
Submitted:
03 April 2024
Posted:
04 April 2024
You are already at the latest version
Abstract
Research on retinoid-based cancer prevention, spurred by the effects of vitamin A deficiency on gastric cancer and subsequent clinical studies on digestive tract cancer, unveils novel avenues for chemoprevention. Acyclic retinoids like 4,5-didehydrogeranylgeranoic acid (4,5-didehydroGGA) emerged as potent agents against hepatocellular carcinoma (HCC), distinct from natural retinoids such as all-trans retinoic acid (ATRA). Mechanistic studies reveal GGA's unique induction of pyroptosis, a rapid cell death pathway, in HCC cells. GGA triggers mitochondrial superoxide hyperproduction and ER stress responses through Toll-like receptor 4 (TLR4) signaling and modulates autophagy, ultimately activating pyroptotic cell death in HCC cells. Unlike ATRA-induced apoptosis, GGA and palmitic acid (PA) induce pyroptosis, underscoring their distinct mechanisms. While all three fatty acids evoke mitochondrial dysfunction and ER stress responses, GGA and PA inhibit autophagy, leading to incomplete autophagic responses and pyroptosis, whereas ATRA promotes autophagic flux. In vivo experiments demonstrate GGA's potential as an anti-oncometabolite, inducing cell death selectively in tumor cells and thus suppressing liver cancer development. This review provides a comprehensive overview of the molecular mechanisms underlying GGA's anti-HCC effects and underscores its promising role in cancer prevention, highlighting its importance in HCC prevention.
Keywords:
acyclic retinoid
; acyclic diterpenoid
; cancer chemoprevention
; lipotoxicity
; palmitic acid
; pyroptosis
; regulated cell death
; retinoic acid
1. Introduction
Research on cancer prevention by retinoids has a long history. Reports of gastric cancer occurrence in animals deficient in vitamin A [1] and studies observing decreased serum vitamin A levels in patients with digestive tract cancer [1,2] were pioneering works in the early days following the discovery of vitamin A. Particularly notable is the study by Saffiotti [3], which demonstrated the suppression of chemical carcinogenesis in tracheal epithelium by excessive administration of vitamin A, laying the foundation for the concept of cancer chemoprevention by retinoids [4,5].
Significant research on the prevention of hepatocellular carcinoma (HCC) by retinoids includes the double-blind clinical trial conducted by Muto et al. using a placebo-controlled "acyclic retinoid" [6]. Administration of 4,5-didehydrogeranylgeranoic acid (4,5-didehydroGGA or peretinoin) at 600 mg/day for one year in patients after hepatic cancer resection significantly inhibited the occurrence of second primary liver cancer for up to five years post-administration. Based on studies investigating the relationship between ligand activity on retinoid receptors and the differentiation-inducing effects on HCC cells, the concept of "acyclic retinoids" was proposed by Muto, Moriwaki, and Omori. Specifically, geranylgeranoic acid (GGA) and its derivative 4,5-didehydroGGA, which possess ligand activity for cellular retinoic acid-binding protein (CRABP) [7] and retinoid receptors (RAR and RXR) [8], as well as differentiation-inducing activity on HCC cell lines [8], were defined as acyclic retinoids. On the other hand, we demonstrated that GGA, unlike all-trans retinoic acid (ATRA), is an endogenous fatty acid synthesized from mevalonic acid in mammalian hepatocytes [9]. Therefore, although GGA is an acyclic retinoid, it cannot be considered a vitamin.
GGA and ATRA belong to the long-chain polyunsaturated branched-chain fatty acids with 4 to 5 unsaturated bonds and a carbon number of 20. On the other hand, palmitic acid (PA), a long-chain saturated fatty acid, is well known for inducing cell death as a lipotoxic effect on various cells along with cholesterol and lysophospholipids [10]. Interestingly, PA is also a fatty acid considered for HCC prevention. However, when the hypoxia-dependent cell death-inducing effect of PA on HCC cells was first discovered, it was believed to be related to the pathogenesis of non-alcoholic fatty liver disease [11]. However, recently, studies proposing the application of PA-induced cell death for cancer treatment have also been reported [12].
Therefore, in this review, we first summarize the inhibitory effect of GGA on HCC as an induction of cell death in HCC cells from the perspective of pyroptosis. Subsequently, we compare the differences in the cell death-inducing effects of GGA, ATRA, and PA on HCC cells based on the latest literature and discuss the molecular targets of GGA on HCC cells.
2. Mechanism of Cell Death Induction by GGA in Hepatocellular Carcinoma (HCC) Cells
GGA, similarly to ATRA, has been confirmed to induce a significant increase in the expression of neurotrophic receptor kinase 2 (NTRK2 or TrkB) and RARβ protein in human neuroblastoma cell line SH-SY5Y, indicating that GGA possesses a "retinoid effect" like ATRA [13]. However, the cell death-inducing effect of GGA on HCC cells, as described below, is a unique action of GGA that is not found in natural retinoids such as ATRA.
Given the detailed historical background of research on the cell death induction mechanism of GGA in HCC cells [9], here we will summarize all the findings obtained to date and present the proposed mechanism.
2.1. Timeline of intracellular Events Related to Cell Death in Human Hepatocellular Carcinoma (HCC)-Derived Cell Lines Induced by GGA
Describing the intracellular events occurring in HCC cells after GGA treatment along a timeline can provide an approximate mechanism of GGA-induced cell death. Of course, the observation of events depends on the analysis technique (detection sensitivity, complexity of the technique, etc.), so the actual sequence may sometimes be reversed. Below, we describe the phenomena observed over time after treatment of human HCC-derived cell lines (mainly HuH-7) with GGA (5-20 µM):
- 15 minutes: The earliest observed phenomena upon adding 10 µM GGA to the culture medium include three main events. Firstly, increased production of superoxide in mitochondria is observed using the MitoSox fluorescence staining technique [14]. Secondly, the splicing of XBP1 mRNA in the endoplasmic reticulum (ER) is detected using the RT-qPCR technique [15,16]. Thirdly, an increase in LC3β-II protein, a marker of autophagosomes, is observed using western blotting [14]. Although these events are observed simultaneously in different cellular organelles (mitochondria, ER, and autophagosomes), it is unlikely that these events occur independently and simultaneously. Since suppressing the increased production of superoxide by GGA with antioxidants does not suppress XBP1 mRNA splicing [16], it appears that at least the increased production of superoxide does not trigger XBP1 mRNA splicing.
- 20 minutes: Monitoring the cytosolic Ca2+ concentration using Fluo-4 AM as a probe reveals a transient peak 16-20 minutes after GGA treatment, which quickly declines ([16]; https://doi.org/10.1042/BSR20194118; see Supplementary Movie S1). This peak is observed, albeit slightly delayed, even when experiments are conducted in a medium without Ca2+, suggesting leakage of Ca2+ from intracellular Ca2+ storage sites such as the ER.
- 30 minutes: Introduction of GFP-LC3 expression vector into HuH-7 cells followed by GGA treatment results in the appearance of green, fluorescent autophagosomes 30 min later [14], consistent with the detection of LC3β-II protein 15 min earlier. An increase in Beclin-1 (BECN1) and Sequestosome 1 (p62/SQSTM1), which are involved in promoting autophagosome formation and cargo protein for autophagosomes, respectively, is also detected [14]. Meanwhile, although there is no change in the transcription level of the Cyclin D1 (CCND1) gene, its translation is significantly suppressed [17].
- 1 hour: Loss of mitochondrial membrane potential (ΔΨm) is observed 1 h after GGA treatment when stained with Rhodamine 123 [18] and 2 h after GGA treatment when stained with MitoTracker® Red CMXRos [14]. Regardless of the staining method, the loss of Δψm is observed after the increased superoxide production in mitochondria. Active caspase-4 (CASP4) is detected by western blotting and observed up to 5 h after GGA addition but disappears thereafter [16]. Concurrently with the detection of active CASP4, the N-terminal fragment of Gasdermin D (GSDMD) is detected, with its amount peaking at 3 h and then decreasing, reaching its maximum at 8 h after GGA treatment when cell death is observed.
- 2 hours: A transient increase in lysophosphatidylcholine containing PA and palmitoleic acid (lysoPC [16:0]; lysoPC [16:1]) is observed. In contrast, a significant increase in lysoPC (lysoPC [20:4]) and lysophosphatidylethanolamine (lysoPE [20:4]) containing arachidonic acid is observed, and then these lysophospholipids (lysoPLs) remain at high levels and continue to increase gradually until 24 h [19].
- 3 hours: Immunofluorescence staining of GSDMD reveals signals mainly in the nucleus in control cells, but in GGA-treated cells, the signals are also detected in the plasma membrane [16]. Immunofluorescence staining of NF-κB, one of the inflammatory transcription factors, shows that its signal is observed granularly in the cytoplasm in control cells, but most of it translocates into the nucleus after GGA treatment [16]. Morphological changes in HuH-7 cells are first observed, with loss of cell adhesion and contraction of cells away from each other. Rod-shaped protrusions or blebs emerge from each contracted cell into the intercellular space ([16]; https://doi.org/10.1042/BSR20194118; see Supplementary Movie S1).
