
Submitted:
05 April 2024
Posted:
09 April 2024
You are already at the latest version
Abstract
LPA3 receptors were expressed in TREx HEK 293 cells, and their signaling and phosphorylation were studied. The agonist, lysophosphatidic acid (LPA), increased intracellular calcium and ERK phosphorylation through pertussis toxin-insensitive processes. Phorbol myristate acetate, but not LPA, desensitizes LPA3-mediated calcium signaling. The agonists and the phorbol ester-induced LPA3 internalization. Pitstop 2 (clathrin heavy chain inhibitor) markedly reduced LPA-induced receptor internalization; in contrast, phorbol ester-induced internalization was only delayed. LPA induced rapid β-arrestin-LPA3 receptor association.
The agonist and the phorbol ester induced marked LPA3 receptor phosphorylation, and phosphorylation sites were detected using mass spectrometry. Phosphorylated residues were detected in the intracellular loop 3 (S221, T224, S225, and S229) and in the carboxyl terminus (S321, S325, S331, T333, S335, Y337, and S343). Interestingly, phosphorylation sites are within sequences predicted to constitute β-arrestin binding sites. These data provide insight into LPA3 receptor signaling and regulation.
Keywords:
Keywords: Lysophosphatidic acid: LPA
; Lysophosphatidic acid receptor 3
; LPA3
; phosphorylation sites
; signaling.
1. Introduction
Lysophosphatidic acid (LPA) is a simple lipid containing a glycerol moiety esterified to a phosphate group at position 3 and a fatty acid at position 2. It is considered a “bioactive lipid”, implying that, in addition to its metabolic roles, it modulates a wide range of cellular, organ, and whole animal responses through a series of six G protein-coupled receptors, i.e., the LPA receptor family [1,2,3]. They have the characteristic seven transmembrane hydrophobic domains connected by three intracellular loops and three extracellular loops, with an extracellular amino-terminal group and an intracellular carboxyl terminus. These receptors are subdivided into those belonging to the lysophospholipid receptors (LPA1-3) and those related to the purinergic family (LPA4-6) [1,2,4].
The present work is focused on the LPA3 receptor (previously named EDG 7 [5]), which is known to interact mainly with two different G protein types, Gαi/o and Gαq/11, likely involved in adenylyl cyclase inhibition, and calcium mobilization, triggering different signaling processes via their α and βγ subunits (reviewed in [1,2,3,4,5,6]); other G proteins might also be involved in LPA3 actions [7,8]. It is generally accepted that in addition to the immediate membrane signaling, many G protein-coupled receptors exert an endosomal second wave of signaling (reviewed in [9,10,11,12,13]; to our knowledge, information on the molecular events in LPA3 endosomal signaling has not been documented. In contrast, there is essential and abundant information on the participation of LPA3 receptors in many physiological functions and pathological processes. These include chemotaxis [14], migration [15], proliferation [16], differentiation [17], embryo implantations [18], determination of vertebrate left-right patterning during embryogenesis [19], and cardiac dysfunction [20,21], among many others (see also [6,22]). The relevant role of LPA and its receptors in cancer has been recently reviewed (see [23] and references therein). It is worth mentioning that an increase in the LPA3 receptor action has been observed in some cancer cell lines, such as rat neuroblastoma cells [24] or rat hepatoma [25]. In addition, LPA3 receptor expression seems to be directly related to resistance to chemotherapy, metastases, and, therefore, aggressiveness in some cancers, such as lung cancer [26], breast cancer [27], or pancreatic cancer [28]. In ovarian cancer, LPA3 expression is considered a therapeutic target and a poor prognosis marker [16,29].
Despite its physiological and pathophysiological relevance, many aspects of the signaling and regulation of the function of this receptor remain poorly explored or unknown. Receptor phosphorylation seems to be an early event participating in the desensitization of G protein-mediated signaling, endocytosis, endosomal traffic, and triggering of the second wave of signaling [9,10,11,12,13,30]. Phosphorylation of LPA3 receptors in response to the agonist, LPA, and to activation of protein kinase C (PKC) by phorbol esters has already been reported [31]. However, the key elements determining the consequences of receptor phosphorylation seem to be the individual sites or clusters phosphorylated; the process is commonly called the “phosphorylation barcode hypothesis” [13,30,32,33,34]. In silico analysis has allowed the suggestion of putative phosphorylation sites [6,35], but no experimental evidence is available. In the present work, we provide evidence to fill the mentioned knowledge gaps. We expressed the human LPA3 receptor employing a cellular model system that allows abundant receptor expression in an inducible fashion [36]. We characterized various signaling events using these cells, immunopurified the receptors, and used mass spectrometry to detect LPA3 phosphorylation sites experimentally.
2. Results
The effect of LPA on intracellular calcium was tested in non-transfected HEK 293 TREx cells. As shown in Figure 1A, a very small (≈ 20 nM) increase in calcium concentration was observed. A similar effect was observed in transfected cells in which receptor expression was not induced (Figure 1A). In contrast, a robust increase in the intracellular calcium concentration (≈ 200-300 nM) was induced by 1 µM LPA in transfected cells in which LPA3 receptor expression was induced (Figure 1A); these data indicate that the LPA3 receptors were functional.
The role of calcium entry in the action of LPA was tested. When cells were incubated in buffer without calcium, the LPA-induced increase in the calcium response was only marginally decreased (352 ± 61 nM and 328 ± 92 nM in cells incubated in buffer with and without calcium, respectively; n = 4, using cells from different cell cultures). The data indicate a predominant role of intracellular calcium mobilization in the effect of the lysophospholipid (Figure 1B). Preincubation for 30 min with the phospholipase C inhibitor, U73122 (10 µM) [37,38], essentially abolished the action of LPA (the increases in calcium concentration were: 306 ± 46 nM without inhibitor and 23 ± 13 with U73122; n = 3, p < 0.001), but thapsigargin was still able to increase intracellular calcium Figure 1C). The inactive analog, U73343 [37,38], did not alter the LPA action under the same conditions (increase 363 nM).
Afterward, we examined the effect of pertussis toxin, a well-known blocker of Gi/o. Surprisingly, the LPA-induced increase in intracellular calcium concentration was not diminished even using preincubation for 24 h with 300 ng/ml (usually we employ 100 ng/ml) (Figure 1D). As a control, we examined pertussis toxin action in cells expressing the LPA1 receptor. Supplementary Figure S1A shows that LPA1-expressing cells have a robust calcium response to the lysophospholipid and that such effect was completely blocked by preincubation with 300 ng/ml pertussis toxin. These data suggest that these two LPA receptor subtypes exhibit distinct preferences in their coupling to the various types of G proteins. To further characterize the G protein(s) involved, we employed YM-254890, a cyclic depsipeptide Gαq inhibitor [39,40]. Surprisingly, cell preincubation for 1 min with 1 µM YM-254890 [40] essentially eliminated the calcium response to LPA in cells expressing either LPA1 or LPA3 receptors; in both cases, thapsigargin was able to increase intracellular calcium (Supplementary Figure S2). Lower YM-254890 concentrations (100-300 nM) only marginally diminished LPA-induced calcium response in cells expressing LPA1 or LPA3 receptors (data not shown).
