
Submitted:
01 April 2024
Posted:
02 April 2024
You are already at the latest version
Abstract
The study of rare diseases is important not only for the individuals affected but also for the advancement of medical knowledge and a deeper understanding of human biology and genetics. The wide repertoire of structural information now available from reliable and accurate prediction methods provides the opportunity to investigate the molecular origins of most of the rare diseases reviewed in the Orpha.net database. Thus, it has been possible to analyze the topology of the missense mutations found in the 2,535 proteins involved in Mendelian rare diseases (MRD), which form the database for our structural bioinformatics study. The amino acid substitutions responsible for MRD show different mutation site distributions at different three-dimensional protein depths. We then predicted the depth-dependent effects of mutations for the 20,248 pathogenic variants that are present in our database. The results of this structural bioinformatics investigation are relevant, as they provide additional clues to mitigate the damage caused by MRD.
Keywords:
rare diseases
; missense mutations
; protein structure
; databank analysis
; structural bioinformatics
1. Introduction
Rare diseases (RD) are defined by the World Health Organization as affecting fewer than 65 per 100,000 people, a characteristic that is mainly responsible for the lack of knowledge, expertise, and therefore effective treatments. Today, RD is emerging as a public health priority, and an increasing number of international networks are active to increase its visibility at the global level and to expand and share research, medical, and social care strategies. The fact that more than 70% of RDs are of genetic origin [1], and therefore the same DNA mutation is present in each cell type, means that a wide variety of effects occur in the affected human body. As a result, Mendelian diseases (MRD) are almost impossible to cure, although there are approaches to treat or manage some of the associated signs and symptoms [2]. If the molecular origins of MRD can be attributed to missense variants, and thus to well-defined changes at the protein level, we could in principle explore the correlations between the protein structural changes that occur, and the abnormal functions observed to develop rational therapeutic strategies. It is interesting to note that missense mutations are very common, as they occur in about half of the items that are present in the ClinVar genomic variant database [3]. The assignment of protein mutation sites for genomic missense variants to surface, core, or interaction regions has been proposed [4] when structural information is available from the Protein Data Bank [5]. However, despite the large number of known protein sequences, the limited number of experimentally resolved protein structures is a significant barrier to studying the structure-function correlation of proteins involved in MRD. Artificial intelligence (AI) has recently partially overcome the problem of limited structural information for investigating the effects of molecular changes on protein function [6]. Millions of reliably calculated protein structure models are currently available in the freely accessible AlphaFold database (https://alphafold.ebi.ac.uk/), providing broad coverage of the entire content of UniProtKB, the standard repository for protein sequences and annotations [7]. In addition, AI has recently developed AlphaMissense, a new powerful tool for predicting pathogenicity scores for all observed missense genomic variants [8]. Thus, structural bioinformatics can operate efficiently to provide powerful shortcuts for suggesting the protein basis of pathologies at the atomic level and possible remedies for MRD, as we describe in the present report with the implementation of a procedure to scan the database provided by Orpha.net, which correlates each MRD point with the corresponding mutated gene [9]. Orpha.net is therefore a suitable starting point for a structural investigation routine, which we have named Orphanetta (Orpha.net topological analysis). Orphanetta provides a general network of molecule-based information about the structural features of MRD mutations. It can therefore be a powerful tool to guide the search for new potential treatments of any pathology, present in the Orpha.net database, having structurally defined missense mutation sites.
2. Results
As of 10 October 2023, the Orpha.net database listed 4,338 genes associated with MRD, which were the starting point for our investigation. ClinVar incorporated all the latter genes, adding the relevant information on the molecular consequence of their reported variants. Thus, we have delineated 3,145 missense-mutated proteins involved in MRD that represent the target of our structural analysis.
2.1. Deriving Structural Information from the Orpha.net Database
Among all the 3,145 proteins that were involved in MRD, several imply impossible double or triple nucleotide codon changes, as we observed in their amino acid replacement matrix, see Figure 1. Then, as we have previously done in the general case of all the ClinVar missense variants [10], we performed a preliminary removal of the latter anomalous items. Thus, for our structural Bioinformatics analysis, we have obtained a final data set containing 2,797 proteins undergoing MRD mutations.
