
Submitted:
05 April 2024
Posted:
05 April 2024
You are already at the latest version
Abstract
Neurodegenerative diseases (NDs) are a set of progressive, chronic, and incurable diseases characterized by the gradual loss of neurons, culminating in the decline of cognitive and/or motor functions. Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the most common NDs and represent an enormous burden both in terms of human suffering and economic cost. The available therapies for AD and PD only provide symptomatic and palliative relief for a limited period and are unable to modify the diseases’ progression. Over the last decades, research efforts have been focused on developing new pharmacological treatments for these NDs. However, to date, no breakthrough treatment has been discovered. Hence, the development of disease-modifying drugs able to halt or reverse the progression of NDs remains an unmet clinical need. This review summarizes the major hallmarks of AD and PD and the drugs available for pharmacological treatment. It also sheds light on potential directions that can be pursued to develop of new, disease-modifying drugs to treat AD and PD, thereby decreasing the social and economic burdens linked to these NDs.
Keywords:
neurodegenerative diseases
; Parkinson’s disease
; Alzheimer’s disease
; drug discovery
; disease-modifying drugs
1. Introduction
Neurodegenerative diseases (NDs) are a set of progressive, chronic, and incurable neurological disorders characterized by the loss of neurons and synaptic connections, which irreversibly produce a series of events commonly related to motor disability, cognitive impairment, and dementia [1]. They represent an enormous disease burden, both in terms of human suffering and economic costs [2], being the foremost contributors of incapacity and dependence due to their debilitating nature [3]. The most common NDs include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and Amyotrophic Lateral Sclerosis (ALS).
The etiology of NDs is not completely understood and the onset of neurodegeneration may precede the clinical symptoms by many years. However, it is generally accepted that the pathogenesis of NDs is multifactorial, involving a complex combination of genetic, environmental, and endogenous factors acting cooperatively or independently [4,5]. Although each disease presents its particular molecular mechanisms and clinical manifestations (Figure 1), NDs share common pathogenic events [6].
Despite the intensive research performed so far, to date, no breakthrough treatment has yet been discovered. The NDs available therapies only provide symptomatic and palliative relief for a limited period [7] and are unable to modify the disease progression [8,9]. Therefore, the development of disease-modifying drugs able to prevent, halt or reverse the progression of NDs remains an unmet clinical need. In this review, we summarize the major hallmarks of AD and PD, the drugs available for pharmacological treatment, and future directions for the development of new and disease-modifying drugs to treat these NDs.
2. Alzheimer’s and Parkinson’s Diseases: The Epidemiologic Forecast
The average human lifespan of the worldwide population has been rising in the last decades [10]. Several factors seem to contribute to this success, namely the cumulative progress in sanitation and medical care, rising living standards, and the decline in child mortality [10,11]. Although the increased life expectancy must be celebrated, a proportional rise in the frequency and prevalence of NDs is expected [10,12]. Therefore, the rise of elderly populations has been soaring with the increased incidence of age-related degenerative diseases, reaching epidemic proportions in high-income countries [13]. Several genetic risk factors, lifestyles, and environmental exposure to a diversity of pollutants are implicated in the neurodegeneration process [14]. In 2020, the worldwide population with age higher than 65 years was estimated to be 727 million, a 195 million increase since 2010. Over the next three decades, the number of worldwide elderly is projected to more than double, reaching over 1.5 billion in 2050. By mid-century, one in six people globally will be aged 65 years or older [15]. Although the increase in longevity represents a progress per se, it can become a social, economic, and medical burden when it is not associated with the maintenance of the quality of life. The World Health Organization indicated that central nervous system (CNS) diseases are the major medical challenge of the 21st Century. Among them, NDs, namely AD and PD are the most prevalent CNS disorders [16].
The World Health Organization recognized AD as the most common form of dementia in the elderly, accounting for 50–56%, and a major cause of death, being considered one of the greatest global public health challenges [17,18]. Currently, the number of people aged 65 and older affected by AD dementia is more than 55 million worldwide and it is expected that this number will rapidly increase to 132 million by 2050 [19,20].
The overall number of people diagnosed with PD has also been growing progressively at a global level. In 2019, approximately 8.5 million individuals had received a PD diagnosis. Estimates suggest that, in 2019, PD resulted in 5.8 million disability-adjusted life years, an increase of 81% since 2000, and caused 329,000 deaths, an increase of over 100% since 2000. This estimation is expected to increase to 12 million people in 2050 [21,22].
3. Alzheimer’s and Parkinson’s Diseases Major Hallmarks
3.1. Alzheimer’s Disease
Alzheimer’s disease was first diagnosed in 1906 by Dr. Alois Alzheimer, when he noticed changes in the brain tissue of a woman who had died of an unusual mental illness [23].
AD is an irreversible, complex, and progressive ND that results in cognitive impairment and memory injury. Despite its prevalence among the elderly, AD dementia is distinct from a normal aging process [24]. The progression of AD can be divided into three stages. The first is often mistakenly attributed to age-related upsets or manifestations of stress [25]. In this stage, the patient has memory lapses such as forgetting familiar words or the location of everyday objects, which denotes its lack of ability to produce new memories and skills [26,27]. The second stage of AD is typically the longest one and can last for many years. Herein, the progressive deterioration of neurons can lead to problems with speech and severe difficulties in reading and writing. During this phase, memory problems worsen, and the patient may fail to recognize close relatives [28]. In the most advanced phase, AD patients show loss of cognitive and motor functions, confusion, and disorientation, having most patients have mobility problems, hallucinations, and delirium, leading to absolute dependence on 24-hour supervision, hospital care, and unavoidable death [29,30].
Although the specific cause of AD is still unknown, it is well recognized that a multiplicity of pathological stimuli can play a key role, which causes an increased risk of disease development [31,32]. Age and family history of the disease are considered the strongest risk factors for familial and sporadic AD [33]. The presence of ε4 allele of the apolipoprotein E4 (ApoE4) genotype, found on chromosome 19, appears to be a primarily risk factor for patients with sporadic AD [34,35,36]. In addition, genetic mutations in APP on chromosome 21, presenilin-1 (PSEN-1) on chromosome 14, and presenilin-2 (PSEN-2) on chromosome 1 can also cause familial AD [37,38]. Other putative risk factors include head trauma, depression, diabetes mellitus, hypothyroidism, and a series of vascular factors [39,40].
A conclusive diagnosis of AD requires a detailed post-mortem microscopic examination of the brain [37]. However, AD can be currently diagnosed with more than 95% accuracy in living patients by carefully analyzing the patients’ family history, assessing cognitive function with neuropsychological tests, and evaluating AD biomarkers, namely with high-tech neuroimaging data or cerebrospinal fluid analysis [41,42].
The progressive cognitive impairment observed in AD patients can be associated with the significant reduction of brain size [43]. The brain atrophy arises from the loss of synapses and from the selective neuronal death in the hippocampus and in the cerebral cortex [43,44,45,46]. The most prominent losses are observed in neurons with long projections, such as cholinergic neurons in the basal forebrain (Figure 2A) [47]. These neurons innervate the hippocampus, thalamus, amygdala, and neocortex, and play key roles in attention, cognitive flexibility, and learning [48]. Although the neurons that degenerate in AD are mostly cholinergic [49], glutamatergic neurons are also affected [50].
AD is characterized by extensive atrophy of the brain caused by two main neuropathologic changes – the formation of amyloid plaques (also called senile plaques) and the appearance of neurofibrillary tangles (NFTs) – that lead to neuronal loss and synaptic changes in brain-specific areas essential for cognitive and memory functions (Figure 2B) [51].
The amyloid plaques result from the abnormal extracellular accumulation and deposition of insoluble aggregates of fibrillar β-amyloid (Aβ) [53]. Sequential cleavage of the amyloid precursor protein (APP) in the cell membrane by the enzymes -β and Ƴ-secretases gives rise to the family of Aβ peptides (most commonly 40–42 amino acids in length) [54,55]. Although the formation of Aβ is believed to be a physiological process in normal aging [56], Aβ1-42 isoform was identified as a major contributor to the disease process [57,58].
Intracellular NFTs are formed by aggregated misfolded tau protein (tau-P), the major microtubule-associated protein predominantly found in the axons of mature neurons [59]. In AD, tau hyperphosphorylation induces a loss of function that hampers its ability to bind to microtubules, leading to microtubule depolymerization that compromises the axonal trafficking and the dendrite structure [60]. When tangle-bearing neurons die, NFTs become extraneuronal and activate a series of neurotoxic processes that can cause synaptic dysfunction and neuronal death [61]. Overall, AD brains show a decline in neuronal mass in regions related to cognition and memory, which leads to a depletion of cholinergic neurons and acetylcholine (ACh), resulting in synaptic dysfunction [47,62].
Other pathological features also play a crucial role in the progress of AD, including cholinergic deficit, enhanced brain oxidative stress and overproduction of free radical, mitochondrial dysfunction, and disruption of metal homeostasis [63]. The downstream consequences of neuropathological processes contribute to neurodegeneration with extensive neuronal loss, synaptic changes, and brain neurotoxic events leading to macroscopic atrophy [18,64].
3.2. Parkinson’s Disease
Initially described by the English surgeon James Parkinson in 1817, Parkinson’s disease is the second most prevalent ND and the most common movement disorder [65].
Clinical manifestations of PD include four cardinal motor symptoms: bradykinesia, resting tremor, rigidity, and postural instability [66,67]. Patients with PD may also experience numerous non-motor symptoms, such as autonomic deficiency, cognitive impairment, neuropsychiatric problems (mood, cognition, behavior, or thought alterations), sensory (especially altered sense of smell), and sleep disorders [7,67,68,69]. Non-motor symptoms are common in the PD early stages (pre-motor/prodromal phase) and frequently precede the onset of motor symptoms [68,69]. Motor dysfunction worsens with the disease progression and is managed with symptomatic treatments [69]. However, long-term therapy is associated with gradual loss of efficacy and the emergence of adverse effects such as motor fluctuations, dyskinesia, and psychosis [69,70]. Late-stage PD is characterized by treatment-resistant motor and non-motor symptoms that substantially contribute to the patient’s disability [69]. The median age of onset of PD is 60 years, and the mean duration from diagnosis to death is 15 years [67].
Parkinson’s disease is mostly sporadic, resulting from a complex interplay between genetic susceptibility and environmental factors [66]. However, approximately 5-10 % of PD cases are caused by familial genetic mutations [71,72] that usually result in early-onset PD [73]. Mutations in SNCA, LRRK2 and VPS35 genes were associated with autosomal dominant PD, while mutations in PINK1, PARK7/DJ-1, PARK2/PARKIN, PLA2G6, ATP13A2, and FBXO7 cause autosomal recessive PD and/or parkinsonism [74]. Despite being extensively studied, the gun trigger that causes PD remains unknown. An appraisal of the literature points towards a complex multifactorial etiology, in which a multiplicity of pathological stimuli contributes to the neurodegenerative cascade. So far, the main causes include impaired calcium homeostasis, iron overload, inflammation, protein aggregation, and defective metabolism [75]. In addition, several studies showed that oxidative stress can cause neuronal death and mitochondrial dysfunction [76].
The motor dysfunction observed in PD is linked to the loss of dopaminergic neurons in specific areas of the substantia nigra pars compacta (SNpc) region of the midbrain, which contributes to severe dopamine (DA) deficiency in the putamen and the caudate nucleus [77]. The cell bodies of nigrostriatal neurons are in the SNpc and their axon terminals projected to the dorsal striatum (i.e., putamen and caudate nucleus) (Figure 3A) [5,72]. Dopamine synthesized in this brain region is directed to the striatum and frontal cortex, allowing control of the musculoskeletal system and movement. Therefore, the degeneration of dopaminergic neurons leads to a decrease in DA levels. Symptoms of PD only develop after the loss of 50-60 % of nigral neurons and the depletion of 70-85 % of DA levels [67,78]. Although the neuropathology of PD is primarily characterized by dopaminergic neuron loss, neurodegeneration also extends to other neurotransmitter systems [68]. Indeed, cholinergic (nucleus basalis of Meynert, dorsal nucleus of vagus), serotonergic (raphe), and noradrenergic (locus coeruleus) neurons are also affected [79].
One pathological hallmark of PD is the formation of Lewy bodies, which are lamellated and fibrillated aggregates that include α-synuclein (αSyn) and ubiquitin [82]. Accumulation of αSyn results in the death of dopaminergic neurons [83]. In dopaminergic neurons, αSyn regulates the synthesis, storage, and release of DA [84]. αSyn is prone to form oligomeric and fibrillar bodies in the cytosol or associate to the cellular membrane [85]. The formation of αSyn inclusions begins in the lower brainstem nuclei [86,87], spreads through the pons to the midbrain and basal forebrain and reaches the neocortex [86]. These inclusions may accumulate in neuronal perikarya (Lewy bodies) and neuronal processes (Lewy neurites) (Figure 3B) [88,89]. The presence of these aggregates is associated with the accumulation of synaptic vesicles, decreased DA release, impairment of degradation pathways, and increased oxidative stress [84].
4. Alzheimer’s and Parkinson’s Diseases Pharmacotherapy
Neurodegeneration is a complex process resulting from multiple defects [90]. The most obvious pathological features of AD and PD include the selective loss of neuronal populations with a consequent decrease of neurotransmitter levels, and the formation of protein aggregates [66,91]. These observations led to the identification of the primary brain enzymatic targets (e.g.: cholinesterases (ChEs), monoamine oxidases (MAOs), catechol-O-methyltransferase (COMT)) [90], which are related to the regulation of neurotransmitter levels, and to the subsequent development of the currently available therapeutic agents [92,93].
4.1. Targeting Neurotransmitter Depletion in Alzheimer’s Disease
Cholinergic neurons are widely distributed in both the central and the peripheral nervous systems [48]. Although they represent less than 1 % of neurons in the nervous system, almost every brain region and peripheral target receives cholinergic innervation [94]. Cholinergic neurons in the basal forebrain contain extensive cortical projections that are involved in the modulation of other neurotransmitter systems [95].
Studies focusing on the cholinergic system have received particular attention since the decline of cholinergic function was linked to age-related learning impairments and memory loss in AD [96]. The damage or the presence of abnormalities in cholinergic pathways, especially in the basal forebrain neurons, was correlated with the level of cognitive decline in late-stage AD patients [97]. Together with the loss of cholinergic markers, such as choline acetyltransferase (ChAT) and AChE, these observations led to the formulation of the “cholinergic hypothesis” [98], which states that the dysfunction of the cholinergic system contributes to the cognitive deficits in AD [99].
Acetylcholine (ACh) was the first neurotransmitter to be identified [100,101] and is widely distributed in the nervous system, playing important functional roles in attention, memory, learning, stress response, wakefulness and sleep, and sensory information [102]. The hippocampal and cortical levels of ACh in the brain of AD patients are decreased by approximately 90 % [103]. It has also a very important role in the structural and functional remodeling of cortical circuits by establishing synaptic contacts in networks of cells [104,105].
The synthesis of ACh is catalyzed by ChAT in the cytosol of presynaptic cholinergic neurons in a single-step reaction, in which choline and acetyl-coenzyme A (acetyl-CoA) are used as substrates (Figure 4) [106]. While acetyl-CoA is synthesized by mitochondria, choline is taken up from the extracellular space since it is not synthesized in neurons [107]. The rate-limiting step for the synthesis of ACh is the uptake of choline by the Na+-dependent, high-affinity choline transporter (ChT1) [108]. The neurotransmitter ACh is then accumulated in synaptic vesicles by the vesicular acetylcholine transporter (VAT). This transporter uses an electrochemical gradient generated by a proton adenosine triphosphate (ATP)ase to perform the uptake of one ACh molecule in exchange for two protons [107,108,109]. During neurotransmission, ACh is released from the presynaptic neuron into the synaptic cleft, where it binds to cholinergic receptors (muscarinic or nicotinic) in the postsynaptic and presynaptic membranes [106,110].
The action of ACh may persist for a long time due to the chemical stability of the neurotransmitter [111]. Therefore, the rapid hydrolysis of ACh by cholinesterases is a process to prevent cholinergic overactivation [112]. Two ChEs are present in mammals and can metabolize Ach: acetylcholinesterase (AchE) and butyrylcholinesterase (BchE) [98]. While the Ch obtained from Ach inactivation is taken up by pre-synaptic neuron via ChT1 [98,106,111], acetic acid is further decomposed [111].
Figure 4.
Enzymes and transporters involved in the synthesis, storage, and metabolism of acetylcholine. Abbreviations: Ach, acetylcholine; AchE, acetylcholinesterase; BchE, butyrylcholinesterase; ChT1, high-affinity choline transporter; VAT, vesicular acetylcholine transporter. Adapted from [110,113].
Figure 4.
Enzymes and transporters involved in the synthesis, storage, and metabolism of acetylcholine. Abbreviations: Ach, acetylcholine; AchE, acetylcholinesterase; BchE, butyrylcholinesterase; ChT1, high-affinity choline transporter; VAT, vesicular acetylcholine transporter. Adapted from [110,113].
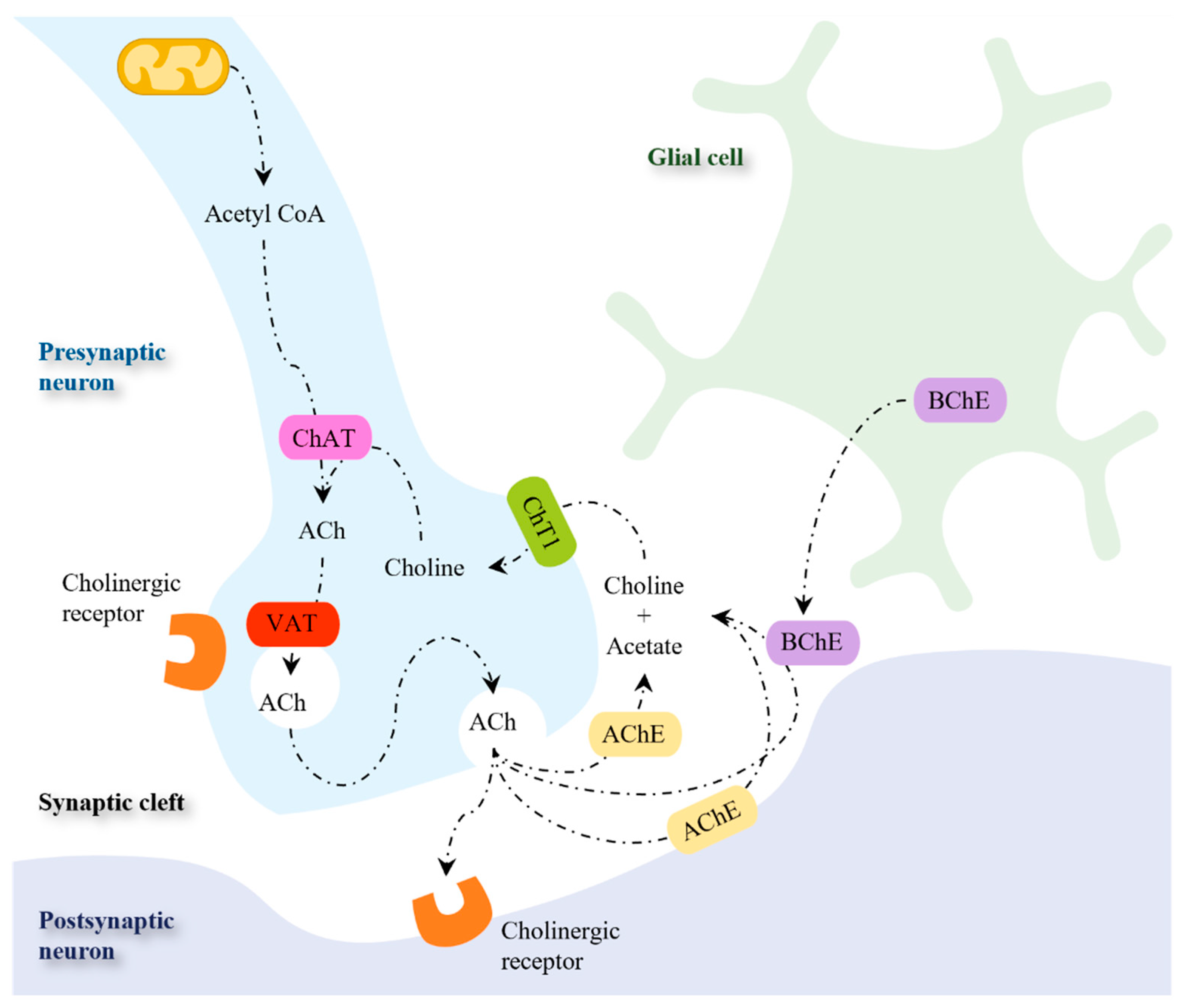
Although different approaches have been investigated to improve cholinergic neurotransmission by modulating ACh release [114,115], cholinesterase inhibitors are the only pharmacological strategy approved so far. Cholinesterase inhibitors enhance cholinergic neurotransmission through the inhibition of ChEs, thereby decreasing the breakdown of ACh and increasing its levels at the synaptic cleft. AChE inhibitors are used to treat cognitive and behavioral symptoms of AD patients. Currently available AChE inhibitors used in AD therapy include donepezil, rivastigmine, and galantamine (Figure 5) [116].
