
Submitted:
05 April 2024
Posted:
08 April 2024
You are already at the latest version
Abstract
Mast cells are tissue-resident immune cells distributed in all tissues and strategically located close to blood and lymphatic vessels and nerves. Thanks to the expression of a wide array of receptors, mast cells act as tissue sentinels, able to detect the presence of bacteria and parasites and to respond to different environmental stimuli.
Mast cells originate from bone marrow progenitors that enter the circulation and mature in peripheral organs under the influence of microenvironment factors, thus differentiating in heterogeneous tissue-specific subsets. Even though mast cell activation has been traditionally linked to IgE-mediated allergic reactions, a role for these cells in other pathological conditions including tumor progression has recently emerged. However, several aspects of mast cell biology remain to be clarified.
The advent of single-cell RNA sequencing platforms has provided the opportunity to understand mast cell origin and differentiation as well as the phenotype and functions within different tissues, including the gut.
This review recapitulates how single cell transcriptomic studies provided insight into mast cell development as well as into the functional role of intestinal MC subsets in health and disease.
Keywords:
Intestinal mast cells
; gut inflammation
; colorectal cancer
1. Introduction
Mast cells (MCs) arise from bone marrow (BM) progenitors that enter the circulation and mature in peripheral tissues under the influence of microenvironment factors [1,2,3].
Mature MCs are tissue-resident innate immune cells that are present in all organs, particularly in skin, in lung and intestinal mucosa and are distributed close to blood and lymphatic vessels and nerves. Thanks to their strategical localization and to the expression of a wide array of receptors, mature MCs act as tissue sentinels, able to firstly detect the presence of bacteria and parasites and to respond to different microenvironmental stimuli [4,5,6,7].
Their functions are mediated by the secretion of a vast array of biologically active molecules, including histamine and proteases that are stored in secretory granules and immediately released upon activation [8,9]. A plethora of newly synthesized lipid inflammatory mediators are secreted within hours [9]. Moreover, by releasing various cytokines and chemokines, MCs orchestrate the recruitment and activation of immune cells to the site of infection and regulate innate and adaptive immunity [10].
Among the main surface receptors, mature MCs are characterized by the expression of c-Kit (CD117) that upon interaction with its ligand (stem cell factor, SCF) regulates MC migration and activation [11], and the high affinity receptor for immunoglobulin E (FcεRI) that orchestrates the IgE-mediated allergic reactions [12]. Indeed, cross-linking of FcεRI-bound IgE by multivalent antigens results in the release of granule-stored mediators such as histamine, accompanied by the generation of newly synthetized soluble mediators [12,13,14] and high quantities of extracellular vesicles, emerging as important players in intercellular communication [15,16].
Similarly, activation by anaphylatoxins or neuropeptides including substance P, results in degranulation of preformed mediators and de-novo synthesis of chemokines/cytokines [17,18].
However, MCs also express a wide range of receptors that are pivotal in the host's defense against pathogens, such as Toll-like receptors [7,19]. More recently, a selective expression of human mas-related G protein-coupled receptor X2 (MRGPRX2) and its mouse homologue, Mrgprb2, have been also reported [20]. This receptor can promote IgE-independent pseudo-allergic reactions by binding an array of host and microbial peptides, often generated from proteolytic cleavage of inactive precursors solely in inflamed tissue [20].
Thus, MC activation has been linked not only to allergy but also to other inflammatory conditions within different tissues, including the gut where a cross-talk between MCs and nerves can also provide a neuroimmune network necessary to control local responses [21,22].
Of note, the presence of MCs has also been reported in several solid cancers accompanied by MC ability to shape tumor microenvironment [23,24]. However, MCs can both orchestrate anti-tumoral responses, promoting the recruitment of other immune cells, or tumor progression favoring angiogenesis, lymphoangiogenesis, fibrosis and metastasis [23,24,25].
More recently, several aspects of MC biology have been solved thanks to the development of single-cell transcriptomic profiling technologies [26,27,28], as depicted in Figure 1.
This novel approach was able to differentiate MCs from other immune cells, including basophils and eosinophils, and to reveal a unique mouse and human MC identity [26,27,28]. Moreover, the presence of distinct MC subsets into different connective tissues has been elucidated [29,30], revealing a high degree of MC heterogeneity [31,32].
However, MC phenotype and functions between and within different organs remain to be clarified. Moreover, how MC plasticity is shaped in different physiological and pathological conditions is largely unexplored.
This review recapitulates data obtained from recent single-cell-based studies mainly focusing on intestinal MC subsets and their roles in health and disease.
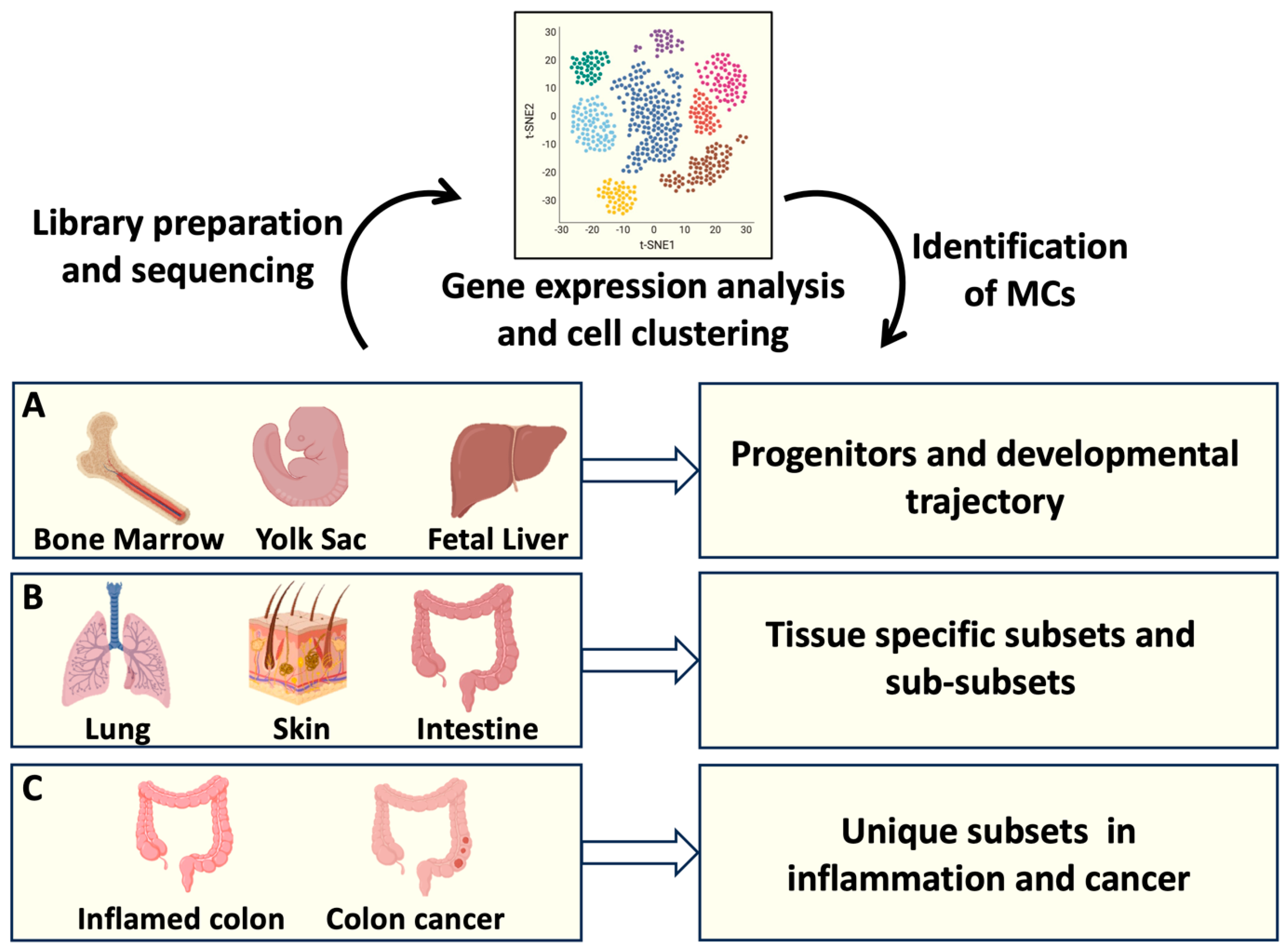
Figure 1.