It is known that either receptor activation by agonists or stimulation of unrelated receptors or signaling proteins, such as some protein kinases, can induce receptor desensitization. They are usually named homologous and heterologous desensitization, respectively. To explore this, cells were incubated with 1 µM LPA for 5 min and washed 3 times with the Krebs-Ringer solution (Section 2.3) to remove the LPA present. The cells were resuspended in the same buffer and stimulated with 1 µM LPA. As shown in Figure 1E, preincubation with the agonist did not induce any detectable homologous desensitization. On the contrary, pretreatment with the PKC activator, PMA (1 µM) for 5 minutes markedly diminished the effect of 1 µM LPA (≈ 80% reduction) (Figure 1E,F); i.e., the data evidenced heterologous desensitization. Data in Supplementary Figure S1B,C showed that cells expressing the LPA1 receptors are subjected to marked homologous and heterologous desensitization.
We considered the possibility that low expression of G protein-coupled receptor kinases (GRKs) could be responsible for the essentially absent, homologous desensitization of the LPA3 receptors. However, in experiments in which GRK2 or GRK5 were overexpressed, agonist activation did not induce any significant desensitization of calcium signaling (Supplementary Figure S3). Similarly, in cells where LPA3 expression was decreased by induction with only 10 ng/ml doxycycline hyclate, absence of agonist-induced LPA3 desensitization was observed (data not shown).
The increase in intracellular calcium is a transient but immediate response to LPA3 receptor stimulation. ERK 1/2 phosphorylation, a lengthier LPA action, was also studied to explore signaling further. LPA3 activation with 1µM LPA resulted in a rapid increment in ERK 1/2 phosphorylation, which was maximal at 2 min and progressively decreased to near-baseline values (Figure 2). In contrast, 1 µM PMA increased ERK phosphorylation more slowly, reaching a near maximum level at 5 min, and remaining at this phosphorylation state during the incubation (60 min) (Figure 2); PMA is known to activate the mitogen-activated protein kinase pathway through PKC [41].
Intrigued by the insensitivity to pertussis toxin of the calcium response to LPA, we assessed the effect of this toxin in the ERK phosphorylation response. In parallel experiments, LPA3-expressing cells were preincubated with or without 300 ng/ml pertussis toxin and then challenged with 1 µM LPA for the times indicated; samples were also run in the same gel and Western blot analysis performed in the same membranes (Supplementary Figure S4). As observed, treatment with pertussis toxin decreased baseline (time 0 min) ERK 1/2 phosphorylation, but the time courses of LPA action were very similar (i.e., essentially identical if the respective baselines were normalized to 100%). Therefore, the data indicate that Gαi blockade with pertussis toxin barely affects the agonist LPA3 receptor-mediated ERK phosphorylation response.
The possibility that internalization of the LPA3 receptor could participate in ERK phosphorylation was explored employing Pitstop 2, an inhibitor of the terminal domain of clathrin heavy chains [42,43]. Pretreatment for 15 min with 10 µM Pitstop 2 markedly decreased (almost eliminated) baseline ERK 1/2 phosphorylation and the effect of LPA at times studied (Supplementary Figure S5). In contrast, the effect of Pitstop 2 on PMA-mediated action showed a different pattern. The marked decrease in baseline ERK phosphorylation was confirmed in these experiments, but PMA markedly and consistently increased ERK phosphorylation in cells pretreated with Pitstop 2, although the effect was noticeably delayed (Supplementary Figure S6). The effect of EGF was also tested; it was observed in parallel experiments that the action of the peptide growth factor was only marginally affected by Pitstop 2 at the time studied (15 min) (Supplementary Figure S8).
The effect of the Gαq inhibitor, YM-254890, on LPA-induced ERK phosphorylation was explored in cells expressing LPA1 or LPA3 receptors. As shown in Supplementary Figure S8, this agent decreased the action of LPA on ERK phosphorylation in cells expressing LPA1 or LPA3 receptors; however, the inhibition was more noticeable in the LPA3-expressing cells.
Accumulation of fluorescence in intracellular vesicles in response to the agonist, LPA, or the PKC activator, PMA, was observed, indicating receptor internalization (Figure 3). These actions took place rapidly (detected as early as 2 min) and were sustained during the 60 min incubations (Figure 3). Supplementary videos show the effects of LPA (video 1) and PMA (video 2) on receptor internalization. These videos allowed observation of cell displacement (LPA) and marked changes in form, including cell contraction (roundness). They also allow the detection of clusters of fluorescence forming “pearl-necklace”-like structures, formation of lamellipodia, blebs, and other changes in membrane appearance. Experiments were also performed inducing lower LPA3 expression (1 ng/ml doxycycline), and under these conditions, both LPA and PMA induced also marked receptor internalization. The nucleus was stained with DAPI in these cells, and fluorescent vesicles occupied most of the cytoplasm in LPA- or PMA-stimulated cells (Supplementary Figure S9).
Cells expressing the LPA3 receptors preincubated with Pitstop 2 notedly exhibit decreased intracellular fluorescence before stimulation (Supplementary Figs S10 and S11). When cells not preincubated with Pitstop 2 were stimulated with LPA, internalization was observed as early as 2 min and maintained throughout the experiment (Supplementary Figure S10). As indicated, Pitstop 2 decreased baseline fluorescence; LPA increased such baseline value, but the effect was smaller than that observed in the absence of the inhibitor (Supplementary Figure S10). PMA-induced LPA3 receptor internalization was quick and sustained in cells preincubated without any agent. In cells preincubated with Pitstop 2, internalization was similar to that observed without the inhibitor but departing from a different baseline (Supplementary Figure S11). These data suggest that a clathrin-independent process might also be involved in receptor internalization, pointing to differences between LPA-mediated and PMA-mediated internalization.
It is well-known that β-arrestins play a role in GPCR desensitization, internalization and signaling [9,10,11,12,13]. Therefore, LPA3-β-arrestin 2 interaction was studied utilizing FRET. It was observed (Figure 4A,B) that LPA induced a rapid receptor-arrestin interaction (maximal at 2 min), which decreased progressively but maintained the signal clearly above the baseline during the experiment (60 min). Supplementary Figure S12 shows the images obtained exciting the LPA3-eGFP construct (green channel), the β-arrestin-mCherry construct (red channel), raw FRET, and the FRET channel images (these are images of the same cells presented in Fig 4). Images of LPA3-β-arrestin colocalization are also presented (Figure 4C); Pearson coefficient indicates that in the baseline condition, there was no clear colocalization (< 0.5), whereas after stimulation with LPA colocalization was evident (Pearson coefficients > 0.9). Videos showing raw LPA-induced FRET (not FRET channel) (Video 3) and colocalization (Video 4) are presented as Supplementary Material.
LPA3-β-arrestin FRET was also studied using 1 µM PMA. The protein kinase C activator also induced a rapid increase in signal followed by a slow decrease (Figure 5A,B). Supplementary Figure S13 shows the images obtained exciting the LPA3-eGFP construct (green channel), the β-arrestin-mCherry construct (red channel), raw FRET, and the FRET channel images (these are images of the same cells presented in Figure 5). In Figure 5C, LPA3-β-arrestin colocalization images are presented; it can be observed that PMA also induced colocalization. We explored, afterward, LPA3 receptor phosphorylation in response to 1 µM LPA or 1 µM PMA. A representative whole membrane autoradiograph is presented in Supplementary Figure S14.
The time courses of the action of these agents are presented in Figure 6. It can be observed that both LPA and PMA induced a rapid increase in LPA3 phosphorylation; the effects were of similar magnitude and reached their maximum at 5-10 min of stimulation, after that decreased progressively, maintaining phosphorylation states above baseline up to the end of the incubation (60 min). These data are consistent with previous observations [31,44]. In silico analyses of putative phosphorylation sites in LPA receptors have already been reported [6,35]. Nevertheless, we repeated such analysis using updated versions of available software systems: GPS, PhosphoSVM, NetPhos, and MusiteDeep (see Section 2.9), and the results are presented in Table 1. Putative phosphorylation sites with high scores include 1 site in the intracellular loop 2, 5 sites in the intracellular loop 3, and 10 in the carboxyl terminus. The table indicates putative protein kinases potentially participating in the phosphorylation of these residues. Not surprisingly, isoforms of G protein-coupled receptor kinases (GRKs) and PKC, as well as other kinases, might play a role.
LPA3 receptors from cells not treated with any agent, 1 µM LPA or 1 µM PMA for 15 min, were immunopurified, and samples were sent to the Taplin Mass Spectrometry Facility (Harvard Medical School). A representative gel and blot is presented in Supplementary Figure S15. Table 2 shows the summary of data from distinct experiments and conditions. It should be noted that the mass spectrometry analyses were not quantitative and only indicated sites that were detected phosphorylated but not their abundance in the samples. In our studies 4 sites were detected in the intracellular loop 3 (S221, T224, S225, and S229), and 7 in the carboxyl terminus (S321, S325, S331, T333, S335, Y337, and S343). Many, but not all, the experimentally-detected phosphorylation sites were predicted by the in silico analyses (Table 2), and similarly, not all predicted sites were experimentally detected. The prediction programs identified, with high score, the following number of sites: GPS, 10 of the 11 experimentally detected sites (90 %, S331 not predicted); PhosphoSVM also 10 (90 %, S331 not predicted); NetPhos, 9 sites (81 %, S331 and Y337 not predicted); and MusiteDeep, only 6 of them (S229, S331, T333, S335, and S343 not predicted). A cartoon showing the putative, in silico, and experimentally found sites in the LPA3 receptor is presented in Figure 7. It is worth mentioning that the coverage of peptide detection was 100% for the intracellular loop 3 in all the experiments, and that of the carboxyl terminus ≈70-75% in four, and only 40% in one, of the five separate experiments performed. The carboxyl terminus tripeptide (STS) was not detected in any experiment.
Sequence alignment of the intracellular loop 3 and carboxyl termini of LPA1-3 receptors is presented in Figure 8. It can be observed that only S225 (intracellular loop 3) of the human LPA3 is conserved in all three of the human lysophospholipid LPA receptor subtypes. Two residues are conserved in two of these receptors: a) S221 (intracellular loop 3, LPA3, and LPA1) and b) S325 (carboxyl terminus, LPA3, and LPA2) (Figure 8). In contrast, the alignment of the available LPA3 receptor ortholog sequences revealed that in the intracellular loop 3, (Figure 9) three phosphorylated residues are conserved in all species studied, and S229, which is not conserved in the feline orthologs, was substituted by T (a phosphorylatable amino acid, conserved substitution). All 7 phosphorylated residues in the carboxyl terminus are conserved among the species studied (Figure 9).
To validate that the phosphorylation sites detected in the mass spectroscopy studies were those phosphorylated in cellulo, we employed cells expressing a mutant receptor in which the amino acids found phosphorylated were substituted by non-phosphorylatable residues (i.e., serines were substituted with alanines and threonines with valines). The mutant receptor construct was fluorescent and functional, as evidenced by the ability of LPA to increase intracellular calcium (data not shown). As anticipated, baseline receptor phosphorylation was markedly diminished (≈ 70%) in cells expressing the mutant receptor compared to those expressing the wild-type, and no significant effect of LPA or PMA was detected (Supplementary Figure S16). It should be mentioned that the mutant receptor migrated more in the electrophoresis experiments, which could be due to proteolysis (that we cannot discard) or to the substitutions made to the sequence.
3. Discussion
LPA receptors can be coupled to several G proteins; in particular, LPA1 and LPA3 receptors can stimulate the Gαq and Gαi/o proteins [6,22]. Activation of LPA receptors, including LPA3, increase intracellular calcium in cells endogenously expressing these receptors and in transfected cells (see, for example, [2,4,5,22,31,44]). LPA1 receptor stimulation elevates intracellular calcium concentration mainly through the stimulation of Gi/o proteins, as reflected by the marked sensitivity to pertussis toxin [44,45], a control employed in the present work. Interestingly, we observed that the LPA3 receptor-mediated increase in intracellular calcium was poorly sensitive to pertussis toxin, indicating that Gi/o proteins are not the primary mediators of this response. These data are consistent with the early observation of Bandoh et al. [5], who reported that the LPA3-mediated calcium response in insect Sf9 cells was insensitive to pertussis toxin but blocked by the phospholipase C blocker U73122. We attempted to define the role of Gq in this action by using the inhibitor YM-254890 [40,46], which prevents Gq protein-coupled receptor signaling by blocking GTP/ GDP exchange [40,46]. However, this agent blocked the LPA calcium response in cells expressing either LPA1 or LPA3 receptors, which precluded a more precise definition of the G protein involved. Consistent with our previous work using transfected C9 cells [31], we observed that LPA-activated LPA3 receptors induced marked but transient ERK phosphorylation, and we show here that this response was also insensitive to pertussis toxin, which indicates that it is not mainly mediated through Gi. The action of YM-254890 was also assessed on ERK phosphorylation using LPA1- and LPA3-expressing cells. The inhibitor decreased the LPA-induced ERK response in both cells, but the effect was more robust in cells expressing the LPA3 receptors. The selectivity of the Gq blocker has been questioned [47], and we cannot discard the possibility that YM-254890 might have actions at levels other than G proteins. Therefore, our present data suggest a role of Gq in these LPA3 effects (intracellular calcium and ERK phosphorylation) but did not permit a clear experimental demonstration. Obviously, our data do not exclude roles of Gi in other LPA3 receptor-mediated actions. Figure 10 (panel A) depicts some of these data interpretations.
