
Submitted:
05 April 2024
Posted:
08 April 2024
You are already at the latest version
Abstract
Misfolding of superoxide dismutase-1 (SOD1) is a pathological hallmark of amyotrophic lateral sclerosis (ALS) with SOD1 mutations. The development of antibodies specific for misfolded SOD1 deepens our understanding of how the protein participates in ALS pathogenesis. Since the term “misfolding” refers to various disordered conformers other than the natively folded one, which misfolded species are recognized by specific antibodies should be determined. Here, we molecularly characterized the recognition by MS785-MS27, an antibody cocktail experimentally confirmed to recognize over 100 ALS-linked SOD1 mutants. Indirect ELISA revealed that the antibody cocktail recognized Zn-deficient wild-type and mutated SOD1 species. It also recognized conformation-disordered wild-type and mutated SOD1 species, such as unfolded and oligomeric forms, but had less affinity for the aggregated form. Antibody-reactive SOD1 exhibited cytotoxicity to a motor neuron cell model, which was blocked by Zn treatment with Zn-deficient SOD1. Immunohistochemistry revealed antibody-reactive SOD1 mainly in spinal motor neurons of SOD1G93A mice throughout the disease course, and the distribution after symptomatic stages differed from that of other misfolded SOD1 species. This suggested that misfolded SOD1 species exist as heterogeneous populations. In conclusion, MS785-MS27 recognizes various misfolded SOD1 lacking the incorporated Zn ion and should be useful for monitoring Zn availability in SOD1.
Keywords:
amyotrophic lateral sclerosis
; MS785-MS27 antibody cocktail
; Zn-deficient SOD1
; recombinant proteins
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the loss of both upper and lower motor neurons in the central nervous system, resulting in muscle weakness, paralysis, and eventually death within 2 to 5 years of diagnosis [1]. While the majority of ALS cases (90% to 95%) are sporadic, the remaining cases are familial [1]. So far, over 30 causative genes have been identified in familial ALS [2], one of which is the gene encoding superoxide dismutase-1 (SOD1); mutations in this gene contribute to approximately 20% of familial cases [3]. Wild-type SOD1 (WT SOD1) is an antioxidant that catalyzes the conversion of superoxide anion to hydrogen peroxide and molecular oxygen [4]. However, the enzymatic activity of the ALS-linked SOD1 mutants varies depending on the variant, from 0% to 100% compared with that of WT SOD1 [5,6]. It is now accepted that the cytotoxicity of ALS-linked SOD1 mutants does not arise from a loss of their enzymatic activity [7]. A common property of ALS-linked SOD1 mutations is that they compromise the stability of the protein’s quaternary structure, which promotes misfolding or disordered conformations [8,9]. Natively folded SOD1 is conformationally characterized by a homodimer containing one Cu and one Zn ion in each subunit, as well as by the formation of a disulfide bond between Cys57 and Cys146, which exhibits high thermostability (Tm >90ºC) [10]. However, once SOD1 loses these metal ions, undergoes dimer dissociation, or undergoes cleavage of the disulfide bond, its stability is remarkably decreased. For instance, metal-free, disulfide bond-cleaved WT SOD1 exhibits lower thermostability (Tm = 42ºC) [10,11].
The concept that misfolded SOD1 participates in the pathogenesis of familial or even sporadic ALS has been reinforced by the development of antibodies specifically targeting misfolded SOD1 [12,13]. To our knowledge, more than 20 specific antibodies have been developed, a subset of which have the potential for use in therapeutic interventions [13,14,15]. Most specific antibodies of this kind are designed to bind to regions that are inaccessible in the natively folded form. In other words, the specific antibodies recognize regions that are exposed at the surface only when SOD1 adopts a non-native conformation initiated by mutations or loss of cofactors involved in the conformational stability (e.g., Zn ions). Unlike for natively folded SOD1, it is difficult to define the inherent conformation of misfolded SOD1 due to the huge number of influential factors acting on this in combination (e.g., monomer/dimer, metal-free/bound, or disulfide bond cleavage/formation). Thus, misfolded SOD1 could consist of a heterogeneous population with distinct conformation. To better understand which misfolded SOD1 species participate in which disease process, it is critical to determine which misfolded SOD1 species are recognized by a specific antibody. However, the mechanism behind the selectivity of most developed antibodies that specifically recognize misfolded SOD1 has not been well characterized at the molecular level. In most cases, it has just been determined by a comparison of specificity between natively folded and unfolded forms treated with chaotropic reagents (e.g., guanidinium ions).
MS785 and MS27 are commercially available antibodies specifically targeting misfolded SOD1 [16,17]. Although each of these antibodies fails to recognize ALS-linked SOD1 mutants with a point mutation in their epitope region (amino acids 8-14 for MS785; amino acids 30-40 for MS27), the use of an MS785 and MS27 cocktail compensates for each of their shortcomings, with this mixture recognizing over 100 ALS-linked SOD1 mutants as determined by immunoprecipitation [17]. Thus, the MS785-MS27 cocktail is the only antibody in which the affinity for a variety of ALS-linked SOD1 mutants has been experimentally confirmed. According to previous cell-based studies [17,18], this antibody cocktail recognizes SOD1 species that show an increase in level upon the treatment of cells with a Zn-chelating reagent, implying that the antibody cocktail recognizes “Zn-deficient SOD1 species.” However, despite substantial efforts, it is unclear which SOD1 species whose misfolding is induced by Zn deficiency can be recognized by this antibody cocktail at the molecular level.
Against the above background, the aim of the present study is to molecularly characterize the recognition by the MS785-MS27 antibody cocktail of various SOD1 species with altered conformational integrity.
2. Results
2.1. MS785-MS27 Antibody Cocktail Recognizes WT SOD1 Species Lacking the Incorporated Zn Ion
To promote rapid analyses of the SOD1 species recognized by the MS785-MS27 antibody cocktail, we developed an indirect enzyme-linked immunosorbent assay (ELISA) coupled with this cocktail. In our ELISA, we used metal-free, disulfide bond-cleaved WT SOD1 (apo-WT SOD1SH) as a representative species of Zn-deficient misfolded SOD1, as well as an internal standard for comparisons among different 96-well plates. We first assessed the consistency and reproducibility of our ELISA. Apo-WT SOD1SH with high purity as confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; Figure 1A) or vehicle buffer was added to 48 wells in the plate and detected by the incubation of the MS785-MS27 antibody cocktail followed by the use of a horseradish peroxidase (HRP) colorimetric system. The coefficient of variation value and the ratio of the signal to the background in the ELISA were 4.5% and 19, respectively (Figure 1B), indicating that our ELISA achieved not only small variation among samples, but also high affinity for apo-WT SOD1SH accompanied by a low background. Moreover, the Z’-factor, an indicator of assay quality, was 0.84 (Figure 1B), implying that our ELISA is suitable for high-throughput analyses because the Z’-factor exceeds the established threshold of 0.50 [19]. We confirmed the antibody cocktail recognition for apo-WT SOD1SH by competitive inhibition ELISA. The antibody cocktail at 1 μg/mL was mixed with apo-WT SOD1SH at different concentrations. The complex of the antibody cocktail and apo-WT SOD1SH was used as a primary antibody in the indirect ELISA. As expected, preincubation of the antibody cocktail with apo-WT SOD1SH led to a decrease of the ELISA signals in a concentration-dependent manner (Figure 1C).
Wild-type SOD1 contains one Cu ion and one Zn ion in its monomeric subunit [10]. We thus next examined the antibody cocktail recognition of WT SOD1 into which these metals were differently incorporated. We treated apo-WT SOD1SH with 1 molar equivalent of either Cu or Zn ions, and investigated the antibody cocktail recognition of the metal-bound forms using indirect ELISA. We found that the antibody cocktail recognized Cu-WT SOD1SH (Zn-deficient form), whereas it did not recognize Zn-WT SOD1SH (Cu-deficient form) (Figure 1D). Moreover, the antibody cocktail did not react with Cu/Zn-WT SOD1, the natively folded and enzymatically active form (Figure 1D), indicating that the antibody cocktail recognizes Zn-deficient WT SOD1SH (metal-free or Cu-binding form).
To determine the role of the Zn coordination to SOD1 in antibody cocktail recognition, we treated apo-WT SOD1SH with 1 to 4 molar equivalents of Zn ions. As shown in Figure 1E, the antibody cocktail reactivity was significantly decreased in a dose-dependent manner upon the Zn coordination to SOD1. This decrease was not observed when the Zn binding site of SOD1, namely, residues His63, His71, His80, and Asp83, was lost by amino acid substitutions (Figure 1E, red). We also performed similar experiments using disulfide bond-forming WT SOD1 (SOD1S-S) with different incorporated metals. The antibody cocktail retained reactivity to Zn-deficient SOD1S-S (Figure 1F). Taking these findings together, the MS785-MS27 antibody cocktail discriminates conformational changes related to lack of Zn ion incorporation in SOD1 regardless of whether the protein forms a disulfide bond.
2.2. MS785-MS27 Antibody Cocktail Reacts with Conformation-Disordered WT SOD1 Species
Conformation-disordered SOD1 species such as unfolded, oligomeric, and aggregated forms are pathological hallmarks of SOD1-ALS or even of a subset of sporadic ALS [7,20,21,22,23]. We determined whether the MS785-MS27 antibody cocktail could recognize conformation-disordered WT SOD1 species. We prepared unfolded, oligomeric, and aggregated forms of SOD1 in a test tube as sources of apo-WT SOD1S-S, as described previously [22,24], and analyzed the antibody cocktail recognition of these disordered species using indirect ELISA. The antibody cocktail strongly reacted with both unfolded and oligomeric SOD1, and the reactivities were of the same intensity as those for apo-WT SOD1SH (Figure 2A). Meanwhile, the antibody cocktail showed less affinity for SOD1 aggregates. This was unsurprising because the epitopes of MS785 and MS27 are amino acids corresponding to residues 8-14 and 30-40 in human SOD1, respectively, which are regions buried within aggregates [25,26]. Such lower reactivity to SOD1 aggregates is unrelated to them being difficult to attach to or easily detached from the 96-well plate because they were detected at the same intensity as other SOD1 species by an anti-SOD1 antibody (Figure 2B) raised against full-length human SOD1 and reacted with all SOD1 conformers [22]. These findings indicate that the antibody cocktail has a broad response to conformation-disordered SOD1 species lacking the incorporated Zn ion.
2.3. MS785-MS27 Recognizes Zn-Deficient SOD1 with ALS-Linked Mutations
Based on previous cell-based studies [17,18], the MS785-MS27 antibody cocktail recognizes over 100 ALS-linked SOD1 mutants. To further molecularly characterize whether the antibody cocktail can recognize ALS-linked SOD1 mutants with distinct biopsychical properties and conformations, we used seven different types of the mutants classified into three groups: (i) WT-like mutants, G37R, D90A, and G93A; (ii) dimer interface mutants, A4V and I113T; and (iii) metal binding region mutants, H46R and G85R (Figure 3A) [10,27]. We first investigated the antibody cocktail recognition of apo-SOD1SH with these mutations, the purity of which was confirmed by SDS-PAGE (Figure 3B). All tested apo-SOD1SH with ALS-linked mutations, except A4V and G37R, exhibited the same signal intensity as that of WT SOD1 (Figure 3C). The antibody cocktail had 30% less affinity for A4V SOD1 than for WT SOD1 (Figure 3C), which was consistent with the findings of a previous cell-based study [17]. Apo-G37R SOD1SH had as much as half of the signal intensity compared with the other mutants and WT SOD1 (Figure 3C). This was reasonable because G37R SOD1 has a point mutation in the epitope region of MS27 (30KVWGSIKGLTE40; the amino acid substitution G37R of SOD1 is shown in bold) [17]. Thus, the reactivity of the antibody cocktail to apo-G37R SOD1SH stems from MS785, but not MS27. Indeed, MS785 alone recognized apo-G37R SOD1SH at a similar intensity to the other mutants, whereas MS27 alone did not (Figure S1). The antibody cocktail exhibited similar affinity patterns to apo-SOD1S-S with the ALS-linked mutations (Figure 3D).
