
Submitted:
30 March 2024
Posted:
03 April 2024
You are already at the latest version
Abstract
The mechanism of immune activation and de-activation is very important. Host should trigger effective immunity against pathogen infection. And, host cannot trigger immunity against self-antigens that will lead to detrimental autoimmune disorders. Spleen and liver have opposite roles in mediating immune responses. Spleen is the organ involving in immune activation, and liver is the organ involving in immune de-activation. Sympathetic nerve system and parasympathetic nerve system have opposite roles in mediating immune responses. Sympathetic nerve system is involving in pro-inflammatory, and parasympathetic nerve system is involving in anti-inflammatory. ACTH and glucocorticosteroid have opposite roles in mediating immune responses. ACTH is involving in activating adaptive immunity with inhibiting innate immunity, and glucocorticosteroid is involving in activating innate immunity with inhibiting adaptive immunity. The molecular chaperons, heat shock proteins, which are induced by fever, have the vital roles in immune activation. On the contrary, IgD B cells and gamma-delta T cells are causing immune de-activation via the mechanism of clonal anergy. These mechanism can help us the know the work of immunological pathways better for better control of infectious diseases and autoimmune disorders.
Keywords:
ACTH
; steroid
; IgD
; gamma-delta T cells
; sympathetic
; parasympathetic
Background
Our immune system is a remarkable defense network, meticulously evolved to protect us from a vast array of threats while maintaining a delicate balance with our own cells and tissues. At its core, this intricate system employs two distinct mechanisms: the eradicable immune response, a powerful offensive against pathogens, and the tolerable immune response, a more measured approach to persistent challenges [1,2,3,4]. Together, these mechanisms orchestrate an intricate dance of cellular interactions, antibody production, and regulatory controls, ensuring our survival while preventing autoimmune disorders.
The Eradicable Immune Response: A Formidable Arsenal
When confronted with harmful invaders such as viruses, bacteria, parasites, or even malignant cells, the body unleashes its most potent defense: the eradicable immune response. This multifaceted approach is driven primarily by a class of antibodies known as Immunoglobulin G (IgG), produced by specialized B cells with the assistance of follicular helper T cells (Tfh)[1,2].
The Tfh cells, distinguished by their expression of the CXCR5 chemokine receptor and their secretion of interleukin-21, play a crucial role in initiating this response. They aid B cells in the germinal centers to undergo class-switching from the initially produced IgM antibodies to the more powerful IgG antibodies, a process mediated by the cytokine interleukin-21. Key transcription factors, such as BCL6 and STAT5B, orchestrate this intricate cellular choreography. Another subset of follicular helper T cells, Tfh13, is involving in inducing Th2b immunological pathway[2,4].
Within the eradicable immune response, there are four distinct branches, each tailored to combat specific types of pathogens:
The Th1 immunity is combating Intracellular Invaders. This pathway is the body's primary defense against intracellular microorganisms, such as certain bacteria, protozoa, and fungi. It involves a coordinated effort among various immune cells, including type 2 myeloid dendritic cells, type 1 innate lymphoid cells, type 1 macrophages, IFN-γ-producing CD4 T cells, CD8 T cells, type 1 invariant natural killer T cells, and IgG3 B cells. Driven by the cytokine interleukin-12 and governed by the transcription factors STAT4 and STAT1, the key effector cytokine, IFN-γ, activates M1 macrophages to generate free radicals that can kill ingested intracellular pathogens. This pathway is associated with type IV delayed-type hypersensitivity reactions.
The Th2 immunity is defending against parasites that is subdivided into Th2a and Th2b. This pathway is the body's primary defense against parasitic infections. The Th2a pathway combats endoparasites like helminths (worms), involving Langerhans cells, type 2 interleukin-25-inducing natural innate lymphoid cells, inflammatory eosinophils, mast cells-tryptase, interleukin-4/interleukin-5-producing CD4 T cells, iNKT2 cells, and IgG4 B cells. The Th2b pathway, on the other hand, targets ectoparasites like parasitic insects, employing Langerhans cells, type 2 interleukin-33-inducing inflammatory innate lymphoid cells, basophils, mast cells-tryptase/chymase, interleukin-4/interleukin-13-producing CD4 T cells, iNKT2 cells, and IgE B cells. Both pathways are driven by interleukin-4, interleukin-5 (Th2a), and interleukin-13 (Th2b), along with the transcription factors STAT6 and STAT1 (Th2a) or STAT3 (Th2b). These pathways are associated with type I allergic hypersensitivity reactions, with Th2a linked to IgG4-dominant allergies and Th2b linked to IgE-dominant allergies.
The Th22 immunity is battling extracellular pathogens. This pathway is the body's line of defense against extracellular microorganisms, such as certain bacteria, fungi, and protozoa. It involves type 1 myeloid dendritic cells as antigen-presenting cells, type 3 NCR+ innate lymphoid cells, neutrophils, interleukin-22-producing CD4 T cells, and IgG2 B cells. Driven by interleukin-1, interleukin-6, and TNF-α, and governed by STAT3 and STAT4, this pathway relies on the effector cytokine interleukin-22 to activate neutrophils, including phagocytosis and necroptosis, to destroy extracellular pathogens through mechanisms like free radical generation and membrane lipid peroxidation. This pathway is associated with type III immune complex-mediated hypersensitivity reactions.
The Thαβ immunity is defending against infectious particles. This pathway is the body's defense against infectious particles, such as viruses and prions. It involves plasmacytoid dendritic cells as antigen-presenting cells, interleukin-10-producing innate lymphoid cells, NK cells, interleukin-10-producing CD4 T cells, CD8 T cells, and IgG1 B cells. Driven by type 1 interferons and interleukin-10, and governed by STAT1, STAT2, and STAT3, this pathway utilizes interleukin-10 as its key effector cytokine. The primary effector mechanism is antibody-dependent cellular cytotoxicity (ADCC), where NK cells, armed with IgG1 antibodies, can induce apoptosis in virus- or prion-infected cells, disrupting the replication and propagation of these infectious particles. This pathway is associated with type II antibody-dependent cellular cytotoxic hypersensitivity reactions[3,5].
The Tolerable Immune Response: A Balanced Approach
While the eradicable immune response is a potent defense mechanism, there are times when complete eradication of a pathogen may cause severe organ damage or failure. In such situations, the body employs a more measured approach known as the tolerable immune response. This response is predominantly driven by regulatory CD4+CD25+ T cells (Treg cells) and is characterized by the production of Immunoglobulin A (IgA) antibodies[1,2,4].
The Treg cells, expressing the transcription factor FOXP3, secrete the cytokine TGF-β, which activates the STAT5 and STAT6 transcription factors, initiating the tolerable immune response. TGF-β also plays a crucial role in inducing B cells to undergo class-switching from IgM to IgA antibodies. Other key players in this response include regulatory dendritic cells, regulatory B cells, and regulatory innate lymphoid cells.
Similar to the eradicable immune response, the tolerable immune response can be further subdivided into four distinct pathways:
The Th1-like immunity is tolerating intracellular pathogens. This pathway is the body's tolerable response to intracellular microorganisms, such as certain bacteria, fungi, and protozoa. It involves type 2 myeloid dendritic cells as antigen-presenting cells, type 1 non-cytotoxic innate lymphoid cells, macrophages, IFN-γ/TGF-β-producing CD4 T cells, CD8 T cells, iNKT1 cells, and IgA1 B cells. Driven by interleukin-12 and TGF-β, this pathway is associated with type IV delayed-type hypersensitivity reactions.
The Th9 immunity is tolerating parasites. This pathway is the body's tolerable response to parasitic infections, encompassing both endoparasites (helminths) and ectoparasites (insects). It involves Langerhans cells as antigen-presenting cells, TSLP-inducing type 2 innate lymphoid cells, regulatory eosinophils, basophils, mast cells, interleukin-9-producing CD4 T cells, iNKT2 cells, and IgA2 B cells. Driven by interleukin-4 and TGF-β, this pathway is associated with type I allergic hypersensitivity reactions.
The Th17 immunity is tolerating extracellular pathogens. This pathway is the body's tolerable response to extracellular microorganisms, such as certain bacteria, fungi, and protozoa. It involves type 1 myeloid dendritic cells as antigen-presenting cells, type 3 non-cytotoxic innate lymphoid cells, neutrophils, interleukin-17-producing CD4 T cells, iNKT17 cells, and IgA2 B cells. Driven by interleukin-6 and TGF-β, this pathway is associated with type III immune complex-mediated hypersensitivity reactions.
The Th3 immunity is tolerating infectious particles. This pathway is the body's tolerable response to infectious particles, such as viruses and prions. It involves plasmacytoid dendritic cells as antigen-presenting cells, interleukin-10-producing innate lymphoid cells, NK cells, interleukin-10/TGF-β-producing CD4 T cells, CD8 T cells, and IgA1 B cells. Driven by TGF-β and interleukin-10, and governed by STAT1, STAT2, STAT3, and STAT5, this pathway is associated with type II antibody-dependent cellular cytotoxic hypersensitivity reactions.
The Spleen and Liver: Opposing Roles in Immune Regulation
The spleen and liver are two crucial abdominal organs that play opposing roles in immune activation and deactivation. Splenectomy, the removal of the spleen, is sometimes performed in severe autoimmune hematological disorders like immune thrombocytopenia. However, this procedure can leave patients more susceptible to infections, especially those caused by encapsulated bacteria such as streptococcus, meningococcus, or Hemophilus.
The spleen is a key organ for immune activation, producing IgM antibodies against bacterial infections. Within the spleen, white pulps contain B lymphocyte germinal centers, while the surrounding red pulps house macrophages that digest old red blood cells and platelets. When bacterial antigens enter the lymph or blood drainage to the spleen, the organ can sense these antigens and activate B cells to produce IgM antibodies.
Immature B lymphocytes are formed in the bone marrow and enter the spleen for further maturation, potentially becoming spleen marginal B-2 lymphocytes that produce T cell-independent IgM antibodies against bacterial polysaccharide or glycolipid antigens. This explains why splenectomy can increase the risk of infections by encapsulated bacteria with polysaccharide antigens. In the spleen's white pulps, follicular B-2 lymphocytes also react to T cell-dependent antigens to produce IgM and later IgG antibodies.
