
Submitted:
04 April 2024
Posted:
05 April 2024
You are already at the latest version
Abstract
Rice, as a staple crop feeding billions, faces constant threats from various diseases jeopardizing global food security. Precise understanding of disease resistance mechanisms is crucial for developing resilient rice varieties. Traditional genetic mapping methods, such as QTL mapping, provide valuable insights into the genetic basis of diseases. However, the complex nature of rice diseases demands a holistic approach to gain accurate knowledge of it. Omics technologies, including genomics, transcriptomics, proteomics, and metabolomics, enable a comprehensive analysis of biological molecules, uncovering intricate molecular interactions within the rice plant. Integration of various mapping techniques using multi-omics data has revolutionized our understanding of rice disease resistance. By overlaying genetic maps with high-throughput omics datasets, researchers can pinpoint specific genes, proteins, or metabolites associated with disease resistance. This integration enhances the precision of disease-related biomarkers with better understanding of their functional roles in disease resistance. Improvement of rice breeding for disease resistance through this integration represents a significant stride in agricultural science because better understanding of the molecular intricacies and interactions underlying disease resistance architecture leads to more precise and efficient development of resilient and productive rice varieties. In this review, we explore how the integration of mapping and omics data can give a transformative impact on rice breeding for enhancing disease resistance.
Keywords:
Oryza sativa
; disease resistance
; QTL mapping
; omics
; integrative approach
1. Introduction
Rice diseases are caused by fungi, bacteria, viruses, and other types of pathogens. Common diseases include blast, sheath blight, bacterial leaf blight, and tungro. Each disease has distinct symptoms and affects different parts of the rice plant, leading to yield losses if not managed effectively [1]. Understanding the intricate interactions between rice plants and pathogens is essential for developing more advanced and sophisticated disease management strategies, targeting specific host or pathogen elements important for susceptibility or resistance. Plant pathogens recognize specific signals from host plants, triggering the initiation of infection process, and they employ diverse strategies to breach the plant's physical barriers. Fungi, for example, produce enzymes to degrade plant cell walls, facilitating penetration. Bacteria use specialized secretion systems to deliver effector molecules into plant cells, modulating host responses and enabling invasion [2]. Secreted effector molecules (mostly proteins) manipulate host cellular processes and interfere with defense responses of host plants, suppressing disease resistance. They may also mimic plant molecules to evade pathogen detection by the host plant. In other words, effectors play a central role in the establishment of infections by subverting plant immunity and creating a conducive environment for pathogen proliferation [3]. In response, plants have evolved sophisticated immune systems to counter pathogen attacks. Pattern Recognition Receptors (PRRs) detect conserved pathogen-associated molecular patterns (PAMPs), initiating PAMP-Triggered Immunity (PTI). Pathogens counter PTI with effectors, leading to Effector-Triggered Immunity (ETI), a robust defense mechanism of plants. The balance between pathogen effectors and plant immune responses determines the outcome of the infection [4]. Rice possesses a variety of genes that confer resistance to specific pathogens. These genes, often referred to as R genes, produce proteins that recognize pathogen molecules, activating the plant's defense mechanisms.
In general, there are two types of resistance in plants. While qualitative resistance is typically controlled by specific R genes and leads to a discrete resistant or susceptible outcome, quantitative resistance involves multiple genes and results in a spectrum of resistance levels. In rice, quantitative resistance is often more durable than qualitative resistance, as it involves a complex interplay of various genes, enhancing the plant's ability to withstand diverse pathogen populations over time [5]. Combining quantitative resistance with major R genes has proven to be a valuable approach for extending the effectiveness of major genes or durability of resistance in rice [6]. In this article, we will discuss how we can achieve a holistic understanding of rice disease resistance through integration of multi-omics approaches and, ultimately, translate it into the innovation of rice breeding for increasing disease resistance.
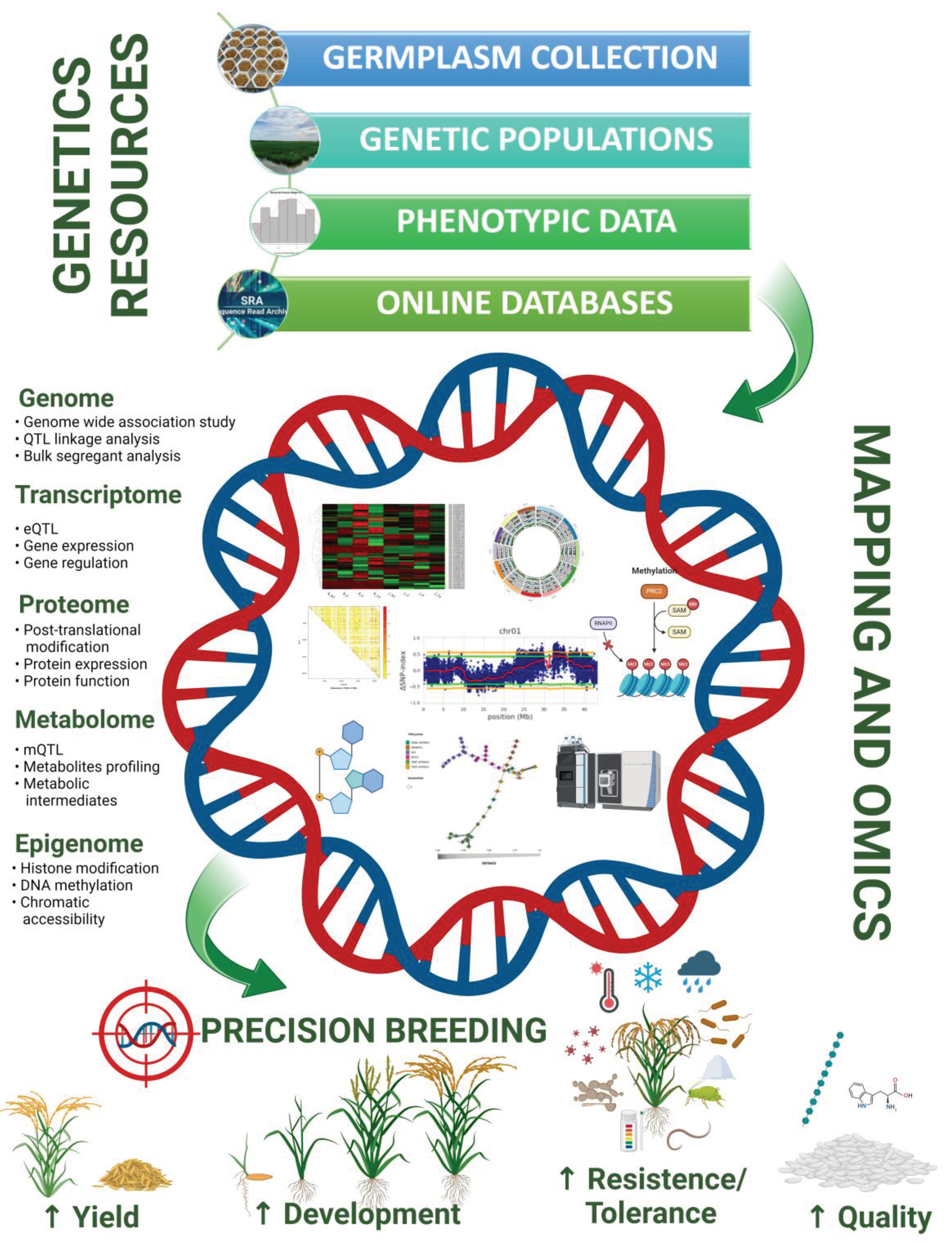
As technology continues to advance, the future holds promising prospects for even more precise and efficient methods in characterizing disease resistance mechanism. As research progresses, the integration of mapping with other omics approaches is poised to further deepen our understanding of the genetic underpinnings of complex disease-related traits. Understanding the complex interactions between plants and pathogens at the molecular level is crucial for developing effective disease-resistant crops. Omics technologies, including genomics, transcriptomics, proteomics, and metabolomics, have revolutionized our ability to decode the intricate mechanisms underlying plant disease resistance in addition to other complex agronomic traits in rice breeding (Figure 1). Hence, we will briefly review how various omics studies have contributed to expanding our knowledge of plant disease resistance and how integrative multi-omics studies have been performed with other plant systems.