We also determined whether the antibody cocktail could recognize conformation-disordered SOD1 with ALS-linked mutations, including unfolded, oligomeric, and aggregated forms. The antibody cocktail had similar affinity to the unfolded SOD1 mutants (Figure 3E), except A4V and G37R, as to unfolded WT SOD1 (Figure 2A). The antibody cocktail also showed similar affinity and intensity to oligomeric SOD1 mutants as to unfolded mutants (Figure 3F). By contrast, the antibody cocktail had lower affinity to mutant SOD1 aggregates (Figure 3G), which was consistent with the findings for WT SOD1 aggregates (Figure 2A).
To address whether the Zn coordination to the ALS-linked SOD1 mutants plays a major role in the antibody cocktail recognition, we treated the seven different types of SOD1 mutants with 1 to 4 molar equivalents of Zn ions, and analyzed the antibody cocktail recognition of these mutants with distinct incorporation of Zn ion using indirect ELISA. Regarding the WT-like mutants (G37R, D90A, and G93A, blue), as well as dimer interface mutants (A4V and I113T, green), the antibody cocktail reactivity significantly decreased in a concentration-dependent manner upon the Zn treatment (Figure 3H), suggesting that the Zn coordination to these mutants is associated with conformational changes, possibly incomplete folding, which inhibits recognition by the antibody cocktail. However, focusing on these Zn-treated mutants, the antibody cocktail still had higher affinity for these mutants than for WT SOD1 (Figure 3H, black), implying that these mutants decrease the capacity to incorporate a Zn ion. With respect to the metal binding region mutants (H46R and G85R, red), the antibody cocktail still recognized the mutants even upon Zn treatment (Figure 3H).
2.4. Zn-Deficient SOD1 Species Have Cytotoxic Effects on NSC-34 Cells
To determine whether the MS785-MS27-reactive SOD1 species could exhibit toxic effects on cultured cells, we first investigated the proliferation of NSC-34 cells, a hybridoma with mouse spinal motor neurons and neuroblastoma cells, by measuring the metabolic activity using Cell Counting Kit-8. The cells were treated with apo-WT SOD1SH or apo-SOD1SH that lacks the Zn binding site, as representatives of MS785-MS27-reactive SOD1 species (Figure 1E), at different concentrations for 48 h. We found that the cell proliferation was significantly decreased in NSC-34 cells treated with apo-WT SOD1SH at the concentration range of 0.25 to 10 μM, compared with that of vehicle controls (Figure 4A, black). Notably, the proliferation of NSC-34 treated with apo-SOD1SH that lacks the Zn binding site decreased markedly compared with that of apo-WT SOD1SH (Figure 4A, red). We confirmed these results via a lactate dehydrogenase (LDH) cytotoxicity assay, measuring the activity of LDH released from damaged cells (Figure 4B). These cytotoxic effects were blocked by the pretreatment of apo-WT SOD1SH with Zn ions (Figure 4C,D, black). This protection was not observed in SOD1SH that lacks the Zn binding site (Figure 4C,D, red), indicating that the cytotoxic effects of apo-SOD1SH are related to lack of Zn ion incorporation in the protein.
2.5. Distribution of MS785-MS27-Reactive SOD1 Species in the Spinal Cord of G93A SOD1 Mice
To clarify whether the MS785-MS27-reactive SOD1 species could localize in motor neurons and how their distribution changes during the disease course of a mouse model of SOD1-ALS, we performed immunofluorescence with the antibody cocktail on the lumbar spinal cord from high-copy G93A SOD1 mice at different disease stages. At the presymptomatic stage (60 days of age), the MS785-MS27-reactive SOD1 species were present as small granules in motor neurons (Figure 5A, inset), and appeared as diffuse staining in axons (Figure 5A, arrows). After the onset of the symptoms (90 days of age), the SOD1-positive granules observed in motor neurons became larger (Figure 5B, inset), and were located around vacuoles in the ventral horn (Figure 5B, arrows). At the terminal stage (120 days of age), the SOD1 species were still detectable in motor neurons (Figure 5C, inset) and exhibited fibril-like structures outside the motor neurons (Figure 5C, arrows).
Notably, in mice overexpressing human WT SOD1 at 120 days of age, the SOD1 species were also observed in motor neurons (Figure 5D, inset) and vacuoles (Figure 5D, arrows), suggesting that WT SOD1 could lack Zn ion incorporation in the protein and misfolding in vivo. By contrast, no fluorescence signals were detected in the ventral horn of non-transgenic mice (Figure 5E). The recognition of murine SOD1 by the MS785-MS27 antibody cocktail was confirmed by indirect ELISA with recombinant murine SOD1 protein, the high purity of which was confirmed by SDS-PAGE (Figure S2A). Indirect ELISA demonstrated that the antibody cocktail reacted with the metal-free form as well as the unfolded form of murine SOD1, but not the natively folded form (Figure S2B). MS27 alone did not recognize murine SOD1 species with which the antibody cocktail reacted (Figure S2C). Thus, the reactivity of MS785-MS27 antibody cocktail to metal-free or unfolded murine SOD1 appears to come from MS785. This is reasonable because the sequence of murine SOD is 100% identical to that of human SOD1 in the epitope region of MS785, whereas there is 54.5% homology between the murine and human SOD1 sequences in the epitope region of MS27 (Figure S2D). Taking these findings together, the endogenous murine SOD1 in non-transgenic mice exists in a conformation that MS785-MS27 antibody cocktail does not recognize, presumably the natively folded form [28,29].
Taking into account the presence of the MS785-MS27-reactive SOD1 species in cell types other than motor neurons throughout the disease course of G93A SOD1 mice (Figure 5A–C), we investigated whether the SOD1 species could localize in astrocytes, microglia, or both. Immunofluorescence studies demonstrated that the SOD1 species localized in a subset of microglia at all stages of the disease (Figure 5I–K, arrows), whereas the SOD1 species were not found in astrocytes (Figure 5F–H, arrowheads). Taking these findings together, the major site of the MS785-MS27-reactive SOD1 species in the ventral horn is the motor neurons.
2.6. MS785-MS27-Reactive SOD1 Species Are Differentially Distributed from the Known Misfolded SOD1 Species in G93A SOD1 Mice
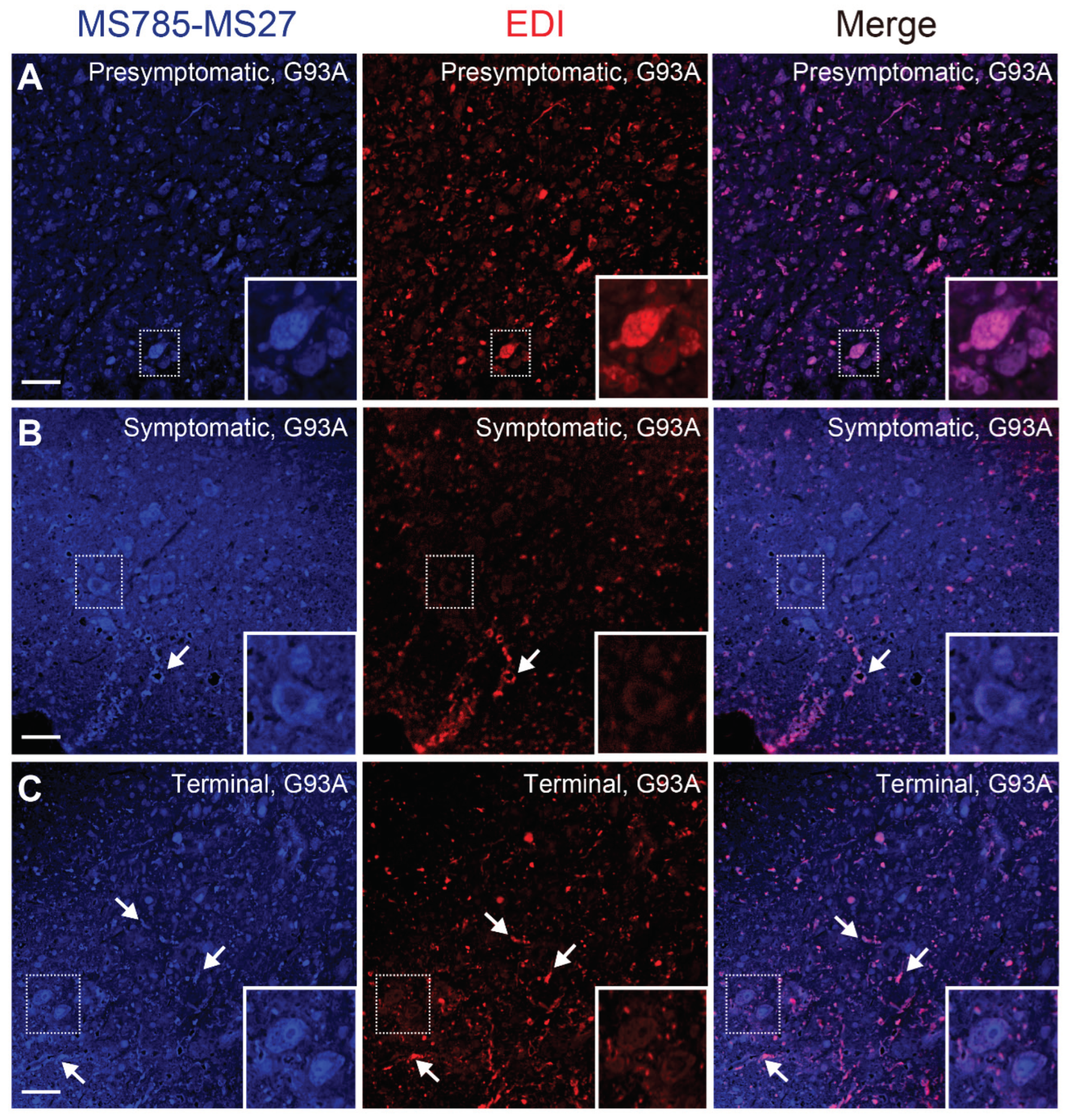
Previous immunohistochemical studies using a panel of specific antibodies for misfolded SOD1 demonstrated that misfolded SOD1 species likely exist as a heterogeneous population in vivo [30]. In other words, misfolded SOD1 could exhibit divergent conformations in the tissues affected by disease, and they could have different implications for the disease pathogenesis. To address the implications of the MS785-MS27-reactive SOD1 species among various misfolded conformers, we performed immunofluorescence on the lumbar spinal cord using the MS785-MS27 antibody cocktail and EDI, a commercially available antibody that specifically recognizes misfolded SOD1 for which the epitope is exposed at the dimer interface [12]. At the presymptomatic stage, the distribution of the MS785-MS27-reactive SOD1 species was identical to that of EDI-reactive misfolded SOD1 (Figure 6A), showing that both species were mainly observed in motor neurons. However, after the onset of symptoms, EDI-reactive misfolded SOD1 disappeared from the motor neurons, whereas the MS785-MS27-reactive SOD1 species were still located in them (Figure 6B). In contrast to the motor neurons, both species were colocalized in the vacuoles (Figure 6B, arrows) as well as in ventral root axons (Figure 6B). These manifestations continued at the terminal stage of the disease, demonstrating that both species were present as fibril-like structures (Figure 6C, arrows). Taking these findings together, there are differences in spatial distribution between MS785-MS27-reactive SOD1 and other species during the disease course of G93A SOD1 mice.