In the red pulps, macrophages provide additional immune stimulation. The degraded products of digested red blood cells, including hemoglobin and heme constituents, act as potent immune stimulants, aiding in the maturation and activation of B-2 cells to produce IgM antibodies[6,7,8]. The spleen is also home to resident memory IgM B cells and abundant heme oxygenase (HO-1) in its macrophages[8].
Heme oxygenase (HO-1) catalyzes the breakdown of heme into ferrous iron, carbon monoxide, and biliverdin, which readily becomes bilirubin[9,10]. These end products – carbon monoxide, biliverdin, and bilirubin – act as immunosuppressants, with carbon monoxide exhibiting anti-inflammatory properties in contrast to the immune-stimulating effects of nitrogen monoxide generated by induced nitric oxide synthase (iNOS)[11].
After leaving the spleen macrophages, bilirubin has a strong affinity for serum albumin and is transported to the liver through the spleen vein. Along with IgM antibodies generated from the spleen's white pulps, these molecules enter the liver's portal vein system, joining the inferior mesenteric vein and superior mesenteric vein, which carry digested food constituents from the stomach and intestines.
Once in the liver, the portal veins become sinusoid vessels containing Kupffer macrophages. These cells digest antigen-IgM antibody complexes, neutralizing bacterial pathogens that may have contaminated food contents. They also clear antigen-IgG antibody complexes produced by spleen follicular B-2 cells, explaining the hypogammaglobulinemia observed in chronic liver disease with Kupffer cell dysfunction.
Safe, digested food contents are not engulfed by Kupffer cells but metabolized into useful molecules within the liver. The liver also contains γδ T cells that mediate clonal anergy, a process that will be discussed later. Additionally, the unconjugated bilirubin becomes water-soluble and conjugated in the liver, aiding in the process of clonal anergy to food molecules.
Thus, the liver serves as the major organ for inducing food antigen tolerance, preventing immune reactions to common food components such as small peptides or glycolipids. Bilirubin, with its immunosuppressive effects, can be stored in the gallbladder and secreted into the intestine, further contributing to clonal anergy of intestinal γδ T cells against food antigens.
Most conjugated bilirubin in the large intestine is metabolized by intestinal bacteria to urobilinogen, which can be reabsorbed and re-secreted in the enterohepatic circulation. Bilirubin's immunosuppressive effects may explain why liver failure patients with elevated bilirubin levels have higher incidences of sepsis, as its normal physiological function involves immune modulation.
The intestine also plays a role in mucosal immune reactions. The Peyer's patch in the intestine allows B lymphocytes to produce tolerable IgA antibodies against bacterial antigens from the intestinal tract. The resulting IgA-bacterial antigen complexes do not enter the inferior mesenteric vein or superior mesenteric vein, preventing an increase in IgA levels in the portal vein.
The Autonomic Nervous System: Striking the Chord of Inflammation
The autonomic nervous system, with its two major components – the sympathetic and parasympathetic branches – plays a vital role in immune regulation, akin to a skilled musician fine-tuning the resonance of a stringed instrument[12,13]. The sympathetic nervous system, fueled by the neurotransmitters dopamine, epinephrine, and norepinephrine, strikes a pro-inflammatory chord, amplifying the body's defensive response[14,15,16,17,18,19].
Dopamine, in particular, takes center stage as the key mediator in triggering follicular helper T cells, the initiators of the eradicable host immune responses[20]. Like a virtuoso conductor, dopamine orchestrates the intricate dance of cellular interactions that culminate in the production of powerful IgG antibodies, the body's frontline soldiers against invading pathogens. Norepinephrine is the next synthesized molecule after dopamine, and it has the potential to promote Thαβ anti-viral immunity while suppressing Th1 anti-intracellular micro-organism immunity[21,22,23]. Finally, epinephrine has the potential to promote Th22/Th17 anti-bacterial immunity while suppressing Th2 anti-parasite immunity[24,25,26,27,28,29,30]. Sympathetic nerve system is activated in emergent situations like virus or extracellular bacterial infection. Thus, Thαβ and Th22/Th17 immunities are up-regulated in sympathetic nerve system activation while sacrificing more chronic Th1 or Th2 immunities.
The sympathetic nervous system's influence extends further, promoting the various arms of the immune response – Th1, Th2, Th17, and Thαβ – akin to a maestro coaxing forth a symphony of defensive melodies[19,31,32,33]. However, like all great compositions, there exists a delicate counterpoint – a negative feedback loop that tempers the exuberance of autocrine catecholamines, preventing unchecked lymphocyte proliferation[12,34].
In contrast, the parasympathetic nervous system, with its cholinergic nerves, takes on the role of the soothing balladeer, tempering the inflammatory fervor with its anti-inflammatory melodies. The cholinergic anti-inflammatory pathway acts as the efferent, or motor arm, of the inflammatory reflex, the neural circuit that responds to and mediates the inflammatory response[35].
The vagus nerve, the tenth cranial nerve, serves as the conductor's baton, innervating the celiac ganglion, the site of origin for the splenic nerve. Its efferent stimulation slows the heart rate, induces gastrointestinal motility, and, perhaps most importantly, inhibits the production of the pro-inflammatory cytokine TNFα in the spleen.
The neurotransmitter acetylcholine, released by the efferent pathway of the vagus nerve, acts as the soothing soloist, interacting with the α7 subunit of the nicotinic acetylcholine receptor (α7 nAChR) expressed on the cell membranes of macrophages and other cytokine-secreting cells. Through this intricate dance of molecular interactions, acetylcholine activates intracellular signal transduction pathways, effectively silencing the release of pro-inflammatory cytokines[36].
Acetylcholine's influence extends further, inhibiting the various arms of the immune response – Th1, Th2, Th17, and Thαβ – in a harmonious counterpoint to the pro-inflammatory melodies of the sympathetic nervous system[37,38]. Moreover, acetylcholine engages in an augmented interaction with TGFβ, a potent immunomodulatory cytokine for initiating tolerable immunities, further reinforcing its role as the soothing balm in the symphony of immunity[39,40].
It is worth noting that the opposing forces of the sympathetic and parasympathetic nervous systems are not mere discord but rather a delicate interplay, a harmonic tension that underpins the exquisite balance of the immune response. The sympathetic nerve employs cyclic AMP (cAMP) as its intracellular second messenger, while the parasympathetic nerve utilizes cyclic GMP (cGMP)[41,42,43]. Though both are derived from purines, the direction of their NH2 groups is opposite, akin to the complementary tones of a major and minor chord.
This natural competition between cAMP and cGMP, mediated by their respective effector proteins – protein kinase A and protein kinase G – governs the intricate interplay between the two autonomic branches[44,45,46]. The substrate amino acid sequence consensus motifs for these kinases, while sharing similarities, bear subtle distinctions, akin to the nuanced phrasing that separates a masterpiece from a mere melody.
Ultimately, the sympathetic nerve system, with its pro-inflammatory "fear-or-fight" posture, and the parasympathetic nerve system, with its anti-inflammatory "rest and digest" disposition, strike a delicate harmony, regulating the body's inflammatory response with the precision of a finely tuned orchestra.
The Endocrine System: Modulating the Tempo of Immunity
Like a skilled composer weaving multiple instruments into a cohesive whole, the endocrine system, through the intricate interplay of hormones, modulates the tempo and intensity of the immune response. At the heart of this intricate symphony lies the hypothalamic-pituitary-adrenal (HPA) axis, a tripartite ensemble that orchestrates the delicate balance between inflammation and immunosuppression.
The hypothalamus, the conductor's podium, initiates the performance by secreting corticotropin-releasing hormone (CRH), a signal that stimulates the pituitary gland to release adrenocorticotropic hormone (ACTH). ACTH, in turn, acts as the rousing crescendo, prompting the adrenal cortex to secrete glucocorticosteroids, the potent immunosuppressants that form the backbone of the stress response.
Glucocorticosteroids, akin to a masterful soloist, exert a profound and far-reaching influence on the immune system. Their primary function is immunosuppression, a carefully modulated restraint that prevents the body's defensive forces from spiraling into unchecked autoimmunity. Yet, like all great performances, there are intricate nuances and counterpoints.
Glucocorticosteroids, while suppressing adaptive immunity, simultaneously upregulate the body's innate defenses, a strategic maneuver akin to a composer shifting the emphasis from one section of the orchestra to another[47,48,49]. This deft touch allows the body to mount a rapid, non-specific response to acute infection, particularly bacterial threats, through the upregulation of neutrophils and other innate immune cells[50].
However, this shift in emphasis comes at a cost – the profound suppression of lymphocytes, the very cells that orchestrate the body's adaptive, antigen-specific defenses. It is a calculated trade-off, a strategic sacrifice of specificity for the sake of immediate survival, a testament to the intricate choreography that underpins the immune response.
Yet, like all great compositions, there exists a delicate balance, a counterpoint that prevents the performance from spiraling into dissonance. The HPA axis employs a negative feedback loop, a self-regulating mechanism akin to a conductor subtly adjusting the tempo and dynamics of the orchestra. Increased glucocorticosteroid levels suppress the production of ACTH, modulating the intensity of the stress response and preventing an unbridled crescendo of immunosuppression.
The interplay between ACTH and glucocorticosteroids is itself a delicate counterpoint, a harmonic tension that underpins the exquisite balance of the immune response. While glucocorticosteroids suppress adaptive immunity, ACTH stimulates lymphocyte activity, upregulating the very defenses that its hormonal counterpart seeks to restrain[51,52]. This opposing force, reminiscent of the tension between major and minor keys, is a testament to the intricate choreography of the endocrine system's influence on immunity.
ACTH's influence extends further, inhibiting Th17-like innate immune reactions, a subtle modulation akin to a skilled composer introducing unexpected harmonic progressions[53,54,55,56]. This delicate interplay between ACTH and glucocorticosteroids, a Yin-Yang regulation of endocrine-mediated immune control, ensures that the body's defensive forces remain poised, neither overreacting to innocuous threats nor succumbing to unchecked autoimmunity.
While CRH, ACTH, and glucocorticosteroids form the core ensemble of the HPA axis, the performance is enriched by the contributions of other hormonal soloists. CRH, the conductor's opening cue, can induce innate immunity through the upregulation of pro-inflammatory cytokines, a stirring prelude that sets the stage for the ensuing immune response[57,58,59,60].
However, like all great compositions, there are intricate nuances and counterpoints that prevent the performance from becoming a mere cacophony of sound. CRH's influence is primarily restricted to the central nervous system, its role in the systemic circulation less pronounced than that of its hormonal counterparts, ACTH and glucocorticosteroids.
