
Submitted:
02 April 2024
Posted:
03 April 2024
You are already at the latest version
Abstract
Achieving effective control over microbial contamination necessitates the precise and concurrent identification of numerous pathogens. In this research, we have devised a remarkably sensitive duplex droplet digital PCR (dddPCR) reaction system to simultaneously detect Pseudomonas aeruginosa (P. aeruginosa) and Pseudomonas fragi (P. fragi). Employing comparative genomics, we identified four genes of P. fragi. By specific analysis, RS22680 gene was selected as the detection target of P. fragi and LasR gene was chosed as P. aeruginosa, which were applied to construct a dddPCR reaction. In terms of specificity, sensitivity and anti-interference ability, the constructed dddPCR detection system was verified and analyzed. The assay showed excellent sensitivity and applicability, as evidenced by a limit of detection of 100 CFU/mL. When the concentration of natural background bacteria in milk or fresh meat was 100 times that of the target detection bacteria, the method was still capable of completing the absolute quantification. In the simulation of actual sample contamination, P. aeruginosa could be detected after 3 h of enrichment culture, and P. fragi could be detected after 6 h. The established ddPCR detection system exhibits exceptional performance, serving as a foundation for the simultaneous detection of various pathogenic bacteria in food products.
Keywords:
Pseudomonas aeruginosa
; Pseudomonas fragi
; simultaneous detection
; droplet digital PCR
1. Introduction
As a common food safety hazard, microbial contamination seriously affect human life and health[1]. There are many kinds of microorganisms with different sources, and the same bacteria include many taxonomically related species. Pseudomonas complex group has been called as a “hodgepodge” for decades, it contains P. aeruginosa, P. fragi, Pseudomonas fluorescens, etc[2,3]. It is well-known that P. aeruginosa is a common bacterium, which is widely found in water and other environments in nature[4]. It has strong drug and high temperature resistance and prefers humid environments[5]. Therefore, it is easy to cause P. aeruginosa contamination during food processing and storage. Studies have shown that the detection rate of P. aeruginosa in drinking water in China can reach 10 %[6]. Legesse Garedew et al.isolated and identified 54 kinds of bacteria in milk containers, of which 18.5% were P. aeruginosa[7]. In addition, due to drug resistance and dense biofilms, P. aeruginosa has a very high growth advantage in animal-derived foods[8,9,10]. Although it does not have a direct fatal hazard to the human body, once infected, the body may appear vomiting, diarrhea, fever and other symptoms. Therefore, the pollution of P. aeruginosa must be strictly controlled.
In contrast, P. fragi was known as“specific spoilage organisms”which is abundant in chilled meat[11]. It can survive for a long time at low temperature and decompose the protein in food, which makes the food lose its original freshness and taste, seriously affecting the appetite of consumers[12,13]. It is reported that 21 % of the huge losses of meat products are caused by microbial spoilage[14]. When microorganisms work together to contaminate food, there is a situation in which some strains dominate. Zhang et al. found P. fragi exhibited a clear predominance in cold chain food[12]. Wang et al. discovered that P. fragi showed the strongest spoilage potential in chilled chicken[15]. In addition, P. fragi is extremely easy to contaminate aquatic products such as salmon due to its ability to form a psychrotrophic biofilm[16,17]. P. fragi CGMCC 1.7759 isolated from the sea surface of the Arctic proved the growth advantage in low temperature environment. It is likely to be polluted during food processing, transportation and storage, especially for fresh, refrigerated and frozen foods that have not been subjected to high temperature treatment or other forms of disinfection or preservation.
At present, the gold standard for the identification of the pathogens is bacteriological culture which is complex, time-consuming and unable to detect those strains that are difficult to culture or lack specificity[18]. More simple and accurate detection methods such as molecular detection, enzyme-linked immuno-sorbent assay, electrochemical detection have been developed[19,20]. Molecular methods have shown great excellence in accurate detection[21]. It uses comparative genomics methods to detect bacteria based on specific nucleic acid sequences. However, the accuracy of this methods for P. aeruginosa is not enough. A large number of specific sequences were used to verify the specificity of molecular experiments to detect P. aeruginosa[22]. Furthermore, Murugan et al. used multiple pairs of primers to detecte P. aeruginosa by mPCR in order to determine the accuracy[23]. So far, there is less reported molecular method for detecting P. fragi in the literature, and most of the studies are about their genes and mechanisms[24]. Therefore, it is necessary to develop more sensitive and accurate molecular detection methods for the identification of microorganisms.
Digital PCR technology is continuously improved on the basis of polymerase chain reaction[25]. It can achieve absolute quantitative detection and shows great superiority in molecular detection. Digital PCR divided reaction system into a large number of independent micro-reaction units, and the nucleic acid copy number was calculated according to the Poisson distribution and the positive ratio[26]. This method can accurately detect the target bacteria, and has a significant advantage in accurately judging the complex flora[27]. P. aeruginosa is a pathogenic bacterium, and P. fragi is a spoilage bacterium. Both of them belong to Pseudomonas Genus and endanger food. To satisfied the need for accurate, sensitive and multiplex detection, this assay established a molecular method for testing P. aeruginosa and P. fragi at the same time in the same device. Using the method of comparative genomics, RS22680 and LasR were identified as detection targets. Primers and probes were designed based on these two genes and dddPCR detection was constructed. The effectiveness of the reaction system was proved by sensitivity, anti-interference ability experiments and artificially simulated contaminated samples. This can help to identify the types and quantities of bacteria in food achieving food quality control and reducing loss.
2. Material and methods
2.1. Sample preparation
Samples(raw chicken and potable water) were purchased in a supermarket in ChuZhou, Anhui, China. Fresh milk was obtained from Dutch dairy cows ( Heping Dairy Ranch, Bengbu City, Anhui Province, China ) through aseptic sampling and sent back to the laboratory for processing as soon as possible at low temperature. The raw chicken was cut into small pieces and frozen at -20℃for subsequent experiments. For the latter experiments of artificial pollution commercial sample, purchased drinking water, sterile milk and raw chicken were identified by microbial culture method without P. aeruginosa or P. fragi.
2.2. Strain culture and DNA extraction
The strains used in this experiment were all from standard strains purchased from formal channels. P. aeruginosa was cultured in a Luria-Bertani (LB) broth at 37 ◦C for 18 h, and P. fragi was cultured at 30 °C. Other bacteria used for specificity analysis were activated according to the culture instructions. DNA was extracted using the bacterial genome extraction kit (Shanghai Sangon Biotech, China), and the concentration was determined under a spectrophotometer (NV3000C VASTECH INC)and stored at-20 °C. Genomic DNA of chilled meat was extracted by modified the direct lysis (DL) method[28]. The sample solution was mixed by ultrasonic treatment for 5 min and incubated 10 min in a boiling water bath. Finally, the sample was centrifuged at 10,000rpm for 5 min and the supernatants were collected as the reaction template.
2.3. Screening of specific genes of P. aeruginosa and P. fragi
Three whole genome sequences of P. fragi (GeneBank: GCA_002128325.1, GCA_02986945.1, GCA_000250595.1)were obtained from NCBI(https://www.ncbi.nlm.nih.gov/). Sequence alignment of P. fragi was performed by NCBI Nucleotide-BLAST(https://blast.ncbi.nlm.nih.gov/Blast.cgi). Each CDS of P. fragi was matched using BLASTN, and those exhibiting low homology with non-Pseudomonas spp. and high homology with all P. fragi (E-value<1e-200, Query Cover≥99%) were used as candidate detection targets. LasR, gyrB and rpoB were finally selected as the P. aeruginosa candidate genes according to the reported literature[29] [30]. The specificity of gene were analyzed using NCBI Primer-BLAST. The genetic information involved in this paper was shown in Table 1.
In order to ensure the accuracy of specific genes,primers were designed according to candidate genes and 20 strains of non-Pseudomonas fragi and non-Pseudomonas aeruginosa were used for comparative analysis. The specificity result was analyzed by PCR experiments and agarose gel electrophoresis imaging. The primers used in the experiment are shown in Table 1 and the PCR reaction was performed in 25 μL amplification mixture containing 1 μL of the DNA templates, 12.5 μL 2×Reaction Mix(Dongsheng Biotechnology Co., Ltd. GuangDong, China), 1 μL each primers F and R(10 μM), 8.5 μL sterilized ultrapure water.