3. Discussion
In this study, we molecularly characterized which misfolded SOD1 species are recognized by the MS785-MS27 antibody cocktail, which binds over 100 ALS-linked SOD1 mutants. Using indirect ELISA with recombinant SOD1, we found that the antibody cocktail recognized WT SOD1 as well as the ALS-linked mutants lacking incorporated Zn ion (Figure 1 and Figure 3). These results build on the earlier cell-based studies [17,18], which demonstrated that treatment of cultured cells overexpressing WT SOD1 with a Zn-chelating reagent increased the range of SOD1 species that are recognized by the antibody cocktail. We also found that the antibody cocktail had strong reactivity to various conformation-disordered SOD1 species, including unfolded and oligomeric forms, but less for the aggregated one (Figure 2 and Figure 3). Thus, the present study deepens our understanding of the antibody cocktail recognition of misfolded SOD1 species at the molecular level.
The present study sheds light on the conformational features of the MS785-MS27-reactive SOD1 species of both WT SOD1 and the ALS-linked mutants. We prepared conformation-disordered SOD1 species including unfolded, oligomeric, and aggregated forms from apo-SOD1S-S as a precursor. The antibody cocktail had similar affinity between apo-SOD1S-S and the conformation-disordered SOD1 species, with the exception of the aggregated form (Figure 2 and Figure 3). This suggested that there could be common conformations among the MS785-MS27-reactive SOD1 species with various disordered conformations regardless of whether SOD1 is mutated or not. Here, we can speculate about the possible conformation that is shared among the MS785-MS27-reactive SOD1 species with regard to the epitope region of each antibody. The epitope region of MS785 is the peptide corresponding to amino acids 8 to 14 in human SOD1 [16], which is located in the small loop between the β1 and β2 strands. Meanwhile, the epitope region of MS27 is the peptide corresponding to amino acids 30 to 40 in human SOD1 [17], which covers the β3 strand, followed by a small loop. It is possible that the apo-form, unfolded form, and oligomeric form share a common disordered conformation around the β1 to β3 strands. In support of this idea, the X-ray crystal structure of apo-SOD1 revealed that the regions of the β1 to β3 strands are exposed to the solvent, in contrast to the case for the natively folded SOD1 (PDB: 3ECU), which provokes the misfolding of SOD1 [31].
Besides determining recognition by the MS785-MS27 antibody cocktail at the molecular level, we also characterized the toxicological features of MS785-MS27-reactive SOD1 species. We found that both apo-WT SOD1SH and apo-SOD1SH that lacks the Zn binding site exhibited toxic effects on NSC-34 cells (Figure 4A,B). It would thus be beneficial to decrease the amounts of Zn-deficient SOD1 species in order to protect motor neurons. One approach to achieve this is to perform Zn coordination to SOD1 species. This approach is promising given the evidence that the disordered conformation and cytotoxicity observed in Zn-deficient SOD1 were reversed by exogenous treatment of these species with Zn ions (Figure 1E, Figure 3H, Figure 4C,D). An earlier in vivo study demonstrated that treatment with ZnII (atsm), a small compound that coordinates Zn ions, improved motor functions of different mouse lines of SOD1-ALS [32]. We have also shown that metallothionein-I, a major Zn binding protein in the cells, suppresses the loss of motor neurons in G93A SOD1 mice [33,34]. Thus, restoration of the Zn bioavailability in SOD1 itself or in the microenvironment surrounding SOD1 would be important for the correction of SOD1 misfolding and suppression of motor neuron death. We propose that the reactivity of the MS785-MS27 antibody cocktail could be used as an indicator of Zn bioavailability in SOD1, since its recognition negatively correlates with the incorporation of Zn ions in SOD1 (Figure 1E and Figure 3H).
We provide evidence of the cell types where the MS785-MS27-reactive SOD1 species were distributed in the spinal cord during the disease course of G93A SOD1 mice. Throughout the disease course, the MS785-MS27-reactive SOD1 species were mainly localized in the motor neurons (Figure 5A–C), suggesting that misfolded SOD1 species present in motor neurons are deficient in Zn ions. These findings are consistent with a study [35] showing that 42% of ALS cases exhibit Zn deficiency in WT or mutated SOD1 localized in the ventral horn. We also provide evidence for there being divergent populations of misfolded SOD1 species that dynamically change over the disease course of G93A SOD1 mice (Figure 6). Co-immunostaining with the antibody cocktail and EDI revealed that the MS785-MS27-reactive SOD1 co-existed with EDI-reactive SOD1 species at the presymptomatic stage (Figure 6A), suggesting that the conformation of misfolded SOD1 at the early stage is likely to be homogeneous. However, as the disease progressed, the EDI-reactive SOD1 species were mainly observed outside of the motor neurons, which decreased the overlap with the MS785-MS27-reactive SOD1 species (Figure 6B,C). These results indicate that misfolded SOD1 species appear to adopt various conformations after the onset of symptoms of the disease. Our histological data are supported by a previous study reporting that there were distinct distribution patterns of misfolded SOD1 species when these species were immunolabeled with a panel of specific antibodies [30]. The divergent populations of misfolded SOD1 species might explain why several passive immunotherapies targeting misfolded SOD1 fail to rescue disease phenotypes in mouse models of SOD1-ALS [36].
4. Materials and Methods
4.1. Expression, Purification, and Demetallation of Recombinant SOD1 Proteins
A bacterial expression plasmid, pET28a(+), encoding human SOD1 with I113T that was N-terminally tagged with hexahistidine, was purchased from Addgene (#117703; Watertown, MA, USA). Plasmids encoding WT, A4V, G37R, H46R, G85R, D90A, or G93A human SOD1 gene were prepared using site-directed mutagenesis, and correct sequences of each construct were confirmed by Azenta (Tokyo, Japan). Human SOD1 with mutations in the Zn binding sites was designed from the WT sequence by replacement of residues H63A, H71A, H80G, and D83A, and subcloned into the pET28a(+) vector. A bacterial expression plasmid, pET28a(+) encoding mouse WT Sod1 that was N-terminally tagged with hexahistidine, was also obtained from Addgene (#117701). Each expression vector was transformed into SHuffle E. coli (New England Biolabs, Ipswich, MA, USA). Colonies isolated from the LB-agar plate were inoculated onto LB medium with 100 μg/mL kanamycin sulfate at 200 rpm and 30ºC for 14 h. Bacterial overexpression was initiated by adding the preculture medium to 500 mL of the LB medium. When turbidity (OD600) of the E. coli reached between 0.60 and 0.80, the cells were cultured with 0.5 mM isopropyl-β-D-thiogalactopyranoside (Bio Medical Science, Tokyo, Japan) at 200 rpm and 20ºC for 20 h. The E. coli was lysed by sonication (Sonifier SFX250; Branson Emerson, Brookfield, CT, USA) in a buffer (pH 7.0) containing 2% (v/v) Triton X-100, 50 mM Tris, 500 mM NaCl, EDTA-free Complete Protease Inhibitor Cocktail (Merck, Darmstadt, Germany), 1 U DNase I (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan), and 5 mM MgSO4. The cell lysates were centrifuged at 20,000 × g and 4ºC for 30 min, and the supernatant was filtered with a 0.22 μm syringe-driven filter unit (Millipore, Burlington, MA, USA). The SOD1 with a hexahistidine tag was isolated using a Ni2+ affinity chromatograph (Complete His-Tag Purification Column; GE Healthcare, Chicago, IL, USA) by an ÄKTA Start (Cytiva, Tokyo, Japan). Pre-equilibration of the column was carried out using a buffer (pH 7.0) containing 50 mM Tris, 1 M NaCl, and 50 mM imidazole. Elution of the fusion protein was performed with a buffer (pH 7.0) containing 50 mM Tris, 100 mM NaCl, and 250 mM imidazole.
The SOD1 with a hexahistidine tag was demetallated by dialysis in a Spectra/Por® (molecular mass cut-off: 6-8 kDa; Repligen, Waltham, MA, USA) against a buffer (pH 4.0) containing 50 mM sodium acetate, 100 mM NaCl, and 5 mM EDTA at 4ºC for 20 h. The apo-SOD1 with a hexahistidine tag was neutralized by dialysis with a buffer (pH 7.0) containing 50 mM Tris, 100 mM NaCl, and 5 mM EDTA for 4 h at 4ºC. The hexahistidine tag was cleaved by treatment with 4 U thrombin, a serine protease (GE Healthcare), at 20ºC for 20 h. The non-cleaved fusion protein and thrombin were removed using a Complete His-Tag Purification Column and Benzamidine Column (Cytiva), respectively. The SOD1 was further purified by size exclusion chromatography (Superdex™ 200 Increase Column 10/300 GL; Cytiva) with an ÄKTA Go (Cytiva). The concentration of SOD1 protein was determined by an Eppendorf BioSpectrometer (Eppendorf, Hamburg, Germany) using a monomeric molar extinction coefficient at 280 nm of 5500 M−1 cm−1. The purity of SOD1 was confirmed by SDS-PAGE followed by InstantBlue® Coomassie Protein Stain (Abcam, Cambridge, UK).
4.2. Preparation of Metal Binding Forms and Conformation-Disordered SOD1
Metal binding forms of WT SOD1 were prepared as described elsewhere with slight modification [22,24]. A buffer (pH 7.0) containing 50 mM Tris and 100 mM NaCl was treated with a Chelex 100 Resin (100-200 mesh; Bio-Rad, Hercules, CA, USA) to remove divalent ions. Apo-WT SOD1S-S was treated with 10 mM (±) dithiothreitol (DTT) at 37ºC for 1 h to obtain the disulfide bond-cleaved form. DTT was removed using a Zeta Spin Desalting Column (molecular mass cut-off: 7 kDa; Thermo Fisher Scientific, Waltham, MA, USA), in accordance with the manufacturer’s protocol. Apo-WT SOD1SH was treated with 1 to 4 molar equivalents of Zn or Cu ions at 4ºC for 3 h. The Cu- and Zn-bound form of WT SOD1 was purchased from Sigma-Aldrich (S9636; St. Louis, MO, USA), which was isolated from human erythrocytes and used as a natively folded form (also known as an enzymatically active form).
Conformation-disordered SOD1 species, including unfolded, oligomeric, and aggregated forms, were prepared as described previously with slight modification [22,24]. For preparation of the unfolded form, Apo-SOD1S-S at 100 μM was treated with 10 mM DTT at 37ºC for 1 h. After removing the DTT using a Zeta Spin Desalting Column (molecular mass cut-off: 7 kDa; Thermo Fisher Scientific), apo-SOD1SH was treated with a buffer (pH 7.0) containing 50 mM Tris, 100 mM NaCl, 5 mM EDTA, and 6 M guanidine hydrochloride for 37ºC for 2 h. For preparation of the oligomeric form, apo-SOD1S-S at 100 μM was incubated at 37ºC for 5 days to induce non-native cross-linking between Cys residues in SOD1 [37]. For preparation of the aggregated form, apo-SOD1SH at 100 μM was added into a ProteoSave 96-well plate (Sumitomo Bakelite Co. Ltd., Tokyo, Japan) and agitated in the presence of a plastic POM ball (φ 2.4 mm; Sanplatec Co. Ltd., Tokyo, Japan) at 1,200 rpm and 37ºC for 5 days. Suspension of the SOD1 aggregates was collected and subjected to centrifugation at 20,000 × g for 30 min at 4ºC. The supernatant was discarded and the pellet was resuspended in a buffer (pH 7.0) containing 50 mM Tris, 100 mM NaCl, and 5 mM EDTA. The SOD1 aggregates were sonicated using a Handy Sonic (UR-21P; Tony Seiko Co. Ltd., Tokyo, Japan). The concentration of SOD1 aggregates was determined by an Eppendorf BioSpectrometer using a monomeric molar extinction coefficient at 280 nm of 5,500 M−1 cm−1.
4.3. ELISA
To analyze the conformation-disordered SOD1 species, EDTA at a final concentration of 5 mM was added to Tris-buffered saline (TBS, pH 7.4) or TBS with 0.05% (v/v) Tween 20 (TBST, pH 7.4) to avoid the metal re-coordination to SOD1. For experiments on the metal binding forms of SOD1, TBS or TBST was treated with a Chelex 100 Resin (100-200 mesh; Bio-Rad) to remove divalent metal ions. SOD1 species at 5 μM were coated onto a Nunc Immuno 96-well Plate with a Maxi Soap (Thermo Fisher Scientific) at 4ºC for 18 h. The plates were blocked with 3% (w/v) bovine serum albumin in TBS (pH 7.4) at room temperature for 1 h. After three washes with TBST, primary antibodies including MS785-MS27 cocktail (0.025 μg/mL, FDV-0021A; Funakoshi, Tokyo, Japan), MS785 (0.025 μg/mL, FDV-0021B; Funakoshi), MS27 (0.025 μg/mL, FDV-0021C; Funakoshi), or pan-SOD1 (0.5 μg/mL, sc-11407; Santa Cruz Biotechnologies, Dallas, TX, USA) were added, and the plates were incubated at 4ºC for 18 h. After three washes with TBST, HRP-conjugated secondary antibody against rat IgG (1:1,000, AS028; AB Clonal, Woburn, MA, USA) or rabbit IgG (1:1,000, A9169; Sigma-Aldrich) was added, and the plates were incubated at room temperature for 1 h. As a chromogenic substrate, 100 mM citrate buffer (pH 5.0) containing 0.03% (v/v) hydrogen peroxide and 1 mg/mL O-phenylenediamine dihydrochloride (Fujifilm Wako Pure Chemical Corporation) was added, and the color reaction was terminated by treatment with 1 M hydrochloric acid. The absorbance at 490 nm was measured on a microplate reader (FLUOstar Omega; BMG LABTECH, Ortenberg, Germany).
For competitive inhibition of ELISA, the MS785-MS27 antibody cocktail at 1 μg/mL was mixed with apo-WT SOD1SH over a concentration range of 10−4 to 101 μΜ at 4ºC for 18 h. The complex of MS785-MS27 and apo-WT SOD1SH was used as a primary antibody for indirect ELISA, where apo-WT SOD1SH at 5 μM was coated onto the Nunc Immuno 96-well plate. The subsequent procedures of competitive ELISA were the same as mentioned above for the indirect ELISA.
4.4. Assessment of Cell Proliferation and Cytotoxicity
The culture and maintenance of NSC-34 cells were performed as described previously [38,39]. In brief, NSC-34 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L D-glucose (Nacalai Tesque, Kyoto, Japan) supplemented with 10% (v/v) fetal bovine serum at 37ºC in a humidified chamber with 5% (v/v) CO2. For experiments on cell proliferation and cytotoxicity after treatment with Zn-deficient SOD1, NSC-34 cells (1.5×103 cells/well) were harvested in 96-well plates (Falcon, Corning, NY, USA). Zn-deficient SOD1 species, such as apo-WT SOD1SH or apo-SOD1SH that lacks the Zn binding site (H63A/H71A/H80G/D83A), were dissolved in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.2) pretreated with a Chelex 100 Resin (100-200 mesh; Bio-Rad) followed by filtered sterilization, and were diluted in DMEM/F-12 with GlutaMAX medium (Thermo Fisher Scientific) containing 1% (v/v) non-essential amino acids (Thermo Fisher Scientific) and 1% (v/v) fetal bovine serum. The cells were exposed to the Zn-deficient SOD1 species at a concentration range from 0.01 to 10 μM for 48 h. In parallel with the exposure of the Zn-deficient SOD1, the cells were also treated with 20 mM HEPES (pH 7.2), which was used as a vehicle control. For experiments on the coordination of Zn ions to the Zn-deficient SOD1 species, 1 μM apo-WT SOD1SH or apo-SOD1SH that lacks the Zn binding site was treated with 1 to 4 molar equivalents of Zn ions at 4ºC for 3 h. The cell proliferation and cytotoxicity were evaluated using the Cell Counting Kit-8 and the Cytotoxicity LDH Assay Kit-WST, in accordance with the manufacturer’s protocol (Dojindo Laboratories, Kumamoto, Japan), respectively.
4.5. Immunofluorescence
Transgenic mice carrying human mutated G93A SOD1 [B6SJL-Tg(SOD1-G93A)1Gur/J, 002726] or human WT SOD1 [B6SJL-Tg(SOD1)2Gur/J, 002297] were purchased from the Jackson Laboratory (Bar Harbor, MA, USA) [40]. Both transgenic lines were maintained through hemizygotes crossing transgenic males with F1 non-transgenic females on a B6SJL background. All animal protocols adhered to the Nihon University Animal Committee guidelines for the care and use of laboratory animals, and were approved by the Institutional Animal Care and Use Committee of Nihon University (#2026).
G93A SOD1 mice were anesthetized with pentobarbital when they reached 60 days (presymptomatic stage), 90 days (symptomatic stage), and 120 days of age (terminal stage), and the mice were transcardially perfused with ice-cold phosphate-buffered saline (PBS, pH 7.4) followed by 4% (v/v) paraformaldehyde in PBS. The spinal cord was postfixed with 4% (v/v) paraformaldehyde in PBS (pH 7.4) at 4 °C for 24 h and embedded in paraffin using a Tissue-Tek VIP5 Jr. (Sakura Finetek, Tokyo, Japan). The lumbar spinal cords of the mice (n=3 per genotype per disease stage) were sliced into 6 μm thick sections. These sections were deparaffinized in xylene, rehydrated in ethanol, and rinsed with deionized water. Epitope retrieval was performed using an autoclave for 20 min in the presence of 10 mM citrate buffer (pH 6.0). To inhibit endogenous mouse immunoglobulins, the lumbar spinal cord sections were treated with an M.O.M. blocking reagent (Vector Labs, Newark, CA, USA) at room temperature for 1 h. Non-specific binding of antibodies was blocked with PBS (pH 7.4) containing 10% (v/v) donkey normal serum (abcam), 3% (w/v) IgG- and protease-free bovine serum albumin (Jackson Immunoresearch, West Grove, PA, USA), and 0.1% (v/v) Trion X-100 at room temperature for 1 h. The following primary antibodies were used: rat monoclonal MS785-MS27 cocktail (2.5 μg/mL, FDV-0021A; Funakoshi, Tokyo, Japan), rabbit polyclonal anti-SOD1 EDI (2.5 μg/mL, SPC-206; StressMarq Bioscences, Victoria, Canada), mouse monoclonal anti-NeuN (2.5 μg/mL, MAB377; Merck Millipore), mouse monoclonal anti-Glial fibrillary acidic protein (GFAP) cocktail (1 μg/mL, 556330; BD Bioscience, Franklin Lakes, NJ, USA), or rabbit polyclonal anti-Ionized calcium-binding adapter molecule 1 (Iba1, 1 μg/mL, 013-27691; Fujifilm Wako Pure Chemical Corporation). As secondary antibodies, Alexa Fluor 405-conjugated donkey anti-rat IgG (5 μg/mL, ab175670; abcam), Alexa Fluor 488-conjugated donkey anti-mouse IgG (5 μg/mL, ab150109; abcam), or Alexa Fluor 594-conjugated donkey anti-rabbit IgG (5 μg/mL, ab150064, abcam) was used. The sections were mounted in ProLong Diamond antifade reagent (Thermo Fisher Scientific). Fluorescence images were acquired using a confocal microscope, Laser Scanning System Z710 (Carl Zeiss, Oberkochen, Germany).
4.6. Statistics
The results are given as the mean ± SD. All statistical tests were performed using Statcel 4 software (OMS Publishing Inc., Saitama, Japan). After determining the data normality, multiple group comparisons were performed using one-way ANOVA followed by Tukey–Kramer’s post hoc test. Statistical significance was defined as P<0.05.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Recognition of G37R SOD1 by MS785 or MS27 alone. Figure S2: Recognition of murine SOD1 species by MS785-MS27 antibody cocktail.
Author Contributions
Conceptualization, E.T.; Methodology, E.T.; Software, E.T.; Validation, E.T., Y.S., and N.T.; Formal Analysis, E.T.; Investigation, E.T., Y.S., N.T., and A.D.; Resources, Y.K.; Data Curation, E.T.; Writing – Original Draft Preparation, E.T.; Writing – Review & Editing, E.T., Y.K., and T.M.; Visualization, E.T.; Supervision, E.T.; Project Administration, E.T.; Funding Acquisition, E.T. and T.M. All authors read and accepted the final version of the manuscript.
Funding
This study was supported by a Grant-in-Aid for Challenging Exploratory Research (21K19456 to E.T.) from the Japan Society for the Promotion of Science. This study was also supported in part by the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to E.T.), the Takeda Science Foundation (to E.T.), the Hamaguchi Biochemical Foundation (to E.T.), the Ichiro Kanehara Foundation for the Promotion of Medical Sciences and Medical Care (to E.T.), the Japan ALS Association (to E.T.), the Serika ALS Foundation (to E.T.), and The Yukihiko Miyata Memorial Trust for ALS Research (to E.T.). This work was also funded in part by a grant to encourage and promote research projects at the School of Pharmacy, Nihon University (to E.T.), a grant for cooperative research at the School of Pharmacy, Nihon University (to E.T.), and a Nihon University Research Grant (to T.M.).
Institutional Review Board Statement
The animal study was approved by the Institutional Animal Care and Use Committee of Nihon University (Permission number: 2026; Date approved: May 8, 2008).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this report are available on request from the corresponding author.
Acknowledgments
The authors would like to thank Dr. Elizabeth Fisher (UCL Institute of Neurology and MRC Centre for Neuromuscular Disease, London, UK) for the kind gift of bacterial expression plasmids (phSOD1-I113T-N’His, Addgene, #117703; pmSod1-WT-N’His, Addgene, #117701). The authors would also like to thank Dr. Neil Cashman (University of British Colombia, Victoria, Canada) for kindly sharing NSC-34 cells. Finally, the authors thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Brown, R. H.; Al-Chalabi, A. , Amyotrophic Lateral Sclerosis. N Engl J Med 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Akçimen, F.; Lopez, E. R.; Landers, J. E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B. J. , Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat Rev Genet 2023, 24, 642–658. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D. R.; Siddique, T.; Patterson, D.; Figlewicz, D. A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O'Regan, J. P.; Deng, H. X.; et al. , Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- McCord, J. M.; Fridovich, I. , Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969, 244, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D. R.; Lee, M. K.; Slunt, H. S.; Guarnieri, M.; Xu, Z. S.; Wong, P. C.; Brown, R. H., Jr.; Price, D. L.; Sisodia, S. S.; Cleveland, D. W. , Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci U S A 1994, 91, 8292–8296. [Google Scholar] [CrossRef] [PubMed]
- Saccon, R. A.; Bunton-Stasyshyn, R. K.; Fisher, E. M.; Fratta, P. , Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136 Pt 8 Pt 8, 2342–2358. [Google Scholar] [CrossRef]
- Bruijn, L. I.; Houseweart, M. K.; Kato, S.; Anderson, K. L.; Anderson, S. D.; Ohama, E.; Reaume, A. G.; Scott, R. W.; Cleveland, D. W. , Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M. J.; Tibell, L.; Oliveberg, M. , Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc Natl Acad Sci U S A 2002, 99, 16607–16612. [Google Scholar] [CrossRef]
- Nordlund, A.; Leinartaite, L.; Saraboji, K.; Aisenbrey, C.; Gröbner, G.; Zetterström, P.; Danielsson, J.; Logan, D. T.; Oliveberg, M. , Functional features cause misfolding of the ALS-provoking enzyme SOD1. Proc Natl Acad Sci U S A 2009, 106, 9667–9672. [Google Scholar] [CrossRef]
- Trist, B. G.; Hilton, J. B.; Hare, D. J.; Crouch, P. J.; Double, K. L. , Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew Chem Int Ed Engl 2021, 60, 9215–9246. [Google Scholar] [CrossRef]
- Furukawa, Y.; O'Halloran, T. V. , Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J Biol Chem 2005, 280, 17266–17274. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Robertson, J.; Vande Velde, C.; Horne, P.; Ruth, D. M.; Griffin, J.; Cleveland, D. W.; Cashman, N. R.; Chakrabartty, A. , An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med 2007, 13, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, M.; Ezzi, S. A.; Julien, J. P. , Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2007, 104, 2495–2500. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.; Welt, T.; Wirth, F.; Montrasio, F.; Preisig, D.; McAfoose, J.; Vieira, F. G.; Kulic, L.; Späni, C.; Stehle, T.; Perrin, S.; Weber, M.; Hock, C.; Nitsch, R. M.; Grimm, J. , A human-derived antibody targets misfolded SOD1 and ameliorates motor symptoms in mouse models of amyotrophic lateral sclerosis. Sci Transl Med 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Marklund, M.; Bolender, A. L.; Bidhendi, E. E.; Zetterström, P.; Andersen, P. M.; Brännström, T.; Marklund, S. L.; Gilthorpe, J. D.; Nordström, U. , Aggregate-selective antibody attenuates seeded aggregation but not spontaneously evolving disease in SOD1 ALS model mice. Acta Neuropathol Commun 2020, 8, 161. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Homma, K.; Yamaguchi, N.; Kadowaki, H.; Tsuburaya, N.; Naguro, I.; Matsuzawa, A.; Takeda, K.; Takahashi, Y.; Goto, J.; Tsuji, S.; Nishitoh, H.; Ichijo, H. , A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann Neurol 2012, 72, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Yamaguchi, N.; Kadowaki, H.; Tsukamoto, Y.; Tsuburaya, N.; Tsubota, A.; Takahashi, H.; Naguro, I.; Takahashi, Y.; Goto, J.; Tsuji, S.; Nishitoh, H.; Homma, K.; Ichijo, H. , A systematic immunoprecipitation approach reinforces the concept of common conformational alterations in amyotrophic lateral sclerosis-linked SOD1 mutants. Neurobiol Dis 2015, 82, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Homma, K.; Fujisawa, T.; Tsuburaya, N.; Yamaguchi, N.; Kadowaki, H.; Takeda, K.; Nishitoh, H.; Matsuzawa, A.; Naguro, I.; Ichijo, H. , SOD1 as a molecular switch for initiating the homeostatic ER stress response under zinc deficiency. Mol Cell 2013, 52, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. H.; Chung, T. D.; Oldenburg, K. R. , A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Shibata, N.; Hirano, A.; Kobayashi, M.; Siddique, T.; Deng, H. X.; Hung, W. Y.; Kato, T.; Asayama, K. , Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol 1996, 55, 481–490. [Google Scholar] [CrossRef]
- Kerman, A.; Liu, H. N.; Croul, S.; Bilbao, J.; Rogaeva, E.; Zinman, L.; Robertson, J.; Chakrabartty, A. , Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol 2010, 119, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.; Anzai, I.; Nomura, T.; Toichi, K.; Watanabe, M.; Ohara, S.; Watanabe, S.; Yamanaka, K.; Morisaki, Y.; Misawa, H.; Furukawa, Y. , Immunochemical characterization on pathological oligomers of mutant Cu/Zn-superoxide dismutase in amyotrophic lateral sclerosis. Mol Neurodegener 2017, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Trist, B. G.; Fifita, J. A.; Hogan, A.; Grima, N.; Smith, B.; Troakes, C.; Vance, C.; Shaw, C.; Al-Sarraj, S.; Blair, I. P.; Double, K. L. , Co-deposition of SOD1, TDP-43 and p62 proteinopathies in ALS: evidence for multifaceted pathways underlying neurodegeneration. Acta Neuropathol Commun 2022, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.; Nomura, T.; Ohara, S.; Watanabe, S.; Yamanaka, K.; Morisaki, Y.; Misawa, H.; Furukawa, Y. , A copper-deficient form of mutant Cu/Zn-superoxide dismutase as an early pathological species in amyotrophic lateral sclerosis. Biochim Biophys Acta Mol Basis Dis 2018, 1864 6 Pt A, 2119–2130. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kaneko, K.; Yamanaka, K.; Nukina, N. , Mutation-dependent polymorphism of Cu,Zn-superoxide dismutase aggregates in the familial form of amyotrophic lateral sclerosis. J Biol Chem 2010, 285, 22221–22231. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Q.; Ma, Y.; Yuan, H. Y.; Zhao, K.; Zhang, M. Y.; Wang, Q.; Huang, X.; Xu, W. C.; Dai, B.; Chen, J.; Li, D.; Zhang, D.; Wang, Z.; Zou, L.; Yin, P.; Liu, C.; Liang, Y. , Cryo-EM structure of an amyloid fibril formed by full-length human SOD1 reveals its conformational conversion. Nat Commun 2022, 13, 3491. [Google Scholar] [CrossRef] [PubMed]
- Hayward, L. J.; Rodriguez, J. A.; Kim, J. W.; Tiwari, A.; Goto, J. J.; Cabelli, D. E.; Valentine, J. S.; Brown, R. H., Jr. , Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem 2002, 277, 15923–15931. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P. A.; Graffmo, K. S.; Andersen, P. M.; Brännström, T.; Lindberg, M.; Oliveberg, M.; Marklund, S. L. , Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 2006, 129 Pt 2 Pt 2, 451–464. [Google Scholar] [CrossRef]
- Tokuda, E.; Okawa, E.; Watanabe, S.; Ono, S.; Marklund, S. L. , Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol Dis 2013, 54, 308–319. [Google Scholar] [CrossRef]
- Pickles, S.; Semmler, S.; Broom, H. R.; Destroismaisons, L.; Legroux, L.; Arbour, N.; Meiering, E.; Cashman, N. R.; Vande Velde, C. , ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol Commun 2016, 4, 43. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Boca, M.; Calderone, V.; Cantini, F.; Girotto, S.; Vieru, M. , Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc Natl Acad Sci U S A 2009, 106, 6980–6985. [Google Scholar] [CrossRef] [PubMed]
- McAllum, E. J.; Roberts, B. R.; Hickey, J. L.; Dang, T. N.; Grubman, A.; Donnelly, P. S.; Liddell, J. R.; White, A. R.; Crouch, P. J. , Zn II(atsm) is protective in amyotrophic lateral sclerosis model mice via a copper delivery mechanism. Neurobiol Dis 2015, 81, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.; Okawa, E.; Watanabe, S.; Ono, S. , Overexpression of metallothionein-I, a copper-regulating protein, attenuates intracellular copper dyshomeostasis and extends lifespan in a mouse model of amyotrophic lateral sclerosis caused by mutant superoxide dismutase-1. Hum Mol Genet 2014, 23, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.; Watanabe, S.; Okawa, E.; Ono, S. , Regulation of Intracellular Copper by Induction of Endogenous Metallothioneins Improves the Disease Course in a Mouse Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Trist, B. G.; Genoud, S.; Roudeau, S.; Rookyard, A.; Abdeen, A.; Cottam, V.; Hare, D. J.; White, M.; Altvater, J.; Fifita, J. A.; Hogan, A.; Grima, N.; Blair, I. P.; Kysenius, K.; Crouch, P. J.; Carmona, A.; Rufin, Y.; Claverol, S.; Van Malderen, S.; Falkenberg, G.; Paterson, D. J.; Smith, B.; Troakes, C.; Vance, C.; Shaw, C. E.; Al-Sarraj, S.; Cordwell, S.; Halliday, G.; Ortega, R.; Double, K. L. , Altered SOD1 maturation and post-translational modification in amyotrophic lateral sclerosis spinal cord. Brain 2022, 145, 3108–3130. [Google Scholar] [CrossRef] [PubMed]
- Poulin-Brière, A.; Rezaei, E.; Pozzi, S. , Antibody-Based Therapeutic Interventions for Amyotrophic Lateral Sclerosis: A Systematic Literature Review. Front Neurosci 2021, 15, 790114. [Google Scholar] [CrossRef]
- Toichi, K.; Yamanaka, K.; Furukawa, Y. , Disulfide scrambling describes the oligomer formation of superoxide dismutase (SOD1) proteins in the familial form of amyotrophic lateral sclerosis. J Biol Chem 2013, 288, 4970–4980. [Google Scholar] [CrossRef] [PubMed]
- Takashima, C.; Kosuge, Y.; Inoue, M.; Ono, S. I.; Tokuda, E. , A Metal-Free, Disulfide Oxidized Form of Superoxide Dismutase 1 as a Primary Misfolded Species with Prion-Like Properties in the Extracellular Environments Surrounding Motor Neuron-Like Cells. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Tokuda, E.; Takei, Y. I.; Ohara, S.; Fujiwara, N.; Hozumi, I.; Furukawa, Y. , Wild-type Cu/Zn-superoxide dismutase is misfolded in cerebrospinal fluid of sporadic amyotrophic lateral sclerosis. Mol Neurodegener 2019, 14, 42. [Google Scholar] [CrossRef]
- Gurney, M. E.; Pu, H.; Chiu, A. Y.; Dal Canto, M. C.; Polchow, C. Y.; Alexander, D. D.; Caliendo, J.; Hentati, A.; Kwon, Y. W.; Deng, H. X.; et al. , Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
Figure 1.
