Submitted:
01 April 2024
Posted:
02 April 2024
You are already at the latest version
Abstract
Recent advancements in artificial intelligence (AI) and deep learning have revolutionized the field of protein engineering, particularly in the area of de novo protein design. This review article explores the impact of AI-driven approaches on protein design, with a specific focus on the role of structure prediction neural networks, such as AlphaFold and RoseTTAFold. The article discusses the paradigm shift brought about by these networks, which have enabled the design of proteins with unique structures and functions that are not found in nature. The review covers various aspects of AI-driven protein design, including the use of protein language models to harness evolutionary information, the development of de novo protein design workflows utilizing deep learning, and the application of generative models like RFdiffusion and RoseTTAFold All-Atom. The article also highlights the successes and applications of AI-driven protein design across diverse domains, such as enzyme engineering, antibody design, and vaccine development. Additionally, the review identifies current challenges and future directions in the field, emphasizing the need to address limitations in modeling conformational dynamics and designing proteins for in vivo functionality. The article concludes by underscoring the potential of AI-driven protein design to transform various aspects of science and technology, while also acknowledging the importance of interdisciplinary collaboration and the development of robust pipelines for the validation and optimization of designed proteins. Overall, this comprehensive review serves as a valuable resource for researchers and practitioners interested in understanding the current state and future prospects of AI-driven protein design.
Keywords:
deep learning
; machine learning
; protein design
; biotechnology
Introduction
The field of protein engineering has witnessed a remarkable transformation in recent years, largely driven by rapid advancements in artificial intelligence (AI) and deep learning. Designing proteins with tailored structures and functions is a long-standing goal in bioengineering, holding immense potential to revolutionize various domains, from drug discovery and biotechnology to materials science. Traditionally, de novo protein design relied on energy function-based methods, which aimed to generate proteins that minimize a predefined energy function derived from physical principles and statistical analysis of natural proteins (Chothia, 1984). While this approach yielded success in creating a range of structural scaffolds and functional proteins, it was limited by relatively low success rates, primarily due to inaccuracies in the employed energy functions (Korendovych and DeGrado, 2020).
The landscape of protein design has undergone a paradigm shift with the advent of deep learning-based protein structure prediction networks, such as AlphaFold (Jumper et al., 2021) and RoseTTAFold (Baek et al., 2021). These groundbreaking tools have demonstrated near-experimental accuracy in predicting protein structures from amino acid sequences, surpassing the performance of previous energy function-based methods. The success of these structure prediction networks has sparked a new wave of research aimed at harnessing their power for protein design tasks. By leveraging the latent representations learned by these models, researchers have developed novel approaches to generate proteins with desired structural and functional properties, leading to significant improvements in the efficiency and success rates of protein design endeavors.
Central to these AI-driven protein design efforts is the "central dogma" paradigm (Wang et al., 2022), which involves specifying a desired function, designing a structure to execute that function, and finding a sequence that folds into that structure. Machine learning models for protein design can be broadly categorized into three groups based on the data modalities they utilize: sequence-based models, sequence-label models, and structure-based models (Dou et al., 2018). Sequence-based models, such as protein language models, learn from diverse evolutionary sequences and capture structural and functional properties. Sequence-label models leverage functional data to guide design towards specific properties. Structure-based models, including inverse folding and diffusion-based approaches, enable the design of novel backbone structures and the scaffolding of functional motifs (Notin et al., 2024).
The application of these AI methods has already led to remarkable successes across various domains, including the design of enzymes, antibodies, vaccines, and nanomachines. Moreover, recent advances suggest a move beyond the classical central dogma paradigm, towards more integrative approaches involving sequence-structure co-design and the incorporation of conformational dynamics (Hsu, Fannjiang and Listgarten, 2024).
In this review, we explore the recent advances in AI-driven protein design, focusing on the pivotal role played by structure prediction networks. We discuss the various strategies employed to harness these models for design tasks and highlight the remarkable achievements made possible by these methods. We also examine the challenges and opportunities that lie ahead, emphasizing the importance of continual improvement in the accuracy and generalizability of AI models to unlock the potential of protein design.
Protein Language Models: Harnessing Evolutionary Information for Protein Design
Protein language models (PLMs) have emerged as a powerful tool in the field of protein design. PLMs were trained from diverse protein sequences spanning the tree of life, capturing the complex patterns and dependencies that have been shaped by billions of years of evolution (Hsu, Fannjiang and Listgarten, 2024). At the core of PLMs lies the concept of coevolution, which refers to the correlated changes in amino acid residues across evolutionarily related proteins. These coevolutionary patterns arise due to the constraints imposed by protein structure and function, and PLMs can learn these patterns by training on large datasets of protein sequences. By capturing the evolutionary information encoded in sequences, PLMs can infer the structural and functional properties of proteins, making them valuable for various protein engineering tasks (Jumper et al., 2021).
One of the key architectures used in PLMs is the transformer, which has revolutionized natural language processing and has been successfully adapted for protein sequence modeling. Transformers employ self-attention mechanisms that allow them to capture long-range dependencies between amino acid residues, enabling them to model the complex interactions that govern protein folding and function (Zhang et al., 2023).
PLMs can be broadly categorized into two main types based on their training objectives: autoregressive models and masked language models. Autoregressive PLMs, such as UniRep, ProGen, and ProtGPT2 (Ferruz, Schmidt and Höcker, 2022), are trained to predict the next amino acid in a sequence based on the preceding residues, effectively learning the sequential dependencies within proteins. On the other hand, masked language models, such as the ESM and ProtTrans families of models, are trained to predict the identity of masked amino acids within a sequence, allowing them to learn bidirectional dependencies and capture global structural information (Martínez-Mauricio, García-Jacas and Cordoves-Delgado, 2024).