AChE inhibitors can be prescribed with memantine (Figure 5), an uncompetitive and low-affinity N-methyl-D-aspartate (NMDA) receptor (NMDAR) antagonist approved for the treatment of moderately severe to severe AD [117,118,119]. The NMDAR is an ionotropic receptor of glutamate, the main neurotransmitter in CNS [115]. The activation of NMDAR generates a long-lasting influx of Ca2+ into neurons, which is thought to be involved in the cellular processes that underlie learning and memory [120,121]. In AD, an increase of extracellular glutamate is observed, leading to excessive activation of NMDAR with consequent intracellular accumulation of Ca2+ and neuronal death [122]. By blocking excessive NMDAR activation, memantine antagonizes glutamate-mediated excitotoxicity and prevents neuronal cell death [123].
4.2. Targeting Neurotransmitter Depletion in Parkinson’s Disease
Dopamine is a catecholamine neurotransmitter present in the CNS and in some peripheral areas [124]. In the brain, DA transmission is associated with the control of fine motor movements and with cognitive functions that include learning, reward, attention, and decision-making [124,125]. The DA released in the nigrostriatal pathway is linked to the performance of voluntary movements, as well as the selection and initiation of suitable motor actions [126].
In the cytoplasm of the presynaptic dopaminergic neuron, DA biosynthesis occurs in two steps (Figure 6). The first step involves the hydroxylation of tyrosine into L-3,4-dihydroxyphenylalanine (L-DOPA) by tyrosine hydroxylase (TH), which is followed by a decarboxylation reaction catalyzed by aromatic amino acid decarboxylase (AADC) to afford DA [81,127]. The neurotransmitter is then transported into synaptic vesicles by the vesicular monoamine transporter (VMAT2) or metabolized by intraneuronal monoamine oxidase A (MAO-A) [128]. Following the release into the synaptic cleft, DA binds to the dopaminergic receptors present in the postsynaptic neuron [127]. The transport of the released DA into the presynaptic neuron occurs via DA transporter (DAT) and is followed by DA recycling into the synaptic vesicles or by DA deamination by MAO-A. Alternatively, the DA transported into non-dopaminergic post-synaptic neurons and glial cells is metabolized by MAO-B and COMT [128].
The loss of nigrostriatal dopaminergic neurons is associated with the development of motor symptoms of PD, namely the difficulty in initiating and terminating movements, gait disturbance, and muscular rigidity [126]. Therefore, to reduce the severity of motor handicaps, most PD therapies are based on enhancing the dopaminergic signaling [92,130].
The use of the DA precursor L-DOPA (Figure 7) remains the gold-standard treatment for PD [131]. Unlike DA, L-DOPA can cross the blood-brain barrier (BBB) and increase DA synthesis in the brain [127]. To prevent the peripheral metabolic activation to DA, L-DOPA is commonly administered with decarboxylase inhibitors (e.g.: carbidopa, benserazide) [132]. Despite the efficacy of L-DOPA in ameliorating motor symptoms, its long-term use is associated with progressive loss of efficacy together with motor fluctuations and dyskinesia [70,133].
In addition to L-DOPA, other therapeutic approaches involve the use of selective MAO-B inhibitors (rasagiline, selegiline, safinamide), COMT inhibitors (entacapone, tolcapone and opicapone) [134,135], or agonists of postsynaptic DA receptors (pramipexole, ropinirole, apomorphine) (Figure 7) [136,137].
Due to their pivotal role in neurotransmitter catabolism and their affinity for specific neurotransmitters, MAOs are considered attractive drug targets in the treatment of depression and NDs [138,139]. In particular, MAO-B is the main isozyme involved in DA metabolism in the aged parkinsonian brain [140]. While MAO-A activity is maintained with aging, the activity and expression of MAO-B in the human brain increase approximately 4-fold in most brain areas such as the basal ganglia [140,141], possibly as a result of glial cell proliferation and the concomitant loss of neuronal cells [142]. The amplified MAO-B activity leads to nigrostriatal DA depletion and to a higher production of H2O2 and toxic aldehydes, which contribute to increased oxidative stress and neuronal degeneration [140,141].
MAO-B inhibitors (Figure 7) are currently used in PD therapies to prevent DA catabolism and prolong the action of DA in the basal ganglia [143]. The inhibition of MAO-B may also decrease the formation of dopamine-derived oxidative products and thereby delay the disease progression [144]. Usually, MAO-B inhibitors are prescribed either as monotherapy or in combination with L-DOPA. Their use in monotherapy is more effective at the early stages of PD and may delay the use of L-DOPA [145]. When combined with L-DOPA, MAO-B inhibitors prolong the therapeutic effects of L-DOPA, decrease the dose of L-DOPA required to control the symptoms, and reduce the occurrence of L-DOPA-associated side effects [140,145].
COMT has received considerable attention due to its involvement in the metabolism of L-DOPA [146]. Given adjunctively with L-DOPA, COMT inhibitors (Figure 7) decrease L-DOPA premature inactivation, prolonging its half-life and improving its delivery to the brain [146,147]. In addition, COMT inhibitors enable a decrease in both the dose and administration frequency of L-DOPA, reducing “off” time (i.e., decreasing periods of time when symptoms are more noticeable and movements are more difficult) and increasing “on” time (i.e., increasing periods when PD patients experience good symptom control), thereby improving and prolonging the clinical response to L-DOPA [147].
Inhibitors of peripheral (entacapone, opicapone, Figure 7) and cerebral COMT (tolcapone, Figure 7) were developed and are available for the adjunctive treatment of PD. Peripheral COMT inhibition decreases the systemic decomposition of L-DOPA. Still, COMT inhibition in the CNS has the additional advantage of decreasing the metabolism of both L-DOPA and DA in the brain [128].
4.3. Alzheimer's and Parkinson’s Diseases: Looking for New Targets
Currently, the pharmacotherapy for AD and PD consists of drugs approved by the Food and Drug Administration (FDA) that regulate neurotransmitter levels. Unfortunately, they only provide valuable but modest symptomatic benefits, being unable to modify the course of these diseases [9,148]. These treatments are also accompanied by limitations. For instance, AChE inhibitors offer relatively short-lasting positive effects in AD patients [149] and display cholinomimetic actions on the gastrointestinal tract that result in diarrhea, nausea, and vomiting [150]. The efficacy of PD medicines also decreases over time, and the chronic treatment often culminates in motor complications (e.g.: L-DOPA-induced dyskinesia) [151].
The need of beneficial neuroprotective agents has been the driving force for the development of new and innovative therapeutic strategies, preferably with disease-modifying outcomes. For instance, over the last years, efforts have been made to develop new drug candidates able to tackle increased oxidative stress, metal dyshomeostasis (iron, copper), neuroinflammation, and aggregation of misfolded protein (Table 1). The following subsections will discuss the development of pharmacological agents targeting oxidative stress or adenosine receptors in NDs as representative examples.
4.3.1. Oxidative Stress as a Target in Alzheimer’s and Parkinson’s Diseases
Oxidative stress is one of the major contributors to the pathogenic cascade that leads to neurodegeneration in AD and PD [207,208]. Evidences of reactive species (RS)-mediated injuries, with increased levels of oxidative markers and damaged cell components, were observed in AD and PD brains [209]. A decline in the pool of endogenous antioxidants and a decrease in the activity of antioxidant enzymes were also reported [25,210].
The brain is particularly prone to oxidative stress-induced damage. Although the brain constitutes only 2 % of the total body weight, it is responsible for more than 20 % of the body's oxygen consumption, with a significant amount of oxygen being converted into reactive oxygen species (ROS) [208,211,212]. Despite this massive oxygen consumption, the brain presents a lower content of endogenous antioxidants (e.g.: glutathione, catalase) in comparison to other tissues, thus being more sensitive to cellular redox dyshomeostasis [211,213]. In addition, redox-active metals (e.g.: iron, copper) accumulate in specific brain regions and catalyze the formation of ROS [120,208]. Finally, the high levels in polyunsaturated fatty acids in the brain increase the susceptibility to lipid peroxidation and subsequent formation of toxic compounds [208,211,214].
The increased oxidative stress in NDs is strictly connected to other pathological events, namely mitochondrial dysfunction, dopamine oxidation, neuroinflammation, and accumulation of protein aggregates (e.g. Aβ, α-syn) (Figure 10) [215].
Although RS are generated in several cellular compartments, mitochondria is one of the main sources of the overproduction of ROS [121]. The formation of ROS in mitochondria occurs primarily at the ETC present in the inner mitochondrial membrane (Figure 11) [122,208]. The mitochondrial ETC consists of a series of membrane-bound complexes (complexes I, II, III and IV) [117], which generate a proton gradient across the inner mitochondrial membrane through electron transfers, leading to the production of ATP by ATP synthase (complex V) [208]. Metabolic intermediates formed during the Krebs cycle are used for oxidative phosphorylation [118]. During the ETC, a small proportion of electrons occasionally leak and directly reduce O2 to [118,119], which in turn is converted into other ROS such as H2O2 and HO• [121]. The formation of occurs mainly in complexes I and III [119,216]. Enzymes from the Krebs cycle (e.g.: α-ketoglutarate dehydrogenase, pyruvate dehydrogenase, aconitase) may also generate ROS [216,217].
Mitochondrial ETC is one of the primary targets of the harmful effects inflicted by high levels of ROS [117,219]. The oxidative damage at this level leads to the inhibition of ATP synthesis and the increased production of ROS in a vicious and detrimental cycle, contributing to cell dysfunction and cell death [122,219,220]. Mitochondria contain other components susceptible to oxidative damage, namely several iron-sulfur centers, proteins and unsaturated fatty acids in the inner membrane, and mitochondrial DNA (mtDNA), all of which are important for proper mitochondrial function [122]. Considering that mtDNA encodes some of the subunits of the complexes that constitute the ETC, the oxidative damage of mtDNA leads to the defective production of these proteins and subsequent mitochondrial dysfunction [219].
Since neurons have limited glycolytic capacity, they are particularly dependent on mitochondrial oxidative phosphorylation to meet their high energy requirements [220,221,222]. In addition to ATP synthesis, mitochondria are involved in other crucial cellular functions such as the synthesis of amino acids and steroids, β-oxidation of fatty acids, Ca2+ homeostasis, and regulation of apoptotic cell death [223]. Therefore, improper mitochondrial function compromises neuronal survival and contributes to neurodegeneration [217,223].
In PD, DA oxidation is associated with a selective vulnerability of dopaminergic neurons to oxidative stress [218]. Despite the essential role of DA in neurotransmission, DA contains a catechol group that may participate in the generation of ROS and metal chelation [224]. Dopamine is normally stored in monoaminergic vesicles under a low pH environment that prevents its oxidation [225]. However, DA may undergo enzymatic and non-enzymatic decomposition in the cytosol, which is accompanied by the formation of ROS (Figure 12) [226].
In the presence of O2, DA generates and electron-deficient DA semiquinones and DA quinones (Figure 12) [226,228,229]. The reaction rate of DA semiquinone formation is slow, but it is accelerated by redox-active transition metals [213]. The spontaneous cyclization of DA quinone yields leucoaminochrome whose further autoxidation forms aminochrome and [228]. Aminochrome participates in redox-cycling reactions that results in the formation of and in the depletion of cellular nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH) [230]. Dopamine quinone and aminochrome also form adducts with cellular nucleophiles modifying their function [231,232]. These include DNA, biothiols (e.g.: glutathione), α-synuclein and proteins involved in ATP synthesis (complexes I, III and V of the ETC), proteasomal degradation (parkin), microtubule stabilization (α- and β-tubulin) and axonal transport (actin) [227]. Therefore, the formation of these adducts will contribute to mitochondrial dysfunction, impairment of the axonal transport, inhibition of the proteasomal system, disruption of cytoskeleton architecture, and formation of α-synuclein aggregates in PD [227]. Aminochrome also polymerizes into neuromelanin, a brain pigment that contributes to neurodegeneration by triggering neuroinflammatory processes [208].
The oxidative deamination of DA by MAOs uses O2 and generates H2O2 and ammonia as by-products (Figure 12) [233]. Due to the increased expression with age in neuronal tissue [102,234], MAO-B becomes the predominant isoform involved in DA metabolism [208]. Monoamine oxidase B is mainly found in glial cells [105,235], but the H2O2 produced during DA deamination can permeate cell membranes and induce toxic effects in the neighboring neurons [208]. In fact, compared with astrocytes, neurons are more vulnerable to H2O2 due to the lower content in antioxidants involved in its detoxification (e.g.: GPx and glutathione) [235]. The H2O2 generated from MAO-B activity in astrocytes is also associated with increased amyloid plaque deposition [111].
Neuroinflammation represents a set of inflammatory processes occurring in the central nervous system that involve the action of glial cells in CNS (microglia, oligodendrocytes, astrocytes), non-glial resident myeloid cells (macrophages and dendritic cells) and peripheral leukocytes [236,237]. Neuroinflammation plays an important role in the progression of NDs [226]. For instance, in AD, microglia are activated by the presence of Aβ and co-localize with the plaques [238]. However, instead of efficiently removing the Aβ deposits, microglia release pro-inflammatory mediators that lead to neuronal damage [239]. In PD, extracellular αSYN aggregates can also interact with and activate surrounding glial cells to trigger a deleterious pro-inflammatory response [240]. In NDs, the expression of NADPH oxidases (NOXs) in activated microglia and reactive astrocytes is increased, resulting in the excessive formation of [226,241]. The activation of RS-producing enzymes in glial cells is associated with neurotoxic effects, which arise not only from the direct oxidative damage in neurons, but also from the intracellular redox signaling that exacerbates the pro-inflammatory response [241,242].
4.3.1.1. Targeting Oxidative Stress with Mitochondria-Targeted Antioxidants
Considering the involvement of oxidative stress in the pathophysiology of NDs, the rationale for using exogenous antioxidants to prevent delay, or remove the oxidative damage is evident [212,243]. In fact, several exogenous antioxidants showed promising results in animal and cellular models [211,212]. However, the results obtained in clinical trials were inconclusive, negative, or showed little benefit in NDs [244]. Numerous factors contribute to the discrepancy between pre-clinical and clinical results. In addition to aspects associated with the design of clinical trials (e.g.: posology, duration of treatment, age, and disease stage of the patients), most known dietary antioxidants display poor bioavailability and are unable to cross the BBB, affecting their delivery into the brain [211,212,244,245].
A common strategy used to overcome these pharmacokinetic limitations is the introduction of minor structural modifications on the antioxidant scaffold. The resulting derivatives may improve the targeting and drug-like properties while preserving or enhancing the antioxidant profile of the parent compounds [246,247].
Aside from the pharmacokinetic constraints, the lack of clinical efficacy of antioxidants may also result from the uniform distribution of antioxidants across all tissues and organs following administration, with only a small fraction being taken up by mitochondria [244,248], the main source and the target of ROS. Therefore, the development of antioxidants that selectively accumulate within mitochondria and tackle oxidative damage is of particular interest [249]. Compounds lacking mitochondriotropism but with relevant biological activities towards mitochondrial targets usually need to be directed to mitochondria [250]. In this sense, several approaches were developed to deliver antioxidants and other bioactive molecules to mitochondria, but one of the most widely used is their conjugation with lipophilic cations such as triphenylphosphonium (TPP+) [249,251].
Lipophilic TPP+ cations can diffuse across phospholipid bilayers because their positive charge is surrounded and dispersed over a large hydrophobic surface area, which decreases the activation energy for membrane permeation [252,253,254]. In response to the plasma and mitochondrial membrane potentials (, respectively), these compounds accumulate within the mitochondrial matrix against the concentration gradient [252] (Figure 13A). Then, TPP+ conjugates are taken up from the intracellular space to the mitochondrial matrix in response to the (-140 to -160 mV), leading to 100 to 500-fold accumulation within the mitochondrial matrix [218,254].
The increased accumulation of lipophilic TPP+ conjugates enhances the compounds’ potency and decreases the external dose required, limiting extramitochondrial metabolism that results in inactivation, excretion, or toxicity [122,256]. However, the extensive accumulation of these compounds within the mitochondrial matrix can disrupt membrane integrity and thereby compromise cellular respiration and ATP production (281, 294).
Following oral or intravenous administration, lipophilic TPP+ conjugates are rapidly taken up by the organs most affected by mitochondrial dysfunction (e.g.: liver, heart, brain) [252,257]. Therefore, targeting antioxidants in mitochondria stands out as a promising strategy in the discovery of new therapies for oxidative stress-related disorders.
Over the last decade, TPP+ cations have been conjugated with dietary antioxidants such as hydroxybenzoic [258] and hydroxycinnamic acids [259]. These compounds displayed remarkable antioxidant properties and were able to protect neuroblastoma cells against the oxidative damage induced by 6-hydroxydopamine or H2O2 [260]. Moreover, in studies performed in skin fibroblasts from male sporadic PD patients (sPD), the caffeic acid-based TPP+ conjugate AntiOXCIN4 restored mitochondrial membrane potential and mitochondrial fission, decreased autophagic flux, and enhanced cellular responses to stress by improving the cellular redox state and decreasing ROS levels [261]. To circumvent the drawbacks associated with the use of TPP+ cation, its replacement with nitrogen-based cationic carriers (e.g. isoquinolinium, imidazolium, picolínium) was recently performed (Figure 13B) [262]. This chemical modification resulted in decreased cytotoxicity while maintaining the compounds’ antioxidant properties and their ability to accumulate within mitochondria [262].
4.3.2. Adenosine Receptors as a Target in Alzheimer’s and Parkinson’s Diseases
Adenosine is a purine nucleoside that may act as a neurotransmitter as neuromodulator in the CNS [263]. It is involved in several physiological and pathophysiological processes in the brain, including motor function, sleep/wake cycle, learning and memory, pain, and astrocytic activity [264]. To perform its physiological roles, adenosine binds to four distinct G-coupled protein adenosine receptors (ARs), designated as A1, A2A, A2B and A3. Adenosine receptors (ARs) represent a group of glycoproteins containing seven transmembrane domains and are coupled to different G proteins [265] (Figure 14). While adenosine A1 and A3 receptors are coupled to inhibitory G proteins, A2A and A2B ARs are coupled to stimulatory G proteins. The A2A and A2B ARs preferably interact with members of the Gs family of G proteins, stimulating adenylyl cyclase to produce cyclic AMP (cAMP) and leading to the activation of a series of downstream signaling pathways. In contrast, A1 and A3 ARs inhibit the adenylyl cyclase activity by interaction with Gi proteins (Figure 14) [266].