MS785-MS27 antibody cocktail recognizes Zn-deficient WT SOD1 species. (A) A representative image showing Instant Blue Coomassie staining with human wild-type SOD1 (WT) and human SOD1 that lacks the Zn binding site by the replacement of amino acids, H63A/H71A/H80G/D83A (DZn binding site). Note that SOD1 that lacks the Zn binding site showed faster mobility than WT SOD1. For analysis of SOD1 with the disulfide bond, the protein at 10 mM was treated with 40 mM iodoacetamide at 37ºC for 1 h and was subjected to SDS-PAGE under non-reducing conditions. SH = disulfide bond-cleaved SOD1; S-S = disulfide bond-formed SOD1. (B) Specificity and reproducibility of indirect ELISA coupled with MS785-MS27 antibody cocktail. Apo-WT SOD1SH at 5 mM (black) or vehicle control (Tris-buffered saline with EDTA, gray) was coated onto the 96-well plates. n = 48 per group. (C) Competitive ELISA for MS785-MS27 antibody cocktail. The antibody cocktail at 1 mg/mL was preincubated with apo-WT SOD1SH at a concentration range from 10−4 to 101 mM. The complex of the antibody cocktail and apo-WT SOD1SH was used as a primary antibody in indirect ELISA. n = 6 per concentration. (D) Indirect ELISA with the antibody cocktail for WT SOD1SH with different incorporated metal ions. n = 6 per group. (E) A plot of the ELISA signals of apo-WT SOD1SH or apo-SOD1SH that lacks the Zn binding site treated with 1 to 4 molar equivalents of Zn ions. n = 4 per group. The significance of differences was analyzed using one-way ANOVA followed by Tukey–Kramer’s post hoc test. **P<0.01 (vs. Zn-untreated apo-WT SOD1SH). N.S. = not significant. (F) Indirect ELISA with the antibody cocktail for WT SOD1S-S with different incorporated metal ions. n = 6 per group. All data are given as the mean ± SD.
Figure 1.
MS785-MS27 antibody cocktail recognizes Zn-deficient WT SOD1 species. (A) A representative image showing Instant Blue Coomassie staining with human wild-type SOD1 (WT) and human SOD1 that lacks the Zn binding site by the replacement of amino acids, H63A/H71A/H80G/D83A (DZn binding site). Note that SOD1 that lacks the Zn binding site showed faster mobility than WT SOD1. For analysis of SOD1 with the disulfide bond, the protein at 10 mM was treated with 40 mM iodoacetamide at 37ºC for 1 h and was subjected to SDS-PAGE under non-reducing conditions. SH = disulfide bond-cleaved SOD1; S-S = disulfide bond-formed SOD1. (B) Specificity and reproducibility of indirect ELISA coupled with MS785-MS27 antibody cocktail. Apo-WT SOD1SH at 5 mM (black) or vehicle control (Tris-buffered saline with EDTA, gray) was coated onto the 96-well plates. n = 48 per group. (C) Competitive ELISA for MS785-MS27 antibody cocktail. The antibody cocktail at 1 mg/mL was preincubated with apo-WT SOD1SH at a concentration range from 10−4 to 101 mM. The complex of the antibody cocktail and apo-WT SOD1SH was used as a primary antibody in indirect ELISA. n = 6 per concentration. (D) Indirect ELISA with the antibody cocktail for WT SOD1SH with different incorporated metal ions. n = 6 per group. (E) A plot of the ELISA signals of apo-WT SOD1SH or apo-SOD1SH that lacks the Zn binding site treated with 1 to 4 molar equivalents of Zn ions. n = 4 per group. The significance of differences was analyzed using one-way ANOVA followed by Tukey–Kramer’s post hoc test. **P<0.01 (vs. Zn-untreated apo-WT SOD1SH). N.S. = not significant. (F) Indirect ELISA with the antibody cocktail for WT SOD1S-S with different incorporated metal ions. n = 6 per group. All data are given as the mean ± SD.
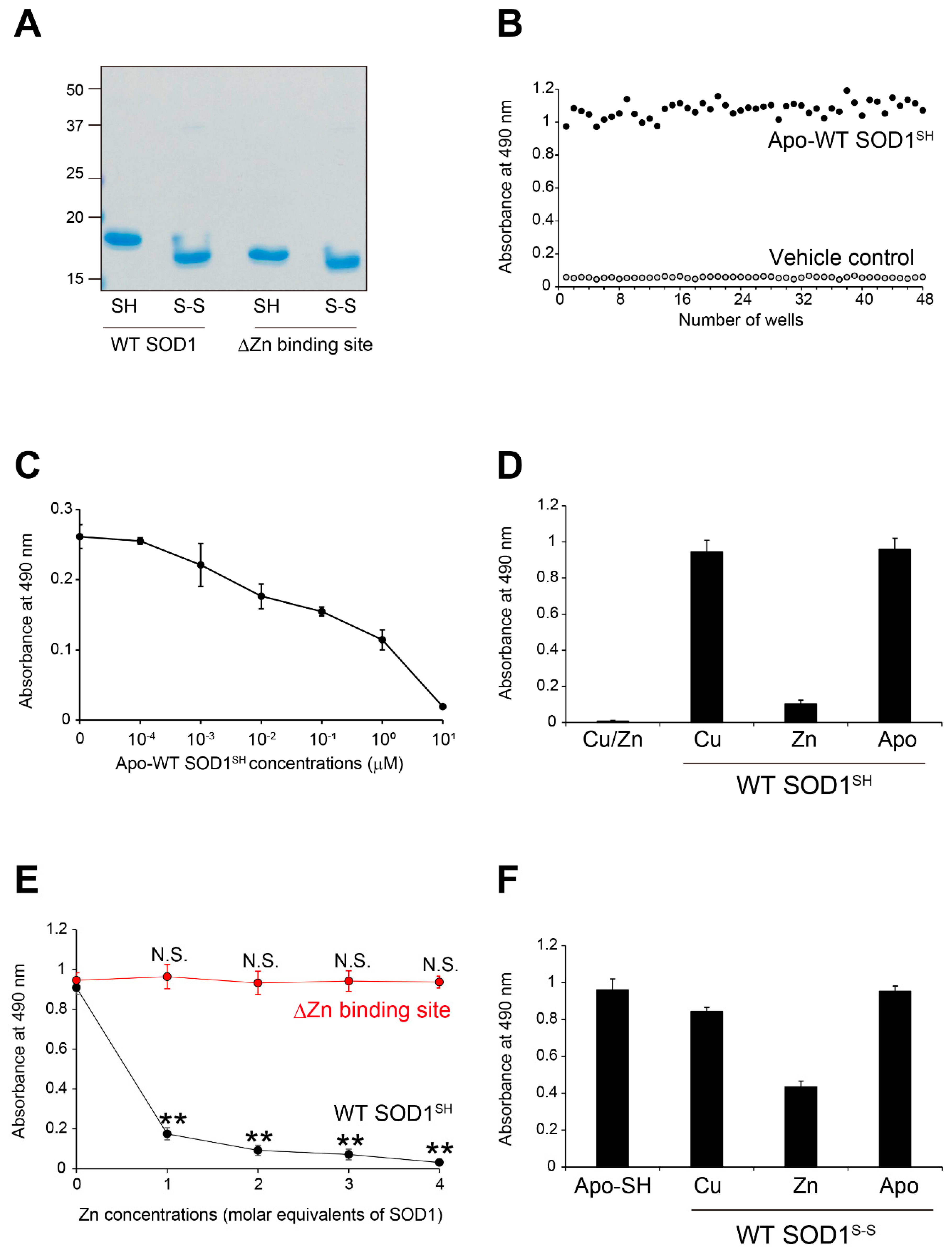
Figure 2.
Recognition by MS785-MS27 antibody cocktail of conformation-disordered WT SOD1 species. Conformation-disordered WT SOD1 species, including unfolded, oligomeric, and aggregated forms, were prepared from apo-WT SOD1S-S as a precursor. (A, B) Indirect ELISA using the conformation-disordered WT SOD1 species analyzed with (A) MS785-MS27 antibody cocktail and (B) anti-pan SOD1 antibody that reacts with various SOD1 conformers. Apo-WT SOD1SH was used as an internal control for the comparisons among different 96-well plates. All data are given as the mean ± SD (n = 6 per group).
Figure 2.
Recognition by MS785-MS27 antibody cocktail of conformation-disordered WT SOD1 species. Conformation-disordered WT SOD1 species, including unfolded, oligomeric, and aggregated forms, were prepared from apo-WT SOD1S-S as a precursor. (A, B) Indirect ELISA using the conformation-disordered WT SOD1 species analyzed with (A) MS785-MS27 antibody cocktail and (B) anti-pan SOD1 antibody that reacts with various SOD1 conformers. Apo-WT SOD1SH was used as an internal control for the comparisons among different 96-well plates. All data are given as the mean ± SD (n = 6 per group).
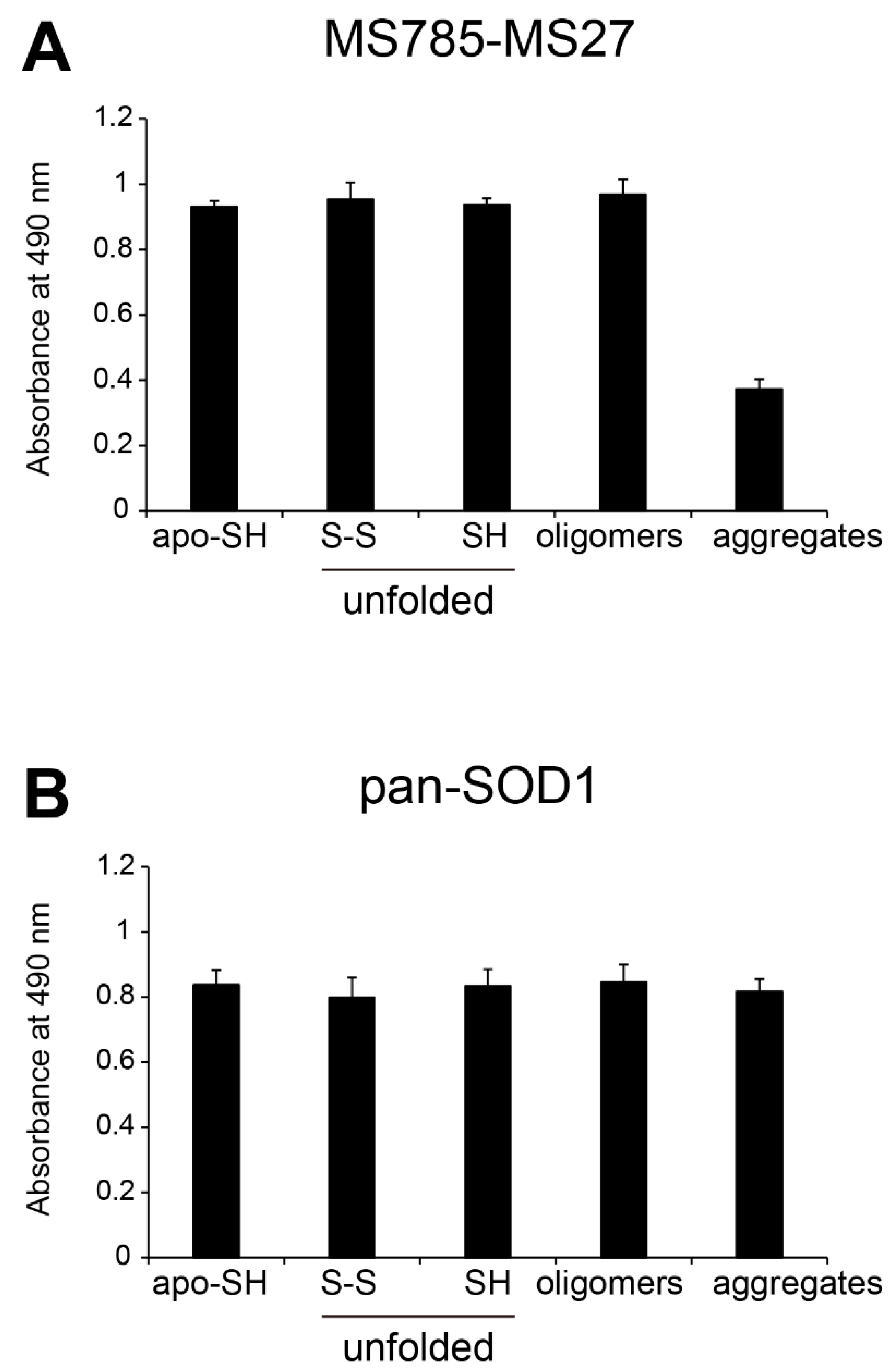
Figure 3.