Immune Activation: The Clarion Call to Arms
When the body encounters a pathogenic threat, a clarion call resonates through its cellular symphony, a signal that mobilizes the intricate machinery of the immune system. This call to arms takes the form of a fever, a carefully orchestrated rise in core body temperature above the normal 37°C, a strategic maneuver that activates a crucial ensemble of molecular players – the heat shock proteins.
At the cellular level, heat shock proteins assume the role of sentinels, their activation triggering a cascade of defensive responses that culminate in the generation of adaptive immunity, including the production of vital antibodies[61]. These molecular chaperones, including HSP60 and HSP70, bind to newly synthesized proteins, facilitating their proper folding and ensuring the generation of suitable antigens to trigger the adaptive immune response. HSP70 can help HSP60 and HSP10 to form a barrel-like structure which aids protein folding. HSP70 also helps to translocate unfolding proteins. The chaperon-assisted protein folding needs ATP energy. Thus, protein folding by chaperon aid occurs in a special situation. Majority of proteins are folded spontaneously in normal situations.
In the face of viral invasion, a particularly insidious threat, the heat shock proteins of the host cell bind to the newly synthesized viral proteins, ensuring their proper three-dimensional structure independent of the viral nucleic acids. This strategic maneuver sets the stage for the next critical step – the activation of the immunoproteasome, a specialized molecular complex that digests the pathogenic proteins, generating the precise peptide antigens necessary for antigen presentation, T cell recognition, and ultimately, antibody production.
The heat shock proteins' influence extends beyond mere chaperoning, assuming a strategic role in regulating the body's hormonal defenses. HSP90, once activated, binds to steroid receptors, preventing their interaction with glucocorticosteroids and their subsequent binding to target DNA. Before the mature complex of HSP90 and steroid receptor, HSP70 and HSP40 can aid to form an immature complex to promote the final mature complex formation[62]. This deft maneuver effectively short-circuits the suppressive effects of steroid signaling on adaptive immunity, allowing the body's defensive forces to mount an unencumbered response.
Moreover, heat shock proteins can bind to Toll-like receptors, initiating a cascade of signaling events that stimulate the host's immune response against a diverse array of pathogens. This multifaceted role, encompassing chaperoning, antigen processing, and immunomodulation, solidifies the heat shock proteins' position as the indispensable maestros of immune activation.
Antibody-Dependent Enhancement: A Cautionary Tale
While the immune system's intricate choreography is a marvel of evolutionary design, there are instances where this finely tuned symphony can veer into dissonance, a phenomenon known as antibody-dependent enhancement (ADE). In several viral infections, the very antibodies meant to neutralize the pathogen can paradoxically exacerbate the disease, a perplexing and potentially dangerous scenario that underscores the delicate balance that underpins immune function.
The underlying mechanisms of ADE have been the subject of intense debate, with hypotheses ranging from antibody engorgement by Fc receptors to complement receptor-mediated uptake by monocytes[63,64]. However, a compelling theory emerges, one that highlights the critical importance of eliciting the appropriate antibody response in the development of effective vaccines against viral pathogens[65].
The crux of this theory lies in the observation that not all antibodies are created equal in their ability to counter viral threats. While IgG1 antibodies are the body's primary defense against viral invaders, other antibody isotypes, such as IgG3, can inadvertently aid the pathogen's entry and propagation within host cells[66].
When monocytes or macrophages encounter these non-neutralizing antibodies bound to viral particles, they engulf the complexes, unwittingly ushering the viral cargo into the very cells meant to destroy it. Once within these immune sentinels, the virus can replicate unimpeded, triggering a cascade of pro-inflammatory cytokines that can exacerbate the disease's severity.
This cautionary tale underscores the critical importance of eliciting the appropriate IgG1 antibody response in the development of effective viral vaccines. The Thαβ immune pathway, distinct from the traditional Th1 pathway, is the body's primary defense against viral pathogens, and it is this pathway that drives the production of the coveted IgG1 antibodies.
In contrast, the Th1 pathway, while formidable against intracellular bacterial and protozoal threats, predominantly elicits IgG3 antibodies, which may inadvertently contribute to ADE in the context of viral infections. It is a subtle yet critical distinction, one that highlights the intricate nuances that underpin the immune system's exquisite choreography.
To develop truly effective vaccines against viral threats, researchers must carefully navigate this intricate symphony, eliciting the appropriate antibody response through the precise modulation of the Thαβ pathway. Only by striking the right chords can we hope to harmonize the body's defenses against these insidious pathogens, averting the dissonance of antibody-dependent enhancement and ensuring a triumphant performance in the ongoing battle against viral diseases.
Clonal Anergy: The Art of Self-Tolerance
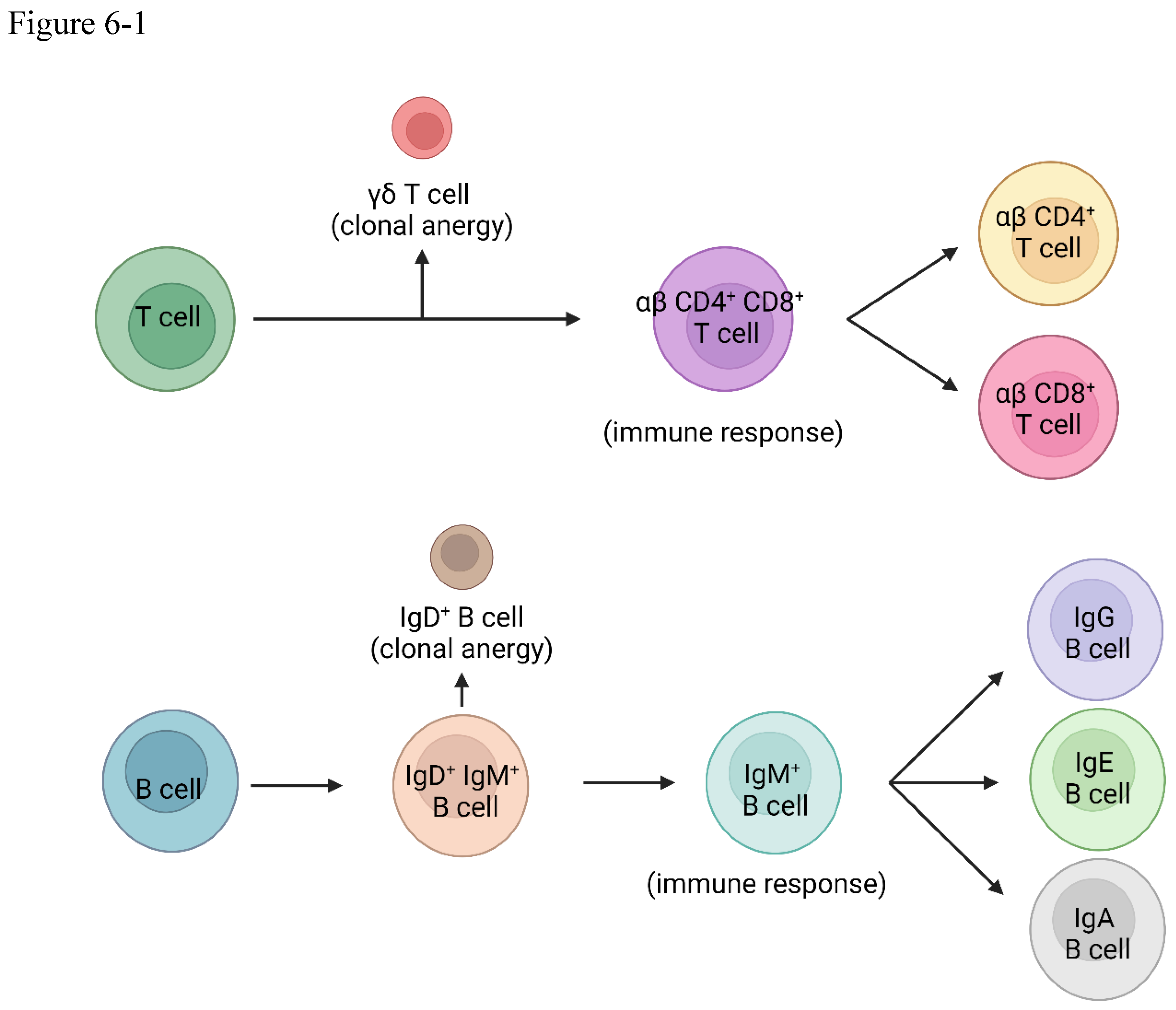
In the grand symphony of immunity, one of the most exquisite movements is the body's ability to distinguish self from non-self, a delicate dance that prevents the immune system from mounting an unnecessary and potentially devastating response against its own tissues. This intricate choreography, known as clonal anergy, is orchestrated by two distinct cell types: gamma-delta (γδ) T cells and IgD B cells, acting as the sentinels of self-tolerance.