Figure 1.
Utilizing genetic and genomic resources for rice breeding enables targeted selection of traits, such as disease resistance, yield potential, quality, and abiotic stress tolerance, leading to the development of improved rice varieties with enhanced productivity and resilience. Some images in this figure are from ‘BioRender.com’.
Figure 1.
Utilizing genetic and genomic resources for rice breeding enables targeted selection of traits, such as disease resistance, yield potential, quality, and abiotic stress tolerance, leading to the development of improved rice varieties with enhanced productivity and resilience. Some images in this figure are from ‘BioRender.com’.
2. Omics for Decoding Resistance Mechanisms
2.1. Harnessing Genomics for Enhancing Rice Disease Resistance
QTL linkage mapping is one of methods for identifying genomic regions involving the construction of genetic maps that link phenotypic traits, such as disease resistance, to molecular markers or genes located on several or specific chromosomes. The breakthrough in the characterization of quantitative traits to select for QTLs was the development of molecular markers used for construction of linkage maps for diverse crop species [7]. Linkage maps have been utilized for identifying chromosomal regions that contain genes controlling simple traits or quantitative traits governed by QTL [8]. Advantages of linkage mapping include high statistical power to effectively identify regions of genome associated with the target trait [9]; trait specific, which allows to map complex traits like disease resistance [10,11]; and identification of linkage with functional variation such as genetic polymorphism, gene expression changes or epigenetic modifications, which can provide valuable information on the underlying mechanisms behind the trait variation [8]. Lastly, the method is suitable for diverse populations such as F2, backcross, and recombinant inbred lines (RILs) [8]. However, QTL studies necessitate large sample sizes due to their reliance on statistical power to detect small genetic effects and can only map differences observed between parents, as it is improbable for every genetic locus contributing to a variation to harbor segregating alleles of major effect within the populations [10,12]. Also, the limited genetic diversity within a biparental population may not fully capture the complexity of trait variation and restrict the detection of QTLs present in a broader genetic backgorund [12].
The transition from QTL mapping to Genome-Wide Association Studies (GWAS) represents a pivotal shift in the field of genetics research. This transition signifies a more comprehensive and high-resolution approach, enabling the detection of subtle genetic variations linked to complex traits. GWAS have emerged as pivotal tools in unravelling the genetic basis of plant disease resistance. It is a powerful genetic investigation aiming to identify associations between specific genetic variations, such as single nucleotide polymorphisms (SNPs), and particular traits, such as disease resistance, within a population [13]. Advanced statistical methods are employed to assess the frequency of genetic variations relative to controls to pinpoint variations that are significantly more prevalent in individuals with the trait of interest [14]. By examining genomic datasets, GWAS enable researchers to identify specific genetic variations associated with resistance traits, paving the way for more precise and effective crop breeding strategies to increase disease resistance. Studies have successfully pinpointed genomic regions associated with disease resistance, aiding in the development of resistant crop varieties. For instance, a study highlighted the power of GWAS in identifying genetic loci for resistance to bacterial leaf streak and rice black-streaked dwarf virus (RBSDV) [15,16]. A comprehensive GWAS was conducted on 236 diverse rice accessions, predominantly indica varieties, revealing 12 QTLs across chromosomes 1, 2, 3, 4, 5, 8, 9, and 11 that confer resistance against five distinct Thai isolates of Xanthomonas oryzae pv. oryzicola (Xoc) [15]. Notably, five QTLs exhibited resistance against multiple Xoc isolates, with qBLS5.1 and qBLS2.3 and xa5 gene being highlighted as a potential candidate gene associated with qBLS5.1, while three genes for, pectinesterase inhibitor (OsPEI), eukaryotic zinc-binding protein (OsRAR1), and NDP epimerase function were proposed as candidate genes for qBLS2.3 [15]. Identifion of these influential genetic factors associated with broad-spectrum resistance potential highlightsit’s the significance of GWAS in rice breeding programs targeting BLS resistance. In addition, a study evaluated RBSDV resistance in 1,953 rice accessions over three years, revealing lower disease incidences in the Xian/indica (XI) subgroup compared to the Geng/japonica (GJ) subgroup, where single-locus GWAS, which scrutinized individual variants at specific genomic sites, identified ten genomic regions [16]. Additionally, a multilocus GWAS which considering multiple genetic variants across genome, pinpointed five genomic regions linked to RBSDV resistance[16]. From reported regions, grRBSDV-6.1 and grRBSDV-6.3, haplotype analysis indicated that specific candidate genes, LOC_Os06g03150 in grRBSDV-6.1 and LOC_Os06g31190 in grRBSDV-6.3 , were associated with resistance differentiation in addition to three novel resistance regions (grRBSDV-1.1, grRBSDV-7.1, and grRBSDV-9.1) identified [16]. These findings provide valuable insights for breeding RBSDV-resistant rice varieties and serves as a compelling demonstration of the efficacy of GWAS in deciphering the genetic architecture of plant defense mechanism and identifying pivotal genes and pathways involved in plant’s response to pathogens.
While QTL mapping is ideal for in-depth studies of a few traits, Bulk Segregant Analysis (BSA) is advantageous for screening multiple traits in large populations, making it a valuable tool in broader genetic studies. BSA is a powerful tool in plant genetics that helps identify genetic markers associated with specific traits, such as disease resistance, with lower cost. This technique accelerates the process of locating genomic regions linked to resistance genes. As described by Majeed et al. in 2022, it is a high-throughput QTL mapping approach that rapidly pinpoints genomic loci regulating a trait of interest, which involves pooling individuals exhibiting extreme trait phenotypes, creating bulks, and then subjecting these bulks to genome-wide analyses [17]. BSA has proven invaluable in various fields, allowing researchers to efficiently unravel the genetic basis of complex traits. Recent advancements in genomic sequencing have given rise to QTL-seq, an innovative next-generation sequencing based BSA technique. Unlike conventional QTL mapping approaches, QTL-seq offers superior resolution and efficiency in pinpointing genetic markers linked to quantitative traits by leveraging high-throughput sequencing technologies to sequence bulks of individuals with contrasting trait phenotypes, enabling researchers to pinpoint candidate QTLs with remarkable precision [18,19]. It has been instrumental in identifying genomic regions associated with resistance to various diseases in rice, such as bacterial panicle blight (BPB) and dirty panicle disease [20,21]. QTL-seq, in combination with traditional mapping, identified a major QTL for BPB resistance on the upper arm of chromosome 3 containing three genes associated with defense (OsMADS50, OsDEF8 and OsCEBiP) [20]. With the same approach, three QTLs (qDP1, qDP9, and qDP10) were identified to be significantly associated with resistance to dirty panicle disease, which contain genes encoding PR-proteins, subtilisin-like protease, and ankyrin repeat proteins [21]. This approach has gained popularity due to its ability to handle larger populations efficiently and its potential to uncover complex trait variations. Also, it is a rapid and effective approach for identifying genetic loci involved in plant disease resistance, facilitating the development of resistant cultivars.