The ability of PLMs to learn from evolutionary information has significant implications for protein design. By capturing the coevolutionary relationships between residues, PLMs can generate novel protein sequences that are likely to fold into stable and functional structures. They can also predict the effects of mutations on protein stability and function, guiding directed evolution experiments and reducing the experimental search space (Schubach et al., 2024).
Moreover, PLMs have been used to generate functional proteins across diverse families, showcasing their ability to learn transferable representations of protein sequences (Ferruz, Schmidt and Höcker, 2022). The success of PLMs in protein design highlights the importance of leveraging evolutionary information and the potential of unsupervised learning from large-scale sequence data.
PLMs can be further categorized into sequence-only models and conditional sequence models . Sequence-only models learn a generative model, P(x) of the primary structure x of a given protein, aiming to implicitly capture the biochemical constraints that characterize the proteins present in the training set. These models can be family-specific, trained on a set of homologous sequences contained in a multiple-sequence alignment, or family-agnostic, trained on unaligned sequences across protein families (Singer et al., 2022).
Conditional sequence models, on the other hand, condition the generative process P(x|t) on broad taxonomic groups or gene ontology annotations t, providing more control over the nature and properties of generated sequences. Several architectures have been proposed for conditional sequence models, based on autoregressive modeling or masked-language modeling (Ferruz et al., 2023).
The choice of PLM architecture depends on the specific protein design task and the available data. For example, if the goal is to generate sequences with specific functional properties, a conditional sequence model trained on gene ontology annotations may be more appropriate. On the other hand, if the aim is to explore the sequence space of a particular protein family, a family-specific sequence-only model may be more suitable.
PLMs have been successfully applied to various protein design tasks, such as generating novel enzymes, optimizing protein stability, and designing antibodies with enhanced affinity and specificity. In one notable example, Madani et al. used a large-scale autoregressive PLM to generate functional enzyme sequences across diverse protein families (Hsu, Fannjiang and Listgarten, 2024). By training the model on a vast dataset of protein sequences and conditioning the generation process on functional annotations, they were able to create novel enzymes with desired catalytic activities (Madani et al., 2023).
Another promising application of PLMs is in the design of protein-protein interfaces. By learning from the evolutionary patterns of interface residues, PLMs can generate sequences that are likely to form specific and stable interactions with target proteins. This approach has been used to design novel protein binders and inhibitors, as well as to optimize the affinity and specificity of existing protein-protein interactions (Gainza et al., 2020).
The objectives of protein design can be categorized into three main areas: redesigning proteins to enhance their existing functions, redesigning proteins to perform new functions, and designing entirely new proteins from scratch (de novo design) (Hsu, Fannjiang and Listgarten, 2024).
Redesigning proteins to enhance their existing functions involves improving properties such as stability, specificity, or activity by modifying the protein's sequence or optimizing the active site. Machine learning models that predict the effects of mutations on protein stability and function are valuable tools in this pursuit. Redesigning proteins to perform new functions involves adapting existing proteins to take on roles they were not originally evolved to perform. This can be accomplished by modifying the protein's sequence or structure to introduce new functional sites or by combining elements from different proteins to create novel chimeric designs. Machine learning models that accurately model protein-protein and protein-ligand interactions are crucial for this type of design (Baek et al., 2021).
De novo protein design aims to create entirely new proteins from scratch, without relying on existing natural proteins as starting points. This approach offers the greatest flexibility in terms of designing proteins with desired structures and functions but also presents significant challenges. De novo design often involves a combination of structure-based and sequence-based methods, leveraging machine learning models to generate novel protein backbones and optimize sequences for stability and function (Chu, Lu and Huang, 2024).
As the field of protein design continues to advance, the integration of PLMs with other computational and experimental techniques holds great promise. PLMs can guide the exploration of the vast sequence space, identify promising candidates for experimental validation, and accelerate the discovery of novel functional proteins.
De Novo Protein Design Workflow Using Deep Learning
The process of de novo protein design begins with identifying the features needed to accomplish the intended function. These functional objectives can range from designing proteins to engage immune cells, creating binders for drugs, nucleic acids, or other proteins, stabilizing the transition state of a reaction for new enzymes, or developing ion-specific transmembrane channels. Regardless of the specific application, the design approaches are built on the principles of energetic stabilization and shape complementarity (Hsu, Fannjiang and Listgarten, 2024).
In earlier de novo design efforts, the design of any foldable protein was already considered a major achievement, and efforts to attain function centered on introducing changes to these scaffolds to accommodate a functional motif in a minimal way (Woolfson, 2021). However, with the rise of increasingly powerful design methods, specifying the functional motif first and then searching for protein scaffolds that are consistent with this motif has become the more common path.
One strategy to derive structural motifs from functional goals is to extract them directly from natural proteins and scaffold them as part of a de novo protein structure. This approach has been successfully applied to scaffold antigen epitopes on the surface of designed immunogens (Sesterhenn et al., 2020), peptide-binding motifs, metal-binding sites, and ligand-binding motifs to accomplish the relevant functional task (Dou et al., 2018; Wang et al., 2022). These motifs can also be extracted from nature to support a designed function, such as the placement of positively charged residues near the membrane–solvent interface in the case of designing transmembrane channels (Scott et al., 2021).
More general approaches to devise yet unknown functional motifs require breaking down the interaction to basic chemical elements and handling the possible combinations and arrangements of these elements accurately. One class of methods solves this problem by considering the chemical properties of the target and enumerating the interactions that a protein might use to bind to the target (Cao et al., 2022). They can also be culled directly from the PDB, relying on statistical enrichment to capture the most effective interactions and perhaps average out noisier information such as side chain flexibility (Polizzi and DeGrado, 2020). This interaction field approach is generalizable to arbitrary binding interactions and has been successfully applied to design de novo binders against conformer-specific small-molecule ligands, miniprotein binders and ultra-high-affinity de novo binders to receptors, monobody binders to nerve toxins, and binding to nucleic acids (Glasscock et al., 2023).