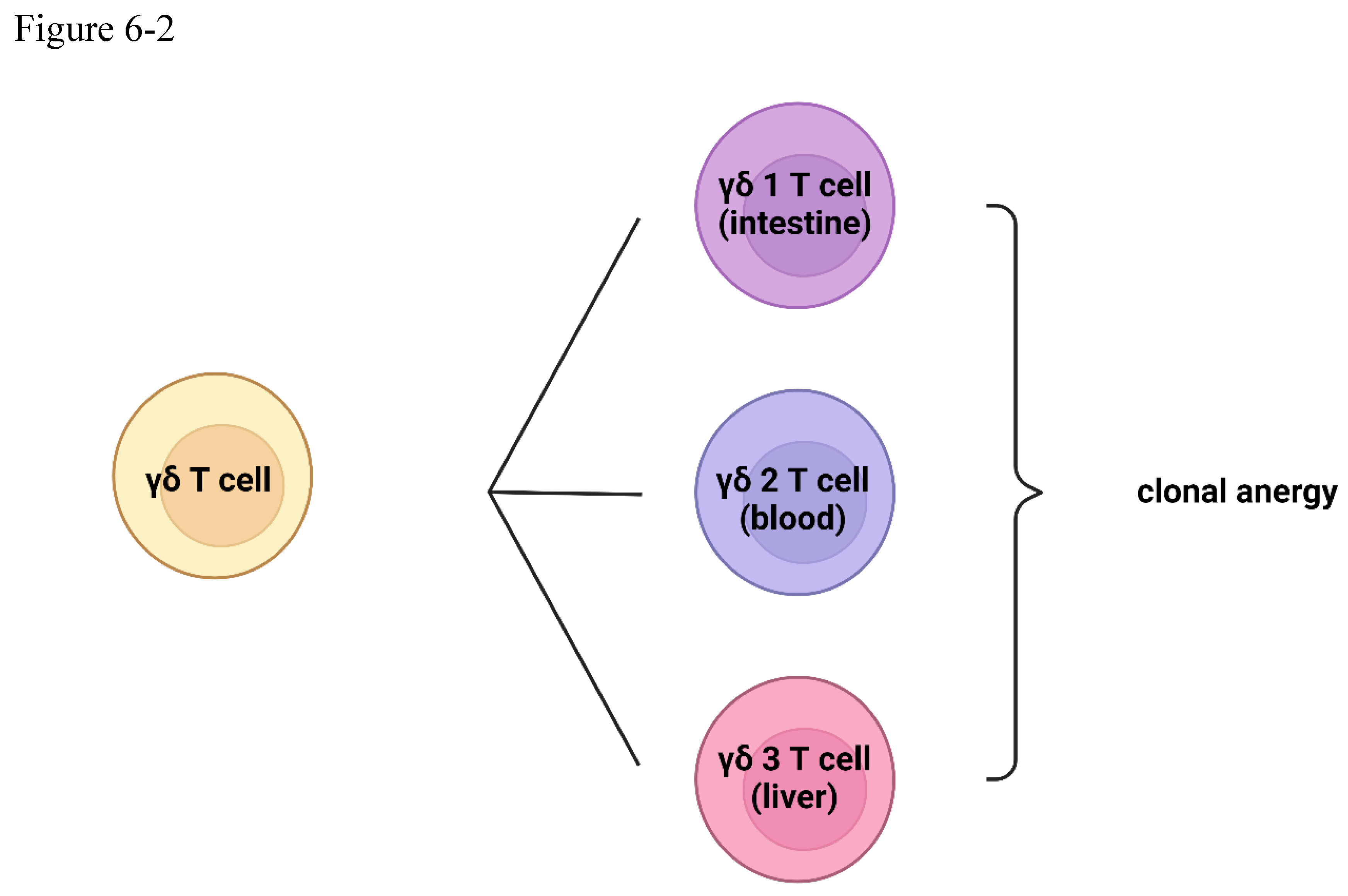
The gamma-delta T cells take center stage early in the immune response, developing in the thymus before the emergence of their more abundant alpha-beta (αβ) counterparts[67]. If, during this formative stage, a T cell clone recognizes a self-antigen, it is not silenced but rather redirected, differentiating into a gamma-delta T cell. This strategic maneuver effectively prevents the subsequent development of alpha-beta T cells that could potentially mount an autoimmune response against the body's own tissues[68,69,70,71,72,73,74,75]. The TCR signaling of alpha-beta T cells and gamma-delta T cells are different, and gamma-delta T cells CD3 confirmation change cannot be triggered by antigens[76,77,78,79,80,81].
In a harmonious counterpoint, the B lymphocytes employ a similar mechanism to maintain self-tolerance[82]. Mature B cells co-express IgD and IgM antibodies on their surface, each isotype serving a distinct purpose in the intricate choreography of immunity[83]. If a self-antigen is recognized by the IgD antibody, it triggers clonal anergy, effectively preventing an immune response against the body's own molecules[84,85,86].
However, should a foreign antigen be detected by the IgM antibody, a different sequence unfolds – one that initiates a robust immune response against the pathogenic invader[87]. This strategic distinction allows the B cell to undergo antibody isotype class-switching, producing IgG, IgE, or IgA antibodies tailored to the specific threat, further refining the body's defensive arsenal. Gamma-delta T cells also can aid B cell tolerance[88].
This intricate system of checks and balances, orchestrated by the gamma-delta T cells and IgD B cells, represents a masterful composition, a delicate interplay of cellular choreography and molecular recognition that ensures the immune system remains vigilant against external threats while maintaining a delicate tolerance towards the body's own tissues.
It is a testament to the exquisite complexity of the human immune system, a masterpiece of evolutionary design that continues to captivate and inspire scientists and medical professionals alike. Like a great symphony, each component, from the lowliest cell to the most intricate signaling pathway, plays an indispensable role in the grand performance, harmonizing to protect us from harm while maintaining the delicate balance that underpins our very existence.
Conclusion: A Symphony of Life
As we stand in awe of the human immune system's intricate choreography, we are reminded of the profound beauty and complexity that underpins the symphony of life. From the delicate interplay of neurotransmitters and hormones that modulate the tempo of inflammation to the exquisite cellular mechanisms that orchestrate self-tolerance, each aspect of this remarkable defense network is a testament to the elegance of evolutionary design. The spleen and the liver have the opposite roles in triggering immune reactions and causing immune tolerance.
The autonomic nervous system, with its sympathetic and parasympathetic branches, strikes the chords of immune activation and deactivation, their opposing forces harmonizing in a delicate counterpoint that maintains the exquisite balance between protection and tolerance. The endocrine system, through the intricate interplay of the HPA axis and its hormonal soloists, modulates the intensity and tempo of the immune response, ensuring that the body's defenses remain poised, neither overreacting to innocuous threats nor succumbing to the perils of autoimmunity.
At the cellular level, a myriad of molecular players take the stage, from the heat shock proteins that sound the clarion call to arms in the face of pathogenic threats to the gamma-delta T cells and IgD B cells that orchestrate the delicate dance of self-tolerance. Each component, each cellular interaction, each signaling pathway, is an indispensable movement in the grand symphony of immunity.
Figure Legends
Figure 1.
The immune activation and tolerance mechanism in the liver and spleen.
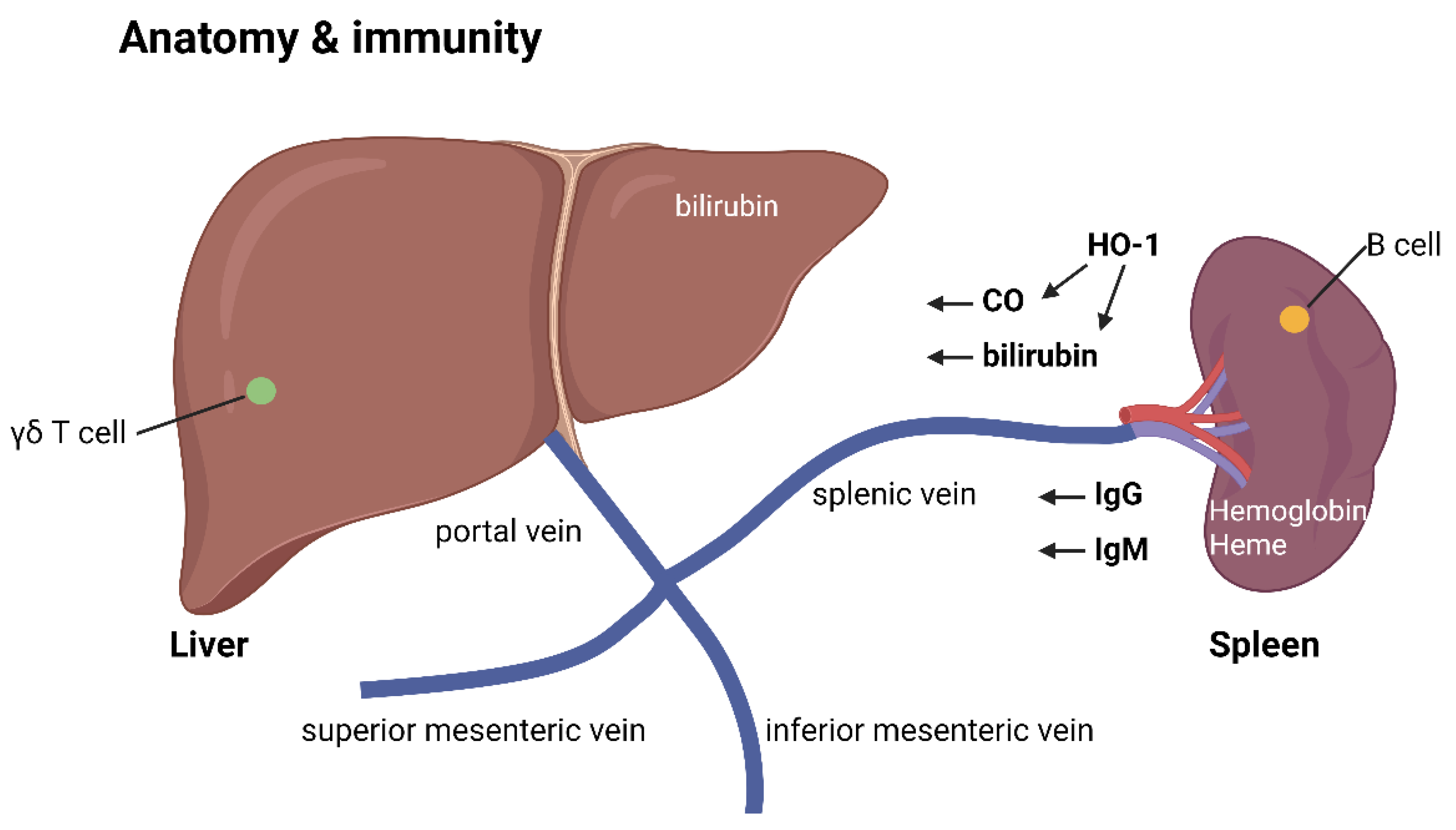
Figure 2.
The inflammation and anti-inflammation relations in autonomic nerve system.