- 6 hours: The transient peak in cytosolic Ca2+ concentration observed 20 min after GGA treatment reappears 6 hours later. When experiments are conducted in a medium without Ca2+, this peak is completely absent, suggesting that it is due to Ca2+ influx into the cytosol from the medium through a ruptured cell membrane by GSDMD translocated to the membrane. Intracellular levels of NLRP3 mRNA and IL1B mRNA increase. Blebs protruding from contracted cells disappear, and spherical balloons appear, gradually increasing in size [16].
- 8 hours: Significant activation of CASP1 is observed by enzyme activity measurement. Detection of the N-terminal fragment of GSDMD also peaks. Leakage of lactate dehydrogenase (LDH) into the medium is observed. The balloons that emerge from contracted cells become larger than the cell diameter [16]. (For chronological morphological changes, [16]; https://doi.org/10.1042/BSR20194118; see Supplementary movie S1)
2.2. Inhibition of GGA-Induced Cell Death by Various Inhibitors
- CASP Inhibitory Peptides: Induction of cell death in HuH-7 cells by GGA is completely inhibited by co-treatment with an active site inhibitor peptide of CASP1 (ac-YVAD-cmk). Co-treatment with an active site inhibitor peptide of CASP3 (ac-DEVD-CHO) delays GGA-induced cell death by several hours but does not inhibit it [18]. Co-treatment with an active site inhibitor peptide of CASP4 (Z-LEVD-fmk) inhibits GGA-induced activation of CASP1 [16].
- TLR4 Inhibitors: Pretreatment of HuH-7 cells with TLR4 (Toll-like receptor 4) siRNA completely inhibits GGA-induced cell death. Co-treatment with VIPER, a peptide that specifically inhibits signaling from TLR4, completely inhibits GGA-induced cell death as well as other processes induced by GGA, such as increased superoxide production in mitochondria, splicing of XBP1 mRNA, nuclear translocation of the cytosolic NF-κB, upregulation of TLR2 and NLRP3 mRNAs, and activation of CASP1 [16].
- Lipotoxicity Inhibitor: Co-treatment with oleic acid (OA), known to inhibit PA-induced lipotoxicity, completely inhibits GGA-induced cell death in HuH-7 cells. Additionally, OA co-treatment inhibits various processes induced by GGA, including splicing of XBP1 mRNA, upregulation of TLR2 mRNA and DDIT3 (CHOP) mRNA, increase in LC3β-II levels, accumulation of autophagosomes, superoxide hyperproduction in mitochondria, translocation of cytoplasmic NF-κB to the nucleus, and activation of CASP1 [16]. The inhibitory effect of OA on GGA is observed only when co-treated simultaneously; treatment with OA before GGA treatment does not exhibit any inhibitory effect [15], suggesting that the inhibitory point of action of OA on GGA is extracellular.
- Antioxidant: Pretreatment with α-tocopherol, a lipid-soluble antioxidant vitamin, dose-dependently inhibits GGA-induced cell death [18]. Co-treatment with α-tocopherol also inhibits other processes induced by GGA, such as dissipation of ΔΨm, activation of CASP1, upregulation of NLRP3 mRNA, translocation of cytoplasmic NF-κB to the nucleus, and increased superoxide production in mitochondria [15,16]. However, co-treatment with α-tocopherol does not inhibit activation of unfolded protein response in the ER (UPRER: splicing of XBP1 mRNA and upregulation of DDIT3 mRNA) by GGA [16].
- Kinase Inhibitors: Co-treatment with wortmannin, a non-specific inhibitor of PI3 kinase, inhibits GGA-induced increase in superoxide production in mitochondria and the appearance of green, fluorescent puncta in GGA-treated GFP-LC3 transfected HuH-7 cells [14]. Each co-treatment with BAY 11 7082, an inhibitor of NF-κB activation, or BI605906, an IKKβ inhibitor, inhibits the translocation of cytoplasmic NF-κB to the nucleus induced by GGA [16].
2.3. The Mechanism of GGA-Induced Cell Death
Based on the timeline of intracellular events after GGA treatment described above, along with observations of the effects of various inhibitors, the mechanism of cell death induction by GGA in HCC cells is currently understood as follows (Figure 1).
Inhibition of TLR4 gene expression using specific siRNA or inhibition of intracellular signaling of TLR4 by the virus-derived peptide VIPER suppresses GGA-induced cell death. Cotreatment with VIPER affects almost all processes associated with GGA-induced cell death. These include enhanced production of superoxide in mitochondria, activation of UPRER, translocation of cytoplasmic NF-κB to the nucleus, priming of the inflammasome (upregulation of NLRP3 and IL1B mRNAs), localization of GSDMD to the cell membrane, and activation of CASP1 [16], suggesting that TLR4 serves as the sole initiator of GGA-induced cell death.
The signal of TLR4 stimulated by GGA appears to bifurcate into two major pathways within the cell. One involves the UPRER, detected as the splicing of XBP1 mRNA and upregulation of DDIT3 mRNA, along with the consequent activation of ER-resident CASP4, leading to the formation of GSDMD pores through the cleavage of GSDMD by active CASP4 and the translocation of the GSDMD N-terminal fragment to the cell membrane (Figure 1, left). Although the precise mechanism linking TLR4 signaling to UPRER is not yet clear, several reports suggest that stimulation of TLR4 induces UPRER, particularly the splicing of XBP1 mRNA [20,21,22,23].
It is believed that the mechanism by which UPRER activates CASP4 involves the release of Ca2+ from the ER. Indeed, treatment solely with thapsigargin, which decreases Ca2+ concentration in the ER by inhibiting the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase known as an ER stress inducer, activates CASP4 similarly to GGA treatment [16]. Hitomi et al. [24] first reported the activation of CASP4 by UPRER, and since then, the activation of ER-resident CASP4 via UPRER has been confirmed in numerous studies [25,26,27,28]. Recently, it has been demonstrated that calpain 5 (CAPN5), localized in mitochondria, is activated by ER stress, and cleaves pro-CASP4 into its active form, serving as a mechanism for CASP4 activation [29]. Conversely, treatment with tunicamycin, an inhibitor of protein N-glycosylation and another ER stress inducer, alone did not activate CASP4 [16]. Therefore, GGA may activate CASP4 through a putative calpain (maybe CAPN5) activated by the initial Ca2+ peak at 16-20 minutes [16]. Activated CASP4 cleaves GSDMD to generate N-terminal fragments, which translocate to the cell membrane and form GSDMD pores. The translation inhibition of the CCND1 gene observed 30 min after GGA treatment [17] is also considered a downstream signal of UPRER [30].
The other pathway of TLR4 signaling involves the enhancement of superoxide production in mitochondria (Figure 1, right). Upon TLR4 activation, the TLR4 adapter protein TRAF6 (TNF receptor-associated factor 6) translocates to the mitochondria and interacts with ECSIT (Evolutionarily conserved signaling intermediate in Toll pathway, mitochondrial), promoting its ubiquitination. ECSIT, a subunit of the Mitochondrial respiratory chain Complex I Assembly complex (MCIA), is thought to be one of the chaperones for complex I [31,32,33]. When ECSIT is ubiquitinated, MCIA undergoes degradation, leading to the increased production of reactive oxygen species (ROS) in mitochondria [34]. Excessive ROS generated in mitochondria promote the translocation of cytoplasmic NF-κB to the nucleus, increasing the transcription of NLRP3 and IL1B genes (priming of inflammasomes). Activation of NLRP3 inflammasome by extracellular Ca2+ influx through GSDMD pores formed by CASP4 leads to the activation of CASP1 [35], which, in turn, cleaves GSDMD, resulting in the formation of GSDMD pores in the plasma membrane and subsequent pyroptotic cell death.
In summary, TLR4 signaling plays an essential role in GGA-induced cell death and UPRER-associated activation of CASP4 occurs within 1 h after GGA addition. The cleavage of GSDMD leading to the formation of GSDMD-N-terminal fragments and their localization to the plasma membrane is observed at 3 h, suggesting activation of the non-canonical pathway of pyroptosis [16]. However, cell death is not observed at 3 h after GGA addition. Subsequently, activation of CASP1 by NLRP3 inflammasome is observed, along with dramatic upregulation of IL1B gene expression, suggesting the possibility of activation of canonical pathway-mediated pyroptosis [16]. Regarding this, the inhibition of CASP1 activation by co-treatment with a CASP4 inhibitor during GGA treatment suggests that activation of the non-canonical pathway may trigger the activation of the canonical pathway. Indeed, recent reviews [36,37] suggesting that the non-canonical pathway's efflux of K+ to the extracellular space and increased mitochondrial ROS production serve as triggers for the secondary activation of the canonical pathway support our findings.
3. GGA-Induced Cell Death in Hepatocellular Carcinoma (HCC) Cells - A Comparison with Palmitic Acid (PA)-Induced Cell Death and All-trans Retinoic Acid (ATRA)-Induced Cell Death
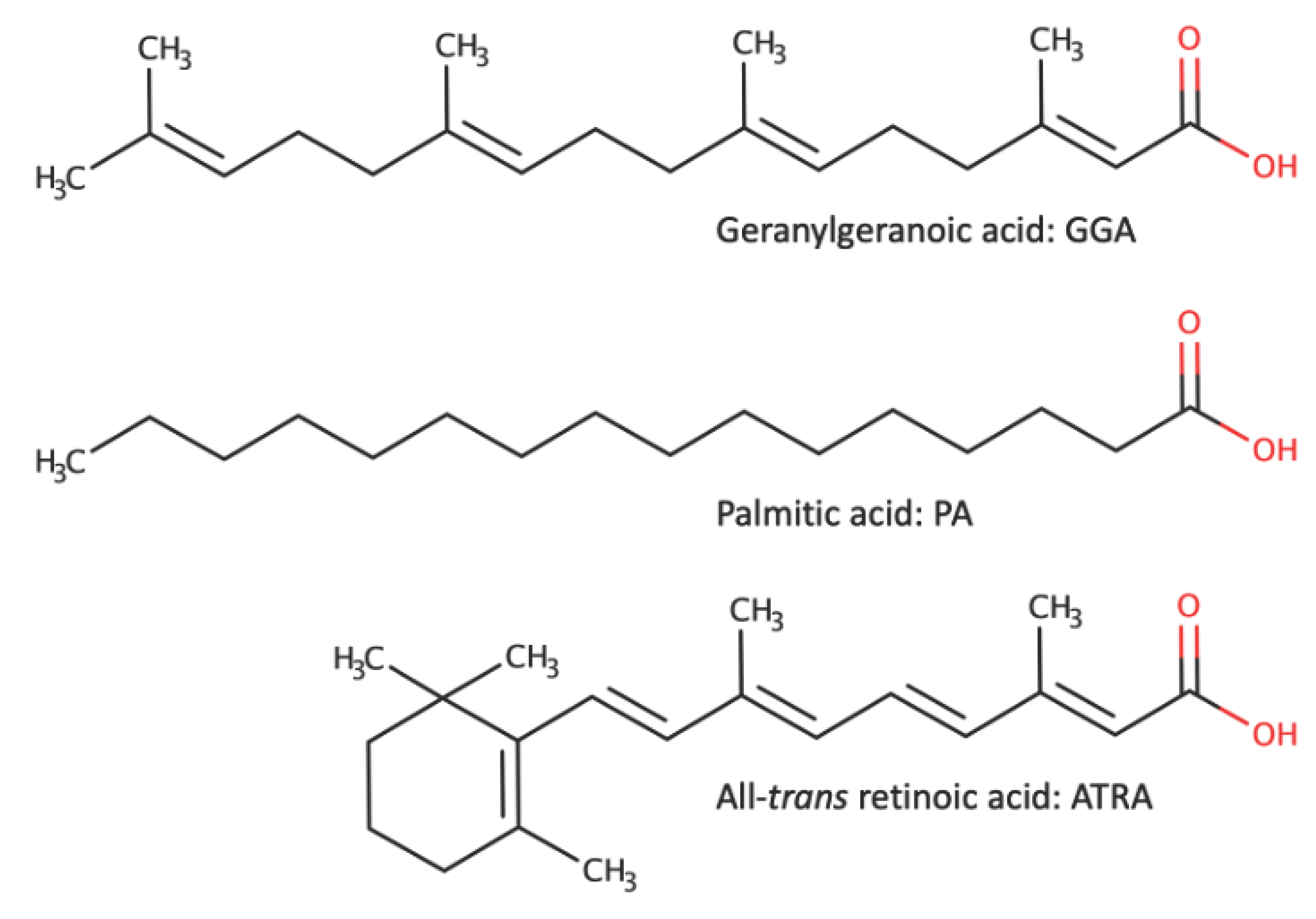
GGA is a branched-chain fatty acid derived from mevalonic acid, with 20 carbon atoms and 4 unsaturated bonds [9]. PA, a saturated fatty acid with 16 carbon atoms, is well known for its lipotoxicity to cells such as hepatocytes [10]. While the molecular lengths of both molecules are almost the same, the major difference lies in the presence of unsaturated bonds and branched chains. On the other hand, ATRA, shown in the lower panel of Figure 2, is a 20-carbon, branched-chain fatty acid with 5 unsaturated bonds and is believed to be one of the active forms of vitamin A, converted in the body from β-carotene and vitamin A esters found in food. ATRA has also been reported to induce cell death in HCC-derived cell lines [38,39]. Therefore, comparing the differences between cell death induced by GGA in HCC-derived cell lines and that induced by PA or ATRA, we can evaluate the significance of GGA-induced cell death.
3.1. Effective Concentrations and Observation Time for Inducing Cell Death (Table 1)
Since the targeted cell lines and experimental conditions vary, a direct comparison may not be meaningful, but citations using human HCC cell lines are compared as much as possible. Experiments inducing cell death in human HCC cell lines by GGA were conducted at final concentrations of 0.5-20 µM, and cell death was observed within 8 to 16 h after GGA addition.
For ATRA, the concentrations added to the medium range from 0.1 to 20 µM, like GGA, but it does not always induce cell death. Although we conducted experiments under the same conditions as with GGA, no cell death was observed in ATRA-treated cells even after 24 h [40]. Lin et al. reported that treatment with 0.1-10 µM ATRA for 3 days did not affect the proliferation of HepG-2 and Hep3B cells [41], and Wang et al. found that 5-20 µM ATRA for HepG-2 and HuH-7 cells and 1-5 µM ATRA for Hep3B cells protected them against cell death occurring 5-10 days after serum removal [42]. Although reports of ATRA treatment inhibiting cell proliferation exist, significant effects are observed after 24 h to more than 5 days [38,39].
On the other hand, there are numerous reports of cell death induction by PA, mainly in the analysis of lipotoxicity to immune cells. Here, only reports using human HCC-derived cell lines are discussed. The final concentrations of PA added to the medium range from 200 to 3000 µM, which is 10 to several hundred times higher than GGA or ATRA. Although there are many reports, cell death is confirmed within 24 h after PA treatment [11,12,43,44,45,46]. Molecules that exert effects at concentrations of several µM like GGA or ATRA are thought to act as signaling molecules. In contrast, PA, which only exerts effects at concentrations of several hundred µM to several mM, may have its metabolites acting as effector molecules rather than itself being a signaling molecule. In other words, it may be possible to consider the cell death induction effect of PA because of changes in membrane fluidity or compactness caused by its metabolites.
In summary, cell death induced by GGA occurs at considerably lower concentrations and more rapidly compared to PA, and even more rapidly compared to ATRA.
3.2. Mode of Regulated Cell Death
All three fatty acids - GGA, PA, and ATRA - appear to induce cell death in HCC-derived cells accompanied by the activation of CASP3, as shown in Table 1. However, the typical intrinsic or extrinsic mechanisms of apoptosis have not been consistently observed. Activation of NF-κB, CASP1, and CASP4 has been observed with GGA treatment, and activation of NF-κB and CASP1 has been noted with PA treatment, thus suggesting the involvement of pyroptosis mechanisms in cell death induction. Conversely, ATRA treatment has been reported to increase the expression of the OTUD7B gene, an NF-κB inhibitory factor [47], indicating that inflammatory cell death, as observed with GGA or PA treatment, is unlikely to occur. ATRA has been shown to induce apoptotic cell death in human HCC cell lines PLC/PRF/5 and HLE [48], and ferroptosis cell death in HepG2 and Hep3B cells [49]. However, there are also reports suggesting that ATRA inhibits ferroptosis in neuronal cell lines [50].
3.3. Action on plasma Membrane and intracellular Organelles
- Plasma Membrane: When fatty acids like GGA, PA, and ATRA are added to the medium, they are expected to encounter the plasma membrane. Both PA and GGA are believed to induce cell death via signaling mediated by the cell surface receptor TLR4, which is localized on the plasma membrane [16,51]. However, few reports are suggesting that these lipids directly act as ligands for TLR4. While computational simulations and isothermal titration calorimetry have demonstrated the docking of five PA molecules with the hydrophobic pocket of TLR4/MD-2, experimental evidence confirming PA as a direct ligand for TLR4 is lacking [52].
On the other hand, certain lipids are known to activate TLR4 through lipid rafts on the cell membrane. Lipid rafts are enriched with saturated lipids (such as sphingomyelin and glycerophospholipids containing saturated fatty acids) and cholesterol. The addition of these lipids increases the lipid phase order, leading to the dimerization of TLR4 and enhanced signaling [53]. Therefore, it is speculated that the increase in glycerophospholipids containing saturated fatty acids, such as PA, upon PA addition may lead to activation of TLR4 through lipid rafts. However, unlike lipopolysaccharide (LPS), a known ligand for TLR4/MD-2, treatment with PA does not induce TLR4 dimerization or internalization as observed with LPS treatment [51].
Since GGA is not known to be incorporated into sphingomyelin or glycerophospholipids, it might be inserted into the plasma membrane as unchanged GGA. In that case, GGA might promote an increase in phase order of the plasma membrane, like cholesterol, as a rod-shaped isoprenoid, leading to activation of TLR4 through lipid rafts. However, further experimental studies are essential to establish this. There are currently no reports suggesting that TLR4 is essential for ATRA-induced cell death in HCC cells or that ATRA activates hepatic TLR4.
- Mitochondria: The effects on mitochondrial morphology and function are relatively similar among GGA, PA, and ATRA. Adding GGA to cultured HCC-derived cell lines leads to mitochondrial accumulation around the nucleus and fragmentation within 2 h [14]. Similarly, mitochondrial fragmentation is observed within 2 h of PA treatment [54]. ATRA treatment results in decreased mitochondrial number and average volume, as well as disappearance of mitochondrial cristae after 48 h [49]. Additionally, loss of ΔΨm is observed after 1 h of GGA (10 µM) treatment [18], 24-96 h of ATRA (1 µM) treatment [55], and 24 h of PA (200 µM) treatment [46], indicating mitochondrial permeability transition (mPT) (Table 1). Furthermore, hyperproduction of superoxide in mitochondria is confirmed after treatment with GGA (10µM, 15 min) [14], PA (500 µM, 3-24 h) [46,56], or ATRA (10 µM, 48 h) [49]. Free fatty acids like PA are proposed to induce superoxide production by directly binding to complexes I and III of the respiratory chain, inhibiting electron transfer and leading to increased superoxide production [57]. Although the signaling from TLR4 to ECSIT resulting in an impaired electron transport chain in mitochondria has been proposed for GGA (as mentioned earlier), the mechanism of increased superoxide production by ATRA remains unclear in the literature.
- Endoplasmic reticulum (ER): UPRER is a common response of HCC cells to treatment with these three fatty acids. Treatment with GGA (10-20 µM, 15-30 min) [15,16], PA (400-500 µM, 8-24 h) [44,56,58], or ATRA (10-20 µM, 1-8 h) [15] results in splicing of XBP1 mRNA, translocation of XBP1 protein to the nucleus, and increased expression of DDIT3 (CHOP) mRNA. Induction of UPRER by ATRA has also been observed in mouse embryonal carcinoma cell line P19, not limited to HCC cells [59]. Membrane phospholipids derived from PA are believed to affect the fluidity of the ER membrane and induce UPRER through the dimerization of IRE1 [60]. Since effective concentrations of GGA and ATRA are in the order of 10 µM and induce relatively rapid responses, they are likely mediated through signaling, although detailed mechanisms require further investigation.
- Nucleus: Translocation of NF-κB to the nucleus due to increased production of reactive oxygen species (ROS) is a well-known phenomenon [61]. Indeed, treatment with GGA induces translocation of NF-κB to the nucleus, which can be inhibited by co-treatment with the antioxidant α-tocopherol, or suppression of increased superoxide production in the mitochondria [16]. Additionally, GGA treatment promotes rapid translocation of the cytoplasmic p53 to the nucleus and increases expression of the PUMA gene, one of the p53 target genes [62]. Like GGA, PA also induces nuclear NF-κB activity in HepG2 [63,64] and HepaRG cells [64].
- Cytosol: An increase in cytosolic Ca2+ concentration is another common response of HCC cells to treatment with these three fatty acids (Table 1). However, the effective concentrations and time of occurrence of this effect differ. Treatment with 10 µM GGA leads to two increases in cytosolic Ca2+ concentration at 20 min and 6 h [16], while PA requires concentrations of 500-1000 µM, with an increase observed 6 h after treatment [46]. Similarly, ATRA requires 24-48 h after treatment to observe an increase in cytosolic Ca2+ concentration, despite effective concentrations being in the order of 10 µM [65]. The mechanism of increased cytosolic Ca2+ concentration by PA has been most extensively analyzed. PA enhances mitochondrial ROS (mtROS) production, which releases Ca2+ from lysosomes, resulting in increased cytosolic Ca2+ concentration and leading to mPT and cell death [46].
- Inflammatory extracellular vesicles: When hepatocytes and HCC cells are treated with PA, they secrete inflammatory extracellular vesicles [66,67]. It is believed that lysoPC, one of the intracellular metabolites of PA, is directly involved in the release of inflammatory extracellular vesicles [68]. It has been reported that one-tenth to one-twentieth lysoPC induces cell death in 24 h in primary human hepatocytes to the same extent as cell death by PA [69]. Considering that lysoPC increases upon GGA treatment of HCC cells, it is hypothesized that GGA treatment may lead to the release of inflammatory extracellular vesicles. In contrast, since lysoPC is reduced upon ATRA treatment, the release of extracellular vesicles is unlikely after ATRA treatment [70].
3.4. Role of Mitochondrial ROS Hyperproduction, UPRER, and Autophagy in GGA-Induced Cell Death
As mentioned earlier, the effects of the three fatty acids on human HCC cells can be broadly categorized into two types: pyroptosis induced by GGA and PA, and apoptosis induced by ATRA. Regarding increased mitochondrial ROS (mtROS) production and UPRER, similar phenomena occur with all three fatty acids. However, the most significant difference lies in the occurrence of autophagy. In the case of ATRA treatment, there is an increase in the flux of autophagy, accompanied by an increase in LC3-II and degradation of p62/SQSTM, which is the cargo of autophagosomes [48]. Moreover, intriguingly, the knockdown of the essential autophagy gene ATG7 leads to the induction of apoptosis by ATRA [48].
On the other hand, while the marker of autophagosomes, LC3-II increases with GGA and PA treatment, autophagic flux is inhibited, and p62/SQSTM increases, opposite to ATRA [14,71]. Data indicating abnormal accumulation of early autophagosomes and impaired formation of autolysosomes due to GGA treatment suggest an "incomplete response" of autophagy (or impaired autophagy) is involved in GGA-induced cell death [14]. Subsequently, autophagy has been shown to suppress pyroptosis. In other words, autophagy inhibits the formation of canonical and non-canonical inflammasomes by sequestering damaged mitochondria generating ROS and Gram-negative bacteria into autophagosomes, which are then cleared via autolysosomes [72]. Therefore, in the cases of GGA and PA treatment, incomplete autophagic responses or cessation of autophagy progression may lead to the inability to regulate inflammasomes, resulting in the execution of cell death by pyroptosis. Recent studies have reviewed the involvement of incomplete autophagic responses in PA lipotoxicity in hepatocytes [73] and adipocytes [74] other than HCC cells. In neurons, incomplete autophagic responses induced by PA are reported to lead to decreased insulin sensitivity [75].
How is autophagy progression impaired by GGA or PA treatment? Reports are suggesting that the treatment of macrophages primed by LPS, a TLR4 ligand, with PA, induces lysosomal dysfunction, which plays a crucial role in the lipotoxicity of PA [76]. Reports of decreased Ca2+ concentration in lysosomes upon PA treatment [46] are intriguing in this regard. MtROS activate the Ca2+ efflux channel of lysosomes. Since cytosolic Ca2+ concentration increases with GGA treatment, and autophagy halts at the stage of autophagosomes, the increase in cytosolic Ca2+ concentration is presumed to be related to lysosomal dysfunction. However, since mtROS production is also increased by ATRA treatment, it is unlikely that lysosomal dysfunction is caused solely by mtROS.
Additionally, Karasawa et al. [77] reported that lysosomal dysfunction caused by PA is due to the precipitation of PA-derived crystals within cells, and that lysosomal dysfunction induces inflammasome activation. Considering that the melting point (62.9°C) of PA is much higher than body temperature, they may be crystals of the PA itself. Co-treatment of PA with oleic acid, which has a much lower melting point (13.4°C), prevents the observation of intracellular crystals. However, the phenomenon of intracellular crystallization by PA treatment is observed in primary cultures of macrophages or macrophage-like cell lines but not in hepatocellular carcinoma cell lines [77]. Moreover, since GGA remains liquid even at room temperature, it is unlikely that GGA crystallizes within living cells after GGA treatment.
In any case, in the case of GGA or PA treatment, inhibition of autolysosome formation due to lysosomal dysfunction results in an incomplete autophagic response, causing autophagy to halt midway, and consequently, control over pyroptosis is lost.

Table 1.
Intracellular events of GGA-induced cell death in HCC cells vs. PA and ATRA.
| GGA | PA | ATRA | |
|---|---|---|---|
| Cytotoxicity |
|
|
|
| Mitochondrial morphology |
- a fragmentation of mitochondria (unpublished, Shidoji). |
|
- reduced mitochondrial volume (Sun 2022) [49]. - reduced or eliminated mitochondrial cristae (Sun 2022) [49]. ✧1 µM ATRA did not alter the mitochondrial mass for 24-72 h in the acute-promyelocytic-leukemia (APL)-derived NB4 cell line (Gianni 2022) [55]. |
| Mitochondrial membrane potential (ΔΨm) |
- a dissipation of MitoTracker Red fluorescence in 2 h (Okamoto 2011) [14]. |
|
✧1 µM ATRA reduced Mito-Tracker Red/Green fluorescence in NB4 cells for 24-96 h (Gianni 2022) [55]. |
| Mitochondrial ROS production |
- MitoSox Red fluorescence of hepatoma cells in 2 h, which was prevented with either 50 µM oleic acid (OA), 100 µM α-tocopherol or VIPER (Yabuta 2020) [16]. |
|
- lipid peroxides (C11-BODIPY fluorescence) (Sun 2022) [49]. |
| ER stress response (UPRER) |
- splicing of XBP1 mRNA in 15 min (Iwao 2015) [15], which was prevented by cotreatment with either 50 µM OA or VIPER, but not with 100 µM α-tocopherol (Yabuta 2020) [16]. - accumulation of XBP1 protein in the nucleus in 8 h (Iwao 2015) [15]. - upregulation of DDIT3 (CHOP) mRNA in 8 h (Iwao 2015), which was prevented by cotreatment with 50 µM OA or VIPER, but not with 100 µM α-tocopherol (Yabuta 2020) [16]. |
- upregulation of DDIT3 (CHOP) protein in 16 h (Qi 2015) [58]. - phosphorylation of IRE1α in 8 h (Qi 2015) [58].
|
|
| Pyroptosis |
- upregulation of TLR2 expression in 3 h, which was prevented with 50 µM OA or VIPER (Yabuta 2020) [16]. - upregulation of NLRP3 expression in 3 h, which was prevented with 50 µM OA, 100 µM α-tocopherol or VIPER (Yabuta 2020) [16]. - appearance of GSDMD-N terminal fragment in 1 h (Yabuta 2020) [16]. - localization of GSDMD to the plasma membrane in 3 h, which was prevented by co-treatment with VIPER (Yabuta 2020) [16].
|
|
|
| Cytoplasmic Ca2+ |
|
|
|
| LysoPLs |
- the most rapid (2 h) upregulation of lysoPC (C20:4) and lysophosphatidylethanolamine (C20:4) (Shidoji 2021) [19]. |
|
✧Liver lysoPC (16:1 or 18:1) was decreased by oral administration (10 mg/kg, 1 wk) of a synthetic ligand (AM580) for RAR to mice and increased by oral administration (30 mg/kg, 1 wk) of a synthetic ligand for RXR (LG268) to female C57BL6 mice (Weiss 2011) [70]. |
| Autophagy |
- time-dependent accumulation of LC3β-II and p62/SQSTM (Okamoto 2011) [14], which was not prevented by co-treatment with 4μ8C (IRE1 Inhibitor III) that inhibits XBP1 mRNA splicing (Iwao 2015) [15]. - upregulation of ATG4B and BECN1 in 30 min (Okamoto 2011) [14]. - nuclear translocation of cytoplasmic p53 in 3 h (Iwao 2014) [62]. |
|
|
| CASP3 |
|
|
|
➢: Human hepatoma-derived cell lines or rodent hepatocytes. Unless otherwise noted in the GGA section, all observations were obtained using human hepatoma cell lines such as HuH-7, PLC/PRF5, HepG2, and Hep3B. ✧: Cell lines other than those derived from hepatoma or rodent liver.
4. GGA as an Anti-Oncometabolite
Thus far, we have summarized the mechanism of GGA-induced cell death in HCC cells from the perspective of pyroptosis. Pyroptosis has traditionally been considered a cell-lytic and highly inflammatory form of regulated cell death, particularly studied in immune cells such as macrophages, serving as regulated cell death that activates the innate immune system [81,82,83]. However, studies have increasingly implicated pyroptosis as a major mechanism in the hepatocyte lipotoxicity induced by cholesterol and saturated fatty acids, highlighting the sterile pyroptosis mediated by these endogenous lipids as a mechanism of hepatocyte injury [78,84].
From our studies on HCC prevention, we found that GGA, classified as an acyclic retinoid, induces cell death via pyroptosis in HCC cell lines [16]. Furthermore, we demonstrated that GGA is an endogenous lipid de novo synthesized in the liver from mevalonic acid, rather than a vitamin-like acyclic retinoid [9,85]. Therefore, in this review, we have discussed the mechanism of cell death induction by endogenous lipid GGA, comparing it with saturated fatty acid PA and retinoid ATRA. As a result, GGA, like PA, induces cell death in HCC cells via pyroptosis, which is distinctly different from ATRA-induced apoptosis.
While this review has predominantly focused on data obtained from in vitro experiments regarding GGA, there are intriguing observations from in vivo experiments with GGA. In C3H/HeN male mice, liver GGA content begins to decrease from 7 months of age, decreases further by around 15 months of age, and becomes undetectable by 23 months of age [86]. By this time, almost all mice can be detected with spontaneous liver cancer [87,88]. However, when GGA or 4,5-didehydroGGA is administered orally as a single dose (50 µg/mouse) at 11-12 months of age, the detection rate of liver cancer at 24 months of age is significantly suppressed [86,88]. Since tumors are observed in the liver of C3H/HeN male mice from the first year of life {87}, it is conceivable that GGA administered at this time induces pyroptotic cell death in tumor cells, sweeping them away from the liver. In other words, GGA administered at this time may prevent the onset of liver cancer by plucking out its buds [9].
Metabolites that abnormally accumulate in cancer cells and promote tumorigenesis are called oncometabolites, and substances related to the tricarboxylic acid cycle such as 2-hydroxyglutarate, succinate, and fumarate are known examples [89]. These oncometabolites contribute to cancer-specific signatures such as epigenetic remodeling of tumor cells, abnormal DNA repair, and redox imbalance. On the other hand, although GGA is synthesized as a metabolite derived from mevalonic acid, in certain strains of mice, liver GGA content decreases with age, and eventually liver cancer spontaneously occurs. As already mentioned, GGA does not induce pyroptotic cell death or other cell death in normal hepatocytes [18]. Therefore, if the hepatic GGA content does not decrease with age, physiological hepatic GGA induces pyroptotic cell death only in tumor cells that are in the process of carcinogenesis and suppress the development of HCC. Compounds like GGA could be termed anti-oncometabolites (or tumor-suppressive metabolites).
5. Conclusions
Initially studied as one of the acyclic retinoids with HCC preventive effects [90], the mechanism of cell death induction in HCC cells by GGA appears to be more like the action of saturated fatty acid PA via UPRER and mitochondrial dysfunction rather than the action of retinoids mediated by nuclear retinoid receptors [91]. Induction of pyroptosis by PA occurs not only in HCC cells but also to a similar extent in hepatocytes, known as hepatic lipotoxicity [10]. However, induction of pyroptosis by GGA is selectively observed in HCC cells, and mitochondrial dysfunction or cell death is not observed in primary hepatocytes after treatment with GGA [18].
How does GGA induce cell death selectively in HCC cells without inducing cell death in hepatocytes? Originally studied in immune cells such as macrophages as an inflammatory form of cell death primarily in response to foreign substances, pyroptosis has recently gained attention as a sterile response in hepatocytes to the abnormal accumulation of endogenous lipids such as cholesterol, saturated fatty acids, and lysophospholipids. GGA, as an endogenous lipid that selectively induces pyroptosis in tumor cells, occupies a slightly different position from saturated fatty acids. If tumor cells are considered foreign substances, further detailed research on factors determining the selectivity of tumor cells in the liver, such as analyzing changes in the expression levels of the TLR4 genes accompanying the development of liver cancer [92], is essential for understanding the tumor-selective action of GGA.
Author Contributions
Conceptualization, Y.S.; methodology, Y.S.; software, Y.S.; validation, Y.S.; formal analysis, Y.S.; investigation, Y.S.; resources, Y.S.; data curation, Y.S.; writing—original draft preparation, Y.S.; writing—review and editing, Y.S.; visualization, Y.S.; supervision, Y.S.; project administration, Y.S.; funding acquisition, Y.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Acknowledgments
The author would like to appreciate Drs. Kyoko Okamoto, Takashi Muraguchi, Maiko Mitake, Chiharu Sakane, Shohei Shimonishi, Haruka Kamiyama, Chieko Iwao, Toshiya Yonekura, Suemi Yabuta, and Yuki Tabata for their dedicated contributions to GGA research in the author's laboratory.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fujimaki, Y. Formation of Gastric Carcinoma in Albino Rats Fed on Deficient Diets. J Cancer Res 1926, 10, 469–477. [Google Scholar] [CrossRef]
- Abels, J.C.; Gorham, A.T.; Pack, G.T.; Rhoads, C.P. Metabolic Studies in Patients with Cancer of the Gastro-Intestinal Tract. I. Plasma Vitamin A Levels in Patients with Malignant Neoplastic Disease, Particularly of the Gastro-Intestinal Tract 1. J Clin Invest 1941, 20, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Saffiotti, U.; Montesano, R.; Sellakumar, A.R.; Borg, S.A. Experimental Cancer of the Lung. Inhibition by Vitamin A of the Induction of Tracheobronchial Squamous Metaplasia and Squamous Cell Tumors. Cancer 1967, 20, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.K.; Sporn, M.B. Recent Advances in Chemoprevention of Cancer. Science (1979) 1997, 278, 1073–1077. [Google Scholar] [CrossRef]
- Hansen, L.A.; Sigman, C.C.; Andreola, F.; Ross, S.A.; Kelloff, G.J.; De Luca, L.M. Retinoids in Chemoprevention and Differentiation Therapy. Carcinogenesis 2000, 21, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Muto, Y.; Moriwaki, H.; Ninomiya, M.; Adachi, S.; Saito, A.; Takasaki, K.T.; Tanaka, T.; Tsurumi, K.; Okuno, M.; Tomita, E.; et al. Prevention of Second Primary Tumors by an Acyclic Retinoid, Polyprenoic Acid, in Patients with Hepatocellular Carcinoma. Hepatoma Prevention Study Group. N. Engl. J. Med. 1996, 334, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Muto, Y.; Moriwaki, H.; Omori, M. In Vitro Binding Affinity of Novel Synthetic Polyprenoids (Polyprenoic Acids) to Cellular Retinoid-Binding Proteins. Jpn J Cancer Res 1981, 72, 974–977. [Google Scholar]
- Araki, H.; Shidoji, Y.; Yamada, Y.; Moriwaki, H.; Muto, Y. Retinoid Agonist Activities of Synthetic Geranylgeranoic Acid Derivatives. Biochem Biophys Res Commun 1995, 209, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Shidoji, Y. Geranylgeranoic Acid, a Bioactive and Endogenous Fatty Acid in Mammals: A Review. J Lipid Res 2023, 64, 100396. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the Gut-Liver Axis in NASH Pathogenesis. J Hepatol 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Yu, S.J.; Lee, J.H.; Kim, H.Y.; Kim, Y.J. Reduction of Oxidative Stress Attenuates Lipoapoptosis Exacerbated by Hypoxia in Human Hepatocytes. Int J Mol Sci 2015, 16, 3323–3334. [Google Scholar] [CrossRef] [PubMed]
- Matsufuji, S.; Kitajima, Y.; Higure, K.; Kimura, N.; Maeda, S.; Yamada, K.; Ito, K.; Tanaka, T.; Kai, K.; Noshiro, H. A HIF-1α Inhibitor Combined with Palmitic Acid and L-Carnitine Treatment Can Prevent the Fat Metabolic Reprogramming under Hypoxia and Induce Apoptosis in Hepatocellular Carcinoma Cells. Cancer Metab 2023, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Sakane, C.; Shidoji, Y. Reversible Upregulation of Tropomyosin-Related Kinase Receptor B by Geranylgeranoic Acid in Human Neuroblastoma SH-SY5Y Cells. J Neurooncol 2011, 104, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Sakimoto, Y.; Imai, K.; Senoo, H.; Shidoji, Y. Induction of an Incomplete Autophagic Response by Cancer-Preventive Geranylgeranoic Acid (GGA) in a Human Hepatoma-Derived Cell Line. Biochem J 2011, 440, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Iwao, C.; Shidoji, Y. Polyunsaturated Branched-Chain Fatty Acid Geranylgeranoic Acid Induces Unfolded Protein Response in Human Hepatoma Cells. PLoS One 2015, 10, e0132761. [Google Scholar] [CrossRef] [PubMed]
- Yabuta, S.; Shidoji, Y. TLR4-Mediated Pyroptosis in Human Hepatoma-Derived HuH-7 Cells Induced by a Branched-Chain Polyunsaturated Fatty Acid, Geranylgeranoic Acid. Biosci Rep 2020, 40, BSR20194118. [Google Scholar] [CrossRef] [PubMed]
- Shimonishi, S.; Muraguchi, T.; Mitake, M.; Sakane, C.; Okamoto, K.; Shidoji, Y. Rapid Downregulation of Cyclin D1 Induced by Geranylgeranoic Acid in Human Hepatoma Cells. Nutr Cancer 2012, 64, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Shidoji, Y.; Nakamura, N.; Moriwaki, H.; Muto, Y. Rapid Loss in the Mitochondrial Membrane Potential during Geranylgeranoic Acid-Induced Apoptosis. Biochem Biophys Res Commun 1997, 230, 58–63. [Google Scholar] [CrossRef]
- Shidoji, Y.; Iwao, C. A Rapid Increase in Lysophospholipids after Geranylgeranoic Acid Treatment in Human Hepatoma-Derived HuH-7 Cells Revealed by Metabolomics Analysis. Biochem Biophys Rep 2021, 28, 101176. [Google Scholar] [CrossRef]
- Smith, J.A.; Turner, M.J.; Delay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic Reticulum Stress and the Unfolded Protein Response Are Linked to Synergistic IFN-β Induction via X-Box Binding Protein 1. Eur J Immunol 2008, 38, 1194–1203. [Google Scholar] [CrossRef]
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR Activation of the Transcription Factor XBP1 Regulates Innate Immune Responses in Macrophages. Nat Immunol 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Savic, S.; Ouboussad, L.; Dickie, L.J.; Geiler, J.; Wong, C.; Doody, G.M.; Churchman, S.M.; Ponchel, F.; Emery, P.; Cook, G.P.; et al. TLR Dependent XBP-1 Activation Induces an Autocrine Loop in Rheumatoid Arthritis Synoviocytes. J Autoimmun 2014, 50, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Boukeileh, S.; Darawshi, O.; Shmuel, M.; Mahameed, M.; Wilhelm, T.; Dipta, P.; Forno, F.; Praveen, B.; Huber, M.; Levi-Schaffer, F.; et al. Endoplasmic Reticulum Homeostasis Regulates TLR4 Expression and Signaling in Mast Cells. Int J Mol Sci 2022, 23, 11826. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of Caspase-4 in Endoplasmic Reticulum Stress-Induced Apoptosis and Aβ-Induced Cell Death. Journal of Cell Biology 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Yukioka, F.; Matsuzaki, S.; Kawamoto, K.; Koyama, Y.; Hitomi, J.; Katayama, T.; Tohyama, M. Presenilin-1 Mutation Activates the Signaling Pathway of Caspase-4 in Endoplasmic Reticulum Stress-Induced Apoptosis. Neurochem Int 2008, 52, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.M.; Elner, S.G.; Elner, V.M. Dual Involvement of Caspase-4 in Inflammatory and ER Stress-Induced Apoptotic Responses in Human Retinal Pigment Epithelial Cells. Invest Ophthalmol Vis Sci 2009, 50, 6006–6014. [Google Scholar] [CrossRef] [PubMed]
- Silva Barcelos, E.C.; Rompietti, C.; Adamo, F.M.; Dorillo, E.; De Falco, F.; Del Papa, B.; Baldoni, S.; Nogarotto, M.; Esposito, A.; Capoccia, S.; et al. NOTCH1-Mutated Chronic Lymphocytic Leukemia Displays High Endoplasmic Reticulum Stress Response with Druggable Potential. Front Oncol 2023, 13, 1218989. [Google Scholar] [CrossRef] [PubMed]
- Correia da Silva, D.; Valentão, P.; Pereira, D.M. A Survey of Naturally Occurring Molecules as New Endoplasmic Reticulum Stress Activators with Selective Anticancer Activity. Cancers (Basel) 2022, 15, 293. [Google Scholar] [CrossRef]
- Chukai, Y.; Ito, G.; Konno, M.; Sakata, Y.; Ozaki, T. Mitochondrial Calpain-5 Truncates Caspase-4 during Endoplasmic Reticulum Stress. Biochem Biophys Res Commun 2022, 608, 156–162. [Google Scholar] [CrossRef]
- Brewer, J.W.; Diehl, J.A. PERK Mediates Cell-Cycle Exit during the Mammalian Unfolded Protein Response. Proc Nat Acad Sci, USA 2000, 97, 12625–12630. [Google Scholar] [CrossRef]
- Vogel, R.O.; Janssen, R.J.R.J.; Van Den Brand, M.A.M.; Dieteren, C.E.J.; Verkaart, S.; Koopman, W.J.H.; Willems, P.H.G.M.; Pluk, W.; Van Den Heuvel, L.P.W.J.; Smeitink, J.A.M.; et al. Cytosolic Signaling Protein Ecsit Also Localizes to Mitochondria Where It Interacts with Chaperone NDUFAF1 and Functions in Complex I Assembly. Genes Dev 2007, 21, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F.R.G.; Lepelley, A.; Seeley, J.J.; Hayden, M.S.; Ghosh, S. An Essential Role for ECSIT in Mitochondrial Complex I Assembly and Mitophagy in Macrophages. Cell Rep 2018, 22, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- McGregor, L.; Acajjaoui, S.; Desfosses, A.; Saïdi, M.; Bacia-Verloop, M.; Schwarz, J.J.; Juyoux, P.; von Velsen, J.; Bowler, M.W.; McCarthy, A.A.; et al. The Assembly of the Mitochondrial Complex I Assembly Complex Uncovers a Redox Pathway Coordination. Nat Commun 2023, 14, 8248. [Google Scholar] [CrossRef] [PubMed]