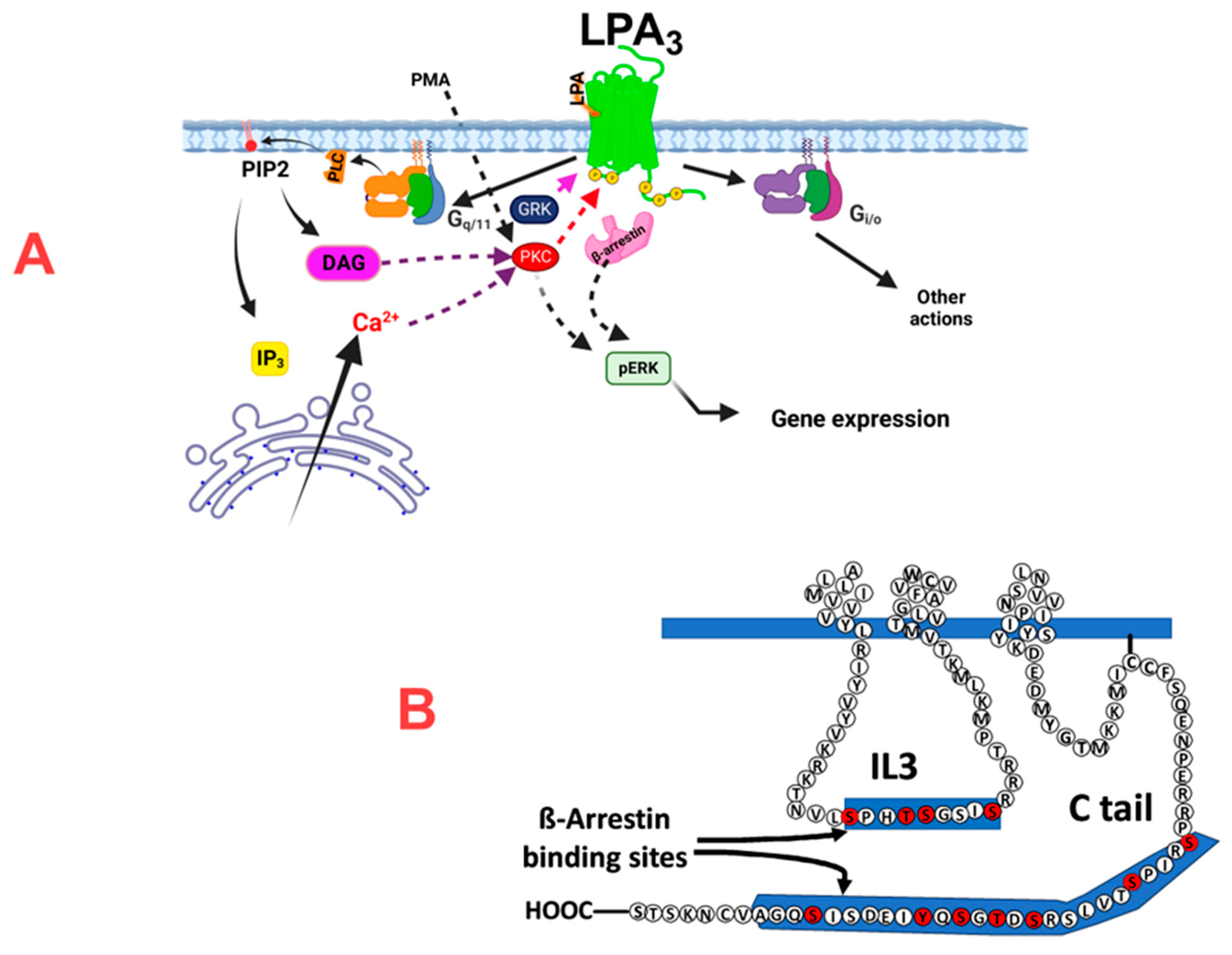
Figure 9.
Cartoon depicting the possible mechanism of action of LPA3 receptors and the possible role of phosphorylation sites in β-arrestin binding. Panel A shows that LPA3 receptors couple to Gq/11 and Gi/o and the finding that it is through Gq/11 that agonist activation of this receptor subtype mainly triggers calcium signaling and ERK phosphorylation. In Panel B, the possible role of the phosphorylation sites detected (marked in red) in the IL3 and Ctail domains in β-arrestin binding is suggested. IL3, intracellular loop 3; Ctail, carboxyl terminus; PLC, phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate, DAG, diacylglycerol, GRK, G protein-coupled re-ceptor kinase, PKC, protein kinase C.
Figure 9.
Cartoon depicting the possible mechanism of action of LPA3 receptors and the possible role of phosphorylation sites in β-arrestin binding. Panel A shows that LPA3 receptors couple to Gq/11 and Gi/o and the finding that it is through Gq/11 that agonist activation of this receptor subtype mainly triggers calcium signaling and ERK phosphorylation. In Panel B, the possible role of the phosphorylation sites detected (marked in red) in the IL3 and Ctail domains in β-arrestin binding is suggested. IL3, intracellular loop 3; Ctail, carboxyl terminus; PLC, phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate, DAG, diacylglycerol, GRK, G protein-coupled re-ceptor kinase, PKC, protein kinase C.
As anticipated by previous work [31], PKC activation with PMA markedly blocked the calcium response observed in cells expressing LPA1 or LPA3 receptors. The possibility that protein kinase activation might alter calcium signaling at other steps in the process cannot be completely ruled out. Our data also confirmed that LPA3 receptors were less sensitive to agonist-induced (homologous) desensitization. In our previous work [31], we observed reduced desensitization, whereas no homologous desensitization was detected in the present work. This difference is likely due to the cellular models used (C9 cells [31] and Flp-In TREx HEK 293 cells in this work) that express different repertoires of signaling elements and the fact that the inducible model expresses a high density of receptors (required for the receptor purification studies). This difference in desensitization is probably relevant to the roles of LPA3 receptors in cancer, particularly in those involving interaction with high concentrations of this lysophospholipid, such as ovarian cancer [6,16,22,29]. It is worth mentioning that the LPA3 receptor has been suggested as a therapeutic target and biomarker for ovarian cancer [29].