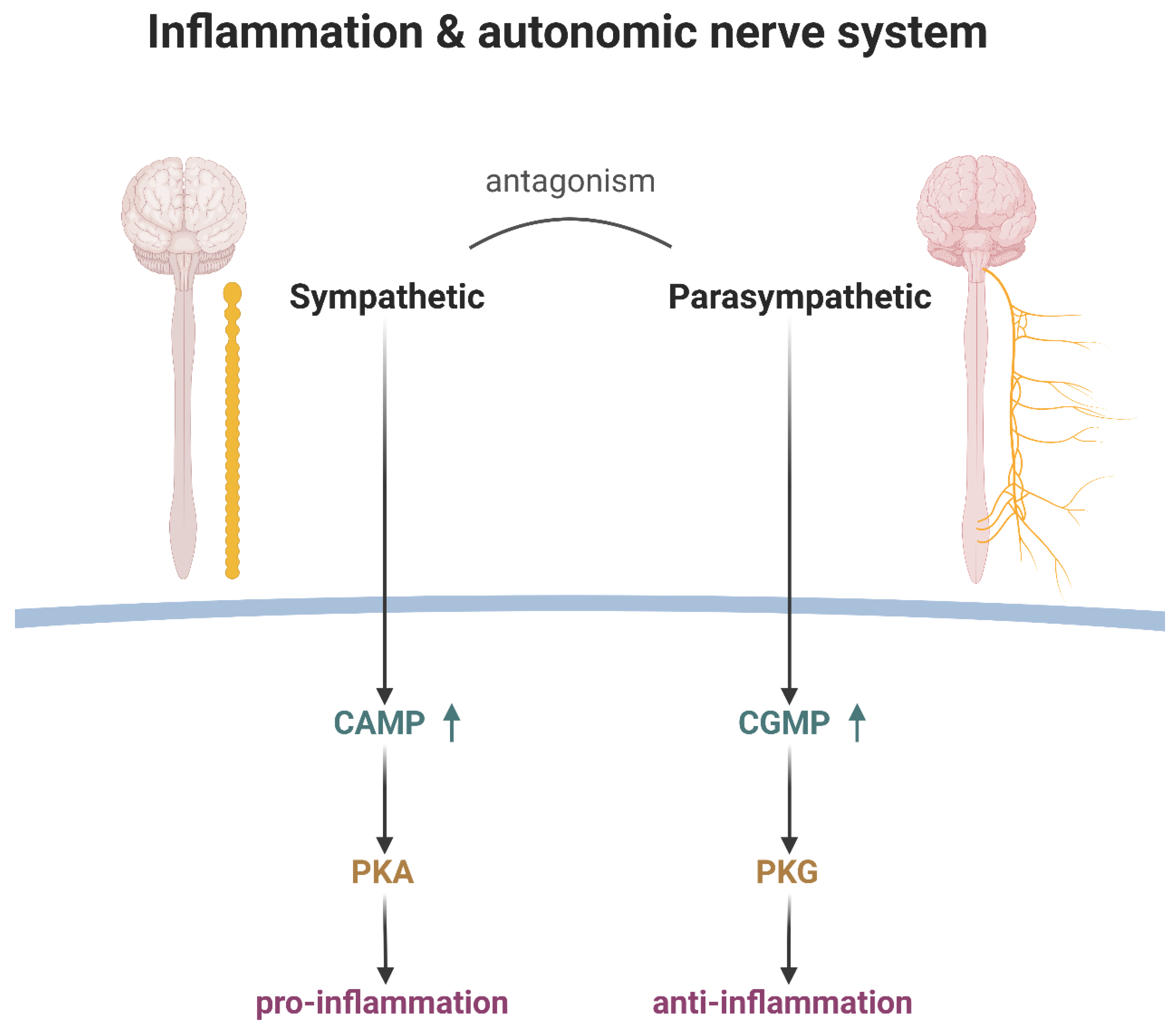
Figure 3.
The HPA axis and immunity.
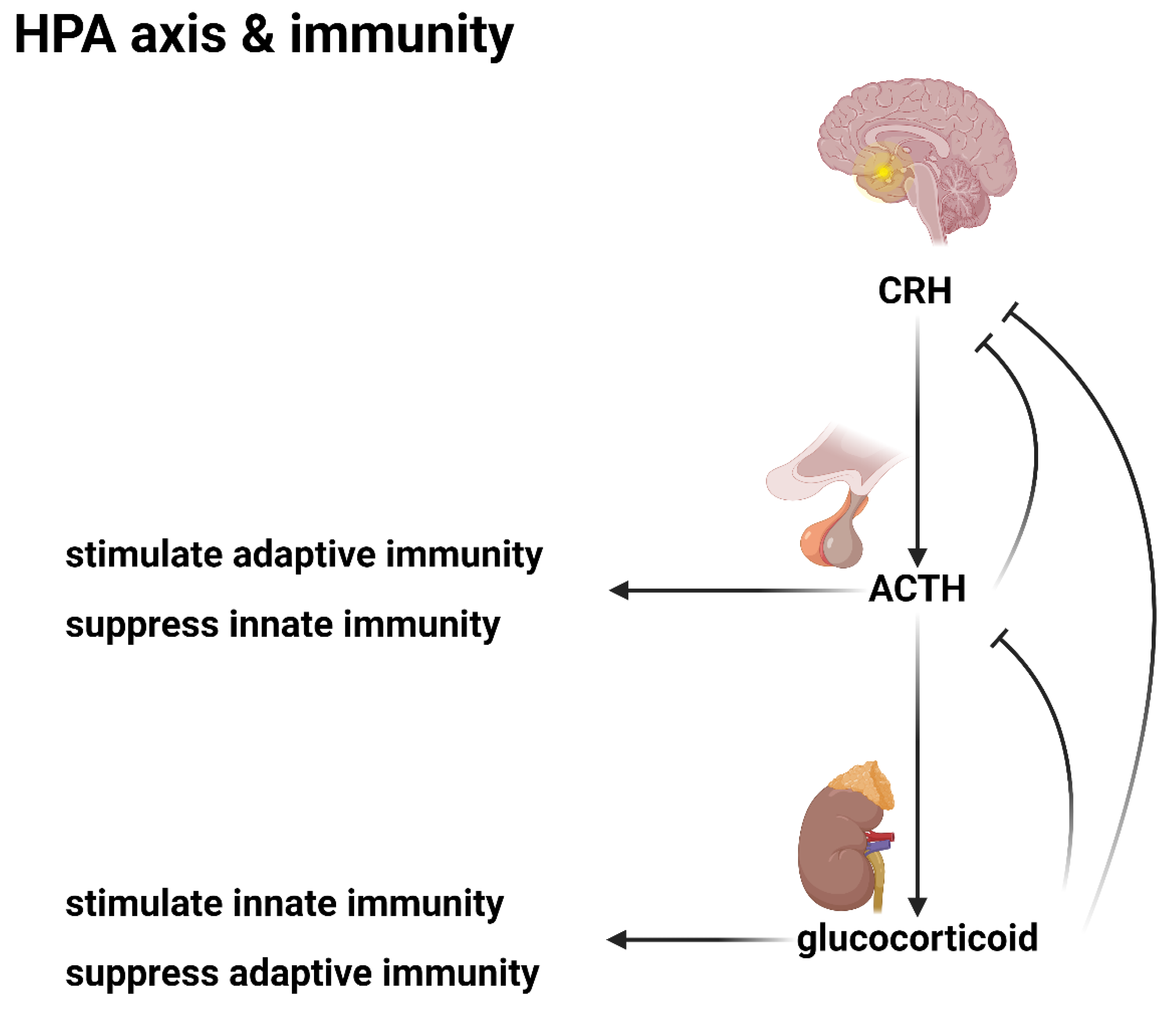
Figure 4.
(4-1) HSP70 and HSP60/HSP10 complex (4-2) HSP70/40 and HSP90/SR (4-3) steroid receptor.
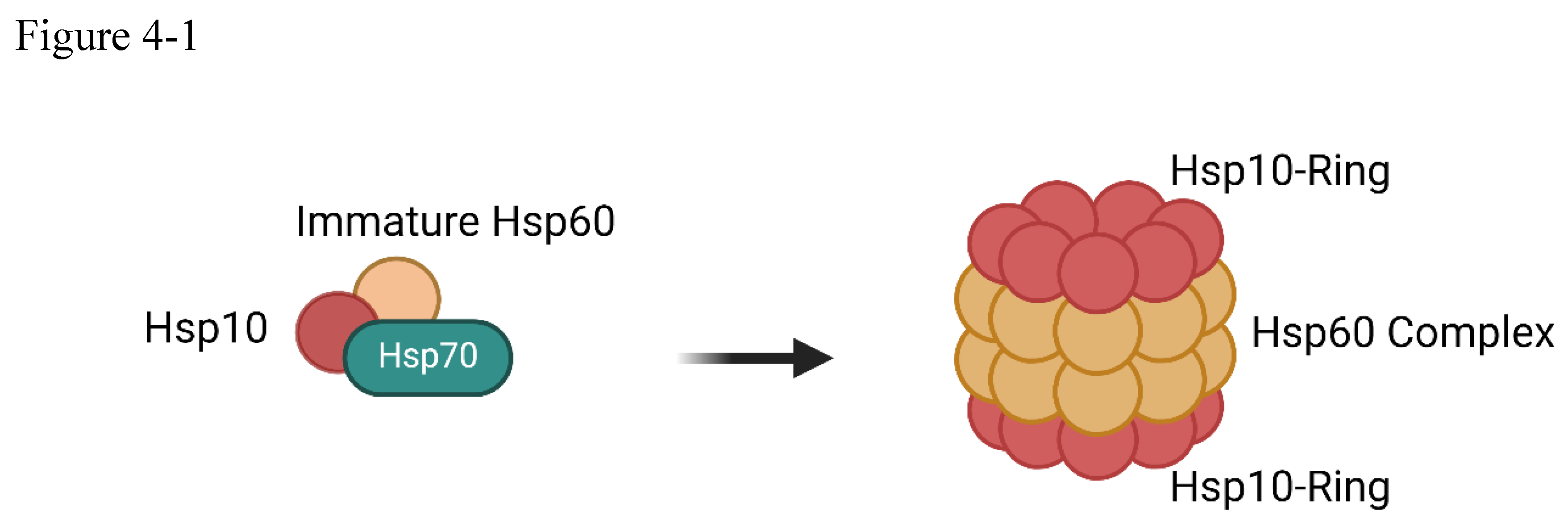
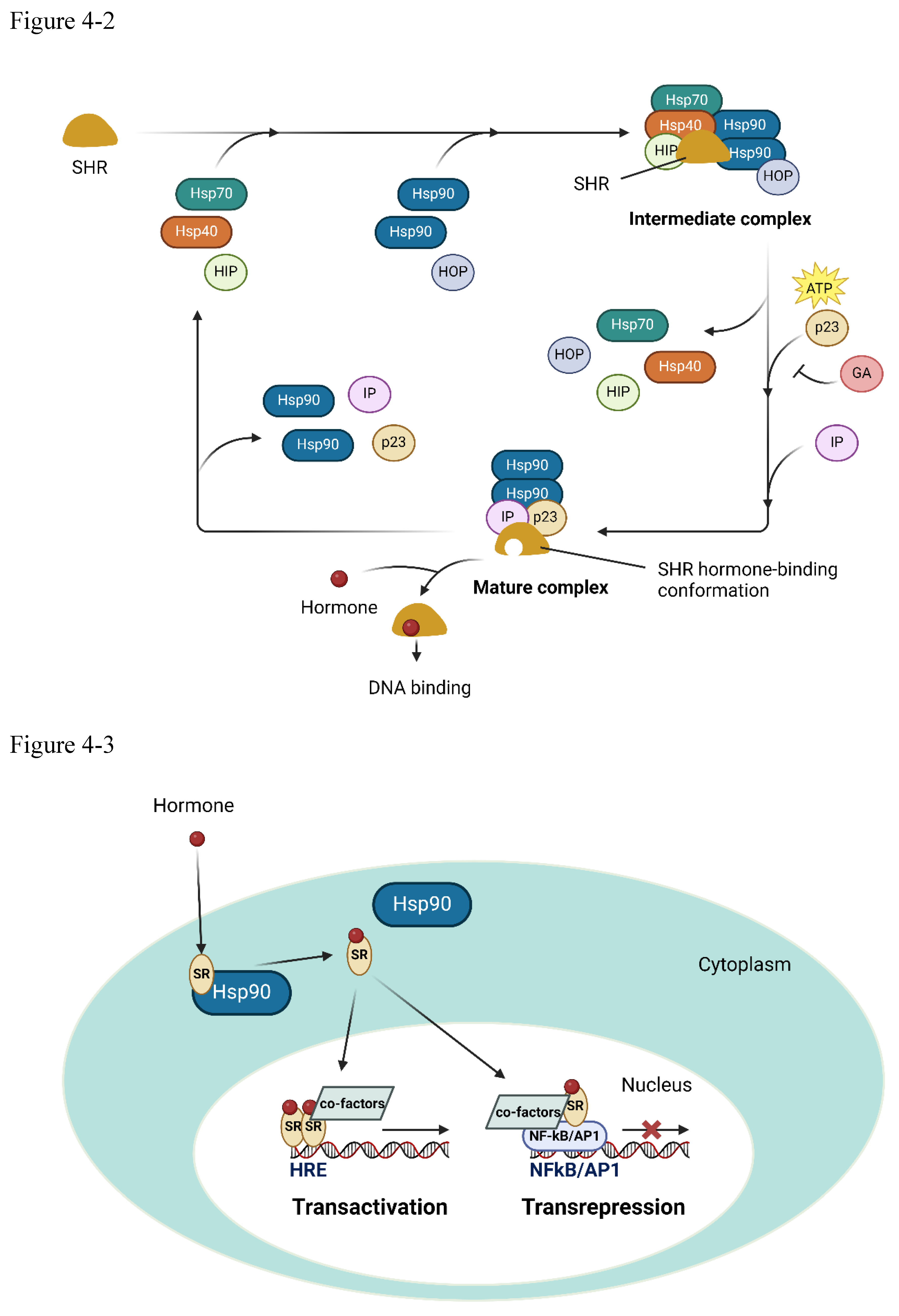