2.4. Primers, probes design for dddPCR and specificity verification
Through the specific genes of P. aeruginosa and P. fragi were screened out, primers and probes were designed according to the experimental requirements based on the highly conserved region, as shown in Table 2. The primers and probes were designed by primer 3.0 and synthesized and purified in Sangon Biotech, Shanghai, China. In order to identify the accuracy of the primers designed in the experiment, the specificity of digital PCR primers was verified by qPCR for common bacteria and other Pseudomonas. This includes 5 other Pseudomonas strains and 5 common bacteria. The genomes of these 10 strains were used as templates for reaction, and the results of qPCR were used to determine the specificity of primers and probes.
2.5. Establishment of the dddPCR assay
The dddPCR mixture composition was list in the Table 3 and operation protocol used was as follows. After all the solutions were fully mixed, 14 μL admixture was sucked into the sample port of the chip which formed a water-in-oil reaction system. The instrument introduces the reaction mixture and mineral oil into the microfluidic chip by negative pressure method, and then absolute quantitative analysis was performed by PCR amplification. The thermocycling protocol for the quantification included a 10 min hot start at 95 ℃ and 40 cycles of PCR (96 ℃ for 20 s and 60 ℃ for 60 s). The whole step is completed in a closed environment in the machine, and the test results are obtained by Poisson distribution calculation.
2.6. Establishment of standard curve
With the purpose of evaluating the reliability of dual reaction system on the chip, the ddPCR method was used to generate the standard curves for the detection of P. fragi and P. aeruginosa. The linear relationship between the detection of P. fragi and P. aeruginosa by digital PCR was calculated by adding 2, 4, 8 and 16 times template concentration. The copy value of the sample detection is obtained by the following calculation formula.
where P is the mean software output value, V is the total reaction volume , V1 is the amount of nucleic acid added and D is the dilution multiple.
2.7 Sensitivity test of dddPCR detection
Genomic DNA sensitivity and bacterial suspension sensitivity of the ddPCR method were test. The whole genome DNA template of P. aeruginosa and P. fragi strains were extracted and determined, which the concentration were serially diluted 106 to 101 fg/μL. P. aeruginosa and P. fragi cells were continuously diluted to the final concentration of 100-105 CFU / mL after plant counting. These DNA templates are used for subsequent sensitivity evaluations.
2.8. Anti-interference ability evaluation
Bacteria usually coexist in a mixed population in food and environmental samples. In order to evaluate the accuracy of the reaction system in the presence of other interfering bacteria, different concentrations of P. fragi and P. aeruginosa were mixed with the natural background flora in the collected food samples. To obtain the natural background flora of milk,25 mL of untreated fresh milk collected from pastures was cultured in 225 mL of LB at 37 ℃ for 18 h, and the natural background flora of cold fresh chicken was also enriched by this method. Plate counts were performed on all selected bacteria to determine the concentration of cells in the mix and gradiently diluted to a concentration of N×102-107 CFU / mL ( 1 < N < 10 ). The counting results showed that the concentration of natural background bacteria in raw milk was 5.4×107 CFU / mL, and the concentration of natural background bacteria in chicken was 1.72×108 CFU / mL. The genome of the mixed bacteria extracted from the gradient diluted flora were used for the template of dddPCR reaction.
2.9. Evaluation of artificial simulated contamination of actual samples
To evaluate the applicability of the proposed methods, several foods with contamination rates of P. aeruginosa and P. fragi were selected as samples for simulation analysis. P. aeruginosa and P. fragi were inoculated in drinking water, sterile milk and cold fresh chicken, respectively( Initial concentration of inoculation:102 CFU / mL,inoculation proportion: 10 % ). The genomes were extracted for dddPCR detection after 0, 3, 6, 9 and 12 h of culture, respectively. All samples through the traditional method of microbial culture to ensure that there is no target gene to be detected. The reaction system and conditions according to the above instructions, the presence of contamination was evaluated by a sterile double distillation water without template control ( NTC ) reaction.
3. Results and discussions
3.1. Analysis of candidate gene selection
A total of 13613 genes were screened according to the whole genome sequence of three groups of P. fragi uploaded from the database, and 38 specific genes were obtained. The homology and coverage of 38 genes of P. fragi were 100 %, and the homology with other bacteria was very weak. Furthermore, four highly specific regions were found which could be used as selectable targets. The primers designed were predicted no cross-reactivity with other species by the Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). The primers were designed according to the specific gene, and the specificity of the primers was verified by PCR as a target gene to determine whether the gene could be used as a quasi-specific gene for subsequent dddPCR reaction. The specificity results showed in Table 4 that RS22680 gene of P. fragi, and LasR gene of P. aeruginosa had high accuracy and could be used for subsequent dddPCR experiments.
3.2. Evaluation of dddPCR reaction system construction results
In this experiment, two luminescence channels, FAM and VIC were selected for dddPCR assay. The experimental results were shown in Figure 1. The effective droplet generation was greater than 20000, the negative droplet and the positive droplet were evenly distributed, and the counting was effective. There was no interference in the absolute quantitative detection between the two, and the constructed dual reaction system had excellent detection results. Zhang et al. successfully detected Salmonella and Shigella by ddPCR[31]. Luo et al. detected Staphylococcus aureus in the mixture by digital PCR[32]. More researchers have focused on the improvement of digital PCR technology. Yin et al. established a multiplex digital PCR method without extracting ctDNA to reduce the reaction steps[33]. Xie Tengbao et al. avoided the interference of cross primers and the overlap of fluorescence in a single-tube by physical separation[34]. Simpler and more practical multiple detection methods still urgent to be developed. The dual channel designed in this assay could accurately detect both microorganisms at the same time, and the luminescent groups used had no interference with each other.
3.3. Linear relationship analysis of the reaction system
The linear relationship of the established dual detection system showed excellent superiority as Figure 2 shown. A linear correlation of P. aeruginosa between the detected and theoretical ratios was obtained with R2 of 0.9989. Additionally, the obtained and expected values of P. fragi showed a good linear correlation (R2 = 0.9995) . The standard curve for detecting P. fragi did not cross the origin. According to the analysis of the reported literature, the reason for this phenomenon may be : different types of fluorescein dyes have different signal intensities when they are accepted during luminescence and quenching, or there is an advance or delay in the machine acceptance signal[35]. Fluorescein amidites (FAM), Cyanine (Cy) and carboxy-X-rhodamine (ROX) are the most common fluoresceins, which are received by signals by releasing reporters to change the emission wavelength[36]. By utilizing these changes, the luminescence sensors about “off-on”,”on-off” could be used to measure the concentration of the target analyte. Liu et al. designed a dual-channel sensor, each channel marked distinctly by FAM and ROX[37]. The FAM fluorescein exhibits particularly excellent sensitivity in detection. However, this also leads to background signal interference in the strong fluorescence signal. In terms of accuracy, it is necessary to increase the quality control point as the basis for judging the positive test results. After multiple template-free parallel experiments,the experimental results about FAM channel were less than 30 copies was judged to be negative . In contrast, the VIC channel could achieve absolutely accurate detection. When there is no template, the luminescence signal cannot be detected. Veronica Bolzon et al. distinguished Listeria spp. and Listeria monocytogenes by using VIC as the internal control of reaction[38]. Therefore, in the development of multiple detection methods, the selection of fluorescein is also very important. Different types of fluorescein need high accuracy and no interference with each other. In the future, multiple high-sensitivity detection will surely have better development in food safety and public health[39].
3.4. Analysis of specific primers for dddPCR
In the previous target gene screening experiment, we knew that the RS22680 gene of P. fragi and the LasR gene of P. aeruginosa had good specificity. The primer and probe of the dddPCR detection system was designed by these two genes, and the results are shown in Figure 3. P. fragi and P. aeruginosa could be well detected, and other bacteria had no positive reaction. The experimental results showed that the primer Pa-2 designed according to the gene LasR had significantly excellent specificity for the detection of P. aeruginosa. No cross-reactivity was observed with Escherichia coli, Listeria monocytogenes and other Pseudomonas. In order to detect the accuracy, researchers have developed a lot of methods. By designing targeted crRNA , P. aeruginosa was determined combining with CRISPR technology[6]. Xiang yong et al. used cross priming amplification to detect P. aeruginosa to determine the accuracy of the results[40]. A fluorescent biosensor combining with the DNAzyme and a new approach using pseudopaline-based probes were designed for the effective detection of P. aeruginosa[41,42]. Researchers have always been committed to identifying hazards more accurately through various methods.
3.5. Sensitivity analysis of genome and colony detection by dddPCR
The accuracy of this established dddPCR platform for simultaneous detection of P. aeruginosa and P. fragi was evaluated by comparing the measured concentration for each genomic DNA and colony. For the sensitivity evaluation of genomic DNA, the result showed that a weak signal value was generated when the DNA template concentration was lower than 5.4×103 fg /μL. Through the previous determination of the control point of FAM fluorescein detection, more than 30 copies values were judged to be positive. Therefore, the lowest detection limit of P. fragi was 5.4×103 fg /μL. The minimum detection limit of P. aeruginosa was 3.6×102 fg /μL.(Figure 4.)
In the sensitivity evaluation of bacterial suspension template, the detection limits of P. fragi and P. aeruginosa were all in single-digit (Figure 5.). At present, CRISPR technology is used to detect P. aeruginosa, with a minimum of 50 CFU / mL[6].Wang et al.detected Salmonella by digital PCR with a sensitivity of 10−4 ng /μL or 102 CFU / mL which sensitivity is lower than the results detected in this assay[43]. According to the specific genes screened, the primers designed in this experiment showed a particularly good sensitivity. However, the high sensitivity of digital PCR limits the detection range to a certain extent. When the number of bacterial colonies exceeded 104 CFU / mL, the statistical results were invalid due to sufficient luminescent points when calculating the positive results. Combining with DNAzyme sensor, Qin et al. detected P. aeruginosa can reach 1.2 CFU / mL[41]. The detection results are comparable to the results of P. aeruginosa detection in this paper, and the detection range is wider. However, dddPCR is easier to achieve multiplex detection and lower cost.
3.6. Analysis of anti-interference ability
The purpose of this test was to validate the accuracy of simultaneous detection for P. fragi and P. aeruginosa under the background microbiota. In this experiment, the natural background flora of fresh milk and chilled meat were selected as the interference factor to explore the accuracy of this method in detecting P. aeruginosa and P. fragi in the case of rich microbial species. The bacterial concentration of P. aeruginosa was 1.5×108 CFU/mL after culture, and that of P. fragi was 1.8×108 CFU/mL. After ten-fold gradient dilution, it was used for anti-interference experimental analysis. The results showed that different natural background flora have no effect on the detection of P. aeruginosa. As the Figure 6 showing, the sensitivity of P. fragi was slightly affected under the natural background flora concentration of milk at 103 CFU / mL. This result might be due to the existence of strains in fresh milk that have a greater growth advantage than P. fragi, and there was a phenomenon of competitive inhibition. Previous studies had shown that E.coli was able to coexist with spoilage Pseudomonas, which would lead to meat food spoilage and has a clear leading role than other microorganisms[44,45]. We guess microorganisms in fresh milk may have more dominant strains,which may affect the detection of the P. fragi. Whether the concentration of the background flora will affect the sensitivity of the detection was also verified. When the concentration of natural background bacteria was higher or lower than 103 CFU / mL, the detection results were stable. This indicates that the detection method we established still has a great sensitivity even under the interference of other background bacteria.
3.7. Analysis of test results of artificially contaminated food
To evaluate the feasibility and reliability of the dddPCR assay, detection of P. fragi and P. aeruginosa in artificially spiked samples was performed. P. aeruginosa and P. fragi obtained from the overnight culture was inoculated of nutrient broth at a ratio of 10% to achieve artificially contaminated samples with an initial contamination level of N×100. The results showed that the target bacteria could not be detected without enrichment culture. After 9 h of culture in milk, the growth of P. aeruginosa and P. fragi was significantly higher than that in chilled chicken or drinking water. This result suggests that nutritious and uniform food is more conducive to the growth of microorganisms, and this type of food should be regularly controlled and monitored. The results showed that cold fresh chicken was more susceptible to microbial infection between 0-6 h after inoculation with Pseudomonas, which also proved that the water activity of raw meat was more likely to nourish bacteria. Longer detection time may reflect the growth status of Pseudomonas in different substrates, but the high sensitivity of ddPCR limits the wide range of detection. The experimental results also showed that there were great differences in the growth status of the two bacteria, even if both bacteria belonged to Pseudomonas genus. P. fragi has the advantage of long-term survival in the environment. In the other hand, P. aeruginosa is easier to achieve early control.
Although the recent detection methods have made great progress, there is still a lot of room for development in the detection and analysis of hazardous substances in food. The composition of food will still greatly affect the accuracy and sensitivity of the detection. Future research will still focus on stability, sample pretreatment and mutual interference. The development of multiple detection methods that can meet the current detection needs is of great significance for food safety monitoring.
Figure 7.
Analysis of the detection results of artificially P. fragi and P. aeruginosa in food.
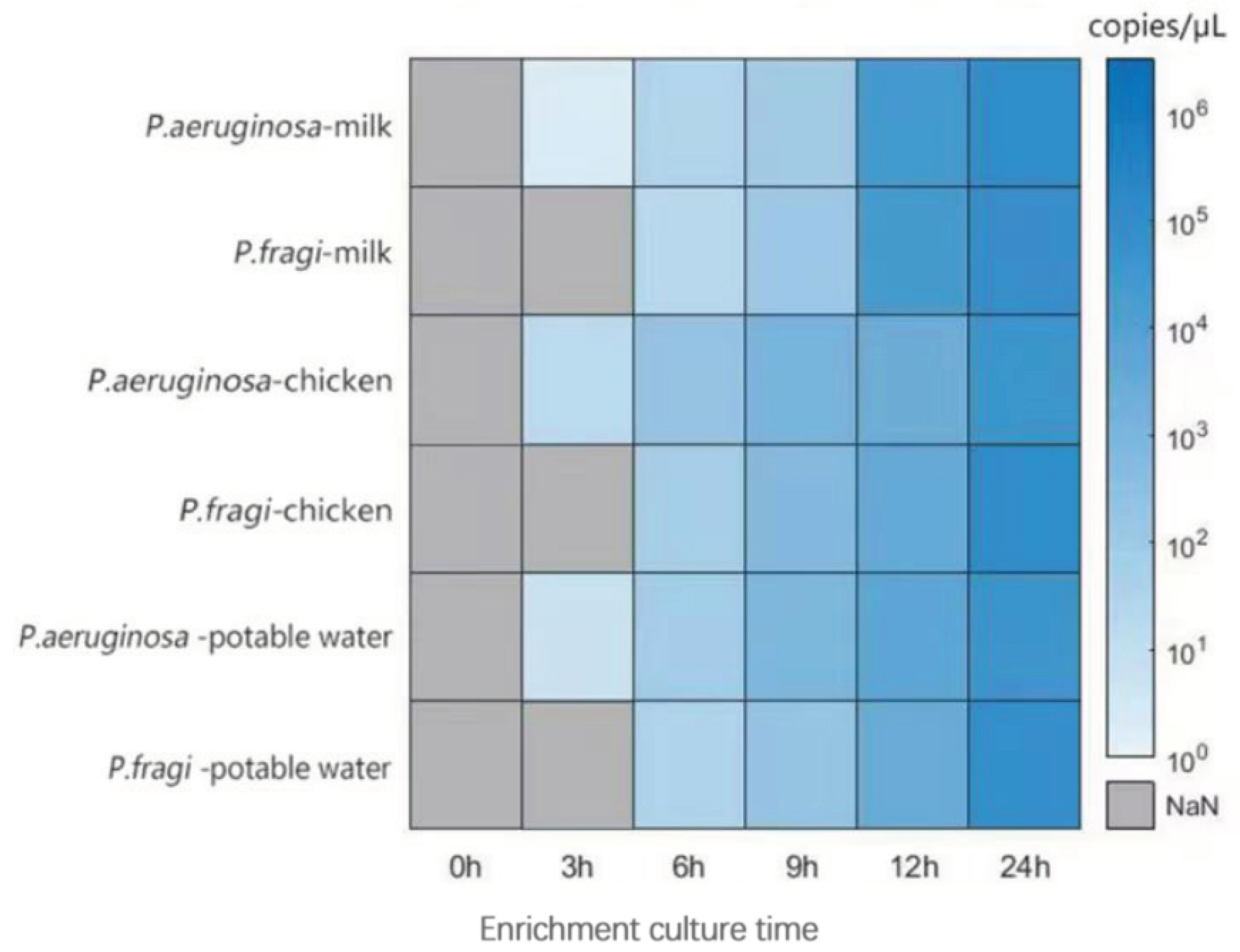
4. Conclusion
Based on two-color fluorescent probes, we developed a dddPCR detection system that detected P. aeruginosa and P. fragi simultaneously. Both bacteria were accurately detected using the duplex ddPCR method, and the mean R2 values were greater than 0.95. Furthermore, the sensitivity was higher than other reported PCR techniques. No significant effect on the test results was observed in the presence of natural background bacteria in chicken or milk. Moreover, this method can detect 100 CFU/mL of bacterial DNA , and the detection genomics DNA limit was 102 fg/μL. The applicability of artificially infected drinking water, milk and chicken samples was evaluated. Both pathogens were successfully detected after 3 h of contamination. The method has great potential for detecting food safety and ensuring product quality. It is necessary to encourage factories to use this method for product supervision. However, as the method has only been tested under laboratory conditions, there is still much room for optimization for the detection of multiple pathogens on the market. In the future, a digital PCR reaction system would be established for multiple pathogenic bacteria detection based on different products in order to achieve rapid and accurate detection of actual products on site.
Author Contributions
Conceptualization, L.Z. and J.H.; Methodology, J.H.; Validation, L.Z. , B.W. and Z.W.; Formal Analysis, J.W.; Investigation, X.S.; Resources, L.Z. and Z.W.; Data Curation, J.H. and J.W; Writing – Original Draft Preparation, J.W.; Writing – Review & Editing, L.Z; Supervision, L.Z. and J.H.; Project Administration, L.Z.; Funding Acquisition, L.Z. and J.H.
Funding
This work was carried out under research activities of the Graduate Academic Innovation Project (No. cxcysj198) funded by Anhui Provincial Department of Education- Graduate Education Quality Engineering. This research was funded by The Natural Science Foundation Project of Anhui Province, grant number 2008085MC89.
Data Availability Statement
Data is contained within the article, more details are available from the corresponding author.
Acknowledgements
We would like to thank the support of Anhui Provincial Department of Education.
Conflicts of Interest
The authors declare that they have no known competing nancial interests which could influence the work reported in this paper.
References
- Tropea, A. Microbial Contamination and Public Health: An Overview. International Journal of Environmental Research and Public Health 2022, 19. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Barranquero, J.A.; Cazorla, F.M.; de Vicente, A. Pseudomonas syringae pv. syringae Associated With Mango Trees, a Particular Pathogen Within the “Hodgepodge” of the Pseudomonas syringae Complex. Frontiers in Plant Science 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Winsor, G.L.; Griffiths, E.J.; Lo, R.; Dhillon, B.K.; Shay, J.A.; Brinkman Fiona, S.L. Enhanced annotations and features for comparing thousands ofPseudomonasgenomes in the Pseudomonas genome database. Nucleic Acids Research 2016, 44, D646–D653. [Google Scholar] [CrossRef] [PubMed]
- Bilican, I.; Bahadir, T.; Bilgin, K.; Guler, M.T. Alternative screening method for analyzing the water samples through an electrical microfluidics chip with classical microbiological assay comparison of P. aeruginosa. Talanta 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, Y.; Li, M.; Jia, L.; Zhang, L.; Zhu, J. Gelatin-based photonic hydrogels for visual detection of pathogenic Pseudomonas aeruginosa. Sensors and Actuators B: Chemical 2021, 329. [Google Scholar] [CrossRef]
- Huang, S.; Wang, X.; Chen, X.; Liu, X.; Xu, Q.; Zhang, L.; Huang, G.; Wu, J. Rapid and sensitive detection of Pseudomonas aeruginosa by isothermal amplification combined with Cas12a-mediated detection. Scientific Reports 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Garedew, L.; Berhanu, A.; Mengesha, D.; Tsegay, G. Identification of gram-negative bacteria from critical control points of raw and pasteurized cow milk consumed at Gondar town and its suburbs, Ethiopia. BMC Public Health 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Quintieri, Fanelli, Caputo: Antibiotic Resistant Pseudomonas Spp. Spoilers in Fresh Dairy Products: An Underestimated Risk and the Control Strategies. Foods 2019, 8.
- Bloomfield, S.J.; Palau, R.; Holden, E.R.; Webber, M.A.; Mather, A.E. Genomic characterization of Pseudomonas spp. on food: implications for spoilage, antimicrobial resistance and human infection. BMC Microbiology 2024, 24. [Google Scholar] [CrossRef]
- Dong, Q.; Sun, L.; Fang, T.; Wang, Y.; Li, Z.; Wang, X.; Wu, M.; Zhang, H. Biofilm Formation of Listeria monocytogenes and Pseudomonas aeruginosa in a Simulated Chicken Processing Environment. Foods 2022, 11. [Google Scholar] [CrossRef]
- Yang, J.; Liang, R.; Mao, Y.; Dong, P.; Zhu, L.; Luo, X.; Zhang, Y.; Yang, X. Potential inhibitory effect of carbon dioxide on the spoilage behaviors of Pseudomonas fragi in high-oxygen packaged beef during refrigerated storage. Food Microbiol 2023, 112, 104229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, J.; Yuan, Y.; Yue, T. Diversity and characterization of spoilage-associated psychrotrophs in food in cold chain. Int J Food Microbiol 2019, 290, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Quintieri, L.; Caputo, L.; Brasca, M.; Fanelli, F. Recent Advances in the Mechanisms and Regulation of QS in Dairy Spoilage by Pseudomonas spp. Foods 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Chen, S.; Wang, H.; Zhang, J.; Xu, X.; Wang, H. Advances in understanding the predominance, phenotypes, and mechanisms of bacteria related to meat spoilage. Trends in Food Science & Technology 2021, 118, 822–832. [Google Scholar]
- Wang, G.-y.; Wang, H.-h.; Han, Y.-w.; Xing, T.; Ye, K.-p.; Xu, X.-l.; Zhou, G.-h. Evaluation of the spoilage potential of bacteria isolated from chilled chicken in vitro and in situ. Food Microbiology 2017, 63, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Wang, Q.; Liu, J.; Wang, D.; Li, J.; Li, T. Effects of deletion of siderophore biosynthesis gene in Pseudomonas fragi on quorum sensing and spoilage ability. International Journal of Food Microbiology 2023, 396. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, G.; Xue, P.; Dong, X.; Xia, Y.; Regenstein, J.; Du, M.; Sun, L. Spoilage microbes’ effect on freshness and IMP degradation in sturgeon fillets during chilled storage. Food Bioscience 2021, 41. [Google Scholar] [CrossRef]
- Jiang, Y.; Zheng, C.; Jin, M.; Zhou, R.; Wu, Q.; Huang, F.; Lou, Y.; Zheng, L. An Ultrasensitive Colorimetric Foodborne Pathogenic Detection Method Using a CRISPR/Cas12a Mediated Strand Displacement/Hybridization Chain Reaction. J Agric Food Chem 2023, 71, 4193–4200. [Google Scholar] [CrossRef]
- Furet, J.P.; Quenee, P.; Tailliez, P. Molecular quantification of lactic acid bacteria in fermented milk products using real-time quantitative PCR. Int J Food Microbiol 2004, 97, 197–207. [Google Scholar] [CrossRef]
- Li, Z.; Xu, X.; Wang, D.; Jiang, X. Recent advancements in nucleic acid detection with microfluidic chip for molecular diagnostics. TrAC Trends in Analytical Chemistry 2023, 158. [Google Scholar] [CrossRef]
- Hadi, J.; Rapp, D.; Dhawan, S.; Gupta, S.K.; Gupta, T.B.; Brightwell, G. Molecular detection and characterization of foodborne bacteria: Recent progresses and remaining challenges. Comprehensive Reviews in Food Science and Food Safety 2023, 22, 2433–2464. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, Q.; Jiang, A.; Zhang, J.; Shang, Y.; Li, F.; Zhou, B.; Xiang, X.; Gu, Q.; Pang, R.; et al. Pseudomonas aeruginosa Detection Using Conventional PCR and Quantitative Real-Time PCR Based on Species-Specific Novel Gene Targets Identified by Pangenome Analysis. Frontiers in Microbiology 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.; Malathi, J.; Therese, K.L.; Madhavan, H.N. Application of six multiplex PCR's among 200 clinical isolates of Pseudomonas aeruginosa for the detection of 20 drug resistance encoding genes. The Kaohsiung Journal of Medical Sciences 2017, 34, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Ercolini, D.; Casaburi, A.; Nasi, A.; Ferrocino, I.; Di Monaco, R.; Ferranti, P.; Mauriello, G.; Villani, F. Different molecular types of Pseudomonas fragi have the same overall behaviour as meat spoilers. Int J Food Microbiol 2010, 142, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Chen, S.; Zheng, Y.; Zheng, X.; Lin, J.-M. Droplet-based digital PCR (ddPCR) and its applications. TrAC Trends in Analytical Chemistry 2023, 158. [Google Scholar] [CrossRef]
- Zhang, L.; Parvin, R.; Fan, Q.; Ye, F. Emerging digital PCR technology in precision medicine. Biosensors and Bioelectronics 2022, 211. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zou, Z.; Hu, Z.; Zhang, S.; Zhang, F.; Wang, B.; Lv, S.; Mu, Y. A “sample-in-multiplex-digital-answer-out” chip for fast detection of pathogens. Lab on a Chip 2020, 20, 979–986. [Google Scholar] [CrossRef]
- Zhao, G.; Shen, X.; Liu, Y.; Xie, P.; Yao, C.; Li, X.; Sun, Y.; Lei, Y.; Lei, H. Direct lysis-multiplex polymerase chain reaction assay for beef fraud substitution with chicken, pork and duck. Food Control 2021, 129. [Google Scholar] [CrossRef]
- Ruiz-Roldán, L.; Rojo-Bezares, B.; Lozano, C.; López, M.; Chichón, G.; Torres, C.; Sáenz, Y. Occurrence of Pseudomonas spp. in Raw Vegetables: Molecular and Phenotypical Analysis of Their Antimicrobial Resistance and Virulence-Related Traits. International Journal of Molecular Sciences 2021, 22. [Google Scholar] [CrossRef]
- Lee, C.S.; Wetzel, K.; Buckley, T.; Wozniak, D.; Lee, J. Rapid and sensitive detection of Pseudomonas aeruginosa in chlorinated water and aerosols targeting gyrB gene using real-time PCR. Journal of Applied Microbiology 2011, 111, 893–903. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Y.; Xue, P.; Zhan, Z.; Huang, Z.; Li, J.; Diao, B.; Kan, B. A duplex droplet digital PCR assay for Salmonella and Shigella and its application in diarrheal and non-diarrheal samples. International Journal of Infectious Diseases 2022, 120, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, J.; Yang, H.; Yu, J.; Wei, H.; Ledeboer, N.A. Accurate Detection of Methicillin-Resistant Staphylococcus aureus in Mixtures by Use of Single-Bacterium Duplex Droplet Digital PCR. Journal of Clinical Microbiology 2017, 55, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xia, L.; Zou, Z.; Zhuang, J.; Mu, Y. A direct and multiplex digital PCR chip for EGFR mutation. Talanta 2022, 250. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Luo, Y.; Wang, P.; Wu, L.; Cui, X.; Sun, B.; Li, G. Controlled Rehydration of Dried Reagents for Robust Multiplex Digital PCR. Analytical Chemistry 2022, 94, 13223–13232. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kutsanedzie, F.Y.H.; Sun, H.; Wang, M.; Chen, Q.; Guo, Z.; Wu, J. Rapid Pseudomonas Species Identification from Chicken by Integrating Colorimetric Sensors with Near-Infrared Spectroscopy. Food Analytical Methods 2017, 11, 1199–1208. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Tian, D.; Phan, A.; Seididamyeh, M.; Alanazi, M.; Ping Xu, Z.; Sultanbawa, Y.; Zhang, R. Luminescent sensors for residual antibiotics detection in food: Recent advances and perspectives. Coordination Chemistry Reviews 2024, 498. [Google Scholar] [CrossRef]
- Liu, C.; Lu, C.; Tang, Z.; Chen, X.; Wang, G.; Sun, F. Aptamer-functionalized magnetic nanoparticles for simultaneous fluorometric determination of oxytetracycline and kanamycin. Microchimica Acta 2015, 182, 2567–2575. [Google Scholar] [CrossRef]
- Bolzon, V.; Bulfoni, M.; Pesando, M.; Nencioni, A.; Nencioni, E. Verification of a Rapid Analytical Method for the Qualitative Detection of Listeria spp. and Listeria monocytogenes by a Real-Time PCR Assay according to EN UNI ISO 16140-3:2021. Pathogens 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-Y.; Chen, Z.; Cao, X.; Ross, T.D.; Falbel, T.G.; Burton, B.M.; Venturelli, O.S. Programming bacteria for multiplexed DNA detection. Nature Communications 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Yan, L.; Zheng, X.-c.; Li, L.-z.; Liu, P.; Cao, W.-s. Rapid detection of Pseudomonas aeruginosa by cross priming amplification. Journal of Integrative Agriculture 2020, 19, 2523–2529. [Google Scholar] [CrossRef]
- Qin, M.; Ma, X.; Fan, S.; Wu, H.; Yan, W.; Tian, X.; Lu, J.; Lyu, M.; Wang, S. Rapid detection of Pseudomonas aeruginosa using a DNAzyme-based sensor. Food Science & Nutrition 2021, 9, 3873–3884. [Google Scholar]
- Zhao, T.; Zhang, J.; Han, X.; Yang, J.; Wang, X.; Vercruysse, M.; Hu, H.-Y.; Lei, X. A Pseudopaline Fluorescent Probe for the Selective Detection of Pseudomonas aeruginosa. CCS Chemistry 2021, 3, 2405–2417. [Google Scholar] [CrossRef]
- Wang, M.; Yang, J.; Gai, Z.; Huo, S.; Zhu, J.; Li, J.; Wang, R.; Xing, S.; Shi, G.; Shi, F.; et al. : Comparison between digital PCR and real-time PCR in detection of Salmonella typhimurium in milk. International Journal of Food Microbiology 2018, 266, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Kim, J.-H.; Oh, S.-W. Combination of filtration and immunomagnetic separation based on real-time PCR to detect foodborne pathogens in fresh-cut apple. Journal of Microbiological Methods 2022, 201. [Google Scholar] [CrossRef]
- Maier, C.; Hofmann, K.; Huptas, C.; Scherer, S.; Wenning, M.; Lücking, G. Simultaneous quantification of the most common and proteolytic Pseudomonas species in raw milk by multiplex qPCR. Applied Microbiology and Biotechnology 2021, 105, 1693–1708. [Google Scholar] [CrossRef]
Figure 1.
Result of the establishment of a dual detection system. Note: P. fragi reaction positive, P. aeruginosa reaction positive, P. fragi & P. aeruginosa reaction positive, P. f ragi & P. aeruginosa reaction negative.
Figure 1.
Result of the establishment of a dual detection system. Note: P. fragi reaction positive, P. aeruginosa reaction positive, P. fragi & P. aeruginosa reaction positive, P. f ragi & P. aeruginosa reaction negative.
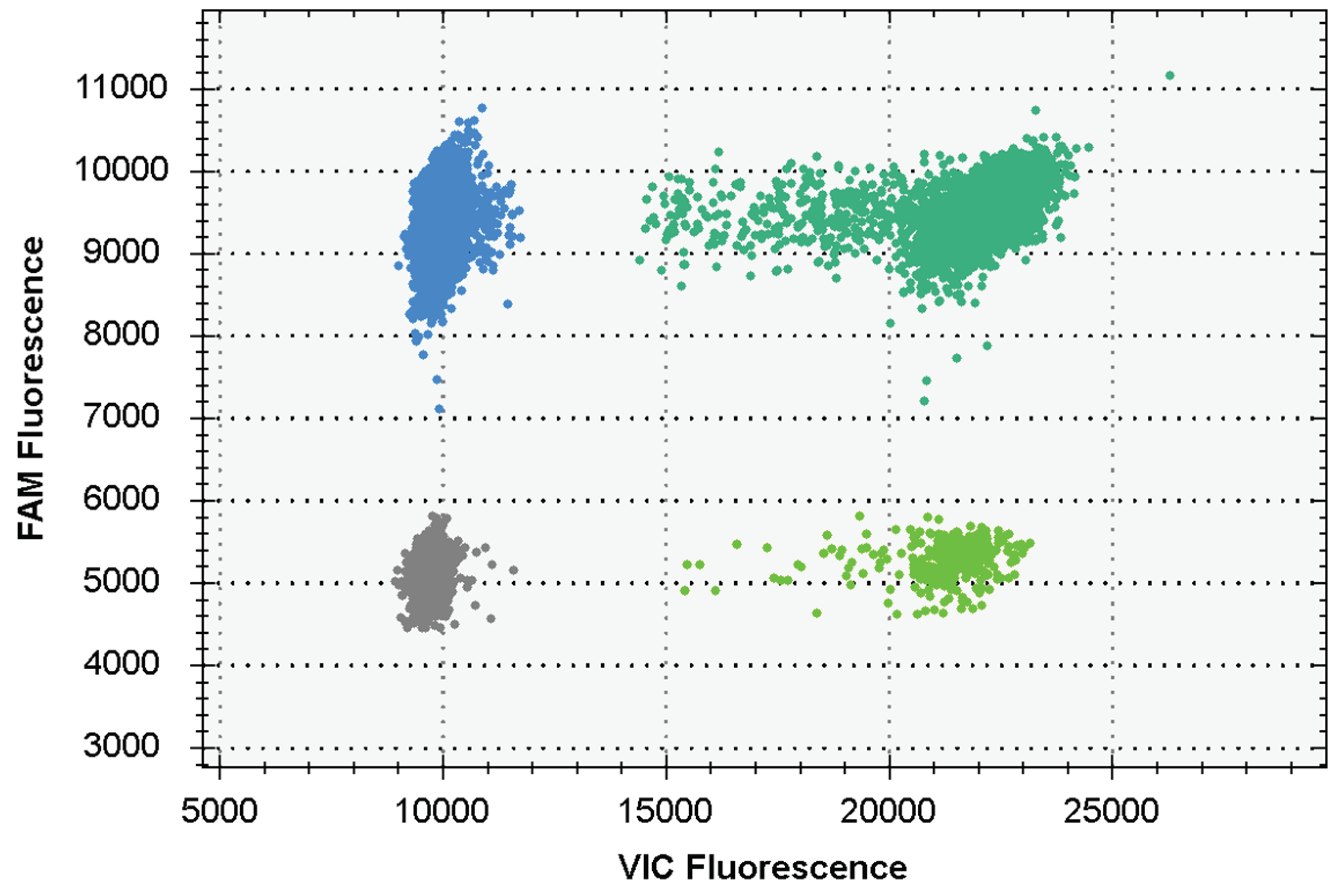
Figure 2.
