
Submitted:
04 April 2024
Posted:
05 April 2024
You are already at the latest version
Abstract
Dysregulation of the Cyclin-dependent kinase 8 (CDK8) activity has been associated to many diseases including the colorectal and breast cancer. As usual in the CDK family, the activity of CDK8 is controlled by a regulatory protein called cyclin C (CycC). But while human CDK family members are generally activated in two steps that is, the binding of the cyclin to CDK and the phosphorylation of a residue in the CDK activation loop, CDK8 does not require the phosphorylation step to be active. Another peculiarity of CDK8 is its ability to be associated to CycC while adopted an inactive form. These specificities raise the question of the role of CycC in the complex CDK8-CycC, which appears to be more complex than for the other members of CDK family. Through MD simulations and binding free energy calculations, we investigated the effect of CycC on the structure and dynamics of CDK8 on the one hand, and the structural molecular basis of the protein-protein interaction between the two partners on the other hand. We found that CycC has a stabilizing effect on CDK8 and identified specific interaction hotspots within its interaction surface compared to other human CDK/Cyc pairs. Targeting these specific interaction hotspots could be a promising approach in terms of specificity, to effectively disrupt the interaction between CDK8. The simulation of the conformational transition from the inactive to the active form of CDK8 suggests that the residue Glu99 of the CycC may assume the role of the missing phosphorylation step in the activation mechanism of CDK8. In a more general view, these results point the importance of keeping the CycC in computational studies when studying the human CDK8 protein in both the active and the inactive form.
Keywords:
CDK8
; Cyclin C
; Protein-protein interaction
; Molecular dynamic simulation
; Free energy calculation
; Drug design
1. Introduction
Cyclin-dependent kinases (CDKs) are serine-threonine kinases that require binding with regulatory proteins called cyclin to be active. CDKs are the main regulators of the cell cycle and gene transcription. The human proteome contains 20 CDKs and 29 cyclins. CDK1 to CDK6 are involved in cell cycle regulation, while CDK7, CDK8, CDK9, CDK11, and CDK20 are primarily involved in transcriptional regulation. More particularly, CDK7, CDK8 and CDK9 control the activity of RNA polymerase II in humans by the phosphorylation of its C-terminus domain, which catalyzes the synthesis of all mRNA precursors [1]. The inhibition of CDK activity by small molecules for the treatment of cancer has been extensively studied [2]. Several CDK inhibitors have undergone clinical trials and in February 2015 palbociclib, a CDK4/6 inhibitor, was the first approved by the FDA [3].
CDK8 is a target of interest that has recently attracted considerable attention after the publication of numerous genetic and biochemical studies highlighting its many key roles in oncogenesis [4,5]. Among its various cellular functions, the most notable is its involvement in regulating transcription trough diverse mechanisms. CDK8 is a part of the mediator complex, which is a large multisubunit protein complex that is central to the regulation of transcription in eukaryotes [6]. The main function of the mediator complex is to transmit regulatory signals from DNA-bound transcription factors to the RNA polymerase II (RNAPII). The complex CDK8-Cyclin C (CDK8-CycC) associates with MED12 and MED13 to form the CDK8 module, a sub-module of the mediator complex [7,8,9]. In humans, it has been demonstrated in vitro that the CDK8 module inhibits the initiation of transcription by deactivating CDK7, which can no longer phosphorylate the carboxy-terminal domain of RNAPII, thereby blocking the transcription [10]. On the other hand, contrary to this transcriptional repression role, it has been observed in vivo, a positive regulatory role for CDK8 via the recruitment of SEC (Super Elongation Complex). In fact, the interaction of the mediator complex with SEC facilitates the elongation and release of certain genes [11,12]. In particular, CDK8-mediated activation of the Wnt-β-catenin signaling pathway [13] and of the transcription of estrogen-inducible genes [14] contribute, respectively, to oncogenesis in colorectal and mammary tumors, making CDK8 an oncogene of interest.
Since Schneider et al. published in 2011 the first crystallographic structure of human CDK8-CycC complexed with sorafenib (PDB id.: 3RGF) [15], a total of 31 experimental structures are currently available. All of these crystal structures present 10 to 20 missing residues within a region that lies outside the active-site cleft called the activation loop. This motif has a central role in regulating the activity of protein kinase by generally adopting a DFG-in conformation in the active form and a DFG-out conformation in the inactive form [16]. In that connection, the first computational study on human CDK8 (with PDB id.: 3RGF) aims at providing insights into two point mutations within the activation loop through 50 ns of all-atom conventional molecular dynamics (cMD) simulation in implicit solvent [17]. Moreover, the theoretical binding free energy between CDK8 and CycC was also determined using MM-PBSA and MM-GBSA on the basis of 2 ns of all-atom cMD simulation in explicit solvent. However, in in silico structural studies, a particular attention should be paid to the building of a relevant model of the protein, especially in this study [17] where the object of the investigation, the activation loop, is missing and has to be reconstructed. Surprisingly, the authors used a template where the activation loop is in DFG-in conformation to model the activation loop of 3RGF (PDB id.), which is in DFG-out conformation. Cholko et al. studied twelve CDK8-CycC systems using 500 ns all-atom cMD simulations in explicit solvent with the aim of elucidating the system motions and the structural determinants that affect protein-ligand interactions [18]. They find that the CycC is important in providing proper interactions for ligand binding whereas the highly flexible activation loop has a little effect. Furthermore, they employed MM-PBSA analysis to characterize protein-ligand interactions from an energetically point of view and discuss the major driving force of protein-ligand binding.
In this study, we investigated the effect of CycC on the structure and dynamics of CDK8 on the one hand, and on the other hand, the structural molecular basis of the protein-protein interaction between the two partners. Indeed, the presence of CycC in the CDK8-CycC complex seems to play a more complex role than for other members of the CDK family [19,20]. CDKs are generally activated in two steps: 1) the binding of the cyclin (Cyc) to CDK and 2) the phosphorylation of a threonine residue in the CDK activation loop (T160 in human CDK2). The binding of the Cyc to CDK induces a conformational change of the αC-helix, which adopts a αC-helix in conformation (shift toward the binding site) from an αC-helix out conformation. The phosphorylated threonine on the activation loop serves as an anchor for adjusting the orientation of three conserved arginine residues, inducing a conformational change in the activation loop that shifts from a DFG-out to a DFG-in conformation [21]. In CDK8 the phosphorylation step has not been observed and is not required to its activation [22,23]. Moreover, the first published crystallographic structures of human CDK8-CycC [15,24] and also a more recent one [25] display a surprising conformation corresponding somehow to the “intermediate state of the activation mechanism”. Indeed, the αC-helix is in αC-helix in conformation, which is expected since CycC is bound to CDK8 in agreement with the activation mechanism. However, the phosphorylation step did not occur due to the lack of the conserved threonine in the CDK8 sequence [22], leading to keeping a DMG-out conformation (in CDK8, a DMG motif replace the well-known DFG motif of protein kinases) for the activation loop. All of these structures are co-crystallized with an inhibitor which is told to be responsible of the conformational change from DMG-in to DMG-out conformation. Protein kinase inhibitors are classified based on their binding to their receptor [26]. Type I inhibitors bind to the ATP binding site, type II inhibitors extend from the ATP binding site into a neighboring pocket, the allosteric pocket (also called “hydrophobic pocket”) which is only accessible by the rearrangement of the DFG motif from the DFG-in to the DFG-out conformation. The type III inhibitors bind only to the allosteric pocket. All co-crystallized inhibitors of CDK8 belong to the type II or type III class of protein kinase inhibitors. As far as we know, CDK8 is the only CDK family member for which the following structure is obtained experimentally: a DFG-out conformation (DMG-out in CDK8) while being associated with CycC. All CDK structures complexed with Cyc are usually in DFG-in conformation in accordance with its activation mechanism. Alexander et al. try to reproduce this particular conformation with the complex CDK2-CycB. They incubate the CDK2-CycB complex with a type II inhibitor and also observe a DFG-out conformation. However, they find that binding of a type II inhibitor to CDK2-CycB results in the dissociation of cyclin B from CDK2 in a competitive manner [27]. All those observations raise the question about the role of CycC in the complex CDK8-CycC in the inactive conformation (DMG-out). Particularly, it is interesting to investigate whether the CycC has an impact on the structure and dynamics of CDK8. In addition, this impact is the same in active (DMG-in) and inactive (DMG-out) conformation and in the presence and absence of the ligand has to be explored. Furthermore, in view of this unique feature of CDK8 to bind the CycC in both conformations, it is relevant to study the interaction between CDK8 and CycC in order to decipher the interaction molecular basis and to highlight possible important CDK8-specific interaction hotspots. We may notice that the particular behavior of the CycC among the cyclin family had already been raised before. This led Barette et al. [28] to manage mutagenesis experiments that highlighted a double-point mutation of R65A/E66A in CDK8 that greatly affects its capacity to bind to CycC. This effect was partly explained by the X-ray structure which shows the contacts between Met61 and Arg65 in the human CDK8 and CycC.
Through MD simulations and binding free energy calculations, we found that CycC has a stabilizing effect on CDK8, and the importance for CDK8 to maintain a proper conformation in the active and inactive form of CDK8-CycC. The per residue free energy decomposition method enabled to characterize the CDK8-CycC binding surface, to identify the important residues and to obtain their energy contributions. We found that CDK8-CycC presents specific interaction hotspots within its interaction surface compared to other human CDK/Cyc pairs. Targeting these specific interaction hotspots could be a promising approach in terms of specificity, to effectively disrupt the interaction between CDK8 and CycC and thus, to interfere with the function of CDK8 as an oncogene. The simulation of the conformational transition from the inactive to the active form of CDK8-CycC through TMD simulation, suggests another mechanism that could substitute the missing phosphorylation step in the activation mechanism of CDK8. In a more general view, these results point the importance of keeping the CycC in computational studies when studying the human CDK8 protein in both the active and the inactive form.
2. Results and Discussion
2.1. Effect of CycC Exclusion on Structure and Dynamics of CDK8
In order to evaluate the effect of CycC on the structure and dynamics of CDK8, the trajectories were analyzed in pairs (with/without Cyc) as shown in the Table 1. For the systems in DMG-out conformation, the average RMSD is higher in the absence of the CycC, which means that CDK8 structure deviated more from its crystallographic structure in the absence of the CycC. For the system in DMG-in conformation, the average RMSD are comparable with and without CycC.
The RMSF plots (Figure 1 and Figure S7) indicates that the absence of CycC increases the motions of one or more of these regions of CDK8: 1) the αC-helix in all cases, which is in direct interaction with the CycC, 2) the αB-helix in all cases except in the system 1b; the αB-helix is also in direct interaction with the CycC and, 3) the activation loop in all cases, except in the system in DMG-in conformation (system 9).
In the absence of CycC, the αC-helix has a larger degree of motion and can move toward the region normally occupy by the CycC to adopt an αC-helix out-like conformation (Figure S6). The αB-helix tends to bend toward CDK8 and to interact with it; in system 1b, this leads to its stabilization (Figure S6).
In order to provide a global view of the effect of the presence of CycC on the structure of CDK8, each pair of trajectory system with and without CycC (1a/2a, 1b/2b, 3/4, 6/7 and 8/9) were combined in a single trajectory by extracting the backbone coordinates of CDK8 from both trajectories. Then a PCA was applied using the conditions described in Materiel & Methods section on each combined trajectory to see if the conformations coming from the simulation with CycC differ from those coming from the simulation without CycC.
In all 5 cases, we observe two groups formed along PC1, that correspond to the CDK8 conformations extracted from the simulations in presence and in absence of CycC respectively. An exemple of the PCA projection is presented Figure 2 for 2 combined trajectories among the 5. The PC1 is thus able to separate the CDK8 conformations according to the presence or not of the CycC in the simulation. We also notice that a larger scattering of the CDK8 conformations obtained in absence of CycC compare to the ones generated in presence of CycC. It means an increase of CDK8 conformational sampling in the absence of CycC. Moreover, in all cases the two first PCs capture more than 60% of the variance, and PC1 alone traduce more than 50% of the variance. Considering these results together, it appears that PC1 has captured the regions of CDK8 whose structure is the most affected by the presence/absence of CycC. It is therefore interesting to analyze the contribution of each residue of CDK8 to PC1, commonly called the “loading plot”, to identify these regions (Figure 2).