Furthermore, genome sequencing has transformed our understanding of plant disease resistance, uncovering intricate genetic details that underpin the plant's ability to fend off pathogens. It led to the discovery of various disease resistance genes in other crop systems. In legumes, Kankanala et al. (2019) delves into the genomics of resistance to various plant pathogens at the genomic level [22]. It provides insights into the molecular basis of different levels of host defense observed in both resistant and susceptible interactions by summarizing large-scale genomic studies, shedding light on host genetics changes and enhancing our understanding of plant-pathogen dynamics [22]. It is not only instrumental in identifying existing disease resistance genes, but also in enhancing plant immunity through genome editing technologies like CRISPR-Cas9. Targeted mutagenesis of genes involved in disease resistance has led to the development of crops resistant to various pathogens, ensuring higher yields and reduced dependence on chemical pesticides [23]. In addition, comparative genome analysis among different plant species, scientists gain insights into evolutionary aspects of disease resistance genes, aiding in the development of robust, broad-spectrum resistance [24]. This approach is particularly vital in understanding the diverse responses of organisms to pathogens and environmental pressures [25]. As it offers a robust framework for identifying genetic variations linked to resistance, including precise markers and mutations, this approach leads to a refined understanding of resistance dynamics and in formulating tailored disease management strategies.
2.2. Harnessing Transcriptomics to Safeguard Rice against Disease
Transcriptome profiling involves studying the complete set of RNA transcripts derived from the genome under specific circumstances. During a plant-pathogen interaction, changes in gene expression patterns play a crucial role in defense responses. Various techniques such as RNA sequencing (RNA-Seq) and microarray analysis are employed to profile transcriptomes. These technologies enable researchers to quantify gene expression levels, identify alternative splicing events, and detect non-coding RNAs. RNA-seq has been extensively utilized in studying rice diseases to unravel the molecular mechanisms underlying pathogen resistance and susceptibility. Numerous studies have employed RNA-seq to analyze gene expression changes, identify differentially expressed genes, and uncover pathways involved in rice immunity against various pathogens. By utilizing this approach, defense-related genes, such as PR1b, transcription factor gene OsWRKY30, and PAL genes (OsPAL1 and OsPAL6), and pathways like the phenylalanine metabolic pathway, alkaloid biosynthesis pathways (tropane, piperidine, and pyridine), and plant hormone signal transduction pathways were identified from a sheath blight resistant cultivar, suggesting the early activation of a SB-induced defense system [26]. Similarly, a study on the enhanced rice Xa7-mediated bacterial blight resistance at high temperature found that the enhanced Xa7-mediated resistance at high temperature is not dependent on salicylic acid signaling [27]. A DNA sequence motif similar to known abscisic acid-responsive cis-regulatory elements was also identified in the same study, suggesting that the plant hormone abscisic acid is an important node for crosstalk between plant transcriptional response pathways to high temperature stress and pathogen attack [27].
Transcriptome profiling during plant-pathogen interactions reveals the activation of specific signaling pathways. For example, genes encoding cellular components associated with defense mechanisms, such as pathogenesis-related (PR) proteins, receptor-like kinases (RLKs), and transcription factors, show significant expression changes in the process of a plant-pathogen interaction[28,29]. Transcriptome analysis also unravels complex regulatory networks involved in plant immunity. By identifying key regulatory genes and their targets, scientists can construct intricate networks governing plant defense responses. In addition, like genomics, comparative transcriptomics involves comparing the transcriptomes of different plant varieties, genotypes, or species in response to pathogen attack by highlighting conserved defense mechanisms and revealing unique responses specific to certain plants. A comparative transcriptome analysis revealed that Rhizoctonia solani AG1 IA infection activated numerous resistance pathways in rice, involving diverse genes in defense response and signal transduction highlighting complex regulation of rice's pathogen response by multiple gene networks [30]. Also, it revealed significant activation of metabolic pathways linked to resistance, particularly emphasizing the biosynthesis of jasmonic acid and phenylalanine metabolism [30]. These comparisons enrich our understanding of plant-pathogen coevolution and offer valuable insights for crop breeding programs. This, in return, provides a wealth of information about the molecular mechanisms underlying plant defense responses and aids in deciphering intricate gene regulatory networks and identify potential targets, thereby offering crucial insights for enhancing crop improvement strategies.
2.3. Harnessing Proteomics for Fortifying Rice against Disease
Proteomic studies have also played a pivotal role in unravelling plant immune responses. Analysis of plant proteome makes it possible to identify key proteins involved in defense pathways and elucidate their functional mechanism. It sheds another light on the complex interactions between plants and pathogens. By comparing protein profiles between infected and uninfected plants, pathogen-responsive proteins have been identified, which include defense-related proteins such as pathogenesis-related (PR) proteins, chitinases, and protease inhibitors [31]. Additionally, proteomics has revealed the modification of host proteins by pathogens to facilitate infection, providing valuable insights into the arms race between plants and pathogens [31].
Techniques such as mass spectrometry (MS) and gel-based methods have been instrumental in characterizing plant defense mechanisms. Advancement in MS and protein isolation techniques have advanced the understanding of subcellular proteomes during plant-pathogen interactions [32]. Proteomic analyses have revealed key proteins pivotal in pathogen recognition, signaling pathways, and metabolic adjustments to combat plant diseases. Notably, receptor-like kinases (RLKs), mitogen-activated protein kinases (MAPKs), and proteins associated with reactive oxygen species (ROS) signaling, hormone modulation, photosynthesis, secondary metabolism, protein degradation, and defense responses have been identified in a rice-Magnaporthe oryzae interaction [32]. Furthermore, proteomics has been employed to identify proteins involved in the lesion mimic associated with program cell death in rice upon biotic or abiotic stimulus [33]. The study by Yong et al. (2021) revealed several differentially expressed proteins, mainly associated with metabolic and cellular processes, notably including resistance-related proteins such as 14-3-3 proteins, OsPR10, and antioxidases in lesion mimic leaves [33]. This study also elucidated the autoimmunity mechanism in rice [33].
Proteomics has also facilitated the exploration of plant-microbe crosstalk. A recent study delved into the identification and profiling of low-abundant proteins in both compatible and incompatible interactions between rice and Xanthomonas oryzae pv. oryzae (Xoo), utilizing a protamine-sulfate-based method to enrich these proteins, which was followed by their identification and quantification through label-free quantitative proteomics [34]. In incompatible interactions, there was a notable increase in the accumulation of protein kinases, including calcium-dependent protein kinases, PTI1-like tyrosine-protein kinase 1, protein kinase domain-containing protein, and serine/threonine-protein kinase, suggesting their pivotal role in signal transduction for the initiation of immunity in rice [34]. Additionally, mitochondrial arginase-1 encoded by OsArg1 demonstrated heightened abundance in the incompatible interaction with Xoo [34]. The elevated expression of OsArg1 significantly bolstered rice resistance against Xoo, enhancing the expression of defense-related genes such as Chitinase II, Glucanase I, and PR1, which indicates the involvement of this protein in Xoo resistance [34]. As a follow-up report, a comprehensive proteome profile was generated elucidating the interaction between rice and Xoo, uncovering the proteome changes in the rice cultivars and highlight the functions of OsARG1 in plant defense against Xoo. [35].