ARs are widely distributed in the human body and participate in a broad range of physiological and pathophysiological processes [196]. While A1Rs and A2Rs can be predominantly found in specific parts of the CNS, A2BRs and A3Rs are mainly located in peripheral tissues [267] .
In the CNS, A1Rs are widely distributed in neocortical and limbic systems and are linked to cognitive functions [196,268,269]. A2ARs are highly expressed in striatal areas [196,268] and participate in the regulation of motor behavior and the management of dopamine-mediated responses [197]. A2ARs co-localize with dopamine D2 receptors (D2Rs) on GABAergic striatopallidal output neurons, where they form heteromer complexes [270]. These receptors within the heteromeric complex exert opposite effects on motor behavior, in which A2A AR agonism induces antagonistic effects on D2Rs. For instance, stimulation of dopamine D2Rs enhances motor activity, while A2A ARs decrease this effect by decreasing the affinity and response of D2Rs to their ligands [269,271].
Excessive A2A AR function has been linked to neuronal damage [272], and increased A2A AR expression is a characteristic feature of PD progression [273]. The cellular mechanisms responsible for A2A AR-mediated neurodegeneration remain elusive. However, evidence suggests that the activation of A2A ARs leads to increased glutamate release, increased Ca2+ entry, and enhanced long-term potentiation, all of which may culminate in excitotoxic damage [271]. The localization of A2A ARs at the basal ganglia, coupled with their pathophysiological role in PD, makes these receptors attractive drug targets to treat this disease [271]. A2A AR antagonism decreases motor impairment by enhancing dopamine D2R-mediated signaling. Moreover, A2A AR antagonism modulates cholinergic, glutamatergic, and GABAergic functions in the CNS [269]. Blockade of A2A AR signaling with selective A2A AR receptor antagonists was shown to be beneficial, not only by enhancing the therapeutic effects of L-DOPA, but also by reducing dyskinesia from long-term L-DOPA treatment [270].
Recent studies have also been disclosing a close association between A2A ARs and cognitive impairment in AD. For instance, abnormally high levels of A2A AR were detected in the hippocampus and in the cortex of AD patients [262,274] and in APP/PS1 transgenic AD mice [275]. Remarkably, activation of A2A ARs with agonists and optogenetic agents led to severe impairments in spatial discrimination in wild-type mice [276]. The involvement of A2A ARs in in hippocampal-dependent spatial reference memory was also shown in A2A AR knock-out studies in an Aβ1-42-based mice model of AD [277]. The memory deficits in APP/PS1 mice were reverted by the blockade of A2A ARs with a selective antagonist or by downregulation driven with shRNA interference [275]. Finally, recently it was shown that the improvement of spatial memory deficits by A2A AR antagonists in APP/PS1 mice results from the promotion of synaptic plasticity of adult-born granule cells [278]. Thus, the blockade of A2A AR activation with selective antagonists can be of great therapeutic benefit to AD patients.
4.3.2.1. A2A Adenosine Receptor Antagonists
The knowledge acquired over the last decades concerning the involvement of adenosine on motor functions, mainly through modulation of A2A AR, makes A2A AR antagonists promising non-dopaminergic agents for the treatment of PD motor symptoms. Over the last decades, the development of potent and selective ligands for ARs has been a dynamic area. Excellent reviews were recently published on this topic [279,280]. A small number of selective A2A AR antagonists reached advanced clinical trials for the treatment of motor symptoms in PD, namely the xanthine derivative istradefylline (KW-6002) and the non-xanthine derivatives Tozadenant (SYN115), Preladenant, and KW-6356 (Figure 15) [268,273].
Istradefylline was approved for the adjunctive treatment of PD in Japan in 2013 and by the FDA in 2019, being the first non-dopaminergic drug approved by FDA for PD in the last two decades [281]. Preladenant and Tozadenant underwent clinical evaluation for the treatment of PD (Preladenant: NCT00406029, NCT01227265; Tozadenant: NCT02453386, NCT03051607) [282]. Unfortunately, the clinical evaluation for both drug candidates were discontinued due to the lack of efficacy (Preladenant) or safety (Tozadenant) in phase 3 clinical trials [283].
KW-6356 is a new, selective, nonxanthine A2A receptor antagonist/inverse agonist. Compared to istradefylline, KW-6356 exhibits approximately 100-times higher affinity for the human A2A receptor and a prolonged drug residence time [284]. In a phase 2b clinical study in patients with PD, KW-6356 was safe and effective in the adjunctive treatment with L-DOPA (NCT03703570) [285]. Moreover, in a phase 2a clinical trial, KW-6356 monotherapy was well tolerated and more effective than placebo in patients with early, untreated PD (NCT02939391) [273].
5. Conclusions
The discovery of new drugs for NDs remains an enormous unmet medical need [286]. The available treatments for AD and PD provide valuable symptomatic relief, but only reduce the symptoms for a short period before the cognitive or motor functions continue to deteriorate [287]. Given the lack of therapeutic efficacy of the current treatments, the use of single-target drugs may be insufficient to address the multiple pathological aspects of NDs [288]. Treatment of AD and PD may thus require the manipulation of several targets to restore the physiological balance and thereby attain significant therapeutic efficacy [289].
Traditionally, the “one-drug, one-target” paradigm is the mainstay drug discovery concept in the pharmaceutical industry [91]. This paradigm is mainly focused in generating drugs that selectively bind to a single biological target, avoiding potential adverse side effects associated with mistargeting other biological entities [290]. The current therapy for AD and PD management is based on this paradigm. However, the currently single-target drugs address the diseases’ symptomatology, without halting or modifying the disease progression [290,291]. Therefore, drugs that can simultaneously manipulate multiple targets may provide therapeutic benefits in AD and PD diseases due to their multifactorial nature and complexity [292]. The limited clinical efficacy and the lack of disease-modifying effects of the available drugs shifted the research focus from single-target agents to multitarget-directed drugs [235,293]. The field of multitarget approach may thus provide innovative therapeutic solutions to feed the pipeline of disease-modifying drugs for AD and PD.
Author Contributions
Conceptualization: L.S., F.B. and D.C.; Writing – original draft preparation: L.S., S.B., C.F., A.G., and D.C.; Writing – review and editing: L.S., S.B., C.F., I.L., J.P., C.A., C.S.M., A.G., F.B. and D.C. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This work was funded by FEDER funds through the Operational Programme Competitiveness Factors COMPETE and national funds by the Foundation for Science and Technology (FCT), I.P., under research grants PT-OPENSCREEN-NORTE-01-0145-FEDER-085468, UIDP/00081/2020, LA/P/0058/2020. Grants are supported by FCT, MCTES, Fundo Social Europeu (FSE), and UE. L. Sequeira, S. Benfeito, C. Fernandes (project reference 2021.04016.CEECIND/CP1655/CT0004 and DOI identifier 10.54499/2021.04016.CEECIND/CP1655/CT0004), A. Gaspar and D. Chavarria grants are supported by FCT and FEDER/COMPETE. C. Fernandes thanks the FCT for the financial support of his work contract through the Scientific Employment Stimulus—Individual Call (2021.04016.CEECIND/CP1655/CT0004).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Moutinho, M.; Codocedo, J.F.; Puntambekar, S.S.; Landreth, G.E. Nuclear Receptors as Therapeutic Targets for Neurodegenerative Diseases: Lost in Translation. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Vos, T.; Nichols, E.; Owolabi, M.O.; Carroll, W.M.; Dichgans, M.; Deuschl, G.; Parmar, P.; Brainin, M.; Murray, C. The global burden of neurological disorders: translating evidence into policy. Lancet Neurol. 2020, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Fereshtehnejad, S.M.; Vosoughi, K.; Heydarpour, P.; Sepanlou, S.G.; Farzadfar, F.; Tehrani-Banihashemi, A.; Malekzadeh, R.; Sahraian, M.A.; Vollset, S.E.; Naghavi, M. Burden of Neurodegenerative Diseases in the Eastern Mediterranean Region, 1990-2016: Findings from the Global Burden of Disease 2016 Study. Eur. J. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- van Bokhoven, P.; de Wilde, A.; Vermunt, L.; Leferink, P.S.; Heetveld, S.; Cummings, J.; Scheltens, P.; Vijverberg, E.G.B. The Alzheimer’s disease drug development landscape. Alzheimer's Res. Ther. 2021, 13, 186. [Google Scholar] [CrossRef] [PubMed]
- Lengyel-Zhand, Z.; Puentes, L.N.; Mach, R.H. PARkinson's: From cellular mechanisms to potential therapeutics. Pharmacol. Ther. 2022, 230, 107968. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. The potential of epigenetic therapies in neurodegenerative diseases. Front Genet 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Burns, S.; Selman, A.; Sehar, U.; Rawat, P.; Reddy, A.P.; Reddy, P.H. Therapeutics of Alzheimer’s Disease: Recent Developments. Antioxidants 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.R.; Huters, A.D.; Towne, T.B.; Reddy, R.E.; Fogle, J.L.; Voight, E.A.; Kym, P.R. Parkinson’s Disease: Advances in Treatment and the Syntheses of Various Classes of Pharmaceutical Drug Substances. Chem. Rev. 2023, 123, 13693–13712. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L. Intervening in ageing to prevent the diseases of ageing. Trends Endocrinol. Metab. 2014, 25, 555–557. [Google Scholar] [CrossRef]
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Urso, D.; Savica, R. Descriptive Epidemiology of Neurodegenerative Diseases: What Are the Critical Questions? Neuroepidemiology 2022, 56, 309–318. [Google Scholar] [CrossRef]
- Zaib, S.; Javed, H.; Khan, I.; Jaber, F.; Sohail, A.; Zaib, Z.; Mehboob, T.; Tabassam, N.; Ogaly, H.A. Neurodegenerative Diseases: Their Onset, Epidemiology, Causes and Treatment. ChemistrySelect 2023, 8, e202300225. [Google Scholar] [CrossRef]
- Yuan, L.; Guo, Y.; Wen, S.; Deng, H. Editorial: Genetic and Epigenetic Basis of Neurodegenerative Diseases. Front Aging Neurosci 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- United, N. World Population Ageing 2020 Highlights: Living Arrangements of Older Persons; UN: 2021.
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Ye, S.; Huangfu, J.; Calimag, D.P. Music therapy is a potential intervention for cognition of Alzheimer’s Disease: a mini-review. Transl. Neurodegener. 2017, 6, 2. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s Disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Wimo, A.; Winblad, B.; Aguero-Torres, H.; von Strauss, E. The Magnitude of Dementia Occurrence in the World. Alz Dis Assoc Dis 2003, 17, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Cahill, S. WHO's global action plan on the public health response to dementia: some challenges and opportunities. Aging Ment. Health 2019, 24, 1–3. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Fiske, B.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson's Disease Drug Therapies in the Clinical Trial Pipeline: 2023 Update.
- Soilemezi, D.; Palmar-Santos, A.; Navarta-Sánchez, M.V.; Roberts, H.C.; Pedraz-Marcos, A.; Haahr, A.; Sørensen, D.; Bragstad, L.K.; Hjelle, E.G.; Haavaag, S.B.; et al. Understanding support systems for Parkinson's disease management in community settings: A cross-national qualitative study. Health Expect. 2023, 26, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer's disease and other dementias: a priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Hemnani, T. Alzheimer’s Disease pathogenesis and therapeutic interventions. J Clin Neurosci 2004, 11, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Waldemar, G.; Dubois, B.; Emre, M.; Georges, J.; McKeith, I.G.; Rossor, M.; Scheltens, P.; Tariska, P.; Winblad, B. Recommendations for the diagnosis and management of Alzheimer's disease and other disorders associated with dementia: EFNS guideline. Eur. J. Neurol. 2007, 14, e1–e26. [Google Scholar] [CrossRef] [PubMed]
- Taler, V.; Phillips, N.A. Language performance in Alzheimer's disease and mild cognitive impairment: A comparative review. J. Clin. Exp. Neuropsychol. 2008, 30, 501–556. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer's-Association. Alzheimer’s Disease Facts and Figures. Alzheimer's Dement. 2017, 13, 325–373. [Google Scholar]
- López, O.L.; DeKosky, S.T. Clinical symptoms in Alzheimer's disease. In Handbook of Clinical Neurology, Elsevier: 2008; Vol. 89, pp. 207–216.
- Scarmeas, N.; Brandt, J.; Albert, M.; Hadjigeorgiou, G.; Papadimitriou, A.; Dubois, B.; Sarazin, M.; Devanand, D.; Honig, L.; Marder, K.; et al. Delusions and Hallucinations Are Associated With Worse Outcome in Alzheimer Disease. Arch. Neurol. 2005, 62, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Tarawneh, R.; Holtzman, D.M. The Clinical Problem of Symptomatic Alzheimer Disease and Mild Cognitive Impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Wang, K.; Dong, J.; Tang, L. Alzheimer's disease: Updated multi-targets therapeutics are in clinical and in progress. Eur. J. Med. Chem. 2022, 238, 114464. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer's disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Sloane, P.D.; Zimmerman, S.; Suchindran, C.; Reed, P.; Wang, L.; Boustani, M.; Sudha, S. The Public Health Impact of Alzheimer's Disease, 2000–2050: Potential Implication of Treatment Advances. Annu Rev Publ Health 2002, 23, 213–231. [Google Scholar] [CrossRef]
- Corder, E.; Saunders, A.; Strittmatter, W.; Schmechel, D.; Gaskell, P.; Small, G.; Roses, A.; Haines, J.; Pericak-Vance, M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Blacker, D.; Haines, J.; Rodes, L.; Terwedow, H.; Go, R.; Harrell, L.; Perry, R.; Bassett, S.; Ga, C.; Meyers, D.; et al. ApoE-4 and Age at Onset of Alzheimer's Disease: The NIMH Genetics Initiative; 1997; Vol. 48, pp. 139–147.
- Rubinsztein, D.C. The genetics of Alzheimer's disease. Prog. Neurobiol. 1997, 52, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Moceri, V.M.; Kukull, W.A.; Emanuel, I.; van Belle, G.; Larson, E.B. Early-life risk factors and the development of Alzheimer’s disease. Neurology 2000, 54, 415–415. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Helkala, E.-L.; Laakso, M.P.; Hänninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and Alzheimer's disease in later life: longitudinal, population based study. BMJ 2001, 322, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L. Alzheimer’s Disease. Nature 2009, 461, 895. [Google Scholar] [CrossRef]
- Bachurin, S.O.; Gavrilova, S.I.; Samsonova, A.; Barreto, G.E.; Aliev, G. Mild cognitive impairment due to Alzheimer disease: Contemporary approaches to diagnostics and pharmacological intervention. Pharmacol Res 2017. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer's disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Schwarzenbacher, R.; Lipton, S.A. Molecular pathways to neurodegeneration. Nat. Med. 2004, 10, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Venkateshappa, C.; Harish, G.; Mahadevan, A.; Srinivas Bharath, M.M.; Shankar, S.K. Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: implications for neurodegeneration in Alzheimer's disease. Neurochem. Res. 2012, 37, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer's disease: therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef] [PubMed]
- Kljakic, O.; Janickova, H.; Prado, V.F.; Prado, M.A.M. Cholinergic/glutamatergic co-transmission in striatal cholinergic interneurons: new mechanisms regulating striatal computation. J. Neurochem. 2017, 142 (Suppl. 2), 90–102. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Biswas, A. Molecular diagnostics of neurodegenerative disorders. Front. Mol. Biosci. 2015, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ehret, M.J.; Chamberlin, K.W. Current Practices in the Treatment of Alzheimer Disease: Where is the Evidence After the Phase III Trials? Clin. Ther. 2015, 37, 1604–1616. [Google Scholar] [CrossRef]
- Kovacs, G.G. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int. J. Mol. Sci. 2016, 17, 189. [Google Scholar] [CrossRef] [PubMed]
- Pepeu, G.; Grazia Giovannini, M. The fate of the brain cholinergic neurons in neurodegenerative diseases. Brain Res. 2017, 1670, 173–184. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. : 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Ghiso, J.; Frangione, B. Biology of Aβ Amyloid in Alzheimer's Disease. Neurobiol Dis 1997, 4, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Stuchbury, G.; Münch, G. Alzheimer’s associated inflammation, potential drug targets and future therapies. J. Neural Transm. 2005, 112, 429–453. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Frequency of Stages of Alzheimer-Related Lesions in Different Age Categories. Neurobiol. Aging 1997, 18, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Amyloid β-Protein and the Genetics of Alzheimer's Disease. J. Biol. Chem. 1996, 271, 18295–18298. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Alzheimer’s Disease: strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Kalra, J.; Khan, A. Reducing Aβ load and tau phosphorylation: Emerging perspective for treating Alzheimer's disease. Eur. J. Pharmacol. 2015, 764, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Pietrzik, C.; Behl, C. Concepts for the treatment of Alzheimer's disease: molecular mechanisms and clinical application. Int. J. Exp. Pathol. 2005, 86, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.K.; Grossberg, G.T. Diagnosis and treatment of Alzheimer’s disease. Neurology 2005, 64, S34–S39. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.; Chand, K.; Chaves, S. Recent progress in multifunctional metal chelators as potential drugs for Alzheimer's disease. Coord. Chem Rev 2016, 327-328, 287–303. [Google Scholar] [CrossRef]
- Cotman, C.W.; Su, J.H. Mechanisms of Neuronal Death in Alzheimer's Disease. Brain Pathol 1996, 6, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Costa, H.N.; Esteves, A.R.; Empadinhas, N.; Cardoso, S.M. Parkinson's Disease: A Multisystem Disorder. Neurosci. Bull. 2023, 39, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Elmabruk, A.; Das, B.; Yedlapudi, D.; Xu, L.; Antonio, T.; Reith, M.E.A.; Dutta, A.K. Design, Synthesis, and Pharmacological Characterization of Carbazole Based Dopamine Agonists as Potential Symptomatic and Neuroprotective Therapeutic Agents for Parkinson's Disease. ACS Chem. Neurosci. 2019, 10, 396–411. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.; Diederich, N.J.; Kruger, R.; Balling, R. The hallmarks of Parkinson's disease. Febs J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson's disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, X.; Zhen, X. Development of Adenosine A2A Receptor Antagonists for the Treatment of Parkinson's Disease: A Recent Update and Challenge. ACS Chem. Neurosci. 2019, 10, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson's disease. EMBO Mol Med 2012, 4, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson's disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139 (Suppl. 1), 59–74. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Recent advances in our understanding of neurodegeneration. J. Neural Transm. 2009, 116, 1111–1162. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 0, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Oertel, W.; Schulz, J.B. Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J. Neurochem. 2016, 139 (Suppl. 1), 325–337. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Adami, H.O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson's disease: a review of the evidence. Eur. J. Epidemiol. 2011, 26 (Suppl. 1), S1–S58. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson's disease: mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Swanson, J.; Posner, M.; Fusella, J.; Wasdell, M.; Sommer, T.; Fan, J. Genes and attention deficit hyperactivity disorder. Curr. Psychiatry Rep. 2001, 3, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Brunelin, J.; Fecteau, S.; Suaud-Chagny, M.-F. Abnormal striatal dopamine transmission in schizophrenia. Curr. Med. Chem. 2013, 20, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Nakata, Y.; Mochizuki, H. α-Synuclein and Neuronal Cell Death. Mol. Neurobiol. 2013, 47, 466–483. [Google Scholar] [CrossRef] [PubMed]
- Melo, T.Q.; Copray, S.; Ferrari, M.F.R. Alpha-Synuclein Toxicity on Protein Quality Control, Mitochondria and Endoplasmic Reticulum. Neurochem. Res. 2018, 43, 2212–2223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Xia, Y.; Wan, F.; Ma, K.; Guo, X.; Kou, L.; Yin, S.; Han, C.; Liu, L.; Huang, J.; et al. New Perspectives on Roles of Alpha-Synuclein in Parkinson's Disease. Front Aging Neurosci 2018, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.; Liu, L.; Bennett, J.P., Jr. Chronic, low-dose rotenone reproduces Lewy neurites found in early stages of Parkinson's disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol. Neurodegener. 2008, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Parkinson's disease and parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M.Y. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Majekova, M.; Medina, M.; Valoti, M. Key Targets for Multi-Target Ligands Designed to Combat Neurodegeneration. Front. Neurosci. 2016, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; ckowska, A.W.; Panek, D.; Malawska, B. Recent Development of Multifunctional Agents as Potential Drug Candidates for the Treatment of Alzheimer's Disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef]
- Shook, B.C.; Jackson, P.F. Adenosine A2A Receptor Antagonists and Parkinson's Disease. ACS Chem. Neurosci. 2011, 2, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.R.; Rockwood, K.; Martin, E.; Darvesh, S. Cholinesterase inhibition in Alzheimer's disease: is specificity the answer? J. Alzheimers Dis. 2014, 42, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Woolf, N.J.; Butcher, L.L. Cholinergic systems mediate action from movement to higher consciousness. Behav. Brain Res. 2011, 221, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17213–17218. [Google Scholar] [CrossRef]
- Lendvai, B. Cholinergic Transmission. In Handbook of Neurochemistry and Molecular Neurobiology: Neurotransmitter Systems, Lajtha, A., Vizi, E.S., Eds. Springer US: Boston, MA, 2008; pp. 113-127. [CrossRef]
- Terry, A.V., Jr.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer's disease-related cognitive deficits: recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.G.; Francis, P.T.; Schwam, E.; Payne-Parrish, J. Cholinesterase inhibitors used in the treatment of Alzheimer's disease: the relationship between pharmacological effects and clinical efficacy. Drugs Aging 2004, 21, 453–478. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Z.M.; Wu, J.J.; Wang, J.; Xie, S.S.; Lan, J.S.; Xu, W.; Kong, L.Y.; Wang, X.B. Synthesis and pharmacological evaluation of donepezil-based agents as new cholinesterase/monoamine oxidase inhibitors for the potential application against Alzheimer's disease. J. Enzym. Inhib. Med. Chem. 2016, 31, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A. The history of the cholinergic hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Dale, H.H. The action of certain esters and ethers of choline, and their relation to muscarine. J Pharmacol Exp Ther 1914, 6, 147–190. [Google Scholar]
- Du, X.; Wang, X.; Geng, M. Alzheimer's disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, S.; Borloz, A.; Urbain, A.; Marston, A.; Hostettmann, K.; Carrupt, P.A.; Reist, M. In vitro screening assays to identify natural or synthetic acetylcholinesterase inhibitors: thin layer chromatography versus microplate methods. Eur. J. Pharm. Sci. 2008, 33, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Berger-Sweeney, J. The cholinergic basal forebrain system during development and its influence on cognitive processes: important questions and potential answers. NeurosciI Biobehav R 2003, 27, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Haga, T. High-affinity choline transporter. Neurochem. Res. 2003, 28, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Amenta, F.; Tayebati, S.K. Pathways of acetylcholine synthesis, transport and release as targets for treatment of adult-onset cognitive dysfunction. Curr. Med. Chem. 2008, 15, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Prado, M.A.; Reis, R.A.; Prado, V.F.; de Mello, M.C.; Gomez, M.V.; de Mello, F.G. Regulation of acetylcholine synthesis and storage. Neurochem. Int. 2002, 41, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer's disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Alpha7 nicotinic acetylcholine receptor is a target in pharmacology and toxicology. Int. J. Mol. Sci. 2012, 13, 2219–2238. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Costantini, E.; Di Nicola, M.; D'Angelo, C.; Franchi, S.; D'Aurora, M.; Di Bari, M.; Orlando, V.; Galizia, S.; Ruggieri, S.; et al. Butyrylcholinesterase and Acetylcholinesterase polymorphisms in Multiple Sclerosis patients: Implication in peripheral inflammation. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Scarpini, E.; Scheltens, P.; Feldman, H. Treatment of Alzheimer's disease: current status and new perspectives. Lancet. Neurol. 2003, 2, 539–547. [Google Scholar] [CrossRef]
- Winkler, J.; Thal, L.J.; Gage, F.H.; Fisher, L.J. Cholinergic strategies for Alzheimer’s disease. J Mol Med 1998, 76, 555–567. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; González-Acedo, A.; Rivas-Domínguez, A.; García-Morales, V.; García-Cozar, F.J.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Onor, M.L.; Trevisiol, M.; Aguglia, E. Rivastigmine in the treatment of Alzheimer’s disease: an update. Clin. Interv. Aging. 2007, 2, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Poritis, N. Memantine in severe dementia: results of the 9M-best study (benefit and efficacy in severly demented patients during treatment with memantine). Int J Geriar Psych 1999, 14, 135–146. [Google Scholar] [CrossRef]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine in Moderate-to-Severe Alzheimer's Disease. New Engl J Med 2003, 348, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Bassil, N.; Thaipisuttikul, P.; Grossberg, G.T. Memantine ER, a once-daily formulation for the treatment of Alzheimer's disease. Expert Opin Pharm. 2010, 11, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.P.; Collingridge, G.L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 1993, 361, 31. [Google Scholar] [CrossRef] [PubMed]
- Sucher, N.J.; Awobuluyi, M.; Choi, Y.-B.; Lipton, S.A. NMDA receptors: from genes to channels. Trends Pharmacol. Sci. 1996, 17, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Greenamyre, J.T.; Porter, R.H. Anatomy and physiology of glutamate in the CNS. Neurology 1994, 44, S7–S13. [Google Scholar] [PubMed]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sánchez-López, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesús, G.; Beas-Zarate, C.; et al. Memantine for the Treatment of Dementia: A Review on its Current and Future Applications. J. Alzheimer's Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Barajas, C.; Coronel, I.; Florán, B. Dopamine Receptors and Neurodegeneration. Aging Dis. 2015, 6, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Tritsch, N.X.; Sabatini, B.L. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 2012, 76, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Lester, D.B.; Rogers, T.D.; Blaha, C.D. Acetylcholine-dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci. Ther. 2010, 16, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R.; Moussaud, S.; McLean, P. Targeting heat shock proteins to modulate α-synuclein toxicity. Ther. Adv. Neurol. Disord. 2014, 7, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Finberg, J.P.M. Inhibitors of MAO-B and COMT: their effects on brain dopamine levels and uses in Parkinson's disease. J. Neural Transm. (Vienna) 2018, 126, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Godar, S.C.; Bortolato, M. Gene-sex interactions in schizophrenia: focus on dopamine neurotransmission. Front Behav Neurosci 2014, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Morales-Garcia, J.A.; Susin, C.; Alonso-Gil, S.; Perez, D.I.; Palomo, V.; Perez, C.; Conde, S.; Santos, A.; Gil, C.; Martinez, A.; et al. Glycogen synthase kinase-3 inhibitors as potent therapeutic agents for the treatment of Parkinson disease. ACS Chem. Neurosci. 2013, 4, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.C.; Rassnick, S.; Wallace, N.; Crooke, J.; Ault, M.; Chakravarty, D.; Barbay, J.K.; Wang, A.; Powell, M.T.; Leonard, K.; et al. Design and characterization of optimized adenosine A(2)A/A(1) receptor antagonists for the treatment of Parkinson's disease. J. Med. Chem. 2012, 55, 1402–1417. [Google Scholar] [CrossRef] [PubMed]
- Zanforlin, E.; Zagotto, G.; Ribaudo, G. The Medicinal Chemistry of Natural and Semisynthetic Compounds against Parkinson's and Huntington's Diseases. ACS Chem. Neurosci. 2017, 8, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Rusiecka, I.; Ruczyński, J.; Kozłowska, A.; Backtrog, E.; Mucha, P.; Kocić, I.; Rekowski, P. TP10-Dopamine Conjugate as a Potential Therapeutic Agent in the Treatment of Parkinson's Disease. Bioconjugate Chem. 2019, 30, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Stefano, E.; Francesca, M.; Damiana, L.; Tatyana, D.S.; Raul, R.G. Role of Catechol-O-Methyltransferase (COMT)-Dependent Processes in Parkinson’s Disease and L-DOPA Treatment. CNS Neurol. Disord. - Drug Targets 2012, 11, 251–263. [Google Scholar] [CrossRef]
- Berger, A.A.; Winnick, A.; Izygon, J.; Jacob, B.M.; Kaye, J.S.; Kaye, R.J.; Neuchat, E.E.; Kaye, A.M.; Alpaugh, E.S.; Cornett, E.M.; et al. Opicapone, a Novel Catechol-O-methyl Transferase Inhibitor, for Treatment of Parkinson's Disease "Off" Episodes. 2022.
- Huleatt, P.B.; Khoo, M.L.; Chua, Y.Y.; Tan, T.W.; Liew, R.S.; Balogh, B.; Deme, R.; Goloncser, F.; Magyar, K.; Sheela, D.P.; et al. Novel arylalkenylpropargylamines as neuroprotective, potent, and selective monoamine oxidase B inhibitors for the treatment of Parkinson's disease. J. Med. Chem. 2015, 58, 1400–1419. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Bao, X.Q.; Xu, S.; Yu, W.W.; Cao, S.N.; Hu, J.P.; Li, Y.; Wang, X.L.; Zhang, D.; Yu, S.S. A Novel Parkinson's Disease Drug Candidate with Potent Anti-neuroinflammatory Effects through the Src Signaling Pathway. J. Med. Chem. 2016, 59, 9062–9079. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Hammuda, A.; Shalaby, R.; Rovida, S.; Edmondson, D.E.; Binda, C.; Khalil, A. Design and synthesis of novel chalcones as potent selective monoamine oxidase-B inhibitors. Eur. J. Med. Chem. 2016, 114, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Booysen, H.P.; Moraal, C.; Terre'Blanche, G.; Petzer, A.; Bergh, J.J.; Petzer, J.P. Thio- and aminocaffeine analogues as inhibitors of human monoamine oxidase. Bioorg. Med. Chem. 2011, 19, 7507–7518. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, N.T.; Stammler, H.G.; Neumann, B.; Hristova, S.; Antonov, L.; Gastreich, M. Crystal structures, binding interactions, and ADME evaluation of brain penetrant N-substituted indazole-5-carboxamides as subnanomolar, selective monoamine oxidase B and dual MAO-A/B inhibitors. Eur. J. Med. Chem. 2017, 127, 470–492. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.K.P.; Ayyannan, S.R. Monoamine oxidase-B inhibitors as potential neurotherapeutic agents: An overview and update. Med. Res. Rev. 2019, 1–104. [Google Scholar] [CrossRef] [PubMed]
- Robakis, D.; Fahn, S. Defining the Role of the Monoamine Oxidase-B Inhibitors for Parkinson's Disease. CNS Drugs 2015, 29, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Rathitharan, G.; Meyer, J.H.; Furukawa, Y.; Ang, L.C.; Boileau, I.; Guttman, M.; Hornykiewicz, O.; Kish, S.J. Brain monoamine oxidase B and A in human parkinsonian dopamine deficiency disorders. Brain 2017, 140, 2460–2474. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Ali, M.; Moustafa, A.A. Effects of combined MAO-B inhibitors and levodopa vs. monotherapy in Parkinson's disease. Front Aging Neurosci 2014, 6, 180. [Google Scholar] [CrossRef] [PubMed]
- Palma, P.N.; Rodrigues, M.L.; Archer, M.; Bonifacio, M.J.; Loureiro, A.I.; Learmonth, D.A.; Carrondo, M.A.; Soares-da-Silva, P. Comparative study of ortho- and meta-nitrated inhibitors of catechol-O-methyltransferase: interactions with the active site and regioselectivity of O-methylation. Mol. Pharmacol. 2006, 70, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Solla, P.; Cannas, A.; Marrosu, F.; Marrosu, M.G. Therapeutic interventions and adjustments in the management of Parkinson disease: role of combined carbidopa/levodopa/entacapone (Stalevo). Neuropsychiatr. Dis. Treat. 2010, 6, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Barbosa, D.J.; Remião, F.; Silva, R. Alzheimer’s disease: Insights and new prospects in disease pathophysiology, biomarkers and disease-modifying drugs. Biochem. Pharmacol. 2023, 211, 115522. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H. Reconsideration of Anticholinesterase Therapeutic Strategies against Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [PubMed]
- Aradi, S.D.; Hauser, R.A. Medical Management and Prevention of Motor Complications in Parkinson's Disease. Neurotherapeutics 2020, 17, 1339–1365. [Google Scholar] [CrossRef] [PubMed]
- Fasae, K.D.; Abolaji, A.O.; Faloye, T.R.; Odunsi, A.Y.; Oyetayo, B.O.; Enya, J.I.; Rotimi, J.A.; Akinyemi, R.O.; Whitworth, A.J.; Aschner, M. Metallobiology and therapeutic chelation of biometals (copper, zinc and iron) in Alzheimer’s disease: Limitations, and current and future perspectives. J. Trace Elem. Med. Biol. 2021, 67, 126779. [Google Scholar] [CrossRef] [PubMed]
- Esmieu, C.; Guettas, D.; Conte-Daban, A.; Sabater, L.; Faller, P.; Hureau, C. Copper-Targeting Approaches in Alzheimer’s Disease: How To Improve the Fallouts Obtained from in Vitro Studies. Inorg. Chem. 2019, 58, 13509–13527. [Google Scholar] [CrossRef]
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef]
- Cukierman, D.S.; Lázaro, D.F.; Sacco, P.; Ferreira, P.R.; Diniz, R.; Fernández, C.O.; Outeiro, T.F.; Rey, N.A. X1INH, an improved next-generation affinity-optimized hydrazonic ligand, attenuates abnormal copper(i)/copper(ii)-α-Syn interactions and affects protein aggregation in a cellular model of synucleinopathy. Dalton Trans. 2020, 49, 16252–16267. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Wang, X.; Pan, X.; Xie, J.; Xu, H. Mitochondrial iron dyshomeostasis and its potential as a therapeutic target for Parkinson's disease. Exp. Neurol. 2024, 372, 114614. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Xu, X.; Xia, H. Targeting MCL1 to induce mitophagy is a potential therapeutic strategy for Alzheimer disease. Autophagy 2021, 17, 818–819. [Google Scholar] [CrossRef] [PubMed]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; Angus McQuibban, G. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- xu, Y.; Zhi, F.; Mao, J.; Peng, Y.; Shao, N.; Balboni, G.; Yang, Y.; Xia, Y. δ-opioid receptor activation protects against Parkinson’s disease-related mitochondrial dysfunction by enhancing PINK1/Parkin-dependent mitophagy. Aging 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Maia, M.A.; Sousa, E. BACE-1 and γ-Secretase as Therapeutic Targets for Alzheimer’s Disease. Pharmaceuticals 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. γ-Secretase: once and future drug target for Alzheimer’s disease. Expert Opin. Drug. Discov. 2024, 19, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer's disease. Biochim. Et Biophys. Acta (BBA) - Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Reddy, V.P.; Aryal, P.; Soni, P. RAGE Inhibitors in Neurodegenerative Diseases. Biomedicines 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, J.; Smeralda, W.; Jouanne, M.; Sopkova-de Oliveira Santos, J.; Catto, M.; Voisin-Chiret, A.S. Tau protein aggregation: Key features to improve drug discovery screening. Drug Discov. Today 2022, 27, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.; Kumar, P.; Ambasta, K.R. Discovery of Novel Compounds Targeting DJ-1 as Neuroprotectants for Parkinson’s Disease by Virtual Screening and In Silico Method. Curr. Comput. -Aided Drug Des. 2021, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, J.M.; Zwirek, M.; Sarhan, A.R.; Vatsan, P.S.; Tonelli, F.; Alessi, D.R.; Davies, P.; Gray, N.S. Development of a highly potent and selective degrader of LRRK2. Bioorg. Med. Chem. Lett. 2023, 94, 129449. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y. Parkinson’s Disease: Are PINK1 Activators Inching Closer to the Clinic? ACS Med. Chem. Lett. 2023, 14, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yao, S.; Yang, Z.-x.; Zhou, C.; Zhang, Y.; Zhang, Y.; Zhang, L.; Li, J.-t.; Xu, Z.-j.; Zhu, W.-l.; et al. Pharmacological characterization of the small molecule 03A10 as an inhibitor of α-synuclein aggregation for Parkinson’s disease treatment. Acta Pharmacol. Sin. 2023, 44, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Cescon, E.; Bolcato, G.; Federico, S.; Bissaro, M.; Valentini, A.; Ferlin, M.G.; Spalluto, G.; Sturlese, M.; Moro, S. Scaffold Repurposing of in-House Chemical Library toward the Identification of New Casein Kinase 1 δ Inhibitors. ACS Med. Chem. Lett. 2020, 11, 1168–1174. [Google Scholar] [CrossRef]
- Redenti, S.; Marcovich, I.; De Vita, T.; Pérez, C.; De Zorzi, R.; Demitri, N.; Perez, D.I.; Bottegoni, G.; Bisignano, P.; Bissaro, M.; et al. A Triazolotriazine-Based Dual GSK-3β/CK-1δ Ligand as a Potential Neuroprotective Agent Presenting Two Different Mechanisms of Enzymatic Inhibition. ChemMedChem 2019, 14, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Barczuk, J.; Siwecka, N.; Lusa, W.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. Targeting NLRP3-Mediated Neuroinflammation in Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Załuski, M.; Łażewska, D.; Jaśko, P.; Honkisz-Orzechowska, E.; Kuder, K.J.; Brockmann, A.; Latacz, G.; Zygmunt, M.; Kaleta, M.; Greser, B.A. Anti-Inflammatory Activities of 8-Benzylaminoxanthines Showing High Adenosine A2A and Dual A1/A2A Receptor Affinity. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, S.S.; Mustafa, S.M.; Moore Ii, B.M. Synthesis and biological evaluation of a ring analogs of the selective CB2 inverse agonist SMM-189. Bioorg. Med. Chem. 2021, 33, 116035. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.-C.; Fung, K.-M.; Hsu, H.-M.; Tseng, T.-S. Discovery of 8-prenylnaringenin from hop (Humulus lupulus L.) as a potent monoacylglycerol lipase inhibitor for treatments of neuroinflammation and Alzheimer's disease. RSC Adv. 2021, 11, 31062–31072. [Google Scholar] [CrossRef]
- Harrison, D.; Bock, M.G.; Doedens, J.R.; Gabel, C.A.; Holloway, M.K.; Lewis, A.; Scanlon, J.; Sharpe, A.; Simpson, I.D.; Smolak, P.; et al. Discovery and Optimization of Triazolopyrimidinone Derivatives as Selective NLRP3 Inflammasome Inhibitors. ACS Med. Chem. Lett. 2022, 13, 1321–1328. [Google Scholar] [CrossRef]
- Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Chigurupati, S.; Alrashdi, I.; Bungau, S.G. Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, B.; Gupta, P.; Laha, J.K.; Roy, I.; Sharma, S.S. Amelioration of Parkinson's disease by pharmacological inhibition and knockdown of redox sensitive TRPC5 channels: Focus on mitochondrial health. Life Sci. 2023, 328, 121871. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Singh, T.G.; Singh, S.; Garg, N.; Dhiman, S. Apoptotic Pathways and Alzheimer’s Disease: Probing Therapeutic Potential. Neurochem. Res. 2021, 46, 3103–3122. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yang, K.; Guo, H.; Zeng, J.; Wang, S.; Xu, H.; Ge, A.; Zeng, L.; Chen, S.; Ge, J. Mechanisms of ferroptosis in Alzheimer's disease and therapeutic effects of natural plant products: A review. Biomed Pharmacather 2023, 164, 114312. [Google Scholar] [CrossRef] [PubMed]
- Malar, D.S.; Thitilertdecha, P.; Ruckvongacheep, K.S.; Brimson, S.; Tencomnao, T.; Brimson, J.M. Targeting Sigma Receptors for the Treatment of Neurodegenerative and Neurodevelopmental Disorders. CNS Drugs 2023, 37, 399–440. [Google Scholar] [CrossRef]
- Reudhabibadh, R.; Binlateh, T.; Chonpathompikunlert, P.; Nonpanya, N.; Prommeenate, P.; Chanvorachote, P.; Hutamekalin, P. Suppressing Cdk5 Activity by Luteolin Inhibits MPP+-Induced Apoptotic of Neuroblastoma through Erk/Drp1 and Fak/Akt/GSK3β Pathways. Molecules 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, Y.; Yu, X.; Lu, J.; Jia, W.; Song, J.; Liu, L.; Wang, Y.; Huang, Y.; Xie, J.; et al. Corynoxine Protects Dopaminergic Neurons Through Inducing Autophagy and Diminishing Neuroinflammation in Rotenone-Induced Animal Models of Parkinson’s Disease. Front Pharmacol 2021, 12. [Google Scholar] [CrossRef]
- Kulesskaya, N.; Bhattacharjee, A.; Holmström, K.M.; Vuorio, P.; Henriques, A.; Callizot, N.; Huttunen, H.J. HER-096 is a CDNF-derived brain-penetrating peptidomimetic that protects dopaminergic neurons in a mouse synucleinopathy model of Parkinson’s disease. Cell Chem. Biol. 2024, 31, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.-s.; Gao, L.; Han, Z.; Eleuteri, S.; Shi, W.; Shen, Y.; Song, Z.-y.; Su, M.; Yang, Q.; Qu, Y.; et al. Ferroptosis in Parkinson's disease: Molecular mechanisms and therapeutic potential. Ageing Res Rev 2023, 91, 102077. [Google Scholar] [CrossRef]
- Vietor, J.; Gege, C.; Stiller, T.; Busch, R.; Schallmayer, E.; Kohlhof, H.; Höfner, G.; Pabel, J.; Marschner, J.A.; Merk, D. Development of a Potent Nurr1 Agonist Tool for In Vivo Applications. J. Med. Chem. 2023, 66, 6391–6402. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wan, C.; He, T.; Han, C.; Zhu, K.; Waddington, J.L.; Zhen, X. Sigma-1 receptor regulates mitophagy in dopaminergic neurons and contributes to dopaminergic protection. Neuropharmacology 2021, 196, 108360. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Hammad, M.; Raftari, M.; Cesário, R.; Salma, R.; Godoy, P.; Emami, S.N.; Haghdoost, S. Roles of Oxidative Stress and Nrf2 Signaling in Pathogenic and Non-Pathogenic Cells: A Possible General Mechanism of Resistance to Therapy. Antioxidants 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Elmazoglu, Z.; Prnova, M.S.; Santamaria, A.; Stefek, M.; Karasu, C. Combatting Nitrosative Stress and Inflammation with Novel Substituted Triazinoindole Inhibitors of Aldose Reductase in PC12 Cells Exposed to 6-Hydroxydopamine Plus High Glucose. Neurotox Res 2021, 39, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kim, S.; Yoo, J.S.; Kim, H.J.; Kim, H.J.; Kim, B.E.; Lee, E.H.; Lee, Y.S.; Park, J.-H.; Park, K.D. Development and optimization of halogenated vinyl sulfones as Nrf2 activators for the treatment of Parkinson’s disease. Eur. J. Med. Chem. 2021, 212, 113103. [Google Scholar] [CrossRef] [PubMed]
- Burns, L.H.; Pei, Z.; Wang, H.-Y. Targeting α7 nicotinic acetylcholine receptors and their protein interactions in Alzheimer's disease drug development. Drug Dev. Res. 2023, 84, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A Review of Butyrylcholinesterase as a Therapeutic Target in the Treatment of Alzheimer’s Disease. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E. Targeting Synaptic NMDA Receptor Co-agonism as a Therapy for Alzheimer’s Disease? Cell Metab. 2020, 31, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Yang, Y.; Du, G.; Dai, Y.; Zhu, X.; Wang, W.; Xu, H.; Zhang, J.; Zheng, L.; Zou, F.; et al. Improving the treatment of Parkinson's disease: Structure-based development of novel 5-HT2A receptor antagonists/inverse agonists. Eur. J. Med. Chem. 2022, 234, 114246. [Google Scholar] [CrossRef] [PubMed]
- Matthee, C.; Terre’Blanche, G.; Janse van Rensburg, H.D.; Aucamp, J.; Legoabe, L.J. Chalcone-inspired rA1/A2A adenosine receptor ligands: Ring closure as an alternative to a reactive substructure. Chem. Biol. Drug Des. 2022, 99, 416–437. [Google Scholar] [CrossRef]
- Rohilla, S.; Bansal, R.; Kachler, S.; Klotz, K.-N. Synthesis, biological evaluation and molecular modelling studies of 1,3,7,8-tetrasubstituted xanthines as potent and selective A2A AR ligands with in vivo efficacy against animal model of Parkinson’s disease. Bioorganic Chem. 2019, 87, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Christie, L.A.; Brice, N.L.; Rowland, A.; Dickson, L.; Anand, R.; Teall, M.; Doyle, K.J.; Narayana, L.; Mitchell, C.; Harvey, J.R.M.; et al. Discovery of CVN417, a Novel Brain-Penetrant α6-Containing Nicotinic Receptor Antagonist for the Modulation of Motor Dysfunction. J. Med. Chem. 2023, 66, 11718–11731. [Google Scholar] [CrossRef] [PubMed]
- de Beer, J.; Petzer, J.P.; Lourens, A.C.U.; Petzer, A. Design, synthesis and evaluation of 3-hydroxypyridin-4-ones as inhibitors of catechol-O-methyltransferase. Mol. Divers. 2021, 25, 753–762. [Google Scholar] [CrossRef] [PubMed]
- García-Cárceles, J.; Vázquez-Villa, H.; Brea, J.; Ladron de Guevara-Miranda, D.; Cincilla, G.; Sánchez-Martínez, M.; Sánchez-Merino, A.; Algar, S.; Teresa de los Frailes, M.; Roberts, R.S.; et al. 2-(Fluoromethoxy)-4′-(S-methanesulfonimidoyl)-1,1′-biphenyl (UCM-1306), an Orally Bioavailable Positive Allosteric Modulator of the Human Dopamine D1 Receptor for Parkinson’s Disease. J. Med. Chem. 2022, 65, 12256–12272. [Google Scholar] [CrossRef] [PubMed]
- Sampaio-Dias, I.E.; Reis-Mendes, A.; Costa, V.M.; García-Mera, X.; Brea, J.; Loza, M.I.; Pires-Lima, B.L.; Alcoholado, C.; Algarra, M.; Rodríguez-Borges, J.E. Discovery of New Potent Positive Allosteric Modulators of Dopamine D2 Receptors: Insights into the Bioisosteric Replacement of Proline to 3-Furoic Acid in the Melanostatin Neuropeptide. J. Med. Chem. 2021, 64, 6209–6220. [Google Scholar] [CrossRef]
- Tolentino, K.T.; Mashinson, V.; Vadukoot, A.K.; Hopkins, C.R. Discovery and characterization of benzyloxy piperidine based dopamine 4 receptor antagonists. Bioorg. Med. Chem. Lett. 2022, 61, 128615. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Monenschein, H.; Schiffer, H.H.; Reichard, H.A.; Kikuchi, S.; Hopkins, M.; Macklin, T.K.; Hitchcock, S.; Adams, M.; Green, J.; et al. First-Time Disclosure of CVN424, a Potent and Selective GPR6 Inverse Agonist for the Treatment of Parkinson’s Disease: Discovery, Pharmacological Validation, and Identification of a Clinical Candidate. J. Med. Chem. 2021, 64, 9875–9890. [Google Scholar] [CrossRef] [PubMed]
- Rehuman, N.A.; Oh, J.M.; Abdelgawad, M.A.; Beshr, E.A.M.; Abourehab, M.A.S.; Gambacorta, N.; Nicolotti, O.; Jat, R.K.; Kim, H.; Mathew, B. Development of Halogenated-Chalcones Bearing with Dimethoxy Phenyl Head as Monoamine Oxidase-B Inhibitors. Pharmaceuticals 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Kent, C.N.; Fulton, M.G.; Stillwell, K.J.; Dickerson, J.W.; Loch, M.T.; Rodriguez, A.L.; Blobaum, A.L.; Boutaud, O.; Rook, J.L.; Niswender, C.M.; et al. Discovery and optimization of a novel CNS penetrant series of mGlu4 PAMs based on a 1,4-thiazepane core with in vivo efficacy in a preclinical Parkinsonian model. Bioorg. Med. Chem. Lett. 2021, 37, 127838. [Google Scholar] [CrossRef]
- Tang, L.; Huang, C.; Zhong, J.; He, J.; Guo, J.; Liu, M.; Xu, J.-P.; Wang, H.-T.; Zhou, Z.-Z. Discovery of arylbenzylamines as PDE4 inhibitors with potential neuroprotective effect. Eur. J. Med. Chem. 2019, 168, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Learmonth, D.A.; Palma, P.N.; Vieira-Coelho, M.A.; Soares-da-Silva, P. Synthesis, biological evaluation, and molecular modeling studies of a novel, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem 2004, 47, 6207–6217. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, S.D.; Serrao, M.P.; Silva, T.; Borges, F.; Soares-da-Silva, P. Pharmacodynamic evaluation of novel Catechol-O-methyltransferase inhibitors. Eur. J. Pharmacol. 2019, 847, 53–60. [Google Scholar] [CrossRef]
- Vianello, R.; Domene, C.; Mavri, J. The Use of Multiscale Molecular Simulations in Understanding a Relationship between the Structure and Function of Biological Systems of the Brain: The Application to Monoamine Oxidase Enzymes. Front. Neurosci. 2016, 10, 327–327. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Ferreira, J.J.; Lees, A.; Stocchi, F.; Poewe, W.; Tolosa, E.; Rascol, O. Opicapone for the treatment of Parkinson's disease: A review of a new licensed medicine. Mov. Disord. 2018, 33, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.; Rocha, J.F.; Falcao, A.; Palma, P.N.; Loureiro, A.I.; Pinto, R.; Bonifacio, M.J.; Wright, L.C.; Nunes, T.; Soares-da-Silva, P. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet 2013, 52, 139–151. [Google Scholar] [CrossRef]
- Lees, A.J.; Ferreira, J.; Rascol, O.; Poewe, W.; Rocha, J.F.; McCrory, M.; Soares-da-Silva, P. Opicapone as Adjunct to Levodopa Therapy in Patients With Parkinson Disease and Motor Fluctuations: A Randomized Clinical Trial. JAMA Neurol 2017, 74, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Parashos, S.A.; Wielinski, C.L.; Kern, J.A. Frequency, reasons, and risk factors of entacapone discontinuation in Parkinson disease. Clin. Neuropharmacol. 2004, 27, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Käenmäki, M.; Tammimäki, A.; Garcia-Horsman, J.A.; Myöhänen, T.; Schendzielorz, N.; Karayiorgou, M.; Gogos, J.A.; Männistö, P.T. Importance of membrane-bound catechol-O-methyltransferase in L-DOPA metabolism: A pharmacokinetic study in two types of Comt gene modified mice. Br. J. Pharmacol. 2009, 158, 1884–1894. [Google Scholar] [CrossRef]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: a critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, M.A.; Wenk, G.L. The Neuropharmacological Basis for the Use of Memantine in the Treatment of Alzheimer's Disease. CNS Drug Rev. 2003, 9, 275–308. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L.; Parsons, C.G.; Danysz, W. Potential role of N-methyl-D-aspartate receptors as executors of neurodegeneration resulting from diverse insults: focus on memantine. Behav Pharmacol 2006, 17, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.-Y.; Tanaka, K.; S Dawe, G.; Ittner, L.; A Farooqui, A. Slow Excitotoxicity in Alzheimer's Disease. J. Alzheimers Dis. 2013, 35. [Google Scholar] [CrossRef] [PubMed]
- Parsons, C.G.; Danysz, W.; Dekundy, A.; Pulte, I. Memantine and Cholinesterase Inhibitors: Complementary Mechanisms in the Treatment of Alzheimer’s Disease. Neurotox Res 2013, 24, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.N.; Farlow, M.R.; Grossberg, G.T.; et al. Memantine treatment in patients with moderate to severe alzheimer disease already receiving donepezil: A randomized controlled trial. JAMA 2004, 291, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Lopez, O.L.; Becker, J.T.; Wahed, A.S.; Saxton, J.; Sweet, R.A.; Wolk, D.A.; Klunk, W.; DeKosky, S.T. Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. J Neurol Neurosur Ps 2009, 80, 600. [Google Scholar] [CrossRef] [PubMed]
- Atri, A.; Molinuevo, J.L.; Lemming, O.; Wirth, Y.; Pulte, I.; Wilkinson, D. Memantine in patients with Alzheimer's disease receiving donepezil: new analyses of efficacy and safety for combination therapy. Alzheimer's Res. Ther. 2013, 5, 6–6. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L. Memantine ER/Donepezil: A Review in Alzheimer’s Disease. CNS Drugs 2015, 29, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Rösler, M.; Bayer, T.; Anand, R.; Cicin-Sain, A.; Gauthier, S.; Agid, Y.; Dal-Bianco, P.; Stähelin, H.B.; Hartman, R.; Gharabawi, M. Efficacy and safety of rivastigmine in patients with Alzheimer's disease: international randomised controlled trial. BMJ 1999, 318, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Raskind, M.A.; Peskind, E.R.; Wessel, T.; Yuan, W. Galantamine in AD. Neurology 2000, 54, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.; Harvey, R.J. Donepezil for dementia due to Alzheimer's disease. Cochrane Db Syst Rev 2003. [Google Scholar] [CrossRef]
- Parsons, C.G.; Stöffler, A.; Danysz, W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system - too little activation is bad, too much is even worse. Neuropharmacology 2007, 53, 699–723. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.A.; Gartlehner, G.; Webb, A.P.; Morgan, L.C.; Moore, C.G.; Jonas, D.E. Efficacy and Safety of Donepezil, Galantamine, and Rivastigmine for the Treatment of Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Clin. Interv. Aging. 2008, 3, 211–225. [Google Scholar] [PubMed]
- Di, L.; Kerns, E.H. Blood-Brain Barrier in Drug Discovery: Optimizing Brain Exposure of CNS Drugs and Minimizing Brain Side Effects for Peripheral Drugs; Wiley: 2015.