First of all, we remark that the PCA loading curves of DMG-out conformation systems display a good alignment (Figure 3B). Second, a common point emerges from all PCA loading curves: the αB-helix and the activation loop contribute greatly in both cases (DMG-in and DMG-out conformation systems) to separate the structures coming from the simulations performed with and without CycC. It means that the activation loop and the αB-helix adopt different conformations depending on whether CycC has been kept or not. Note that this observation does not mean that the activation loop and the αB-helix adopt each of them a unique conformation in the absence of CycC. As seen previously through the analysis of the RMSF, the activation loop and the αB-helix are more flexible in the absence of CycC. In average, their conformations sampled in the absence of CycC are significantly different from those adopted in the presence of CycC. Other regions contribute at varying level in DMG-in and DMG-out conformation systems to separate the two groups (with & without CycC) such as: the αF-αG loop that contributes greatly in the DMG-in conformation system and not in the DMG-out conformation systems, and the contrary for αD-αE loop. It is also interesting to note that the αC-helix, which is more flexible in the absence of CycC (cf. Figure 1 and Figure S7), does not show a significant different conformation in the absence of CycC for the DMG-out conformation systems (Figure 3).
In conclusion of this part, RMSF plots show that CycC stabilizes the αC-helix in both DMG-in and DMG-out conformation systems and the activation loop of CDK8 in DMG-out conformation system. It also reduces the fluctuations of the αB-helix, but in some cases no difference was observed between systems with/without CycC because the αB-helix bend toward CDK8 and stabilizes itself (Figure S6). The PCA analysis was able to separate CDK8 structures coming from the simulation performed with and without CycC, which highlights an effect of the CycC on the conformation of CDK8. Particularly, the CycC greatly affects the conformation adopted by the αB-helix and the activation loop. CycC also impacts the dynamics of CDK8 since the greatest amplitude motions within CDK8 are not the same depending on whether CycC is present or not (Figure S8 and S9). In the literature, Cholko et al. also pointed out the importance of the CycC for maintaining a proper structure and dynamics of CDK8. Through MD simulation on 12 CDK8-CycC systems (6 of DMG-in conformation and 6 of DMG-out conformation), they observed that CycC stabilizes CDK8 by reducing the fluctuations of the αB-helix, αC-helix and the activation loop. They also mentioned that αC-helix adopt a αC-helix out conformation in the absence of the CycC and pointed the importance of the CycC for maintaining proper protein-ligand interaction. Concerning this last point, we also find that the CycC stabilizes the ligand in the binding site (Figure S10). In a more general view, these results highlight the importance of keeping the CycC in computational studies.
2.2. Understanding the Molecular Basis of the Interaction between CDK8 and CycC
2.2.1. CDK8-CycC Binding Free Energy
To compute the binding free energy of CycC to CDK8 and gain insights into the binding interaction surface, the MM-GBSA approach was applied on the 9500 snapshots extracted from the trajectories in the range of 50ns-1μs (i.e., one snapshot every 100 ps). We want to know whether CycC has a stabilizing effect in term of binding free energy in: 1) the active form of CDK8-CycC complex (with CDK8 in DMG-in conformation), 2) the inactive form of the complex (with CDK8 in DMG-out conformation) and, 3) the mutated form of the complex CDK8R65A-E66A-CycC. In the presence of Cyc, the active form of CDK-Cyc complex is the form commonly observed in the crystallographic structures of human CDK family members, in agreement with the general activation mechanism of CDKs. In contrast, the inactive form of the CDK8-CycC complex is the first experimental structure exhibiting such conformation. The mutant CDK8R65A-E66A-CycC was designed based on experimental mutagenesis data published on CDK8-CycC complex and CDK4-CycD1 complex. A R55A-E56A double point mutation in the αC-helix of CDK4, corresponding to R65A-E66A in CDK8, decreased its binding activity towards cyclin D1 by 85 % [29]. On the basis of these results, Barette et al. introduce the R65A-E66A double point mutation in CDK8 and find that similarly to CDK4, this double point mutation greatly affects the capacity of CDK8 to bind to CycC. However, for the formed complex, they find that CDK8R65A-E66A is still able to stabilize the complex CDK8R65A-E66A-CycC [28]. We therefore calculated the binding energies for the different CDK8-CycC complexes (system 1a, 1b, 3, 5, 6, 8) and summarized the results in Table 2.
Only the enthalpy part of the binding energy was calculated here. Indeed, the relative contribution of the entropic term to the ΔΔG is considered to be negligible when comparing two similar systems for example in mutational studies or when comparing ligands that bind to the same binding site (as it is the case here), since both contributions are supposed to cancel each other [30]. Therefore, in this study, ΔG corresponds to the binding free energy without the entropic term. In agreement with our structural and dynamical observations, the binding free energy values range from -141.0 ± 0.2 to -124.6 ± 0.2 kcal.mol-1, which confirms the stabilizing effect of CycC. In particular, the result for the mutated system CDK8R65A-E66A is consistent with the experimental observations, which report that the double point mutation does not affect the stabilization of the complex. The non-polar part of the free energy, composed of the Van der Waals term in gas-phase (ΔEVDW) and the non-polar part of the solvation energy term (ΔEnp), is the major favorable component of the CycC binding. Its value is comprised between -171.5 kcal.mol-1 and -200.7 kcal.mol-1 depending on the system. The highly favorable non-polar part of the free energy might come from the desolvation of the non-polar groups at the binding interface between CDK8 and CycC, as well as the hydrophobic interactions formed between the two partners. Such phenomenon has been seen in several protein-protein interactions where the main interactions that are responsible for the binding of proteins are hydrophobic in nature [31,32]. On the other hand, the very favorable electrostatic term in gas phase (ΔEeel) is completely compensated by the unfavorable contribution of the polar part of the solvation free energy (ΔEGB), resulting in a unfavorable total electrostatics interaction comprised between 40.0 kcal.mol-1 and 69.5 kcal.mol-1 depending on the system. This compensation phenomenon due to the desolvation penalty of polar groups upon complex formation was discussed in several studies of protein-protein interactions [33].
2.2.2. CDK8-CycC Binding Free Energy: Decomposition Per Residue
The method of per-residue binding free energy decomposition can reveal the energy contribution of key residues involved in the protein-protein interaction interface. The total of 9500 snapshots extracted from the trajectories in the range of 50 ns - 1 μs (i.e., one snapshot every 100 ps) were decomposed by the MM-GBSA method. We first identify the common list of residues that significantly contribute to the CDK8-CycC binding in all the studied complexes (system 1a, 1b, 3, 5, 6, 8).
Common Hotspots to All Studied CDK8-CycC Complexes
For each of these systems, the important CDK8-CycC binding residues were extracted using the following condition as cut-off: the absolute value of ΔGtotal of the residue has to be superior to 1 kcal.mol-1. In the supporting information, the list of the extracted important residues of each system is represented as barplot (Figure S11). To extract the common list of important residues shared by all the studied complexes, we took the intersection of these different lists. The heat map presented in Figure 4 contains the common list of important residues (26 in total) and their binding free energy contributions.
The first obvious result is that no great difference in free energy values is seen between the different studied systems. Second, all the residues present a favorable contribution to CDK8-CycC binding. Moreover, the 26 residues are uniformly distributed on the interaction surface. These first observations suggest that the studied complexes share a large and similar surface of interaction.
Common Hotspots to CDK Family
The members of human CDK family share a conserved common interaction surface with their Cyc partner. This common interaction surface includes the β3–αC region, the αC-helix, and the post-αC region (β4–β5) of the CDK protein in contact with the H5-helix, H5-H1′ loop and the residues on both sides of the H3-helix of the Cyc [34,35]. 73.1 % of the identified common important residues of the CDK8-CycC interaction belong to this conserved core as we can see on the heat map (Figure 4). We subsequently analyzed the interactions between CDK8 and CycC involving these common important residues of the CDK family conserved core.
This conserved core is located at the center of the interaction surface and is mainly composed of hydrophobic residues. Among them, the central Phe140CycC situated on the CycC H5-helix seems to have a crucial role in CDK8-CycC binding. Indeed, in a parallel stacking, Phe140CycC establishes a cation-π interaction with Arg91CDK8 of the β4–β5 loop, both characterized by a high ΔG absolute value (Figure 4). This interaction has an average occupancy of about 78.8% ± 8.3 along all the simulations. It has been reported in the literature that a planar cation-π stacking between an arginine and an aromatic side chain may be a critical interaction for the function of a protein, particularly to allow the arginine to form other hydrogen bonds [36]. This is precisely the case here, since Arg91CDK8 also establishes a hydrogen bond with Glu137CycC in the H5-helix with occupancy of 75.6 % ± 12.2. Another residue of the CycC H5-helix, the Leu143CycC does hydrophobic contact with Cys64CDK8 of the αC-helix with occupancy of 55.7% ± 13.0. Concerning the H5-H1′ loop, a hydrogen bond is formed between Cys148CycC and Arg71CDK8 with occupancy of 87% ± 7.8 and a water bridge between Ile151CycC and Glu72CDK8 of the αC-helix with occupancy of 74 % ± 13.3%. Finally, in the C-terminus of the H3-helix, Lys96CycC interacts with Ile59CDK8 localized in β3–αC loop through a hydrogen bond with occupancy of 92.4% ± 7.6%.
In summary, the studied complexes (system 1a, 1b, 3, 5, 6, 8) display a large common binding surface composed of 26 residues distributed uniformly along the interaction surface. This common binding surface is also very similar since the free energy values present few variations from a system to another. All of the 26 residues contributed favorably to CDK8-CycC binding with free energy values ranging from -9.4 kcal.mol-1 to -1.0 kcal.mol-1. 73.1 % of those residues (19/26 residues) belong to the conserved common interaction interface in the human CDK/Cyc family. Interestingly, we found that the remaining 9 residues belong to regions that are specific to CDK8.
Specific Hotspots to CDK8-CycC
- involving the N-terminus segment of CycC
Although the cyclins are less similar in sequence among themselves compared with the CDKs, they share a common fold constituted of two cyclin boxes comprising five helices each (H1-H5 and H1′-H5′), which are generally associated with two additional helices at the N-terminus and at the C-terminus segments noted HN and HC, respectively (Figure 5). Unlike cell cycle cyclins (cyclin A/B/D/E), in transcriptional cyclins (cyclin C/T/K/H) [35], the HN is located on the side opposite to the CDK binding surface and is not involved in kinase recognition. However, in this case, the N-terminus of CycT is still able to maintain some contacts with CDK9. CDK8-CycC appears as an exception since the CycC N-terminus segment is part of the interaction surface positioned below the αC-helix and between CDK8 αE-helix and CycC H5-H1′ loop (Figure 5). A strong hydrogen bond interaction is observed between the Glu72CDK8 and the Ser9CycC with occupancy of 87 ± 9.3% along all the simulations.
- involving the CDK8-specific N-terminus helix (αB-helix)
CDK8 exhibits an additional N-terminus αB-helix (residues 1-12) preceding the αC-helix, which is unique within human CDK family members [15]. Other CDKs display a shorter N-terminus segment by 5-10 residues, except CDK9 where the segment is of equal length, but unstructured (random coil). Among the identified common important binding residues (Figure 5), many of them interact with the αB-helix. Particularly, we observed interactions between the proline rich C-terminus segment of CycC and the αB-helix. The Pro260CycC and the Ser80CycC establish both a hydrogen bond with Asp2CDK8 with an average occupancy of 82.1% ± 10.3 and 79.1% ± 10.4, respectively. The Lys261CycC interacts with Tyr3 CDK8 and Asp4CDK8 with an average occupancy of 83.4% ± 9.9 and 73.3% ± 11.1, respectively. The CDK8 αB-helix also forms a hydrophobic interaction particularly the Leu9CDK8 with Phe140CycC with occupancy of 88.2% ± 7.5.
