
Submitted:
29 March 2024
Posted:
02 April 2024
You are already at the latest version
Abstract
Legionella pneumophila can cause a large panel of symptoms beside the classic pneumonia presentation. We present here a case of fatal nosocomial cellulitis in an immunocompromised patient followed, a year later, by a second case of legionnaires’ disease in the same ward. While the first one was easily assumed as nosocomial based on the date of symptoms onset, the other case needed to rely on clear typing results to be either assigned as nosocomial and related to the same environmental source as the first one, or community acquired. To untangle this specific question, we applied Core-genome MultiLocus Typing (MLST), whole-genome Single Nucleotide Polymorphism and whole-genome MLST methods to a collection of 36 Belgian and 41 international sequence-type 1 (ST1) isolates using both thresholds recommended in the literature and tailored threshold based on local epidemiological data. Based on the thresholds applied to cluster isolates together, the three methods gave different results and no firm conclusion about the nosocomial setting of the second case could been drawn. Our data highlight that despite promising results in the study of outbreaks and for large scale epidemiological investigations, NGS typing methods applied to ST1 outbreak investigation still need standardization regarding both wet-lab protocols and bioinformatics. A deeper evaluation of L. pneumophila evolutionary clock is also required to increase our understanding of genomic differences between isolates sampled during a clinical infection and in environment.
Keywords:
Legionella pneumophila
; Legionella pneumophila ST1
; WGS
; genomic typing
; wgMLST
; cgMLST
; wgSNP
; nosocomial
; hygiene investigation
1. Case report/Background
Legionella pneumophila (L. pneumophila) is a waterborne pathogen, ubiquitously infecting patients via water reservoirs and mostly known to cause mild to severe pneumonia. It can also be involved in extra-pulmonary infections such as cardiologic or neurological, hematopoietic disorders, hepatitis or cellulitis [1,2]. Infection episodes are mostly community-acquired and sporadic, but clusters and epidemics often happen despite a tight control of L. pneumophila ratio in water systems [3,4]. In Belgium, each case leads to an environmental investigation by public health institutions to identify the source of contamination and prevent epidemics.
1.1. Case 1
A patient (P1) was hospitalized for deterioration of general status, in a secondary hospital of Wallonia, Belgium in October 2019, further mentioned as H1. The patient was also treated for a metastatic cancer for two years, recently complicated by an episode of febrile neutropenia. At day 16, the patient developed signs of sepsis and large spectrum antibiotics (glycopeptides and third generation cephalosporin) were initiated. Within 24 hours, they were admitted in intensive care unit as they became hypoxemic, developed a spontaneous and rapidly progressing abdominal wall cellulitis and signs of systemic shock. The X-ray performed at that time showed bilateral condensing infiltrates with right pleurisy. In this context, a urinary L. pneumophila antigen test was performed and turned out to be positive. Interestingly it was also the case of a liquid sample punctured in a small third space close to the cellulitis. Moxifloxacin was immediately added to treatment, but the patient died a few hours later.
Four clinical samples were sent to the Belgian National reference center (NRC) for Legionella pneumophila for culture and PCR detection: an endotracheal aspirate (ETA), a pleural fluid, an ascites fluid and a cutaneous biopsy. All turned out positive for L. pneumophila both by PCR and culture. The isolates were all serogroup 1 (Sg1) and sequence-based typing (SBT) sequence-type (ST) 1.
As the patient had no recent travel history, no activity suggesting a contamination via a recreational water system and was hospitalized for more than two weeks before the beginning of the symptoms, a diagnostic of nosocomial infection was retained.
The water distribution system of the patient’s room was sampled. The culture of the sink’s hot and cold-water systems samples reached 50 000 unit forming colonies (UFC)/L and 20 000 UFC/L of L. pneumophila Sg1, respectively, and confirmed to be ST1 by the NRC. The water system was then decontaminated following up-to-date protocols.
1.2. Case 2
One year later, another immunocompromised patient (P2) was hospitalized in the same ward. At day 10 after hospitalization, he was diagnosed with a clinical pneumonia caused by L. pneumophila, which was ST1 according to SBT (BAL isolate). As 10 days is the usual breakpoint used for nosocomial onset, an environmental investigation was set up. P2 did not stay in the same bedroom than P1 and two isolates of L. pneumophila ST1 were cultured from the water systems of this bedroom. While the link between environmental and clinical isolates was, here, difficult to clarify, the additional question of a link between the two clinical cases was triggered as the persistence of a strain in the hospital environment is a well described possibility.
Nowadays, when an investigation is set up, SBT is the reference technique for comparing L. pneumophila isolates. But, as illustrated by the two cases described above, when the isolates investigated belong to ST1, the epidemiological link can rarely be confirmed [9]. In Belgium, 19% of L. pneumophila infections are related to Sg1 ST1, one of the five major STs found worldwide [3,4,5,6]. As opposed to the four other major lineages of L. pneumophila (ST23, ST37, ST47 and ST62), ST1 is ubiquitously found in the environment and very often isolated from environmental investigations following both community-acquired (CA) and nosocomial L. pneumophila cases [5,6]. In addition, Legionella forms biofilms and lives at slow pace until factors favoring its multiplication occur [7,8], then the epidemiological link between the source and the patient P2 was only assumed. We thus decided to investigate the correlation between the isolates from both episodes by core-genome (cg) MultiLocus Sequence Typing (MLST), to extend the number of alleles included in the comparison, using the pattern designed by Moran-Gilad et al. [10] that serves as a reference for many European studies on Legionella (Table 1).
Three out of four clinical samples from P1, the two environmental matching isolates and the isolates found in the environmental investigations of P2 clustered together when applying a maximum 4 allelic differences (AD) between two isolates [10] (Figure 1A). The clinical isolate from P2 is 5 allelic differences away from the closest isolates, putting it at the limit of being included the cluster. Interestingly, the P1 isolate LEG1116 is 6 allelic differences away from the closest isolate, 9 to 10 AD from isolates sampled from the same patient the very same day and 8AD away from the clinical isolate of P2. Consequently, along with the practical question of a possible link between these two specific cases trough the persistence of a strain in the ward environment [7], the question of the adequate technique and the adequate threshold to infer reliable conclusions regarding ST1 isolates of L. pneumophila relationship was raised.
Since NGS has been developed, its use as high-resolution typing tool to distinguish closely related isolates has contributed to improve infection control and outbreak management for a wide range of bacterial pathogens [9,10,11,12,13,14,15]. Applied to L. pneumophila , core-genome, whole-genome (wg) Single Nucleotide Polymorphism (SNP) and wg and cg-Multi-Locus Sequence Typing have been reported as showing good discrimination performances [9,10,15,16,17]. Nevertheless, issues remain for highly represented STs like ST1 [7,10,13,14]. Indeed, ST1 is known to be both genetically highly conserved and ubiquitous, but its population structure remains poorly described [18]. We carried out a mini review of L. pneumophila genomic studies conducted since 2015 in order to explore the methods and threshold for clustering used to investigate both outbreaks and long-term surveillances to identify which to apply to our specific question (Table 1).

Table 1.
Mini review of the literature about L. pneumophila genomic studies from 2015.
