted Disruption of V600E-Mutant BRAF Gene by CRISPR-Cpf1. Molecular Therapy - Nucleic Acids 8, 450–458 (2017). [CrossRef]
- Zeng, G. et al. ; Sirna-mediated beta-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia (New York, N.Y.). [CrossRef]
- Moon, Randall T. “Wnt/β-Catenin Pathway | Science's STKE.” Wnt/β-Catenin Pathway. Science Signaling. https://www.science.org/doi/10.1126/stke.2712005cm1 (2005).
Figure 1.
Illustration of the canonical WNT Pathway. A representation of the pathway in which WNT ligands are not present (left) where the destruction complex consisting of APC, CK1, Axin, and GSK3β surrounds β-Catenin and guides it through the degradation pathway. A representation of the pathway in which WNT ligands are present (right). When WNT ligands are present, the destruction complex detaches from and releases β-Catenin, allowing nuclear accumulation. β-Catenin then infiltrates the nucleus of the cell, replacing Groucho and activating the TCF. The activation of TCF transcribes the WNT target genes .[19] Created using Biorender.com. .
Figure 1.
Illustration of the canonical WNT Pathway. A representation of the pathway in which WNT ligands are not present (left) where the destruction complex consisting of APC, CK1, Axin, and GSK3β surrounds β-Catenin and guides it through the degradation pathway. A representation of the pathway in which WNT ligands are present (right). When WNT ligands are present, the destruction complex detaches from and releases β-Catenin, allowing nuclear accumulation. β-Catenin then infiltrates the nucleus of the cell, replacing Groucho and activating the TCF. The activation of TCF transcribes the WNT target genes .[19] Created using Biorender.com. .
Figure 2.
Illustration of the canonical WNT Pathway with a mutation in exon 3 of CTNNB1. CTNNB1, a gene that codes for β-Catenin, contains an exon known as exon 3, which codes for the residues vital for the phosphorylation of β-Catenin. Without these residues, β-Catenin cannot be properly degraded. As seen in the illustration above, β-Catenin is attached to the destruction complex but cannot be phosphorylated and ubiquitinated due to the mutation of exon 3 in CTNNB1, leading to an overaccumulation of β-Catenin. β-Catenin continues to infiltrate the nucleus and activate the TCF, transcribing WNT genes and leading to uncontrolled cell proliferation.[21] Created using BioRender.com.
Figure 2.
Illustration of the canonical WNT Pathway with a mutation in exon 3 of CTNNB1. CTNNB1, a gene that codes for β-Catenin, contains an exon known as exon 3, which codes for the residues vital for the phosphorylation of β-Catenin. Without these residues, β-Catenin cannot be properly degraded. As seen in the illustration above, β-Catenin is attached to the destruction complex but cannot be phosphorylated and ubiquitinated due to the mutation of exon 3 in CTNNB1, leading to an overaccumulation of β-Catenin. β-Catenin continues to infiltrate the nucleus and activate the TCF, transcribing WNT genes and leading to uncontrolled cell proliferation.[21] Created using BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Advanced Radiation Techniques in the Treatment of Esthesioneuroblastoma: A 7-Year Single-Institution’s Clinical Experience
Jakob Liermann
et al.
Cancers,
2018
Gamma Knife Radiosurgery as a Salvage Treatment for Nasopharyngeal Carcinoma with Skull Base and Intracranial Invasion (T4b)
Shao-Ang Chu
et al.
Life,
2022
The Impact of an Ultra-Early Postoperative MRI on Treatment of Lower Grade Glioma
Andrej Pala
et al.
Cancers,
2021



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated