
Submitted:
02 April 2024
Posted:
03 April 2024
You are already at the latest version
Abstract
Triple-negative breast cancer (TNBC) is an aggressive and highly metastatic type of tumor. TNBC is often enriched in tumor-infiltrating neutrophils (TINs), which support cancer growth in part by counteracting tumor-infiltrating lymphocytes (TILs). Prior studies identified the enhancer of zeste homolog 2 (EZH2) as a pro-tumor methyltransferase in primary and metastatic TNBC. We hypothesized that EZH2 inhibition in TNBC cells per se would exert anti-tumor activity by altering the tumor immune microenvironment. To test this hypothesis, we used CRISPR to generate EZH2-knockout (KO) and overexpressing lines from parent (wild-type -WT) 4T1 cells, an established murine TNBC model. In vitro, EZH2 KO and OE cells showed little change in replicative capacity and invasiveness, but marked changes in surface marker profile and cytokine/chemokine secretion compared to WT cells. On in vivo injection, EZH2 KO cells showed significantly reduced primary tumor growth and a 10-fold decrease in lung metastasis compared to WT cells, while EZH2 OE cells were unchanged. Compared to WT tumors, TIN:TIL ratios were greatly reduced in EZH2 KO tumors but unchanged in EZH2 OE tumors. Our findings show a tumor-intrinsic role of EZH2 in the 4T1 TNBC model in regulating TIN/TIL poise, primary tumor progression and lung metastasis in vivo.
Keywords:
CD4+ T-cell
; CD8+ T-cell
; invasion
; myeloid-derived suppressor cell
; triple-negative breast cancer
; tumor-infiltrating neutrophils
1. Introduction
Breast cancer is a global health problem affecting 2.3 million individuals globally [1]. Defined as estrogen- and progesterone-receptor negative and lacking HER2 overexpression, triple-negative breast cancer (TNBC) holds the poorest prognosis among breast cancer types [2]. This aggressive form occurs in 15-20% of patients, accounting for ~170,000 cases worldwide [3]. TNBC is comprised of different subtypes, characterized by distinct molecular signatures. Common treatments include chemotherapy, radiation, and surgical resection. Patients with T-cell rich or non-refractory (“hot”) tumors also benefit from newly developed immune checkpoint inhibitor (ICI) and chimeric antigen receptor T-cell therapies [4]. Unfortunately, a large proportion of TNBC patients are refractory to ICI therapy (designated as “cold”) [3,5]. Consequently, primary tumors and metastatic outgrowth from chemotherapy-resistant TNBC are a major cause of mortality [6].
Genetic and epigenetic alterations in TNBC are some of the main obstacles for successful response to therapy [7]. Among TNBC markers identified as potential therapeutic targets, the methyltransferase enhancer of zeste homolog 2 (EZH2) holds significant promise, as its overexpression is associated with poor prognosis and short-disease-free survival in patients [8]. EZH2 contributes to tumor development, progression and metastasis via multiple pathways, including, but not limited to, modulation of transforming growth factor β (TGF β) and stimulator of interferon genes (STING) signaling [9,10]. Thus, EZH2 inhibitors have been tested in combination with ICIs and other chemotherapies [11]. A confounding factor, however, is that expression of EZH2 occurs in both tumor cells and in tumor-associated innate and adaptive immune cells (e.g., tumor-infiltrating neutrophils -TINs- and lymphocytes -TILs-, respectively) [7].
Here, we probed the intrinsic role of EZH2 in primary and metastatic TNBC [12] using the 4T1 murine TNBC model. To this end, we used CRISPR technology to drive EZH2 knockout (KO) and overexpression (OE) in stable cell lines derived from parent wild-type (WT) 4T1 cells. While replicative and invasive capacities of EZH2 KO and OE cells did not broadly differ from those of WT 4T1 cells in vitro, the former but not the latter showed significantly decreased primary growth and lung metastasis in vivo, along with dramatic reductions in the ratios of TINs to both CD4+ and CD8+ T-cells.
2. Results
2.1. EZH2 Knockout and Overexpressing Lines Derived from the 4T1 TNBC Model Behave Similarly to the Parent Line In Vitro
We generated multiple EZH2 KO and EZH2 OE clones from parent WT 4T1 cells using CRISPR/Cas9 and gene amplification followed by a round of sorting. Lysates were prepared from each EZH2 KO and EZH2 OE clones and analyzed by western blot, using α-tubulin as a normalization control. All KO and OE clones demonstrated successful elimination and overexpression of EZH2, respectively (Figure 1).
To characterize the effect of altered cell-intrinsic EZH2 expression on 4T1 behavior in vitro, we selected 4T1 EZH2 KO11 and EZH2 OE6 clones for further investigation. These two clones were selected because of grossly normal morphology and viability compared to WT. They were plated in parallel with the parent WT 4T1 cell line at 5x105 cells and counts were performed at 24, 48, and 72 hours post-plating, revealing no significant difference in replication (Figure 2A). Next, we used a 3D spheroid invasion assay to evaluate the invasive capacity of EZH2 KO, EZH2 OE and WT 4T1 cells at 0, 24, 48, and 72 hours post-platingd (Figure 2B). Spheroid circularity was similar at time 0 across WT, EZH2 KO and EZH2 OE lines but slightly higher for EZH2 OE at 24, 48, and 72 hours and lower for EZH2 KO at 24 hours, compared to WT (Figure 2C). Invasive chains emerging from spheroids were quantified and found to be higher at 0, 24 hours for EZH2 KO and lower at 24 hours for EZH2 OE lines, compared to WT, and higher for EZH2 KO than EZH2 OE at 0, 24, and 48 hours, but showed no difference across all lines at 72 hours (Figure 2D).
2.2. EZH2 Expression Impacts Surface Phenotype and Secreted Mediators of 4T1 Cells In Vitro
Using flow cytometry (Figure S1A), expression of relevant markers on EZH2 KO, EZH2 OE and WT 4T1 cells was assessed against unstained controls for positivity (Figure S1B), and positively expressed markers were then quantified over multiple repeats across lines (Figure 3A). The expression levels of CD24, CD44, ICAM-1, MHC-I and checkpoint inhibitor PD-L1 differed across 4T1 WT, EZH2 KO and EZH2 OE cells. Specifically, expression of CD24 and CD44 were lower on EZH2 KO compared to both WT and EZH2 OE cells, and expression of ICAM-1 and PD-L1 were lower EZH2 KO than in EZH2 OE cells, while EZH2 OE cells were similar to WT for all markers except for lower MHC-I expression. When assessing their profile of twelve secreted mediators (Figure 3B), we observed no significant difference between WT and either EZH2 KO or EZH2 OE cells. However, EZH2 KO cells secreted higher levels of several mediators in comparison to EZH2 OE cells, with significant >4-fold increases for GM-CSF and MCP-1 and >2-fold increase for IL1β. Similar trends for increased secretion in EZH2 KO vs EZH2 OE cultures were observed for IFNα, IL-10, IP-10, and TNFα secretion, albeit non-significant.
2.3. EZH2 Knockout Reduces Primary Tumor Growth and Lung Metastasis of 4T1 Cells In Vivo
Next, to test the impact of EZH2 knockout and overexpression on 4T1 TNBC primary tumor growth and metastasis in vivo, we challenged mice with WT, EZH2 KO and EZH2 OE cells. Cells were injected subcutaneously in the flank of BALB/c mice and allowed to grow over 21 days (Figure 4A). While WT and EZH2 OE cells showed similar primary tumor growth across all timepoints, EZH2 KO cells grew significantly slower than both, resulting in a 2-fold smaller size at day 21 (Figure 4B). Since the 4T1 TNBC model is spontaneously metastatic, the lungs of tumor-bearing mice were isolated at the end of the in vivo challenge and processed for quantification of metastatic cells. Strikingly, EZH2 KO showed significantly reduced (approximately 10-fold lower) lung metastatic burden compared to WT and EZH2 OE cells (Figure 4C). To confirm these results, we repeated the in vivo challenge comparing WT and EZH2 KO cells and extended the duration by a week. Again, EZH2 KO cells grew significantly slower than WT, reaching 4-fold smaller size at day 28 (Figure S2A). The difference in primary tumor size between the two groups was evident macroscopically by day 18 (Figure S2B). A 10-fold lower lung metastatic burden was again observed between EZH2 KO and WT cells (Figure S2C).
2.4. Tumor-Intrinsic EZH2 Knockout Alters the Balance of Neutrophils, CD4+ and CD8+ T-Cells in Primary 4T1 Tumors
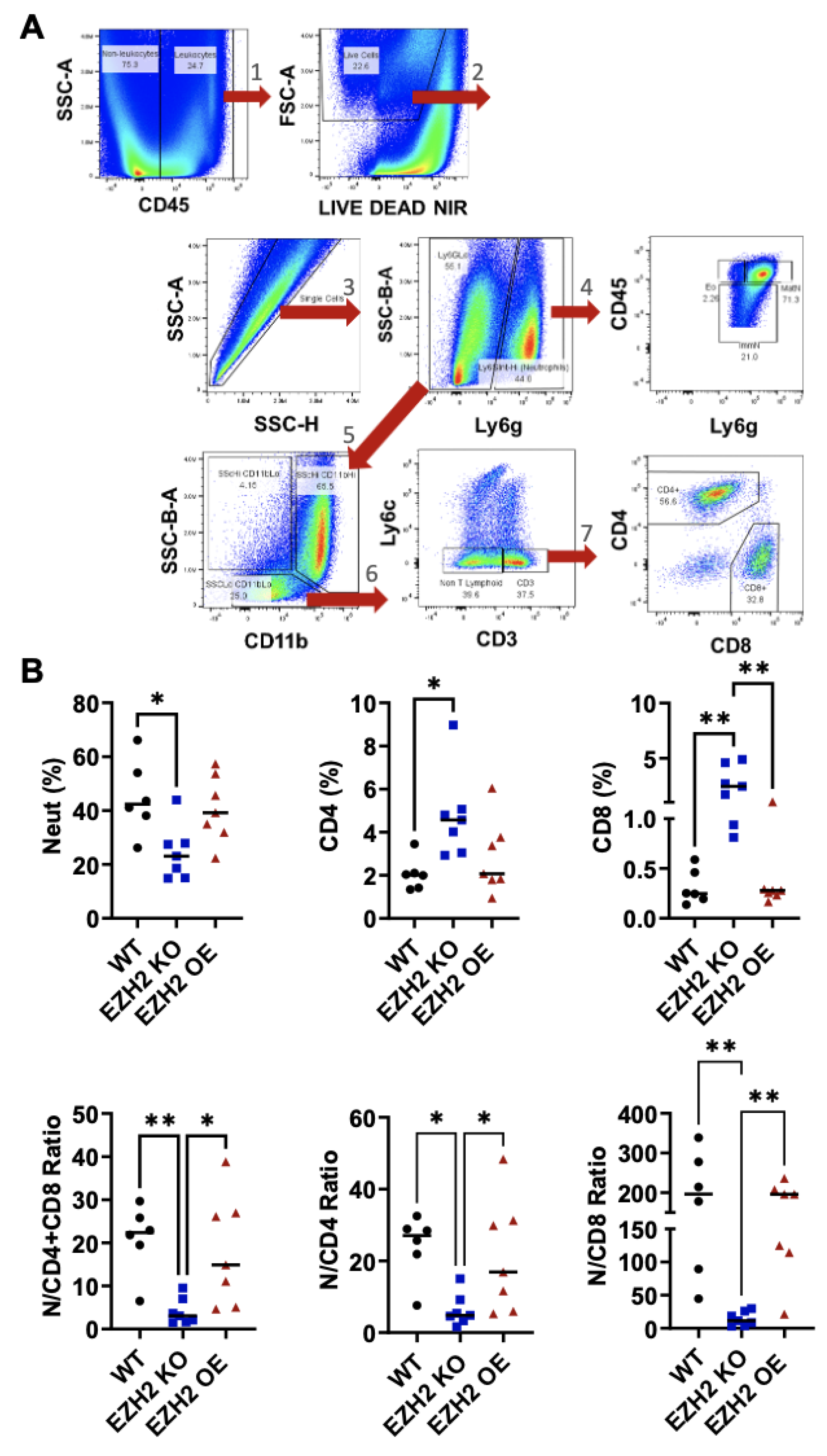
To determine whether altering EZH2 expression affects immune cell infiltration into the tumors, we prepared single-cell suspensions from WT, EZH2 KO, and EZH2 OE primary tumors and quantified live infiltrating leukocytes through flow cytometry analysis and sequential gating of relevant subsets (Figure 5A). While WT and EZH2 OE tumors had similar proportions across all leukocyte subsets measured, EZH2 KO tumors showed on average 2-fold lower proportion of neutrophils, and 2- and 10-fold higher proportions of CD4+ and CD8+ T-cells, respectively, than WT and EZH2 OE tumors (Figure 5B). Consistently, EZH2 KO tumors showed on average 3- to 4-fold higher proportion of CD3+ T-cells (encompassing both CD4+ and CD8+), while their lower proportion of infiltrated neutrophils included both mature and immature cells (Figure S3). The paradoxical effect of tumor-intrinsic EZH2 knockout on infiltrated neutrophils (decrease) as well as CD4+ and CD8+ T-cells (increase) was even more striking when expressed as ratios, revealing a >20-fold decrease in the neutrophil:CD8+ T-cell ratio in EZH2 KO vs WT and EZH2 OE tumors (Figure 5B).
3. Discussion
Here we report the successful CRISPR-aided generation of several EZH2 KO and EZH2 OE clones derived from the 4T1 murine TNBC cell line. Based on grossly normal morphology and viability compared to WT, one EZH2 KO line and EZH2 OE line were selected for further phenotypic and evaluation in vitro and in vivo. Analysis of in vitro proliferation on 2D plates, and invasiveness in a 3D spheroid assay [17] showed no major differences between EZH2 KO, EZH2 OE and WT 4T1 cell lines, suggesting little regulatory impact of EZH2 on these properties. These results are consistent with prior data on siRNA-aided knockdown of EZH2 in 4T1 cells, also reporting no apparent effect on cell proliferation or invasiveness [10]. Our study is the first to introduce EZH2 OE cells and shows that they behave very similarly to WT cells both in vitro and in vivo, suggesting that EZH2 expression may be already saturating in parent WT 4T1 cells.