Figure 5.
Antibody dependent enhancement.
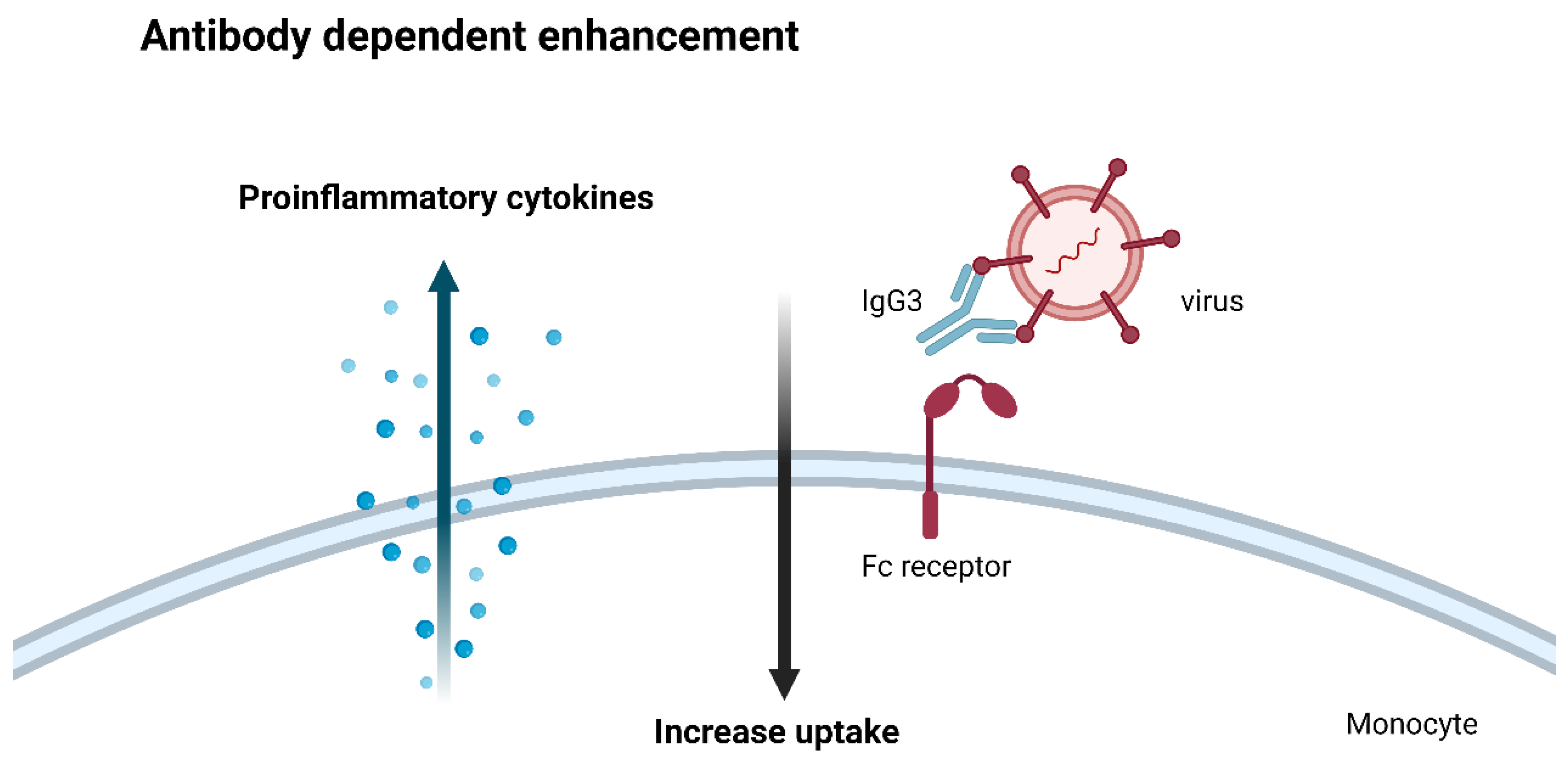
Figure 6.
(6-1) gamma-delta T cells and IgD B cells (6-2) the classification of gamma-delta T cells.
Figure 6.
(6-1) gamma-delta T cells and IgD B cells (6-2) the classification of gamma-delta T cells.