Linear relationship analysis of P. fragi and P. aeruginosa detected by ddPCR method.
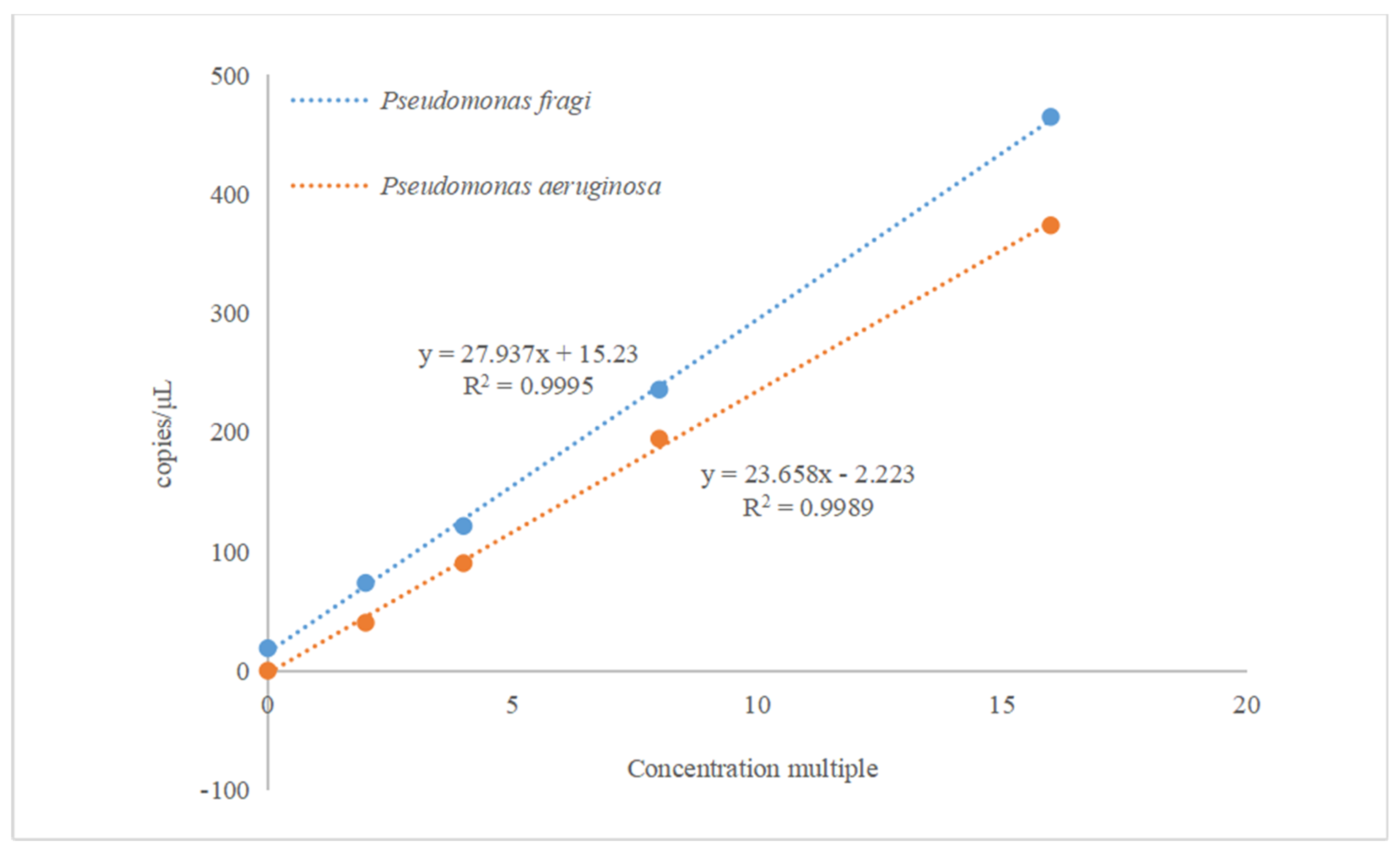
Figure 3.
Specific results of dddPCR detection of P. aeruginosa and P. fragi Note: A: Specific results of P. aeruginosa; B: Specific results of P. fragi.
Figure 3.
Specific results of dddPCR detection of P. aeruginosa and P. fragi Note: A: Specific results of P. aeruginosa; B: Specific results of P. fragi.
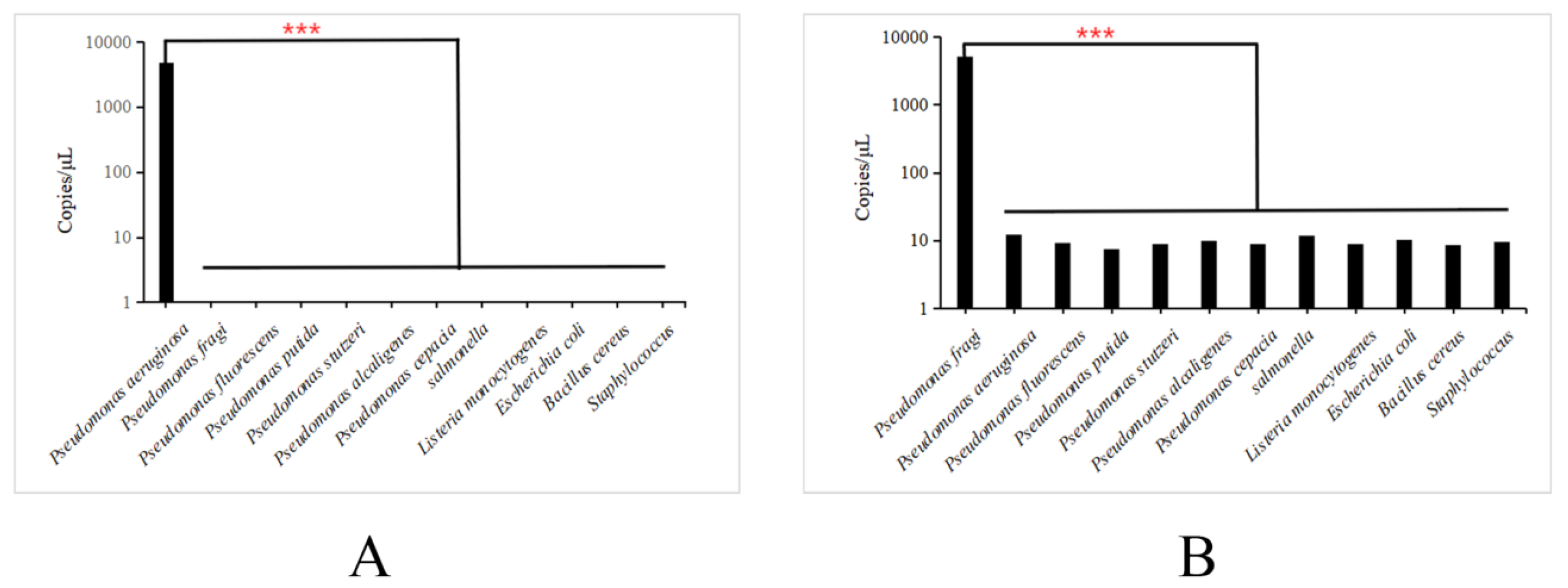
Figure 4.