Based on the corresponding UniProtKBKB accession codes, we searched for the presence of each of these 2,797 proteins in the Protein Data Bank [5]. As this search yielded only 1,738 non-redundant experimentally resolved protein structures, we investigated for the presence of the additional structural information in the database of AlphaFold predicted structures [6]. We have considered only those files that ensured reliable AlphaFold models, i.e., the ones having pLDDT scores higher than 0.8. Accordingly, we have analyzed the structural features of 2,535 missense-mutated proteins that form the complete repertoire of our structural Bioinformatics investigation. Multiple MRD pathogenic variants are associated with the latter proteins and the complete list of the structurally defined 20,248 mutations is given in Table S1.
2.2. Topological Assignments of Protein Mutants Responsible for MRD
The effect of amino acid replacements on protein evolution has been considered since the times when protein structural information was limited to a handful of experimentally resolved examples [11,12,13]. Nowadays, the wealth of the available protein structures, both experimentally obtained and high-quality predicted, allows us to discuss amino acid replacements also in terms of their 3D location. Thus, as the structure-based analysis can yield powerful information for understanding the molecular mechanisms of diseases [14], we carried out the topological analysis of mutation sites that are present in each of the 2,535 proteins of our data set to distinguish outer and inner locations of the amino acid replacements by using POPScomp [15]. Hence, the Q(SASA) parameter has been used to assign the mutation site topology from protein cores to their most external regions [4]. Accordingly, Q(SASA) values lower than 0.15 have been considered diagnostic of inner positions of the missense mutation site, and the topology of all the variants related to MRD diseases are distributed as reported in Figure 2 and Table S1.
All the internal mutation sites related to MRD represent the very large majority of all the variants, in agreement with previous suggestions for the incidence of inner residues in general on pathogenicity [16]. Data reported in Figure 3, clearly show how the replacement profiles of amino acids are quite different in the case of buried or exposed mutation sites.
Furthermore, from inspection of the amino acid replacement matrices of the latter two groups of variants, shown in Figure 4, several features are worth a preliminary discussion. As far as fully buried mutation sites are concerned, Gly and Arg are equally abundant and much more frequent than all the other replaced amino acids, see Figure 4b, and Cys, despite its low occurrence in proteins, exhibits a very frequent involvement in pathogenicity. In the case of fully exposed mutation sites, see Figure 2 and Figure 3, a main signal arises from the observation that very frequent substitutions occur for Met and Arg, representing respectively 53 and 16 % of the total ones. The relevance of these findings in relation to MRD onset is underlined in Section 3 of the present report.
2.3. Predicting the Structural Effects of Specific Amino Acid Replacements
The mutation matrices shown in Figure 4 confirm that MRD pathogenicity arising from amino acid substitutions in the proteins of our dataset is not univocal, but it is determined by the topology of the mutation site. For instance, replacements of amino acids bearing hydrophobic bulky side chains with other ones having electric charges, in the case they occur in the protein interior, determine a disruption of the folding nucleus, and the unfolded protein undergoes a fast proteolytic digestion. Whenever the same type of event occurs in the solvent-exposed protein surface, the protein folding process is fully conserved, but this feature causes a strong change in the protein interaction pattern with its molecular environment, ranging from the inhibition of protein quaternary assemblies to changes of protein-ligand interactions. In general, the effects of an amino acid substitution can determine i) reduction of the structural stability, ii) inhibition of folding nucleus formation, iii) interference in protein-protein, protein-nucleic acids, or protein-ligand interactions. To predict the effect of amino acid replacements they are usually grouped into four main categories, which must undergo further subdivision to account for their hydrophobicity, polarity, electric charge, and side chain size. Hence, we have considered 10 different subgroups of amino acids, as reported in Table 1.
Thus, possible effects of the amino acid variations in the structurally characterized proteins of our Orpha. net-derived dataset are predicted and listed in Table S1. In Table S2 are listed all the 5221 MRD reviewed from Orpha.net, whose structural origins can be tracked with the Orphanetta procedure.
3. Discussion
The genetic information offered by the Orpha.net database has been our starting point for collecting additional clues at a molecular level for possible MRD remediation. Our topological analysis of all the amino acid replacements that are correlated to MRD, summarized in Figure 5, clearly confirms what has been already observed in general [17], i.e., low solvent exposure of mutation sites is mainly correlated to the onset of genetic diseases.