Arrestins are a family of proteins that bind to phosphorylated receptors and participate in desensitization, signaling, internalization, and many other functions [9,10,12]. The LPA3 receptors-β-arrestin 2 interaction observed in the FRET and colocalization experiments suggests that in the baseline state, β-arrestin is distributed in the cytoplasm; upon stimulation with LPA, β-arrestin interacts with plasma membrane-located LPA3 receptors, after a short time the FRET signal diminished (although remained above baseline during the 60 min of incubation), and the colocalization images suggest that some receptors internalize colocalizing with β-arrestin. G protein-coupled receptors have been divided into two classes based on the stability of the interaction with arrestins [48,49]. Class A receptors have a transient interaction with β-arrestin and translocate with this protein to clathrin-coated pits; once internalized, the receptor and β-arrestin dissociate. Class B receptors’ interaction with β-arrestin is more stable, and the receptors internalize accompanied by β-arrestin [48,49]. Our data indicate that the LPA3 receptors have many characteristics of Class A receptors but also have a feature of Class B receptors (i.e., apparently, some receptors maintain interaction with β-arrestin after internalization and FRET and colocalization signals decrease but do not vanish). The possibility that classification might not be binary but that receptors with intermediate characteristics might exist is suggested. It is worth reminding that colocalization and FRET are different parameters reflecting distinct aspects. Colocalization has a resolution of ≈ 200 nm, and objects could be at distinct optical planes, whereas to obtain a FRET signal, a distance of < 10 nm should exist between fluorophores.
We confirmed [31] the ability of LPA and PMA to induce LPA3 receptor internalization. Pitstop 2 inhibits the terminal domain of clathrin-heavy chains [42,43] and markedly decreases LPA-induced LPA3 internalization. The action of PMA on receptor internalization was delayed but essentially followed the same pattern of cells incubated without the inhibitor. Similarly, Pitstop 2 markedly decreased LPA-induced ERK phosphorylation but only delayed the action of PMA. The parallelism of these processes is consistent with the idea that complexes involving receptors, β-arrestin, clathrin, the endocytic machinery, and MAP kinases are involved in both events [50].
LPA3 receptor phosphorylation was confirmed in the present study. This work’s central objective was to characterize the phosphorylated LPA3 residues. We detected sites in the intracellular loop 3 and carboxyl terminus with reproducibility and high Ascore [51]. Determination of these sites opens the possibility to study their functional impact. Nevertheless, further work will be required to define this in the two domains. This final goal will help better understand this receptor’s function and regulation, which is relevant considering its roles in health and disease. It is worth mentioning that phosphorylation codes for β-arrestin recruitment by G protein-coupled receptors have been discovered ( [52] see also (http://tools.vai.org/phoscofinder/downloads.php), which allows finding putative β-arrestin binding sites in receptors. Several of these sites were reported for the LPA3 receptors [52] located in the carboxyl terminus within the region S321-A346. Other sites were detected, using the “phoscofinder” webpage, in the intracellular loop 3, within the sequence S221-S229. All phosphorylated residues in the mass spectrometry studies were within these regions. These aspects are depicted in Figure 10 (panel B).
It is worth noticing that the lack of conservation of the phosphorylation sites was far from surprising. Previous work has shown that phosphorylation sites are not conserved among the α -adrenergic subtypes [53,54,55]. In contrast, the phosphorylation sites were conserved among receptor orthologs, which is also similar to what has been observed for α1-adrenoceptor subtypes [53,54,55].
The present experiments provide insight into the actions and regulation of LPA3 receptors. Limitations of this work include using a single cellular model, i.e., Flp-In TREx HEK293 cells. We previously studied LPA3 receptor phosphorylation, internalization, and desensitization in C9 cells [31]; some differences in these studies were observed, which is not unexpected [33,34], since receptor action and regulation depend on the proteins expressed which is cell-specific. However, in both cellular systems, the LPA3 receptor was overexpressed. Unfortunately, to our knowledge, there is no suitable cellular model to study LPA3 action and regulation since most cells express various receptor subtypes for lysophosphatic acid.
Another limitation is the use of PMA to study heterologous desensitization and internalization. One advantage of PMA is that most of its actions are mediated through activation of PKC isoforms; however, there are other targets for active phorbol esters and diacylglycerol [56,57]. Nevertheless, we have previously shown that PKC inhibitors and down-regulation of some isoforms block the ability of PMA to induce LPA3 phosphorylation and block LPA3-mediated increases in intracellular calcium [31]. These data strongly indicate that these actions are mediated through PKC but do not allow us to exclude the roles of other phorbol ester-targets completely; similarly, actions secondary to PKC cascade activation might alter signaling processes (such as ERK phosphorylation studied in this work) [41]. In addition, sustained activation of PKC is induced by phorbol esters, whereas diacylglycerol action seems to be transient under physiological conditions. Despite these limitations, unavoidable due to pharmacological and molecular biological strategies, the present finding provides new insights into LPA3–triggered signaling pathways, actions, and regulation.
In our opinion, the phosphorylation experiments with the mutant receptor validate that the sites detected were those mainly phosphorylated in cellulo. However, defining the functional roles of the phosphorylation sites detected in the intracellular loop 3 and carboxyl terminus is an experimental challenge. Understanding the roles of different phosphorylation domains, clusters of phosphorylation sites, and even individual phosphor amino acids seems relevant, considering the importance of this receptor in physiology and pathophysiology. Experiments on this aspect are currently being performed in our laboratory.