- Domínguez, A.; Álvarez, A.; Hilario, E.; Suarez-Merino, B.; Goñi-de-Cerio, F. Central nervous system diseases and the role of the blood-brain barrier in their treatment. Neurosci. Discov. 2013, 1. [Google Scholar] [CrossRef]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. In Peptide Transport and Delivery into the Central Nervous System, Prokai, L., Prokai-Tatrai, K., Eds. Birkhäuser Basel: Basel, 2003; pp. 39-78. [CrossRef]
- Pardridge, W.M. Molecular biology of the blood-brain barrier. Mol Biotechnol 2005, 30, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood–brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K. Pathophysiology of the blood-brain barrier. In Immunoneurology, Chofflon, M., Steinman, L., Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 1996; pp. 175-191. [CrossRef]
- Rankovic, Z. CNS drug design: balancing physicochemical properties for optimal brain exposure. J Med Chem 2015, 58, 2584–2608. [Google Scholar] [CrossRef] [PubMed]
- Deeken, J.F.; Löscher, W. The Blood-Brain Barrier and Cancer: Transporters, Treatment, and Trojan Horses. Clin Cancer Res 2007, 13, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Kasinathan, N.; Jagani, H.V.; Alex, A.T.; Volety, S.M.; Rao, J.V. Strategies for drug delivery to the central nervous system by systemic route. Drug Deliv 2015, 22, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; George, A.M. The ABC transporter structure and mechanism: perspectives on recent research. Cell. Mol. Life Sci. 2004, 61, 682–699. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Yildirim-Balatan, C.; Fenyi, A.; Besnault, P.; Gomez, L.; Sepulveda-Diaz, J.E.; Michel, P.P.; Melki, R.; Hunot, S. Parkinson’s disease-derived α-synuclein assemblies combined with chronic-type inflammatory cues promote a neurotoxic microglial phenotype. J. Neuroinflammation 2024, 21, 54. [Google Scholar] [CrossRef] [PubMed]
- Bingham, M.; Rankovic, Z. CHAPTER 18 Medicinal Chemistry Challenges in CNS Drug Discovery. In Drug Discovery for Psychiatric Disorders, The Royal Society of Chemistry: 2012; pp. 465-509. [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRx. 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.E.; Soares-da-Silva, P. Medicinal chemistry of catechol O-methyltransferase (COMT) inhibitors and their therapeutic utility. J. Med. Chem. 2014, 57, 8692–8717. [Google Scholar] [CrossRef] [PubMed]
- Strydom, B.; Bergh, J.J.; Petzer, J.P. 8-Aryl- and alkyloxycaffeine analogues as inhibitors of monoamine oxidase. Eur. J. Med. Chem. 2011, 46, 3474–3485. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, S.A.; Pennington, L.D. Structure−Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Tox Met 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.H.; Ibrahim, A.; Acree, W.E. Air to brain, blood to brain and plasma to brain distribution of volatile organic compounds: linear free energy analyses. Eur. J. Med. Chem. 2006, 41, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Goñi, F. Immunotherapy for Alzheimer's disease. Biochem. Pharmacol. 2014, 88, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Solfrizzi, V.; Imbimbo, B.P.; Tortelli, R.; Santamato, A.; Logroscino, G. Amyloid-based immunotherapy for Alzheimer's disease in the time of prevention trials: the way forward. Expert Rev Clin Immu 2014, 10, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer's disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Tayeb, H.O.; Murray, E.D.; Price, B.H.; Tarazi, F.I. Bapineuzumab and solanezumab for Alzheimer's disease: is the ‘amyloid cascade hypothesis' still alive? Expert Opin Biol Th 2013, 13, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Deng, Y.; Qing, H. Potential Therapeutic Strategies for Alzheimer's Disease Targeting or Beyond β-Amyloid: Insights from Clinical Trials. Biomed. Res. Int. 2014, 2014, 837157. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; De Strooper, B. Alzheimer’s Disease: where next for anti-amyloid therapies? Brain 2017, 140, 853–855. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L. BACE1 ([small beta]-secretase) inhibitors for the treatment of Alzheimer's disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef] [PubMed]
- Grüninger, F. Invited review: Drug development for tauopathies. Neuropath Appl Neuro 2015, 41, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Oliveira, C.; Amorim, R.; Cagide, F.; Garrido, J.; Ribeiro, J.A.; Pereira, C.M.; Silva, A.F.; Andrade, P.B.; Oliveira, P.J.; et al. Development of hydroxybenzoic-based platforms as a solution to deliver dietary antioxidants to mitochondria. Sci. Rep. 2017, 7, 6842. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Cagide, F.; Benfeito, S.; Soares, P.; Garrido, J.; Baldeiras, I.; Ribeiro, J.A.; Pereira, C.M.; Silva, A.F.; Andrade, P.B.; et al. Development of a Mitochondriotropic Antioxidant Based on Caffeic Acid: Proof of Concept on Cellular and Mitochondrial Oxidative Stress Models. J. Med. Chem. 2017, 60, 7084–7098. [Google Scholar] [CrossRef] [PubMed]
- Benfeito, S.; Oliveira, C.; Fernandes, C.; Cagide, F.; Teixeira, J.; Amorim, R.; Garrido, J.; Martins, C.; Sarmento, B.; Silva, R.; et al. Fine-tuning the neuroprotective and blood-brain barrier permeability profile of multi-target agents designed to prevent progressive mitochondrial dysfunction. Eur. J. Med. Chem. 2019, 167, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Pereira, S.P.; Cunha-Oliveira, T.; Teixeira, J.; Simões, R.F.; Cagide, F.; Benfeito, S.; Borges, F.; Raimundo, N.; Oliveira, P.J. A mitochondria-targeted caffeic acid derivative reverts cellular and mitochondrial defects in human skin fibroblasts from male sporadic Parkinson's disease patients. Redox Biol. 2021, 45, 102037. [Google Scholar] [CrossRef] [PubMed]
- Benfeito, S.; Fernandes, C.; Chavarria, D.; Barreiro, S.; Cagide, F.; Sequeira, L.; Teixeira, J.; Silva, R.; Remião, F.; Oliveira, P.J.; et al. Modulating Cytotoxicity with Lego-like Chemistry: Upgrading Mitochondriotropic Antioxidants with Prototypical Cationic Carrier Bricks. J. Med. Chem. 2023, 66, 1835–1851. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-P.; Wu, K.-C.; Lin, C.-Y.; Chern, Y. Emerging roles of dysregulated adenosine homeostasis in brain disorders with a specific focus on neurodegenerative diseases. J. Biomed. Sci. 2021, 28, 70. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gil, M.; Camici, M.; Allegrini, S.; Pesi, R.; Tozzi, M.G. Metabolic Aspects of Adenosine Functions in the Brain. Front Pharmacol 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Mazziotta, C.; Rotondo, J.C.; Lanzillotti, C.; Campione, G.; Martini, F.; Tognon, M. Cancer biology and molecular genetics of A3 adenosine receptor. Oncogene 2022, 41, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Comeo, E.; Kindon, N.D.; Soave, M.; Stoddart, L.A.; Kilpatrick, L.E.; Scammells, P.J.; Hill, S.J.; Kellam, B. Subtype-Selective Fluorescent Ligands as Pharmacological Research Tools for the Human Adenosine A2A Receptor. J. Med. Chem. 2020, 63, 2656–2672. [Google Scholar] [CrossRef] [PubMed]
- Candito, D.A.; Simov, V.; Gulati, A.; Kattar, S.; Chau, R.W.; Lapointe, B.T.; Methot, J.L.; DeMong, D.E.; Graham, T.H.; Kurukulasuriya, R.; et al. Discovery and Optimization of Potent, Selective, and Brain-Penetrant 1-Heteroaryl-1H-Indazole LRRK2 Kinase Inhibitors for the Treatment of Parkinson’s Disease. J. Med. Chem. 2022, 65, 16801–16817. [Google Scholar] [CrossRef] [PubMed]
- Hagenow, S.; Affini, A.; Pioli, E.Y.; Hinz, S.; Zhao, Y.; Porras, G.; Namasivayam, V.; Müller, C.E.; Lin, J.-S.; Bezard, E.; et al. Adenosine A2AR/A1R Antagonists Enabling Additional H3R Antagonism for the Treatment of Parkinson’s Disease. J. Med. Chem. 2021, 64, 8246–8262. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.D.; Legoabe, L.J.; Terre'Blanche, G.; Aucamp, J. Synthesis and evaluation of methoxy substituted 2-benzoyl-1-benzofuran derivatives as lead compounds for the development adenosine A1 and/or A2A receptor antagonists. Bioorganic Chem. 2020, 94, 103459. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Barawkar, D.A.; Thorat, S.; Shejul, Y.D.; Patel, M.; Naykodi, M.; Jain, V.; Salve, Y.; Prasad, V.; Chaudhary, S.; et al. Design, Synthesis of Novel, Potent, Selective, Orally Bioavailable Adenosine A2A Receptor Antagonists and Their Biological Evaluation. J. Med. Chem. 2017, 60, 681–694. [Google Scholar] [CrossRef]
- Prasad, K.; de Vries, E.F.J.; van der Meiden, E.; Moraga-Amaro, R.; Vazquez-Matias, D.A.; Barazzuol, L.; Dierckx, R.A.J.O.; van Waarde, A. Effects of the adenosine A2A receptor antagonist KW6002 on the dopaminergic system, motor performance, and neuroinflammation in a rat model of Parkinson's disease. Neuropharmacology 2024, 247, 109862. [Google Scholar] [CrossRef] [PubMed]
- Cervetto, C.; Maura, G.; Guidolin, D.; Amato, S.; Ceccoli, C.; Agnati, L.F.; Marcoli, M. Striatal astrocytic A2A-D2 receptor-receptor interactions and their role in neuropsychiatric disorders. Neuropharmacology 2023, 237, 109636. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Kimura, T.; Sugiyama, K.; Yamada, K.; Hiraiwa, R.; Nishi, M.; Hattori, N.; Abe, T.; Deguchi, K.; Fujimoto, K.; et al. Randomized controlled trial of KW-6356 monotherapy in patients with early untreated Parkinson's disease. Park. Relat. Disord. 2023, 117, 105907. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.G.; Hsiao, E.C.; Wang, M.M.; Ho, K.; Kim, D.H.; Wang, X.; Guo, W.; Kang, J.; Yu, G.-Q.; Adame, A.; et al. Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat. Neurosci. 2015, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Viana da Silva, S.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Gonçalves, N.; Gorlewicz, A.; Malezieux, M.; Gonçalves, F.Q.; Grosjean, N.; et al. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 2016, 7, 11915. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Rial, D.; Canas, P.M.; Yoo, J.H.; Li, W.; Zhou, X.; Wang, Y.; van Westen, G.J.P.; Payen, M.P.; Augusto, E.; et al. Optogenetic activation of intracellular adenosine A2A receptor signaling in the hippocampus is sufficient to trigger CREB phosphorylation and impair memory. Mol. Psychiatry 2015, 20, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, Â.R.; Ferreira, S.G.; Canas, P.M.; Rial, D.; et al. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer's disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol Dis 2019, 132, 104570. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Yang, Y.; Xiong, Y.; Zhang, Y.-J.; Jiang, J.; Zhou, L.-P.; Du, X.-H.; Wang, C.-X.; Zhu, Z.-R. Blockade of adenosine A2A receptors reverses early spatial memory defects in the APP/PS1 mouse model of Alzheimer’s disease by promoting synaptic plasticity of adult-born granule cells. Alzheimer's Res. Ther. 2023, 15, 187. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.; Patel, R.; Gaba, S.; Singh, G.; Gupta, G.D.; Monga, V. Adenosine receptor antagonists: Recent advances and therapeutic perspective. Eur. J. Med. Chem. 2022, 227, 113907. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Pasquini, S.; Contri, C.; Cappello, M.; Nigro, M.; Travagli, A.; Merighi, S.; Gessi, S.; Borea, P.A.; Varani, K. Pharmacology of Adenosine Receptors: Recent Advancements. Biomolecules 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Cunha, R.A. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.A.; Olanow, C.W.; Kieburtz, K.D.; Pourcher, E.; Docu-Axelerad, A.; Lew, M.; Kozyolkin, O.; Neale, A.; Resburg, C.; Meya, U.; et al. Tozadenant (SYN115) in patients with Parkinson's disease who have motor fluctuations on levodopa: a phase 2b, double-blind, randomised trial. Lancet Neurol. 2014, 13, 767–776. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A.; Aradi, S.D.; Hauser, R.A.; Rascol, O. The challenge of developing adenosine A2A antagonists for Parkinson disease: Istradefylline, preladenant, and tozadenant. Park. Relat. Disord. 2020, 80, S54–S63. [Google Scholar] [CrossRef] [PubMed]
- Yutaro, O.; Michihiko, S.; Hidetsugu, A.; Tomoyuki, K.; Mayumi, S.; Hikaru, M.; Mai, Y.; Chiyo, S.; So, I.; Jun-ichi, S.; et al. In Vitro Pharmacological Profile of KW-6356, a Novel Adenosine A<sub>2A</sub> Receptor Antagonist/Inverse Agonist. Mol. Pharmacol. 2023, 103, 311. [Google Scholar] [CrossRef]
- Ntetsika, T.; Papathoma, P.-E.; Markaki, I. Novel targeted therapies for Parkinson’s disease. Mol. Med. 2021, 27. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, R.J.; Hoppin, J.; Sevigny, J.; Patel, S.; Chiao, P.; Klimas, M.; Verma, A. Optimizing central nervous system drug development using molecular imaging. Clin. Pharmacol. Ther. 2015, 98, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer's Disease, Parkinson's Disease, and Amyotrophic Lateral Sclerosis in the Last Decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Cavalli, A.; Bolognesi, M.L. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer's Disease. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist's Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Grosso, C.; Silva, A.; Delerue-Matos, C.; Barroso, M.F. Single and Multitarget Systems for Drug Delivery and Detection: Up-to-Date Strategies for Brain Disorders. Pharmaceuticals 2023, 16. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, S.A.; Pennington, L.D. Structure-brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Primary brain regions affected in the most common neurodegenerative diseases. Adapted from [6].
Figure 1.
Primary brain regions affected in the most common neurodegenerative diseases. Adapted from [6].
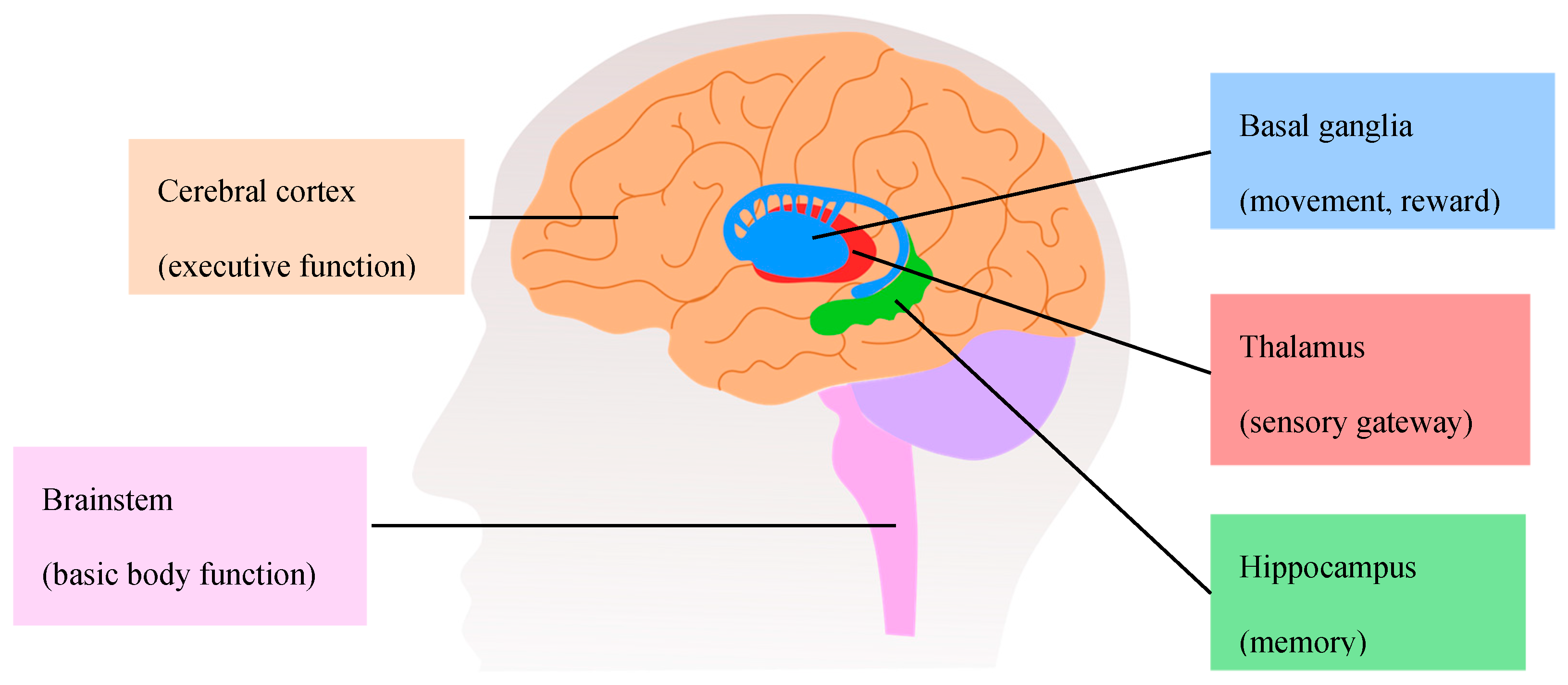
Figure 2.
Major hallmarks of AD. (A) Degeneration of cholinergic neurons in the basal forebrain. (B) Formation of senile plaques and neurofibrillary tangles. Adapted from [52].
Figure 2.
Major hallmarks of AD. (A) Degeneration of cholinergic neurons in the basal forebrain. (B) Formation of senile plaques and neurofibrillary tangles. Adapted from [52].
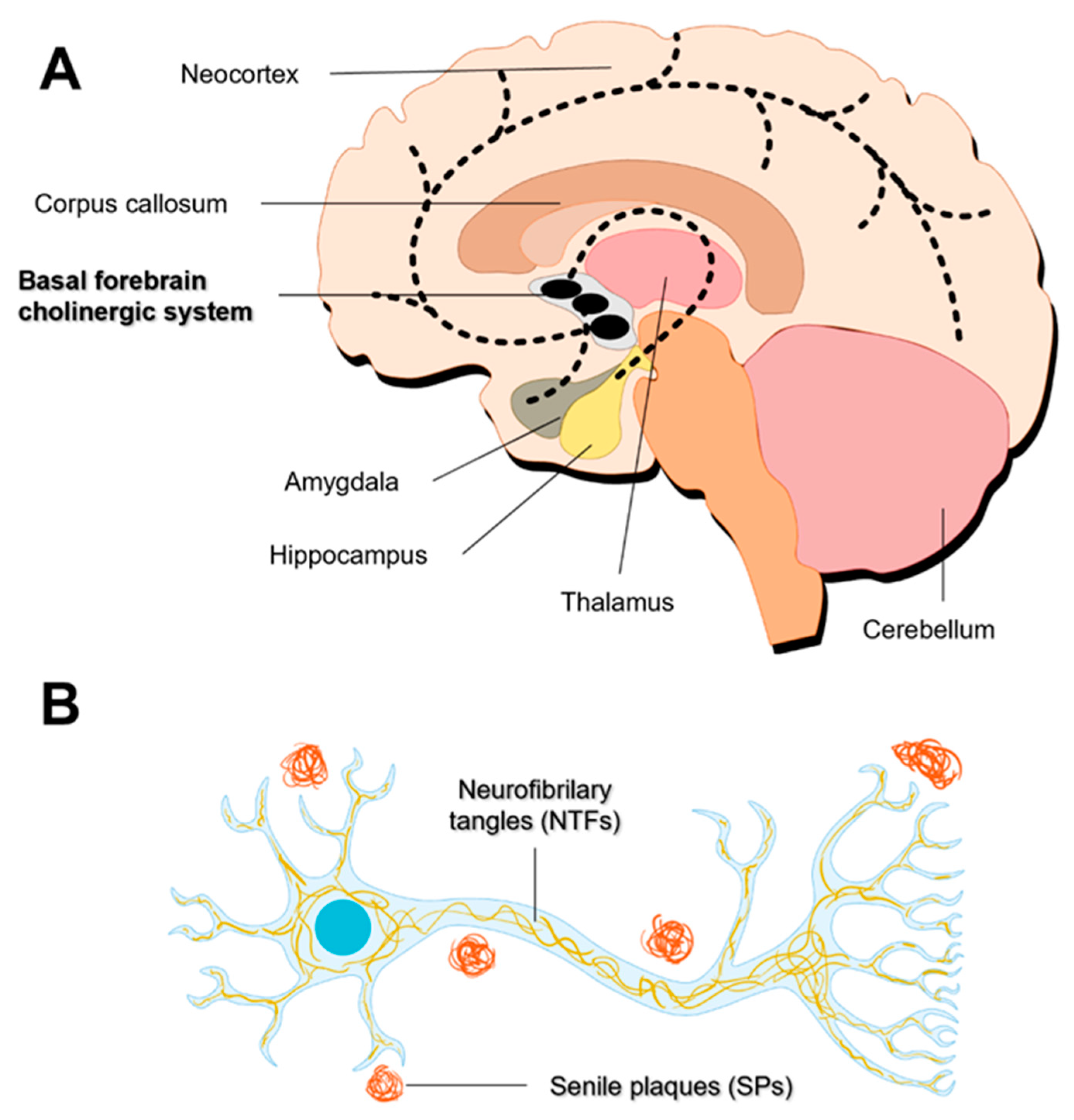
Figure 3.
Major hallmarks of Parkinson’s disease. (A) Degeneration of nigrostriatal dopaminergic neurons. (B) Formation of Lewy inclusions. Adapted from [80,81].
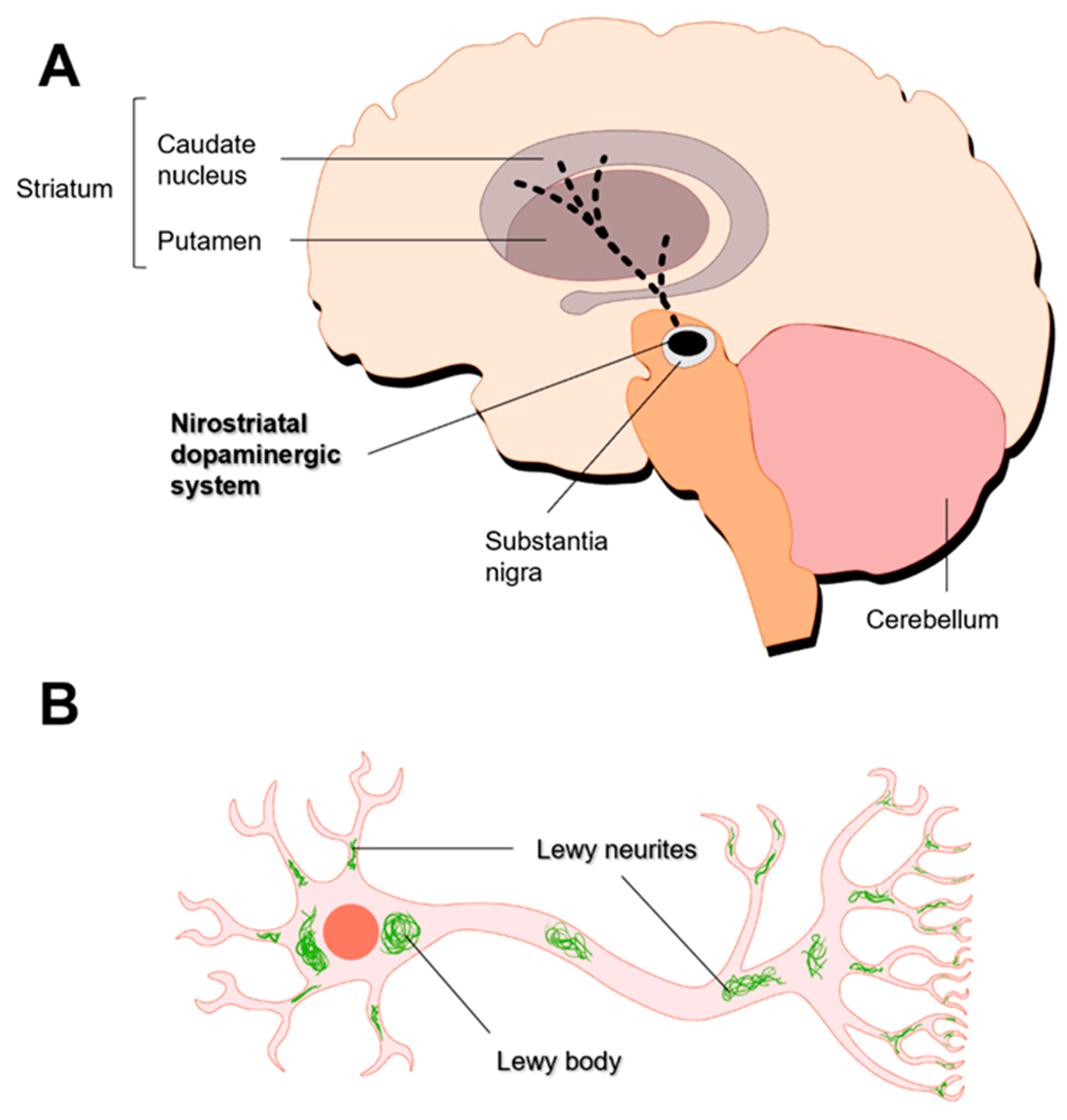
Figure 5.