References
- Hu, W.C. A Framework of All Discovered Immunological Pathways and Their Roles for Four Specific Types of Pathogens and Hypersensitivities. Front Immunol 2020, 11, 1992. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Tsai, K.W.; Lu, K.C.; Shih, L.J.; Hu, W.C. Cancer as a Dysfunctional Immune Disorder: Pro-Tumor TH1-like Immune Response and Anti-Tumor THαβ Immune Response Based on the Complete Updated Framework of Host Immunological Pathways. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Hu, W.C. Human immune responses to Plasmodium falciparum infection: molecular evidence for a suboptimal THαβ and TH17 bias over ideal and effective traditional TH1 immune response. Malar J 2013, 12, 392. [Google Scholar] [CrossRef]
- Chu, Y.T.; Liao, M.T.; Tsai, K.W.; Lu, K.C.; Hu, W.C. Interplay of Chemokines Receptors, Toll-like Receptors, and Host Immunological Pathways. Biomedicines 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.C. The Central THαβ Immunity Associated Cytokine: IL-10 Has a Strong Anti-Tumor Ability Toward Established Cancer Models In Vivo and Toward Cancer Cells In Vitro. Front Oncol 2021, 11, 655554. [Google Scholar] [CrossRef] [PubMed]
- Hedblom, A.; Hejazi, S.M.; Canesin, G.; Choudhury, R.; Hanafy, K.A.; Csizmadia, E.; Persson, J.L.; Wegiel, B. Heme detoxification by heme oxygenase-1 reinstates proliferative and immune balances upon genotoxic tissue injury. Cell Death Dis 2019, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Montecinos, L.; Eskew, J.D.; Smith, A. What Is Next in This "Age" of Heme-Driven Pathology and Protection by Hemopexin? An Update and Links with Iron. Pharmaceuticals (Basel) 2019, 12. [Google Scholar] [CrossRef]
- Wu, B.; Wu, Y.; Tang, W. Heme Catabolic Pathway in Inflammation and Immune Disorders. Front Pharmacol 2019, 10, 825. [Google Scholar] [CrossRef]
- Brusko, T.M.; Wasserfall, C.H.; Agarwal, A.; Kapturczak, M.H.; Atkinson, M.A. An integral role for heme oxygenase-1 and carbon monoxide in maintaining peripheral tolerance by CD4+CD25+ regulatory T cells. J Immunol 2005, 174, 5181–5186. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, M.; Ishimura, Y. The heme oxygenase-carbon monoxide system: a regulator of hepatobiliary function. Hepatology 2000, 31, 3–6. [Google Scholar] [CrossRef]
- Mackern-Oberti, J.P.; Obreque, J.; Mendez, G.P.; Llanos, C.; Kalergis, A.M. Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus. Clin Exp Immunol 2015, 182, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Elkhatib, S.K.; Case, A.J. Autonomic regulation of T-lymphocytes: Implications in cardiovascular disease. Pharmacol Res 2019, 146, 104293. [Google Scholar] [CrossRef] [PubMed]
- Bucsek, M.J.; Giridharan, T.; MacDonald, C.R.; Hylander, B.L.; Repasky, E.A. An overview of the role of sympathetic regulation of immune responses in infectious disease and autoimmunity. Int J Hyperthermia 2018, 34, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Contreras, F.; Prado, C.; Gonzalez, H.; Franz, D.; Osorio-Barrios, F.; Osorio, F.; Ugalde, V.; Lopez, E.; Elgueta, D.; Figueroa, A. , et al. Dopamine Receptor D3 Signaling on CD4+ T Cells Favors Th1- and Th17-Mediated Immunity. J Immunol 2016, 196, 4143–4149. [Google Scholar] [CrossRef] [PubMed]
- Crary, B.; Hauser, S.L.; Borysenko, M.; Kutz, I.; Hoban, C.; Ault, K.A.; Weiner, H.L.; Benson, H. Epinephrine-induced changes in the distribution of lymphocyte subsets in peripheral blood of humans. The Journal of Immunology 1983, 131, 1178–1181. [Google Scholar] [CrossRef] [PubMed]
- Flierl, M.A.; Rittirsch, D.; Nadeau, B.A.; Chen, A.J.; Sarma, J.V.; Zetoune, F.S.; McGuire, S.R.; List, R.P.; Day, D.E.; Hoesel, L.M. , et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 2007, 449, 721–725. [Google Scholar] [CrossRef]
- Flierl, M.A.; Rittirsch, D.; Nadeau, B.A.; Sarma, J.V.; Day, D.E.; Lentsch, A.B.; Huber-Lang, M.S.; Ward, P.A. Upregulation of phagocyte-derived catecholamines augments the acute inflammatory response. PLoS One 2009, 4, e4414. [Google Scholar] [CrossRef] [PubMed]
- Scanzano, A.; Cosentino, M. Adrenergic regulation of innate immunity: a review. Front Pharmacol 2015, 6, 171. [Google Scholar] [CrossRef]
- Sharma, D.; Farrar, J.D. Adrenergic regulation of immune cell function and inflammation. Semin Immunopathol 2020, 42, 709–717. [Google Scholar] [CrossRef]
- Papa, I.; Saliba, D.; Ponzoni, M.; Bustamante, S.; Canete, P.F.; Gonzalez-Figueroa, P.; McNamara, H.A.; Valvo, S.; Grimbaldeston, M.; Sweet, R.A. , et al. T(FH)-derived dopamine accelerates productive synapses in germinal centres. Nature 2017, 547, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Frohman, E.M.; Vayuvegula, B.; Gupta, S.; van den Noort, S. Norepinephrine inhibits gamma-interferon-induced major histocompatibility class II (Ia) antigen expression on cultured astrocytes via beta-2-adrenergic signal transduction mechanisms. Proc Natl Acad Sci U S A 1988, 85, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, M.; Rogovskii, V.; Sviridova, A.; Lopatina, A.; Pashenkov, M.; Boyko, A. The Dual Role of the beta(2)-Adrenoreceptor in the Modulation of IL-17 and IFN-gamma Production by T Cells in Multiple Sclerosis. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Moriuchi, M.; Yoshimine, H.; Oishi, K.; Moriuchi, H. Norepinephrine inhibits human immunodeficiency virus type-1 infection through the NF-kappaB inactivation. Virology 2006, 345, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Koch-Weser, J. Beta adrenergic blockade and circulating eosinophils. Arch Intern Med 1968, 121, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Engstad, C.S.; Lund, T.; Osterud, B. Epinephrine promotes IL-8 production in human leukocytes via an effect on platelets. Thromb Haemost 1999, 81, 139–145. [Google Scholar] [PubMed]
- Carvajal Gonczi, C.M.; Tabatabaei Shafiei, M.; East, A.; Martire, E.; Maurice-Ventouris, M.H.I.; Darlington, P.J. Reciprocal modulation of helper Th1 and Th17 cells by the beta2-adrenergic receptor agonist drug terbutaline. FEBS J 2017, 284, 3018–3028. [Google Scholar] [CrossRef] [PubMed]
- Kesici, S.; Demirci, M.; Kesici, U. Antibacterial effects of lidocaine and adrenaline. Int Wound J 2019, 16, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.; Osterud, B. The promoting effect of epinephrine on lipopolysaccharide-induced interleukin-8 production in whole blood may be mediated by thromboxane A2. J Thromb Haemost 2003, 1, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Mattingly, A.J.; Laitano, O.; Clanton, T.L. Epinephrine stimulates CXCL1 IL-1alpha, IL-6 secretion in isolated mouse limb muscle. Physiol Rep 2017, 5. [Google Scholar] [CrossRef]
- Zhou, J.; Yan, J.; Liang, H.; Jiang, J. Epinephrine enhances the response of macrophages under LPS stimulation. Biomed Res Int 2014, 2014, 254686. [Google Scholar] [CrossRef]
- Kradin, R.; Rodberg, G.; Zhao, L.H.; Leary, C. Epinephrine yields translocation of lymphocytes to the lung. Exp Mol Pathol 2001, 70, 1–6. [Google Scholar] [CrossRef]
- Platzer, C.; Döcke, W.; Volk, H.; Prösch, S. Catecholamines trigger IL-10 release in acute systemic stress reaction by direct stimulation of its promoter/enhancer activity in monocytic cells. J Neuroimmunol 2000, 105, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.H.; Cheng, C.; Dai, L.; Peng, Y.P. Effect of endogenous catecholamines in lymphocytes on lymphocyte function. J Neuroimmunol 2005, 167, 45–52. [Google Scholar] [CrossRef]
- Bergquist, J.; Tarkowski, A.; Ekman, R.; Ewing, A. Discovery of endogenous catecholamines in lymphocytes and evidence for catecholamine regulation of lymphocyte function via an autocrine loop. Proc Natl Acad Sci U S A 1994, 91, 12912–12916. [Google Scholar] [CrossRef]
- Zhou, L.; Lin, X.; Ma, X.; Liu, Y.; Ma, L.; Chen, Z.; Chen, H.; Si, L.; Chen, X. Acetylcholine regulates the development of experimental autoimmune encephalomyelitis via the CD4+ cells proliferation and differentiation. Int J Neurosci 2020, 130, 788–803. [Google Scholar] [CrossRef]
- Malin, S.G.; Shavva, V.S.; Tarnawski, L.; Olofsson, P.S. Functions of acetylcholine-producing lymphocytes in immunobiology. Curr Opin Neurobiol 2020, 62, 115–121. [Google Scholar] [CrossRef]
- Kanauchi, Y.; Yamamoto, T.; Yoshida, M.; Zhang, Y.; Lee, J.; Hayashi, S.; Kadowaki, M. Cholinergic anti-inflammatory pathway ameliorates murine experimental Th2-type colitis by suppressing the migration of plasmacytoid dendritic cells. Sci Rep 2022, 12, 54. [Google Scholar] [CrossRef]
- Nizri, E.; Irony-Tur-Sinai, M.; Lory, O.; Orr-Urtreger, A.; Lavi, E.; Brenner, T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J Immunol 2009, 183, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Oenema, T.A.; Smit, M.; Smedinga, L.; Racke, K.; Halayko, A.J.; Meurs, H.; Gosens, R. Muscarinic receptor stimulation augments TGF-beta1-induced contractile protein expression by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2012, 303, L589–597. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.; Backman, L.J.; Alfredson, H.; Scott, A.; Danielson, P. The effects of substance P and acetylcholine on human tenocyte proliferation converge mechanistically via TGF-beta1. PLoS One 2017, 12, e0174101. [Google Scholar] [CrossRef]
- Ahluwalia, A.; Foster, P.; Scotland, R.S.; McLean, P.G.; Mathur, A.; Perretti, M.; Moncada, S.; Hobbs, A.J. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. Proc Natl Acad Sci U S A 2004, 101, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Flores-Costa, R.; Duran-Guell, M.; Casulleras, M.; Lopez-Vicario, C.; Alcaraz-Quiles, J.; Diaz, A.; Lozano, J.J.; Titos, E.; Hall, K.; Sarno, R. , et al. Stimulation of soluble guanylate cyclase exerts antiinflammatory actions in the liver through a VASP/NF-kappaB/NLRP3 inflammasome circuit. Proc Natl Acad Sci U S A 2020, 117, 28263–28274. [Google Scholar] [CrossRef] [PubMed]
- Wollberg, P.; Söderqvist, H.; Nelson, B.D. Mitogen activation of human peripheral T lymphocytes induces the formation of new cyclic AMP response element-binding protein nuclear complexes. Journal of Biological Chemistry 1994, 269, 19719–19724. [Google Scholar] [CrossRef] [PubMed]
- Aley, K.O.; Levine, J.D. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci 1999, 19, 2181–2186. [Google Scholar] [CrossRef] [PubMed]