Genomic sensitivity analysis of dddPCR assay
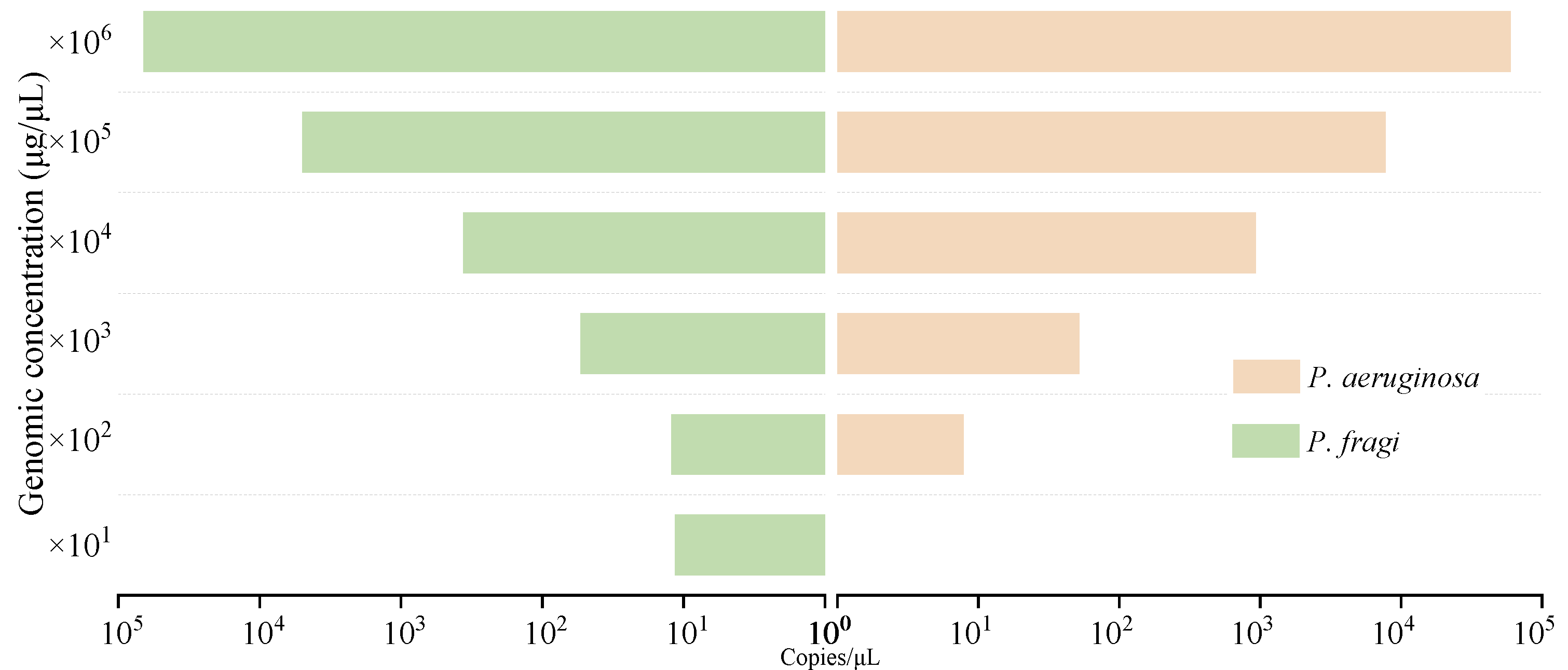
Figure 5.
Colonial sensitivity analysis of dddPCR assay
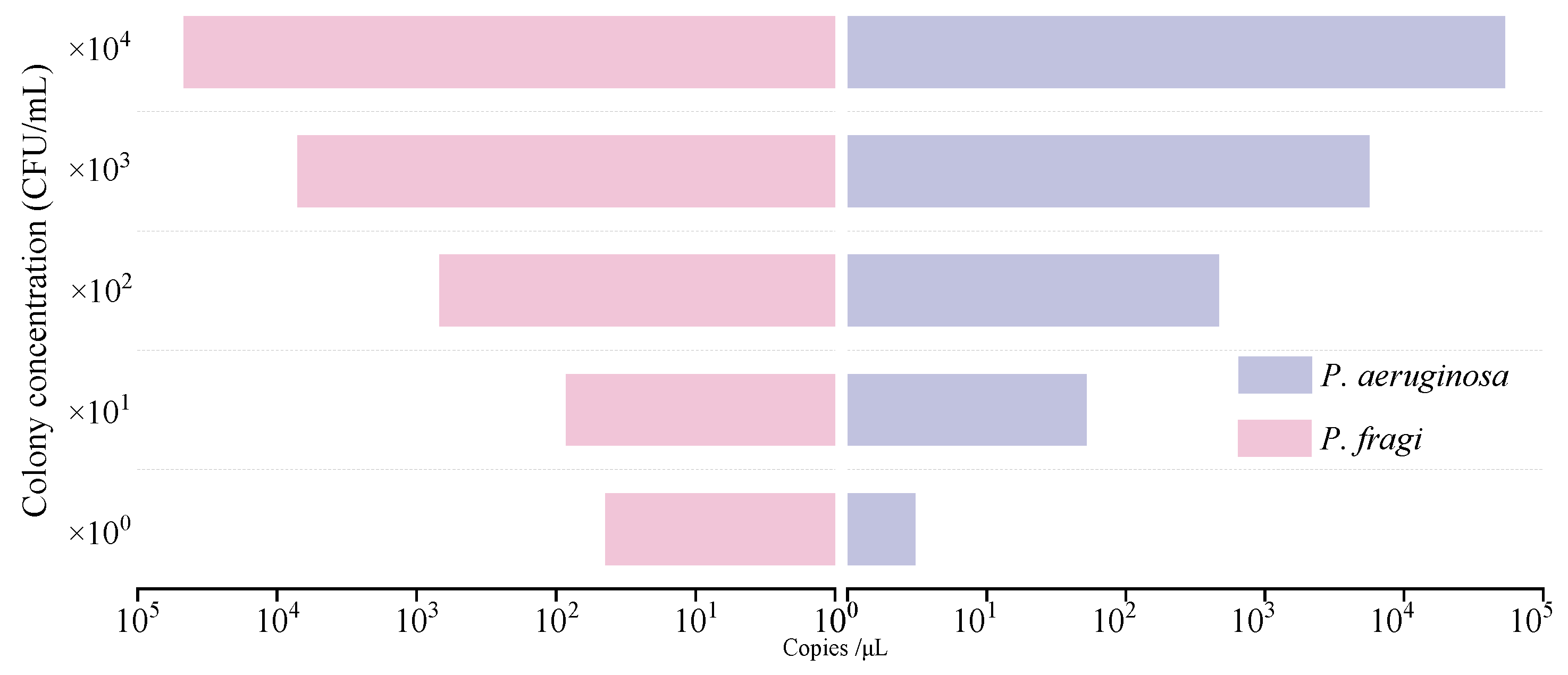
Figure 6.
Sensitivity evaluation of ddPCR method in the presence of food natural background flora. Note: Ⅰ:The concentration of natural background bacteria in milk was 5.4×103 CFU/mL;Ⅱ:The concentration of natural background bacteria in chicken was 1.72×103 CFU/mL; NaN: Invalid result
Figure 6.