4. Materials and Methods
4.1. Reagents
1-Oleyl lysophosphatidic acid (LPA) was from Cayman Chemical Co. (Ann Arbor, MI, USA). Phorbol 12-myristate-13-acetate (PMA), cyanogen bromide-activated agarose, thapsigargin, the phospholipase C inhibitor U73122 and its inactive analog U73343, as well as Pitstop 2 (N-[5-[(4-Bromophenyl) methylene]-4,5-dihydro-4-oxo-2-thiazolyl]-1-naphthalene-sulfonamide), and free fatty acid-free bovine serum albumin (fraction V) were obtained from Sigma-Aldrich (St.Louis, MO, USA). YM 254890 was obtained from Wako Chemicals (Richmond, VA, USA) (lot: WDK3168). Dulbecco’s modified Eagle’s medium, trypsin, streptomycin, penicillin, amphotericin B, blasticidin, hygromycin B, doxycycline hyclate, and Fura-2 AM were purchased from Invitrogen-Life Technologies (Carlsbad, CA, USA). Fetal bovine serum was obtained from BioWest (Nuaillé, France). Pertussis toxin was purified from vaccine concentrates [58]. [32P]Pi (8500–9120 Ci/mmol) was obtained from Perkin-Elmer Life Sciences-New England Nuclear (Boston, MA, USA). Polyvinylidene difluoride membranes were purchased from BioRad, Lipofectamine 2000 (catalog number 11668-019, lot 1854318), and SuperSignal West Pico Chemiluminescence kits were purchased from Thermo Fisher Scientific (Waltham, MA, USA). DAPI (4′,6-diamidino-2-fenilindol), anti-GRK2 (G protein-coupled receptor kinase 2) rabbit polyclonal antibody (catalog number SC 562, Lot F248), and anti-GRK5 (G protein-coupled receptor kinase 2) rabbit polyclonal antibodies (catalog number SC 565, lot D1912) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and employed as indicated by the supplier. Agarose-coupled protein A was obtained from Merck-Millipore (Burlington, MA, USA). Anti-phospho-ERK 1/2 (Thr202/Tyr204) (catalog number 9101S, Lot: 30) and anti-total ERK (p42/44) antibodies (catalog number 4695S, Lot: 21) were from Cell Signaling Technology (Danvers, MA, USA), monoclonal anti-GFP (green fluorescent protein) antibodies were from Clontech (JL-8; catalog number 632381, Lot: A8034133) (Mountain View, CA, USA), and polyclonal anti-GFP antibody was generated in our laboratory [31,53,54]. Antibodies were obtained from a polyclonal anti-GFP antisera by ammonium sulfate salting-out. Following the supplier indications, the dialyzed IgGs were coupled to cyanogen bromide-activated agarose beds. This anti-GFP coupled agarose was used for the immunopurification procedure. Primary antibodies were used at a dilution of 1:2,000, whereas secondary antibody dilution was 1:10,000. The peroxidase affiniPure Goat anti-mouse IgG light-high chain antibody and other secondary antibodies were purchased from Zymed (Thermo Fisher Scientific; (Waltham, MA, USA) or Jackson ImmunoResearch (West Grove, PA, USA. Human embryonic kidney (HEK) 293 cells were obtained from the American Type Culture Collection (HEK293; ATCC CRL-1573) (Manassas, VA, USA). Parental Flp-In T-Rex HEK293 cells and the plasmid, pOG44, were obtained from Invitrogen (Carlsbad, California, USA). The plasmid for the expression of the LPA1 receptor fused to the mCherry red fluorescent protein (plasmid ID EX-Z7377-M56) was obtained from Genecopoeia (Rockville, MD, USA). The plasmid for the expression of β-arrestin 2 mCherry-tagged was generously provided by Dr. Adrian J. Butcher (University of Leicester, UK) [59]. Plasmids for the expression of GRK2 and GRK5 were generously provided by Dr. Jeffrey Benovic (Thomas Jefferson University) [60].
LPA (10 mM) was dissolved in culture medium supplemented with 0.1 % fatty acid-free bovine serum albumin, and subsequent dilutions were made in water. PMA (10 mM) was dissolved in dimethyl sulfoxide (DMSO), and subsequent dilutions were also made in water (concentration of DMSO in contact with the cells 0.01%). The absence of an effect of the vehicle was checked in all experiments. In the FRET experiments, we notice that DMSO, per se, induced some transient changes in the signal. Therefore, for these experiments, PMA was dissolved in absolute ethanol (PMA 10 mM), and subsequent dilutions were made in water containing 10% ethanol (use of water or culture medium resulted in turbid suspensions). Ethanol in contact with the cells was 0.11% and was devoid of any effect on FRET; PMA dissolved this way was fully effective, as evidenced by its effect on LPA-induced increases in intracellular calcium (data not shown).
4.2. Cell Lines and LPA1 and LPA3 Receptor Expression
The LPA3 receptor sequence was fused at the carboxyl terminus (Ctail) with the GFP and cloned into the pCDNA5/FRT/TO plasmid (Bioinnovatise, Inc., Rockville, MD) to employ the inducible Flp-In TREx expression system [36]. A plasmid to express a mutant LPA3 receptor in which the residues found phosphorylated in the mass spectrometry analysis were substituted by non-phosphorylatable residues was obtained commercially, and proper substitution was confirmed by sequencing (Bioinnovatise, Inc., Rockville, MD). The plasmids were transfected into parental Flp-In TREx HEK293 cells with the pOG44 plasmid using lipofectamine 2000. These cells were subjected to selection for one month with 10 µg/ml blasticidin and 100 µg/ml hygromycin B and grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 μg/ml streptomycin, 100 U/ml penicillin, and 0.25 μg/ml amphotericin B. Unless otherwise indicated, LPA3 receptor expression was induced with 10 µg/ml doxycycline hyclate for 12 h and confirmed expression using fluorescence microscopy and calcium signaling
In some experiments, cells expressing the LPA1 receptor were used for comparison. HEK 293 cells were cultured as previously described [61] and transfected with the plasmid to express the LPA1 receptor fused to the mCherry red fluorescent protein indicated above (2.1 Reagents). Cells were subjected to selection with G418 (initially, 900 µg/ml was used and subsequently decreased, for maintenance, to 300 µg/ml). A cell line was selected based on expression (fluorescence microscopy) and robust functional response (rise in intracellular calcium concentration).
4.3. Intracellular Calcium Concentration
Determinations were performed as previously described ([31,61]; see [62] for a detailed description). In brief, the cells (80-90 % confluence) were serum-starved and treated for 12 h with 10 µg/ml doxycycline hyclate to induce LPA3 expression. After confirming the expression of this receptor construct (epifluorescence microscopy), the cells were loaded with 2.5 µM Fura-2 AM in Krebs–Ringer–Hepes containing 0.05% bovine serum albumin (pH 7.4) for 1 h. Cells were carefully detached from the Petri dish, washed to eliminate unincorporated dye, and maintained in suspension [62]. In the experiments to determine if extracellular calcium entry was involved in LPA action, the cell suspension was divided into two halves; one half was washed in the regular buffer and the other half in the same buffer but without calcium. Determinations were carried out in an AMINCO-Bowman Series 2 luminescence spectrometer equipped to maintain the temperature at 37 °C and gentle, constant stirring of the cell suspension. Two excitation wavelengths (340 and 380 nm) and an emission wavelength of 510 nm were employed with a chopper interval of 0.5 s. Intracellular calcium levels were calculated as described by Grynkiewicz et al. [63]; maximal fluorescence by determined by lysing the cells with Triton X-100 (0.1 %, final), and the minimal fluorescence signal by adding EGTA (5 mM, final) [62]. Only the intracellular calcium determinations were made in cells in suspension, and all other studies were performed in cells attached to the Petri dishes.