In regard to cell surface markers, only MHC class I was altered (lowered) in EZH2 OE cells compared to WT, while EZH2 KO cells showed decreased expression of multiple immune activation markers (CD24, CD44, ICAM-1, PD-L1) compared to EZH2 OE cells, with intermediate levels in WT cells. Significantly altered secretion of GM-CSF, MCP-1 and IL1β (all myeloid mediators) was observed in EZH2 KO culture supernatants, suggesting that EZH2 may impact immune crosstalk by 4T1 cells. Indeed, EZH2 KO tumors grown in vivo decreased the proportion of infiltrated neutrophils and increased those of infiltrated CD4+ and CD8+ T-cells (culminating in a 20-fold reduction of the neutrophil:CD8+ T-cell ratio). Concomitantly, EZH2 KO tumors displayed significantly reduced growth in the primary tumor site (by 2 to 4-fold) and lung metastatic potential (by 10-fold). Together, our findings suggest that while baseline EZH2 expression in 4T1 TNBC cells does not seem to play essential roles in vitro, it is necessary to maintain high TIN:TIL ratios and their growth and metastatic potential in vivo.
There is ample evidence that EZH2 is involved in the epigenetic control of critical immune regulatory pathways [7], notably including the STING pattern recognition receptor [9,19]. STING plays multiple crucial roles in danger signaling, interferon secretion and leukocyte infiltration in solid tumors [20]. Prior research on the 4T1 model demonstrated altered levels pro-inflammatory and interferon-related cytokines in the serum [21,22]. Future studies in our EZH2 KO and EZH2 OE lines will evaluate the relative roles of STING and other transcriptional regulators in immune signaling by 4T1 cells.
Our in vivo findings are consistent with previous studies demonstrating that cells with high EZH2 expression have an advantage in metastasizing, while EZH2 KO 4T1 tumor-bearing mice have significantly longer survival and decreased occurrence of metastatic colonies [23]. Previous work showed that metastasis in the WT 4T1 model is impacted by the capacity of circulating cancer cells to undergo epithelial-mesenchymal transition in order to successfully establish micrometastases at distant organ sites [24]. Since EZH2 KO primary tumors likely have to undergo similar processes to WT cells to metastasize, our results suggest that EZH2 deficiency in the former may not only negatively affect tumor cell mobilization and survival but also the processes leading to metastasis.
TINs have emerged as key modulators of primary tumor growth and metastatic progression in TNBC [25]. As the most abundant leukocyte in human bone marrow and blood, neutrophils can infiltrate tumors in high numbers and acquire novel activities therein, promoting anti- and/or pro-tumorigenic functions [26]. In the context of TNBC patients, high levels of TINs are predictive of poor treatment responses and decreased survival [27,28]. TILs and TINs often play antagonistic roles; for example, TINs may inhibit recruitment and/or activation of TILs via metabolic (e.g., arginase-mediated amino acid depletion) or cell-cell (e.g., PD-1/PD-L1, TIM-3/Gal-9) interactions [29,30]. Because of these activities, TINs, (whether derived from developmentally mature or immature neutrophils) are often categorized under the functional term “myeloid-derived suppressive cells” (MDSCs) [31].
Current therapeutic approaches face the challenge of alleviating cold tumor progression in the absence of T-cell activation. Although conventional anticancer treatments like chemotherapy and radiotherapy still have important roles to play in tumor burden reduction and preventing selection of immune-resistant clones, incorporation of improved therapies that suit the mutational burden in patients are much needed. In this context, increasing tumor sensitivity to ICI therapy by converting them from a “cold” to a “hot” phenotype may lead to better outcomes. Overall, limiting TIN infiltration to enable TIL antitumor activity (i.e., making cold tumors hot) is a major goal of current research [4]. Since EZH2 can be expressed in both TINs [32] and TILs [33], further understanding its role and impact as a tumor-intrinsic and/or immune-associated factor is critical to overcoming TNBC resistance and improving patient outcomes.
4. Materials and Methods
4.1. Cell Lines
The parent WT 4T1 (CRL-2539) cell line [12] was purchased from ATCC, and cultured at 37°C and 5% CO2 in DMEM-high glucose medium (Sigma Aldrich), supplemented with 10% fetal bovine serum (FBS, HyClone), 1% HEPES, 1% L-glutamine, and penicillin-streptomycin (100 U mL−1). Expression constructs were made using a modified Golden Gate Assembly protocol [13]. Murine EZH2 was amplified from pINTO-NFH:mEZH2 (Addgene #65925; gift from Roberto Bonasio [14]) and cloned into pBD170abc, a level 0 destination vector for downstream cloning of an open reading frame. Then, a level 1 expression construct (pBD320) was cloned with a CMV promoter, BetaGlobin 3’UTR into a position 1 destination vector (pTW324; Addgene #115955; gift from Ron Weiss) using Bsa1 and T4 DNA Ligase. Puromycin alone (pBD324) or mScarlet-IRES[EMCV]-puromycin (pBD332) expression vectors under the control of a PGK promoter were cloned into a position 2 destination vector (pTW325; Addgene #115956; gift from Ron Weiss). Then, pBD320 was combined with a minimal linker and pBD324 or pBD332, respectively, to generate level 2 dual expression constructs pBD321 and pBD333. For KO plasmids, the murine EZH2 sequences from AOI-WT-Cas9-sq-mouse Ezh2-E18-GFP and AOI-WT-Cas9-sq-mouse Ezh2-E10-GFP (Addgene #91880 and Addgene #91879; gift from Martine Roussel [15]) were cloned into pSPCas9(BB)-2A-Puro (Addgene #62988; gift from Feng Zhang [16]). Plasmids were transfected into 4T1 cells using lipofectamine 3000 (ThermoFisher Scientific) following the manufacturer’s protocol and selected using puromycin at concentration 5 µg/ml. Stable, OE clones were continually grown in puromycin; KO clones were only exposed to puromycin for up to a week. Cells were then isolated into 96-well plates and clones selected.
4.2. Western Blot
Cell lysates were prepared using the Minute Total Protein Extraction Kit (Invent Biotechnologies). The provided denaturing buffer was supplemented with Halt’s protease and phosphatase inhibitor cocktail at a 3x concentration (ThermoFisher Scientific). Lysates were passed through spin columns to remove viscosity. Total protein concentration was measured using the Pierce Rapid Gold BCA protein assay kit, concentration was normalized across treatments with denaturing buffer, then Laemmli sample buffer (Bio-Rad) supplemented with β-mercaptoethanol was added. Samples were then boiled at 95°C for 5 minutes and lysates were separated by SDS-PAGE on Any-KD gels (Bio-Rad). Protein transfer to nitrocellulose membranes (LI-COR) was done at 4°C by wet transfer in Towbin buffer [25 mM Tris, 192 mM glycine, 20% MeOH (v/v) without SDS]. After transfer, the membrane was rinsed three times with double-distilled H2O. Blocking was done using Intercept (TBS) blocking buffer (LI-COR) for one hour at room temperature in motion. Primary antibodies to EZH2 and α-tubulin (from Cell Signaling Technologies, used at 1:1000) were added in Intercept T20 antibody diluent (LI-COR) at 4°C overnight in motion. Secondary IRDye 800CW donkey anti-rabbit IgG antibody (1:15,000 LI-COR) was added in Intercept antibody diluent (LI-COR) for 1 hour at 37°C on the shaker. Membranes were analyzed using an Odyssey CLx imager and Image Studio software (LI-COR).