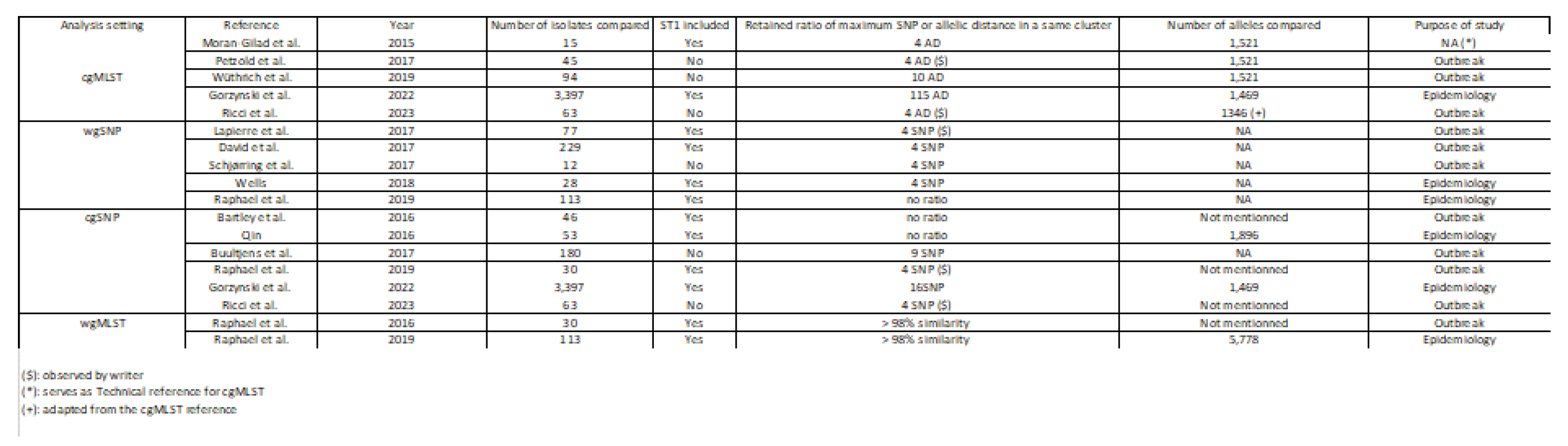
cgMLST of a panel of ST1.
As the pattern for cgMLST with the proposed threshold of 4 AD used as reference in most studies, did not give conclusive results using the 9 isolates described above (hospital 1 - H1), a larger analysis was performed (Figure 1B,C). A panel of 26 ST1 well documented Belgian isolates collected between 1985 and 2020 was added. These isolates were related to two nosocomial ST1 epidemics that took place in the 1980’s in two other Belgian hospitals: Hospital 2 (H2) (6 isolates of which 2 clinical) and Hospital 3 (H3) (13 isolates; 11 clinical) but also isolates from another hospital (H4) (2 clinically related). Five unrelated (1 nosocomial, 4 community-acquired) and an environmental isolate from H3 were also used. To complete the panel, 41 ST1 isolates from 9 countries collected between 1992 and 2018 (Supplementary Table S1a).
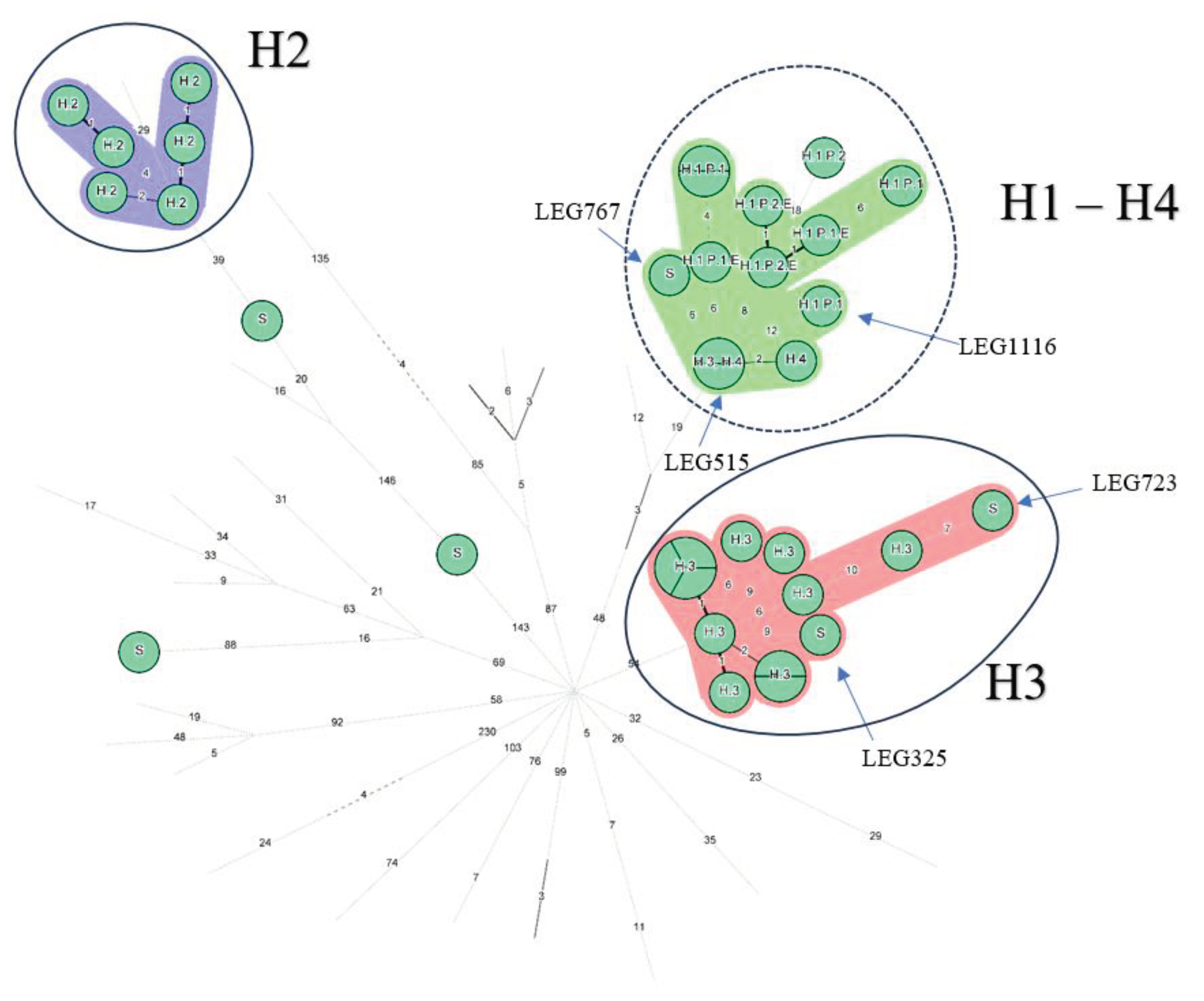
Most isolates grouped together by country (Figure 1B). Regarding Belgian isolates, three groups appeared, the well-identified epidemics from H2 and H3 were at maximum separated by 5 allelic differences except for the H3 isolate LEG515, (> 50 AD from the H3 closest isolate) which is opposed to the conclusion made by the initial investigation but consistent with the monoclonal subtyping performed at the time (Philadelphia versus Benidorm) [19]. However, in the same study LEG517, also subtyped Philadelphia belonged to the H3 cluster [19] (Supplementary Table S1). To cluster together isolates known to be epidemiologically linked (H3 epidemic isolates and H2 epidemic isolates, and P1 isolates), a threshold of 6 AD should be applied (Figure 1C). Then, all the 9 clinical and environmental isolates from H1 would also cluster together. However, the H1 cluster would also involve LEG767, an epidemiologically unrelated 2017 community-acquired (CA) sporadic case from the same region, the H3 LEG515 isolate and the two H4 isolates (located in the same city as H3) from 15 years earlier (2003-2004). Similarly, the H3 epidemic cluster would englobe an environmental isolate from the same hospital (LEG723), but from 30 years later (2016 - Supplementary Table S1) and a sporadic community isolate acquired in the same city than H3 (LEG325).