4.4. ERK 1/2 Phosphorylation
The cells were serum-starved for 4 h and then stimulated with 1 µM LPA or 1 µM PMA (these concentrations were determined in preliminary experiments) for the times indicated; after this incubation, cells were washed twice with ice-cold phosphate-buffered saline and lysed [61] for 1 h on an ice bath; the lysates were centrifuged at 12,700xg for 15 minutes and proteins contained in supernatants were denatured with Laemmli sample buffer [64] and separated by SDS-polyacrylamide gel electrophoresis. Proteins were electrotransferred onto polyvinylidene difluoride membranes, and immunoblotting was performed. Total- and phospho-ERK 1/2 levels were determined in the same membranes for each experiment; the baseline value was considered 100% for normalization.
4.5. LPA3 Receptor-β-Arrestin 2 Interaction
Cells expressing the LPA3-GFP construct were transfected with the β-arrestin 2-mCherry plasmid described above (2.1 Reagents) (500 ng/ 6 cm Petri dish). After 24 h, the cells were collected and seeded on glass-bottomed Petri dishes, and after an additional 48 h in culture, protein-protein interactions were studied in these cells. The LPA3-β-arrestin interaction was analyzed using Föster Resonance Energy Transfer (FRET), employing an FV10i Olympus microscope with an automated laser spectral scan. The GFP was excited at 488 nm, and the emitted fluorescence was detected at 510 nm, whereas mCherry was excited at 580 nm and emitted fluorescence was detected at 610 nm; this was routinely performed, and images were obtained to check the expression of these proteins. For FRET channel analysis, GFP (but not mCherry) was excited, and fluorescence was detected at 610 nm; such fluorescence indicated that the proximity among the fluorescent proteins was enough to allow energy transfer (i.e., 1-10 nm) [65]. The FRET index was quantified using ImageJ software (version 1.49v) [66], which removes bleed-through and false FRET. The images were analyzed using 8 bits that permit pixel-by-pixel supervised computational FRET index analysis. The average FRET index obtained with the vehicle (time 0 min) was normalized as 100%. Individual cells (not clusters) expressing both fluorescent proteins were randomly selected; 10-14 cells were analyzed for each experimental condition in all the experiments.
4.6. Video Experiments
The video experiments employed a confocal Zeiss LSM800 microscope with a temperature- and atmosphere (CO2 and humidity)-controlled chamber. The expression of eGFP- and mCherry-tagged proteins was confirmed for each experiment. The eGFP was excited at 488 nm, and the emitted fluorescence was detected at 490-570 nm, whereas mCherry was excited at 564 nm, and the fluorescence emitted was detected at 580-700 nm. In the receptor internalization experiments, only eGFP was excited, and emission was detected. In these experiments, recording (1.27 frames per second) was for 5 min (176 frames, no interval) during the baseline conditions, and then cells were stimulated with 1 μM LPA or 1 μM PMA. Recording under stimulated conditions was for 60 min (2182 frames, no interval). In the colocalization experiments, eGFP and mCherry were excited, and their fluorescence was recorded. Image merge was performed using the software included in the microscope. In these experiments, recording “Frame mode” (2.53 frames per second) was for 5 min (134 frames, no interval) during the baseline, and then cells were stimulated for 15 min (599 frames, no interval). In the FRET experiments, eGFP (but not mCherry) was excited, and fluorescence was detected in the red channel (580-700 nm); fluorescence at this range was considered raw FRET. Conditions (gain, contrast. brightness) were determined by allowing a minimum “background noise” that permits cell and nucleus delimitation. Such background was maintained constant in all and during all the experiments. In these studies, recording “Line mode” (1.27 frames per second) was for 5 min (185 frames, no interval) during the baseline, and under-stimulated conditions were for 15 min (556 frames, no interval). In all the video experiments, processing was performed with the FIJI program (a distribution of ImageJ) [67]. The video was saved in the “mp4” format, and the letterings “baseline”, “LPA”, or “PMA” were included to specify the condition being studied. The fact that cells and organelles move (i.e., migrate and change their form) during the experiments and can leave the plane of observation should be reminded. In addition, LPA is known to induce cell contraction and migration (see, for example, [68] and references therein).
4.7. Receptor Internalization
Cells seeded at a low density were cultured on glass-bottomed Petri dishes for 12 h. After this, LPA3 receptor expression was induced by incubation with doxycycline (10 µg/ml) for an additional 12 h. Before the experiment, the cells were serum-fasted for 1 h (prolonged serum fasting (i.e., 4 h or more) resulted in diminished internalization). After this incubation, cells were stimulated with LPA or PMA for the times indicated. Cells were washed with phosphate-buffered saline and immediately fixed with 4% paraformaldehyde for 10 minutes. The images were obtained using a Fluoview Confocal model FV10i microscope (Olympus); GFP was excited at 488 nm and emitted fluorescence registered at 515-540 nm. The plasma membrane was delineated using differential interference contrast images to determine receptor internalization. Each cell’s intracellular fluorescence (i.e., excluding the plasma membrane) was quantified as “integrated density”, employing the ImageJ software [66]. The procedure is described in detail for “Corrected total cell fluorescence” (in [67] and in The Open Lab Book (https://theolb.readthedocs.io/en/latest/imaging/measuring-cell-fluorescence-using-imagej.html)). Usually, 10-14 images were taken for each condition from 3 or 4 different cultures obtained on different days. In some experiments, doxycyclin was employed at a concentration of 10 ng/ml to reduce the amount of receptor expressed in the cells and allow a more evident detection of internalization.
4.8. Receptor Phosphorylation
The cells were incubated for 1 h in phosphate-free Dulbecco’s Modified Eagle’s media and 3 h in phosphate-free media supplemented with 50 μCi/ml [32P]Pi. Labeled cells were treated with vehicle, LPA, or PMA, washed with ice-cold phosphate-buffered saline, and solubilized for 1 h in the lysis buffer [53,54,55]. The extracts were centrifuged, and the supernatants were incubated overnight with protein A-agarose and the anti-GFP antiserum generated in our laboratory. Samples were washed five times, and the pellets were denaturalized with sample buffer [64]. Proteins were separated using SDS-polyacrylamide gel electrophoresis, electrotransferred onto nitrocellulose membranes, and exposed for 24 h. The amount of phosphorylated receptor was assessed by PhosphorImager analysis using the ImageQuant program. Western blotting for loading controls was performed utilizing a commercial monoclonal anti-GFP antibody.