4.3. Cell Proliferation Assay
EZH2 KO, EZH2 OE and WT 4T1 cells were harvested, resuspended in DMEM-complete medium and counted. Then 5x105 cells were plated on a T25 flask in 5 mL of medium and cultured at 37°C and 5% CO2. At 24, 48, and 72 hours, cells were harvested and stained with propidium iodide to determine cell count and viability using a hemocytometer.
4.4. 3D Spheroid Invasion Assay
To generate EZH2 KO, EZH2 OE and WT 4T1 cell spheroids, 3,000 cells were plated in 200 µL on a Spheroid Nunclon 96-well plate (Thermo Scientific) and centrifuged at 450gs for 5 minutes at 4°C and incubated at 37°C and 5% CO2. After 48-72 hours of incubation, spheroids were collected embedded in 3 mg/mL collagen type I (Corning) and then plated in a 35 mm glass bottom dish (In Vitro Scientific) for incubation overnight at 37°C. After collagen polymerized, complete DMEM (Invitrogen) was added to cover the collagen matrix and spheroids. An Olympus IX51 at 10X magnification (equal to 1.5 pixels/μm) with an Infinity2 charge-coupled device camera was used for 3D spheroid imaging. Spheroid circulatory and invasiveness was measured by ImageJ as described before [17].
4.5. Extracellular Mediator Assay
Supernatants from in vitro cultures of EZH2 KO, EZH2 OE and WT 4T1 cells were collected and stored at -80°C until use. Extracellular mediators were quantified using a U-PLEX multiplexed chemiluminescent ELISA assay (Meso Scale Discovery), following the manufacturer’s protocol. Plates were acquired on the QuickPlex SQ 120MM reader and later analyzed using the Discovery Workbench 4.0 software (both from Meso Scale Discovery).
4.6. Animals
BALB/c mice (female, 6-8 weeks old) were purchased from Jackson Laboratories and maintained in the Division of Animal Resources (DAR) facilities at Emory University. Experiments were performed in accordance with Emory University Institutional Animal Care and Use Committee (IACUC) approved protocol (DAR-2017-00-504). Female mice were chosen to match the strain and sex of origin of the parent WT 4T1 cell line. To establish the model, 5x105 cells were injected subcutaneously (s.c.) in the right flank as the location of the primary tumor (Munoz et al., 2021). Tumor size (mm2) was measured in two dimensions with Vernier calipers every 3 days.
4.7. Lung Metastasis Assay
Lungs were isolated under sterile conditions from tumor-bearing mice 21-28 days post-injection, then minced and digested in 1 mg/mL collagenase IV (Millipore Sigma) for 2 hours at 37°C under rotating motion. After digestion, single-cell suspensions were filtered through a 70 μm strainer and washed twice in selection medium consisting of complete DMEM with 6-thioguanine (Millipore Sigma) at 60 μM. Cells were resuspended in 8 mL of selection medium, and 1 mL was plated per well in a 6-well plate for each lung digestion. After 7 to 14 days of incubation in selection medium (to kill lung fibroblasts without affecting tumor cells), as soon as one of the wells reached confluency, all wells were harvested and counted on a Cellometer T4 Automated Counter (Nexcelom) using trypan blue to discriminate dead cells.
4.8. Flow Cytometry Staining and Data Acquisition
For in vitro analyses, EZH2 KO, EZH2 OE and WT 4T1 cells were thawed, resuspended in DMEM-complete medium and cultured in T75 flasks. Before reaching confluency, cells were harvested and counted using propidium iodide. For ex vivo analyses, tumors grown in the flank of mice were harvested, weighed, minced, and digested in liberase TL (Roche) and DNAse (Roche) for 30 minutes at 37°C in motion. Cell suspensions were filtered through a 70 μm strainer and washed with PBS. Total cell count was determined using a Cellometer T4 Automated Counter and trypan blue. All cells were pre-incubated with Fc receptor blocking antibody (Clone 24G2, BioLegend) in FACS buffer at room temperature for 10 minutes. Then, cells were incubated with fluorochrome-conjugated antibodies for 30 minutes at 4°C. The in vitro staining panel included antibodies to CD24 (clone M1/69), CD44 (clone IM7), CD80 (clone 16-10A1), ICAM-1 (clone YN1/1.7.4), MHC class I (clone M1/42), MHC class II (clone M5/114.15.2), and PD-L1 (clones 10F.9G2), all purchased from BioLegend. The ex vivo staining panel included, in addition to the above, antibodies to CD3 (clone 17A2), CD4 (clone GK15), CD8a (clone 53-6.7), CD11b (clone M1/70), CD11c (clone N418), CD19 (clone 6D5), CD45 (clone 30-F11), CD69 (clone H1.2F3), CD107a (clone 1D4B), F4/80 (clone BM8), Ly6C (clone HK1.4), Ly6G (clone 1A8), NK1.1 (clone PK136), and PD-1 (clone 29F.1A12), also purchased from BioLegend. The live dead fixable NIR (1:400 in PBS) was obtained from Thermo Fisher Scientific. After incubation, cells were washed three times with FACS buffer and analyzed using the Aurora Spectral Flow Cytometer (Cytek). Data was analyzed using FlowJo software (BD Biosciences).
4.9. Statistical Analysis
All statistical analysis and graphs were done using Prism software (GraphPad). Non-parametric methods were used for descriptive statistics (box plots with median line and interquartile range forming outside boundaries to illustrate distributions), and comparisons between conditions and or timepoints. One-way ANOVA was used to analyze differences between the three groups (EZH2 KO, EZH2 OE and WT 4T1 cells) at fixed timepoints. Two-way ANOVA was used to test group differences across timepoints, e.g., for invasiveness in 3D spheroid assay over 3 days in vitro or primary tumor growth over 21 days in vivo. Values of p<0.05 were considered significant (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: In vitro surface phenotype of EZH2 KO and EZH2 OE compared to parent WT 4T1 cells; Figure S2. Experiment repetition of in vivo primary tumor growth and lung metastasis by EZH2 KO compared to parent WT 4T1 cells to corroborate significant reduction of tumor growth and decreased lung metastasis; Figure S3. In vivo primary tumor infiltration by CD3+ T cells, mature and immature neutrophils for EZH2 KO and EZH2 OE compared to parent WT 4T1 cells.
Author Contributions
Conceptualization, L.M., P.S., and R.T.; methodology, L.M., S.B.Y., B.S.D., P.S., and R.T.; validation, P.S., R.B., and R.T.; formal analysis, L.M., J.H., R.B., P.S., and R.T.; investigation, L.M., M.M.P., S.G.S., and D.E.R.R.; resources, L.C.B., A.M., P.S., and R.T.; data curation, L.M., J.H., and R.T.; writing—original draft preparation, L.M., P.S., and R.T.; writing—review and editing, L.M., M.M.P., S.G.S., D.E.R.R., S.B.Y., R.B., P.S., and R.T.; visualization, L.M., M.M.P., and R.T.; supervision, P.S. and R.T.; project administration, L.M., R.B., P.S. and R.T.; funding acquisition, P.S. and R.T. All authors have read and agreed to the published version of the manuscript.