MC origin and tissue heterogeneity analysed by Single cell RNAseq. scRNAseq offers the possibility to identify the transcriptomic profiles of several cells from a tissue of interest. Transcripts associated to individual cells are sequenced and analysed, resulting in cell clustering based on gene expression. A: Identification of MC progenitors in bone marrow, fetal liver and Yolk sac has defined MC developmental trajectories; B: Gene expression profiles of MCs resident in different organs has clarified MC tissue heterogeneity; C: Transcript analysis of intestinal MCs has provided information on MC phenotypical and functional plasticity in health and disease. Created using BioRender.com.
Figure 1.
MC origin and tissue heterogeneity analysed by Single cell RNAseq. scRNAseq offers the possibility to identify the transcriptomic profiles of several cells from a tissue of interest. Transcripts associated to individual cells are sequenced and analysed, resulting in cell clustering based on gene expression. A: Identification of MC progenitors in bone marrow, fetal liver and Yolk sac has defined MC developmental trajectories; B: Gene expression profiles of MCs resident in different organs has clarified MC tissue heterogeneity; C: Transcript analysis of intestinal MCs has provided information on MC phenotypical and functional plasticity in health and disease. Created using BioRender.com.
2. Transcriptomic Analysis and MC Development
A first transcriptomic study on MC differentiation by Saito and coauthors has been performed using progenitors derived from human umbilical cord blood and adult peripheral blood and has revealed a series of MC-specific genes, including TPSAB1/2 (tryptase α1 and β1), HDC (L-histidine decarboxylase), and CPA3 (carboxypeptidase A) [33].
More recently, a single-cell transcriptomic analysis revealed the identity of human MC progenitors showing a temporal association between the appearance of FcɛRI and the MC signature in hematopoietic progenitors isolated from adult peripheral blood [34].
Regarding the existence of a common progenitor between basophils and MCs, a first study integrating flow cytometric and transcriptomic data has been performed on primary BM-derived hematopoietic stem cells showing the presence of a cluster of cells expressing a set of common signature genes between basophils, eosinophils and MCs [35]. Similarly, a single-cell RNA sequencing of progenitors from human cord blood identified an intermediate stage progenitor that co-expresses gene modules of basophil, eosinophil and MC lineages [36].
Notably, erythro-myeloid progenitors were found also in yolk sac, suggesting that, as happens in mice, human MC arise from multiple compartments during and after embryogenesis [37].
More recently, by analysing single-cell dataset of human BM, Hamey and coauthors have provided a road map of MC and basophil development supporting the existence of a common progenitor until a bifurcation into the two specific cell lines [38].
However, a transcriptome analysis of mature human skin MCs in relation to other mature cell lineages, demonstrated a unique MC transcriptional landscape delineating a limited relation between MCs and basophils [39]. Moreover, the low mRNA levels in mature human basophils make it difficult to characterize the basophils’ transcriptional profile in-depth.
Thus, although human basophils and MCs express common marker genes (e.g., HDC and FcεRI encoding genes), further studies are needed to explore in-depth the transcriptional differences between them in order to better discriminate their developmental trajectories.
In regard to similarities between distinct lineages, recent human and murine studies have suggested the existence of a hematopoietic progenitor with MC-erythrocyte potential [36,37,40,41]. However, the contributions of these progenitors to the resolution of infection-induced inflammation remain only poorly defined [42], as further discussed in paragraph 4.
3. Insights into Intestinal MC Origin and Phenotype through scRNA-Seq
A tissue compartment in which MCs are particularly abundant is the gut. Intestinal MCs are involved in the maintenance of tissue homeostasis and at the same time act as sentinels of the host defense against different pathogens, orchestrating inflammation [21,43].
In the mouse, the small intestine represents a large reservoir of MC-committed progenitors (MCps) that are recruited by a mechanism that involves α4β7 integrin and the CXC chemokine receptor-2 (CXCR2) [44]. As in all organs, critical signals for homing and recruitment of MCps are also provided by SCF binding to c-Kit [3]. Thanks to the role of c-Kit in MC maturation, migration and proliferation [11], murine models with spontaneous mutations in White spotting locus coding for c-Kit, have been used to identify and understand the contribution of MCs in several biological processes [45].
The study performed by Hamey and coauthors [38], offers valuable insights into the intricate process of MC differentiation, shedding light on the nuances of gene regulation during maturation. Focusing on peritoneal MC, they observed that MC differentiation/maturation is characterized by the downregulation of β7 integrin, as well as the protease genes Mcpt8 and Gzmb (Granzyme B). Of note, they also reported the upregulation of MC specific protease genes including Cpa3, Cma1, Mcpt4, Tpsb2 and Tpsab1 also revealing that their induction occurs in distinct temporal stages (Cpa3 first, followed by Tpsb2, and finally Tpsab1) [38].
Mature MCs in the intestine are heterogeneous and comprise two main subsets that differ for localization and protease content [46]. In rodents, MCs are divided in mucosal MCs (MMC) present into the intestinal lamina propria close to the epithelium and positive for Mctp1 and Mcpt2 proteases, and connective-tissue MCs (CTMCs) that reside in gut submucosa and are characterized by the expression of proteases Mcpt4-7 and carboxypeptidase A3 (Cpa3) as well as a higher amount of histamine and heparin compared to MMCs [47].
In humans, mucosal MCs present in lamina propria contain only tryptase in their granules (MCT), while MCs that predominate in the intestinal submucosa contain tryptase, chymase, the metallopeptidase CPA3, and cathepsin G (MCTC) [48,49]. MCs that exclusively express chymase have also been identified as a rare population that resides in both lamina propria and submucosa [50]. Similarly, an intraepithelial MMC subpopulation has also been described in mice [51]. However, the role of these rare MC populations is still unclear.
Recent advancements in RNAseq profiling technologies and fate mapping revealed different developmental origins between the two main MC populations in mice: MMCs originate from fetal hematopoietic stem cells and depend on adult stem cells for their replacement, while CTMCs originate from yolk sac and can self-maintain independently from BM derived stem cells [41]. A similar conclusion came from the study by Gentek and coauthors revealing that CTMC are maintained independently of adult hematopoietic stem cells [52].
Regarding human MCs, transcriptomic profile obtained by scRNAseq analysis revealed that MCs are characterized by different transcriptomic signatures in diverse organs [53]. In gut, MCs express specific transcripts such as Vascular Endothelial Growth Factor A (VEGFA), the cytoskeleton component utrophin (UTRN), the chemokine receptor CXCR4, the aryl hydrocarbon receptor (AHR), and the interleukin 1 receptor associated kinase 3 (IRAK3). However, this signature is not a unique characteristic of human intestinal MCs but is shared by MCs resident in bladder, lymph nodes, skeletal muscle, trachea and tongue [53].
Furthermore, by integrating different datasets from Mouse cell Atlas derived from different tissues, Tauber and co-author demonstrated that CTMCs and MMCs are characterized by diverse gene signature across organs. In gastrointestinal tract MMCs, besides mucosal Mcpt1 and Mcpt2 protease genes, are characterized by a high expression of genes encoding adhesion molecules (Itgae, Itga2a, Ly6e) and the chemokine receptor Cxcr1, while CTMCs are enriched in Cma1, Mcpt4, Tpsb2 and Cpa3 protease genes, Ccl2 chemokine gene, lipid metabolism genes (Apoe) together with the expression of Mgbrb2 gene [53]. These results are in line with previous studies showing that Mrgprb2 and the human ortholog MRGPRX2 are exclusively expressed on connective tissue-like MCs [47,54].
The origin of the two subsets was further explored comparing mice at different ages. CTMCs positive for Mrgprb2 were found in both neonatal pups and adults, while Mrgprb2- Mcpt1+ MMCs were exclusively detected in adult mice, suggesting that Mrgprb2+ CTMCs originate embryonically, whereas Mrgprb2- MMCs after birth. Moreover, the use of BM chimeras confirmed that the Mrgprb2- MMCs are continuously renewed from BM progenitors, while the Mrgprb2+ CTMC population appears to be independent of BM-derived cells for turnover not only in the gut but also in the skin and peritoneal cavity [53]. Of note, CTMCs isolated by different organs showed a high degree of heterogeneity [26], definitively demonstrating a microenvironment-dependent MC differentiation and suggesting that tissue-specific MC subsets exist beyond the traditional MMC/CTMC classification.
4. Deciphering Intestinal MC Function in Homeostasis and Inflammatory Conditions
Intestinal MCs contribute to homeostasis controlling physiological processes such as mucosal integrity and epithelial barrier activity [43,55]. Indeed, mice deficient in MCs or Mcpt4 protease have a reduced small intestinal permeability and altered epithelial cell migration as well as intestinal morphology and tight junctions [55].