A wgSNP analysis was then run on the same group of strains. The commonly proposed SNPs threshold in the literature is 4 SNPs. The minimum spanning tree (Figure 2A,B) shows that this threshold does not cluster together neither the H3 nor the P1 isolates. The fitting threshold would be 8 SNPs, which would allow more discrimination than cgMLST. Indeed, while still showing clusters by country of origin on the large scale this threshold allows for H2 and H3 epidemics to form well-defined clusters, excluding LEG723 from the H3 cluster but not LEG 325. In this analysis, P2 clinical isolate stays outside the cluster formed by all the other H1 isolates with a difference of 11 SNPs, as opposed to the H4 isolates, H3 LEG515 and the unrelated sporadic LEG767 isolate which remain inside the cluster (Figure 2A,B).
1.3. wgMLST Analysis of the ST1 Isolates
The last analysis was the wgMLST pattern from the Bionumerics software (Figure 3). Of note, only an average of 52% of the supposed 5770 alleles were called (Supplementary Table S1). Lacking references to settle a threshold (Table 1), a threshold of 12 AD was then set up based on the maximum distance between known to be linked H3, H2 and P1 isolates (Figure 3). All isolates from the H2 and H3 epidemics and sporadic cases cluster then together. The P2 isolate, 18 loci away from its closest relative, was clearly distant from the H1 cluster, which, by contrast included, like with wgSNP, sporadic LEG767 and H4 isolates.
2. Discussion
L. pneumophila infections incidence is raising steadily since 2000’s [3,4]. This phenomenon, suspected to be directly or indirectly linked to climate change [20], is expected to keep on increasing in the coming years which stresses the necessity of finding efficient typing methods for environmental investigations and prevention of epidemics. Lpne has a wide genomic diversity and is easily subject to recombination events, mostly within a same serogroup [21,22]. L. pneumophila is also able to spread in several distinct geographical area without acquiring substantial genomic diversity over time [5,14,15,22]. The hypothesis of intermittent replication of L. pneumophila was mentioned after several large epidemiological investigations spanning diverse countries and several decades [15,23]: the cycle of life of L. pneumophila involves a latent phase that would explain both this intermittence phenomenon, and low rate of genomic polymorphism over space and time [15,23,24,25].
All STs of L. pneumophila seem to have evolved separately over time and ST1, one of the best fitted lineages for infections and adaptation in the environmental niches created by humans is one of the two STs that show the slowest genomic evolution [5,26]. This is resulting in the fact that even highly discriminatory typing methods can cluster together isolates known as epidemiologically unrelated.
We report here two cases of LD caused by ST1 isolates in patients hospitalized in the same ward one year apart. One was easily assumed as nosocomial based on the date of symptoms onset, the other case needed to rely on clear typing results to be either assigned as nosocomial and related to the same environmental source as the first one, or community acquired. To untangle this specific question, we applied several previously described NGS-based typing methods and thresholds to a collection of ST1 isolates retrieved either from documented nosocomial epidemics or sporadic community acquired infections in Belgium and ST1 published onto repository online databases. The cgMLST with a maximum 4 AD threshold seemed a little too stringent, as several isolates of the H3 outbreak were separated by 5 AD. According to David et al. ST1 and others major STs are better discriminate by SNP approach using a threshold of 4 SNP than by the cgMLST, although it is slightly variable according to the ST studied [27] (Table 1). In our setting, this threshold was also too stringent, as isolates known to be linked (H2 and H3 outbreaks, P1 clinical isolates) can be separated up to 8 SNPs from their closest relative (Figure 2B).
Regardless of the method used, unrelated isolates, acquired in the same geographical areas but years apart, did cluster together as witnessed for sporadic LEG723 and H3 isolates, or isolates from H4 and LEG767, which are from the same city. These isolates could be linked as a possibility of a long-lasting intermittently replicating reservoir in common water system is considered. For instance, David et al. demonstrated that cases acquired in a community close to an hospital can show the same genotype as nosocomial cases of this hospital [8]. Indeed, the water reservoirs of a city can be contaminated by the same predominant clone of L. pneumophila, and thus distribute it in several geographically close plumbing networks [9]. Similarly, an Australian study demonstrated that outbreak clones had a maximum of 10 SNPs difference within a common cg-SNP ST30 clade in a 30 km2 area around Melbourne [25]. The relatedness of L. pneumophila types and subtypes in both water reservoirs of health-care facilities and their respective municipal water systems should be regularly investigated by quantitative methods and NGS typing in order to better understand and evaluate the L. pneumophila ecology within large water distribution systems over long periods of time.
The unusual availability of more than one sequenced isolate for P1 gives an unexpected perspective to our analysis. These isolates (collected the same day, from the same patients but from different body sites) differed by up to 12 SNPs and 13 AD (cgMLST).
This difference observed between same-day P1 isolates as compared with one-year apart H1 environmental isolates could reflect the differential evolutionary clock of ST1 strain when infecting a patient as compared with dormancy into a pipe. Usually, when an investigation by NGS methods is set up, one isolate per patient is compared to one isolate by suspected source. If the diversity observed among P1 isolates does usually occur in patients, this might induce a bias in analysis using tight discrimination scale. On the other hand, most SNPs found between L. pneumophila isolates are > 90% related to homologous recombination events that occur in a very limited area of the genome [24]: the genes encoding activation factors of the Type 4 Secretion System, which is the case in the 9 P1 and P2 related isolates (data not shown). In a recombination hot spot, a large number of SNP will appear in a single genetic event, thus, the choice of SNP filtering previous to the performance of a wgSNP should be better documented before using this method as a routine tool. The filter we used filter only takes into account a SNP when it is separated by at least 12 bp from another one.
The necessity of standardized quality metrics for NGS typing methods also deserve comments. Raw data found on online repositories can be used by different key users with different approaches. We excluded several isolates from our analysis after bad quality results for de novo assemblies based on N50 < 100 000, which generally was associated to discordant genome length and /or high N bases numbers, nevertheless they were presented by studies considering these isolates of reliable quality after using another de novo alignment protocol (Supplementary Table S1) [15,28].
Using epidemiologically linked H3 and P1 isolate, wgSNP and cgMLST analyses gave contradictory results regarding P2 link to P1 and H1 environmental isolates. wgMLST is described as a good tool for large epidemiological investigations, but less if applied to ST1 [29]. As described by David and al. [27], by taking more genes into account, wgMLST indexes more dissimilarities between isolates than cgMLST, but also need a much better quality of sequencing to be sure of the allelic call. Furthermore, as the number of alleles differ between studies, the results obtained are not comparable. Here, even limited to an average of 52% of alleles called on supposedly 5,770, the results matched wgSNP, in agreement of David’s observations. A tailored threshold based on known epidemiological links between isolates, gave both good discrimination on the large scale and on the clustering of Belgian well described epidemics apart from sporadic isolates (Figure 3) and suggested that P2 isolate is not linked to P1 and H1 environmental isolates.