Funding
Dormarie E. Rivera-Rodriguez is supported by the NIH grant T32AI106699. Lenore Monterroza was partly supported by a NIH Diversity Supplement Award R44CA257278-01A1S1. Study was supported by internal funds from the Selvaraj and Tirouvanziam labs.
Institutional Review Board Statement
All animal experiments were done in accordance with the Emory University Institutional Animal Care and Use Committee (IACUC) approved protocols (DAR-2017-00-504).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are available upon reasonable request to the author.
Acknowledgments
The authors thank Emory University Pediatrics and Winship Flow Cytometry core, Emory Division of Animal Resources (DAR) and Emory University Institutional Animal Care and Use Committee (IACUC). The authors also thank Dr. Curtis Henry from the University of Colorado Anschutz Medical Campus, Rebecca Parker, and Drs. James Lyles and Wei Zhou from Emory University for feedback and critical reading of the manuscript.
Conflicts of Interest
Not applicable.
References
- Arnold, M.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Zagami, P.; Carey, L.A. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Aysola, K.; et al. Triple Negative Breast Cancer - An Overview. Hereditary Genet, 2013. 2013(Suppl 2).
- Wang, L.; et al. Hot and cold tumors: Immunological features and the therapeutic strategies. MedComm (2020) 2023, 4, e343. [Google Scholar] [CrossRef] [PubMed]
- Hong, R. and B. Xu, Breast cancer: an up-to-date review and future perspectives. Cancer Commun (Lond) 2022, 42, 913–936. [Google Scholar] [CrossRef] [PubMed]
- Peddi, P.F., M. J. Ellis, and C. Ma, Molecular basis of triple negative breast cancer and implications for therapy. Int J Breast Cancer 2012, 2012, 217185. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; et al. EZH2, a prominent orchestrator of genetic and epigenetic regulation of solid tumor microenvironment and immunotherapy. Biochim Biophys Acta Rev Cancer 2022, 1877, 188700. [Google Scholar] [CrossRef] [PubMed]
- Adibfar, S.; et al. The molecular mechanisms and therapeutic potential of EZH2 in breast cancer. Life Sci 2021, 286, 120047. [Google Scholar] [CrossRef]
- Duan, D.; et al. EZH2-CCF-cGAS Axis Promotes Breast Cancer Metastasis. Int J Mol Sci. 2022, 23. [Google Scholar] [CrossRef]
- Zhang, L.; et al. EZH2 engages TGFbeta signaling to promote breast cancer bone metastasis via integrin beta1-FAK activation. Nat Commun 2022, 13, 2543. [Google Scholar] [CrossRef]
- Liu, Y. and Q. Yang, The roles of EZH2 in cancer and its inhibitors. Med Oncol 2023, 40, 167. [Google Scholar] [CrossRef]
- Pulaski, B.A. and S. Ostrand-Rosenberg, Mouse 4T1 breast tumor model. Curr Protoc Immunol, 2001. Chapter 20: p. Unit 20 2.
- Wagner, T.E.; et al. Small-molecule-based regulation of RNA-delivered circuits in mammalian cells. Nat Chem Biol 2018, 14, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.B.; et al. In vivo proximity labeling for the detection of protein-protein and protein-RNA interactions. J Proteome Res 2014, 13, 6135–6143. [Google Scholar] [CrossRef] [PubMed]
- Vo, B.T.; et al. Inactivation of Ezh2 Upregulates Gfi1 and Drives Aggressive Myc-Driven Group 3 Medulloblastoma. Cell Rep 2017, 18, 2907–2917. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Summerbell, E.R.; et al. Epigenetically heterogeneous tumor cells direct collective invasion through filopodia-driven fibronectin micropatterning. Sci Adv 2020, 6, eaaz6197. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.E.; et al. Metformin reduces PD-L1 on tumor cells and enhances the anti-tumor immune response generated by vaccine immunotherapy. J Immunother Cancer, 2021. 9(11).
- Mathenge, E.G.; et al. Core needle biopsy of breast cancer tumors increases distant metastases in a mouse model. Neoplasia 2014, 16, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Flood, B.A.; et al. STING pathway agonism as a cancer therapeutic. Immunol Rev 2019, 290, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Walker Ii, W.H.; et al. Mammary Tumors Induce Central Pro-inflammatory Cytokine Expression, but Not Behavioral Deficits in Balb/C Mice. Sci Rep 2017, 7, 8152. [Google Scholar] [CrossRef] [PubMed]
- DuPre, S.A., D. Redelman, and K.W. Hunter, Jr., The mouse mammary carcinoma 4T1: characterization of the cellular landscape of primary tumours and metastatic tumour foci. Int J Exp Pathol 2007, 88, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; et al. Blocking immunosuppressive neutrophils deters pY696-EZH2-driven brain metastases. Sci Transl Med, 2020. 12(545).
- Liu, X.; et al. Epithelial-type systemic breast carcinoma cells with a restricted mesenchymal transition are a major source of metastasis. Sci Adv 2019, 5, eaav4275. [Google Scholar] [CrossRef]
- Zheng, C.; et al. Neutrophils in triple-negative breast cancer: an underestimated player with increasingly recognized importance. Breast Cancer Res 2023, 25, 88. [Google Scholar] [CrossRef] [PubMed]
- Arpinati, L.; et al. Tumor-Derived Factors Differentially Affect the Recruitment and Plasticity of Neutrophils. Cancers (Basel), 2021. 13(20).
- Uribe-Querol, E. and C. Rosales, Neutrophils in Cancer: Two Sides of the Same Coin. J Immunol Res 2015, 2015, 983698. [Google Scholar] [CrossRef] [PubMed]
- Hurt, B.; et al. Cancer-promoting mechanisms of tumor-associated neutrophils. Am J Surg 2017, 214, 938–944. [Google Scholar] [CrossRef]
- Brandau, S., C. A. Dumitru, and S. Lang, Protumor and antitumor functions of neutrophil granulocytes. Semin Immunopathol 2013, 35, 163–176. [Google Scholar] [CrossRef]
- Liu, Y.; et al. Advances in immunotherapy for triple-negative breast cancer. Mol Cancer 2023, 22, 145. [Google Scholar] [CrossRef]
- Kim, I.S.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol 2019, 21, 1113–1126. [Google Scholar] [CrossRef]
- Kitchen, G.B.; et al. The histone methyltransferase Ezh2 restrains macrophage inflammatory responses. Faseb j 2021, 35, e21843. [Google Scholar] [CrossRef] [PubMed]
- Koss, B.; et al. Epigenetic Control of Cdkn2a.Arf Protects Tumor-Infiltrating Lymphocytes from Metabolic Exhaustion. Cancer Res 2020, 80, 4707–4719. [Google Scholar] [CrossRef]
Figure 1.