Sensitivity evaluation of ddPCR method in the presence of food natural background flora. Note: Ⅰ:The concentration of natural background bacteria in milk was 5.4×103 CFU/mL;Ⅱ:The concentration of natural background bacteria in chicken was 1.72×103 CFU/mL; NaN: Invalid result
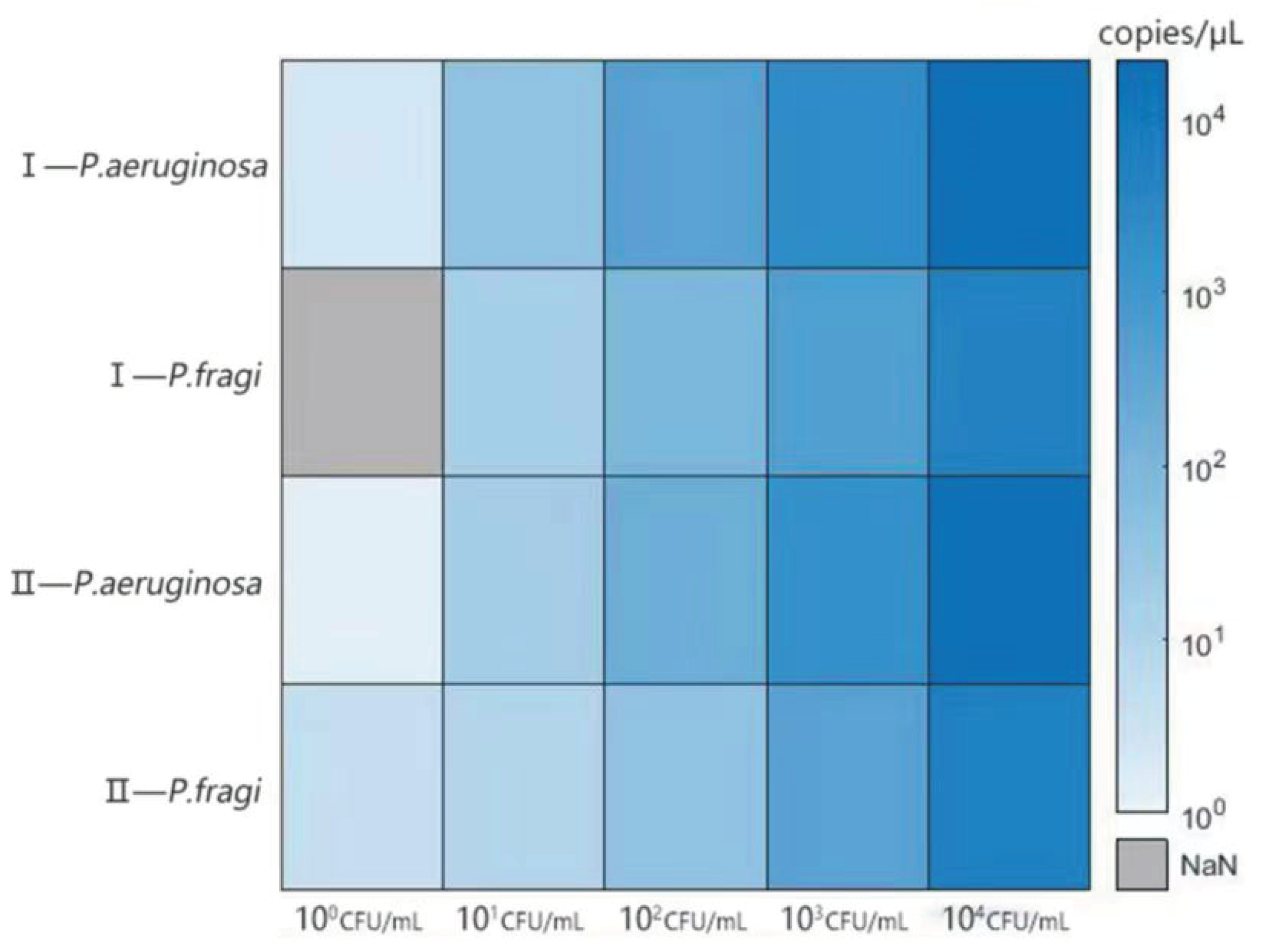

Table 1.
The candidate genes and primers of P. aeruginosa and P. fragi used in PCR specificity verification experiments.
Table 1.
The candidate genes and primers of P. aeruginosa and P. fragi used in PCR specificity verification experiments.
| Source | gene | Annotation | Primer | Sequence(5’-3’) | Source |
|---|---|---|---|---|---|
| P. fragi | RS22665 | Transcriptional regulatory protein | pf1-7 | ATAACGGCAAGAACACCA | In this study |
| CCAAACACGCCTCTGAAC | |||||
| RS22680 | NeuD/PglB/VioB family sugar acetyltransferase | pf1-18 | GGCACAAGTCAATGGTCG | ||
| CACAGTCAGGGCAAGGAT | |||||
| RS10890 | triacylglycerol lipase | Pf3-21 | CCTTGAATGCGCTTAACGCCCTGACCACC | ||
| CGTAGACCCGGTCCAGTAGGCGAGGCTGAT | |||||
| ribA | GTP cyclohydrolase II | Pf3-13 | CGATGTATTCGGGTCCAGACGCTGTGATT | ||
| ATAGTGGTAGTTGTCTTGGGACGGTAGGC | |||||
| P. aeruginosa | LasR | Transcriptional regulatory protein | lasR1 | CGAGAACGCCTTCATCGTCGGCAACTACC | In this study |
| GAAGAACTCGTGCTGCTTTCGCGTCTGGTA | |||||
| gyrB | DNA gyrase subunit B | gyrB2 | ATCCGCACCCTGCTGTTGACCTTCTTCTTCCG | ||
| TGATGTACTGCTCCTGCTTGCCACGCTTGACC | |||||
| rpoB | DNA-directed RNA polymerase beta chain | rpoB3 | TGCCCGATCGAAACCCCTGAAGGTCCGAA | ||
| ATCTCGTCGGTTACCAGGCTGTCCTTGACT |
Table 2.
The primers and probes in the dddPCR assay for detection of P. aeruginosa and P. fragi.
| Bacterial strains | gene | primer | Sequences of primer(5’-3’) | PCR product | source |
|---|---|---|---|---|---|
| P. aeruginosa | LasR | Pa-2F | AGCCGGGAGAAGGAAGTGTT | 80 bp | In this study |
| Pa-2R | TCCGAGCAGTTGCAGATAACC | ||||
| Pa-2P | VIC-TGCGCCATCGGCAAGACCAGT-BHQ1 | ||||
| P. fragi | RS22680 | Pf-2F | GGCCGGCACGCAAGT | 59 bp | In this study |
| Pf-2R | CTTGGACAGTAGCGAAAAACGA | ||||
| Pf-2P | FAM-TGTCGAGAAGCCAGTCTCCGTGTTCC-BHQ1 |
Table 3.
The reaction system of dddPCR assay.
| Component | Addition |
|---|---|
| 5× MIX | 4.5μL |
| Primer 1-F(10μM) | 1μM |
| Primer 1-R(10μM) | 1μM |
| Primer 2-F(10μM) | 1μM |
| Primer 2-R(10μM) | 1μM |
| Probe1(10μM) | 0.25μM |
| Probe2(10μM) | 0.25μM |
| ROX dye | 0.3μL |
| Enzyme | 0.2μL |
| Template 1 | 1μL |
| Template 2 | 1μL |
| Complemented by water to | 15μL |
Table 4.
The results of PCR analysis of species-specific genes.
| Bacterial strains | Source | Results | ||||||
|---|---|---|---|---|---|---|---|---|
| LasR | rpoB | gyrB | RS22665 | RS22680 | RS10890 | ribA | ||
| Pseudomonas fragi | SHBCC D24613 | - | - | - | + | + | + | + |
| Pseudomonas fragi | CGMCC1.3349 | - | - | - | + | + | + | + |
| Pseudomonas fragi | Laboratory isolates | - | - | - | + | + | + | - |
| Pseudomonas aeruginosa | ATCC 15442 | + | + | + | - | - | - | - |
| Pseudomonas aeruginosa | ATCC 27853 | + | + | + | - | - | - | - |
| Pseudomonas aeruginosa | DSM 939 | + | + | + | - | - | - | - |
| Pseudomonas aeruginosa | Laboratory isolates | + | + | + | - | - | - | - |
| Pseudomonas fluorescens | ATCC 13525 | - | - | - | + | - | + | - |
| Pseudomonas putida | ATCC 49128 | - | + | - | - | - | - | + |
| Pseudomonas pseudoalaligenes | CGMCC1.10611 | - | - | - | - | - | - | - |
| Pseudomonas mendocina | ATCC 25411 | - | + | - | - | - | - | + |
| Pseudomonas stutzeri | ATCC 17588 | - | - | - | - | - | - | - |
| Pseudomonas alcaligenes | ATCC 14909 | - | - | - | - | - | - | - |
| Pseudomonas cepacia | SHBCC D 14769 | - | - | - | - | - | - | - |
| Pseudomonas putida | ATCC 17485 | - | - | - | - | - | - | - |
| Pseudomonas fluorescens | GIM1.110 | - | - | - | - | - | + | - |
| Pseudomonas fluorescens | ATCC 17397 | - | - | + | - | - | - | - |
| Staphylococcus | CICC 10788 | - | - | - | - | - | - | - |
| Enterococcus avium | ATCC 14025 | - | - | - | - | - | - | - |
| Bacillus pumilus | CMCC 63202 | - | - | - | - | - | - | - |
| Listeria monocytogenes | CICC 21622 | - | - | - | - | - | - | - |
| salmonella enterica | CICC 21482 | - | - | - | - | - | - | - |
| Cronobacter sakazakii | CICC 21560 | - | - | - | - | - | - | - |
| Cronobacter universalis | NCTC 9529 | - | - | - | - | - | - | - |
| salmonella anatum | CICC 21498 | - | - | - | - | - | - | - |
| Escherichia coli | ATCC 25922 | - | - | - | - | - | - | + |
| Bacillus cereus | CICC 23384 | - | - | - | - | - | - | - |
Note: +: positive result; -: negative result.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