By studying defense responses elicited by bacterial, fungal, and viral pathogens, researchers have unravelled proteins specifically involved in these interactions. Furthermore, comparative proteomics analysis significantly contributes to the study of the intricate molecular mechanisms underlying rice disease resistance. By comparing the protein profiles of susceptible and resistant rice cultivars, researchers have identified various proteins associated with disease resistance pathways, including those involved in signal transduction, defense responses, and metabolic processes [36,37,38] . Proteomics approach also revealed novel insights into the interaction between rice and Xoo. In this study, most of the differentially abundant proteins (DAPs) in Xoo were related to pathogen virulence, which included the outer member proteins, type III secretion system proteins, TonB-dependent receptors, and transcription activator-like effectors [38]. These DAPs were less abundant in the incompatible interaction and, in thi condition, DAPs in rice were mainly involved in secondary metabolic processes, including phenylalanine metabolism and the biosynthesis of flavonoids and phenylpropanoids [38]. This indicates that during incompatible interaction, the rice prevents pathogen invasion and initiate multi-component defense responses.
This knowledge gained from proteomic studies is pivotal for developing strategies to enhance crop resistance against diverse pathogens and provides valuable insights into the dynamic changes occurring at the proteome level during pathogen challenge, shedding light on potential targets for enhancing rice resistance against pathogens. Overall, proteomics serves as a powerful tool for deciphering the molecular basis of rice disease resistance and holds promise for informing strategies to improve technologies for crop protection and agricultural sustainability.
2.4. Harnessing Metabolomics for Bolstering Rice Disease Resistance
Metabolomics studies on rice disease resistance involve the comprehensive analysis of metabolites present in different parts of the plant, such as leaves, roots, and seeds, under various conditions. By employing techniques such as liquid chromatography-mass spectrometry (LC-MS), gas chromatography-mass spectrometry (GC-MS), and nuclear magnetic resonance spectroscopy (NMR) to detect and quantify metabolites profiles of diseased and healthy plants, specific metabolites associated with disease resistance can be identified. These metabolites serve as crucial biomarkers, shedding light on the biochemical pathways involved in plant defense mechanisms [39].
Metabolomics studies have successfully linked changes in primary or specialized metabolism to plant defense responses. For instance, proteomic analysis of Xoo-secreted proteins, in vitro and in planta, sheds light on the diverse functions and expression patterns of these proteins during rice bacterial blight infection [40]. The comprehensive proteomic analysis conducted in this study identified 109 unique proteins, elucidating their diverse roles in crucial biological processes such as metabolism, nutrient uptake, pathogenicity, and host defense mechanisms and the observed correlation between protein and transcript abundances unveils the intricate regulatory mechanisms governing protein secretion during in planta infection [40]. In addition, the investigation reveals the potential of transgenic rice expressing these specific secretory proteins to influence cell death signaling, underscoring their pivotal role in pathogenicity [40]. This research significantly advances our understanding of rice bacterial blight disease and provides valuable insights for the development of disease-resistant rice varieties. Furthermore, a study using metabolomics techniques found differences in metabolite accumulation between resistant and susceptible rice plants when exposed to Xoo infection [41]. Specifically, plants expressing the XA21 gene differed from wild-type plants, exhibiting elevated levels of sugar alcohols, tricarboxylic acid cycle (TCA) intermediates, and various other compounds before treatment [41]. Following the inoculation of Xoo strain PXO99, XA21-expressing plants displayed increased levels of responsive metabolites, such as rutin, pigments, fatty acids, lipids, and arginine, which likely play roles in polyamine biosynthesis and alkaloid metabolism [41]. Additionally, metabolomic analyses have revealed the role of secondary metabolites, such as phenolic compounds and terpenoids, in bolstering plant immunity, which paved the way for developing strategies to enhance the production of disease resistant crops [42]. The same approach was used to identify metabolite levels in rice lines during Rhizoctonia solani infection using CE/TOF-mass spectrophotometry in positive ion mode where alterations in metabolite levels in inoculated resistant and susceptible rice were examined along the tricarboxylic acid and glycolysis pathways, revealing ten metabolites that were differentially regulated [43]. Notably, chlorogenic acid exhibited increased levels in 32R, a resistant line, while 29S, the susceptible line, pipecolic acid exhibited the highest fold change and significance level and eight amino acids (i.e. glutamate, γ-aminobutyric acid, glycine, histidine, phenylalanine, serine, tryptophan, and tyrosine) displayed elevated levels [43]. These metabolomic signatures often include alterations in the levels of amino acids, organic acids, sugars, and secondary metabolites, which play crucial roles in plant defense mechanisms.
Metabolomics studies have provided valuable insights into the metabolic pathways and key metabolites involved in rice disease resistance. Analyses have revealed the accumulation of defense-related metabolites, such as phenolic compounds, flavonoids, and phytoalexins, in response to pathogen attack. Furthermore, metabolomics approaches have facilitated the identification of metabolic quantitative trait loci (mQTLs) associated with disease resistance, providing valuable targets for breeding programs aimed at developing resistant rice varieties [44]. However, data integration, standardization of analytical techniques, and the functional validation of identified metabolites are remaining areas for active research [45]. Moving forward, continued advancements in metabolomics technologies and methodologies hold promise for further elucidation of the complex mechanisms underlying rice disease resistance and acceleration of the development of resilient rice varieties.
3. Integrative Omics: Bridging the Layers
Integrative omics approaches merge diverse datasets from genomics, transcriptomics, proteomics, and metabolomics, providing a comprehensive view of biological systems. By integrating these multi-omics data layers, researchers can construct sophisticated systems biology models that unveil complex molecular interactions within organisms. These models offer valuable insights into how genes, proteins, and metabolites interact and function together, facilitating a deeper understanding of biological processes and disease mechanisms.
3.1. Integrative Studies in Rice for Disease Resistance
Understanding the crosstalk between different biological components provides a holistic view of rice disease resistance, aiding in the development of resistant rice varieties [46]. By deciphering the genetic basis, gene expression patterns, protein functions, and metabolic changes, these integrative approaches can facilitate precise breeding strategies for improving disease resistance.