3. Conclusions
Our data highlight that despite promising results in the study of outbreaks and for large scale epidemiological investigations, NGS typing methods applied to ST1 outbreak investigation still need standardization regarding both wet-lab protocols and bioinformatics, for several important varia like reference strain, quality metrics of de novo alignment, filtering of SNPs and threshold for clustering. A deeper evaluation of L. pneumophila evolutionary clock is also required to increase our understanding of genomic differences between isolates sampled during a clinical infection and in environment.
4. Materials and Methods
4.1. Isolates Preparation, Routine Diagnostic and Typing
In Belgium, all case of Legionnaires disease (LD) must be reported to the federal health institute. An environmental investigation is then undertaken to identify the source. Belgian laboratories can send on voluntary basis both samples and Legionella isolates to the National Reference Centre for Legionella pneumophila either for diagnostic by PCR or SB typing. Cultures are performed on BCYE with GVPC agar in a humid atmosphere at 35°C +/- and incubated for 48-72 hours. Sequence based typing is performed according to the European Legionnaires' Disease Surveillance Network ‘s (ELDSNet) method (http://bioinformatics.phe.org.uk/legionella/legionella_sbt/php/sbt_homepage.php) [30].
4.2. Whole Genome Sequencing
Extraction of total DNA was performed using the Qiagen dneasy blood & tissue kit (Qiagen) from growth colonies of each isolate. DNA concentration was assessed using the Qubit dsDNA HS (or BR) assay kit (ThermoFisher Scientific) and the Qubit 2.0 Fluorometer (ThermoFisher Scientific). Library preparation was done with the Kapa HyperPlus Library Preparation Kit (Kapa Biosystems). Quality control and pooling of the library was performed employing a 2100 BioAnalyzer (Agilent), the Qubit 2.0 Fluorometer (ThermoFisher Scientific) and the KAPA Illumina Library Quantification Kit (Kapa Biosystems). The library was pooled with the addition of a 1% PhiX control library after denaturation with 0,2N NaOH, to a final concentration of 2 nM. MiSeq or Hiseq (Illumina) sequencers were used with the MiSeq Reagent kit v2 (500 cycle; 2 x 250 bp read-length) the expected coverage was of 100 [31].
4.3. Bioinformatic Analysis
The raw data were uploaded as fastq files on the software Bionumerics v8.1 (Biomérieux©, Marcy-l'Étoile, France). De novo assembly was done with a SPAdes algorithm, for one isolate (LEG1116) as quality metrics were not achieved by SPAdes assembly another scheme based also on DeBruijn graph was used: SKESA [32,33]. Reference genome was downloaded from NCBI website: L. pneumophila Paris 1 (Accession number (AN): CR628336). Quality scores of each method for each isolate are found in the Supplementary Table S1. N-50 was superior to 100,000, Q-score at least 30 (32-38) and average de novo covering was superior to 40 (46 -203) [15,28].
wgSNP was performed by remapping the reads to a reference genome and then a strict SNP filtering was applied (inter-SNP distance of min 12 bp, absolute coverage of 5 minimum, removal of non-informative SNPs, ambiguous bases, unreliable bases and gaps). wgMLST was performed by both free-based calls and assembly-based calls with the plugin of the software with default settings and screened 5,770 alleles of the genome. cgMLST was calculated out of the previous analysis and cover 1,521 loci based on the definition by Moran-Gilad’s group [10]. Number of alleles called for both wgMLST and cgMLST was calculated by the statistical plugin of the BioNumerics software.
The phylogenic analyses were made on the BN software by MST for categorical data analysis. Branch were logarithmically scaled according to the distance between each node.
All genomes of Belgian isolates are publicly available on NCBI website in the project PRJNA1073851. Five Fastq files for the isolates from other countries were downloaded from the European Nucleotide Archive from projects listed in the Supplementary Table S1, 4 isolates were excluded because of quality scores.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure Table S1: Dataset, quality scores and clustering.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, C.M. , O.V. and M.H.; methodology, C.M. and M.H.; software, C.M. and F.E.; validation, D.P., F.E. and M.H.; formal analysis, C.M.,F.E. and S.P..; investigation, S.P., V.C. and L.F..; resources, F.E., L.F., V.C. and S.P..; data curation, C.M. and M.H..; writing—original draft preparation, C.M.; writing—review and editing, D.M., O.V., D.P., N.Y., V.C, S.P. and M.H.; visualization, C.M..; supervision, O.V., M.H..; project administration, C.M., O.V. and M.H.. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research received no external funding.
Data Availability Statement
Michel, Charlotte (Forthcoming 2024). From investigating a case of cellulitis to exploring nosocomial infection control of ST1 Legionella pneumophila using genomic approaches. [Dataset]. Dryad. https://doi.org/10.5061/dryad.7m0cfxq35.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Han JH, Harada S, Edelstein PH, Nguyen JC, Baddour LM, Edelstein PH. Relapsing Legionella pneumophila cellulitis: a case report and review of the literature. J Infect Chemother. 2010, 16, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Vaidya T, Schmidt E, Papanicolaou G, Hauser J, Lezcano C, Tang YW, et al. Cutaneous Legionella infections in allogeneic hematopoietic cell transplantation recipients. Dermatol Online J. 2020, 26, 13030–qt05f926n7. [Google Scholar]
- European Centre for Disease Prevnetion and Control - ECDC. Legionnaires’ disease Annual Epidemiological Report for 2021 [Internet]. Stockholm, Sweden; 2023 Jun. Available from: https://www.ecdc.europa.eu/sites/default/files/documents/legionnaires-disease-annual-epidemiological-report-2021.pdf.
- Echahidi F MC. Activity report from 2011 to 2022 Reference centre for Legionella pneumophila UZ Brussel – LHUB-ULB [Internet]. National Reference Centre for Legionella pneumophila, Vrije Universiteit Brussel (VUB), Universitair Ziekenhuis Brussel (UZ Brussel), Department of Microbiology and infection control, Brussels; Available from: https://www.sciensano.be/sites/default/files/legionella_2011-2022_nrc_rapport_english_final.pdf.