The crucial role of MCs in epithelial integrity is confirmed by their involvement in intestinal inflammatory conditions but also in food allergy and nematode infections (Figure 2).
During parasite infections, including Trichinella spiralis and Trichuris muris, MMCs are the main subset that increases in number due to a shift from a connective tissue-like phenotype to a mucosal phenotype characterized by the expression of the proteases Mcpt1 and Mcpt2.
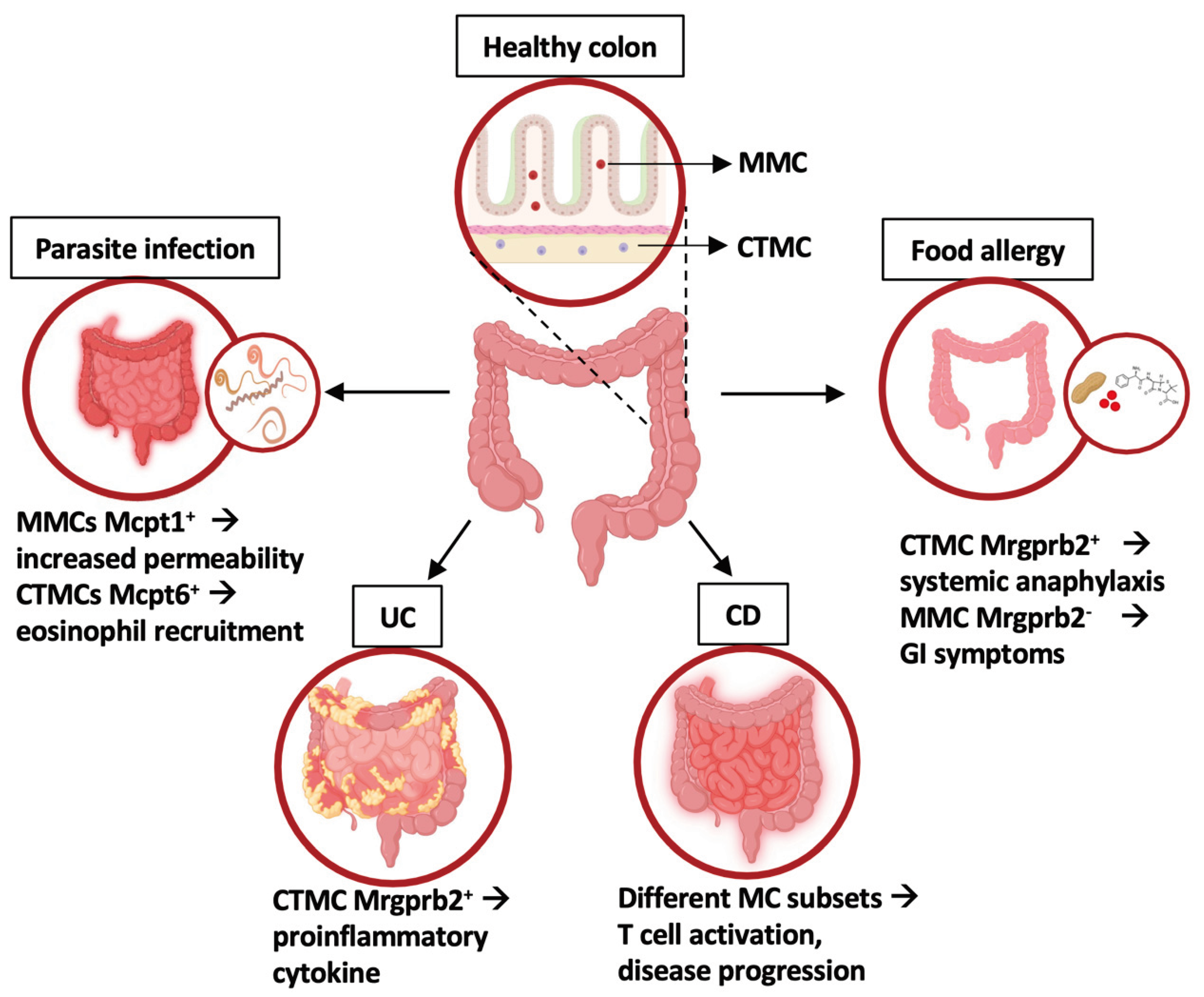
Figure 2.
Intestinal mast cell phenotype and functions in homeostasis and inflammatory conditions. In healthy colon, two MC subsets have been identified: mucosal MC (MMC) and connective tissue-like MC (CTMC). In disease states, distinct MC subsets with unique gene expression profiles contribute to the intestinal inflammation, as highlighted by different transcriptomic approaches. UC: Ulcerative Colitis; CD: celiac disease. Created using BioRender.com.
Figure 2.
Intestinal mast cell phenotype and functions in homeostasis and inflammatory conditions. In healthy colon, two MC subsets have been identified: mucosal MC (MMC) and connective tissue-like MC (CTMC). In disease states, distinct MC subsets with unique gene expression profiles contribute to the intestinal inflammation, as highlighted by different transcriptomic approaches. UC: Ulcerative Colitis; CD: celiac disease. Created using BioRender.com.
In particular, Mcpt1 appeared to be responsible for the degradation of occludin, thus increasing intestinal permeability and facilitating worm expulsion [56,57,58]. On the other hand, the connective tissue MC-specific tryptase Mcpt6 was shown to be required for eosinophil recruitment and the eradication of T. spiralis [59].
Notably, by single cell RNA-sequencing Inclan-Rico and coauthors demonstrated that infection by T. spiralis induces the recruitment into the intestine of a hematopoietic progenitor with dual MC-erythrocyte potential [42], likely contributing to eradicate the infection and to alleviate blood loss.
Besides parasite infections, intestinal MCs are involved in IgE-mediated response to food antigens contributing to both local and systemic development of food allergy.
An increase in MC number, mainly due to the expansion of intestinal MMCs, has been demonstrated both in humans and mice sensitized by food allergens [60,61], and correlates with the severity of symptoms. However, using two common models of IgE-mediated food allergy, Benedé and Berin demonstrated that systemic anaphylaxis was uniquely associated with activation of connective tissue-like MCs, while gastrointestinal manifestations of food allergy were associated with an increase of Mcpt1-expressing MCs together with a clear activation of both mucosal and connective tissue-like MCs [62]. More recently, Tauber and co-authors confirmed these findings demonstrating that depletion of Mrgprb2+ CTMC subset protects murine models from anaphylactic shock, while Mrgprb2- MMCs in gut is not implicated in anaphylaxis, despite being the first population to encounter the allergen [53].
MC ability to rapidly sense and adapt to specific triggers including neuropeptides can explain the activated MC phenotype described in different human gastrointestinal disorders such as celiac disease, irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD) [22]. IBD are complex multifactorial diseases of the gastrointestinal tract, including Ulcerative Colitis (UC), triggered by environmental factors in genetically susceptible individuals [22,63]. Current therapies based on the use of monoclonal antibodies directed against cytokines offer amelioration and prolonged period of remission but have important limitations. Indeed, more than 30% of patients do not initially respond to therapy while others lose response over time [64,65]. Thus, new treatment strategies are needed.
Several studies have reported MC accumulation in patients affected by celiac disease (CD) and UC, but their contribution in disease progression remained unclear.
In this context, Atlasy and co-authors compared transcriptomic profile of immune infiltrate isolated from small intestine of patients affected by active CD. They found four MC subsets differentially enriched in healthy and affected intestine: MCs more abundant in control patients show a profile associated with ‘humoral immune response’ and ‘positive regulation of B cell activation’ biological processes, while MC clusters accumulated in active disease display a transcriptomic profile associated to ‘protein to ER process’, ‘antigen processing and presentation’ and ‘positive regulation of T cell-mediated cytotoxicity’ processes [66], suggesting their active role in disease progression.
Similarly, Smillie and coauthors focused on colonic tissues from UC patients and healthy donors and using single-cell RNA sequencing mapped different cell circuits. They identified 51 cell subsets (epithelial, stromal, and immune cells) revealing an increase of inflammatory-associated genes in UC patients compared with healthy volunteers. Of note, together with cytotoxic and regulatory T cells a selective MC subset expressing the activation marker CD69 increase in inflamed tissues [67]. However, this MC subset was not further characterized in term of protease content.
More recently, Chen and coauthors compared acutely inflamed and uninflamed UC tissue to establish the requirement of MRGPRX2-specific MC activation in inflamed colonic tissues [68]. Using both bulk RNASeq and scRNASeq, they reported a key role for adrenomedullin ADM (ADM) and its proteolytic product, PAMP-12, in perpetuating UC inflammation. Moreover, by single-cell RNA sequencing they were also able to show that both activated fibroblasts and epithelial cells express adrenomedullin and that interferon γ is a key upstream regulator of MC gene expression [68], thus defining a new potential therapeutic target.