Other approaches seek a higher level of abstraction by capturing features of functional interfaces with machine learning. For protein–protein interfaces, machine-learned representations of a surface can be used to propose the binding counterpart. Embeddings of protein surfaces can be learned that capture general biophysical and biochemical properties of an interface region as well as additional information that may be encoded in subtle variations in the sequence but is difficult to explain with energy functions or visual inspection. The patch embeddings from a target protein can then be inverted and mapped to sets of favorably interacting motifs for scaffolding into a designed protein (Gainza et al., 2023).
After identifying the relevant structural motifs, the next step is to design a protein backbone that can accommodate these motifs and support the desired function. This is a critical step in the design process, as the backbone scaffold determines the overall shape and stability of the protein. Deep learning models, such as generative adversarial networks (GANs) and variational autoencoders (VAEs), have been employed to generate novel protein backbones that are compatible with the specified structural motifs (Chu, Lu and Huang, 2024). These models learn from the structural patterns present in natural proteins and can generate diverse and physically realistic backbone conformations. By sampling from the latent space of these generative models, researchers can explore a wide range of backbone designs and select those that are most suitable for the desired function (Hsu, Fannjiang and Listgarten, 2024).
Once the backbone scaffold is designed, the next step is to optimize the protein sequence to ensure proper folding and stability. Protein language models (PLMs) and sequence optimization algorithms come into play at this stage. PLMs, trained on large datasets of protein sequences, can capture the evolutionary relationships between amino acids and generate sequences that are likely to fold into the desired structure (Rives et al., 2021). Sequence optimization algorithms, such as genetic algorithms and Monte Carlo methods, can further refine the generated sequences by searching for mutations that improve the stability and functionality of the protein. These algorithms often incorporate energy functions and structural constraints derived from the designed backbone and structural motifs, ensuring that the optimized sequences are compatible with the intended function (Yang, Wu and Arnold, 2019).
After the sequence optimization step, the designed proteins undergo rigorous computational evaluation to assess their structural and functional properties. This involves a combination of structure prediction, molecular dynamics simulations, and docking studies to predict the folding, stability, and binding interactions of the designed proteins. Deep learning models, such as AlphaFold and RoseTTAFold, have significantly advanced the field of protein structure prediction, enabling the accurate assessment of designed proteins (Jumper et al., 2021). These models can predict the 3D structure of the designed sequences, providing valuable insights into their folding and potential functionality.
Finally, the most promising designed proteins are selected for experimental validation. This involves the synthesis of the designed sequences, followed by structural and functional characterization using a range of experimental techniques, such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and biochemical assays. The experimental validation step is crucial for assessing the success of the design process and identifying areas for further optimization. The feedback from experimental studies can be used to refine the design objectives, structural motifs, and sequence optimization strategies, leading to iterative improvements in the design workflow (Huang, Boyken and Baker, 2016).
The integration of deep learning into the de novo protein design workflow has greatly accelerated the discovery of novel functional proteins. Moreover, the ability of deep learning models to learn from diverse protein families and capture evolutionary relationships has enabled the design of proteins with functions beyond those found in nature. This has opened up new opportunities for the development of novel enzymes, biosensors, and therapeutic proteins, see Figure 1.
Structure-Prediction Neural Networks for Protein Design
The advent of highly accurate protein structure prediction using deep learning, exemplified by AlphaFold and RoseTTAFold, has opened up new avenues for protein design. Beyond structure prediction, these networks have been repurposed for protein design tasks, leveraging their learned representations of protein sequence-structure relationships. In this section, we discuss the use of structure-prediction neural networks for protein design, focusing on approaches such as activation maximization, inpainting, and denoising diffusion.
Activation maximization is a technique that involves optimizing the input to a neural network to maximize the activation of a specific neuron or set of neurons. In the context of protein design, activation maximization can be applied to structure-prediction networks to generate sequences that are predicted to fold into a desired structure. This is achieved by starting with a random or partially specified sequence and iteratively updating it to maximize the predicted probability of the target structure. Anishchenko et al. (Anishchenko et al., 2021) demonstrated the effectiveness of this approach using the trRosetta structure-prediction network, generating sequences that folded into a variety of complex topologies, including β-barrels and α/β folds. The designed proteins were experimentally characterized and found to adopt the intended structures with high accuracy.
Inpainting, another approach borrowed from the field of computer vision, involves filling in missing regions of an image based on the surrounding context (Xie et al., 2022). In the protein design context, inpainting can be used to complete partial protein structures or to design sequences for specific structural motifs. Wang et al. utilized the RoseTTAFold network for protein inpainting, demonstrating its ability to generate sequences that fold into desired structural motifs, such as protein-binding sites and enzyme active sites. By training the network on a large dataset of protein structures and fine-tuning it for specific design tasks, they achieved high success rates in designing functional proteins, including metalloproteins and protein binders (Wang et al., 2022).
Denoising diffusion, a recently introduced generative modeling approach, has shown remarkable success in generating high-quality samples across various domains, including images and audio (Li et al., 2023). In the protein design context, denoising diffusion models learn to generate protein structures by modeling the reverse process of gradually adding noise to a structure until it becomes indistinguishable from random noise. The model is then trained to denoise the corrupted structures, effectively learning to generate realistic protein structures from scratch.
The application of denoising diffusion to protein design has been explored in several recent studies. A denoising diffusion model for protein structure generation, demonstrated its ability to generate novel and diverse protein folds. It was trained on a large dataset of protein structures and showed that it could generate structures with complex topologies and realistic geometric properties (Wu et al., 2024). Trippe et al. extended this approach to the problem of protein scaffold generation, using a denoising diffusion model to generate backbone structures compatible with specific functional motifs. Their model, trained on a diverse set of protein structures, was able to generate scaffolds that accommodated the desired motifs while maintaining structural plausibility (Trippe, 2022).