- Perez-Perez, D.; Santos-Argumedo, L.; Rodriguez-Alba, J.C.; Lopez-Herrera, G. Role of Protein Kinase A Activation in the Immune System with an Emphasis on Lipopolysaccharide-Responsive and Beige-like Anchor Protein in B Cells. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, K.M.; Vang, T.; Abrahamsen, H.; Yaqub, S.; Taskén, K. Molecular mechanisms for protein kinase A-mediated modulation of immune function. Cell Signal 2002, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Busillo, J.M.; Azzam, K.M.; Cidlowski, J.A. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem 2011, 286, 38703–38713. [Google Scholar] [CrossRef] [PubMed]
- de Castro Kroner, J.; Knoke, K.; Kofler, D.M.; Steiger, J.; Fabri, M. Glucocorticoids promote intrinsic human T(H)17 differentiation. J Allergy Clin Immunol 2018, 142, 1669–1673. [Google Scholar] [CrossRef] [PubMed]
- Schleimer, R.P. Glucocorticoids suppress inflammation but spare innate immune responses in airway epithelium. Proc Am Thorac Soc 2004, 1, 222–230. [Google Scholar] [CrossRef]
- Shoenfeld, Y.; Gurewich, Y.; Gallant, L.A.; Pinkhas, J. Prednisone-induced leukocytosis. Influence of dosage, method and duration of administration on the degree of leukocytosis. Am J Med 1981, 71, 773–778. [Google Scholar] [CrossRef]
- Gonsalkorale, W.M.; Dascombe, M.J.; Hutchinson, I.V. Adrenocorticotropic hormone as a potential enhancer of T-lymphocyte function in the rat mixed lymphocyte reaction. Int J Immunopharmacol 1995, 17, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.W.; Hughes, T.K., Jr.; Smith, E.M. ACTH enhancement of T-lymphocyte cytotoxic responses. Cell Mol Neurobiol 2005, 25, 743–757. [Google Scholar] [CrossRef]
- Dittel, L.J.; Dittel, B.N.; Brod, S.A. Ingested ACTH blocks Th17 production by inhibiting GALT IL-6. J Neurol Sci 2020, 409, 116602. [Google Scholar] [CrossRef]
- Dittel, L.J.; Dittel, B.N.; Brod, S.A. Ingested (Oral) Adrenocorticotropic Hormone Inhibits IL-17 in the Central Nervous System in the Mouse Model of Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Immunohorizons 2022, 6, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Cai, D.; Yang, X.; Shang, Y.; Li, X.; Jia, Y.; Yin, C.; Zou, H.; Xu, Y.; Sun, Q. , et al. Stress Response Simulated by Continuous Injection of ACTH Attenuates Lipopolysaccharide-Induced Inflammation in Porcine Adrenal Gland. Front Vet Sci 2020, 7, 315. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhao, L.; Liu, J.; Cao, L.; Zhao, T.; Yu, Y.; Xuan, D.; Wan, W.; Xue, Y.; Zou, H. Natural Adrenocorticotropic Hormone (ACTH) Relieves Acute Inflammation in Gout Patients by Changing the Function of Macrophages. J Healthc Eng 2022, 2022, 9241835. [Google Scholar] [CrossRef]
- Agelaki, S.; Tsatsanis, C.; Gravanis, A.; Margioris, A.N. Corticotropin-releasing hormone augments proinflammatory cytokine production from macrophages in vitro and in lipopolysaccharide-induced endotoxin shock in mice. Infect Immun 2002, 70, 6068–6074. [Google Scholar] [CrossRef]
- Benou, C.; Wang, Y.; Imitola, J.; VanVlerken, L.; Chandras, C.; Karalis, K.P.; Khoury, S.J. Corticotropin-releasing hormone contributes to the peripheral inflammatory response in experimental autoimmune encephalomyelitis. J Immunol 2005, 174, 5407–5413. [Google Scholar] [CrossRef]
- O'Kane, M.; Murphy, E.P.; Kirby, B. The role of corticotropin-releasing hormone in immune-mediated cutaneous inflammatory disease. Exp Dermatol 2006, 15, 143–153. [Google Scholar] [CrossRef]
- Wang, W.; Ji, P.; Dow, K.E. Corticotropin-releasing hormone induces proliferation and TNF-alpha release in cultured rat microglia via MAP kinase signalling pathways. J Neurochem 2003, 84, 189–195. [Google Scholar] [CrossRef]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu Rev Genet 1988, 22, 631–677. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci 2006, 63, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Foo, S.S.; Bruzzone, R.; Dinh, L.V.; King, N.J.; Mahalingam, S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol Rev 2015, 268, 340–364. [Google Scholar] [CrossRef] [PubMed]
- Rothman, A.L.; Ennis, F.A. Immunopathogenesis of Dengue hemorrhagic fever. Virology 1999, 257, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 2020, 584, 353–363. [Google Scholar] [CrossRef]
- Cao, R.Y.; Dong, D.Y.; Liu, R.J.; Han, J.F.; Wang, G.C.; Zhao, H.; Li, X.F.; Deng, Y.Q.; Zhu, S.Y.; Wang, X.Y. , et al. Human IgG subclasses against enterovirus Type 71: neutralization versus antibody dependent enhancement of infection. PLoS One 2013, 8, e64024. [Google Scholar] [CrossRef]
- Fiala, G.J.; Gomes, A.Q.; Silva-Santos, B. From thymus to periphery: Molecular basis of effector gammadelta-T cell differentiation. Immunol Rev 2020, 298, 47–60. [Google Scholar] [CrossRef]
- Fujihashi, K.; Dohi, T.; Kweon, M.N.; McGhee, J.R.; Koga, T.; Cooper, M.D.; Tonegawa, S.; Kiyono, H. gammadelta T cells regulate mucosally induced tolerance in a dose-dependent fashion. Int Immunol 1999, 11, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Xuan, L.; Wu, X.; Fan, Z.; Huang, F.; Yi, Z.; Li, Y.; Liu, Q. Increase of Regulatory γδ T Cells Reduces the Incidence of Acute Graft-Versus-Host Disease after Allogeneic Hematopoietic Stem Cell Transplantation. Blood 2016, 128, 2230–2230. [Google Scholar] [CrossRef]
- Guan, H.; Zu, G.; Slater, M.; Elmets, C.; Xu, H. GammadeltaT cells regulate the development of hapten-specific CD8+ effector T cells in contact hypersensitivity responses. J Invest Dermatol 2002, 119, 137–142. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Chinen, T.; Matsuzaki, G.; Sasaki, A.; Sakamoto, Y.; Hiromatsu, K.; Nakamura-Uchiyama, F.; Nawa, Y.; Yoshimura, A. Mucosal T cells bearing TCRgammadelta play a protective role in intestinal inflammation. J Immunol 2004, 173, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Mengel, J.; Cardillo, F.; Aroeira, L.S.; Williams, O.; Russo, M.; Vaz, N.M. Anti-gamma delta T cell antibody blocks the induction and maintenance of oral tolerance to ovalbumin in mice. Immunol Lett 1995, 48, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Wang, H.Y.; Peng, W.; Kiniwa, Y.; Seo, K.H.; Wang, R.F. Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique toll-like receptor signaling pathway. Immunity 2007, 27, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Rezende, R.M.; Nakagaki, B.N.; Moreira, T.G.; Lopes, J.R.; Kuhn, C.; Tatematsu, B.K.; Boulenouar, S.; Maghzi, A.H.; Rubino, S.; Menezes, G.B. , et al. gammadelta T Cell-Secreted XCL1 Mediates Anti-CD3-Induced Oral Tolerance. J Immunol 2019, 203, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Schilbach, K.; Krickeberg, N.; Kaisser, C.; Mingram, S.; Kind, J.; Siegers, G.M.; Hashimoto, H. Suppressive activity of Vdelta2(+) gammadelta T cells on alphabeta T cells is licensed by TCR signaling and correlates with signal strength. Cancer Immunol Immunother 2020, 69, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Hayday, A.C.; Barber, D.F.; Douglas, N.; Hoffman, E.S. Signals involved in gamma/delta T cell versus alpha/beta T cell lineage commitment. Semin Immunol 1999, 11, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Dopfer, E.P.; Hartl, F.A.; Oberg, H.H.; Siegers, G.M.; Yousefi, O.S.; Kock, S.; Fiala, G.J.; Garcillan, B.; Sandstrom, A.; Alarcon, B. , et al. The CD3 conformational change in the gammadelta T cell receptor is not triggered by antigens but can be enforced to enhance tumor killing. Cell Rep 2014, 7, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Dave, V.P.; Cao, Z.; Browne, C.; Alarcon, B.; Fernandez-Miguel, G.; Lafaille, J.; de la Hera, A.; Tonegawa, S.; Kappes, D.J. CD3 delta deficiency arrests development of the alpha beta but not the gamma delta T cell lineage. Embo j 1997, 16, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Kadlecek, T.A.; van Oers, N.S.C.; Lefrancois, L.; Olson, S.; Finlay, D.; Chu, D.H.; Connolly, K.; Killeen, N.; Weiss, A. Differential Requirements for ZAP-70 in TCR Signaling and T Cell Development. The Journal of Immunology 1998, 161, 4688–4694. [Google Scholar] [CrossRef]
- Laird, R.M.; Hayes, S.M. Roles of the Src tyrosine kinases Lck and Fyn in regulating gammadeltaTCR signal strength. PLoS One 2010, 5, e8899. [Google Scholar] [CrossRef]
- Van Neerven, J.; Coligan, J.E.; Koning, F. Structural comparison of alpha/beta and gamma/delta T cell receptor-CD3 complexes reveals identical subunit interactions but distinct cross-linking patterns of T cell receptor chains. Eur J Immunol 1990, 20, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Goodnow, C.C.; Crosbie, J.; Jorgensen, H.; Brink, R.A.; Basten, A. Induction of self-tolerance in mature peripheral B lymphocytes. Nature 1989, 342, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, L.; Kosco, M.H.; Köhler, G.; Lamers, M.C. Immunoglobulin D-deficient mice can mount normal immune responses to thymus-independent and -dependent antigens. Proc Natl Acad Sci U S A 1993, 90, 1887–1891. [Google Scholar] [CrossRef] [PubMed]
- Duty, J.A.; Szodoray, P.; Zheng, N.Y.; Koelsch, K.A.; Zhang, Q.; Swiatkowski, M.; Mathias, M.; Garman, L.; Helms, C.; Nakken, B. , et al. Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J Exp Med 2009, 206, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Quach, T.D.; Manjarrez-Orduno, N.; Adlowitz, D.G.; Silver, L.; Yang, H.; Wei, C.; Milner, E.C.; Sanz, I. Anergic responses characterize a large fraction of human autoreactive naive B cells expressing low levels of surface IgM. J Immunol 2011, 186, 4640–4648. [Google Scholar] [CrossRef] [PubMed]
- Soulas, P.; Koenig-Marrony, S.; Julien, S.; Knapp, A.M.; Garaud, J.C.; Pasquali, J.L.; Martin, T. A role for membrane IgD in the tolerance of pathological human rheumatoid factor B cells. Eur J Immunol 2002, 32, 2623–2634. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Klaus, G.G. Cross-linking of surface IgM, but not surface IgD receptors, by soluble monoclonal antibodies primes murine B cells to secrete immunoglobulin in response to lymphokines. Eur J Immunol 1993, 23, 574–577. [Google Scholar] [CrossRef]
- Huang, Y.; Heiser, R.A.; Detanico, T.O.; Getahun, A.; Kirchenbaum, G.A.; Casper, T.L.; Aydintug, M.K.; Carding, S.R.; Ikuta, K.; Huang, H. , et al. gammadelta T cells affect IL-4 production and B-cell tolerance. Proc Natl Acad Sci U S A 2015, 112, E39–48. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