By integrating data from genomics, transcriptomics, proteomics, and metabolomics, researchers can perform in-depth analyses, leading to a deeper understanding of plant responses to diseases, and targeted breeding approach based on the integrative data analysis will accelerate the development of disease-resistant rice varieties. For example, through the combination of GWAS and transcriptional analysis, two genes, RNG1 and RNG3 encoding zinc finger protein with a B-box domain and dehydrogenase, respectively, were identified as molecular markers for selecting blast-resistant rice accessions based on their differential expression caused by polymorphisms in 3′-untranslated regions (3′-UTR) [47] (Table 1). Through a combined genetic analysis of leaf blast resistance in upland rice, which included QTL mapping, bulked segregant analysis, and transcriptome sequencing, a novel QTL for blast resistance was fine mapped on Chromosome 11 [48]. This study improved the genetic understanding of the mechanism of blast resistance and led to identification of suitable genotypes with resistance alleles that would be useful genetic resources in rice blast resistance breeding [48] (Table 1). Proteomics study further deepens this kind of molecular genetic knowledge by pinpointing specific proteins involved in the defense mechanisms, while metabolomics complements these findings by identifying metabolites related to disease resistance pathways. For example, molecular interactions between rice and the fungal pathogen R. solani could be characterized in regard to gene expression mechanism by employing proteomic and transcriptomic approaches [49]. This led to the elucidation of defense responses in tolerant and susceptible genotypes of O. sativa against R. solani, which was essential to identify the crucial players of their underlying molecular mechanism [49] (Table 1).
There are also several studies that explored the integration of transcriptomics and metabolomics to understand rice disease resistance. Valuable insights for the defense mechanisms against Xoo were obtained from an integrative analysis of metabolomics and transcriptomics profiling data, which included differential expression of several pathogenesis related genes in PXO99-challenged transgenic Xa21 plants, GAD, PAL, ICL1 and GS10 between resistant and susceptible varieties [37] (Table 1). Though Bph30 gene has been successfully cloned and conferred rice with broad-spectrum resistance to brown plant hopper (BPH), the molecular mechanisms by which Bph30 enhances resistance to BPH remain poorly understood [50]. By utilizing both transcriptomic and metabolomic analysis, Shi et al., (2023) elucidated the response of Bph30 to BPH infestation, using Bph30-transgenic (BPH30T) and BPH-susceptible Nipponbare plants, suggesting that Bph30 might coordinate the movement of primary and secondary metabolites and hormones in plants via the shikimate pathway to enhance the resistance of rice to BPH [50] (Table 1). In case of bacterial panicle blight in rice, by combining QTL-mapping and QTL-seq, one major QTL, qBPB3.1, was found in chromosome 3 that conferred resistance to this disease [20].
This integrated approach helps in the precise identification of key molecular players of resistance mechanism. Integrating omics data also allows to unravel regulatory pathways governing rice disease resistance by understanding the intricate interactions between genes, proteins, and metabolites leading in identifying regulatory elements and signaling pathways involved in the plant's defense mechanisms [51]. This knowledge aids in targeted interventions for enhancing disease resistance. Data generated from this integration can be utilized for precision breeding for rice disease resistance and to devise crop management practices. Breeders can utilize these integrated molecular data to develop rice varieties with enhanced disease resistance traits through targeted breeding strategies, resulting in the development of resilient rice cultivars that can withstand various diseases [46]. Furthermore, it can also offer insights into the plant's response to environmental stressors and diseases, where farmers can adopt informed crop management practices based on these molecular insights implementing strategies such as tailored irrigation and fertilization practices [52]. Nevertheless, the application of integrative omics for bolstering rice disease resistance has not been explored extensively yet. While research has focused more on abiotic stress tolerance and agronomic traits, there's a notable gap in leveraging these approaches for enhancing disease resistance in rice. Closing this gap could unlock novel insights into the molecular mechanisms underlying disease resistance and lead to the development of resilient rice varieties capable of withstanding diverse biotic challenges.
3.2. Integrative Studies in Rice for Tolerance to Abiotic Stresses
This integrative approach has also been transforming the understanding of rice abiotic stress tolerance, leading to remarkable advancements in crop improvement strategies. Several studies exemplify the practical applications of these techniques, showcasing their effectiveness in enhancing rice resilience to various environmental stresses (Table 1). Integration of multi-omics data has been instrumental in deciphering the complex interactions between genes, proteins, and metabolites upon salt stress. Better understanding of salt tolerance mechanism through integration of genomics, transcriptomics, and other omics information can accelerate rice breeding for developing salinity tolerant rice varieties , [53]. Comprehensive multi-omics approaches also have been instrumental in understanding the components of drought tolerance in rice. Comparative mapping within and across species provided a holistic view of the genetic factors contributing to drought resistance [54] (Table 1), and key transcription factors for allontain biosynthesis, OsERF059 and ONAC007, were found to be important for enhancing drought tolerance in rice through combining transcriptomic and metabolic analyses [55]. For heat tolerance, integrating GWAS and transcriptomics has mapped candidate locus, LOC_Os07g48710, responsible for heat tolerance in rice which encodes a VQ domain containing protein, providing valuable genetic markers for molecular breeding programs for enhancement of heat resilience in rice varieties [56] (Table 1). In terms of metal toxicity, combined transcriptomics and metabolomics have provided a comprehensive view of the molecular processes underlying arsenic stress response by identifying arsenic-responsive genes and metabolites aids in understanding the intricate pathways involved in detoxification and tolerance mechanisms [47] (Table 1). For cold tolerance, integration of transcriptomic and metabolic profiling revealed that OsSEH1 encoding a nucleoporin/ WD40 domain containing protein, plays a role in the oxidation-reduction process contributing to cold tolerance in rice [58] (Table 1).
Integrative omics approaches have become increasingly prevalent in enhancing rice tolerance to abiotic stressors, such as salinity and drought, resulting in significant agricultural advancements offering a comprehensive understanding of stress response mechanisms, aiding in the development of stress-tolerant rice cultivars.
4. Challenges and Gaps: Exploring the Intersections
While both genetic mapping and omics studies have significantly contributed to the field of rice disease resistance, integrating these approaches face several challenges and gaps that need to be resolved for efficient and precise characterization of rice resistance against diseases. One major concern is the integration of diverse data types generated from diverse platforms. Genomic data provide information about genetic loci, while transcriptomic data reveal gene expression patterns. Integrating these diverse datasets requires robust computational methods that can align and analyse data from various sources [59]. In addition, the accuracy of functional annotation of genomic variants is critical. Understanding the functional significance of genetic variants detected through mapping studies is essential, and integrating omics data can aid in annotating these variants by correlating them with gene expression profiles and protein functions [60]. This integration is crucial for deciphering the molecular basis of disease resistance. Typically, stress often induces dynamic changes in gene expression, protein abundance, and metabolite levels, so static omics snapshots might miss these dynamic responses [61]. In this context, incorporating time-series omics data during disease progression or dynamic omics profiling is essential to capture the temporal aspects of the host-pathogen interaction. Integrating networks derived from mapping and omics data can provide insights into key regulatory nodes, but existing tools lack robustness in handling large-scale integrated networks [46]. Advanced network analysis algorithms are required to identify central genes, proteins, and metabolites crucial for disease resistance. Lastly, environmental factors significantly influence disease resistance. Integration of environmental parameters like temperature, humidity, and soil quality into multi-omics datasets allows researchers to identify genes and pathways modulated by environmental cues. Understanding genotype-environment interactions can provide valuable insights into the adaptability of rice varieties under varying environmental conditions [62]. As addressing the research gaps related to data integration, functional annotation, dynamic profiling, network analysis, and environmental interactions is crucial, future research should focus on refining computational models, enhancing data integration methods, and exploring emerging omics technologies to further unravel the complexities of rice disease resistance [53]. By overcoming these challenges, we can unravel the intricate molecular mechanisms governing disease resistance, with precision and efficiency, paving the way for the development of disease-resistant rice varieties.