One of the key advantages of denoising diffusion models for protein design is their ability to generate diverse and novel structures. By sampling from the learned distribution, these models can explore a vast space of possible protein folds and discover structures that may not be present in existing databases. This is particularly valuable for de novo protein design, where the goal is to create proteins with new functions and properties. Additionally, denoising diffusion models can be easily conditioned on various design constraints, such as the presence of specific structural motifs or the desired secondary structure composition, enabling targeted protein design for specific applications. Another benefit of denoising diffusion models is their scalability and efficiency. Once trained, these models can generate a large number of diverse protein structures in a matter of seconds, without the need for expensive simulations or extensive sampling. This allows for rapid exploration of the protein design space and facilitates the identification of promising candidates for experimental validation. Furthermore, the generated structures can serve as starting points for further optimization and refinement using other computational tools or experimental techniques.
Despite the promising results achieved by denoising diffusion models in protein design, there are still challenges to be addressed. One important consideration is the physical realizability of the generated structures. While denoising diffusion models can generate structures that are geometrically plausible, they may not always correspond to energetically favorable or stable conformations. Incorporating additional constraints or energy terms into the diffusion process could help ensure that the generated structures are more likely to be experimentally viable. Another challenge is the limited diversity of the training data, as current protein structure databases are biased towards naturally occurring and well-behaved proteins. Expanding the training data to include a broader range of protein structures, including those from de novo designs and engineered variants, could improve the generalization capabilities of the models.
In conclusion, structure-prediction neural networks, such as AlphaFold and RoseTTAFold, have emerged as powerful tools for protein design. Approaches like activation maximization, inpainting, and denoising diffusion leverage the learned representations of these networks to generate novel protein sequences and structures with desired properties. Denoising diffusion models, in particular, have shown great promise in generating diverse and plausible protein structures, enabling efficient exploration of the vast protein design space. As these methods continue to develop and mature, they are expected to play an increasingly important role in advancing the field of de novo protein design and accelerating the discovery of novel functional proteins for a wide range of applications (Chu, Lu and Huang, 2024).
The RFdiffusion Approach
The field of de novo protein design has witnessed a paradigm shift with the advent of structure-prediction neural networks, such as AlphaFold and RoseTTAFold. These networks, originally developed for the task of predicting protein structures from amino acid sequences, have been repurposed and fine-tuned to enable the design of novel proteins with desired structures and functions. In their seminal work, Watson et al. (Watson et al., 2023) introduced RFdiffusion, a generative model for protein design based on the RoseTTAFold structure prediction network. RFdiffusion leverages the power of denoising diffusion probabilistic models (DDPMs) to generate diverse and accurate protein structures, addressing a wide range of design challenges.
The key innovation of RFdiffusion lies in its ability to learn the reverse process of gradually adding noise to a protein structure until it becomes indistinguishable from random noise. By fine-tuning RoseTTAFold on this denoising task, the authors obtained a generative model capable of producing realistic protein structures from scratch. The model operates on a rigid-frame representation of residues, ensuring rotational equivariance and enabling precise control over the generated structures. Importantly, RFdiffusion can be conditioned on various design specifications, such as partial sequences, fold information, or fixed functional motifs, providing flexibility and control over the design process.
The authors demonstrated the versatility and power of RFdiffusion through an extensive set of computational and experimental validations. For unconditional protein monomer generation, RFdiffusion outperformed previous methods, generating diverse and accurate structures up to 600 residues in length. The designs were closely recapitulated by AlphaFold2 structure predictions and exhibited high solubility and thermostability in experimental characterizations. RFdiffusion also excelled in designing higher-order oligomers with various symmetries, including cyclic, dihedral, tetrahedral, and icosahedral architectures. Electron microscopy studies confirmed the close agreement between the designed and experimentally determined structures, highlighting the precision of the generative model.
One of the most impactful applications of RFdiffusion is in the scaffolding of functional motifs. The authors showed that the model can accurately position catalytic sites, binding interfaces, and metal-coordinating residues within de novo designed scaffolds. The successful design of high-affinity MDM2 binders and Ni2+-coordinating tetrameric assemblies exemplifies the potential of RFdiffusion in creating functional proteins for therapeutic and biotechnological applications. Furthermore, the model achieved remarkable success in the de novo design of protein binders, identifying high-affinity binders for multiple targets with orders of magnitude higher efficiency than previous methods. A cryo-EM structure of an influenza hemagglutinin binder in complex with its target provided atomic-level validation of the design accuracy.
The success of RFdiffusion can be attributed to several factors. First, the use of a structure-prediction network as the foundation ensures that the model learns from the vast knowledge of protein structure encoded in these networks. Second, the DDPM framework allows for the generation of diverse and realistic structures by iteratively refining random noise. Third, the ability to condition the generative process on various design specifications enables targeted and controllable protein design. Finally, the rigorous computational and experimental validations provide confidence in the utility and accuracy of the generated designs.
In conclusion, RFdiffusion represents a significant advancement in the field of de novo protein design. By harnessing the power of structure-prediction neural networks and denoising diffusion models, the authors have developed a comprehensive and versatile framework for designing proteins with complex structures and functions. The successful experimental characterization of the designs across a wide range of applications demonstrates the immense potential of this approach. As the field continues to evolve, the integration of structure-prediction networks with generative models is expected to accelerate the discovery of novel functional proteins and revolutionize the fields of bioengineering and therapeutics.
Generalized Biomolecular Modeling and Design with Rosettafold All-Atom
The field of protein design has witnessed remarkable progress in recent years, with the development of powerful machine learning methods capable of generating novel proteins with desired structures and functions. However, most of these approaches have focused on designing single-chain proteins, limiting their applicability to more complex biomolecular systems involving multiple chains, ligands, or other non-protein components. To address this challenge and expand the scope of computational protein design, Krishna et al. (Krishna et al., 2024) introduce RoseTTAFold All-Atom (RFAA), a generalized biomolecular modeling and design framework that enables the design of a wide range of biomolecular systems, including protein-ligand complexes, protein-nucleic acid complexes, and multi-chain assemblies.