Data shown in Figure 4 indicate the very high frequency of pathogenic mutations found for Arg and Gly in protein cores. The presence of Arg in inner protein regions, indeed, is very critical to maintain a positive charge, when is needed in hydrophobic environments or to bind internal water molecules [18]. Thus, MRDs frequently come from mutations of Arg, a residue that has CG (U, C, A, G) codons particularly unstable [16,19]. Then, it is well known that Gly residues are largely conserved, as with their small dimensions they can play unique roles in the structure of folded proteins [20,21]. As shown in Figure 4a, the core glycines of our structural data set are mostly replaced by charged amino acids or residues with larger side chains, in both cases perturbing the folding nucleus formation. Figure 4a also evidentiates how frequently Cys substitutions in the inner protein region are lethal for the folding process, as they interrupt the cysteine-bridge network, which is necessary to stabilize the correct protein structure. Upon defining fully-exposed protein residues the ones exhibiting Q(SASA) above 0.8, we have selected 1.002 mutation sites. They are mainly due to Met and Arg substitutions, respectively at 53 and 17%. It is worth noting that pathogenicity always occurs whenever the mutation involves a surface-exposed Met occupying the amino terminus position. This finding is in total agreement with a recent investigation [22] that underlines how such a mutation in the signal peptide interferes with protein targeting, translocation, processing, and stability. Several very pathogenic effects can arise from replacements of Arg occupying protein surface positions, as the latter amino acid drives most of the protein interactions with other proteins, nucleic acids, and ligands. As reported in Figure 4b, there are 167 cases where Arg is changed into all the possible alternatives given by single nucleotide variations. It would be precious information to know when these Arg mutation sites occur at the interface with their molecular partners for a better understanding of the mechanisms of pathogenicity. Very likely, this task will be possible in the near future thanks to artificial intelligence procedures such as AlphaFold-Multimer [23]. Thus, from any of the MRD listed in Table S2, we can find the structurally defined protein(s) involved in missense mutations. Then, from the UniProtKB code(s) reported in Table S1, the mutation site topology can be delineated to predict the structure and function damage caused by the amino acid replacement.
4. Conclusions
The Orphanetta procedure, summarized in Figure 5, by linking the genomic information provided by the Orpha.net database to the ones available from structural data banks, seems to be well suited for AI developments that can yield fast and automatic answers for deciding the priorities for genomic editing to solve or, at least mitigate, the effects of MRD.
5. Materials and Methods
5.1. Dataset of Missense Variants
As of 10 October 2023, the Orpha.net database (https://www.orpha.net/), providing a standardized classification and coding system for all the known rare diseases, listed 4,338 genes associated with MRD. This information represents the starting point for the present investigation. ClinVar databank [3] incorporated all the latter genes, adding the relevant information on the molecular consequence of the reported variants. Thus, we have delineated 3,145 missense-mutated proteins involved in MRD that represent the target of our structural analysis.
5.2. Structural Analysis
To complete the data set of the present structural analysis, the Protein Data Bank (PDB) [5], a widely used repository for 3D structural data of biological biopolymers, was accessed to retrieve structural information for MRD mutations. In addition, AlphaFold [6] was used to obtain structural information on the proteins that were not present in the PDB. AlphaFold’s algorithm analyzes the amino acid sequence of a protein to predict the distances between pairs of amino acids, which are then used to generate a 3D model of the protein structure. For each residue AlphaFold outputs a predicted Local Distance Difference Test (pLDDT) score, to assess the reliability of specific regions within the structure of interest. In the present investigation, we have taken into account only predicted models possessing very high reliability with a pLDDT >0.8.
5.3. Atom Depth Calculations
The POPScomp program [15] computes the Solvent Accessible Surface Area (SASA) of any structure in a suitable PDB format. Q(SASA), which represents the ratio between the SASA of a generic Xyz amino acid whether inserted in the protein structure and a reference value related to the SASA of the amino acid side chain in a tripeptide GlyXyzGly.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Topologies of the missense mutations responsible for MRD; Table S2: The complete list of structurally characterized MRD.