5. Conclusions
The intersection of traditional genetic mapping and cutting-edge omics technologies has opened new horizons in understanding and enhancing rice disease resistance. The integration of mapping and omics data is not merely a combination of techniques; it represents a paradigm shift in our approach to studying complex biological systems. Regarding the significant advancements, challenges, and prospects of this integration reviewed in this article, it becomes evident that this interdisciplinary approach offers unprecedented precision in characterizing rice disease resistance. Integrative studies encompassing these technologies hold promise in not only characterizing resistance but also in engineering highly resilient rice varieties. This represents a transformative approach in the realm of rice disease resistance research and breeding. Furthermore, the knowledge gained from these studies will provide huge impacts on development of innovative strategies for sustainable agriculture.
Author Contributions
Conceptualization, J.O. and J.H.H; writing—original draft preparation, J.O.; writing—review and editing, J.H.H.; visualization, J.O.; supervision, J.H.H.; project administration, J.H.H.; funding acquisition, J.H.H. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by funding from the USDA National Institute of Food and Agriculture (Grants, 2022-67013-36140 and 2023-68012-39002; Hatch, CT0477).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Singh, P.K.; Nag, A.; Arya, P.; Kapoor, R.; Singh, A.; Jaswal, R.; Sharma, T.R. Prospects of Understanding the Molecular Biology of Disease Resistance in Rice. Int. J. Mol. Sci. 2018, 19, 1141. [Google Scholar] [CrossRef]
- Zhang, S.; Li, C.; Si, J.; Han, Z.; Chen, D. Action Mechanisms of Effectors in Plant-Pathogen Interaction. Int. J. Mol. Sci. 2022, 23, 6758. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Yang, L. Pathogen-triggered changes in plant development: Virulence strategies or host defense mechanism? Front. Microbiol. 2023, 14, 1122947. [Google Scholar] [CrossRef] [PubMed]
- Mapuranga, J.; Zhang, N.; Zhang, L.; Chang, J.; Yang, W. Infection Strategies and Pathogenicity of Biotrophic Plant Fungal Pathogens. Front. Microbiol. 2022, 13, 799396. [Google Scholar] [CrossRef]
- Pilet-Nayel, M.-L.; Moury, B.; Caffier, V.; Montarry, J.; Kerlan, M.-C.; Fournet, S.; Durel, C.-E.; Delourme, R. Quantitative Resistance to Plant Pathogens in Pyramiding Strategies for Durable Crop Protection. Front. Plant Sci. 2017, 8, 1838. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, Y.; Shi, H.; Qiu, J.; Ding, X.; Kou, Y. Recent Progress in Rice Broad-Spectrum Disease Resistance. Int. J. Mol. Sci. 2021, 22, 11658. [Google Scholar] [CrossRef] [PubMed]
- Collard, B.C.Y.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar] [CrossRef]
- Powder, K.E. Quantitative Trait Loci (QTL) Mapping. Methods Mol Biol 2020, 2082, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.B. QTL Mapping. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, MA, USA, 2001; pp. 8–12. [Google Scholar]
- Mohan, M.; Nair, S.; Bhagwat, A.; Krishna, T.G.; Yano, M.; Bhatia, C.R.; Sasaki, T. Genome mapping, molecular markers and marker-assisted selection in crop plants. Mol. Breed. 1997, 3, 87–103. [Google Scholar] [CrossRef]
- Mulualem, T.; Bekeko, Z. Advances in Quantitative Trait Loci, Mapping and Importance of Markers Assisted Selection in Plant Breeding Research. Int. J. Plant Breed. Genet. 2016, 10, 58–68. [Google Scholar] [CrossRef]
- Miles, C.; Wayne, M. Quantitative trait locus (QTL) analysis. Nature Education 2008, 1, 208. [Google Scholar]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Prim. 2021, 1, 1–21. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Sattayachiti, W.; Wanchana, S.; Arikit, S.; Nubankoh, P.; Patarapuwadol, S.; Vanavichit, A.; Darwell, C.T.; Toojinda, T. Genome-Wide Association Analysis Identifies Resistance Loci for Bacterial Leaf Streak Resistance in Rice (Oryza sativa L.). Plants 2020, 9, 1673. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Lan, G.; Zhu, Y.; Chen, K.; Shen, C.; Zhao, X.; Zhang, F.; Xu, J.; Li, Z. Genome-Wide Association Study on Resistance to Rice Black-Streaked Dwarf Disease Caused by Rice black-streaked dwarf virus. Plant Dis. 2021, 105, 607–615. [Google Scholar] [CrossRef]
- Majeed, A.; Johar, P.; Raina, A.; Salgotra, R.K.; Feng, X.; Bhat, J.A. Harnessing the potential of bulk segregant analysis sequencing and its related approaches in crop breeding. Front. Genet. 2022, 13, 944501. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Li, Z.; Chen, X.; Shi, S.; Zhang, H.; Wang, X.; Chen, H.; Li, W.; Li, L. DeepBSA: A deep-learning algorithm improves bulked segregant analysis for dissecting complex traits. Mol. Plant 2022, 15, 1418–1427. [Google Scholar] [CrossRef]
- Ontoy, J.C.; Shrestha, B.; Karki, H.S.; Barphagha, I.; Angira, B.; Famoso, A.; Ham, J.H. Genetic Characterization of the Partial Disease Resistance of Rice to Bacterial Panicle Blight and Sheath Blight by Combined QTL Linkage and QTL-seq Analyses. Plants 2023, 12, 559. [Google Scholar] [CrossRef]
- Riangwong, K.; Aesomnuk, W.; Sonsom, Y.; Siangliw, M.; Unartngam, J.; Toojinda, T.; Wanchana, S.; Arikit, S. QTL-seq Identifies Genomic Regions Associated with Resistance to Dirty Panicle Disease in Rice. Agronomy 2023, 13, 1905. [Google Scholar] [CrossRef]
- Kankanala, P.; Nandety, R.S.; Mysore, K.S. Genomics of Plant Disease Resistance in Legumes. Front. Plant Sci. 2019, 10, 1345. [Google Scholar] [CrossRef]
- Yin, K.; Qiu, J.-L. Genome editing for plant disease resistance: applications and perspectives. Philos. Trans. R. Soc. B: Biol. Sci. 2019, 374, 20180322. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.; Yeom, S.-I.; Kim, Y.-M.; Seo, E.; Kim, K.-T.; Kim, M.-S.; Lee, J.M.; Cheong, K.; Shin, H.-S.; et al. New reference genome sequences of hot pepper reveal the massive evolution of plant disease-resistance genes by retroduplication. Genome Biol. 2017, 18, 210. [Google Scholar] [CrossRef]