- Ginevra C, Chastang J, David S, Mentasti M, Yakunin E, Chalker VJ, et al. A real-time PCR for specific detection of the Legionella pneumophila serogroup 1 ST1 complex. Clin Microbiol Infect. 2020, 26, e1–e514. [Google Scholar]
- Vekens E, Soetens O, De Mendonça R, Echahidi F, Roisin S, Deplano A, et al. Sequence-based typing of Legionella pneumophila serogroup 1 clinical isolates from Belgium between 2000 and 2010. Euro Surveill Bull Eur Sur Mal Transm Eur Commun Dis Bull. 2012, 17, 20302. [Google Scholar]
- David S, Afshar B, Mentasti M, Ginevra C, Podglajen I, Harris SR, et al. Seeding and Establishment of Legionella pneumophila in Hospitals: Implications for Genomic Investigations of Nosocomial Legionnaires’ Disease. Clin Infect Dis. 2017, 64, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Caicedo C, Rosenwinkel KH, Exner M, Verstraete W, Suchenwirth R, Hartemann P, et al. Legionella occurrence in municipal and industrial wastewater treatment plants and risks of reclaimed wastewater reuse: Review. Water Res. 2019, 149, 21–34. [Google Scholar] [CrossRef]
- Wüthrich D, Gautsch S, Spieler-Denz R, Dubuis O, Gaia V, Moran-Gilad J, et al. Air-conditioner cooling towers as complex reservoirs and continuous source of Legionella pneumophila infection evidenced by a genomic analysis study in 2017, Switzerland. Euro Surveill Bull Eur Sur Mal Transm Eur Commun Dis Bull. 2019, 24, 1800192. [Google Scholar]
- Moran-Gilad J, Prior K, Yakunin E, Harrison TG, Underwood A, Lazarovitch T, et al. Design and application of a core genome multilocus sequence typing scheme for investigation of Legionnaires’ disease incidents. Eurosurveillance [Internet]. 2015 Jul 16 [cited 2022 Nov 15];20[28]. Available from: https://www.eurosurveillance.org/content/10.2807/1560-7917.ES2015.20.28.21186.
- Wee BA, Alves J, Lindsay DSJ, Klatt AB, Sargison FA, Cameron RL, et al. Population analysis of Legionella pneumophila reveals a basis for resistance to complement-mediated killing. Nat Commun. 2021, 12, 7165. [Google Scholar] [CrossRef]
- Kozak-Muiznieks NA, Morrison SS, Mercante JW, Ishaq MK, Johnson T, Caravas J, et al. Comparative genome analysis reveals a complex population structure of Legionella pneumophila subspecies. Infect Genet Evol. 2018, 59, 172–185. [Google Scholar] [CrossRef]
- Haas W, Lapierre P, Musser KA. A Bioinformatic Pipeline for Improved Genome Analysis and Clustering of Isolates during Outbreaks of Legionnaires’ Disease. Dekker JP, editor. J Clin Microbiol. 2021, 59, e00967-20. [Google Scholar] [CrossRef]
- Krøvel AV, Bernhoff E, Austerheim E, Soma MA, Romstad MR, Löhr IH. Legionella pneumophila in Municipal Shower Systems in Stavanger, Norway; A Longitudinal Surveillance Study Using Whole Genome Sequencing in Risk Management. Microorganisms. 2022, 10, 536. [Google Scholar] [CrossRef]
- Gorzynski J, Wee B, Llano M, Alves J, Cameron R, McMenamin J, et al. Epidemiological analysis of Legionnaires’ disease in Scotland: a genomic study. Lancet Microbe. 2022, 3, e835-45. [Google Scholar]
- Petzold M, Prior K, Moran-Gilad J, Harmsen D, Lück C. Epidemiological information is key when interpreting whole genome sequence data – lessons learned from a large Legionella pneumophila outbreak in Warstein, Germany, 2013. Eurosurveillance [Internet]. 2017 Nov 9 [cited 2024 Jan 16];22[45]. Available from: https://www.eurosurveillance.org/content/10.2807/1560-7917.ES.2017.22.45.17-00137.
- Ricci ML, Fillo S, Ciammaruconi A, Lista F, Ginevra C, Jarraud S, et al. Genome analysis of Legionella pneumophila ST23 from various countries reveals highly similar strains. Life Sci Alliance. 2022, 5, e202101117. [Google Scholar] [CrossRef] [PubMed]
- Raphael BH, Huynh T, Brown E, Smith JC, Ruberto I, Getsinger L, et al. Culture of Clinical Specimens Reveals Extensive Diversity of Legionella pneumophila Strains in Arizona. Fey PD, editor. mSphere. 2019, 4, e00649-18. [Google Scholar] [CrossRef]
- Struelens MJ, Maes N, Rost F, Deplano A, Jacobs F, Liesnard C, et al. Genotypic and Phenotypic Methods for the Investigation of a Nosocomial Legionella pneumophila Outbreak and Efficacy of Control Measures. J Infect Dis. 1992, 166, 22–30. [Google Scholar] [CrossRef]
- Semenza JC, Ko AI. Waterborne Diseases That Are Sensitive to Climate Variability and Climate Change. Solomon CG, Salas RN, editors. N Engl J Med. 2023, 389, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Cazalet C, Rusniok C, Brüggemann H, Zidane N, Magnier A, Ma L, et al. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat Genet. 2004, 36, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Underwood AP, Jones G, Mentasti M, Fry NK, Harrison TG. Comparison of the Legionella pneumophila population structure as determined by sequence-based typing and whole genome sequencing. BMC Microbiol. 2013, 13, 302. [Google Scholar]
- Wells M, Lasek-Nesselquist E, Schoonmaker-Bopp D, Baker D, Thompson L, Wroblewski D, et al. Insights into the long-term persistence of Legionella in facilities from whole-genome sequencing. Infect Genet Evol. 2018, 65, 200–209. [Google Scholar] [CrossRef]
- David S, Sánchez-Busó L, Harris SR, Marttinen P, Rusniok C, Buchrieser C, et al. Dynamics and impact of homologous recombination on the evolution of Legionella pneumophila. Didelot X, editor. PLOS Genet. 2017, 13, e1006855. [Google Scholar]
- Buultjens AH, Chua KYL, Baines SL, Kwong J, Gao W, Cutcher Z, et al. A Supervised Statistical Learning Approach for Accurate Legionella pneumophila Source Attribution during Outbreaks. Schaffner DW, editor. Appl Environ Microbiol. 2017, 83, e01482-17. [Google Scholar] [CrossRef] [PubMed]
- David S, Rusniok C, Mentasti M, Gomez-Valero L, Harris SR, Lechat P, et al. Multiple major disease-associated clones of Legionella pneumophila have emerged recently and independently. Genome Res. 2016, 26, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Lau KA, Gonçalves Da Silva A, Theis T, Gray J, Ballard SA, Rawlinson WD. Proficiency testing for bacterial whole genome sequencing in assuring the quality of microbiology diagnostics in clinical and public health laboratories. Pathology (Phila). 2021, 53, 902–911. [Google Scholar]
- Raphael BH, Huynh T, Brown E, Smith JC, Ruberto I, Getsinger L, et al. Culture of Clinical Specimens Reveals Extensive Diversity of Legionella pneumophila Strains in Arizona. mSphere. 2019, 4, e00649-18. [Google Scholar] [CrossRef] [PubMed]
- David S, Mentasti M, Tewolde R, Aslett M, Harris SR, Afshar B, et al. Evaluation of an Optimal Epidemiological Typing Scheme for Legionella pneumophila with Whole-Genome Sequence Data Using Validation Guidelines. J Clin Microbiol. 2016, 54, 2135–2148. [Google Scholar] [CrossRef] [PubMed]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J Comput Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Souvorov A, Agarwala R, Lipman DJ. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018, 19, 153. [Google Scholar]
- Gaia V, Fry NK, Harrison TG, Peduzzi R. Sequence-Based Typing of Legionella pneumophila Serogroup 1 Offers the Potential for True Portability in Legionellosis Outbreak Investigation. J Clin Microbiol [Internet]. 2003, 41, 2932–2939, Available from: https://journals.asm.org/doi/10.1128/JCM.41.7.2932-2939.2003. [Google Scholar] [CrossRef]
- Muyldermans A, Crombé F, Bosmans P, Cools F, Piérard D, Wybo I. Serratia marcescens outbreak in a neonatal intensive care unit and the potential of whole-genome sequencing. Journal of Hospital Infection [Internet]. 2021, 111, 148–154, Availablefrom:https://linkinghub.elsevier.com/retrieve/pii/S019567012100061X. [Google Scholar] [CrossRef]
Figure 1.