5. Exploring Intestinal Mast Cell Role in Tumor Biology: Colon Cancer under RNA Seq Microscope
MC physiologic function in tumor biology has raised particular interest for decades since these cells potentially influence different aspects of tumorigenesis including tumor angiogenesis, invasiveness and immunosuppression [69,70,71]. However, the MC contributions in cancer initiation and progression remain controversial. Indeed, several studies have demonstrated both positive and negative correlation of MCs in the development of different types of cancers [72], including colorectal cancer (CRC).
CRC is the third most common type of malignancy that affects the colon or rectum [73]. The majority of CRC cases emerge sporadically, 20% present a familial history and 5% are attributed to inherited syndromes such as Familial Adenomatous Polyposis and Lynch syndrome [74,75]. Moreover, lifestyle as well as chronic inflammation in patients with IBD all represent independent risk factors for CRC development [76].
The tumor microenvironment (TME) and the interactions between cancer cells and surrounding stromal cells, also influences CRC development [77].
The density of tumor-infiltrating cytotoxic and memory T cells, which are associated with a better prognosis [78,79], define the “immunoscore” as an additional parameter to classify CRC. However, the knowledge about innate immune cell infiltration, including MCs, is limited [80,81,82]. Recent advancements in sequencing investigations have provided crucial opportunities to dissect the heterogeneity and functional role of MCs within the CRC microenvironment and adjacent normal tissue.
By examining the transcriptomic profile among wild-type (WT) mice, MC-deficient mice KitW-sh), and KitW-sh mice engrafted with MCs derived from WT mice, Ko and coauthors identified several genes downregulated in MC-deficient mice but recovered by MC engraftment that were named “mast cell–dependent genes” [83].
These genes were found associated to pathways related to cancer progression including immunosuppression, apoptosis and angiogenesis. Interestingly, these pathways are enriched in lung, breast, and colon cancer compared to normal tissues, supporting a pro-angiogenic and anti-apoptotic role for MCs in tumor microenvironment. Moreover, gene associated to lymphocyte cytotoxicity are upregulated in the absence of MCs, suggesting that these cells promote immunosuppression [83]. These results support in vitro and in vivo evidence demonstrating a role for tumor-infiltrating MCs in favoring a suppressive microenvironment and in promoting tumor growth [84,85,86,87,88,89]. In particular, a role for SCF in favoring tumor progression has been envisaged: SCF-activated MCs can release adenosine to directly suppress T cells and NK cells and can contribute to the production of pro-inflammatory cytokines [88,89].
By a RNAseq approach, Sakita and co-authors showed that MC role in colon cancer development and progression is multifaceted and context-dependent [88]. Indeed, in a model of spontaneous CRC, MC deficiency promoted tumor development while in colitis-dependent CRC the absence of MCs reduced tumor burden and increased the frequency of tumor-infiltrating CD8+ T cells [88]. Bulk RNAseq analysis of colitis-dependent tumor masses showed that MC deficiency upregulated the cytokine-cytokine receptor interaction pathway further supporting a role for MCs in suppressing immune responses during tumorigenesis [88].
Thus, a characterization of murine MC infiltrating CRC and their role in tumor progression is still unavailable. Moreover, whether different MC subsets may play an anti-tumorigenic or pro-tumorigenic role in different stages of the disease is still unknown.
Using a murine colitis-dependent model of tumorigenesis we have recently demonstrated that tumor masses are enriched of CTMCs with an activated phenotype [89]. However, a single cell RNAseq approach is necessary to better define tumor-infiltrating MC subsets in different murine models of CRC.
Regarding human CRC, several groups have profiled immune and non-immune cells isolated from tumoral lesions [90,91,92,93,94,95,96,97,98,99,100,101,102]. MCs were identified based on a unique set of genes including those coding for the receptor c-Kit (KIT), the chymase (CMA1), the carboxipeptidase (CPA3), and tryptase (TPSAB1, TPSB2), and compose one of the most represented cell types present in the TME [90,91,92,93,94,95,96,97,98,99,100,101,102].
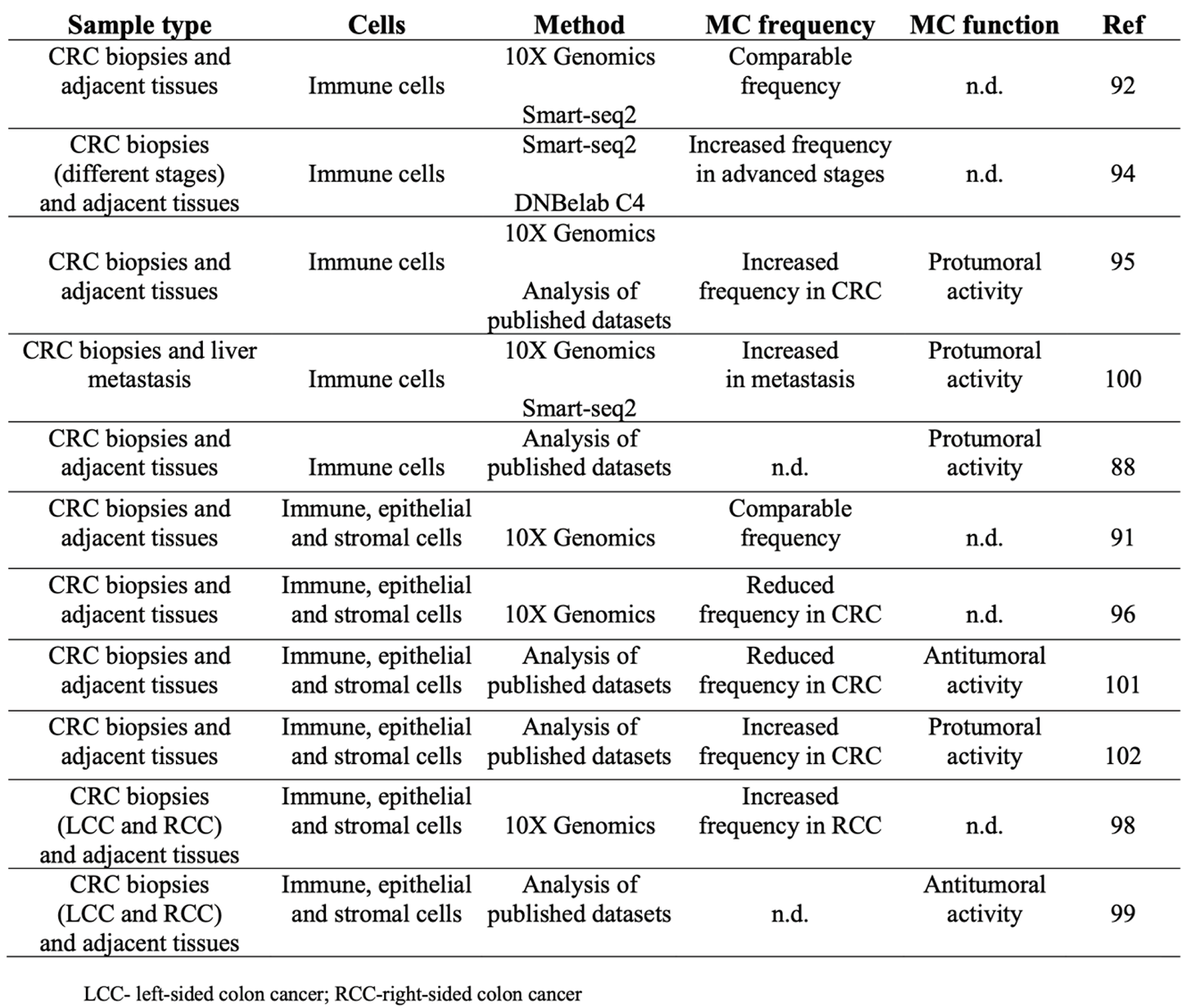
However, there is still inconsistency regarding the frequency of MCs in transformed and not transformed tissue and their pro/anti-tumor activity (Table 1).
Regarding MC frequency, by employing single cell RNAseq approach two different research groups demonstrated a comparable MC enrichment in both tumors and normal mucosa [91,92], while single cell analysis of tumor infiltrating immune cells from different kind of cancers demonstrated accumulation of MCs in several tumors, including CRC, compared to non-tumoral adjacent tissue [95]. Furthermore, a higher number of MCs was reported in advanced CRC stages [94] and in right-sided tumors compared to the left part of the colon [98].