EZH2 knockout (KO) and overexpression (OE) cell clones were generated from the parent wild-type (WT) 4T1 TNBC line. (A) Western blots (L indicates protein ladder) and (B) densitometric analysis comparing 11 clones from each EZH2 KO (upper panel) and EZH2 OE (lower panel) lines to the 4T1 WT line (2 sets of blots for each), with -tubulin as normalization control.
Figure 1.
EZH2 knockout (KO) and overexpression (OE) cell clones were generated from the parent wild-type (WT) 4T1 TNBC line. (A) Western blots (L indicates protein ladder) and (B) densitometric analysis comparing 11 clones from each EZH2 KO (upper panel) and EZH2 OE (lower panel) lines to the 4T1 WT line (2 sets of blots for each), with -tubulin as normalization control.
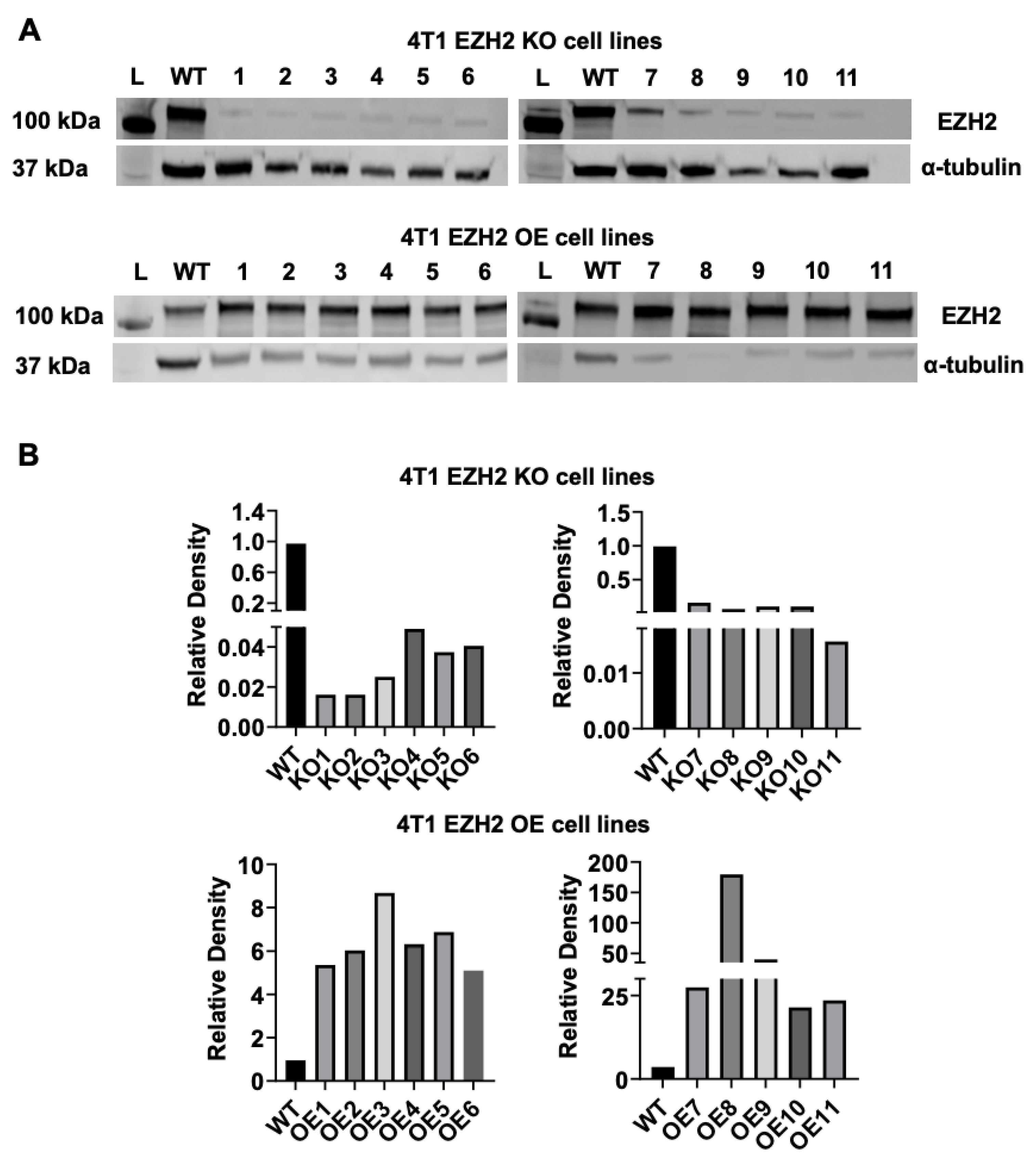
Figure 2.
In vitro replicative and invasive behaviors of EZH2 KO and EZH2 OE compared to parent WT 4T1 cells. (A) Count of 4T1 WT, EZH2 KO and EZH2 OE lines over 72 hours of growth in 2D plates. (B) Representative images of spheroid for 4T1 WT, EZH2 KO and EZH2 OE lines and quantification of (C) circularity and (D) invasive area over 72 hours of growth in a 3D invasion assay (n=8 spheroids per group). Comparison across groups and timepoints is by two-way ANOVA with Tukey post-hoc test and shown as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Figure 2.