Drugs in clinical use for AD treatment.
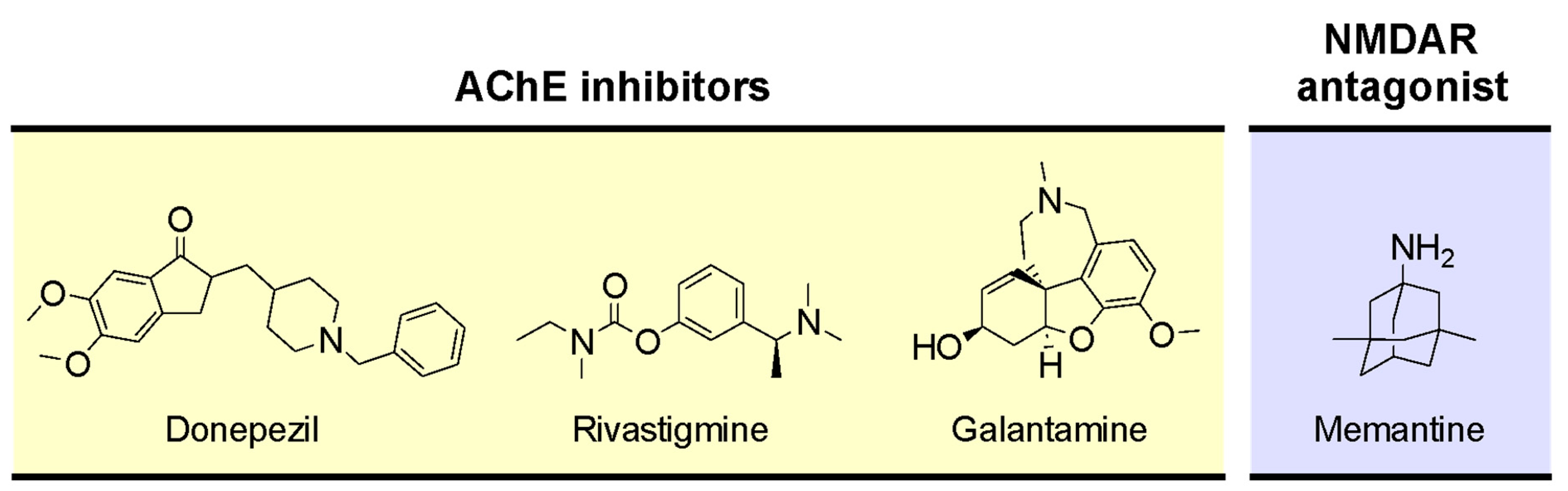
Figure 6.
Enzymes and transporters involved in the synthesis, storage, and metabolism of dopamine. Abbreviations: 3-MT, 3-methoxytyramine; AADC, aromatic amino acid decarboxylase; COMT, catechol-O-methyltransferase; DA, dopamine; DAT, dopamine transporter; DOPAC, 3,4-Dihydroxyphenylacetic acid; HVA, homovanillic acid; L-DOPA, L-3,4-dihydroxyphenylalanine; MAO-A, monoamine oxidase A; MAO-B, monoamine oxidase B; TH, tyrosine hydroxylase; VMAT2, vesicular monoamine transporter.Adapted from [128,129].
Figure 6.
Enzymes and transporters involved in the synthesis, storage, and metabolism of dopamine. Abbreviations: 3-MT, 3-methoxytyramine; AADC, aromatic amino acid decarboxylase; COMT, catechol-O-methyltransferase; DA, dopamine; DAT, dopamine transporter; DOPAC, 3,4-Dihydroxyphenylacetic acid; HVA, homovanillic acid; L-DOPA, L-3,4-dihydroxyphenylalanine; MAO-A, monoamine oxidase A; MAO-B, monoamine oxidase B; TH, tyrosine hydroxylase; VMAT2, vesicular monoamine transporter.Adapted from [128,129].
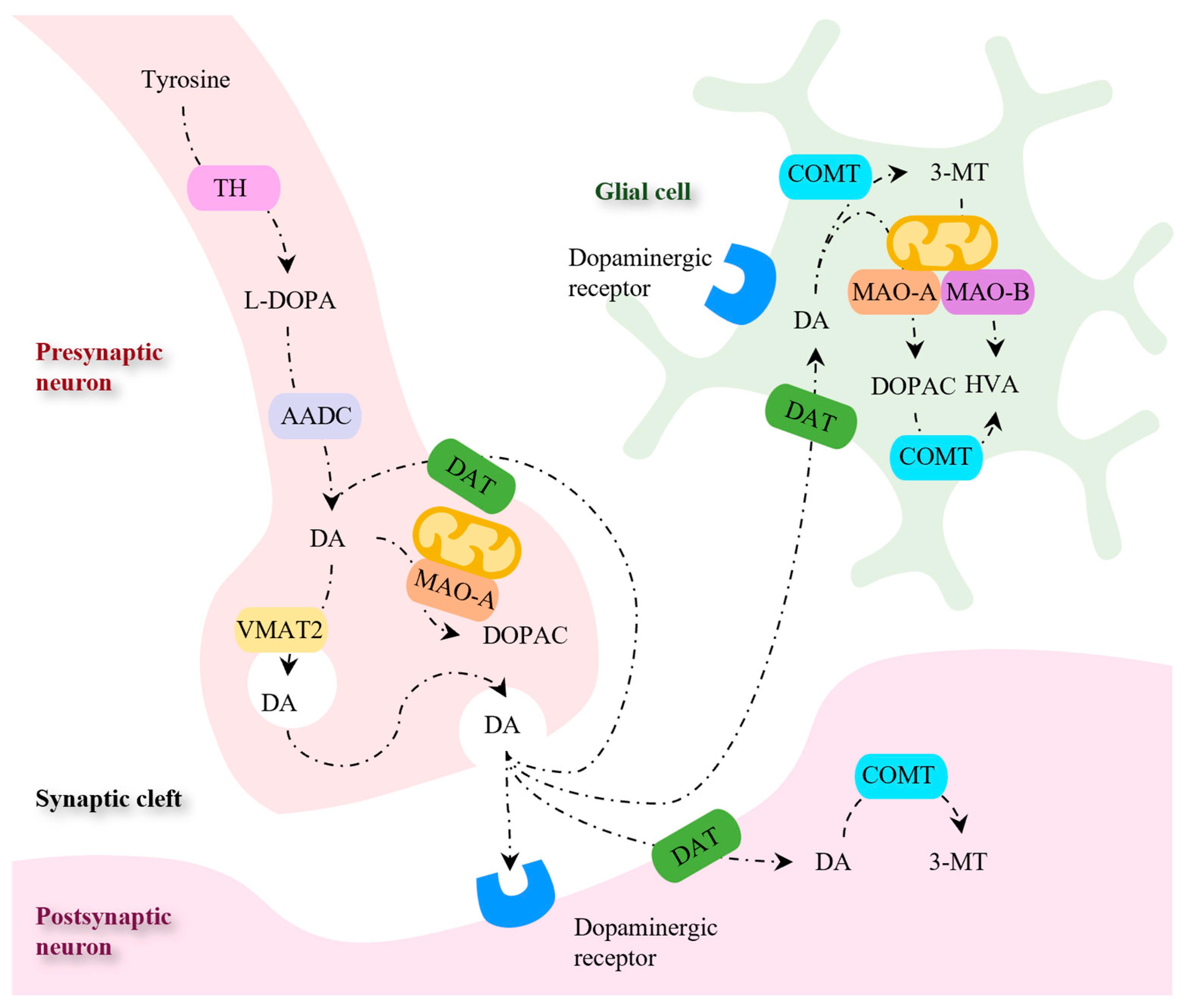
Figure 7.
Drugs in clinical use for PD treatment.
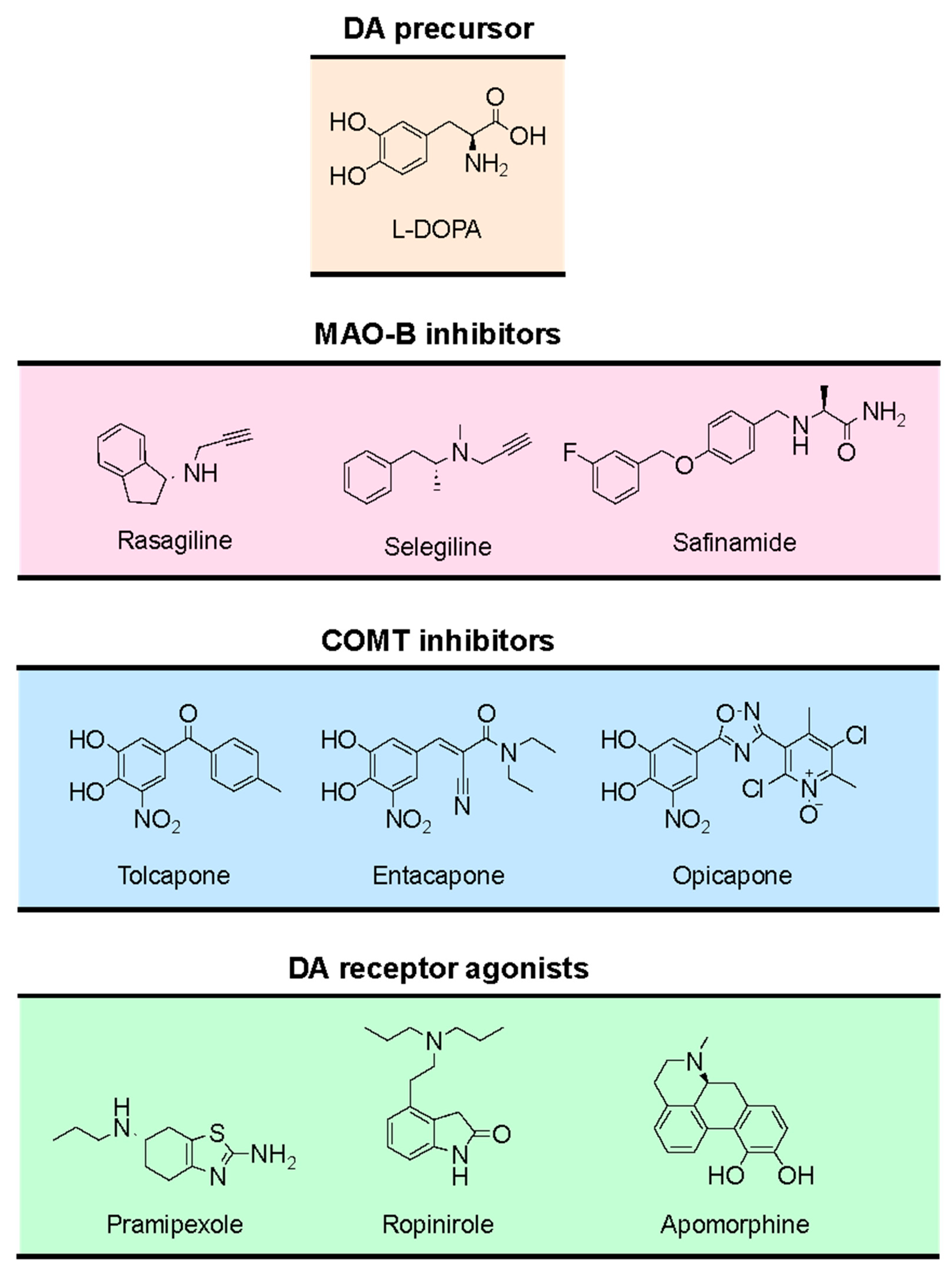
Figure 10.
Pathological events of neurodegenerative diseases associated with increased oxidative stress.
Figure 10.
Pathological events of neurodegenerative diseases associated with increased oxidative stress.
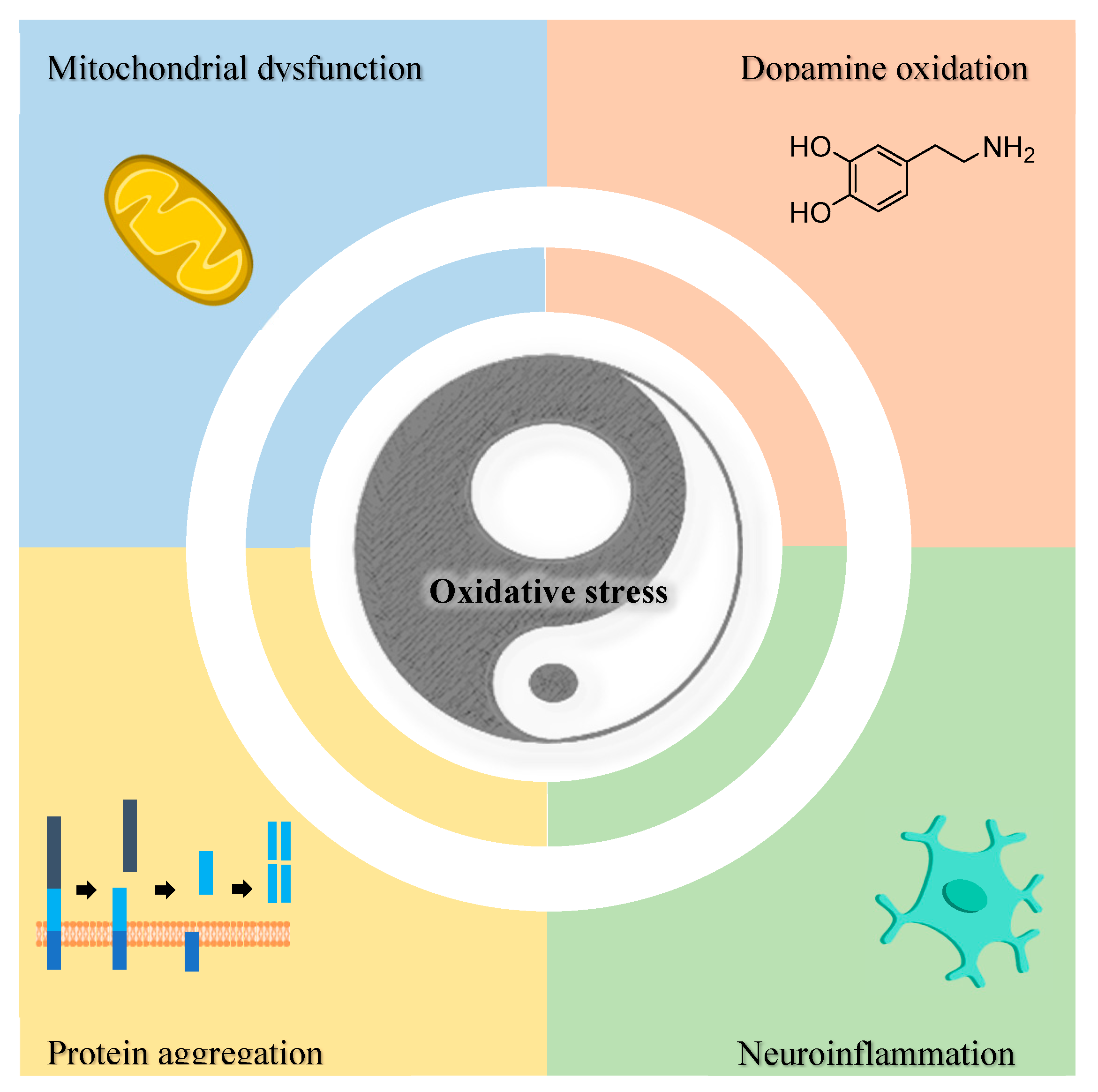
Figure 11.
Formation of ROS in mitochondria. Abbreviations: ADP, adenosine diphosphate; ATP, adenosine triphosphate; CAT, catalase; Cyt C, Cytochrome C; GPx, Glutathione peroxidase; I, complex I; II, complex II; III, complex III; IV, complex IV; NADH, Nicotinamide adenine dinucleotide; Q, coenzyme Q10; SOD1, superoxide dismutase 1; SOD2, superoxide dismutase 2; V, complex V (ATP synthase). Adapted from [117,218].
Figure 11.
Formation of ROS in mitochondria. Abbreviations: ADP, adenosine diphosphate; ATP, adenosine triphosphate; CAT, catalase; Cyt C, Cytochrome C; GPx, Glutathione peroxidase; I, complex I; II, complex II; III, complex III; IV, complex IV; NADH, Nicotinamide adenine dinucleotide; Q, coenzyme Q10; SOD1, superoxide dismutase 1; SOD2, superoxide dismutase 2; V, complex V (ATP synthase). Adapted from [117,218].
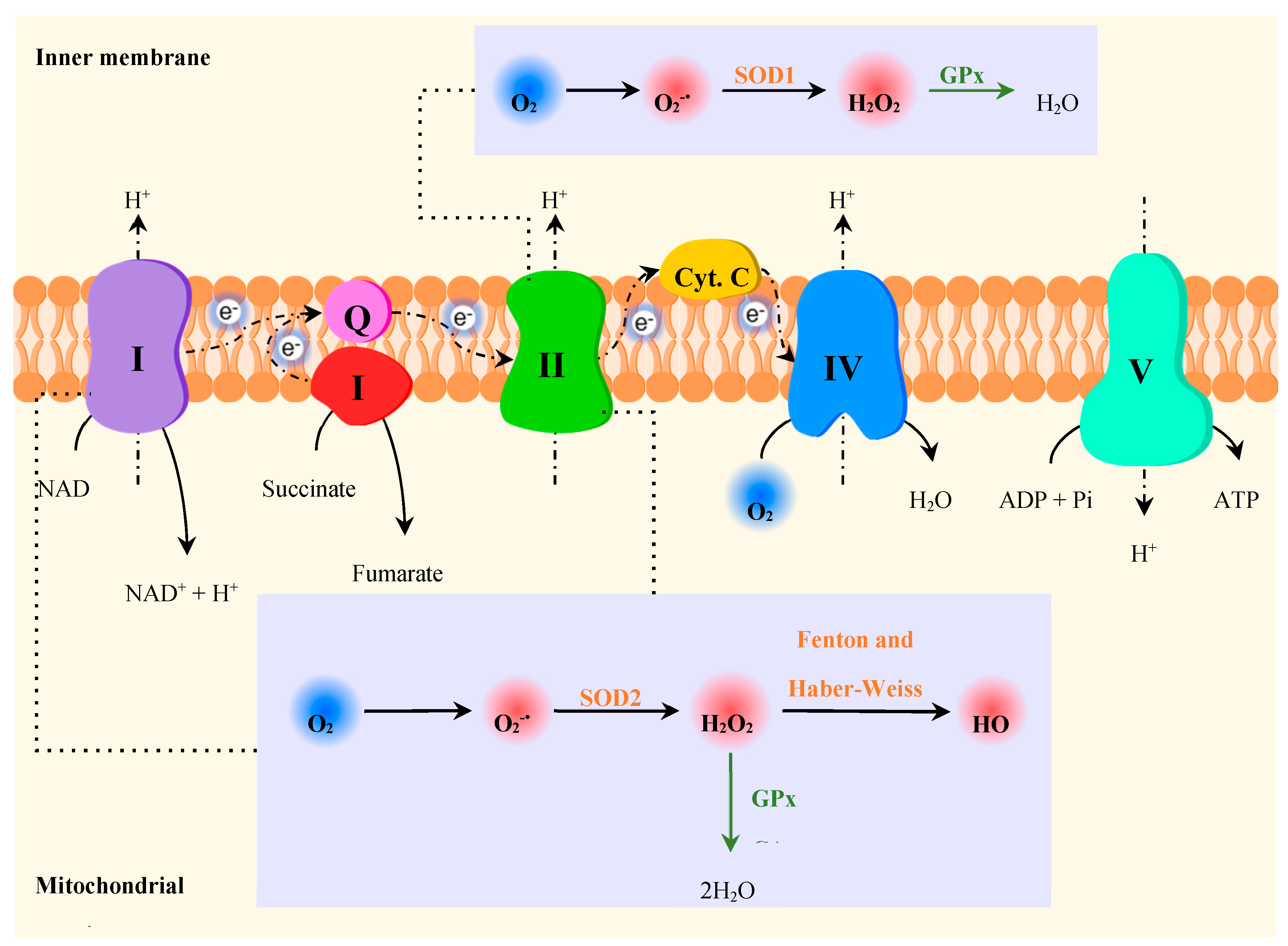
Figure 12.