On the other hand, a reduced number of MCs in tumor lesions in respect to matched unaffected tissue was observed by transcriptomic profiling of five non metastatic CRC patients [96] and in the bioinformatic analysis of three single cell RNAseq datasets of both tumor and tumor-free tissue [101].
Few data are currently available also regarding the role of MCs in human CRC (Table 1).
Indeed, the majority of single-cell RNAseq analysis have been mainly focused on tumor microenvironment (epithelial and stromal cells) or on macrophages and adaptive immune infiltrate.
Sakita and co-workers found a negative correlation in CRC between the number of activated MCs and infiltrating CD8+ T lymphocytes, supporting a pro-tumoral role for MCs [88].
Cheng and coauthors performed a meta-analysis by combined previously published and newly generated sc-RNA-seq data set to compare transcriptomic signature associated to MCs infiltrating different cancer types [95].
Focusing on CRC patients, they found a down-modulation of TNFA transcript and an upregulation of VEGFA in respect to adjacent healthy tissue and suggest association of this signature with decreased patient survival. However, whether this signature is associated to a selective MC subset (mucosal vs connective) is not clear.

Table 1.
MC characterization by scRNAseq analysis of human CRC samples.
 |
The integrated analysis of different CRC datasets revealed with more details the potential function of tumor-infiltrating MCs [101].
Based on the expression level of several markers, MCs were clustered into distinct subpopulations and their relative abundance was compared in tumor versus adjacent tissue.
In healthy control tissue, the most representing subset expresses high levels of the protease CMA1 and upregulates the angiogenesis pathway. In TME, activated MCs express abundant amounts of KIT and FcεRI subunits (FCER1A, FCER1G, MS4A2), high levels of genes for Th2 cytokines, and transcripts for proteases and enzymes involved in histamine and lipid mediator synthesis [101]. However, these results are in apparent contrast with the MC signature reported by Cheng and coauthors [95].
All these discrepant results may depend on the different isolation procedures (whole tissue vs immune infiltrate) or by different protocols used to prepare the sequencing library. Moreover, special considerations should be taken into account when linking genomic data, for instance the heterogeneity of patients in terms of tumor stages. To this regard, a clear CRC stratification of patients in different tumor stages could help to understand whether during the onset of intestinal transformation and in later stages of CRC progression different MC subsets are involved. It could be also possible that in the transcriptomic analysis of whole tissue the expression of epithelial and stromal genes affects the relative abundance of MCs and their related genes.
Considering the very low frequency of MCs in the whole CRC immune infiltrate, single cell RNAseq analysis of sorted MCs could help to better discriminate different clusters and subclusters associated to CRC development and progression.
Regarding a potential interplay between MCs and other cells in TME, Wang and coauthors conducted cell-cell communication analysis mapping the expression of ligand-receptor pairs.
Their finding highlighted a possible MC interaction with B cells, epithelial cells and fibroblasts [102]. Of note, MC co-localization with fibroblasts and endothelial cells was also reported in the stromal region of CRC tissue by spatial transcriptomic analysis [101].
Thus, in future studies the exact localization of MCs within tumor tissue and their interaction with different cell types in CRC could be clarified by integrating single cell with spatial transcriptomic analysis.
6. Conclusions and Future Perspectives
MCs are innate immune cells distributed in all tissue and particularly abundant in the intestine where they play different roles in homeostasis as well as in inflammatory diseases. Moreover, the increase of MCs in different tumors including colonic tumors has been demonstrated in the last years. MCs are characterized by a vast heterogeneity among tissues and their phenotypical and functional plasticity allow them to respond to different environmental stimuli. However, whether distinct MC subsets are involved in intestinal diseases and their functions are poorly understood.
The advent of single-cell RNA sequencing platforms has provided a step forward in the understanding of many biological processes and in the definition of cell functions. Several aspects of MC origin and differentiation into peripheral tissues have been elucidated.
Even though MCs represent an abundant population in healthy intestines, their number appeared increased during inflammation. It could be interesting to clarify whether and how the recruitment of new progenitors contributes to expansion of MCs during inflammation. Moreover, the role of classical MMC and CTMC subsets in different inflammatory states including allergy to food antigens, parasite infections or autoinflammatory diseases is still poorly investigated. It is also largely unknown whether MC populations with unique phenotype and functions arise during inflammation.
In regard to MC role during colonic transformation, it is still unknown how tumor microenvironment shapes MC plasticity in term of phenotype and function and whether unique MC subset(s) differentiate in diverse stages of progression.
Finally, MCs are located near nerves and the bidirectional interaction of MCs with the enteric nervous system plays an important role in gastrointestinal inflammation. It could be interesting to investigate whether these interactions are also involved in tumor progression.
Spatial transcriptomic combined with single cell RNA-seq could help to decipher MC cross-talk with nervous system as well as additional MC interactions in the tumor microenvironment and construct an immune landscape for CRC.
A better characterization of intestinal MCs at various stages of gut inflammation and tumorigenesis would help to define novel potential targets for a therapeutic intervention.
Author Contributions
All authors contributed to write the manuscript and prepared the figures. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by grants of the Italian Association for Cancer Research (AIRC IG-24955) and Istituto Pasteur Italia-Fondazione Cenci Bolognetti (2020-366).
Acknowledgments
We apologize to all our colleagues whose important work could not be cited directly. Most of these references can be found in the review articles cited in the manuscript.
Conflicts of Interest
The authors declare no commercial or financial conflicts of interest.
References
- Chen CC, Grimbaldeston MA, Tsai M, Weissman IL, Galli SJ. Identification of mast cell progenitors in adult mice. Proc Natl Acad Sci U S A. 2005 Aug 9;102(32):11408-13. [CrossRef]
- Gurish MF, Boyce JA. Mast cells: ontogeny, homing, and recruitment of a unique innate effector cell. J Allergy Clin Immunol. 2006 Jun;117(6):1285-91. [CrossRef]
- Tsai M, Valent P, Galli SJ. KIT as a master regulator of the mast cell lineage. J Allergy Clin Immunol. 2022 Jun;149(6):1845-1854. [CrossRef]
- Chan CY, St John AL, Abraham SN. Plasticity in mast cell responses during bacterial infections. Curr Opin Microbiol. 2012 Feb;15(1):78-84. [CrossRef]
- Voehringer D. Protective and pathological roles of mast cells and basophils. Nat Rev Immunol. 2013 May;13(5):362-75. [CrossRef]