- Silwal, P.; Kim, J.K.; Kim, Y.J.; Jo, E.K. Mitochondrial Reactive Oxygen Species: Double-Edged Weapon in Host Defense and Pathological Inflammation during Infection. Front Immunol 2020, 11, 1649. [Google Scholar] [CrossRef] [PubMed]
- Horng, T. Calcium Signaling and Mitochondrial Destabilization in the Triggering of the NLRP3 Inflammasome. Trends Immunol 2014, 35, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Li, M.; Yangzhong, X.; Zhang, X.; Zu, A.; Hou, Y.; Li, L.; Sun, S. Pyroptosis in Inflammation-Related Respiratory Disease. J Physiol Biochem 2022, 78, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cai, J.; Tang, Y.; Lu, B. The Noncanonical Inflammasome-Induced Pyroptosis and Septic Shock. Semin Immunol. 2023, 70, 101844. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-L.; Wu, W.-S.; Tyan, Y.-S.; Chou, C.-K. Retinoic Acid-Induced Apoptosis Is Prevented by Serum Albumin and Enhanced by Lipiodol in Human Hepatoma Hep3B Cells. Cancer Letter 1998, 129, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Hu, C.; Xu, L.; Cui, J.; Tao, L.; Gong, M.; Wang, Y.; He, Y.; He, T.; Bi, Y. All-Trans-Retinoic Acid Inhibits the Malignant Behaviors of Hepatocarcinoma Cells by Regulating Autophagy. Am J Transl Res 2020, 12, 6793–6810. [Google Scholar]
- Nakamura, N.; Shidoji, Y.; Yamada, Y.; Hatakeyama, H.; Moriwaki, H.; Muto, Y. Induction of Apoptosis by Acyclic Retinoid in the Human Hepatoma-Derived Cell Line, HuH-7. Bioche. Biphys. Res. Commun. 1995, 270, 382–388. [Google Scholar] [CrossRef]
- Lin, L.-M.; Li, B.-X.; Xiao, J.-B.; Lin, D.-H.; Yang ELSEVIER, B.-F.; Yang, B.-F. Synergistic Effect of All-Trans-Retinoic Acid and Arsenic Trioxide on Growth Inhibition and Apoptosis in Human Hepatoma, Breast Cancer, and Lung Cancer Cells in Vitro. World J Gastroenterol 2005, 11, 5633–5637. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, G.; Ding, C.L.; Zhao, L.J.; Zhao, P.; Ren, H.; Qi, Z.T. All-Trans Retinoic Acid Protects Hepatocellular Carcinoma Cells against Serum-Starvation-Induced Cell Death by Upregulating Collagen 8A2. FEBS Journal 2013, 280, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zeng, X.; Liu, Z.; Chen, X.; Li, L.; Luo, R.; Liu, X.; Zhang, J.; Liu, J.; Lu, Y.; et al. Mesenchymal Stromal Cells Protect Hepatocytes from Lipotoxicity through Alleviation of Endoplasmic Reticulum Stress by Restoring SERCA Activity. J Cell Mol Med 2021, 25, 2976–2993. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhu, M.; Liu, X.; Chen, X.; Yuan, Y.; Li, L.; Liu, J.; Lu, Y.; Cheng, J.; Chen, Y. Oleic Acid Ameliorates Palmitic Acid Induced Hepatocellular Lipotoxicity by Inhibition of ER Stress and Pyroptosis. Nutr Metab (Lond) 2020, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Verma, R.; Kumar, C.; Kumar, B.; Dey, A.K.; Surjit, M.; Mylavarapu, S.V.S.; Maiti, T.K. Proteomic Analysis Reveals USP7 as a Novel Regulator of Palmitic Acid-Induced Hepatocellular Carcinoma Cell Death. Cell Death Dis 2022, 13, 563. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Hwang, Y.; Hur, K.Y.; Lee, M.S. Lysosomal Ca2+ as a Mediator of Palmitate-Induced Lipotoxicity. Cell Death Discov 2023, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Kanki, K.; Akechi, Y.; Ueda, C.; Tsuchiya, H.; Shimizu, H.; Ishijima, N.; Toriguchi, K.; Hatano, E.; Endo, K.; Hirooka, Y.; et al. Biological and Clinical Implications of Retinoic Acid-Responsive Genes in Human Hepatocellular Carcinoma Cells. J Hepatol 2013, 59, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Feng, R.; Shi, Y.; Chen, D.; Weng, H.; Ding, H.; Zhang, C. Intracellular Alpha-Fetoprotein Interferes with All-Trans Retinoic Acid Induced ATG7 Expression and Autophagy in Hepatocellular Carcinoma Cells. Sci Rep 2021, 11, 2146. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; He, Y.; Tong, J.; Liu, D.; Zhang, H.; He, T.; Bi, Y. All-Trans Retinoic Acid Inhibits the Malignant Behaviors of Hepatocarcinoma Cells by Regulating Ferroptosis. Genes Dis 2022, 9, 1742–1756. [Google Scholar] [CrossRef]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Vitamin A Metabolites Inhibit Ferroptosis. Biomedicine and Pharmacotherapy 2023, 164. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence That TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab 2018, 27, 1096–1110. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, D.A.; Zhang, K.; Hung, C.; Glasgow, S.; Aruni, A.W.; Unternaehrer, J.; Payne, K.J.; Langridge, W.H.R.; De Leon, M. Palmitic Acid Is a Toll-like Receptor 4 Ligand That Induces Human Dendritic Cell Secretion of IL-1β. PLoS One 2017, 12, e0176793. [Google Scholar] [CrossRef] [PubMed]
- Gianfrancesco, M.A.; Paquot, N.; Piette, J.; Legrand-Poels, S. Lipid Bilayer Stress in Obesity-Linked Inflammatory and Metabolic Disorders. Biochem Pharmacol 2018, 153, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Eynaudi, A.; Díaz-Castro, F.; Bórquez, J.C.; Bravo-Sagua, R.; Parra, V.; Troncoso, R. Differential Effects of Oleic and Palmitic Acids on Lipid Droplet-Mitochondria Interaction in the Hepatic Cell Line HepG2. Front Nutr 2021, 8, 775382. [Google Scholar] [CrossRef] [PubMed]
- Gianni, M.; Goracci, L.; Schlaefli, A.; Di Veroli, A.; Kurosaki, M.; Guarrera, L.; Bolis, M.; Foglia, M.; Lupi, M.; Tschan, M.P.; et al. Role of Cardiolipins, Mitochondria, and Autophagy in the Differentiation Process Activated by All-Trans Retinoic Acid in Acute Promyelocytic Leukemia. Cell Death Dis 2022, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Chen, H.-W.; Lo, C.-W.; Wang, Y.-R.; Li, C.-C.; Liu, K.-L.; Lii, C.-K. Luteolin Ameliorates Palmitate-Induced Lipotoxicity in Hepatocytes by Mediating Endoplasmic Reticulum Stress and Autophagy. Food and Chemical Toxicology 2023, 171, 113554. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Wojtczak, L. Fatty Acids as Modulators of the Cellular Production of Reactive Oxygen Species. Free Radic Biol Med 2008, 45, 231–241. [Google Scholar] [CrossRef]
- Qi, Y.; Wang, W.; Chen, J.; Dai, L.; Kaczorowski, D.; Gao, X.; Xia, P. Sphingosine Kinase 1 Protects Hepatocytes from Lipotoxicity via Down-Regulation of IRE1α Protein Expression. Journal of Biological Chemistry 2015, 290, 23282–23290. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Mimori, S.; Okuma, Y.; Kawada, K. Sel1l May Contributes to the Determinants of Neuronal Lineage and Neuronal Maturation Regardless of Hrd1 via Atf6-Sel1l Signaling Affiliations. Neurochem. Res. 2023, 48, 263–272. [Google Scholar] [CrossRef]
- Kitai, Y.; Ariyama, H.; Kono, N.; Oikawa, D.; Iwawaki, T.; Arai, H. Membrane Lipid Saturation Activates IRE1α without Inducing Clustering. Genes to Cells 2013, 18, 798–809. [Google Scholar] [CrossRef]
- Gloire, G.; Piette, J. Redox Regulation of Nuclear Post-Translational Modifications during NF-ΚB Activation. Antioxid Redox Signal 2009, 11, 2209–2222. [Google Scholar] [CrossRef] [PubMed]