- Feng, Y.; Neme, R.; Beh, L.Y.; Chen, X.; Braun, J.; Lu, M.W.; Landweber, L.F. Comparative genomics reveals insight into the evolutionary origin of massively scrambled genomes. eLife 2022, 11. [Google Scholar] [CrossRef]
- Yang, X.; Gu, X.; Ding, J.; Yao, L.; Gao, X.; Zhang, M.; Meng, Q.; Wei, S.; Fu, J. Gene expression analysis of resistant and susceptible rice cultivars to sheath blight after inoculation with Rhizoctonia solani. BMC Genom. 2022, 23, 1–16. [Google Scholar] [CrossRef]
- Cohen, S.P.; Liu, H.; Argueso, C.T.; Pereira, A.; Cruz, C.V.; Verdier, V.; Leach, J.E. RNA-Seq analysis reveals insight into enhanced rice Xa7-mediated bacterial blight resistance at high temperature. PLOS ONE 2017, 12, e0187625. [Google Scholar] [CrossRef]
- Stokes, T. Transcriptional responses to plant pathogen interactions. Trends Plant Sci. 2001, 6, 50–51. [Google Scholar] [CrossRef]
- Tyagi, P.; Singh, D.; Mathur, S.; Singh, A.; Ranjan, R. Upcoming progress of transcriptomics studies on plants: An overview. Front. Plant Sci. 2022, 13, 1030890. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, H.; Wang, H.; Xiang, Z.; Wei, S.; Zheng, W. Comparative transcriptome analysis of rice cultivars resistant and susceptible to Rhizoctonia solani AG1-IA. BMC Genom. 2022, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, S.; Liu, K.; Wang, S.; Huang, L.; Guo, L. Proteomics: a powerful tool to study plant responses to biotic stress. Plant Methods 2019, 15, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Gupta, R.; Min, C.W.; Kwon, S.W.; Wang, Y.; Je, B.I.; Kim, Y.-J.; Jeon, J.-S.; Agrawal, G.K.; Rakwal, R.; et al. Proteomics of Rice—Magnaporthe oryzae Interaction: What Have We Learned So Far? Front. Plant Sci. 2019, 10, 1383. [Google Scholar] [CrossRef] [PubMed]
- Yong, Y.; Qiujun, L.; Xinyu, C.; Weifang, L.; Yuwen, F.; Zhengjin, X.; Yuanhua, W.; Xuming, W.; Jie, Z.; Chulang, Y.; et al. Characterization and Proteomic Analysis of Novel Rice Lesion Mimic Mutant with Enhanced Disease Resistance. Rice Sci. 2021, 28, 466–478. [Google Scholar] [CrossRef]
- Gupta, R.; Min, C.W.; Son, S.; Lee, G.H.; Jang, J.W.; Kwon, S.W.; Park, S.R.; Kim, S.T. Comparative proteome profiling of susceptible and resistant rice cultivars identified an arginase involved in rice defense against Xanthomonas oryzae pv. oryzae. Plant Physiol. Biochem. 2021, 171, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Min, C.W.; Park, S.-R.; Kim, S.T. Label-free proteome data of susceptible and resistant rice cultivars in response to Xanthomonas oryzae pv. oryzae inoculation. Data Brief 2022, 41, 107890. [Google Scholar] [CrossRef]
- Tian, D.; Yang, L.; Chen, Z.; Chen, Z.; Wang, F.; Zhou, Y.; Luo, Y.; Yang, L.; Chen, S. Proteomic analysis of the defense response to Magnaporthe oryzae in rice harboring the blast resistance gene Piz-t. Rice 2018, 11, 47. [Google Scholar] [CrossRef]
- Wei, L.; Wang, D.; Gupta, R.; Kim, S.T.; Wang, Y. A Proteomics Insight into Advancements in the Rice–Microbe Interaction. Plants 2023, 12, 1079. [Google Scholar] [CrossRef]
- Zhang, F.; Huang, L.; Zeng, D.; Cruz, C.V.; Li, Z.; Zhou, Y. Comparative proteomic analysis reveals novel insights into the interaction between rice and Xanthomonas oryzae pv. oryzae. BMC Plant Biol. 2020, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Gupta, P.; Priscilla, K.; Kumar, S.; Hangargi, B.; Veershetty, A.; Ramrao, D.P.; Suresh, S.; Narasanna, R.; Naik, G.R.; et al. Metabolomics Intervention Towards Better Understanding of Plant Traits. Cells 2021, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kim, S.G.; Wu, J.; Huh, H.-H.; Lee, S.-J.; Rakwal, R.; Agrawal, G.K.; Park, Z.-Y.; Kang, K.Y.; Kim, S.T. Secretome analysis of the rice bacteriumXanthomonas oryzae(Xoo) using in vitro and in planta systems. Proteomics 2013, 13, 1901–1912. [Google Scholar] [CrossRef]
- Sana, T.R.; Fischer, S.; Wohlgemuth, G.; Katrekar, A.; Jung, K.-H.; Ronald, P.C.; Fiehn, O. Metabolomic and transcriptomic analysis of the rice response to the bacterial blight pathogen Xanthomonas oryzae pv. oryzae. Metabolomics 2010, 6, 451–465. [Google Scholar] [CrossRef]
- Castro-Moretti, F.R.; Gentzel, I.N.; Mackey, D.; Alonso, A.P. Metabolomics as an Emerging Tool for the Study of Plant–Pathogen Interactions. Metabolites 2020, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Suharti, W.S.; Nose, A.; Zheng, S.-H. Metabolite profiling of sheath blight disease resistance in rice: in the case of positive ion mode analysis by CE/TOF-MS. Plant Prod. Sci. 2016, 19, 279–290. [Google Scholar] [CrossRef]
- Gong, L.; Chen, W.; Gao, Y.; Liu, X.; Zhang, H.; Xu, C.; Yu, S.; Zhang, Q.; Luo, J. Genetic analysis of the metabolome exemplified using a rice population. Proc. Natl. Acad. Sci. 2013, 110, 20320–20325. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bohra, A.; Pandey, A.K.; Pandey, M.K.; Kumar, A. Metabolomics for Plant Improvement: Status and Prospects. Front. Plant Sci. 2017, 8, 1302. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Saand, M.A.; Huang, L.; Abdelaal, W.B.; Zhang, J.; Wu, Y.; Li, J.; Sirohi, M.H.; Wang, F. Applications of Multi-Omics Technologies for Crop Improvement. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Bai, L.; Liu, M.; Liu, Y.; Peng, S.; Hu, P.; Wang, D.; Liu, Q.; Yan, S.; Gao, L.; et al. Identification of two novel rice S genes through combination of association and transcription analyses with gene-editing technology. Plant Biotechnol. J. 2023, 21, 1628–1641. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; He, H.; Chen, W.; Huang, L.; Zhao, D.; Chen, X.; Li, J.; Yang, X. Integrated genetic analysis of leaf blast resistance in upland rice: QTL mapping, bulked segregant analysis and transcriptome sequencing. AoB PLANTS 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Prathi, N.B.; Palit, P.; Madhu, P.; M, R.; Laha, G.; Balachandran, S.; Madhav, M.S.; Sundaram, R.; Mangrauthia, S.K. Proteomic and transcriptomic approaches to identify resistance and susceptibility related proteins in contrasting rice genotypes infected with fungal pathogen Rhizoctonia solani. Plant Physiol. Biochem. 2018, 130, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zha, W.; Yu, X.; Wu, Y.; Li, S.; Xu, H.; Li, P.; Li, C.; Liu, K.; Chen, J.; et al. Integrated transcriptomics and metabolomics analysis provide insight into the resistance response of rice against brown planthopper. Front. Plant Sci. 2023, 14, 1213257. [Google Scholar] [CrossRef]