In vitro replicative and invasive behaviors of EZH2 KO and EZH2 OE compared to parent WT 4T1 cells. (A) Count of 4T1 WT, EZH2 KO and EZH2 OE lines over 72 hours of growth in 2D plates. (B) Representative images of spheroid for 4T1 WT, EZH2 KO and EZH2 OE lines and quantification of (C) circularity and (D) invasive area over 72 hours of growth in a 3D invasion assay (n=8 spheroids per group). Comparison across groups and timepoints is by two-way ANOVA with Tukey post-hoc test and shown as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
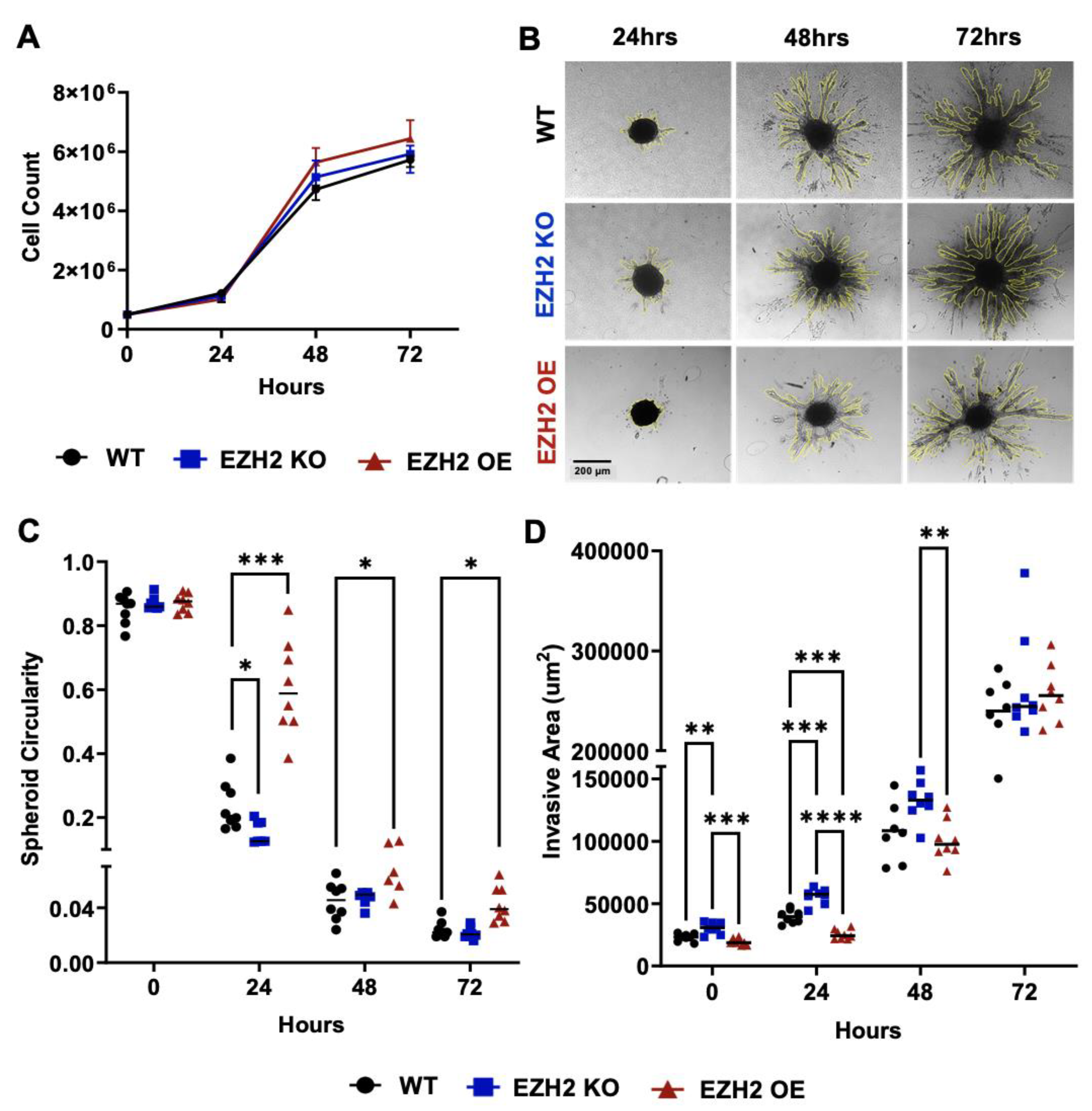
Figure 3.
In vitro surface phenotype and secreted factors by EZH2 KO and EZH2 OE compared to parent WT 4T1 cells. (A) 4T1 WT, EZH2 KO and EZH2 OE lines were cultured in DMEM for 24h and analyzed for surface expression of relevant surface markers by flow cytometry (six repeats, see Methods and Figure S1 for details). (B) Culture supernatants were screened for relevant extracellular mediators via Mesoscale assay (four repeats). Comparison between groups is by one-way ANOVA with Tukey post-hoc test and shown as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Figure 3.
In vitro surface phenotype and secreted factors by EZH2 KO and EZH2 OE compared to parent WT 4T1 cells. (A) 4T1 WT, EZH2 KO and EZH2 OE lines were cultured in DMEM for 24h and analyzed for surface expression of relevant surface markers by flow cytometry (six repeats, see Methods and Figure S1 for details). (B) Culture supernatants were screened for relevant extracellular mediators via Mesoscale assay (four repeats). Comparison between groups is by one-way ANOVA with Tukey post-hoc test and shown as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
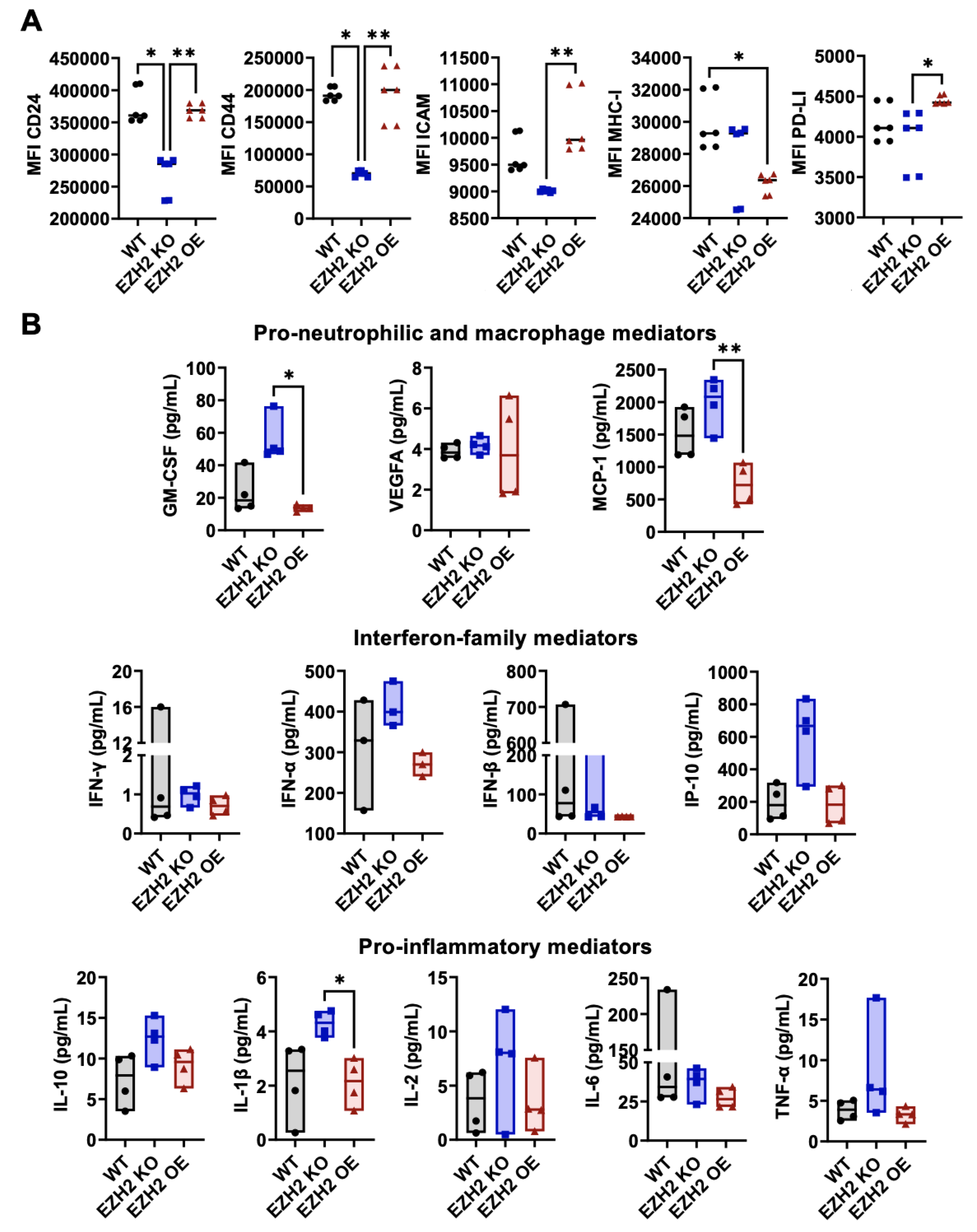
Figure 4.