RFAA builds upon the success of RoseTTAFold (Wang et al., 2022). The key innovation in RFAA is the extension of the RoseTTAFold architecture to model arbitrary biomolecular systems by incorporating non-protein components, such as small molecules, nucleic acids, and covalently modified amino acids. This is achieved through a generalized representation of biomolecules, where each component is treated as a graph node, and edges represent chemical interactions between nodes. By learning from a diverse set of biomolecular structures in the Protein Data Bank (PDB), RFAA acquires a comprehensive understanding of the structural principles governing protein-ligand, protein-nucleic acid, and multi-chain interactions.
To showcase the capabilities of RFAA, the authors demonstrate its application in various protein design tasks. In the realm of protein-ligand design, RFAA is used to generate novel proteins that bind to specific small molecules, such as digoxigenin, heme, and bilin. Remarkably, the designed proteins exhibit high binding affinities, with a digoxigenin binder achieving a dissociation constant (KD) of 10 nM, comparable to a previously designed binder that had undergone directed evolution. This highlights the potential of RFAA to streamline the design process by eliminating the need for extensive experimental optimization.
RFAA is also applied to the design of protein-nucleic acid complexes, a crucial step towards engineering novel DNA- and RNA-binding proteins with desired specificities. The authors demonstrate the design of a de novo protein that binds to a specific DNA sequence, showcasing the ability of RFAA to capture the intricate interactions between proteins and nucleic acids. Furthermore, RFAA is used to design multi-chain protein assemblies, such as homodimers and heterodimers, with atomic-level accuracy. These designed assemblies exhibit high stability and predicted structures that closely match the design models, highlighting the precision of the RFAA framework.
The success of RFAA in designing protein-ligand, protein-nucleic acid, and multi-chain complexes opens up new avenues for protein engineering. For example, the ability to design proteins that bind to specific small molecules could accelerate the development of biosensors, drug delivery systems, and catalytic enzymes. Similarly, the design of DNA- and RNA-binding proteins with tailored specificities could enable the engineering of novel transcriptional regulators, genome editing tools, and RNA-targeting therapeutics. The precise control over multi-chain assembly afforded by RFAA could also facilitate the design of artificial protein nanomachines and supramolecular structures with desired functions.
In conclusion, RoseTTAFold All-Atom represents a significant advancement in the field of computational protein design, extending the capabilities of deep learning-based methods to model and design complex biomolecular systems. By leveraging a generalized representation of biomolecules and learning from diverse structural data, RFAA enables the design of protein-ligand, protein-nucleic acid, and multi-chain complexes with atomic-level accuracy. The successful experimental characterization of designed proteins across various applications demonstrates the potential of RFAA to accelerate the development of novel biomolecular systems with tailored functions. As the field of protein design continues to evolve, the integration of RFAA with other computational and experimental techniques holds great promise for unlocking new frontiers in biomolecular engineering and expanding the functional repertoire of designed proteins.
Applications and Successes of Ai-Driven Protein Design
The advent of AI-driven protein design has led to remarkable successes across various application domains, ranging from enzyme engineering and antibody design to the creation of novel vaccines and nanomachines. These successes highlight the transformative potential of integrating machine learning with protein engineering and showcase the ability of AI-driven approaches to solve complex biological problems.
In the field of enzyme design, AI has been leveraged to improve the thermostability, specificity, and activity of enzymes for industrial and biomedical applications. For example, researchers have used machine learning models to optimize the stability of a polyester hydrolase enzyme, PETase, which has the potential to degrade plastic waste (Lu et al., 2022). By training a model on a large dataset of protein sequences and their associated stability measurements, the researchers were able to identify mutations that significantly enhanced the enzyme's thermostability without compromising its activity. Similar approaches have been applied to improve the specificity and activity of enzymes involved in biosynthesis, bioremediation, and other industrially relevant processes.
Antibody design is another area where AI has made significant contributions. Machine learning models have been used to enhance the features of existing antibodies, such as improving binding affinity, reducing polyspecificity, and optimizing developability properties (Notin et al., 2024). For instance, a deep learning model trained on antibody sequences and their corresponding binding affinities was able to predict mutations that increased the binding strength of a therapeutic antibody by up to 160-fold (Hie et al., 2024). AI-driven approaches have also been applied to accelerate the discovery of novel antibodies by designing targeted libraries enriched for specific binding properties, reducing the need for extensive experimental screening .
Beyond enzymes and antibodies, AI has enabled the design of proteins for a wide range of applications. In the field of vaccine development, machine learning has been used to design self-assembling protein nanoparticles that efficiently display antigenic epitopes, leading to improved immune responses. These nanoparticle vaccines have shown promise in eliciting protective immunity against various pathogens, including SARS-CoV-2 (Walls et al., 2020). AI-driven protein design has also been applied to create novel nanomachines, such as protein-based molecular motors and sensors, which have the potential to revolutionize fields like targeted drug delivery and diagnostic testing.
One of the most exciting developments in AI-driven protein design is the ability to create proteins with entirely new functions, not found in nature. A prime example of this is the de novo design of a functional β-barrel protein that can catalyze a retro-aldol reaction, a feat that had previously only been achieved through extensive directed evolution (Jiang et al., 2008). By leveraging deep learning models to design the protein's backbone and optimize its sequence for stability and activity, the researchers were able to create a highly efficient enzyme from scratch. This demonstrates the power of AI to expand the functional repertoire of proteins beyond what is found in the natural world.
As the field of AI-driven protein design continues to advance, it is expected that more complex and ambitious design challenges will be tackled. The integration of deep learning with structure prediction, generative modeling, and high-throughput experimental validation will enable the creation of proteins with novel functions, tailored for specific applications in medicine, biotechnology, and materials science (Chu, Lu and Huang, 2024). The success stories highlighted above are just the beginning of what is possible with AI-driven protein design, and it is clear that this approach will play a crucial role in shaping the future of bioengineering and synthetic biology.