Author Contributions
Conceptualization, N.N.; methodology, A.V. and R.F.; formal analysis, N.N., A.V. and R.F.; investigation, N.N., A.V. and R.F., O.S.; resources, A.V. and A.S.; writing—original draft preparation, N.N.; writing—review and editing, N.N., A.V., R.F., O.S. and A.S.; supervision, N.N; project administration, O.S. and A.S.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank Dr. F. Fraternali for helpful suggestions and Dr. A. Trezza for encouraging this work.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- S. Nguengang Wakap et al., ‘Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database’, European Journal of Human Genetics, vol. 28, no. 2, pp. 165–173, Feb. 2020. [CrossRef]
- T. P. O’Connor and R. G. Crystal, ‘Genetic medicines: Treatment strategies for hereditary disorders’, Nature Reviews Genetics, vol. 7, no. 4. pp. 261–276, Apr. 2006. [CrossRef]
- M. J. Landrum et al., ‘ClinVar: Public archive of interpretations of clinically relevant variants’, Nucleic Acids Res, vol. 44, no. D1, pp. D862–D868, 2016. [CrossRef]
- A. Laddach, J. C. Fung Ng, and F. Fraternali, ‘Pathogenic missense protein variants affect different functional pathways and proteomic features than healthy population variants’, PLoS Biol, vol. 19, no. 4, Apr. 2021. [CrossRef]
- H. M. Berman et al., ‘The Protein Data Bank’, 2000. [Online]. Available: http://www.rcsb.org/pdb/status.html.
- J. Jumper et al., ‘Highly accurate protein structure prediction with AlphaFold’, Nature, vol. 596, no. 7873, pp. 583–589, Aug. 2021. [CrossRef]
- A. Bateman et al., ‘UniProt: the Universal Protein Knowledgebase in 2023′, Nucleic Acids Res, vol. 51, no. D1, pp. D523–D531, Jan. 2023. [CrossRef]
- J. Cheng et al., ‘Accurate proteome-wide missense variant effect prediction with AlphaMissense’, Science (1979), vol. 381, no. 6664, Sep. 2023. [CrossRef]
- S. Pavan, K. Rommel, M. E. M. Marquina, S. Höhn, V. Lanneau, and A. Rath, ‘Clinical practice guidelines for rare diseases: The orphanet database’, PLoS One, vol. 12, no. 1, Jan. 2017. [CrossRef]
- P. Bongini et al., ‘Structural Bioinformatic Survey of Protein-Small Molecule Interfaces Delineates the Role of Glycine in Surface Pocket Formation’, IEEE/ACM Trans Comput Biol Bioinform, vol. 19, no. 3, pp. 1881–1886, 2022. [CrossRef]
- Grantham R, ‘Amino acid difference formula to help explain protein evolution.’, Science (1979), pp. 862–864, 1974.
- Epstein C, ‘Non-randomness of Ammo-acid Changes in the Evolution of Homologous Proteins.’, Nature, vol. 215, pp. 355–359, 1967.
- T. Miyata, S. Miyazawa, and T. Yasunaga, ‘Journal of Molecular Evolution Two Types of Amino Acid Substitutions in Protein Evolution’, 1979.
- S. Teng et al., ‘Structural assessment of the effects of Amino Acid Substitutions on protein stability and protein-protein interaction’, 2010.
- L. Cavallo, J. Kleinjung, and F. Fraternali, ‘POPS: A fast algorithm for solvent accessible surface areas at atomic and residue level’, Nucleic Acids Res, vol. 31, no. 13, pp. 3364–3366, Jul. 2003. [CrossRef]
- D. Vitkup, C. Sander, and G. M. Church, ‘The amino-acid mutational spectrum of human genetic disease’, 2003. [Online]. Available: http://genomebiology.com/2003/4/11/R72.
- M. Hackel, H.-J. Hinz, and G. R. Hedwig, ‘Partial molar volumes of proteins: amino acid side-chain contributions derived from the partial molar volumes of some tripeptides over the temperature range 1090C’, 1999.
- M. J. Harms, J. L. Schlessman, G. R. Sue, and B. García-Moreno, ‘Arginine residues at internal positions in a protein are always charged’, Proc Natl Acad Sci U S A, 2011. [CrossRef]
- S. E. Antonarakis, M. Krawczak, and D. N. Cooper, ‘Disease-causing mutations in the human genome’. [Online]. Available: http://www.kazusa.or.jp/codon/.