Recognition by MS785-MS27 antibody cocktail of ALS-linked SOD1 mutants with distinct biophysical properties. (A) The location of the ALS-linked SOD1 mutations, including A4V, G37R, H46R, G85R, D90A, G93A, and I113T. They are categorized into three groups: (i) wild-type-like mutants (blue; G37R, D90A, and G93A), (ii) dimer interface mutants (green; A4V and I113T), and (iii) metal binding region mutants (red; H46R and G85R). Each mutation is highlighted in the X-ray crystal structure of human WT SOD1 (PDB: 2C9V). Cu and Zn ions are represented by cyan and pink colors, respectively. (B) A representative image showing Instant Blue Coomassie staining with WT SOD1 and the ALS-linked SOD1 mutants. For the analysis of SOD1 with a disulfide bond, the protein at 10 mM was treated with 40 mM iodoacetamide at 37ºC for 1 h and subjected to SDS-PAGE under non-reducing conditions. SH = disulfide bond-cleaved SOD1; S-S = disulfide bond-formed SOD1. (C–G) Indirect ELISA of the recognition by MS785-MS27 antibody cocktail of (C) apo-SOD1SH, (D) apo-SOD1S-S, (E) unfolded SOD1SH, (F) SOD1 oligomers, and (G) SOD1 aggregates with the ALS-linked mutantions. Apo-WT SOD1SH (black, WT-SH) was used as an internal control. Data are given as the mean ± SD (n = 6 per mutant). (H) A plot of the ELISA signals of apo-SOD1SH with the ALS-linked mutations treated with 1 to 4 molar equivalents of Zn ions. Data are given as the mean ± SD (n = 3 per mutant). The significance of differences was analyzed using one-way ANOVA followed by Tukey–Kramer’s post hoc test. *P<0.05, **P<0.01 (vs. the same Zn-untreated mutant). N.S. = not significant.
Figure 3.
Recognition by MS785-MS27 antibody cocktail of ALS-linked SOD1 mutants with distinct biophysical properties. (A) The location of the ALS-linked SOD1 mutations, including A4V, G37R, H46R, G85R, D90A, G93A, and I113T. They are categorized into three groups: (i) wild-type-like mutants (blue; G37R, D90A, and G93A), (ii) dimer interface mutants (green; A4V and I113T), and (iii) metal binding region mutants (red; H46R and G85R). Each mutation is highlighted in the X-ray crystal structure of human WT SOD1 (PDB: 2C9V). Cu and Zn ions are represented by cyan and pink colors, respectively. (B) A representative image showing Instant Blue Coomassie staining with WT SOD1 and the ALS-linked SOD1 mutants. For the analysis of SOD1 with a disulfide bond, the protein at 10 mM was treated with 40 mM iodoacetamide at 37ºC for 1 h and subjected to SDS-PAGE under non-reducing conditions. SH = disulfide bond-cleaved SOD1; S-S = disulfide bond-formed SOD1. (C–G) Indirect ELISA of the recognition by MS785-MS27 antibody cocktail of (C) apo-SOD1SH, (D) apo-SOD1S-S, (E) unfolded SOD1SH, (F) SOD1 oligomers, and (G) SOD1 aggregates with the ALS-linked mutantions. Apo-WT SOD1SH (black, WT-SH) was used as an internal control. Data are given as the mean ± SD (n = 6 per mutant). (H) A plot of the ELISA signals of apo-SOD1SH with the ALS-linked mutations treated with 1 to 4 molar equivalents of Zn ions. Data are given as the mean ± SD (n = 3 per mutant). The significance of differences was analyzed using one-way ANOVA followed by Tukey–Kramer’s post hoc test. *P<0.05, **P<0.01 (vs. the same Zn-untreated mutant). N.S. = not significant.
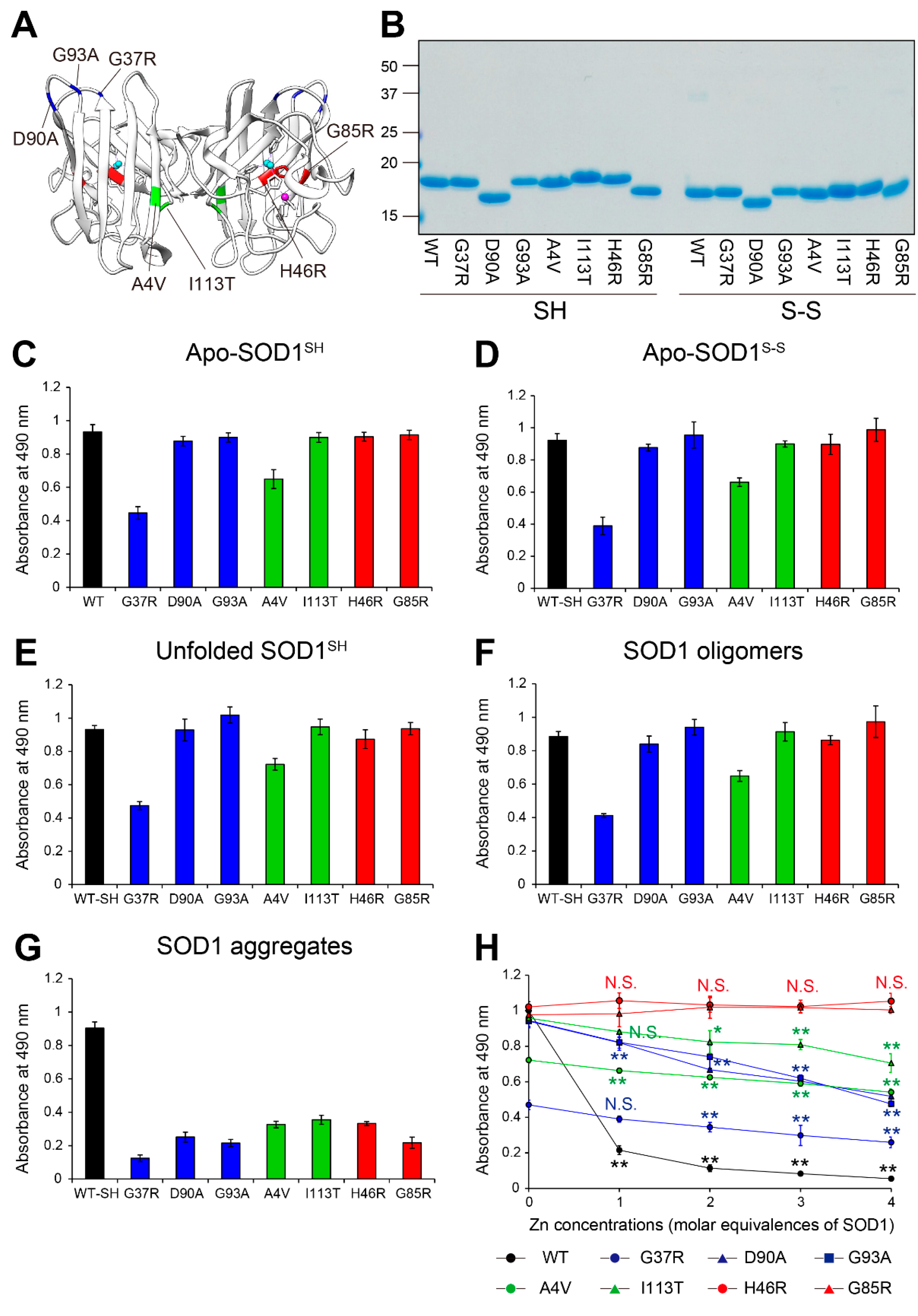
Figure 4.
Toxicological features of MS785-MS27-reactive SOD1 species against NSC-34 cells. (A, B) NSC-34, a cell model of motor neurons, was treated with apo-WT SOD1SH (black) or apo-SOD1SH that lacks the Zn binding site (red) at the indicated concentrations (0.01 mM to 10 mM) for 48 h. HEPES buffer was used as a vehicle control. (A) Cell proliferation and (B) cytotoxicity were assessed by CCK-8 and LDH assays, respectively. The right panels in A and B represent enlargements of the dotted area in the left panels. The significance of differences was analyzed using one-way ANOVA followed by Tukey–Kramer’s post hoc test. *P<0.05, **P<0.01 vs. HEPES-treated NSC-34. N.S. = not significant. (C, D) Apo-WT SOD1SH (black) or apo-SOD1SH that lacks the Zn binding site at 1 mM (red) was treated with 1 to 4 molar equivalents of Zn ions. NSC-34 cells were exposed to the Zn-pretreated SOD1 species. NSC-34 cells were also exposed to ZnSO4 resolved in HEPES buffer. (C) Cell proliferation and (B) cytotoxicity were assessed by CCK-8 and LDH assays, respectively. The significance of differences was analyzed using one-way ANOVA followed by Tukey–Kramer’s post hoc test. **P<0.01 vs. HEPES-treated NSC-34. N.S. = not significant. All data are given as the mean ± SD (n = 6 per group).
Figure 4.
Toxicological features of MS785-MS27-reactive SOD1 species against NSC-34 cells. (A, B) NSC-34, a cell model of motor neurons, was treated with apo-WT SOD1SH (black) or apo-SOD1SH that lacks the Zn binding site (red) at the indicated concentrations (0.01 mM to 10 mM) for 48 h. HEPES buffer was used as a vehicle control. (A) Cell proliferation and (B) cytotoxicity were assessed by CCK-8 and LDH assays, respectively. The right panels in A and B represent enlargements of the dotted area in the left panels. The significance of differences was analyzed using one-way ANOVA followed by Tukey–Kramer’s post hoc test. *P<0.05, **P<0.01 vs. HEPES-treated NSC-34. N.S. = not significant. (C, D) Apo-WT SOD1SH (black) or apo-SOD1SH that lacks the Zn binding site at 1 mM (red) was treated with 1 to 4 molar equivalents of Zn ions. NSC-34 cells were exposed to the Zn-pretreated SOD1 species. NSC-34 cells were also exposed to ZnSO4 resolved in HEPES buffer. (C) Cell proliferation and (B) cytotoxicity were assessed by CCK-8 and LDH assays, respectively. The significance of differences was analyzed using one-way ANOVA followed by Tukey–Kramer’s post hoc test. **P<0.01 vs. HEPES-treated NSC-34. N.S. = not significant. All data are given as the mean ± SD (n = 6 per group).