- Roychowdhury, R.; Das, S.P.; Gupta, A.; Parihar, P.; Chandrasekhar, K.; Sarker, U.; Kumar, A.; Ramrao, D.P.; Sudhakar, C. Multi-Omics Pipeline and Omics-Integration Approach to Decipher Plant’s Abiotic Stress Tolerance Responses. Genes 2023, 14, 1281. [Google Scholar] [CrossRef]
- Naik, B.; Kumar, V.; Rizwanuddin, S.; Chauhan, M.; Choudhary, M.; Gupta, A.K.; Kumar, P.; Kumar, V.; Saris, P.E.J.; Rather, M.A.; et al. Genomics, Proteomics, and Metabolomics Approaches to Improve Abiotic Stress Tolerance in Tomato Plant. Int. J. Mol. Sci. 2023, 24, 3025. [Google Scholar] [CrossRef]
- Ullah, M.A.; Abdullah-Zawawi, M.-R.; Zainal-Abidin, R.-A.; Sukiran, N.L.; Uddin, I.; Zainal, Z. A Review of Integrative Omic Approaches for Understanding Rice Salt Response Mechanisms. Plants 2022, 11, 1430. [Google Scholar] [CrossRef]
- Zargar, S.M.; Mir, R.A.; Ebinezer, L.B.; Masi, A.; Hami, A.; Manzoor, M.; Salgotra, R.K.; Sofi, N.R.; Mushtaq, R.; Rohila, J.S.; et al. Physiological and Multi-Omics Approaches for Explaining Drought Stress Tolerance and Supporting Sustainable Production of Rice. Front. Plant Sci. 2022, 12, 803603. [Google Scholar] [CrossRef]
- Lu, S.; Jia, Z.; Meng, X.; Chen, Y.; Wang, S.; Fu, C.; Yang, L.; Zhou, R.; Wang, B.; Cao, Y. Combined Metabolomic and Transcriptomic Analysis Reveals Allantoin Enhances Drought Tolerance in Rice. Int. J. Mol. Sci. 2022, 23, 14172. [Google Scholar] [CrossRef]
- Li, P.; Jiang, J.; Zhang, G.; Miao, S.; Lu, J.; Qian, Y.; Zhao, X.; Wang, W.; Qiu, X.; Zhang, F.; et al. Integrating GWAS and transcriptomics to identify candidate genes conferring heat tolerance in rice. Front. Plant Sci. 2023, 13, 1102938. [Google Scholar] [CrossRef]
- Ma, L.; Zeng, J.; Zhang, R.Q.; Wang, L.; Zhang, F.; Zhao, X.; Yuan, Y.; Li, L. Integrated transcriptomic and metabolomic analysis the variation of rice cultivars response to arsenite stress. Environ. Technol. Innov. 2023, 31. [Google Scholar] [CrossRef]
- Gu, S.; Zhuang, J.; Zhang, Z.; Chen, W.; Xu, H.; Zhao, M.; Ma, D. Multi-omics approach reveals the contribution of OsSEH1 to rice cold tolerance. Front. Plant Sci. 2023, 13, 1110724. [Google Scholar] [CrossRef]
- Dai, L.; Li, P.; Li, Q.; Leng, Y.; Zeng, D.; Qian, Q. Integrated Multi-Omics Perspective to Strengthen the Understanding of Salt Tolerance in Rice. Int. J. Mol. Sci. 2022, 23, 5236. [Google Scholar] [CrossRef]
- Zaghum, M.J.; Ali, K.; Teng, S. Integrated Genetic and Omics Approaches for the Regulation of Nutritional Activities in Rice (Oryza sativa L.). Agriculture 2022, 12, 1757. [Google Scholar] [CrossRef]
- Geng, L.; Zhang, W.; Zou, T.; Du, Q.; Ma, X.; Cui, D.; Han, B.; Zhang, Q.; Han, L. Integrating linkage mapping and comparative transcriptome analysis for discovering candidate genes associated with salt tolerance in rice. Front. Plant Sci. 2023, 14, 1065334. [Google Scholar] [CrossRef] [PubMed]
- McCouch, S.R.; Wright, M.H.; Tung, C.-W.; Maron, L.G.; McNally, K.L.; Fitzgerald, M.; Singh, N.; DeClerck, G.; Agosto-Perez, F.; Korniliev, P.; et al. Open access resources for genome-wide association mapping in rice. Nat. Commun. 2016, 7, 10532. [Google Scholar] [CrossRef]
- Iqbal, Z.; Iqbal, M.S.; Khan, M.I.R.; Ansari, M.I. Toward Integrated Multi-Omics Intervention: Rice Trait Improvement and Stress Management. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef]

Table 1.
List of studies utilizing integrative mapping and omics to identify and characterize rice resilience to biotic and abiotic challenges.
Table 1.
List of studies utilizing integrative mapping and omics to identify and characterize rice resilience to biotic and abiotic challenges.
| Abiotic/ Biotic Factors | QTL/ loci/ genes (gene products) | Methods | Reference |
| Drought tolerance | OsERF059 (ethylene response factor 59) and ONAC007 (NAC domain-containing protein 7) | Transcriptomics and metabolomics | Lu et al. (2022) [55] |
| Heat tolerance | qHT7/ LOC_Os07g48710 (VQ motif-containing protein 30) | GWAS and transcriptomics | Li et al. (2023) [56] |
| Arsenic toxicity | DEGs and DAMs ion transporters, ROS, etc. | Transcriptomics and metabolomics | Ma et al. (2023) [57] |
| Cold tolerance | OsSEH1 (nucleoporin SEH1) | Transcriptomics and metabolomics | Gu et al. (2023) [58] |
| Blast resistance (Magnaporthe grisea) | RNG1 (Zinc finger protein with B-box-domain) and RNG3 (Dehydrogenase) | GWAS and transcriptomics | Xu et al. (2023) [47] |
| Blast resistance (Magnaporthe grisea) | Os11g0700900 (glycoside hydrolase), Os11g0704000 (SelT selenoprotein family), Os11g0702400 (zinc finger, C2H2-type domain containing protein) and Os11g0703600 (hypothetical protein) | BSA, QTL-mapping, and transcriptomics | Tan et al. (2022) [48] |
| Sheath blight resistance (Rhizoctonia solani) | LOC_Os12g44010.1 (purple acid phosphatase 10b), LOC_Os04g43290.3 (actin-related protein (ARP) C2 subunit), LOC_Os11g48000.1 (EPF zinc-finger), LOC_Os09g29480.2 (2-aminoethanethiol dioxygenase), LOC_Os06g45890.1 (MYB-like transcription factor), LOC_Os04g46980.1 (cis-zeatin-O-glucosyltransferase), and LOC_Os09g12790.1 (potassium channel protein) | Proteomics and transcriptomics | Prathi et al. (2018) [49] |
| Bacterial blight resistance (Xanthomonas oryzae pv oryzae) | GAD (Glutamate decarboxylase), PAL (Phenylalanine ammonia-lyase), ICL1 (Isocitrate lyase) and GS10 (Glutathione-S-transferase) | Transcriptomics and metabolomics | Sana et al. (2010) [41] |
| Brown planthopper resistance (Tungro virus) | Bph30 (Leucine rich repeat (LRR) family protein) | Transcriptomics and metabolomics | Shi et al. (2023) [50] |
| Bacterial panicle blight resistance (Burkholderia glumae) | qBPB3.1 | QTL-mapping and QTL-seq | Ontoy et al. (2023) [20] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