- T. J. Branden C, Introduction to Protein Structure Second Edition.
- Parrini et al., ‘Glycine residues appear to be evolutionarily conserved for their ability to inhibit aggregation’, Structure, vol. 13, no. 8, pp. 1143–1151, Aug. 2005. [CrossRef]
- S. A. G. Guarnizo, M. K. Kellogg, S. C. Miller, E. B. Tikhonova, Z. N. Karamysheva, and A. L. Karamyshev, ‘Pathogenic signal peptide variants in the human genome’, NAR Genom Bioinform, vol. 5, no. 4, Dec. 2023. [CrossRef]
- R. Evans et al., ‘Protein complex prediction with AlphaFold-Multimer’, 2022. [CrossRef]
Figure 1.
Amino acid distributions of missense mutations reviewed by Opha.net data set. Rows describe how each of the natural amino acids has been replaced by column residues. Colors refer to the number of codon nucleotides involved in mutations: green, yellow, and red indicate respectively one, two, and three nucleotide changes.
Figure 1.
Amino acid distributions of missense mutations reviewed by Opha.net data set. Rows describe how each of the natural amino acids has been replaced by column residues. Colors refer to the number of codon nucleotides involved in mutations: green, yellow, and red indicate respectively one, two, and three nucleotide changes.
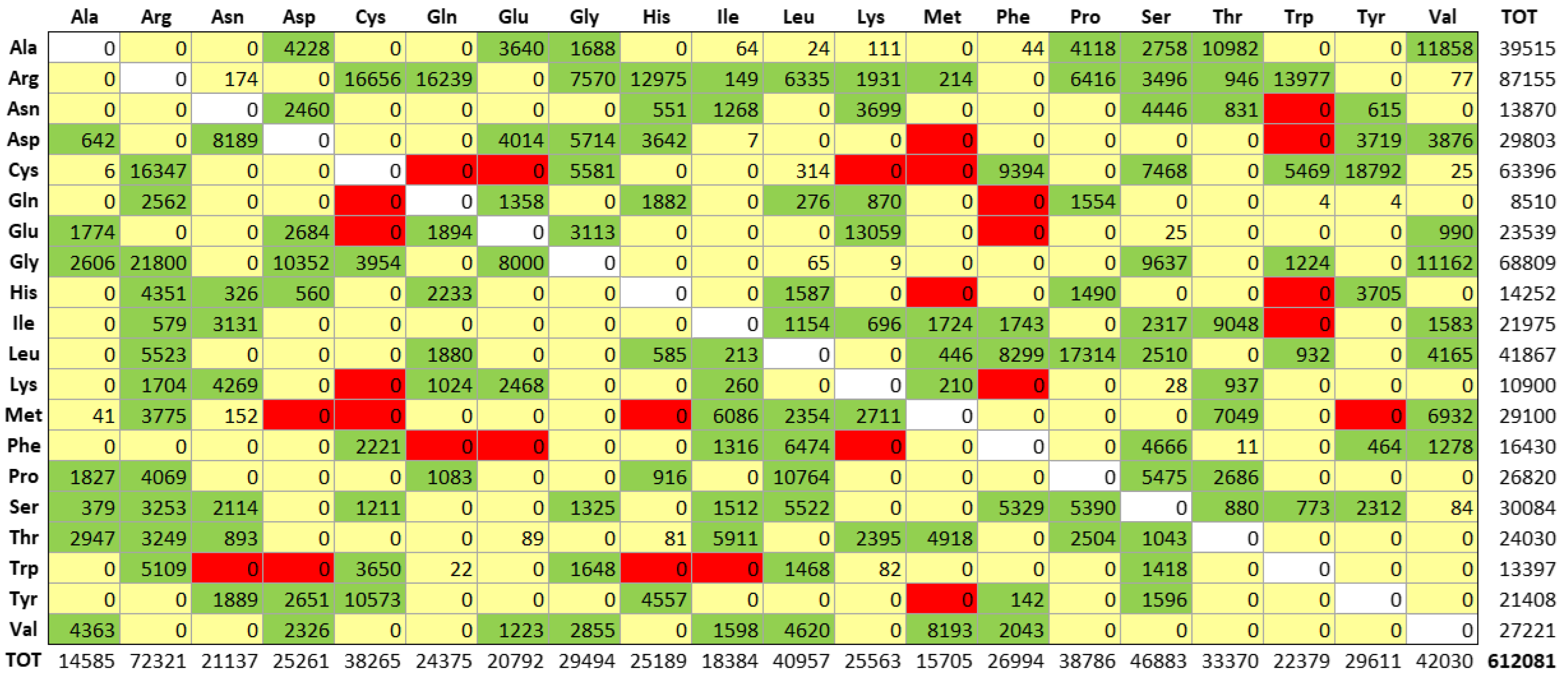
Figure 2.