Taking these results together, it appears that strong and favorable interactions are formed between the proline rich C-terminus segment, which shows a dramatic divergence in length and in orientation among CycC partners, and the CDK8 specific αB-helix. Together with the contacts involving the N-terminus segment of CycC, these strong interactions are specific to CDK8-CycC complex and could be one of the mechanisms explaining the selectivity of CDK8 against CycC. Indeed, unlike CDK2 which can bind different Cyc partners (Cyc A/B/E) [37], CDK8 is specific to CycC. Moreover, experimental mutational studies converge with our observations since the mutant CDK8-CycC complex missing the αB-helix (first 22 residues in the N-terminus segment of CDK8) has an affinity of 300.71 nM against 7.05 nM for the native complex [15]. Thus, in addition to mediating a specific interaction between the CDK8 and CycC, the αB-helix contributes also to ensure a tight binding between CDK8 and CycC. For comparison, the affinities of native CDK9-CycT1, CDK2-CycA and CDK7-CycH are weaker by at least 1 order of magnitude: 300 nM [38], 52 nM and 57 nM respectively [39]. It is generally assumed that a high affinity to a partner compared to other homologous partners, leads to a highly specific binding to the considered partner. It may be achieved by small structural variations, as it seen to be here, with the CDK8-CycC recognition αB-helix. Targeting the highlighted specific interaction hotspots between CDK8 and CycC could be a promising approach to design a peptide inhibiting specifically the CDK8-CycC activity by preventing the binding of CDK8 to CycC. Two peptides targeting the CDK2-CycA interface were reported but none of them has yet made it to the clinic. The first one binds at the core of the common binding surface, at the αC-helix/H5-helix interface [40]. The second one targets a surface pocket in CycA, which is a structurally conserved domain comprising H3, H4 and H5 helix of cyclin A [41].
Difference in Binding Surface between the Different Complexes
After deciphering the common molecular features of the CDK8-CycC interaction surface, we want to assess now if a significant difference exists between the binding surfaces of the studied complexes. In order to highlight possible differences in energy contribution of the residues, we extract the list of the residues that form at least one significant interaction (using the same cutoff as above: absolute (ΔG) > 1 kcal.mol-1) in one of the studied complexes. In other words, instead of taking the intersection of the lists of important residues of each system, as we did previously to get the common molecular features, we took the union of these lists. The resulted matrix has been attached in the supporting information (Figure S12). To compare the contributions of the residues of each system with each other in a convenient way, we calculated a correlation matrix from the contribution matrix and present the results as a scatterplot matrix (Figure 5).
DMG-Out CDK8-CycC Complexes
The residues of DMG-out conformation complexes (1a, 1b, 3, 5, 6) display very similar energy contribution values since the correlation coefficients are comprised between 0.74 and 0.90. The mutated DMG-out conformation complex (system 5) does not exhibit significant difference with the others native DMG-out conformation complexes (1a, 1b, 3, 6) in terms of energy contribution of residues. Indeed, it presents a correlation coefficient always superior to 0.74 against them. It is particularly close to system 6 (correlation coefficient = 0.88). Moreover, the double-point mutation (CDK8R65A_ E66A) does not significantly affect the binding interaction network between CDK8 and CycC. Therefore, the mutant complex presents a similar stability (Table 2) associated with a similar binding interaction network compared to native systems. Together, these results indicate that the DMG-out conformation complexes share a similar binding interaction surface.
Difference in Binding Surface between DMG-in and DMG-Out
Although the studied CDK8-CycC complexes share a large common interaction surface, as we detailed previously, the distribution of the energy contribution of the residues of the DMG-in complex is the least correlated with that of others complexes, with a correlation coefficient comprised between 0.51 and 0.63. In the DMG-in conformation complex, the CycC is slightly shifted towards CDK8 as shown in Figure 6. This shift increases the contacts between CycC H3-H4 loop and the CDK8 activation loop, which is folded towards the CycC in DMG-in conformation.
As a consequence, Arg178CDK8 and Pro183CDK8 of the activation loop that did not contribute to CDK8-CycC binding in the DMG-out conformation complexes are now close to CycC and present a favorable contribution (Figure S12). Moreover, the shift of the CycC modifies the interaction network at CDK8-CycC interface which might explain for some residues a change in their energy contribution: Arg13CDK8 of the αB-helix, Leu86CDK8 on the β4 strand, Asn145CDK8, Trp146CDK8 at the C-terminus of the αE-helix, Ala2CycC and Gly3CycC of the N-terminus segment and three residues at the N-terminus of CycC H3-helix (Ile81, Asp82 and leu85) (Figure S12). Interestingly, others residues that are far from the interaction surface but part of the binding site also display a difference in their energy contribution in the DMG-in conformation system compared to the DMG-out one: the Val27CDK8 and the Val35CDK8 which are part of the P-loop, Tyr99CDK8 and Ala100CDK8 in the hinge region and Arg356CDK8 of the C-terminus of CDK8.
2.3. Activation Mechanism of CDK8
In the DMG-in conformation complex, we observe that the shift of the CycC toward CDK8 allow the Glu99CycC to be closer to Arg65CDK8. Glu99CycC establishes hydrogen bonds with Arg65CDK8, Arg178CDK8 and to a lesser extent with Arg150CDK8. The three arginines also interact with each other through water-mediated hydrogen bonds. This interaction network is maintained over time (Figure S13) and could therefore have a role in the stabilization of the activation loop in the DMG-in conformation. In this context, we turn to literature to find a possible known role of Arg65CDK8, Arg150CDK8 and Arg178CDK8 in the activation mechanism of CDK8. In that regard, it was reported that these three arginines are conserved within human CDK members and are involved in the second step of the activation mechanism [22]. As mentioned earlier in the introduction, the second step of the general activation mechanism of CDKs is the phosphorylation of a residue within the activation loop. The phospho-residue serves as anchor to adjust the orientation of three conserved arginines, thereby inducing a DMG-in conformation of the activation loop. In CDK8, these three conserved arginines are Arg65CDK8, Arg150CDK8 and Arg178CDK8. However, since in CDK8 there is no phosphorylation, on the basis of crystallographic structure analysis, Glu99CycC was hypothesized to mimic the missing phospho-residue within CDK8 and serves as anchor to adjust the orientation of the three important arginines, Arg65CDK8, Arg150CDK8 and Arg178CDK8 in CDK8 [22]. The stable interaction network formed by the three arginines and Glu99CycC observed during the MD simulation supports this hypothesis.
To further investigate this hypothesis and brought a dynamical view of the process, we simulate through targeted molecular dynamic simulation the conformational transition from a DMG-out conformation complex to a DMG-in one. The restraint was applied only on the activation loop (and not on the whole complex) because we want to verify if a relationship exists between the shift of the CycC and the conformational change of the activation loop (residue 171 to 182). We first check on the stability of the protein structure over time during the TMD simulation, by verifying the RMSF, the RMSD of the protein and the restraint potential over time (Figure S14). The DMG-in conformation obtained through TMD simulation followed by 50 ns of cMD is in agreement with the one of system 8 (Figure S15).
To monitor the shift of the CycC toward CDK8, we measure the distance between Glu99CycC and a stable residue of CDK8, the Lys153CDK8 (according to its RMSF, cf Figure 1). As the activation loop gets closer to the CycC, the CycC shifts toward CDK8 as shown by the Lys153CDK8 - Glu99CycC distance curve over time (Figure 7). At the beginning of the TMD simulation, Arg178CDK8 first interacts with the Arg150CDK8 and at this stage the CycC already undergoes a small shift (≈ 2.5 Å). This displacement of the CycC enables the Glu99CycC to become closer to Arg65CDK8 and to optimize its interaction with it. Then we observe that the interaction of Arg178CDK8 with Arg65CDK8 and Glu99CDK8 occurs at the same time as the second shift (≈ 6 Å) of the CycC toward CDK8. Therefore, the displacement of the CycC might be an important event to adjust the orientation of the three conserved arginine residues. During the following 50 ns of cMD production, the Glu99CycC–mediated hydrogen bond interaction network stabilizes and the same interaction network as in the system 8 is formed (Figure S13): Arg178CDK8 becomes sandwiched between Arg150CDK8 and Arg65CDK8 and the three arginines forms hydrogen bonds with Glu99CycC (Figure 7). The three arginines also interact with each other through water-mediated hydrogen bonds. It may be noted that finding this network is not trivial since only the activation loop (residue 171 to 182), and therefore only Arg178CDK8 was submitted to the restraint potential (Arg150CDK8, Arg65CDK8 and Glu99CycC were not under restraint).
From these results, it appears that the Glu99CycC and the shift of the CycC are important to orient and stabilize the three conserved arginines known to be involved in the second step of the general activation mechanism of other CDK members. Therefore, our observations support the hypothesis that the Glu99CycC in CDK8 mimics the missing phospho-residue whose role is to adjust the orientation of three conserved arginines thereby inducing a DFG-in conformation of the activation loop. In addition to that, our results suggest that a shift of the CycC toward the CDK8 is also required to obtain the active form of CDK8-CycC.
3. Materials and Methods
3.1. Material Description
The catalytic site of CDK8 lies between the N- and C-terminal lobes as in other kinase proteins. Two conformations of CDK8 exist in the PDB that differentiate by the conformation of the activation loop that adopts either a DMG-in or a DMG-out conformation. CycC interacts mainly with the N-terminal lobe (Figure 8). The studied systems are summarized in Table 3. The corresponding crystallographic structures are all coming from the paper of Schneider et al. [24]. The structure 4F6U (PDB id.) presents the best resolution among all DMG-out structures resolved up to now. This structure is co-crystallized with a type II inhibitor (system 1a and 1b). To be sure that the results obtained are not ligand-dependent, the apo form of 4F6U (system 3) and another type II inhibitor (PDB id.: 4F7L) (system 6) with a slightly different binding mode (Figure S1) were also simulated. Then, to compare our results with experimental mutagenesis results, two residues of the αC-helix were mutated in the structure 4F6U (system 5). Finally, a DMG-in conformation of the complex (PDB id.: 4F7S), which is the conformation usually observed in the presence of CycC, has also been simulated in order to compare the behavior of CycC in the complexes DMG-in and DMG-out conformation (system 8). These systems were also modeled without the CycC in order to investigate the effect of the CycC (except the system 5).
3.2. Model Building
The structure of PDB id. 4F6U presents 3 missing loops: the activation loop containing the key DMG-motif (residues 177 to 193) and the loops from residues 116 to 120 and residues 240 to 244. In order to reconstruct these missing residues, we aligned the UniProt canonical sequence of CDK8 on the PDB database to retrieve the most homologous template structures having the missing regions resolved and the activation loop in the DMG-out conformation. Two crystallographic structures of the human homologous CDK6 (PDB id.: 1BI8 and 1G3N), were retained and used as template structures. The sequence alignment was performed with Clustal Omega [42] with a particular care on the alignment of domain kinase conserved motifs. CDK6 shares 37 % of identity and 63 % of similarity with CDK8 (Figure S2 and S3). Only missing regions in the target structure were rebuilt in order to keep the coordinates of the resolved parts of the protein unchanged. The sequence of CycC and the information of the presence of crystallographic molecules of water and of a ligand (ligand id. 0SR) were conserved during the modelling of the missing part of CDK8. Finally, MODELLER version 9.16 [43] was used in order to generate the model. We thus obtain a model of CDK8 (residues 1 to 359) complexed to CycC (residues 1 to 264) and to the ligand. The missing C-terminus segments of CDK8 (residues 360 to 464) and of CycC protein (residues 265 to 283) were not reconstructed. The complete model was subjected to structural validation through PROCHECK [44] and ProSA-web tools [45] (Figure S4). We did not build another model for the structure of PDB id. 4F7L but rather derive the model by replacing the inhibitor 0SO in that model (chemical replacement). Chemical replacement was considered sufficient because the orientation of the binding site residues is highly conserved in the two structures (PDB id.: 4F6U and 4F7L); and their respective inhibitors (ligand id.: 0SR and 0SO) share a same scaffold bound in the same orientation within the binding site (Figure S1). Therefore, the full structure of CDK8-CycC complexed with the ligand 0SO was obtained by, first, aligning the crystallographic structure 4F7L to the model, and then, by placing the ligand and the crystallographic molecules of water inside. We manually adjust some residues to be in agreement with protein-ligand interactions observed in the crystallographic structure of PDB id. 4F7L using Molecular Operating Environment (MOE) version 2016.0802 from the Chemical Computing Group. The same procedure than the one described above was followed to fill the 3 missing loops of the structure of PDB id. 4F7S (which are the activation loop residues from 187 to 195 and the loops from residues 116 to 121 and from residues 238 to 242). The crystallographic structures of the human homologous CDK1 (PDB id. 1P5E) and CDK2 (PDB id. 1P5E) were retained and used as template structures. CDK1 and CDK2 share respectively 37.8%, 38.3% of identity and 54.5%, 55.9% of similarity with CDK8.
3.3. System Preparation
In total, 9 systems were prepared (all described in Table 3). The AmberTools 14 suite [46] was employed to protonate, solvate, neutralize and generate topology and coordinate files of the systems. Ligands were prepared by using the Antechamber tool and the GAFF force field after adding hydrogen atoms with the reduce utility [47,48]. The three inhibitors were modeled in their neutral state. Further analysis was carried out for the protonation state of the inhibitor 0SR (ligand id.) (Figure S5), since the pKa of alkylmorpholines is about 7.4 [49]. The morpholine of the inhibitor 0SR was finally modeled in its unprotonated state. Partial charges on the ligands were generated with the AM1/BCC method [50]. PROPKA version 3.0 [51] was used to check the protonation state of ionizable residue side-chains at pH = 7. The protein force field ff14SB parameters were assigned [52]. Then, the system was solvated in a rectangular TIP3P water box, the side of the box being at least 10 Å away from any solute atom. Finally, Cl- ions were added to neutralize the positively charged system for a total number of atoms around 110 000 atoms.
3.4. Conventional MD Simulation (cMD)
A four-cycle minimization was performed with 2000 steps each cycle, minimizing first the solvent, second the residue side-chains, then the solute and finally the entire system. The SHAKE algorithm was applied to constrain bonds involving hydrogen atoms, allowing a time increment of 2 fs. Temperature regulation at 300 K was ensured through Langevin dynamics with a collision frequency of 2 ps-1. The long-range electrostatic interactions were computed by the Particle Mesh Ewald (PME) method beyond 10 Å distance. The system was slowly heated in NVT ensemble from 0 to 300 K over a period of 50 ps, where a harmonic restraint on the solute (20 kcal.mol-1.Å-1 force-field constant) prevents the system from structural distortion. The system was then equilibrated during 10 ns MD simulation in the NPT ensemble at 300 K and 1 atm, through which the harmonic restraint is gradually decreased from 20 kcal.mol-1.Å-1 to 3 kcal.mol-1.Å-1 in 1.3 ns and then, totally relaxed during 8.7 ns. The pressure relaxation time was set to 1 ps. cMD calculations were performed using the PMEMD.cuda module of the AMBER14 program [46]. We performed 1 μs of cMD production on each system presented in Table 3 and save the coordinate every 10 ps.
3.5. Targeted Molecular Dynamics (TMD)
The TMD is a simulation technique for determining the pathway of a conformational transition between two states: (un)bound, (un)folded, open/close conformation etc. [53]. It consists in constraining the root mean square deviation (RMSD) between the current structure (which is the starting structure at the beginning of the simulation) and a reference structure (RMSDcurrent) to a user-defined value, namely the RMSDtarget. This value of RMSDtarget is slowly varied from an initial value to a targeted final value (RMSDtarget_final), which results in the simulation of the process leading to the final desired state. In AMBER14 program, a harmonic restraining potential (Vrestraint) is added to the force field, to help the RMSDcurrent reaching the successive values of RMSDtarget until the final value (RMSDtarget_final).
where f is the harmonic force constant, Natoms is the number of restrained atoms, that is, the number of atoms on which the RMSD is calculated. Note that the atomic coordinates are mass weighted in the calculation of RMSD. It exists two approaches of TMD: direct TMD and reverse TMD (TMD-1). We apply direct TMD. In direct TMD, the reference structure corresponds to the final targeted structure, so that the value of RMSDtarget is decreased from the RMSD between the initial and target structure to a value close to 0. In this study, the initial structure is the complex CDK8-CycC in DMG-out conformation and the target structure is that in DMG-in conformation. The RMSD is calculated on the residues 171 to 182 of the activation loop, after aligning the current and the target structure on the backbone of the less flexible residues of the active site (90 residues in total: residue 26 to 105 and 148 to 158). The spring constant f was set to 2 kcal.mol-1. The RMSDtarget is changed by step of 0.12 Å every 50 ps from the value of 12.3 Å to 0.01 Å during a total simulation time of 5 ns. TMD runs were performed with the parallelized version of the SANDER module from the AMBER14 program. The TMD simulation was then continued by 50 ns of cMD simulation following the same parameters as described above.
3.6. RMSD, RMSF
The trajectories were aligned on the corresponding crystallographic structures using the heavy atoms of CDK8 as mask. The root mean square deviation (RMSD) and the root mean square fluctuation were calculated using the same mask.
3.7. PCA
When applying MD simulations on biological system, some questions are often raised: i) are the sampled conformations in one MD replicate similar to those extracted from a second replicate ii) does the conformational sampling vary over time within a same trajectory, iii) what are the protein regions whose movements contribute the most to explain the conformational diversity. To answer such questions, the principal component analysis method (PCA) is a suitable method. PCA is a linear dimensionality reduction technique that linearly combines a set of variables (here the coordinates of CDK8 backbone residues) into a reduced number of uncorrelated variables called principal components (PCs). The PCs correspond to the directions of largest variance that is the largest-amplitude fluctuations. To obtain the PCs, we first extract the CDK8 backbone of the last 500 ns of a trajectory by selecting one snapshot every 2.5 ns (200 snapshots in total). Trajectories of the system simulated in absence and presence of cycC are concatenated, leading to a total of 400 snapshots. It is important to align the trajectories to be analyzed on a same referential. Then, a covariance matrix is calculated from the atomic coordinate matrix of the trajectory. The eigenvectors of the covariance matrix are the PCs. The PCs are ordered with PC1 the direction of largest variance, PC2 the direction of second largest variance etc. To visualize the largest amplitude motions, a PDB format trajectory has been produced that interpolates between the most dissimilar structures in the distribution along PC1. PCA analysis were performed with bio3d package [54].
3.8. MM-GBSA
The molecular mechanics generalized Born surface area continuum solvation (MM-GBSA) method supplied with AMBER was used to calculate protein-protein free energy [55]. 9500 snapshots were extracted from the trajectories in the range of 50ns-1μs (i.e., one snapshot every 100 ps). The binding free energy is calculated as follows:
where corresponds to the average of the total free energy of the component x over snapshots taken from the MD trajectory. The total free energy of each molecule is computed from the following equation:
where is the molecular mechanical energy, is the solvation free energy and the term is the entropic contribution. The solvation free energy is the sum of polar and non-polar contributions. The non-polar contribution is attributed to cavity formation in the solvent and to van der Waals interactions between the solute and the solvent, which are typically calculated from the solvent-accessible surface area. The polar contribution of is obtained following the generalized Born model [56] available in AMBER.
While the molecular mechanics energy term can be easily obtained from the results of a molecular dynamics simulation, the entropic term is often difficult to achieve. It can be approximated by a quasi-harmonic approximation or calculated through a normal mode analysis. However, the calculation is time-consuming and can be affected by large errors. Such a calculation was not considered in this study.
3.9. Others Analysis Tools
4. Conclusion
Theoretical studies were conducted on the human CDK8-CycC complex in order to provide more understanding about the binding of CycC to CDK8 that is an important target in cancer therapy. We first investigated the role of CycC on the structure and dynamics of CDK8. We found that the CycC is crucial for maintaining a proper structure and dynamics of CDK8 in both the active (DMG-in) and inactive (DMG-out) form of the complex. Unlike CDK2, where the binding of a type II inhibitor to CDK2-CycB results in the dissociation of CycB from CDK2 in a competitive manner [27], Schneider et al. have shown that the binding of a type II inhibitor to CDK8-CycC does not dissociate CycC [24]. Our findings converge to this result since the presence of a type II inhibitor does not affect the stabilizing effect of the CycC toward CDK8. The free energy values of CDK8-CycC binding calculated through the MM-GBSA method confirm these results, and show that the CycC stabilizes both CDK8 forms (active and inactive) to the same extent.
The analysis of the interaction between CDK8 and CycC, through the method of per-residue binding free energy decomposition, highlighted 26 hotspot residues uniformly distributed on the interaction surface, that strongly and favorably (ΔGtotal < -1kcal.mol-1) contribute to CDK8-CycC binding in all studied CDK8-CycC complexes. 19 of the 26 important residues belong to the conserved common interaction surface in the human CDK family. On the contrary, the remaining 7 hotspot residues are situated in two binding sites of the interaction surface that are specific to CDK8-CycC complex and involve the proline rich C-terminus segment, the CDK8 αB-helix and the N-terminus segment of CycC. These key amino acids proposed in this work are valuable information to design an inhibitor, that will effectively prevent the binding of the CycC to CDK8, which will block the activation of the complex, thereby interfering with the function of CDK8 as an oncogene. The active and the inactive forms display some differences in their CDK8-CycC binding energy contribution values. These differences might be explained by the flip of the activation loop from a DMG-out to a DMG-in conformation and the displacement of the CycC toward CDK8 in the active form.
The simulation of the conformational transition from the inactive to the active form through TMD simulation showed that this displacement of the CycC toward CDK8 occurs during the conformational change. This displacement is an important event to adjust the orientation of three conserved arginine residues (Arg65CDK8, Arg178CDK8 and Arg150CDK8), which is meditated by the Glu99CycC, thereby inducing a DMG-in conformation (active form). The active form is maintained through a hydrogen bond interaction network involving the three arginines and the Glu99CycC. In human CDK family, the three conserved arginine residues, together with a phosphorylated residue, are known to have a role in the conformational change of CDK and in the stabilization of the active form. Our TMD simulation suggests that Glu99CycC assumes the role of the missing phosphorylated residue in CDK8.
Our study provides interesting molecular insights, describing the interaction between CDK8 and CycC in terms of structure and energy. Since this interaction is essential to the activity of CDK8, the particular characteristics of this interaction and of its mechanism of activation highlighted in this study, are valuable information to design specific compounds targeting the CDK8/CycC interface. In a more general view, these results point the importance of keeping the CycC in computational studies when studying the human CDK8 protein in both the inactive and active form.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Binding site of the crystal structure of human CDK8 in complex with inhibitors 0SR and 0SO from respectively PDB id. 4F6U and 4F7L; Figure S2: Alignment of the UNIPROT sequence of the human Cyclin C with the Cyclin C sequence of 4F6U (PDB id.); Figure S3: Alignment of the human sequences of CDK8 (canonical UNIPROT sequence and 4F6U (PDB id.) sequence) with the template sequences of human CDK6 (1BI8 and 1G3N (PDB id.)); Figure S4: Model validation; Figure S5: Difference in protein-ligand interaction between the form non-protonated and protonated of the morpholine of inhibitor 0SR; Figure S6: Conformational changes observed in the absence of CycC; Figure S7: Comparison of the root mean square atomic fluctuations of CDK8 in the presence/absence of CycC; Figure S8: Largest fluctuation motions (PC1) in the DMG-out conformation complex in the presence (left) and absence (right) of CycC; Figure S9: Largest fluctuation motions (PC1) in the DMG-in conformation complex in the presence (left) and absence (right) of CycC; Figure S10: Distribution of the ligand RMSD in the presence and absence of CycC; Figure S11: Energy contribution of important residues of each studied CDK8-CycC complex; Figure S12: Matrix of the per-residue energy contribution (ΔG without entropy in kcal.mol-1) of the residues that present at least one significant energy contribution (absolute(ΔG) > 1 kcal.mol-1) in one of the studied CDK8-CycC complexes; Figure S13: Interaction network between the three conserved arginines of CDK8 (Arg65CDK8, Arg150CDK8 and Arg178CDK8) and the glutamate 99 of the CycC in the DMG-in conformation system; Figure S14: Analysis of the stability of the protein structure over time during the TMD simulation through the investigation of the RMSF, RMSD and the restraining potential (Vrestraint); Figure S15: Comparison of the structures of CDK8-CycC in DMG-in conformation obtained from TMD and cMD simulation.
Author Contributions
Conceptualization, S.Z., P.B. and S.A.S.; methodology, S.Z., J.D. and S.A.S.; validation, S.Z., J.D. and S.A.S.; formal analysis, D.S. and S.Z.; investigation, D.S. and S.Z.; resources, P.B.; data curation, D.S. and S.Z.; writing—original draft preparation, S.Z.; writing—review and editing, S.Z., J.D., P.B. and S.A.S.; visualization, S.Z., J.D., P.B. and S.A.S.; supervision, P.B. and S.A.S.; project administration, P.B..; funding acquisition, P.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Institut de Recherche Servier.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All PDB structures used to build the initial models of MD simulations were downloaded from the RCSB protein data bank (https://www.rcsb.org). Homology modeling was realized by using the Modeler software (https://salilab.org/ modeller/). The licensed Amber14 was used to perform MD simulation (https://ambermd.org). The publicly available AmberTools15 was used to analyze the MD trajectories, in addition with VMD (https://www.ks.uiuc.edu/Research/ vmd/). Data simulations are available upon demand by mailing the corresponding author (S.A.S).