Challenges and Future Directions
Despite the remarkable progress in AI-driven protein design, several challenges remain to be addressed to fully realize the potential of this field. One of the primary limitations of current design methods is the difficulty in modeling and predicting the conformational dynamics of proteins. Most design approaches focus on generating static structures, treating proteins as rigid molecules. However, proteins are dynamic entities that undergo conformational changes, which are often crucial for their function. Incorporating conformational flexibility and multiple structural states into the design process is a significant challenge that requires the development of more sophisticated modeling techniques and the integration of molecular dynamics simulations with AI-driven design.
Another challenge lies in designing proteins that can function effectively in the complex cellular environment. Many current design methods focus on optimizing proteins for specific in vitro conditions, which may not translate well to the in vivo setting (Papaleo et al., 2016). Factors such as cellular localization, interactions with other biomolecules, and the effects of post-translational modifications need to be considered to design proteins that can operate robustly in living systems. Addressing this challenge will require a deeper understanding of the cellular context and the development of multiscale modeling approaches that can bridge the gap between the molecular and cellular scales (Feig and Sugita, 2019).
To tackle these challenges, several emerging approaches hold promise. One such approach is the development of sequence-structure co-design methods, which aim to simultaneously optimize both the sequence and structure of proteins. By iteratively refining the sequence and structure in a coupled manner, these methods can potentially capture the complex interplay between the two and generate designs that are more likely to fold and function as intended. Another promising direction is the incorporation of deep learning techniques that can learn and model the conformational landscape of proteins from large-scale molecular dynamics simulations. These methods could enable the design of proteins with desired dynamical properties and facilitate the exploration of the vast conformational space.
In addition to methodological advancements, the future of AI-driven protein design will also benefit from the integration of diverse data sources and the establishment of standardized benchmarks and evaluation metrics. Incorporating data from high-throughput experiments, such as deep mutational scanning and proteomics, can provide valuable information on the functional effects of mutations and guide the design process (Rocklin et al., 2017). Establishing well-defined benchmarks and evaluation criteria will be crucial for assessing the performance of different design methods and facilitating the comparison and reproducibility of results across studies.
As the field of AI-driven protein design continues to mature, it is expected to have a transformative impact on various domains, from basic research to biotechnology and medicine. The design of novel proteins with tailored functions could revolutionize fields such as biocatalysis, biomaterials, and synthetic biology. In the realm of healthcare, de novo designed proteins could lead to the development of innovative therapies for a wide range of diseases, including cancer, infectious diseases, and genetic disorders. However, realizing these potential applications will require close collaboration between computational scientists, experimental biologists, and clinicians, as well as the development of robust pipelines for the rapid validation and optimization of designed proteins.
In conclusion, while AI-driven protein design has made remarkable strides in recent years, significant challenges remain in modeling conformational dynamics, designing proteins for in vivo functionality, and integrating diverse data sources. Addressing these challenges will require the development of more sophisticated computational methods, the establishment of standardized evaluation frameworks, and close collaboration between multidisciplinary teams. As the field continues to advance, it holds immense promise for unlocking the full potential of de novo protein design and transforming various aspects of science and technology.
References
- Anishchenko, I. et al. (2021) ‘De novo protein design by deep network hallucination’, Nature, 600(7889), pp. 547–552. [CrossRef]
- Baek, M. et al. (2021) ‘Accurate prediction of protein structures and interactions using a three-track neural network’, Science, 373(6557), pp. 871–876. [CrossRef]
- Cao, L. et al. (2022) ‘Design of protein-binding proteins from the target structure alone’, Nature, 605(7910), pp. 551–560. [CrossRef]
- Chothia, C. (1984) ‘Principles that determine the structure of proteins’, Annual review of biochemistry, 53, pp. 537–572. [CrossRef]