The sterical distribution of mutation sites in MRD proteins of our dataset. Structural data are clustered according to the POPS algorithm (4), and the Q(SASA) parameter categories the 20.248 mutation sites as follows: i) fully buried < 0.15; ii) internal > 0.15 and < 0.24; iii) intermediate > 0.24 and < 0.60; iv) external > 0.60 and < 0.80; v) fully solvent-exposed> 0.8.
Figure 2.
The sterical distribution of mutation sites in MRD proteins of our dataset. Structural data are clustered according to the POPS algorithm (4), and the Q(SASA) parameter categories the 20.248 mutation sites as follows: i) fully buried < 0.15; ii) internal > 0.15 and < 0.24; iii) intermediate > 0.24 and < 0.60; iv) external > 0.60 and < 0.80; v) fully solvent-exposed> 0.8.
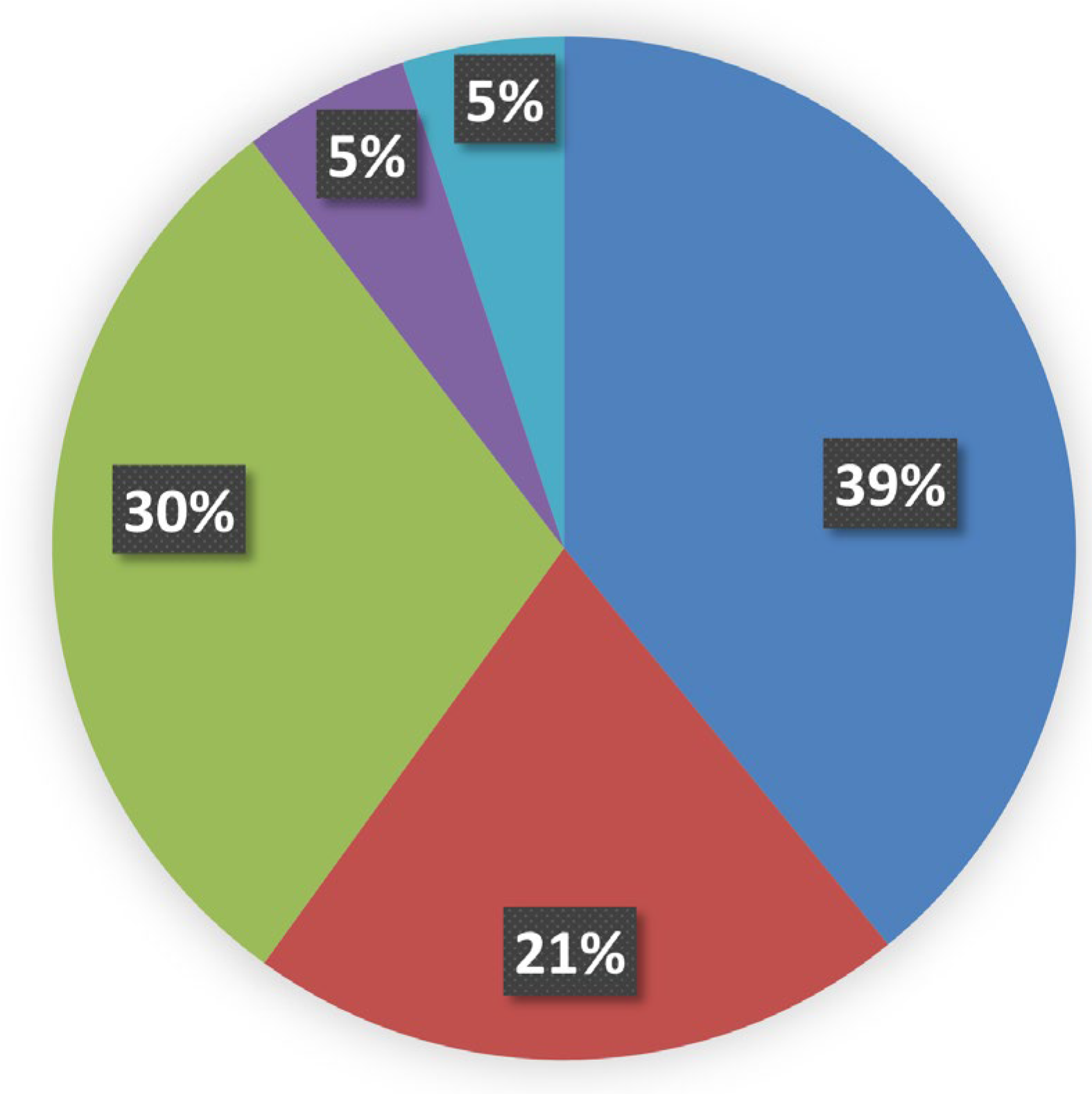
Figure 3.
Profile of amino acid replacements in the structurally defined proteins that are responsible of MRD. Histograms refer to percentual amino acid variations in the fully buried and fully exposed regions, see Figure 3, respectively in black and grey colors.
Figure 3.
Profile of amino acid replacements in the structurally defined proteins that are responsible of MRD. Histograms refer to percentual amino acid variations in the fully buried and fully exposed regions, see Figure 3, respectively in black and grey colors.
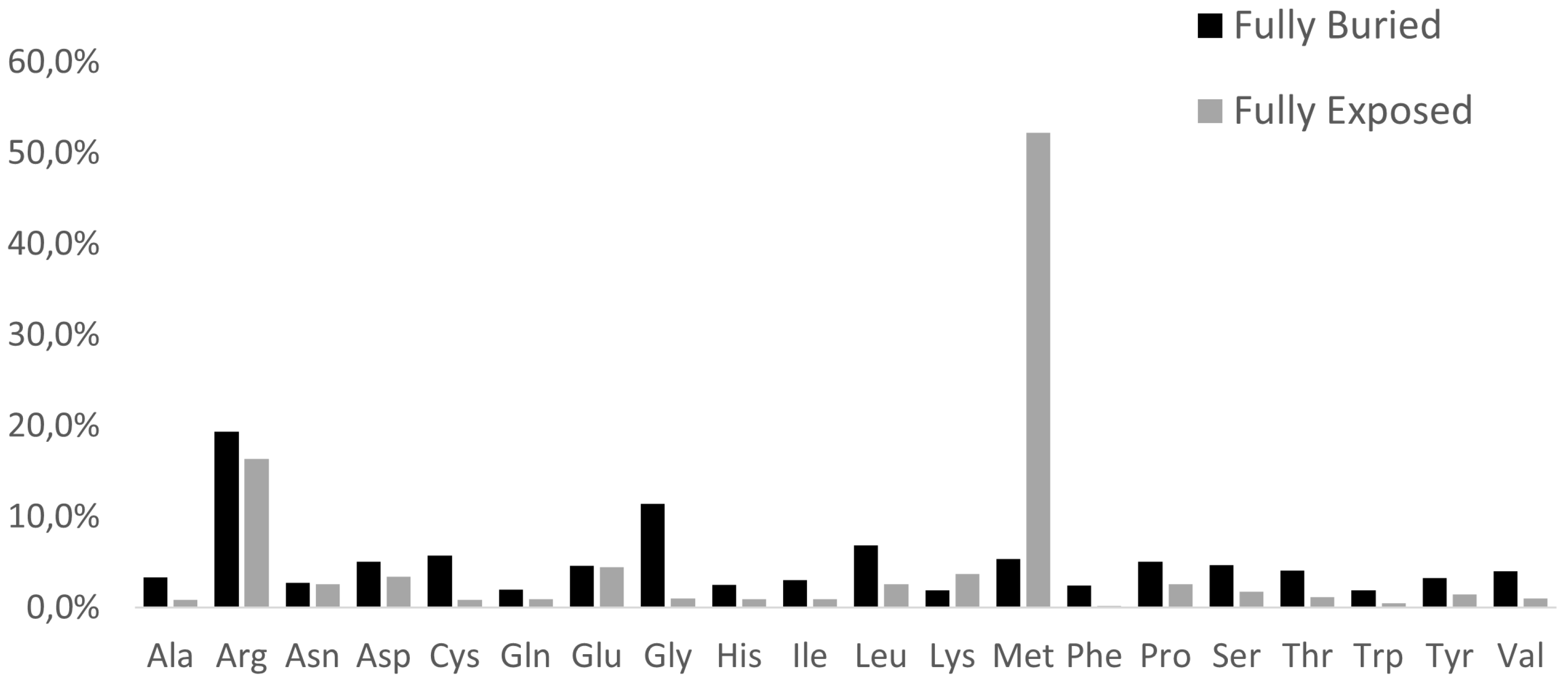
Figure 4.