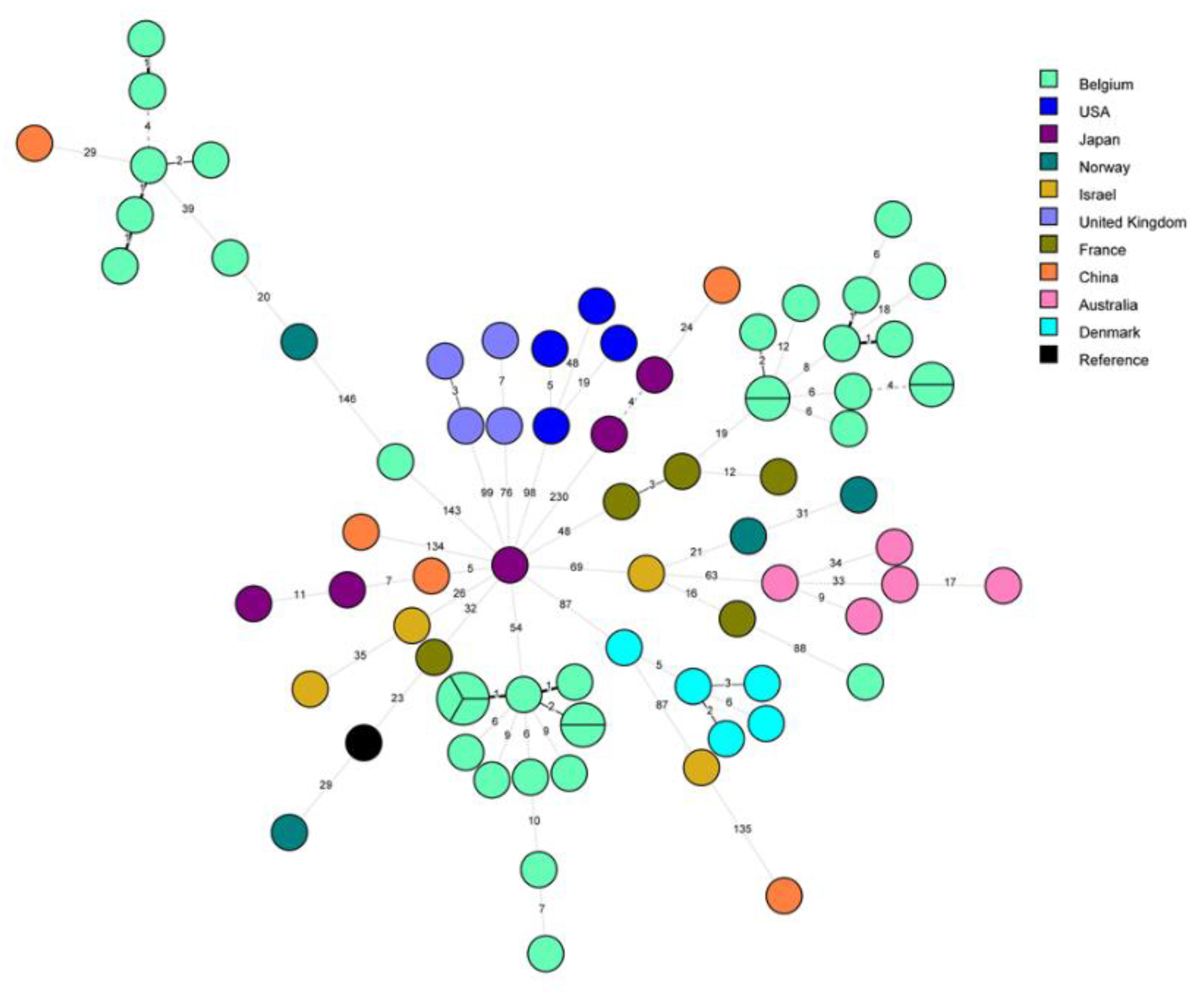
Minimum Spanning Tree for categorical data of the cgMLST analysis based on the call of 1,521 alleles [1] performed on Bionumerics software v8.1. A. Isolates from the hospital 1, including related isolates to Patients 1 and 2. A cluster analysis of maximum 4 allelic differences between two isolates is highlighted in red. Hospital 1: H1, Patient 1 and 2: P1 and P2 respectively; Environmental linked isolates: E. B: 77 ST1 isolates of L. pneumophila are represented by nodes colored according to their country of origin. Belgian isolates are coloured in light green. C: Nodes representing non-Belgian isolates were reduced to uncover the relationship between the 36 Belgian ST1 isolates. The setting of each isolate is written above each node: Hospital 1,2, 3 and 4: H1, H2, H3 and H4 respectively; Sporadic cases: S; Patient 1 and 2: P1 and P2 respectively; Environmental linked isolates: E. As an indication, two groups are circled in solid blue line which include well characterized clusters corresponding to the epidemics in H2 and H3 and their closely related isolates. The suspected cluster, corresponding to H1 and H4, is circled in a blue dashed line. Clustering performed using a 4 allelic differences distance between isolates appears in colorful background. wgSNP analysis of the ST1 isolates.
Figure 1.
Minimum Spanning Tree for categorical data of the cgMLST analysis based on the call of 1,521 alleles [1] performed on Bionumerics software v8.1. A. Isolates from the hospital 1, including related isolates to Patients 1 and 2. A cluster analysis of maximum 4 allelic differences between two isolates is highlighted in red. Hospital 1: H1, Patient 1 and 2: P1 and P2 respectively; Environmental linked isolates: E. B: 77 ST1 isolates of L. pneumophila are represented by nodes colored according to their country of origin. Belgian isolates are coloured in light green. C: Nodes representing non-Belgian isolates were reduced to uncover the relationship between the 36 Belgian ST1 isolates. The setting of each isolate is written above each node: Hospital 1,2, 3 and 4: H1, H2, H3 and H4 respectively; Sporadic cases: S; Patient 1 and 2: P1 and P2 respectively; Environmental linked isolates: E. As an indication, two groups are circled in solid blue line which include well characterized clusters corresponding to the epidemics in H2 and H3 and their closely related isolates. The suspected cluster, corresponding to H1 and H4, is circled in a blue dashed line. Clustering performed using a 4 allelic differences distance between isolates appears in colorful background. wgSNP analysis of the ST1 isolates.