In vivo primary tumor growth and lung metastasis by EZH2 KO and EZH2 OE compared to parent WT 4T1 cells. (A) Experimental timeline of 4T1 WT, EZH2 KO and EZH2 OE injection in mice. (B) Growth of 4T1 WT, EZH2 KO and EZH2 OE primary tumors over 21 days post-injection (n=6-7 mice per group). Comparison across groups and timepoints is by two-way ANOVA with Tukey post-hoc test and shown as *p<0.05, **p<0.01. (C) Lung metastasis assay for 4T1 WT, EZH2 KO and EZH2 OE lines. Comparison between groups is by one-way ANOVA with Tukey post-hoc test and shown as **p<0.01, ***p<0.001.
Figure 4.
In vivo primary tumor growth and lung metastasis by EZH2 KO and EZH2 OE compared to parent WT 4T1 cells. (A) Experimental timeline of 4T1 WT, EZH2 KO and EZH2 OE injection in mice. (B) Growth of 4T1 WT, EZH2 KO and EZH2 OE primary tumors over 21 days post-injection (n=6-7 mice per group). Comparison across groups and timepoints is by two-way ANOVA with Tukey post-hoc test and shown as *p<0.05, **p<0.01. (C) Lung metastasis assay for 4T1 WT, EZH2 KO and EZH2 OE lines. Comparison between groups is by one-way ANOVA with Tukey post-hoc test and shown as **p<0.01, ***p<0.001.
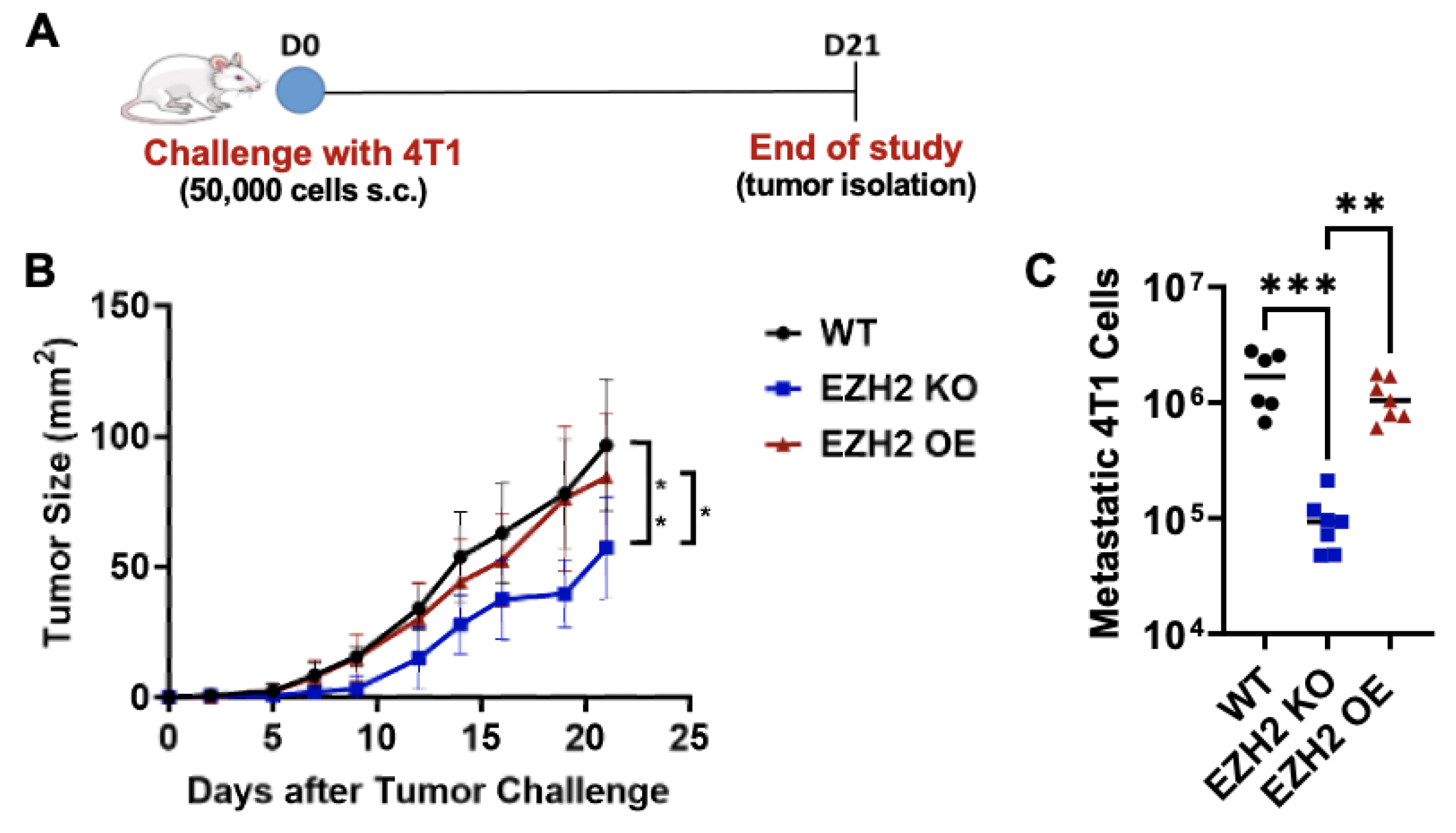
Figure 5.
In vivo primary tumor infiltration by neutrophils, CD4+ and CD8+ T cells for EZH2 KO and EZH2 OE compared to parent WT 4T1 cells. (A) Flow cytometry strategy for gating of infiltrating leukocyte subsets in 4T1 WT, EZH2 KO and EZH2 OE primary tumors, with sequential steps 1 (leukocytes), 2 (live cells), 3 (singlets), 4 (granulocytes, including mature neutrophils, immature neutrophils, and eosinophils), 5 (non-granulocytes), 6 (T-cells), 7 (CD4+ and CD8+). (B) Relative percentages of infiltrating neutrophils, CD4+ and CD8+ cells among live leukocytes (top), and ratios between these subsets (bottom) in 4T1 WT, EZH2 KO and EZH2 OE primary tumors. Comparison between groups is by one-way ANOVA with Tukey post-hoc test and shown as *p<0.05, **p<0.01.
Figure 5.
In vivo primary tumor infiltration by neutrophils, CD4+ and CD8+ T cells for EZH2 KO and EZH2 OE compared to parent WT 4T1 cells. (A) Flow cytometry strategy for gating of infiltrating leukocyte subsets in 4T1 WT, EZH2 KO and EZH2 OE primary tumors, with sequential steps 1 (leukocytes), 2 (live cells), 3 (singlets), 4 (granulocytes, including mature neutrophils, immature neutrophils, and eosinophils), 5 (non-granulocytes), 6 (T-cells), 7 (CD4+ and CD8+). (B) Relative percentages of infiltrating neutrophils, CD4+ and CD8+ cells among live leukocytes (top), and ratios between these subsets (bottom) in 4T1 WT, EZH2 KO and EZH2 OE primary tumors. Comparison between groups is by one-way ANOVA with Tukey post-hoc test and shown as *p<0.05, **p<0.01.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