- Reber LL, Sibilano R, Mukai K, Galli SJ. Potential effector and immunoregulatory functions of mast cells in mucosal immunity. Mucosal Immunol. 2015 May;8(3):444-63. [CrossRef]
- Jiménez M, Cervantes-García D, Córdova-Dávalos LE, Pérez-Rodríguez MJ, Gonzalez-Espinosa C, Salinas E. Responses of Mast Cells to Pathogens: Beneficial and Detrimental Roles. Front Immunol. 2021 Jun 15;12:685865. [CrossRef]
- Pejler G, Rönnberg E, Waern I, Wernersson S. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood. 2010 Jun 17;115(24):4981-90. [CrossRef]
- Elieh Ali Komi D, Wöhrl S, Bielory L. Mast Cell Biology at Molecular Level: a Comprehensive Review. Clin Rev Allergy Immunol. 2020 Jun;58(3):342-365. [CrossRef]
- Mukai K, Tsai M, Saito H, Galli SJ. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev. 2018 Mar;282(1):121-150. [CrossRef]
- Rönnstrand L. Signal transduction via the stem cell factor receptor/c-Kit. Cell Mol Life Sci. 2004 Oct;61(19-20):2535-48. [CrossRef]
- Siraganian RP. Mast cell signal transduction from the high-affinity IgE receptor. Curr Opin Immunol. 2003 Dec;15(6):639-46. [CrossRef]
- Kraft S, Kinet JP. New developments in FcepsilonRI regulation, function and inhibition. Nat Rev Immunol. 2007 May;7(5):365-78. [CrossRef]
- Nagata Y, Suzuki R. FcεRI: A Master Regulator of Mast Cell Functions. Cells. 2022 Feb 11;11(4):622. [CrossRef]
- Molfetta R, Lecce M, Quatrini L, Caracciolo G, Digiacomo L, Masuelli L, Milito ND, Vulpis E, Zingoni A, Galandrini R, Santoni A, Paolini R. Immune complexes exposed on mast cell-derived nanovesicles amplify allergic inflammation. Allergy. 2020 May;75(5):1260-1263. [CrossRef]
- Shefler I, Salamon P, Mekori YA. Extracellular Vesicles as Emerging Players in Intercellular Communication: Relevance in Mast Cell-Mediated Pathophysiology. Int J Mol Sci. 2021 Aug 25;22(17):9176. [CrossRef]
- Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology (2008) 123:398–410. [CrossRef]
- West PW, Bahri R, Garcia-Rodriguez KM, Sweetland G, Wileman G, Shah R, et al.. Interleukin-33 amplifies human mast cell activities induced by complement anaphylatoxins. Front Immunol (2021) 11:615236. [CrossRef]
- Agier J, Pastwińska J, Brzezińska-Błaszczyk E. An overview of mast cell pattern recognition receptors. Inflamm Res. 2018 Sep;67(9):737-746. [CrossRef]
- Subramanian H, Gupta K, Ali H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J Allergy Clin Immunol. 2016 Sep;138(3):700-710. [CrossRef]
- Buhner S, Schemann M. Mast cell-nerve axis with a focus on the human gut. Biochim Biophys Acta. 2012 Jan;1822(1):85-92. [CrossRef]
- Bischoff SC. Mast cells in gastrointestinal disorders. Eur J Pharmacol. 2016 May 5;778:139-45. [CrossRef]
- Varricchi G, Galdiero MR, Loffredo S, Marone G, Iannone R, Marone G, Granata F. Are Mast Cells MASTers in Cancer? Front Immunol. 2017 Apr 12;8:424. [CrossRef]
- Rigoni A, Colombo MP, Pucillo C. Mast cells, basophils and eosinophils: From allergy to cancer. Semin Immunol. 2018 Feb;35:29-34. [CrossRef]
- Komi DEA, Redegeld FA. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin Rev Allergy Immunol. 2020 Jun;58(3):313-325. [CrossRef]
- Dwyer DF, Barrett NA, Austen KF; Immunological Genome Project Consortium. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol. 2016 Jul;17(7):878-87. [CrossRef]
- Dahlin JS, Malinovschi A, Öhrvik H, Sandelin M, Janson C, Alving K, Hallgren J. Lin- CD34hi CD117int/hi FcεRI+ cells in human blood constitute a rare population of mast cell progenitors. Blood. 2016 Jan 28;127(4):383-91. [CrossRef]
- Huang H, Li Y, Liu B. Transcriptional regulation of mast cell and basophil lineage commitment. Semin Immunopathol. 2016 Sep;38(5):539-48. [CrossRef]
- Varricchi G, Raap U, Rivellese F, Marone G, Gibbs BF. Human mast cells and basophils-How are they similar how are they different? Immunol Rev. 2018 Mar;282(1):8-34. [CrossRef]
- Iuliano C, Absmaier-Kijak M, Sinnberg T, Hoffard N, Hils M, Köberle M, Wölbing F, Shumilina E, Heise N, Fehrenbacher B, Schaller M, Lang F, Kaesler S, Biedermann T. Fetal Tissue-Derived Mast Cells (MC) as Experimental Surrogate for In Vivo Connective Tissue MC. Cells. 2022 Mar 8;11(6):928. [CrossRef]
- Cildir G, Yip KH, Pant H, Tergaonkar V, Lopez AF, Tumes DJ. Understanding mast cell heterogeneity at single cell resolution. Trends Immunol. 2021 Jun;42(6):523-535. [CrossRef]
- Derakhshan T, Boyce JA, Dwyer DF. Defining mast cell differentiation and heterogeneity through single-cell transcriptomics analysis. J Allergy Clin Immunol. 2022 Oct;150(4):739-747. [CrossRef]
- Saito H, Nakajima T, Matsumoto K. Human mast cell transcriptome project. Int Arch Allergy Immunol. 2001 May;125(1):1-8. [CrossRef] [PubMed]
- Wu C, Boey D, Bril O, Grootens J, Vijayabaskar MS, Sorini C, Ekoff M, Wilson NK, Ungerstedt JS, Nilsson G, Dahlin JS. Single-cell transcriptomics reveals the identity and regulators of human mast cell progenitors. Blood Adv. 2022 Aug 9;6(15):4439-4449. [CrossRef]
- Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, Hirche C, Lutz C, Buss EC, Nowak D, Boch T, Hofmann WK, Ho AD, Huber W, Trumpp A, Essers MA, Steinmetz LM. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. 2017 Apr;19(4):271-281. [CrossRef]
- Zheng S, Papalexi E, Butler A, Stephenson W, Satija R. Molecular transitions in early progenitors during human cord blood hematopoiesis. Mol Syst Biol. 2018 Mar 15;14(3):e8041. [CrossRef]
- Popescu DM, Botting RA, Stephenson E, Green K, Webb S, Jardine L, Calderbank EF, Polanski K, Goh I, Efremova M, Acres M, Maunder D, Vegh P, Gitton Y, Park JE, Vento-Tormo R, Miao Z, Dixon D, Rowell R, McDonald D, Fletcher J, Poyner E, Reynolds G, Mather M, Moldovan C, Mamanova L, Greig F, Young MD, Meyer KB, Lisgo S, Bacardit J, Fuller A, Millar B, Innes B, Lindsay S, Stubbington MJT, Kowalczyk MS, Li B, Ashenberg O, Tabaka M, Dionne D, Tickle TL, Slyper M, Rozenblatt-Rosen O, Filby A, Carey P, Villani AC, Roy A, Regev A, Chédotal A, Roberts I, Göttgens B, Behjati S, Laurenti E, Teichmann SA, Haniffa M. Decoding human fetal liver haematopoiesis. Nature. 2019 Oct;574(7778):365-371. [CrossRef]
- Hamey FK, Lau WWY, Kucinski I, Wang X, Diamanti E, Wilson NK, Göttgens B, Dahlin JS. Single-cell molecular profiling provides a high-resolution map of basophil and mast cell development. Allergy. 2021 Jun;76(6):1731-1742. [CrossRef]
- Motakis E, Guhl S, Ishizu Y, Itoh M, Kawaji H, de Hoon M, Lassmann T, Carninci P, Hayashizaki Y, Zuberbier T, Forrest AR, Babina M; FANTOM consortium. Redefinition of the human mast cell transcriptome by deep-CAGE sequencing. Blood. 2014 Apr 24;123(17):e58-67. [CrossRef] [PubMed] [PubMed Central]
- Tusi BK, Wolock SL, Weinreb C, Hwang Y, Hidalgo D, Zilionis R, Waisman A, Huh JR, Klein AM, Socolovsky M. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature. 2018 Mar 1;555(7694):54-60. [CrossRef]
- Li Z, Liu S, Xu J, Zhang X, Han D, Liu J, Xia M, Yi L, Shen Q, Xu S, Lu L, Cao X. Adult Connective Tissue-Resident Mast Cells Originate from Late Erythro-Myeloid Progenitors. Immunity. 2018 Oct 16;49(4):640-653.e5. [CrossRef]
- Inclan-Rico JM, Hernandez CM, Henry EK, Federman HG, Sy CB, Ponessa JJ, Lemenze AD, Joseph N, Soteropoulos P, Beaulieu AM, Yap GS, Siracusa MC. Trichinella spiralis-induced mastocytosis and erythropoiesis are simultaneously supported by a bipotent mast cell/erythrocyte precursor cell. PLoS Pathog. 2020 May 18;16(5):e1008579. [CrossRef]