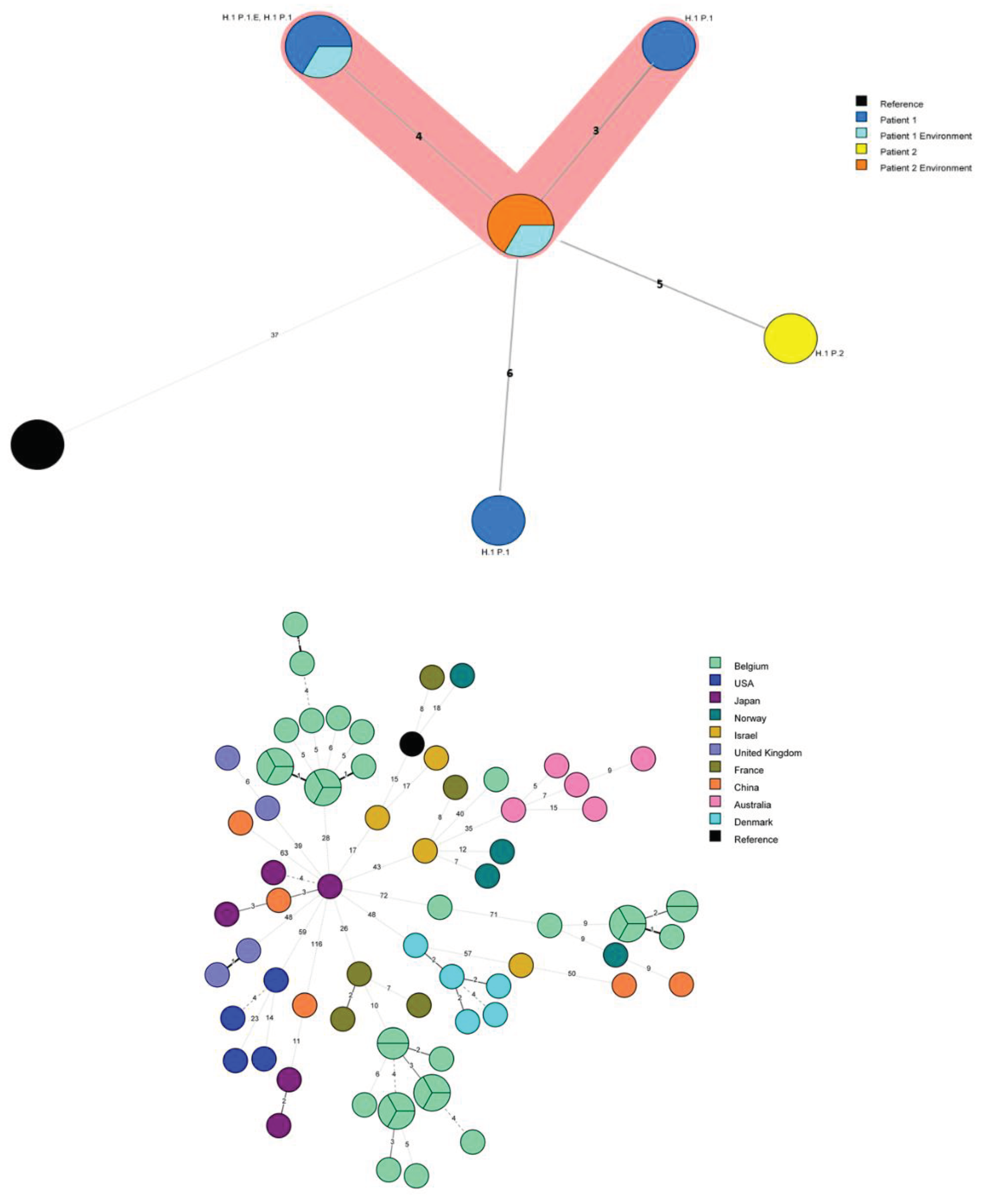
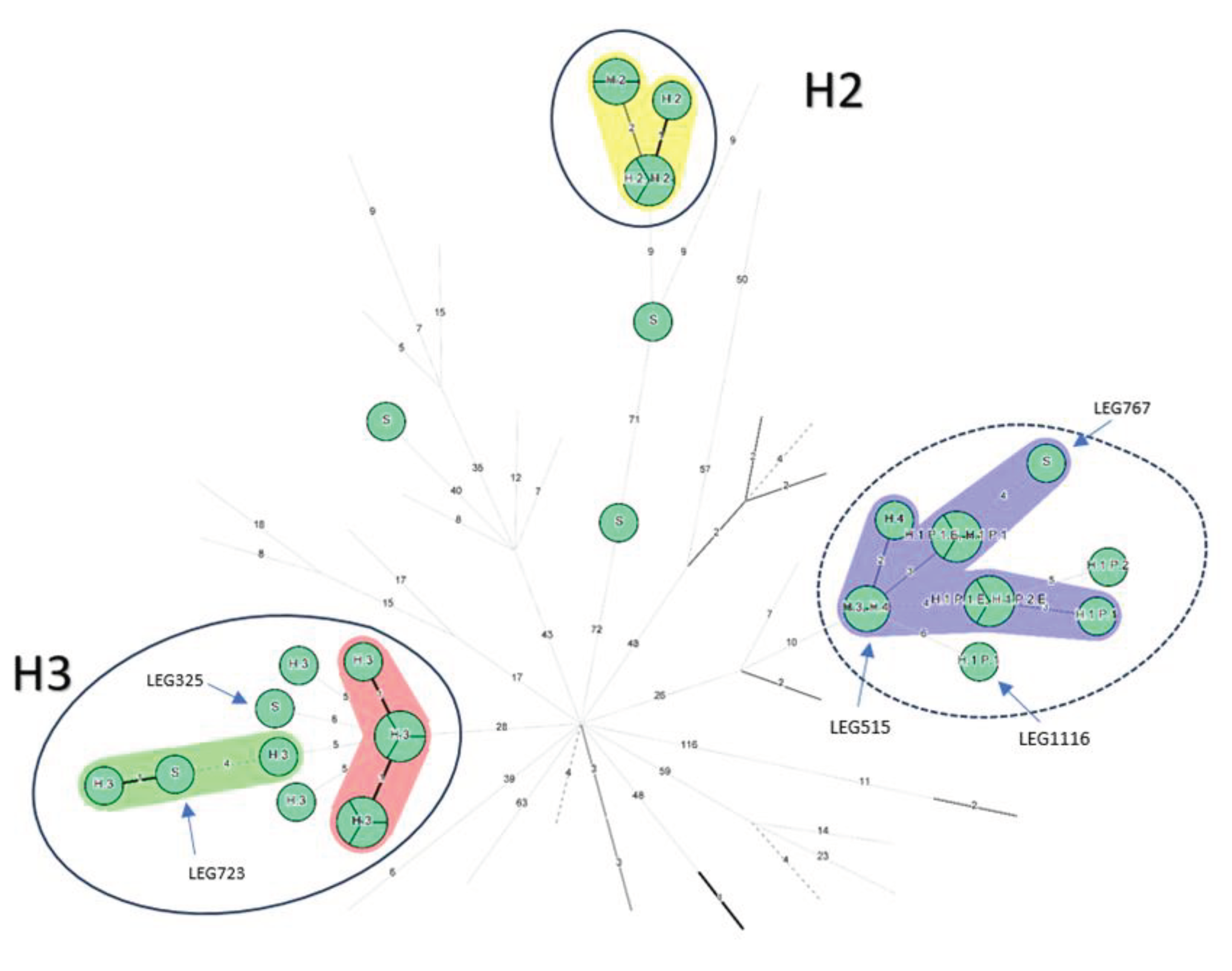
Figure 2.