- Chu, A.E., Lu, T. and Huang, P.-S. (2024) ‘Sparks of function by de novo protein design’, Nature biotechnology, 42(2), pp. 203–215. [CrossRef]
- Dou, J. et al. (2018) ‘De novo design of a fluorescence-activating β-barrel’, Nature, 561(7724), pp. 485–491. [CrossRef]
- Feig, M. and Sugita, Y. (2019) ‘Whole-Cell Models and Simulations in Molecular Detail’, Annual review of cell and developmental biology, 35, pp. 191–211. [CrossRef]
- Ferruz, N. et al. (2023) ‘From sequence to function through structure: Deep learning for protein design’, Computational and structural biotechnology journal, 21, pp. 238–250. [CrossRef]
- Ferruz, N., Schmidt, S. and Höcker, B. (2022) ‘ProtGPT2 is a deep unsupervised language model for protein design’, Nature communications, 13(1), p. 4348. [CrossRef]
- Gainza, P. et al. (2020) ‘Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning’, Nature methods, 17(2), pp. 184–192. [CrossRef]
- Gainza, P. et al. (2023) ‘De novo design of protein interactions with learned surface fingerprints’, Nature, 617(7959), pp. 176–184. [CrossRef]
- Glasscock, C.J. et al. (2023) ‘Computational design of sequence-specific DNA-binding proteins’, bioRxiv : the preprint server for biology [Preprint]. [CrossRef]
- Hie, B.L. et al. (2024) ‘Efficient evolution of human antibodies from general protein language models’, Nature biotechnology, 42(2), pp. 275–283. [CrossRef]
- Hsu, C., Fannjiang, C. and Listgarten, J. (2024) ‘Generative models for protein structures and sequences’, Nature biotechnology, 42(2), pp. 196–199. [CrossRef]
- Huang, P.-S., Boyken, S.E. and Baker, D. (2016) ‘The coming of age of de novo protein design’, Nature, 537(7620), pp. 320–327. [CrossRef]
- Jiang, L. et al. (2008) ‘De novo computational design of retro-aldol enzymes’, Science, 319(5868), pp. 1387–1391. [CrossRef]
- Jumper, J. et al. (2021) ‘Highly accurate protein structure prediction with AlphaFold’, Nature, 596(7873), pp. 583–589. [CrossRef]
- Korendovych, I.V. and DeGrado, W.F. (2020) ‘protein design, a retrospective’, Quarterly reviews of biophysics, 53, p. e3. [CrossRef]
- Krishna, R. et al. (2024) ‘Generalized biomolecular modeling and design with RoseTTAFold All-Atom’, Science, p. eadl2528. [CrossRef]
- Li, Y. et al. (2023) ‘Denoising Diffusion Probabilistic Models and Transfer Learning for citrus disease diagnosis’, Frontiers in plant science, 14, p. 1267810. [CrossRef]
- Lu, H. et al. (2022) ‘Machine learning-aided engineering of hydrolases for PET depolymerization’, Nature, 604(7907), pp. 662–667. [CrossRef]
- Madani, A. et al. (2023) ‘Large language models generate functional protein sequences across diverse families’, Nature biotechnology, 41(8), pp. 1099–1106. [CrossRef]
- Martínez-Mauricio, K.L., García-Jacas, C.R. and Cordoves-Delgado, G. (2024) ‘Examining evolutionary scale modeling-derived different-dimensional embeddings in the antimicrobial peptide classification through a KNIME workflow’, Protein science: a publication of the Protein Society, 33(4), p. e4928. [CrossRef]
- Notin, P. et al. (2024) ‘Machine learning for functional protein design’, Nature biotechnology, 42(2), pp. 216–228. [CrossRef]
- Papaleo, E. et al. (2016) ‘The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery’, Chemical reviews, 116(11), pp. 6391–6423. [CrossRef]
- Polizzi, N.F. and DeGrado, W.F. (2020) ‘A defined structural unit enables de novo design of small-molecule-binding proteins’, Science, 369(6508), pp. 1227–1233. [CrossRef]
- Rives, A. et al. (2021) ‘Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences’, Proceedings of the National Academy of Sciences of the United States of America, 118(15). [CrossRef]
- Rocklin, G.J. et al. (2017) ‘Global analysis of protein folding using massively parallel design, synthesis, and testing’, Science, 357(6347), pp. 168–175. [CrossRef]
- Schubach, M. et al. (2024) ‘CADD v1.7: using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions’, Nucleic acids research, 52(D1), pp. D1143–D1154. [CrossRef]
- Scott, A.J. et al. (2021) ‘Constructing ion channels from water-soluble α-helical barrels’, Nature chemistry, 13(7), pp. 643–650. [CrossRef]
- Sesterhenn, F. et al. (2020) ‘De novo protein design enables the precise induction of RSV-neutralizing antibodies’, Science, 368(6492). [CrossRef]
- Singer, J.M. et al. (2022) ‘Large-scale design and refinement of stable proteins using sequence-only models’, PloS one, 17(3), p. e0265020. [CrossRef]
- Trippe, B.L. et al (2022) ‘Diffusion probabilistic modeling of protein backbones in 3D for the motif-scaffolding problem’, arXiv, 2206.04119. [CrossRef]
- Walls, A.C. et al. (2020) ‘Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for SARS-CoV-2’, Cell, 183(5), pp. 1367–1382.e17. [CrossRef]
- Wang, J. et al. (2022) ‘Scaffolding protein functional sites using deep learning’, Science, 377(6604), pp. 387–394. [CrossRef]
- Watson, J.L. et al. (2023) ‘De novo design of protein structure and function with RFdiffusion’, Nature, 620(7976), pp. 1089–1100. [CrossRef]