Acknowledgments
Authors thanks the Orléans-Tours CaSciModOT at the Centre de Calcul Scientique de la Région Centre Val de Loire and the Centre Régional Informatique et d’Applications Numériques de Normandie (CRIANN) for providing computer facilities, ChemAxon for providing the academic license free of charge and also the projects CHemBio (FEDER-FSE 2014-2020-EX003677), Valbiocosm (FEDER-FSE 2014-2020-EX003202), Techsab (FEDER-FSE 2014-2020-EX011313), QUALICHIM (APR-IA-PF 2021-00149467), the RTR Motivhealth (2019-00131403) and the Labex programs SYNORG (ANR-11-LABX-0029) and IRON (ANR-11-LABX-0018-01) for their financial support of ICOA, UMR 7311, University of Orléans, CNRS.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Malumbres, M. Cyclin-Dependent Kinases. Genome Biol 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK Inhibitors in Cancer Therapy, an Overview of Recent Development. Am J Cancer Res 2021, 11, 1913–1935. [Google Scholar] [PubMed]
- Canavese, M.; Santo, L.; Raje, N. Cyclin Dependent Kinases in Cancer: Potential for Therapeutic Intervention. Cancer Biol. Ther. 2012, 13, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Philip, S.; Kumarasiri, M.; Teo, T.; Yu, M.; Wang, S. Cyclin-Dependent Kinase 8: A New Hope in Targeted Cancer Therapy? J. Med. Chem. 2018, 61, 5073–5092. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, T.; Mikula, M.; Wiklik, K.; Brzózka, K. CDK8 Kinase—An Emerging Target in Targeted Cancer Therapy. Biochim. Et Biophys. Acta (BBA) - Proteins Proteom. 2015, 1854, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Taatjes, D.J. The Mediator Complex: A Central Integrator of Transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Harper, T.M.; Taatjes, D.J. The Complex Structure and Function of Mediator. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Ding, Z.; Ji, J.; Sun, Q.; Cai, G. Structural Flexibility and Functional Interaction of Mediator Cdk8 Module. Protein Cell 2013, 4, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Friedson, B.; Cooper, K.F. Cdk8 Kinase Module: A Mediator of Life and Death Decisions in Times of Stress. Microorganisms 2021, 9, 2152. [Google Scholar] [CrossRef] [PubMed]
- Akoulitchev, S.; Chuikov, S.; Reinberg, D. TFIIH Is Negatively Regulated by Cdk8-Containing Mediator Complexes. Nature 2000, 407, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Furumoto, T.; Tanaka, A.; Ito, M.; Malik, S.; Hirose, Y.; Hanaoka, F.; Ohkuma, Y. A Kinase Subunit of the Human Mediator Complex, CDK8, Positively Regulates Transcriptional Activation. Genes Cells 2007, 12, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kung, C.; Fishburn, J.; Ansari, A.Z.; Shokat, K.M.; Hahn, S. Two Cyclin-Dependent Kinases Promote RNA Polymerase II Transcription and Formation of the Scaffold Complex. Mol. Cell. Biol. 2004, 24, 1721–1735. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 Is a Colorectal Cancer Oncogene That Regulates Beta-Catenin Activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.S.J.; Chumanevich, A.A.; Lim, C.-U.; Liang, J.; Chen, M.; Altilia, S.; Oliver, D.; Rae, J.M.; Shtutman, M.; Kiaris, H.; et al. Inhibition of CDK8 Mediator Kinase Suppresses Estrogen Dependent Transcription and the Growth of Estrogen Receptor Positive Breast Cancer. Oncotarget 2017, 8, 12558–12575. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.V.; Böttcher, J.; Blaesse, M.; Neumann, L.; Huber, R.; Maskos, K. The Structure of CDK8/CycC Implicates Specificity in the CDK/Cyclin Family and Reveals Interaction with a Deep Pocket Binder. J. Mol. Biol. 2011, 412, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kornev, A.P. Protein Kinases: Evolution of Dynamic Regulatory Proteins. Trends Biochem Sci 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Amire-Brahimi, B.; Xie, X.-J.; Huang, L.; Ji, J.-Y. All-Atomic Molecular Dynamic Studies of Human CDK8: Insight into the A-Loop, Point Mutations and Binding with Its Partner CycC. Comput. Biol. Chem. 2014, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cholko, T.; Chen, W.; Tang, Z.; Chang, C.A. A Molecular Dynamics Investigation of CDK8/CycC and Ligand Binding: Conformational Flexibility and Implication in Drug Discovery. J Comput Aided Mol Des 2018, 32, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tang, H.-C.; Tsai, K.-L. Unveiling the Noncanonical Activation Mechanism of CDKs: Insights from Recent Structural Studies. Front. Mol. Biosci. 2023, 10, 1290631. [Google Scholar] [CrossRef] [PubMed]
- Ježek, J.; Smethurst, D.G.J.; Stieg, D.C.; Kiss, Z.A.C.; Hanley, S.E.; Ganesan, V.; Chang, K.-T.; Cooper, K.F.; Strich, R. Cyclin C: The Story of a Non-Cycling Cyclin. Biology 2019, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Pavletich, N.P. Mechanisms of Cyclin-Dependent Kinase Regulation: Structures of Cdks, Their Cyclin Activators, and Cip and INK4 Inhibitors. J. Mol. Biol. 1999, 287, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Hoeppner, S.; Baumli, S.; Cramer, P. Structure of the Mediator Subunit Cyclin C and Its Implications for CDK8 Function. J. Mol. Biol. 2005, 350, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, F.S.; Gnad, F.; Olsen, J.V.; Hornberger, R.; Greff, Z.; Kéri, G.; Mann, M.; Daub, H. Large-Scale Proteomics Analysis of the Human Kinome. Mol Cell Proteom. 2009, 8, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.V.; Bottcher, J.; Huber, R.; Maskos, K.; Neumann, L. Structure-Kinetic Relationship Study of CDK8/CycC Specific Compounds. Proc. Natl. Acad. Sci. 2013, 110, 8081–8086. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, P.; Koehler, M.F.T.; Blackwood, E.M.; Bowman, K.; Clark, K.; Firestein, R.; Kiefer, J.R.; Maskos, K.; McCleland, M.L.; Orren, L.; et al. Design and Development of a Series of Potent and Selective Type II Inhibitors of CDK8. ACS Med. Chem. Lett. 2016, 7, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Classification of Small Molecule Pro-Tein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Alexander, L.T.; Möbitz, H.; Drueckes, P.; Savitsky, P.; Fedorov, O.; Elkins, J.M.; Deane, C.M.; Cowan-Jacob, S.W.; Knapp, S. Type II Inhibitors Targeting CDK2. ACS Chem. Biol. 2015, 10, 2116–2125. [Google Scholar] [CrossRef]
- Barette, C.; Jariel-Encontre, I.; Piechaczyk, M.; Piette, J. Human Cyclin C Protein Is Stabilized by Its Associated Kinase Cdk8, Independently of Its Catalytic Activity. Oncogene 2001, 20, 551–562. [Google Scholar] [CrossRef]
- Coleman, K.G.; Wautlet, B.S.; Morrissey, D.; Mulheron, J.; Sedman, S.A.; Brinkley, P.; Price, S.; Webster, K.R. Identification of CDK4 Sequences Involved in Cyclin D1 and P16 Binding. J. Biol. Chem. 1997, 272, 18869–18874. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Computational Alanine Scanning To Probe Protein−Protein Interactions: A Novel Approach To Evaluate Binding Free Energies. J. Am. Chem. Soc. 1999, 121, 8133–8143. [Google Scholar] [CrossRef]
- Lo Conte, L.; Chothia, C.; Janin, J. The Atomic Structure of Protein-Protein Recognition Sites. J. Mol. Biol. 1999, 285, 2177–2198. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Lin, S.L.; Wolfson, H.J.; Nussinov, R. Studies of Protein-Protein Interfaces: A Statistical Analysis of the Hydrophobic Effect. Protein Sci. 1997, 6, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Kundrotas, P.J.; Alexov, E. Electrostatic Properties of Protein-Protein Complexes. Biophys J 2006, 91, 1724–1736. [Google Scholar] [CrossRef] [PubMed]
- Echalier, A.; Endicott, J.A.; Noble, M.E.M. Recent Developments in Cyclin-Dependent Kinase Biochemical and Structural Studies. Biochim. Biophys. Acta 2010, 1804, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Lolli, G. Structural Dissection of Cyclin Dependent Kinases Regulation and Protein Recognition Properties. Cell Cycle 2010, 9, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Flocco, M.M.; Mowbray, S.L. Planar Stacking Interactions of Arginine and Aromatic Side-Chains in Proteins. J. Mol. Biol. 1994, 235, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.J.; Endicott, J.A. Structural Insights into the Functional Diversity of the CDK–Cyclin Family. Open Biol. 2018, 8, 180112. [Google Scholar] [CrossRef] [PubMed]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.É.; Knapp, S.; Johnson, L.N. The Structure of P-TEFb (CDK9/Cyclin T1), Its Complex with Flavopiridol and Regulation by Phosphorylation. EMBO J 2008, 27, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Heitz, F.; Morris, M.C.; Fesquet, D.; Cavadore, J.-C.; Dorée, M.; Divita, G. Interactions of Cyclins with Cyclin-Dependent Kinases: A Common Interactive Mechanism. Biochemistry 1997, 36, 4995–5003. [Google Scholar] [CrossRef] [PubMed]
- Gondeau, C.; Gerbal-Chaloin, S.; Bello, P.; Aldrian-Herrada, G.; Morris, M.C.; Divita, G. Design of a Novel Class of Peptide Inhibitors of Cyclin-Dependent Kinase/Cyclin Activation. J. Biol. Chem. 2005, 280, 13793–13800. [Google Scholar] [CrossRef]
- Canela, N.; Orzáez, M.; Fucho, R.; Mateo, F.; Gutierrez, R.; Pineda-Lucena, A.; Bachs, O.; Pérez-Payá, E. Identification of an Hexapeptide That Binds to a Surface Pocket in Cyclin A and Inhibits the Catalytic Activity of the Complex Cyclin-Dependent Kinase 2-Cyclin A. J. Biol. Chem. 2006, 281, 35942–35953. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol Syst Biol 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J Appl Cryst J Appl Crystallogr 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T.; Darden, T.; Duke, R.; Gohlke, H.; et al. {Amber 14}; 2014.
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J Comput Chem 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Hall, H.K. Potentiometric Determination of the Base Strength of Amines in Non-Protolytic Solvents. J. Phys. Chem. 1956, 60, 63–70. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J Comput Chem 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Schlitter, J.; Engels, M.; Krüger, P. Targeted Molecular Dynamics: A New Approach for Searching Pathways of Conformational Transitions. J. Mol. Graph. 1994, 12, 84–89. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R Package for the Comparative Analysis of Protein Structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD – Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossvary, I.; Moraes, M.; Sacerdoti, F.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06); IEEE: Tampa, FL, USA, November, 2006; pp. 43–43. [Google Scholar]
Figure 1.
Root mean square fluctuations (RMSF) per residue of CDK8 in presence (black) and absence (blue) of the CycC during 1 μs of simulation.
Figure 1.
Root mean square fluctuations (RMSF) per residue of CDK8 in presence (black) and absence (blue) of the CycC during 1 μs of simulation.

Figure 2.
PCA projection of the structural evolution of CDK8 in DMG-in conformation (systems 8/9, left) and DMG-out conformation (systems 1a/2a, right) in presence (black) and absence (blue) of the CycC during the MD simulations. One snapshot of a trajectory is represented by a dot in the individual map of PC1 against PC2.
Figure 2.
PCA projection of the structural evolution of CDK8 in DMG-in conformation (systems 8/9, left) and DMG-out conformation (systems 1a/2a, right) in presence (black) and absence (blue) of the CycC during the MD simulations. One snapshot of a trajectory is represented by a dot in the individual map of PC1 against PC2.