Minimum spanning tree of wgSNP with strict SNP filtering performed on Bionumerics software v8.1. A: 77 ST1 isolates of L. pneumophila are represented by nodes colored according to their country of origin. B: Nodes representing non-Belgian isolates were reduced to undercover only the relationship between the 35 Belgian ST1. The setting of each isolate is written above each node. Hospital 1,2, 3 and 4: H1, H2, H3 and H4 respectively; Sporadic cases: S; Patient 1 and 2: P1 and P2 respectively; Environmental linked isolates: E As an indication, two groups are circled in solid blue line which include well characterized clusters corresponding to the epidemics in H2 and H3 and closely related isolates. The suspected cluster, corresponding to H1 is circled in a blue dashed line. Clustering was performed using a 4 SNPs distance between isolates and appears in colorful background within the group.
Figure 2.
Minimum spanning tree of wgSNP with strict SNP filtering performed on Bionumerics software v8.1. A: 77 ST1 isolates of L. pneumophila are represented by nodes colored according to their country of origin. B: Nodes representing non-Belgian isolates were reduced to undercover only the relationship between the 35 Belgian ST1. The setting of each isolate is written above each node. Hospital 1,2, 3 and 4: H1, H2, H3 and H4 respectively; Sporadic cases: S; Patient 1 and 2: P1 and P2 respectively; Environmental linked isolates: E As an indication, two groups are circled in solid blue line which include well characterized clusters corresponding to the epidemics in H2 and H3 and closely related isolates. The suspected cluster, corresponding to H1 is circled in a blue dashed line. Clustering was performed using a 4 SNPs distance between isolates and appears in colorful background within the group.
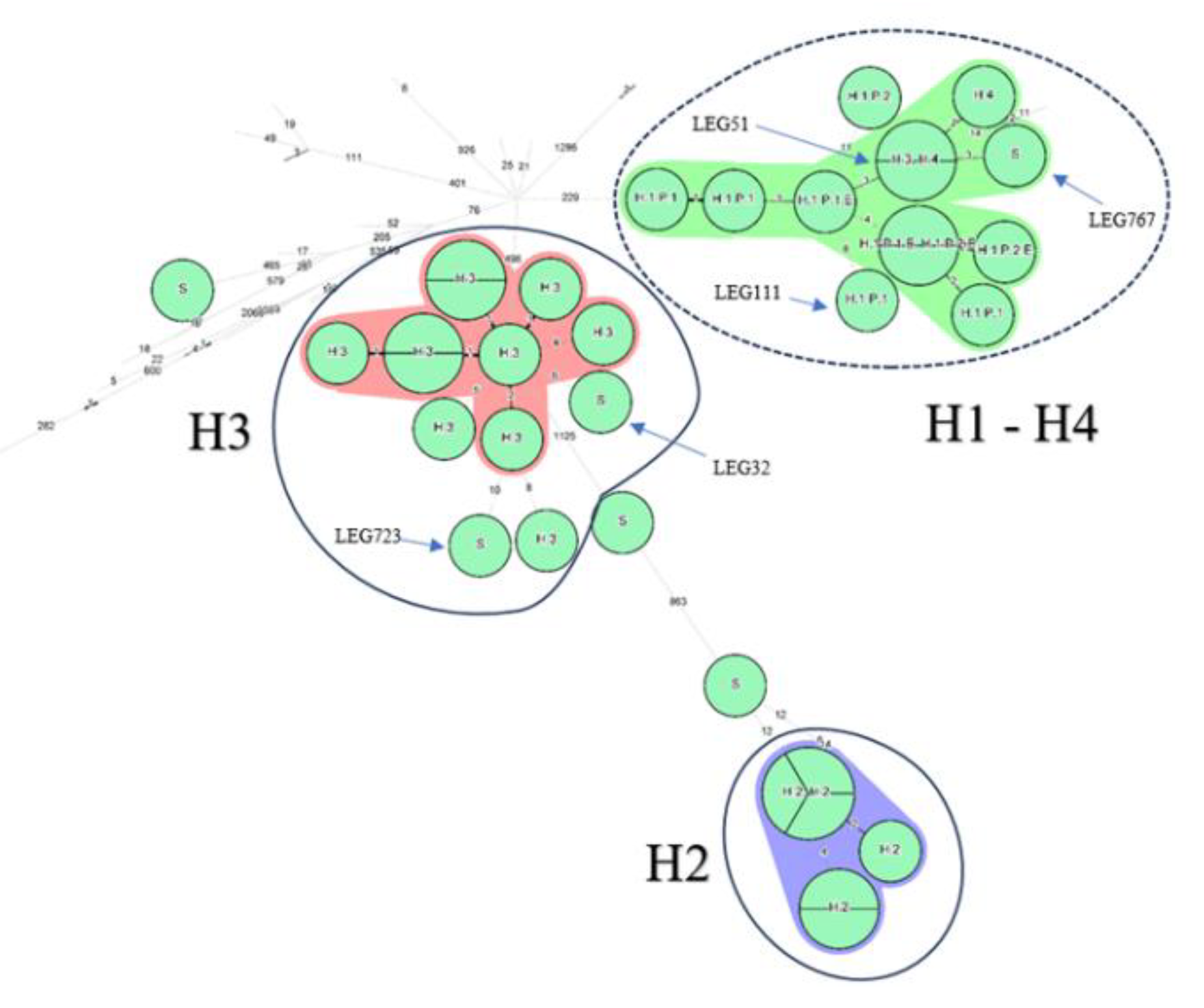
Figure 3.
Minimum spanning tree of categorical data analysis for wgMLST performed on Bionumerics software v8.1. A: 77 ST1 isolates of L. pneumophila are represented by nodes colored according to their country of origin. The allele call is on average 52% for a total alignment of 5,770 alleles. B: Nodes representing non-Belgian isolates were reduced to undercover only the relationship between the 35 Belgian ST1. The setting of each isolate is written above each node. Hospital 1,2, 3 and 4: H1, H2, H3 and H4 respectively; Sporadic cases: S; Patient 1 and 2: P1 and P2 respectively; Environmental linked isolates: E. As an indication, two groups are circled in solid blue line which include well characterized clusters corresponding to the epidemics in H2 and H3 and closely related isolates. The suspected cluster, corresponding to H1 is circled in a blue dashed line. Clustering was performed using a 12 allelic differences distance between isolates and appears in colorful background within the groups.
Figure 3.
Minimum spanning tree of categorical data analysis for wgMLST performed on Bionumerics software v8.1. A: 77 ST1 isolates of L. pneumophila are represented by nodes colored according to their country of origin. The allele call is on average 52% for a total alignment of 5,770 alleles. B: Nodes representing non-Belgian isolates were reduced to undercover only the relationship between the 35 Belgian ST1. The setting of each isolate is written above each node. Hospital 1,2, 3 and 4: H1, H2, H3 and H4 respectively; Sporadic cases: S; Patient 1 and 2: P1 and P2 respectively; Environmental linked isolates: E. As an indication, two groups are circled in solid blue line which include well characterized clusters corresponding to the epidemics in H2 and H3 and closely related isolates. The suspected cluster, corresponding to H1 is circled in a blue dashed line. Clustering was performed using a 12 allelic differences distance between isolates and appears in colorful background within the groups.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