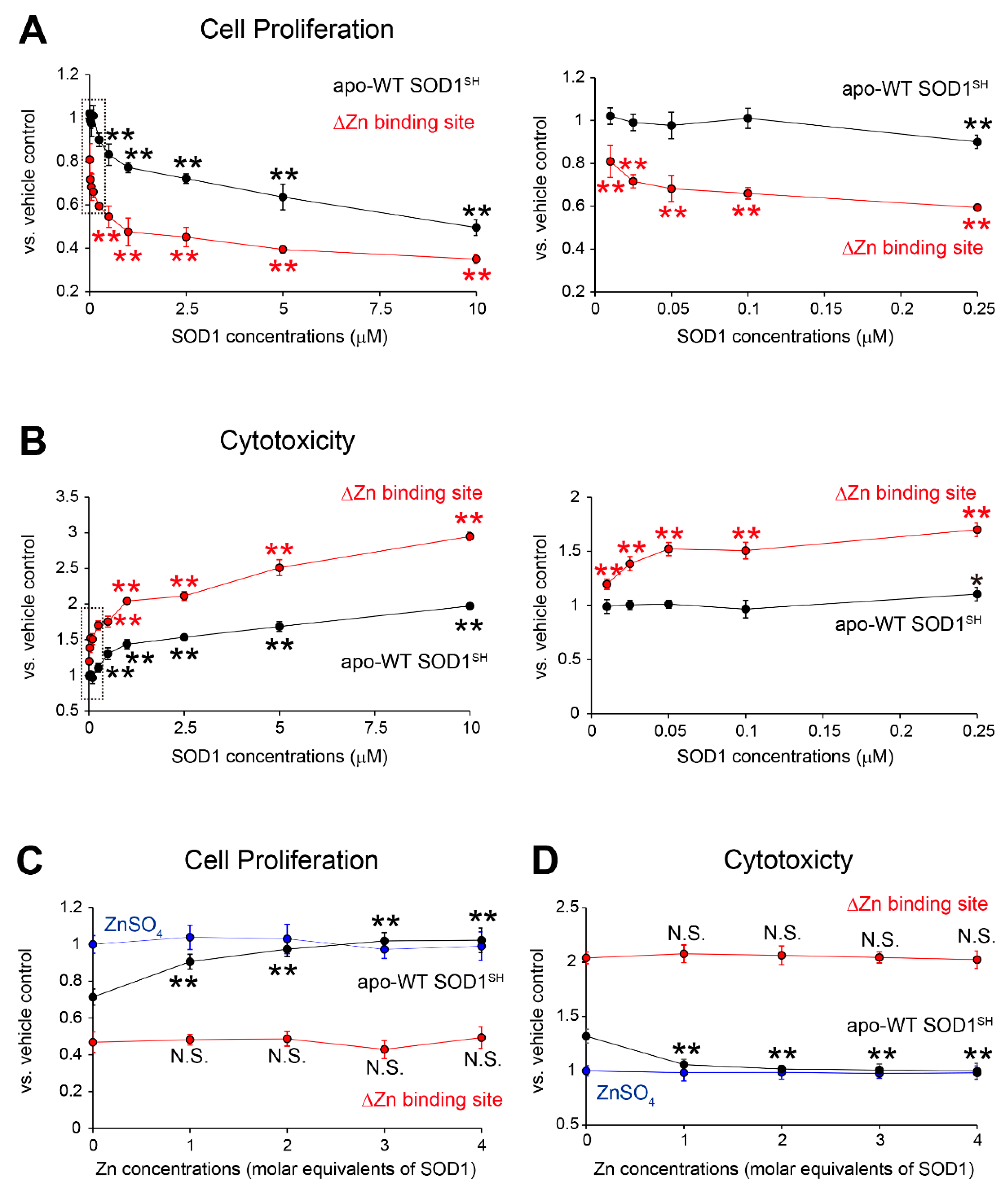
Figure 5.
MS785-MS27-reactive SOD1 species are distributed in motor neurons throughout the disease course of G93A SOD1 mice. (A–E) Confocal imaging of the lumbar spinal cord sections dually immunostained with MS785-MS27 (blue) and NeuN (green), a marker of neurons. G93A SOD1 mice at different disease stages, namely, (A) presymptomatic (60 days), (B) symptomatic (90 days), and (C) terminal (120 days) stages, were used. (D) Mice expressing human WT SOD1 and (E) non-transgenic (Non-Tg) mice at 120 days of age were also analyzed. n = 3 per genotype. Insets represent enlargements of the dotted area. (F–H) Confocal imaging of the lumbar spinal cord from G93A SOD1 mice at (F) presymptomatic, (G) symptomatic, and (H) terminal stages. The sections were immunostained with the MS785-MS27 cocktail (blue) and GFAP (green), a marker of astrocytes. (I–K) Confocal imaging of the lumbar spinal cord sections immunostained with the MS785-MS27 cocktail (blue) and Iba1 (red), a marker of microglia. G93A SOD1 mice at different disease stages, namely, (I) presymptomatic, (J) symptomatic, and (K) terminal stages, were used. n = 3 per disease stage. Scale bars = 50 mm.
Figure 5.
MS785-MS27-reactive SOD1 species are distributed in motor neurons throughout the disease course of G93A SOD1 mice. (A–E) Confocal imaging of the lumbar spinal cord sections dually immunostained with MS785-MS27 (blue) and NeuN (green), a marker of neurons. G93A SOD1 mice at different disease stages, namely, (A) presymptomatic (60 days), (B) symptomatic (90 days), and (C) terminal (120 days) stages, were used. (D) Mice expressing human WT SOD1 and (E) non-transgenic (Non-Tg) mice at 120 days of age were also analyzed. n = 3 per genotype. Insets represent enlargements of the dotted area. (F–H) Confocal imaging of the lumbar spinal cord from G93A SOD1 mice at (F) presymptomatic, (G) symptomatic, and (H) terminal stages. The sections were immunostained with the MS785-MS27 cocktail (blue) and GFAP (green), a marker of astrocytes. (I–K) Confocal imaging of the lumbar spinal cord sections immunostained with the MS785-MS27 cocktail (blue) and Iba1 (red), a marker of microglia. G93A SOD1 mice at different disease stages, namely, (I) presymptomatic, (J) symptomatic, and (K) terminal stages, were used. n = 3 per disease stage. Scale bars = 50 mm.
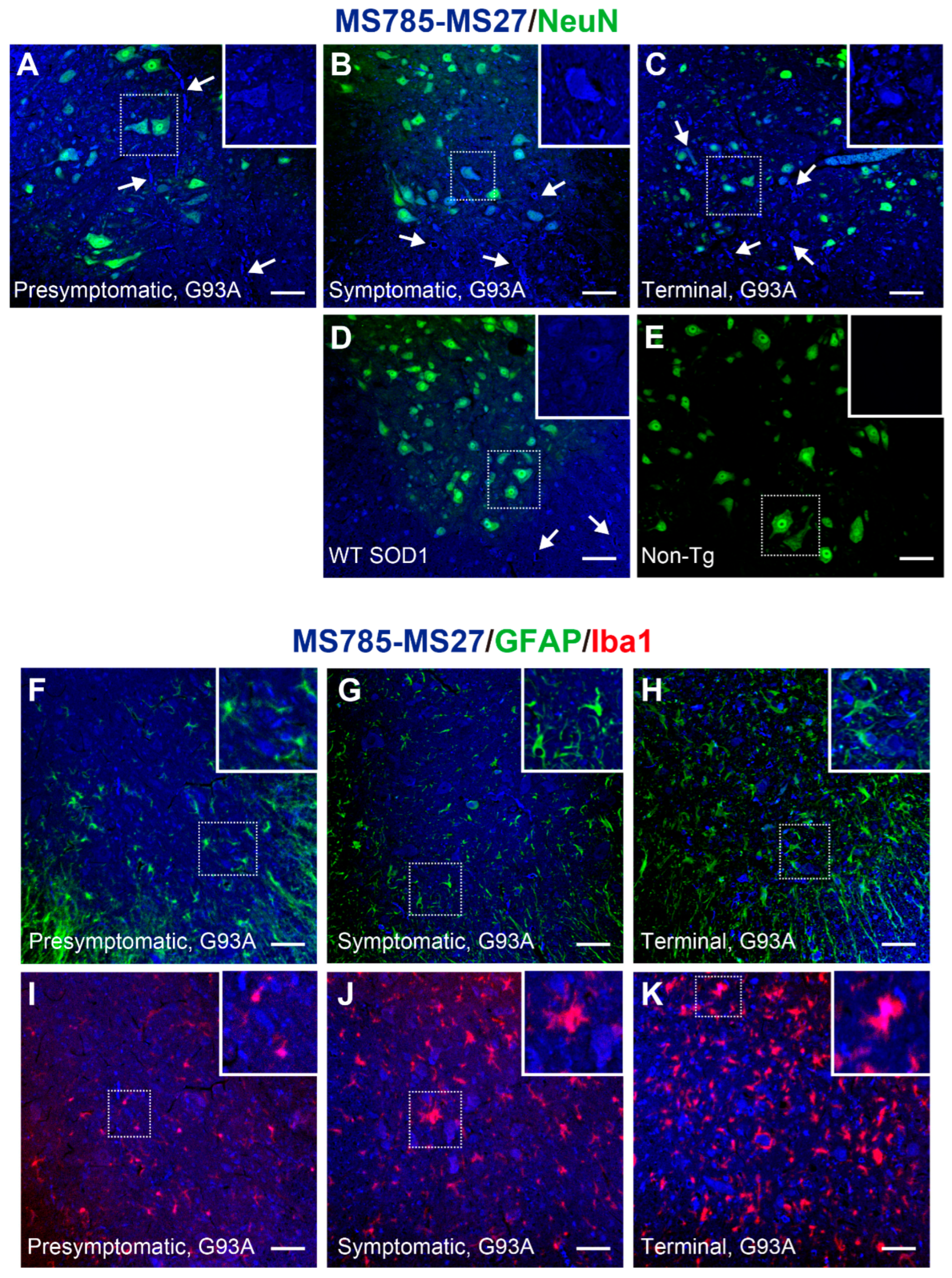
Figure 6.
Conformational heterogeneity of misfolded SOD1 species in G93A SOD1 mice revealed by co-immunostaining with MS785-MS27 cocktail and EDI (A–C) Confocal imaging of lumbar spinal cord sections from G93A SOD1 mice at (A) presymptomatic (60 days), (B) symptomatic (90 days), and (C) terminal (120 days) stages. The sections were dually immunostained with the MS785-MS27 cocktail (blue) and EDI (red), an antibody specific to misfolded SOD1 species for which the epitope is exposed at the dimer interface. n = 3 per disease stage. Scale bars = 50 mm.
Figure 6.
Conformational heterogeneity of misfolded SOD1 species in G93A SOD1 mice revealed by co-immunostaining with MS785-MS27 cocktail and EDI (A–C) Confocal imaging of lumbar spinal cord sections from G93A SOD1 mice at (A) presymptomatic (60 days), (B) symptomatic (90 days), and (C) terminal (120 days) stages. The sections were dually immunostained with the MS785-MS27 cocktail (blue) and EDI (red), an antibody specific to misfolded SOD1 species for which the epitope is exposed at the dimer interface. n = 3 per disease stage. Scale bars = 50 mm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