Generation of ROS during enzymatic (dashed line) and non-enzymatic (plain line) DA decomposition. Abbreviations: ALDH, aldehyde dehydrogenase; COMT, catechol O-methyltransferase; DA, dopamine; DA-quinone, dopamine quinone; MAO, monoamine oxidase, NAD(P)H, nicotinamide adenine dinucleotide (phosphate). Adapted from [227].
Figure 12.
Generation of ROS during enzymatic (dashed line) and non-enzymatic (plain line) DA decomposition. Abbreviations: ALDH, aldehyde dehydrogenase; COMT, catechol O-methyltransferase; DA, dopamine; DA-quinone, dopamine quinone; MAO, monoamine oxidase, NAD(P)H, nicotinamide adenine dinucleotide (phosphate). Adapted from [227].
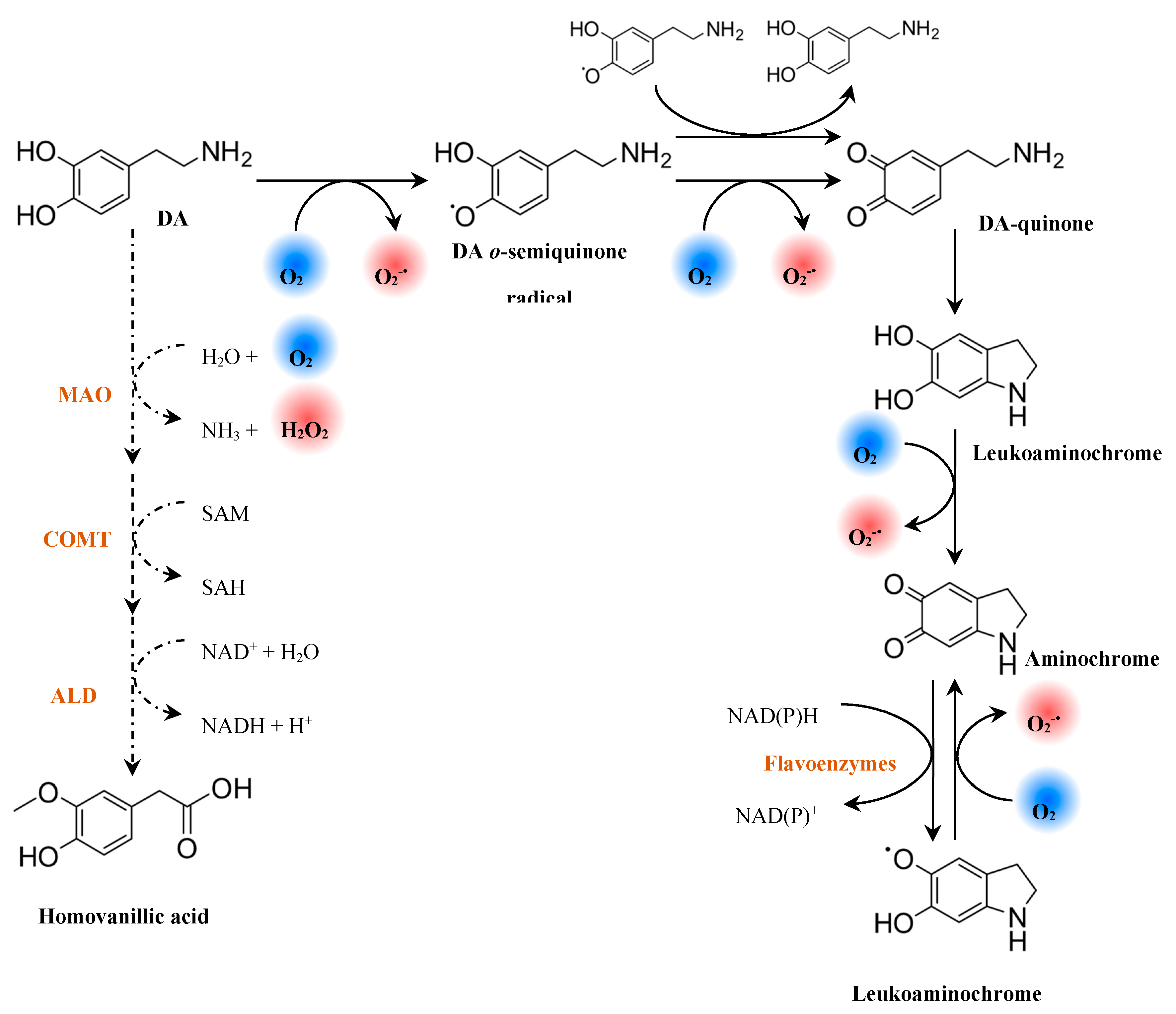
Figure 13.
(A) Mitochondrial uptake of lipophilic cations. (B) Representative examples of lipophilic cations to target bioactive molecules to mitochondria. Adapted from [253,255].
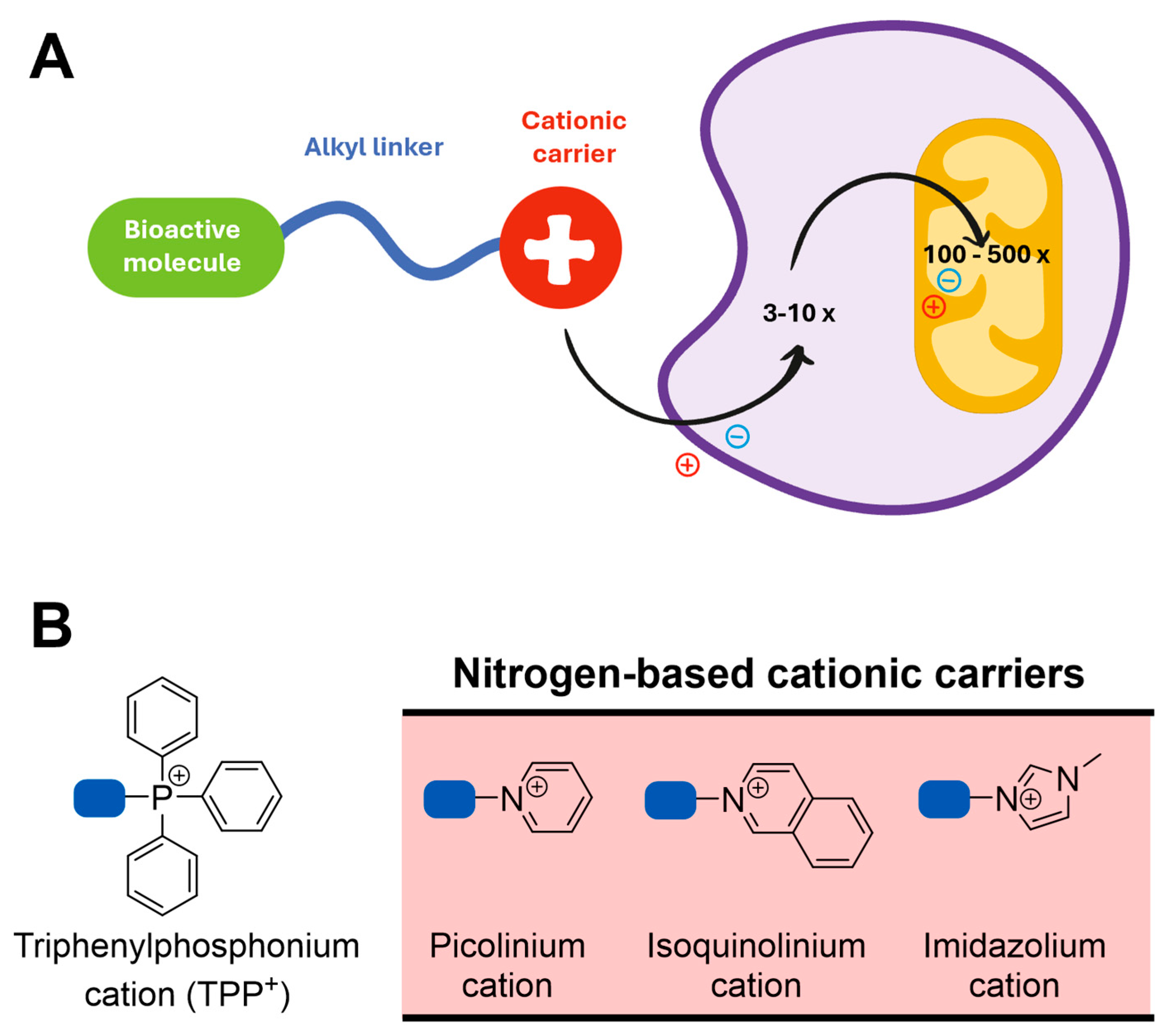
Figure 14.
Schematic representation of G protein-coupled adenosine receptors.
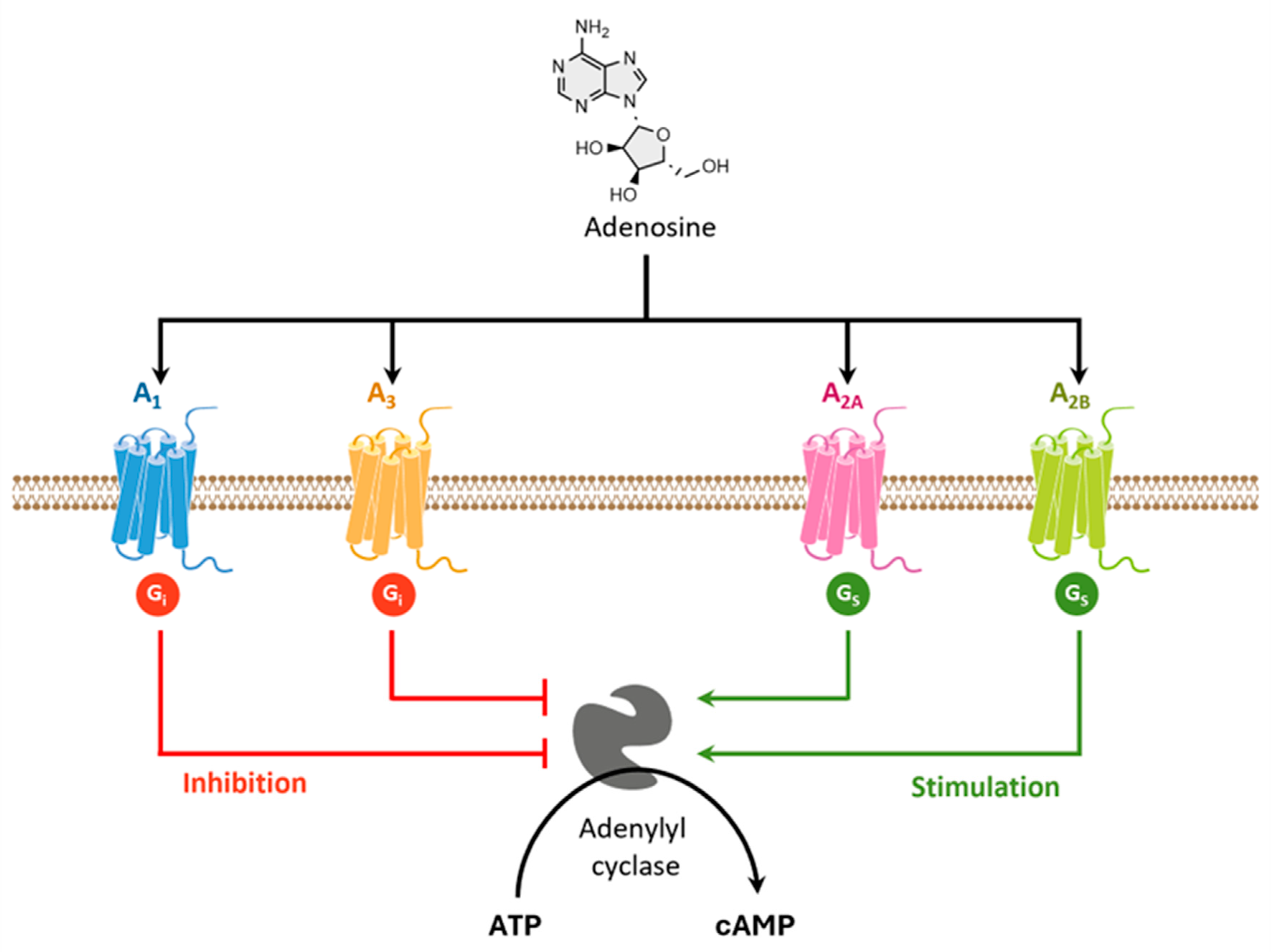
Figure 15.
Chemical structures of selective A2A AR antagonists evaluated in clinical trials.
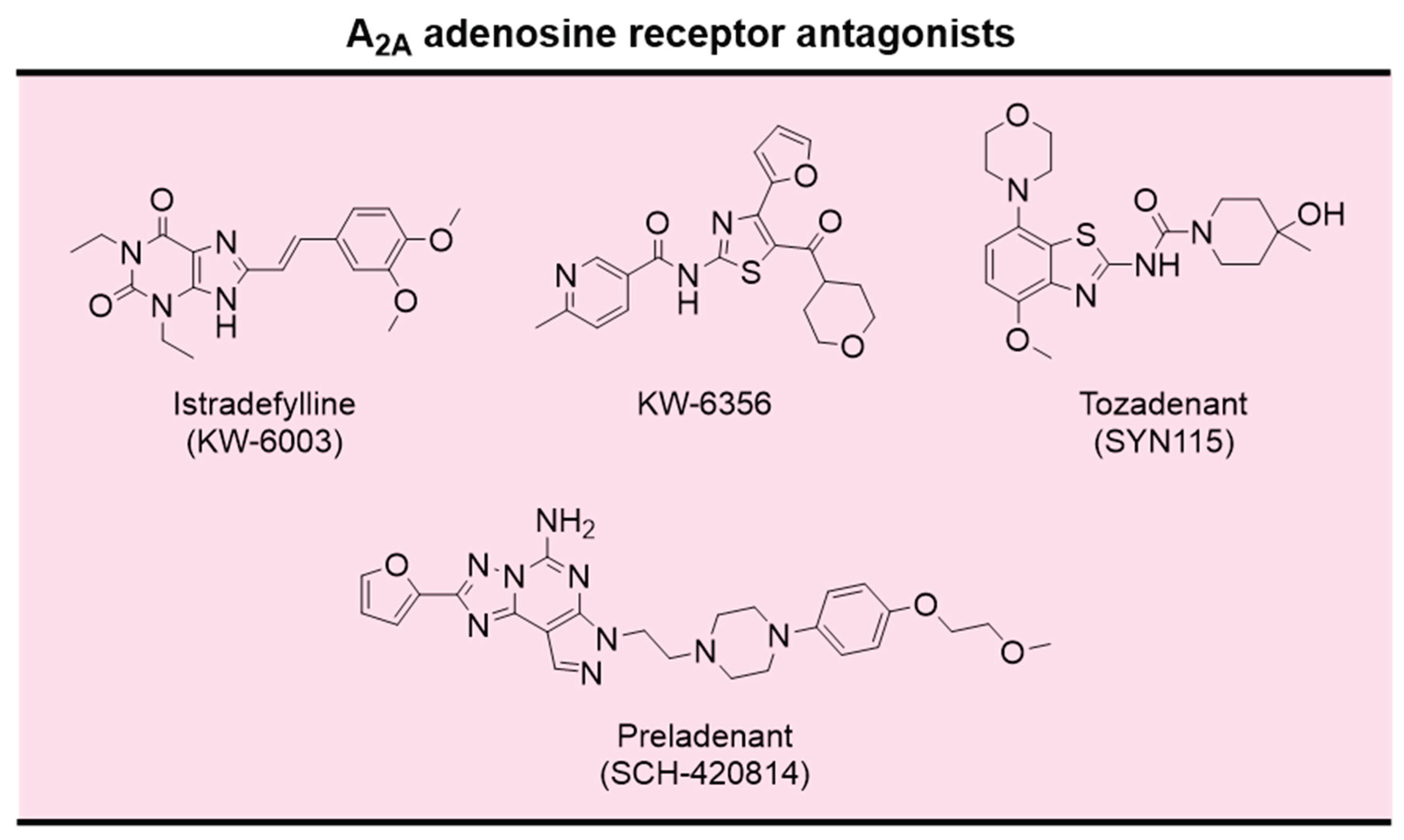

Table 1.
Examples of therapeutic targets looked for drug discovery and development for AD and PD.
| (Patho)physiological process | Target | |
|---|---|---|
| AD | PD | |
| Metal dyshomeostasis | copper [152]; iron [153]; zinc [154] | Copper [155]; iron [156](ref) |
| Mitochondria and metabolic functions | MCL1 [157] | ROCK [158], δ-opioid receptor [159] |
| Mutated/misfolded proteins | Amyloid-β [160]; β-secretase [161]; γ-secretase [162]; GSK3-β [163]; RAGE [164]; Tau [165] | DJ1 [166]; LRRK2 [167]; Pink1 [168]; α-synuclein [169]; CK1δ [170]; CK1δ+GSK3b [171] |
| Neuroinflammation | NRLP3 inflammasome [172] | Adenosine receptors [173]; cannabinoid receptor 2 [174]; monoacylglycerol lipase [175]; NRLP3 Inflammasome [176]; PPAR [177]; TRPC5 [178] |
| Neuroprotection | Apoptosis [179]; ferroptosis [180]; sigma 1 and 2 receptors [181] | Apoptosis [182]; autophagy/neuroinflammation [183]; CDNF peptidomimetic [184]; ferroptosis [185]; nurr1 [186]; sigma 1 and 2 receptors [187] |
| Oxidative stress | Antioxidants [188]; NRF2 signaling pathway [189] | Aldose reductase [190]; NRF2 signaling pathway [191] |
| Synaptic activity | AChE [110]; α7nAChR [192]; butyrylcholinesterase [193]; NMDA receptor [194] | 5-HT2A [195]; adenosine receptors [196,197]; α6AChR [198]; COMT [199]; dopaminergic D1-D4 receptors [200,201,202]; GPR6 [203]; MAO-B [204]; mGlu4 [205]; PDE4 [206] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