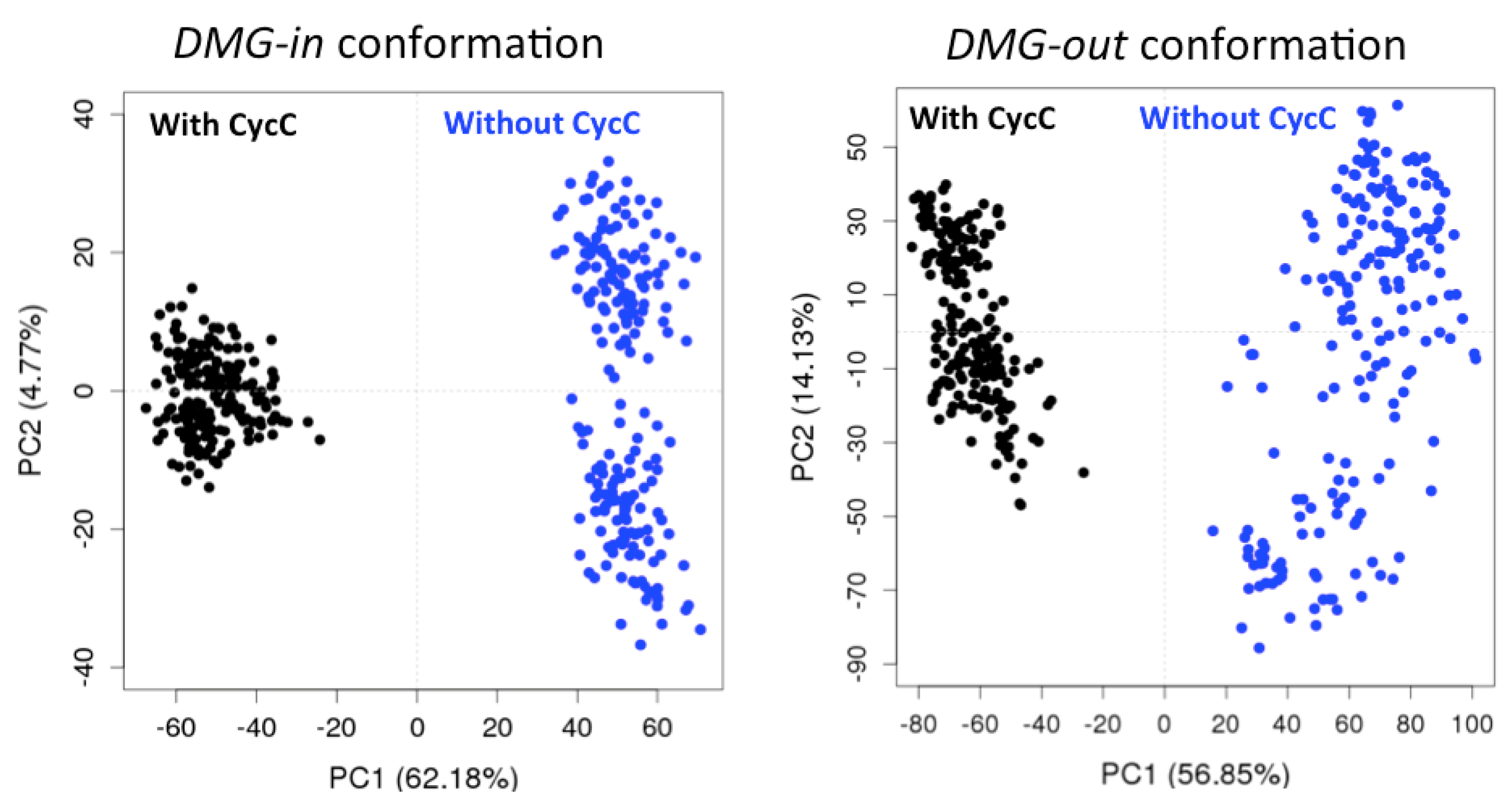
Figure 3.
Impact of the CycC on the structure of CDK8: contribution of each residue of CDK8 to PC1.Loading plot of the PCA done on the combined trajectory (with/without CycC) in DMG-in conformation system (top left) and DMG-out conformation systems (bottom left). On right representation of CDK8 in dark gray ribbon except the regions of the kinase domain containing the conserved motifs.
Figure 3.
Impact of the CycC on the structure of CDK8: contribution of each residue of CDK8 to PC1.Loading plot of the PCA done on the combined trajectory (with/without CycC) in DMG-in conformation system (top left) and DMG-out conformation systems (bottom left). On right representation of CDK8 in dark gray ribbon except the regions of the kinase domain containing the conserved motifs.
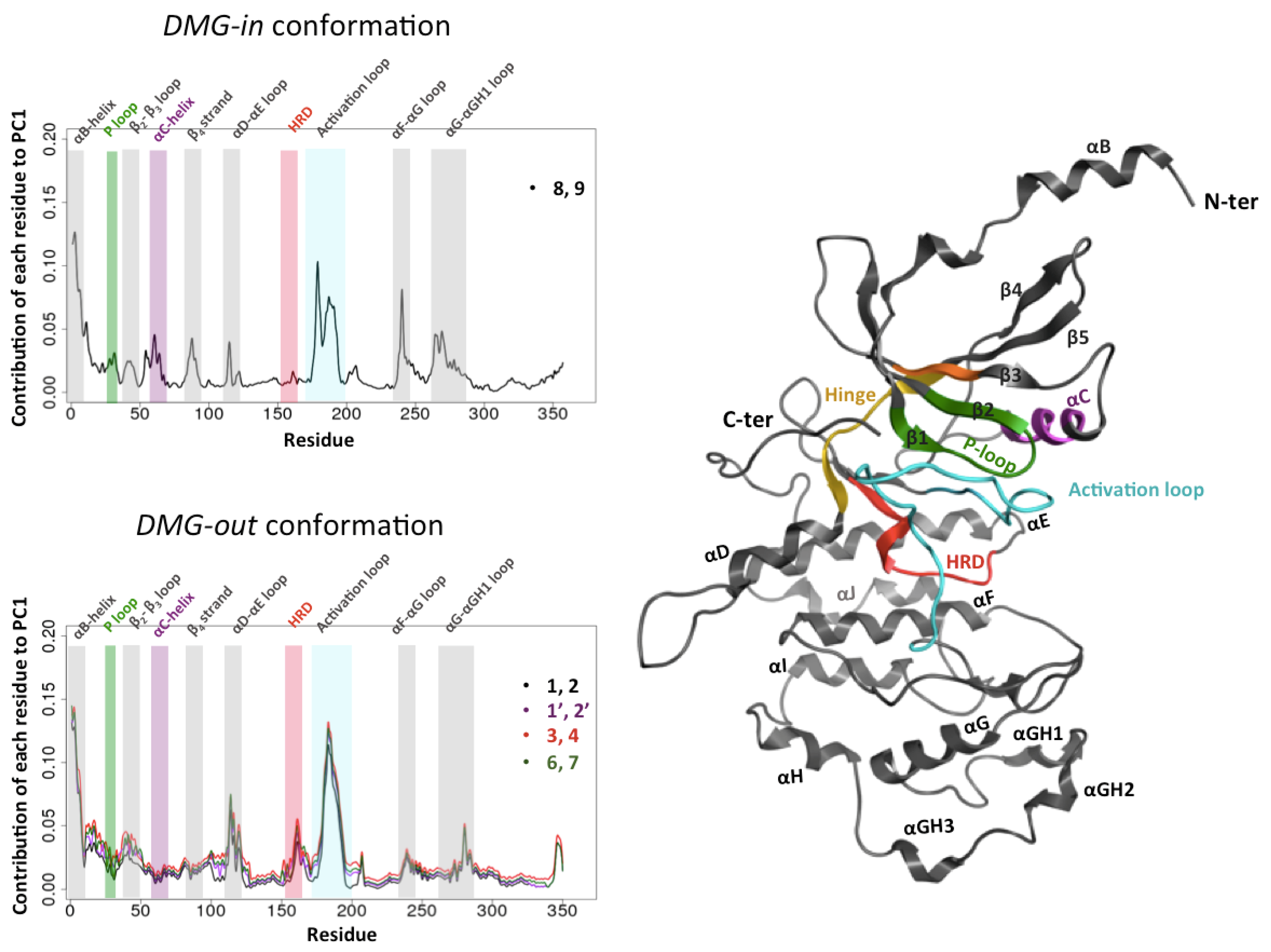
Figure 4.
CDK8-CycC binding interface: (a) Matrix of the per-residue energy contribution (ΔG without entropy). The residues are those that highly contribute to CDK8-CycC binding (absolute (ΔG) > 1 kcal.mol-1) in all studied CDK8-CycC complexes. Residues are tagged according to the secondary structure they belong to. The residues colored in pink-purple tones are those belonging to CDK8 specific binding sites. While those colored in green-yellow tones are those belonging to human CDK common binding sites; (b) CDK8/CycC structure and the binding sites at the interface CDK8-CycC. CDK8 in dark gray ribbon except the secondary structures having residues that highly contribute to CDK8-CycC binding in all studied CDK8-CycC complexes. Idem for CycC, which is colored in light gray. Besides the common binding area (represented by a dashed green box), CDK8/CycC complex forms additional contacts mediated by the CDK8 N-terminus αB-helix and the CycC N-terminus, including the HN helix (highlighted by dashed pink boxes).
Figure 4.
CDK8-CycC binding interface: (a) Matrix of the per-residue energy contribution (ΔG without entropy). The residues are those that highly contribute to CDK8-CycC binding (absolute (ΔG) > 1 kcal.mol-1) in all studied CDK8-CycC complexes. Residues are tagged according to the secondary structure they belong to. The residues colored in pink-purple tones are those belonging to CDK8 specific binding sites. While those colored in green-yellow tones are those belonging to human CDK common binding sites; (b) CDK8/CycC structure and the binding sites at the interface CDK8-CycC. CDK8 in dark gray ribbon except the secondary structures having residues that highly contribute to CDK8-CycC binding in all studied CDK8-CycC complexes. Idem for CycC, which is colored in light gray. Besides the common binding area (represented by a dashed green box), CDK8/CycC complex forms additional contacts mediated by the CDK8 N-terminus αB-helix and the CycC N-terminus, including the HN helix (highlighted by dashed pink boxes).
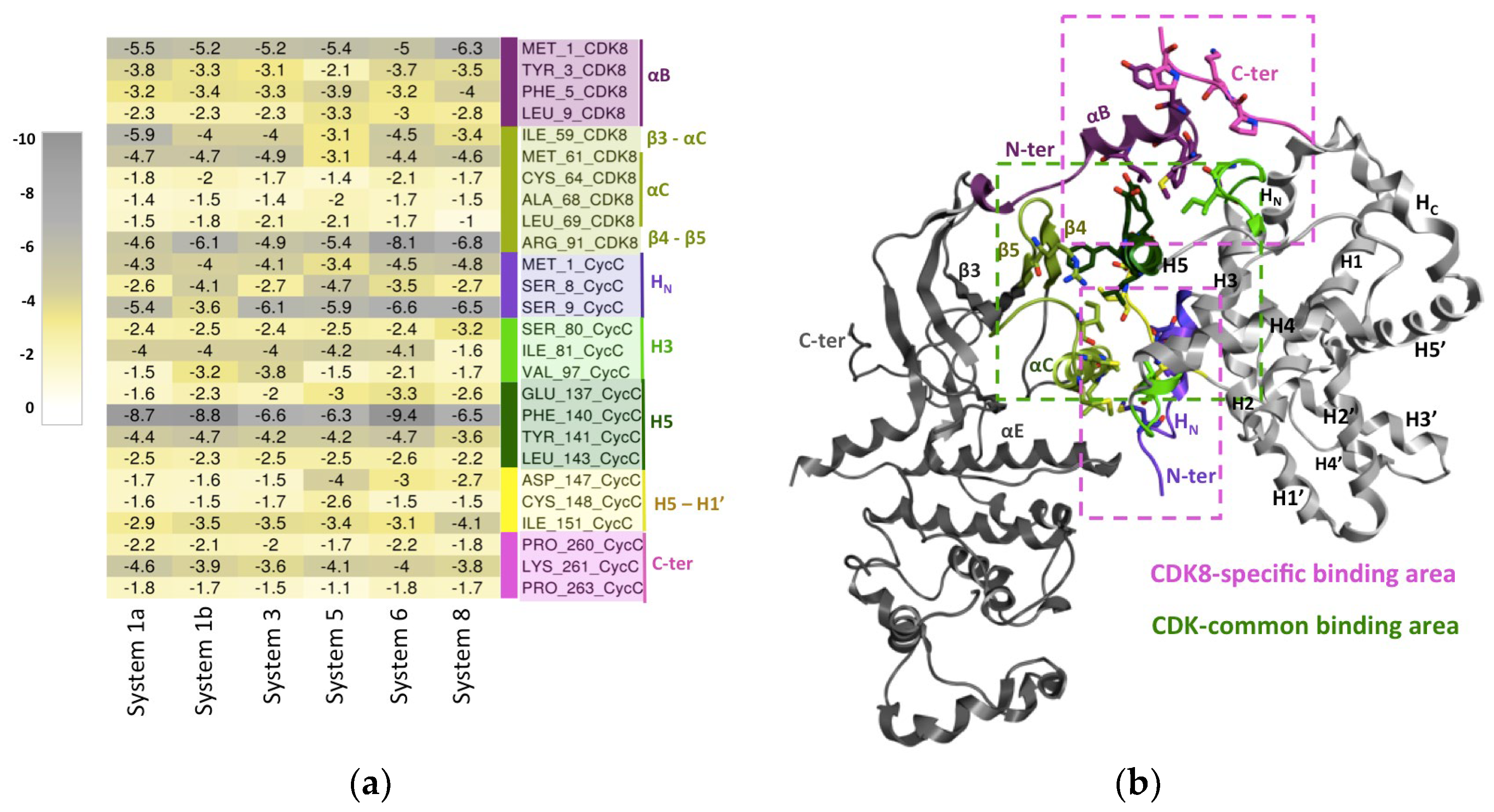
Figure 5.