- Woolfson, D.N. (2021) ‘A Brief History of De Novo Protein Design: Minimal, Rational, and Computational’, Journal of molecular biology, 433(20), p. 167160. [CrossRef]
- Wu, K.E. et al. (2024) ‘Protein structure generation via folding diffusion’, Nature communications, 15(1), p. 1059. [CrossRef]
- Xie, K. et al. (2022) ‘Inpainting the metal artifact region in MRI images by using generative adversarial networks with gated convolution’, Medical physics, 49(10), pp. 6424–6438. [CrossRef]
- Yang, K.K., Wu, Z. and Arnold, F.H. (2019) ‘Machine-learning-guided directed evolution for protein engineering’, Nature methods, 16(8), pp. 687–694. [CrossRef]
- Zhang, Y. et al. (2023) ‘Attention is all you need: utilizing attention in AI-enabled drug discovery’, Briefings in bioinformatics, 25(1). [CrossRef]
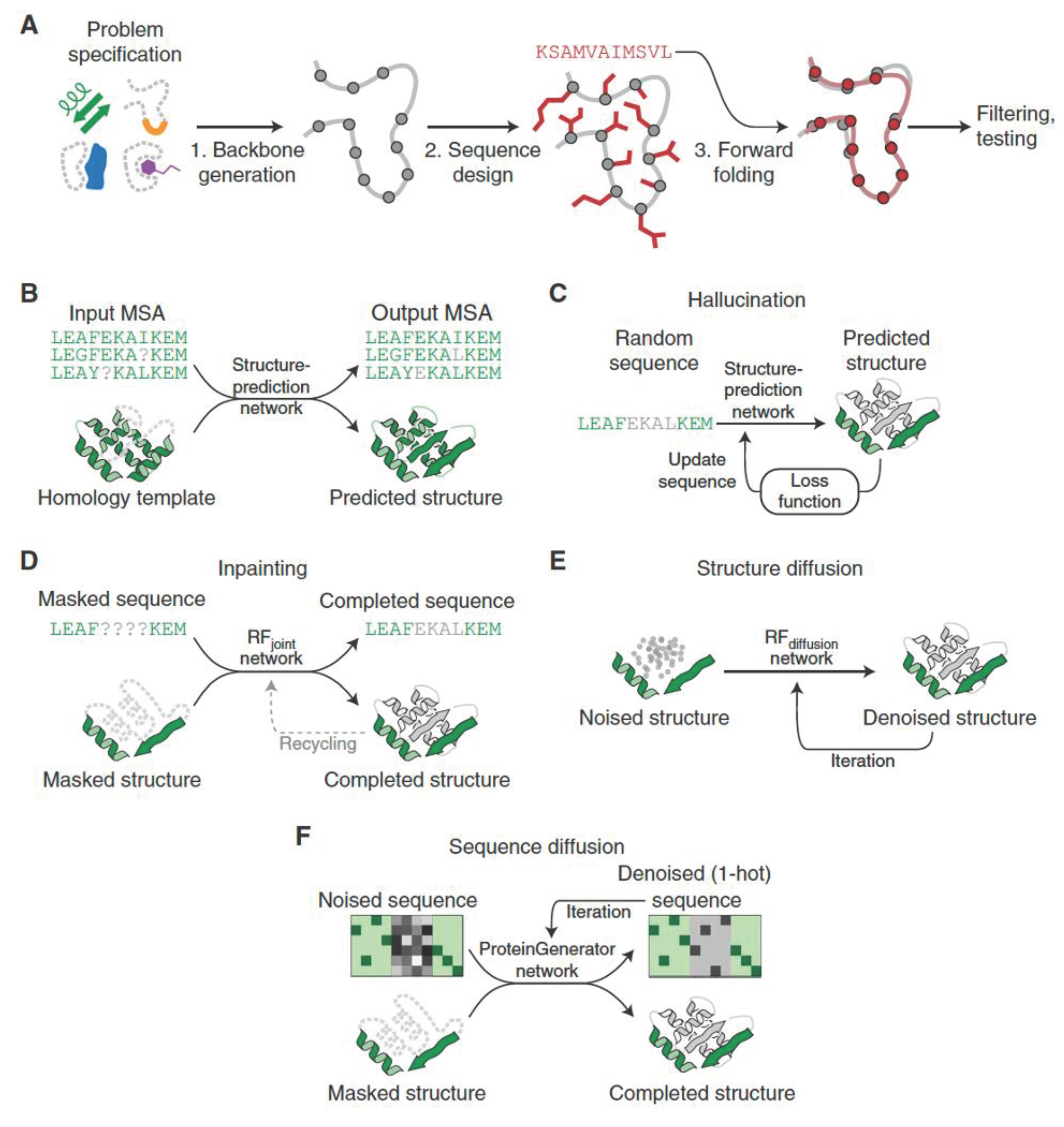
Figure 1.
Modified from Wang et al. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a041472. Overview of de novo protein design techniques. (A) Common workflow for designing proteins from scratch. First, the design objective is defined, such as targeting specific secondary structures (green), incorporating a motif (orange), or binding to a protein (blue) or ligand (purple). Next, step 1 involves generating a protein backbone that aligns with the design goals. In step 2, a sequence is designed to adopt the desired backbone fold (sequence and sidechains depicted in red). Step 3 involves validating that the designed sequence folds into the intended backbone using an independent structure-prediction approach (predicted structure shown in red). Lastly, designs are chosen based on various computational metrics and experimentally validated. (B) Inputs and outputs of structure-prediction networks like AlphaFold and RoseTTAFold. During training, random residues in the input multiple-sequence alignment (MSA) are masked (substituted with a special token) and re-predicted, allowing sequence information to be output by the network for fine-tuning tasks. Structures of "template" proteins homologous to the query sequence aid structure prediction, enabling structure information to be input during fine-tuning. (C–E) Structure-prediction network-based protein design methods: (C) hallucination, (D) inpainting with RFjoint, (E) structure diffusion with RFdiffusion, and (F) sequence diffusion with ProteinGenerator. An example motif-scaffolding task is illustrated, with green representing a predefined structural motif and gray indicating what is generated by the design method. While hallucination and diffusion are inherently iterative, RFjoint inpainting can perform one-shot design. However, iteration or "recycling" of outputs is necessary for effective design in practice (gray dotted arrow in D). In F, sequence is schematically represented as a logit matrix, where the region to be generated is initialized with random real-numbered values and denoised until it becomes 1-hot.
Figure 1.
Modified from Wang et al. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a041472. Overview of de novo protein design techniques. (A) Common workflow for designing proteins from scratch. First, the design objective is defined, such as targeting specific secondary structures (green), incorporating a motif (orange), or binding to a protein (blue) or ligand (purple). Next, step 1 involves generating a protein backbone that aligns with the design goals. In step 2, a sequence is designed to adopt the desired backbone fold (sequence and sidechains depicted in red). Step 3 involves validating that the designed sequence folds into the intended backbone using an independent structure-prediction approach (predicted structure shown in red). Lastly, designs are chosen based on various computational metrics and experimentally validated. (B) Inputs and outputs of structure-prediction networks like AlphaFold and RoseTTAFold. During training, random residues in the input multiple-sequence alignment (MSA) are masked (substituted with a special token) and re-predicted, allowing sequence information to be output by the network for fine-tuning tasks. Structures of "template" proteins homologous to the query sequence aid structure prediction, enabling structure information to be input during fine-tuning. (C–E) Structure-prediction network-based protein design methods: (C) hallucination, (D) inpainting with RFjoint, (E) structure diffusion with RFdiffusion, and (F) sequence diffusion with ProteinGenerator. An example motif-scaffolding task is illustrated, with green representing a predefined structural motif and gray indicating what is generated by the design method. While hallucination and diffusion are inherently iterative, RFjoint inpainting can perform one-shot design. However, iteration or "recycling" of outputs is necessary for effective design in practice (gray dotted arrow in D). In F, sequence is schematically represented as a logit matrix, where the region to be generated is initialized with random real-numbered values and denoised until it becomes 1-hot.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