- Kurashima Y, Goto Y, Kiyono H. Mucosal innate immune cells regulate both gut homeostasis and intestinal inflammation. Eur J Immunol. 2013 Dec;43(12):3108-15. [CrossRef]
- Abonia JP, Austen KF, Rollins BJ, Joshi SK, Flavell RA, Kuziel WA, Koni PA, Gurish MF. Constitutive homing of mast cell progenitors to the intestine depends on autologous expression of the chemokine receptor CXCR2. Blood. 2005 Jun 1;105(11):4308-13. [CrossRef]
- Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. 2005 Sep;167(3):835-48. [CrossRef]
- Gurish MF, Austen KF. Developmental origin and functional specialization of mast cell subsets. Immunity. 2012 Jul 27;37(1):25-33. [CrossRef]
- Xing W, Austen KF, Gurish MF, Jones TG. Protease phenotype of constitutive connective tissue and of induced mucosal mast cells in mice is regulated by the tissue. Proc Natl Acad Sci U S A. 2011 Aug 23;108(34):14210-5. [CrossRef]
- Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci U S A. 1986 Jun;83(12):4464-8. [CrossRef]
- da Silva EZ, Jamur MC, Oliver C. Mast cell function: a new vision of an old cell. J Histochem Cytochem. 2014 Oct;62(10):698-738. [CrossRef]
- Weidner N, Austen KF. Heterogeneity of mast cells at multiple body sites. Fluorescent determination of avidin binding and immunofluorescent determination of chymase, tryptase, and carboxypeptidase content. Pathol Res Pract. 1993 Mar;189(2):156-62. [CrossRef]
- Vogel P, Janke L, Gravano DM, Lu M, Sawant DV, Bush D, Shuyu E, Vignali DAA, Pillai A, Rehg JE. Globule Leukocytes and Other Mast Cells in the Mouse Intestine. Vet Pathol. 2018 Jan;55(1):76-97. [CrossRef]
- Gentek R, Ghigo C, Hoeffel G, Bulle MJ, Msallam R, Gautier G, Launay P, Chen J, Ginhoux F, Bajénoff M. Hemogenic Endothelial Fate Mapping Reveals Dual Developmental Origin of Mast Cells. Immunity. 2018 Jun 19;48(6):1160-1171.e5. [CrossRef]
- Tauber M, Basso L, Martin J, Bostan L, Pinto MM, Thierry GR, Houmadi R, Serhan N, Loste A, Blériot C, Kamphuis JBJ, Grujic M, Kjellén L, Pejler G, Paul C, Dong X, Galli SJ, Reber LL, Ginhoux F, Bajenoff M, Gentek R, Gaudenzio N. Landscape of mast cell populations across organs in mice and humans. J Exp Med. 2023 Oct 2;220(10):e20230570. [CrossRef]
- Forsythe P. Mast Cells in Neuroimmune Interactions. Trends Neurosci. 2019 Jan;42(1):43-55. [CrossRef]
- Groschwitz KR, Ahrens R, Osterfeld H, Gurish MF, Han X, Abrink M, Finkelman FD, Pejler G, Hogan SP. Mast cells regulate homeostatic intestinal epithelial migration and barrier function by a chymase/Mcpt4-dependent mechanism. Proc Natl Acad Sci U S A. 2009 Dec 29;106(52):22381-6. [CrossRef]
- Knight PA, Wright SH, Lawrence CE, Paterson YY, Miller HR. Delayed expulsion of the nematode Trichinella spiralis in mice lacking the mucosal mast cell-specific granule chymase, mouse mast cell protease-1. J Exp Med. 2000 Dec 18;192(12):1849-56. [CrossRef]
- McDermott JR, Bartram RE, Knight PA, Miller HR, Garrod DR, Grencis RK. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc Natl Acad Sci U S A. 2003 Jun 24;100(13):7761-6. [CrossRef]
- Sorobetea D, Holm JB, Henningsson H, Kristiansen K, Svensson-Frej M. Acute infection with the intestinal parasite Trichuris muris has long-term consequences on mucosal mast cell homeostasis and epithelial integrity. Eur J Immunol. 2017 Feb;47(2):257-268. [CrossRef]
- Shin K, Watts GF, Oettgen HC, Friend DS, Pemberton AD, Gurish MF, Lee DM. Mouse mast cell tryptase mMCP-6 is a critical link between adaptive and innate immunity in the chronic phase of Trichinella spiralis infection. J Immunol. 2008 Apr 1;180(7):4885-91. [CrossRef]
- Nakano N, Kitaura J. Mucosal Mast Cells as Key Effector Cells in Food Allergies. Cells. 2022 Jan 19;11(3):329. [CrossRef]
- Oettgen HC. Mast cells in food allergy: Inducing immediate reactions and shaping long-term immunity. J Allergy Clin Immunol. 2023 Jan;151(1):21-25. [CrossRef] [PubMed]
- Benedé S, Berin MC. Mast cell heterogeneity underlies different manifestations of food allergy in mice. PLoS One. 2018 Jan 25;13(1):e0190453. [CrossRef]
- Zhang L., Song J., Hou X. Mast cells and irritable bowel syndrome: From the bench to the bedside. J. Neurogastroenterol. Motil. 2016;22:181–192. [CrossRef]
- Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, de Villiers WJ, Present D, Sands BE, Colombel JF. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005 Dec 8;353(23):2462-76. Erratum in: N Engl J Med. 2006 May 18;354(20):2200. [CrossRef]
- Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, Van Assche G, Axler J, Kim HJ, Danese S, Fox I, Milch C, Sankoh S, Wyant T, Xu J, Parikh A; GEMINI 1 Study Group. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013 Aug 22;369(8):699-710. [CrossRef]
- Atlasy N, Bujko A, Bækkevold ES, Brazda P, Janssen-Megens E, Lundin KEA, Jahnsen J, Jahnsen FL, Stunnenberg HG. Single cell transcriptomic analysis of the immune cell compartment in the human small intestine and in Celiac disease. Nat Commun. 2022 Aug 22;13(1):4920. [CrossRef]
- Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, Sud M, Andrews E, Velonias G, Haber AL, Jagadeesh K, Vickovic S, Yao J, Stevens C, Dionne D, Nguyen LT, Villani AC, Hofree M, Creasey EA, Huang H, Rozenblatt-Rosen O, Garber JJ, Khalili H, Desch AN, Daly MJ, Ananthakrishnan AN, Shalek AK, Xavier RJ, Regev A. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell. 2019 Jul 25;178(3):714-730.e22. [CrossRef]
- Chen E, Chuang LS, Giri M, Villaverde N, Hsu NY, Sabic K, Joshowitz S, Gettler K, Nayar S, Chai Z, Alter IL, Chasteau CC, Korie UM, Dzedzik S, Thin TH, Jain A, Moscati A, Bongers G, Duerr RH, Silverberg MS, Brant SR, Rioux JD, Peter I, Schumm LP, Haritunians T, McGovern DP, Itan Y, Cho JH. Inflamed Ulcerative Colitis Regions Associated With MRGPRX2-Mediated Mast Cell Degranulation and Cell Activation Modules, Defining a New Therapeutic Target. Gastroenterology. 2021 Apr;160(5):1709-1724. [CrossRef]
- Starkey JR, Crowle PK, Taubenberger S. Mast-cell-deficient W/Wv mice exhibit a decreased rate of tumor angiogenesis. Int J Cancer. 1988;42(1):48-52. [CrossRef] [PubMed]
- Crivellato E, Nico B, Ribatti D. Mast cells and tumour angiogenesis: New insight from experimental carcinogenesis. Cancer Lett. 2008;269(1):1-6. [CrossRef]
- Liu J, Zhang Y, Zhao J, Yang Z, Li D, Katirai F, Huang B. Mast cell: Insight into remodeling a tumor microenvironment. Cancer Metastasis Rev. 2011;30(2):177-84. [CrossRef]
- Marichal T, Tsai M, Galli SJ. Mast cells: Potential positive and negative roles in tumor biology. Cancer Immunol Res. 2013;1(5):269-79. [CrossRef]
- Maltby S, Khazaie K, Blatner NR, Khan MW, Gounari F, Gounaris E, Dennis K, Bonertz A, Tsai FN, Strouch MJ, Cheon E, et al.. The significant role of mast cells in cancer. Cancer Metastasis Rev. 2011;30(1):45-60. [CrossRef]
- Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. 2019 Dec;16(12):713-732. [CrossRef]
- Kanth P, Grimmett J, Champine M, Burt R, Samadder NJ. Hereditary Colorectal Polyposis and Cancer Syndromes: A Primer on Diagnosis and Management. Am J Gastroenterol. 2017 Oct;112(10):1509-1525. [CrossRef] [PubMed]
- Nguyen LH, Goel A, Chung DC. Pathways of Colorectal Carcinogenesis. Gastroenterology. 2020 Jan;158(2):291-302. [CrossRef]
- Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med. 2015 Apr 9;372(15):1441-52. [CrossRef]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoué F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pagès F. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006 Sep 29;313(5795):1960-4. [CrossRef]
- Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, Church SE, Lafontaine L, Fischer M, Fredriksen T, Sasso M, Bilocq AM, Kirilovsky A, Obenauf AC, Hamieh M, Berger A, Bruneval P, Tuech JJ, Sabourin JC, Le Pessot F, Mauillon J, Rafii A, Laurent-Puig P, Speicher MR, Trajanoski Z, Michel P, Sesboüe R, Frebourg T, Pagès F, Valge-Archer V, Latouche JB, Galon J. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity. 2016 Mar 15;44(3):698-711. [CrossRef]
- Lanzi A, Pagès F, Lagorce-Pagès C, Galon J. The consensus immunoscore: toward a new classification of colorectal cancer. Oncoimmunology. 2020 Jul 11;9(1):1789032. [CrossRef]
- Fionda C, Scarno G, Stabile H, Molfetta R, Di Censo C, Gismondi A, Paolini R, Sozzani S, Santoni A, Sciumè G. NK Cells and Other Cytotoxic Innate Lymphocytes in Colorectal Cancer Progression and Metastasis. Int J Mol Sci. 2022 Jul 16;23(14):7859. [CrossRef]
- Molfetta R, Paolini R. The Controversial Role of Intestinal Mast Cells in Colon Cancer. Cells. 2023 Jan 31;12(3):459. [CrossRef]
- Liu X, Li X, Wei H, Liu Y, Li N. Mast cells in colorectal cancer tumour progression, angiogenesis, and lymphangiogenesis. Front Immunol. 2023 Jul 11;14:1209056. [CrossRef]
- Ko EA, Sanders KM, Zhou T. A transcriptomic insight into the impacts of mast cells in lung, breast, and colon cancers. Oncoimmunology. 2017 Aug 8;6(11):e1360457. [CrossRef]
- Wedemeyer J, Galli SJ. Decreased susceptibility of mast cell-deficient Kit(W)/Kit(W-v) mice to the development of 1, 2-dimethylhydrazine-induced intestinal tumors. Lab Invest. 2005 Mar;85(3):388-96. [CrossRef]
- Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, Lee DM, Zhang G, Glickman JN, Shin K, Rao VP, Poutahidis T, Weissleder R, McNagny KM, Khazaie K. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci U S A. 2007 Dec 11;104(50):19977-82. [CrossRef]
- Huang B, Lei Z, Zhang GM, Li D, Song C, Li B, Liu Y, Yuan Y, Unkeless J, Xiong H, et al.. SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood. 2008;112(4):1269-79. [CrossRef]
- Rigoni A, Bongiovanni L, Burocchi A, Sangaletti S, Danelli L, Guarnotta C, Lewis A, Rizzo A, Silver AR, Tripodo C, Colombo MP. Mast Cells Infiltrating Inflamed or Transformed Gut Alternatively Sustain Mucosal Healing or Tumor Growth. Cancer Res. 2015 Sep 15;75(18):3760-70. [CrossRef]
- Sakita JY, Elias-Oliveira J, Carlos D, de Souza Santos E, Almeida LY, Malta TM, Brunaldi MO, Albuquerque S, Araújo Silva CL, Andrade MV, Bonato VLD, Garcia SB, Cunha FQ, Cebinelli GCM, Martins RB, Matthews J, Colli L, Martin FL, Uyemura SA, Kannen V. Mast cell-T cell axis alters development of colitis-dependent and colitis-independent colorectal tumours: potential for therapeutically targeting via mast cell inhibition. J Immunother Cancer. 2022 Oct;10(10):e004653. [CrossRef]
- Molfetta R, Lecce M, Milito ND, Putro E, Pietropaolo G, Marangio C, Scarno G, Moretti M, De Smaele E, Santini T, Bernardini G, Sciumè G, Santoni A, Paolini R. SCF and IL-33 regulate mouse mast cell phenotypic and functional plasticity supporting a pro-inflammatory microenvironment. Cell Death Dis. 2023 Sep 20;14(9):616. [CrossRef]
- Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS, Wong M, Choi PJ, Wee LJK, Hillmer AM, Tan IB, Robson P, Prabhakar S. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. 2017 May;49(5):708-718. [CrossRef]
- Lee HO, Hong Y, Etlioglu HE, Cho YB, Pomella V, Van den Bosch B, Vanhecke J, Verbandt S, Hong H, Min JW, Kim N, Eum HH, Qian J, Boeckx B, Lambrechts D, Tsantoulis P, De Hertogh G, Chung W, Lee T, An M, Shin HT, Joung JG, Jung MH, Ko G, Wirapati P, Kim SH, Kim HC, Yun SH, Tan IBH, Ranjan B, Lee WY, Kim TY, Choi JK, Kim YJ, Prabhakar S, Tejpar S, Park WY. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet. 2020 Jun;52(6):594-603. [CrossRef]
- Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O'Brien SA, He Y, Wang L, Zhang Q, Kim A, Gao R, Orf J, Wang T, Sawant D, Kang J, Bhatt D, Lu D, Li CM, Rapaport AS, Perez K, Ye Y, Wang S, Hu X, Ren X, Ouyang W, Shen Z, Egen JG, Zhang Z, Yu X. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell. 2020 Apr 16;181(2):442-459.e29. [CrossRef]
- Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, Chao SX, Zollinger DR, Lieb DJ, Reeves JW, Fuhrman CA, Hoang ML, Delorey T, Nguyen LT, Waldman J, Klapholz M, Wakiro I, Cohen O, Albers J, Smillie CS, Cuoco MS, Wu J, Su MJ, Yeung J, Vijaykumar B, Magnuson AM, Asinovski N, Moll T, Goder-Reiser MN, Applebaum AS, Brais LK, DelloStritto LK, Denning SL, Phillips ST, Hill EK, Meehan JK, Frederick DT, Sharova T, Kanodia A, Todres EZ, Jané-Valbuena J, Biton M, Izar B, Lambden CD, Clancy TE, Bleday R, Melnitchouk N, Irani J, Kunitake H, Berger DL, Srivastava A, Hornick JL, Ogino S, Rotem A, Vigneau S, Johnson BE, Corcoran RB, Sharpe AH, Kuchroo VK, Ng K, Giannakis M, Nieman LT, Boland GM, Aguirre AJ, Anderson AC, Rozenblatt-Rosen O, Regev A, Hacohen N. Spatially organized multicellular immune hubs in human colorectal cancer. Cell. 2021 Sep 2;184(18):4734-4752.e20. [CrossRef]
- Wang W, Zhong Y, Zhuang Z, Xie J, Lu Y, Huang C, Sun Y, Wu L, Yin J, Yu H, Jiang Z, Wang S, Wang C, Zhang Y, Huang Y, Han C, Zhong Z, Hu J, Ouyang Y, Liu H, Yu M, Wei X, Chen D, Huang L, Hou Y, Lin Z, Liu S, Ling F, Yao X. Multiregion single-cell sequencing reveals the transcriptional landscape of the immune microenvironment of colorectal cancer. Clin Transl Med. 2021 Jan;11(1):e253. [CrossRef]
- Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, Zhang L, Ouyang H, Du P, Jiang L, Zhang B, Yang Y, Wang X, Ren X, Bei JX, Hu X, Bu Z, Ji J, Zhang Z. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021 Feb 4;184(3):792-809.e23. [CrossRef]
- Becker WR, Nevins SA, Chen DC, Chiu R, Horning AM, Guha TK, Laquindanum R, Mills M, Chaib H, Ladabaum U, Longacre T, Shen J, Esplin ED, Kundaje A, Ford JM, Curtis C, Snyder MP, Greenleaf WJ. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat Genet. 2022 Jul;54(7):985-995. [CrossRef]
- Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z, Ding X, Bao R, Hong L, Jia W, Fang F, Liu H, Chen L, Zhong J, Zou D, Liu L, Han L, Ginhoux F, Liu Y, Ye Y, Su B. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat Commun. 2022 Apr 1;13(1):1742. [CrossRef]
- Guo W, Zhang C, Wang X, Dou D, Chen D, Li J. Resolving the difference between left-sided and right-sided colorectal cancer by single-cell sequencing. JCI Insight. 2022 Jan 11;7(1):e152616. [CrossRef]
- Guo JN, Chen D, Deng SH, Huang JR, Song JX, Li XY, Cui BB, Liu YL. Identification and quantification of immune infiltration landscape on therapy and prognosis in left- and right-sided colon cancer. Cancer Immunol Immunother. 2022 Jun;71(6):1313-1330. [CrossRef]
- Liu Y, Zhang Q, Xing B, Luo N, Gao R, Yu K, Hu X, Bu Z, Peng J, Ren X, Zhang Z. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell. 2022 Apr 11;40(4):424-437.e5. [CrossRef]
- Xie Z, Niu L, Zheng G, Du K, Dai S, Li R, Dan H, Duan L, Wu H, Ren G, Dou X, Feng F, Zhang J, Zheng J. Single-cell analysis unveils activation of mast cells in colorectal cancer microenvironment. Cell Biosci. 2023 Nov 29;13(1):217. [CrossRef]
- Wang Q, Zhang YF, Li CL, Wang Y, Wu L, Wang XR, Huang T, Liu GL, Chen X, Yu Q, He PF. Integrating scRNA-seq and bulk RNA-seq to characterize infiltrating cells in the colorectal cancer tumor microenvironment and construct molecular risk models. Aging (Albany NY). 2023 Dec 5;15(23):13799-13821. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