Correlation plot matrix of the residues energy contributions to CDK8-CycC binding of each system. The selected residues present at least one significant energy contribution (absolute (ΔG) > 1 kcal.mol-1) in one system.
Figure 5.
Correlation plot matrix of the residues energy contributions to CDK8-CycC binding of each system. The selected residues present at least one significant energy contribution (absolute (ΔG) > 1 kcal.mol-1) in one system.
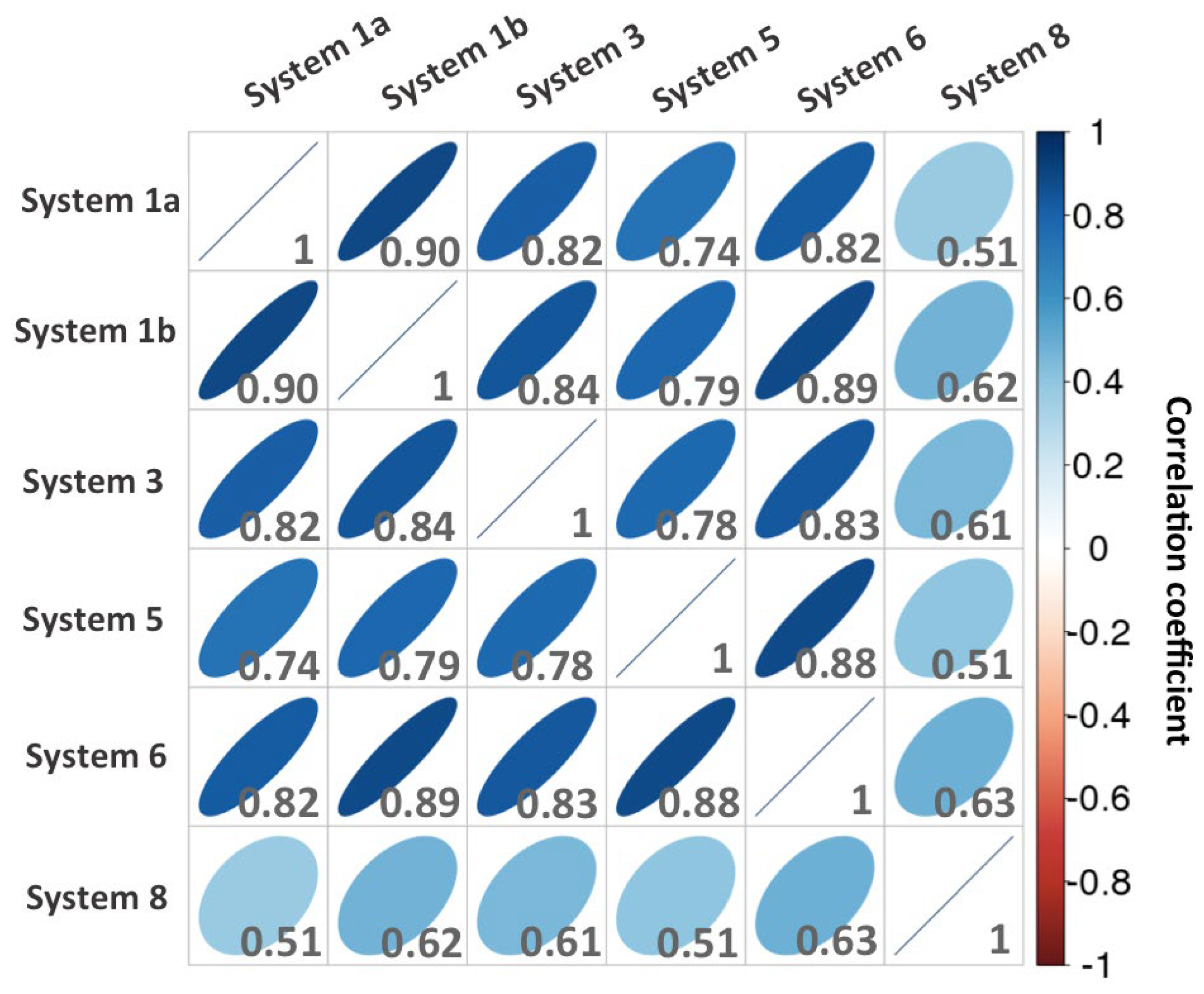
Figure 6.
Pipes and planks representation of the DMG-in and DMG-out conformations of the CDK8-CycC complexes in cartoon. CDK8 structures are colored in gray, except the activation loop. The activation loop and the CycC structures are colored in pink in the DMG-in conformation and in blue in the DMG-out conformation.
Figure 6.
Pipes and planks representation of the DMG-in and DMG-out conformations of the CDK8-CycC complexes in cartoon. CDK8 structures are colored in gray, except the activation loop. The activation loop and the CycC structures are colored in pink in the DMG-in conformation and in blue in the DMG-out conformation.
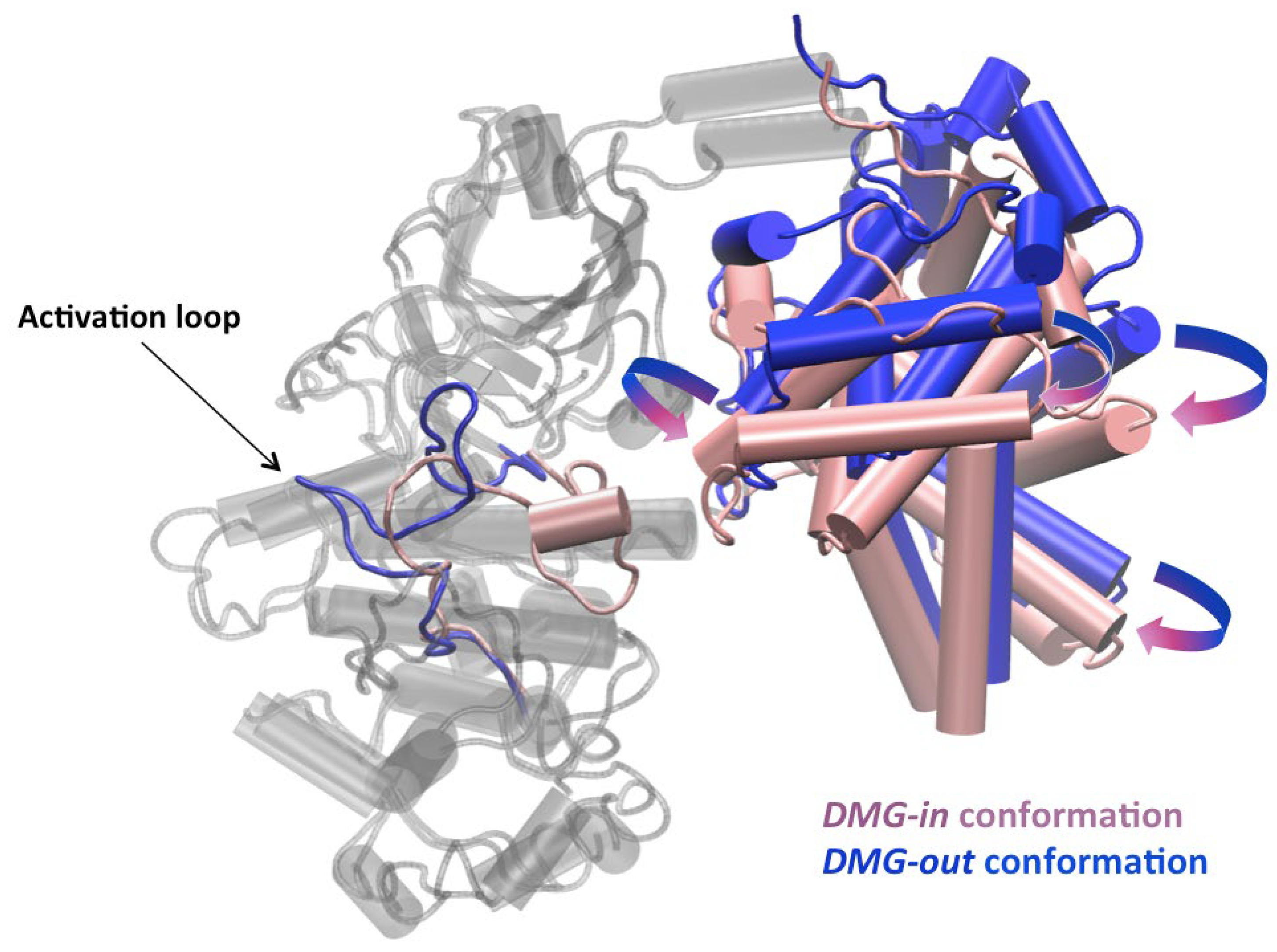
Figure 7.
Conformational transition of the CDK8 activation loop from DMG-out to DMG-in conformation: (a) plots of the measured distance between the pairs Glu99CycC/Arg178CDK8 and Glu99CycC/Lys153CDK8. The distance between Glu99CycC and Arg178CDK8 enables to monitor the transition of the activation loop over the simulation time. The distance between Glu99CycC and Lys153CDK8 enables to monitor the displacement of the CycC toward CDK8; (b) Representation of the three conserved arginines Arg65CDK8, Arg150CDK8 and Arg178CDK8 and of Glu99CycC over the simulation time course. CDK8 is represented in dark gray ribbon except the regions of the kinase domain containing the conserved motifs, particularly the activation loop is in cyan. CycC is in light gray. Arg65CDK8, Arg150CDK8, Arg178CDK8, Glu99CycC and Lys153CDK8 are represented in sticks, arginines and glutamate in light green and Lys153CDK8 in gray.
Figure 7.
Conformational transition of the CDK8 activation loop from DMG-out to DMG-in conformation: (a) plots of the measured distance between the pairs Glu99CycC/Arg178CDK8 and Glu99CycC/Lys153CDK8. The distance between Glu99CycC and Arg178CDK8 enables to monitor the transition of the activation loop over the simulation time. The distance between Glu99CycC and Lys153CDK8 enables to monitor the displacement of the CycC toward CDK8; (b) Representation of the three conserved arginines Arg65CDK8, Arg150CDK8 and Arg178CDK8 and of Glu99CycC over the simulation time course. CDK8 is represented in dark gray ribbon except the regions of the kinase domain containing the conserved motifs, particularly the activation loop is in cyan. CycC is in light gray. Arg65CDK8, Arg150CDK8, Arg178CDK8, Glu99CycC and Lys153CDK8 are represented in sticks, arginines and glutamate in light green and Lys153CDK8 in gray.