- Iwao, C.; Shidoji, Y. Induction of Nuclear Translocation of Mutant Cytoplasmic P53 by Geranylgeranoic Acid in a Human Hepatoma Cell Line. Sci Rep 2014, 4, 4419. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic Acid Induces Production of Proinflammatory Cytokine Interleukin-8 from Hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 Signaling in Obese NAFLD. Am J Physiol Gastrointest Liver Physiol 2015, 309, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ye, C.; Liu, F.; Wang, W. All-Trans Retinoic Acid and Arsenic Trioxide Induce Apoptosis and Modulate Intracellular Concentrations of Calcium in Hepatocellular Carcinoma Cells. Journal of Chemotherapy 2014, 26, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles from Hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, A.; Chen, X.; Zhang, Y.; George, J.; Huang, M. bo; Bond, V.; Thompson, W.; Zhao, X. Saturated Fatty Acid Stimulates Production of Extracellular Vesicles by Renal Tubular Epithelial Cells. Mol Cell Biochem 2019, 458, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Lipotoxicity: Many Roads to Cell Dysfunction and Cell Death Lipotoxic Lethal and Sublethal Stress Signaling in Hepatocytes: Relevance to NASH Pathogenesis. J Lipid Res 2016, 57, 1758–1770. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Park, S.Y.; Shinzawa, K.; Kim, S.; Kun, W.C.; Lee, J.H.; Choon, H.K.; Lee, K.W.; Lee, J.H.; Cheol, K.P.; et al. Lysophosphatidylcholine as a Death Effector in the Lipoapoptosis of Hepatocytes. J Lipid Res 2008, 49, 84–97. [Google Scholar] [CrossRef]
- Weiss, K.; Mihály, J.; Liebisch, G.; Marosvölgyi, T.; Schmitz, G.; Decsi, T.; Rühl, R. Effect of Synthetic Ligands of PPAR α, β/δ, γ, RAR, RXR and LXR on the Fatty Acid Composition of Phospholipids in Mice. Lipids 2011, 46, 1013–1020. [Google Scholar] [CrossRef]
- Frietze, K.K.; Brown, A.M.; Das, D.; Franks, R.G.; Cunningham, J.L.; Hayward, M.; Nickels, J.T. Lipotoxicity Reduces DDX58/Rig-1 Expression and Activity Leading to Impaired Autophagy and Cell Death. Autophagy 2022, 18, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Takahama, M.; Akira, S.; Saitoh, T. Autophagy Limits Activation of the Inflammasomes. Immunol Rev 2018, 281, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.T.; Soh, N.J.H.; Chang, H.C.; Yu, V.C. P62/SQSTM1 in Liver Diseases: The Usual Suspect with Multifarious Identities. FEBS Journal 2023, 290, 892–912. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, K.; Gajewska, M. Fatty Acids as Potent Modulators of Autophagy Activity in White Adipose Tissue. Biomolecules 2023, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Cáceres, M.P.; Toledo-Valenzuela, L.; Díaz-Castro, F.; Ávalos, Y.; Burgos, P.; Narro, C.; Peña-Oyarzun, D.; Espinoza-Caicedo, J.; Cifuentes-Araneda, F.; Navarro-Aguad, F.; et al. Palmitic Acid Reduces the Autophagic Flux and Insulin Sensitivity through the Activation of the Free Fatty Acid Receptor 1 (FFAR1) in the Hypothalamic Neuronal Cell Line N43/5. Front Endocrinol (Lausanne) 2019, 10, 176. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Schilling, J.D. Lysosomes Integrate Metabolic-Inflammatory Cross-Talk in Primary Macrophage Inflammasome Activation. Journal of Biological Chemistry 2014, 289, 9158–9171. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Kasahara, T.; Takahashi, M.; Karasawa, T.; Kawashima, A.; Usui-Kawanishi, F.; Watanabe, S.; Kimura, H.; Kamata, R.; Shirasuna, K.; et al. Saturated Fatty Acids Undergo Intracellular Crystallization and Activate the NLRP3 Inflammasome in Macrophages. Arterioscler Thromb Vasc Biol 2018, 38, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Liu, J.; Cui, Q.; Jiang, T.; Xie, X.; Du, X.; Zhao, Z.; Lai, B.; Xiao, L.; Wang, N. CCN1/Integrin A5β1 Instigates Free Fatty Acid-Induced Hepatocyte Lipid Accumulation and Pyroptosis through NLRP3 Inflammasome Activation. Nutrients 2022, 14, 3871. [Google Scholar] [CrossRef]
- Kakisaka, K.; Cazanave, S.C.; Fingas, C.D.; Guicciardi, M.E.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Gores, G.J. Mechanisms of Lysophosphatidylcholine-Induced Hepatocyte Lipoapoptosis. Am J Physiol Gastrointest Liver Physiol 2012, 302, 77–84. [Google Scholar] [CrossRef]
- Chen, H.; Ma, J.; Liu, J.; Dou, L.; Shen, T.; Zuo, H.; Xu, F.; Zhao, L.; Tang, W.; Man, Y.; et al. Lysophosphatidylcholine Disrupts Cell Adhesion and Induces Anoikis in Hepatocytes. FEBS Lett 2022, 596, 510–525. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic Cell Death Defends against Intracellular Pathogens. Immunol Rev 2015, 265, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Liu, M.; Yang, F.; Liu, G.; Liu, J.; Zhao, W.; Ma, S.; Duan, Z. Programmed Cell Death and Lipid Metabolism of Macrophages in NAFLD. Front Immunol 2023, 14, 1118449. [Google Scholar] [CrossRef]
- Bourne, C.M.; Taabazuing, C.Y. Harnessing Pyroptosis for Cancer Immunotherapy. Cells 2024, 13, 346. [Google Scholar] [CrossRef] [PubMed]
- Horn, C.L.; Morales, A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol Commun 2022, 6, 12–35. [Google Scholar] [CrossRef] [PubMed]
- Shidoji, Y.; Tabata, Y. Unequivocal Evidence for Endogenous Geranylgeranoic Acid Biosynthesized from Mevalonate in Mammalian Cells. J Lipid Res 2019, 60, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Tabata, Y.; Omori, M.; Shidoji, Y. Age-Dependent Decrease in Hepatic Geranylgeranoic Acid Content in C3H/HeN Mice and Its Oral Supplementation Prevents Spontaneous Hepatoma. Metabolites 2021, 11, 634. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Mitsuoka, T. Effect of Dietary Phenobarbital on Spontaneous Hepatic Tumorigenesis in Germfree C3H/He Male Mice. Cancer Lett 1988, 39, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Omori, M.; Shidoji, Y.; Moriwaki, H. Inhibition of Spontaneous Hepatocarcinogenesis by 4,5-Didehydrogeranylgeranoic Acid: Effects of Small-Dose and Infrequent Administration. International Journal of Translational Medicine 2023, 3, 487–495. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, C. Oncometabolites in Cancer: Current Understanding and Challenges. Cancer Res 2021, 81, 2820–2823. [Google Scholar] [CrossRef]
- Yamada, Y.; Shidoji, Y.; Fukutomi, Y.; Ishikawa, T.; Kaneko, T.; Nakagama, H.; Imawari, M.; Moriwaki, H.; Muto, Y. Positive and Negative Regulations of Albumin Gene Expression by Retinoids in Human Hepatoma Cell Lines. Mol Carcinog 1994, 10, 151–158. [Google Scholar] [CrossRef]
- Brown, G. Targeting the Retinoic Acid Pathway to Eradicate Cancer Stem Cells. Int J Mol Sci 2023, 24, 2373. [Google Scholar] [CrossRef] [PubMed]
- Papadakos, S.P.; Arvanitakis, K.; Stergiou, I.E.; Vallilas, C.; Sougioultzis, S.; Germanidis, G.; Theocharis, S. Interplay of Extracellular Vesicles and TLR4 Signaling in Hepatocellular Carcinoma Pathophysiology and Therapeutics. Pharmaceutics 2023, 15, 2460. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Intracellular processes of GGA-induced cell death in HuH-7 cells. Black T-shaped arrows indicate the site of inhibitor action.
Figure 1.
Intracellular processes of GGA-induced cell death in HuH-7 cells. Black T-shaped arrows indicate the site of inhibitor action.
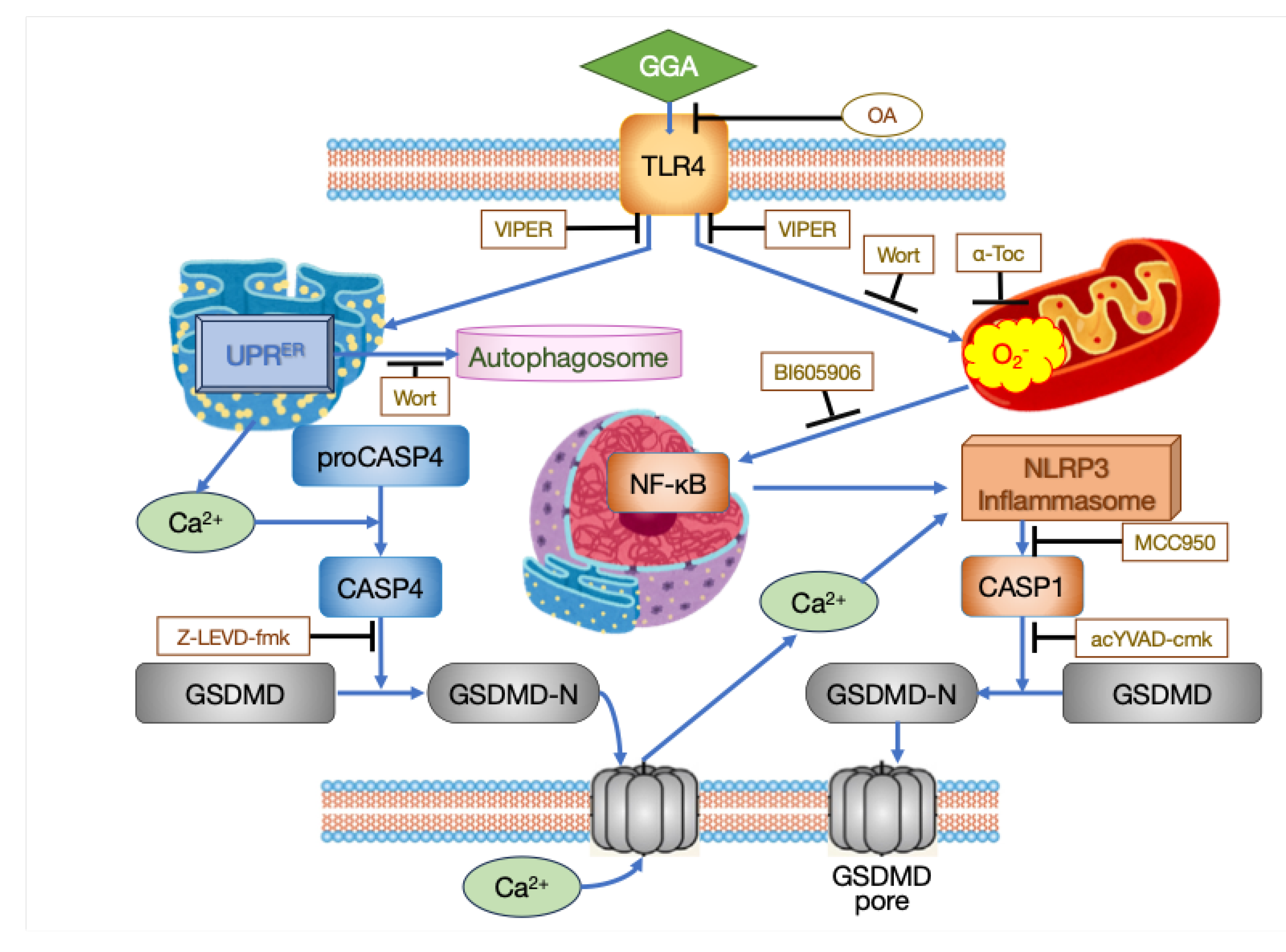
Figure 2.
Three fatty acids reported to induce cell death in hepatocellular carcinoma (HCC)-derived cell lines.
Figure 2.
Three fatty acids reported to induce cell death in hepatocellular carcinoma (HCC)-derived cell lines.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