Amino acid replacement matrices of fully buried (a) and fully exposed (b) mutations. As in Figure 2, rows describe how each of the natural amino acids has been replaced by column residues.
Figure 4.
Amino acid replacement matrices of fully buried (a) and fully exposed (b) mutations. As in Figure 2, rows describe how each of the natural amino acids has been replaced by column residues.
Figure 5.
The Orphanetta workflow.
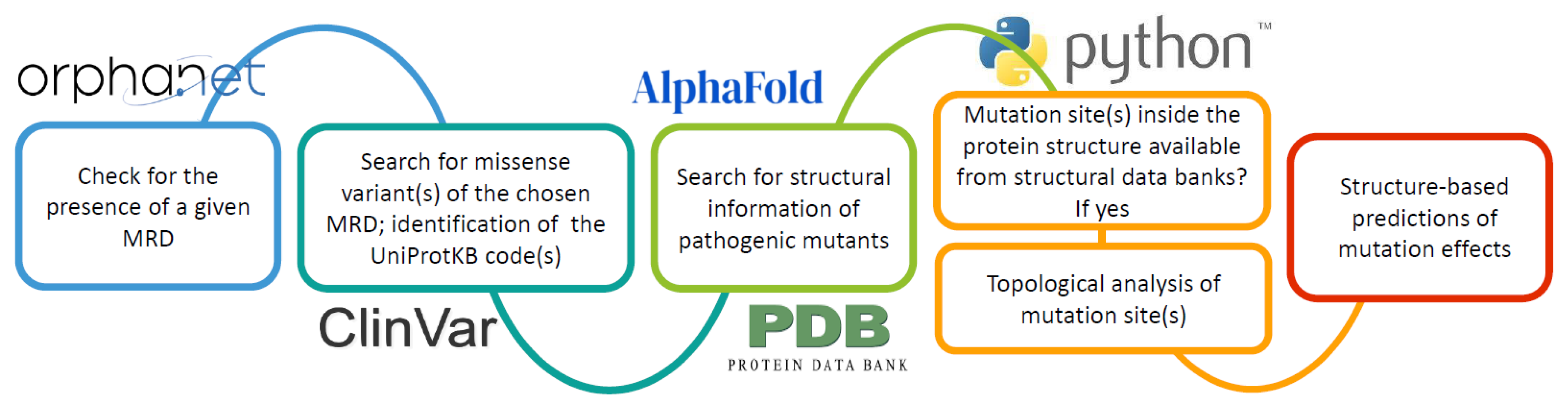

Table 1.
Physico-chemical properties of natural amino acids. Sizes of amino acid side chains are classified according to ref. 19.
Table 1.
Physico-chemical properties of natural amino acids. Sizes of amino acid side chains are classified according to ref. 19.
| Electrically charged side chains |
Polar uncharged side chains |
Hydrophobic side chains |
Special cases |
|---|---|---|---|
| Positive: Arg, His, Lys Negative: Asp, Glu |
Small size: Ser, Thr Large size: Asn, Gln, Tyr |
Small size: Ala, Val Medium size: Ile, Leu, Met Large size: Phe, Trp |
Cys; Gly; Pro |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