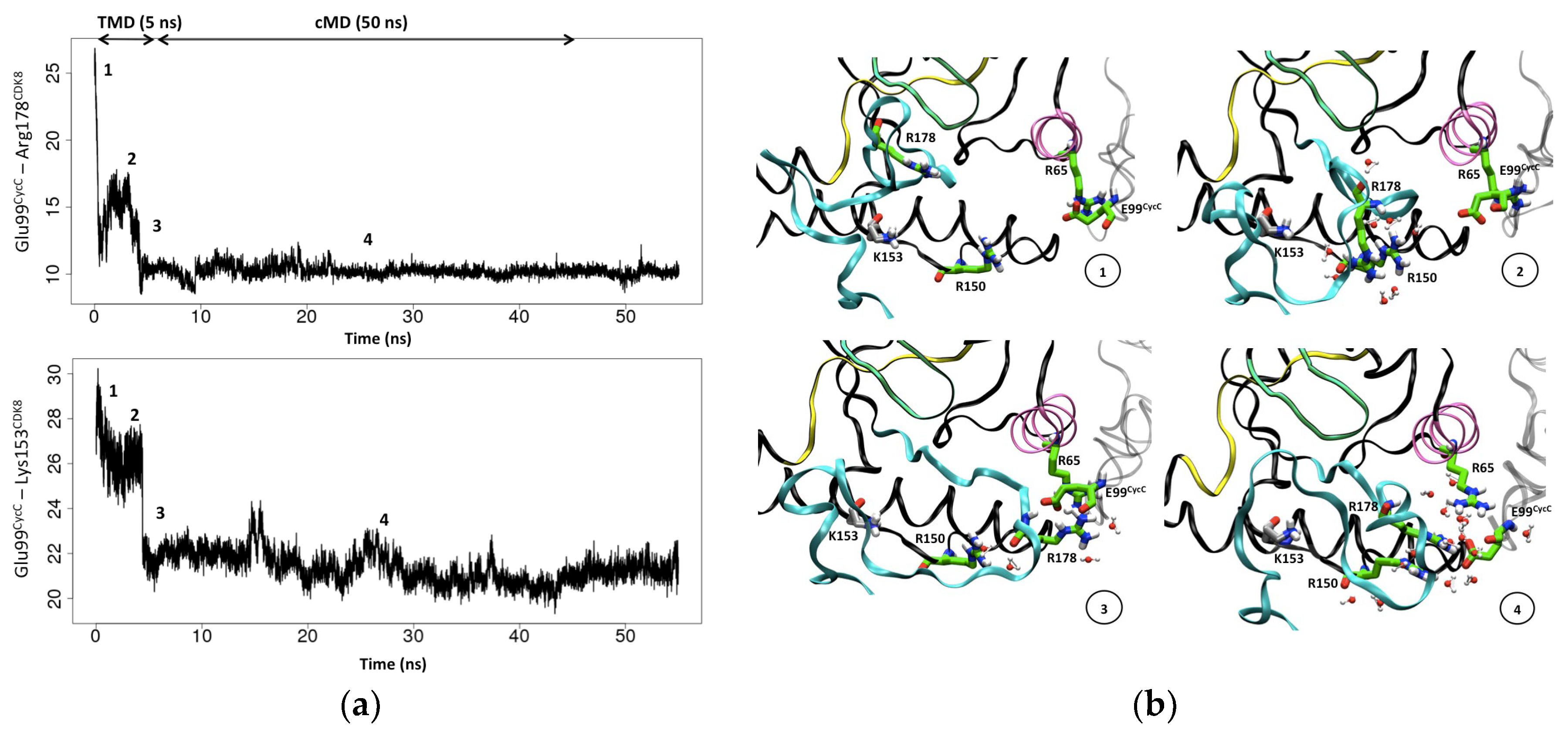
Figure 8.
Ribbon representation of the CDK8-CycC complex. Cyclin C (CycC, PDB id.: 4F6U) is colored in light grey and CDK8 in dark gray, except the conserved motifs of the kinase domain. Among these motifs, the activation loop in DMG-out conformation (inactive form) is colored in cyan (PDB id.: 4F6U) and that in DMG-in conformation (active form) in dark blue (PDB id.: 4F7S).
Figure 8.
Ribbon representation of the CDK8-CycC complex. Cyclin C (CycC, PDB id.: 4F6U) is colored in light grey and CDK8 in dark gray, except the conserved motifs of the kinase domain. Among these motifs, the activation loop in DMG-out conformation (inactive form) is colored in cyan (PDB id.: 4F6U) and that in DMG-in conformation (active form) in dark blue (PDB id.: 4F7S).
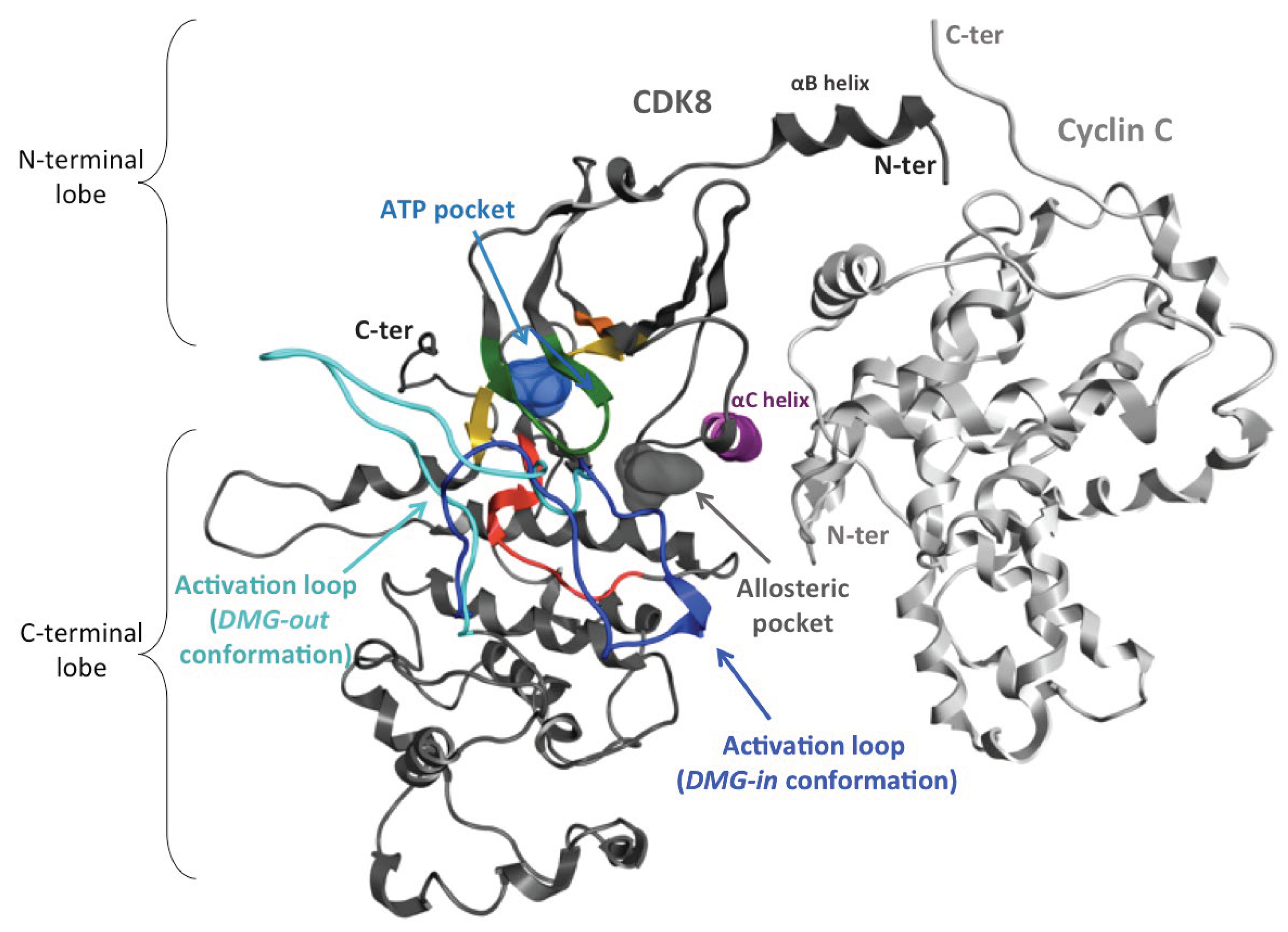

Table 1.
Comparison of the average RMSD calculated on the heavy atoms of CDK8 during 1 μs simulation for the systems with and without CycC.
Table 1.
Comparison of the average RMSD calculated on the heavy atoms of CDK8 during 1 μs simulation for the systems with and without CycC.
| System id. With Cyclin C |
Average RMSD (Å) (± Standard deviation) |
System id. Without Cyclin C |
Average RMSD (Å) (± Standard deviation) |
|---|---|---|---|
| 1a | 3.6 ± 0.2 | 2a | 5.5 ± 1 |
| 1b | 3.9 ± 0.3 | 2b | 5.5 ± 0.3 |
| 3 | 3.8 ± 0.3 | 4 | 5.4 ± 0.9 |
| 6 | 3.2 ± 0.2 | 7 | 5 ± 0.6 |
| 8 | 3.4 ± 0.3 | 9 | 3.7 ± 0.4 |
Table 2.
The binding free energy and energy components of the CDK8-CycC complex calculated with MM-GBSA method and averaged on the simulations. All the energies are reported in kcal.mol-1, with their corresponding standard errors. ΔEeel and ΔEVDW are respectively electrostatic and van der Waals contributions in gas phase. ΔEGB and ΔEnp are respectively electrostatic and non-polar contributions in solvation phase. ΔGtotal is the total binding free energy without considering the entropic term.
Table 2.
The binding free energy and energy components of the CDK8-CycC complex calculated with MM-GBSA method and averaged on the simulations. All the energies are reported in kcal.mol-1, with their corresponding standard errors. ΔEeel and ΔEVDW are respectively electrostatic and van der Waals contributions in gas phase. ΔEGB and ΔEnp are respectively electrostatic and non-polar contributions in solvation phase. ΔGtotal is the total binding free energy without considering the entropic term.
| Systems PDB id. Conformation |
System 1a (4F6U) DMG-out |
System 1b (4F6U-replica) DMG-out |
System 3 (4F6U-apo) DMG-out |
System 5 (4F6UR65A_E66A) DMG-out |
System 6 (4F7L) DMG-out |
System 8 (4F7S) DMG-in |
|---|---|---|---|---|---|---|
| ΔEVDW | -163.0 ± 0.1 | -160.7 ± 0.1 | -160.0 ± 0.2 | -148.7 ± 0.1 | -156.3 ± 0.1 | -176.5 ± 0.1 |
| ΔEeel | -508.4 ± 1.0 | -436.5 ± 1.0 | -490.0 ± 1.5 | -573.9 ± 1 | -500.6 ± 0.9 | -588.5 ± 0.9 |
| ΔEGB | 554.3 ± 0.9 | 491.1 ± 0.9 | 543.2 ± 1.4 | 613.9 ± 0.9 | 541.0 ± 0.8 | 613.9 ± 0.8 |
| ΔEnp | -23.9 ± 0.0 | -23.1 ± 0.0 | -23.2 ± 0.3 | -22.7 ± 0.0 | -22.9 ± 0.0 | -25.4 ± 0.0 |
| ΔGtotal (Without entropy) |
-141.0± 0.2 | -129.2 ± 0.2 | -130.0 ± 0.3 | -131.5 ± 0.2 | -138.8 ± 0.2 | -124.6 ± 0.2 |
Table 3.
Description of the studied systems.
| System id. | PDB id. | Ligand name | DMG conformation | Manipulation |
| 1a | 4F6U | 0SR | DMG-out | (-) |
| 1b (replica) | 4F6U | 0SR | DMG-out | (-) |
| 2a | 4F6U | 0SR | DMG-out | Removal of CycC |
| 2b (replica) | 4F6U | 0SR | DMG-out | Removal of CycC |
| 3 | 4F6U | (-) | DMG-out | Removal of ligand |
| 4 | 4F6U | (-) | DMG-out | Removal of ligand and CycC |
| 5 | 4F6U | 0SR | DMG-out | CDK8 mutations: E66A, R65A |
| 6 | 4F7L | 0SO | DMG-out | (-) |
| 7 | 4F7L | 0SO | DMG-out | Removal of CycC |
| 8 | 4F7S | 0SW | DMG-in | (-) |
| 9 | 4F7S | 0SW | DMG-in | Removal of CycC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