4.9. In Silico Analysis
In silico analyses were performed to determine putative phosphorylation sites in the sequence of the LPA3 receptor. These were carried out using the software Group-based Prediction System, GPS [69], versions 6.0 (http://gps.biocuckoo.cn; last accessed on March 23, 2022), PhosphoSVM [70,71]: (http://sysbio.unl.edu/PhosphoSVM/; last accessed on May 10, 2023) and NetPhos 3.1 (http://www.cbs.dtu.dk; last accessed on April 9, 2023) and MusiteDeep (https://www.musite.net/; last accessed on May 10, 2023). We considered only the sites with the highest phosphorylation score.
4.10. Immunopurification and Mass Spectrometric Analysis
Immunopurification was performed as previously employed for other receptors [53,54,55]. In brief, fifteen Petri dishes (10 cm) containing LPA3 receptor-expressing cells (with 95-100% confluence) were divided for the 3 experimental conditions studied (5 Petri dishes each): baseline, LPA, or PMA. After the stimulation, cultures were washed twice with ice-cold phosphate-buffered saline and solubilized in a lysis buffer containing detergents, proteases, and phosphatase inhibitors [53,54,55], for 1 h in an ice bath. Cell lysates were centrifuged for 15 minutes at 4 °C, and the supernatant was added to a slurry of anti-GFP-agarose beads and incubated with constant movement for 4 hours at 4 °C. The pellets were washed five times with lysis buffer and solubilized with Laemmli sample buffer [64], and the samples were resolved using SDS-polyacrylamide gel electrophoresis. Finally, the bands corresponding to the LPA3-GFP construct (≈70 kDa, identified by Western blotting) were excised and sent to the Taplin Mass Spectrometry Facility (Harvard Medical School. Cambridge, MA, USA), where the analysis was performed. Five independent experiments, including all conditions, were performed and analyzed.
4.11. Statistical Analyses
The data are presented as the means + standard errors of the means. Statistical analysis between comparable groups was performed using ordinary one-way ANOVA with the Bonferroni post-test, employing the included software in the GraphPad Prism program (version 10.1.2). A P value < 0.05 was considered statistically significant
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Material, Legends for the videos, Videos 1, 2,3, and 4.
Author Contributions
K. H. S., Conceptualization, Methodology, Investigation, Formal analysis, Visualization, Writing – Original Draft, Writing - Review & Editing; M. T. R.-Á., Investigation, Formal analysis, Visualization, Writing - Review & Editing; R. R.-H., Investigation, Formal analysis, Visualization, Writing - Review & Editing; and J. A.G.-S., Conceptualization, Formal analysis, Writing - Original Draft, Writing - Review & Editing.
Funding
This research was partially supported by Grants from CONAHCyT (Fronteras 6676) and DGAPA (IN201221 and IN201924).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Acknowledgments
The authors thank Ross Tomaino (Harvard University) for his help with the mass spectrometry analysis. The advice and technical support of the following members of the indicated Service Units of our Institute is gratefully acknowledged: Dr. Héctor Malagón and Dr. Claudia Rivera (Bioterio); Juan Barbosa and Gerardo Coello (Cómputo); and Aurey Galván and Manuel Ortínez (Taller). We thank Nadia Teresa Cedillo-Romero for reading and correcting the manuscript. K. Helivier Solís is a student of the Programa de Doctorado en Ciencias Biomédicas (UNAM) (account 520014983).
Conflicts of Interest
All authors declare no conflicts of interest.
References
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Phar macol. 2014, 171, 3575–94. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; Chun, J. LPA receptors: subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–86. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, L.H.M.; Spohr, T.; Amaral, R.F.D.; Fonseca, A.; Garcia, C.; Mendes, F.A.; Freitas, C.; dosSantos, M.F.; Lima, F.R.S. Role of lysophosphatidic acid and its receptors in health and disease: novel therapeutic strategies. Signal Transduct Target Ther 2021, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Bandoh, K.; Aoki, J.; Hosono, H.; Kobayashi, S.; Kobayashi, T.; Murakami-Murofushi, K.; Tsujimoto, M.; Arai, H.; Inoue, K. Molecular cloning and characterization of a novel human G-protein-coupled receptor, EDG7, for lysophosphatidic acid. J. Biol. Chem. 1999, 274, 27776–85. [Google Scholar] [CrossRef] [PubMed]
- Solís, K.H.; Romero-Ávila, M.T.; Guzmán-Silva, A.; García-Sáinz, J.A. The LPA(3) Receptor: Regulation and Activation of Signaling Pathways. Int J Mol Sci 2021, 22, 6704. [Google Scholar] [CrossRef] [PubMed]
- Hains, M.D.; Wing, M.R.; Maddileti, S.; Siderovski, D.P.; Harden, T.K. Galpha12/13- and rho-dependent activation of phospholipase C-epsilon by lysophosphatidic acid and thrombin receptors. Mol. Pharmacol. 2006, 69, 2068–75. [Google Scholar] [CrossRef]
- Cai, H.; Xu, Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun Signal 2013, 11, 31. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. Plethora of functions packed into 45 kDa arrestins: biological implications and possible therapeutic strategies. Cell Mol Life Sci 2019, 76, 4413–4421. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front Pharmacol 2019, 10, 125. [Google Scholar] [CrossRef]
- Eichel, K.; von Zastrow, M. Subcellular Organization of GPCR Signaling. Trends Pharmacol. Sci. 2018, 39, (2), 200–208. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Lefkowitz, R.J. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32, (9), 521–33. [Google Scholar] [CrossRef]
- Martínez-Morales, J.C.; Romero-Ávila, M.T.; Reyes-Cruz, G.; García-Sáinz, J.A. Roles of Receptor Phosphorylation and Rab Proteins in G Protein-Coupled Receptor Function and Trafficking. Mol. Pharmacol. 2022, 101, (3), 144–153. [Google Scholar] [CrossRef]
- Chan, L.C.; Peters, W.; Xu, Y.; Chun, J.; Farese, R.V., Jr.; Cases, S. LPA3 receptor mediates chemotaxis of immature murine dendritic cells to unsaturated lysophosphatidic acid (LPA). J. Leukoc. Biol. 2007, 82, (5), 1193–200. [Google Scholar] [CrossRef]
- Tanabe, E.; Kitayoshi, M.; Yoshikawa, K.; Shibata, A.; Honoki, K.; Fukushima, N.; Tsujiuchi, T. Loss of lysophosphatidic acid receptor-3 suppresses cell migration activity of human sarcoma cells. J Recept Signal Transduct Res 2012, 32, (6), 328–34. [Google Scholar] [CrossRef]
- Goldsmith, Z.G.; Ha, J.H.; Jayaraman, M.; Dhanasekaran, D.N. Lysophosphatidic Acid Stimulates the Proliferation of Ovarian Cancer Cells via the gep Proto-Oncogene Galpha(12). Genes Cancer 2011, 2, (5), 563–75. [Google Scholar] [CrossRef]
- Chiang, J.C.; Chen, W.M.; Lin, K.H.; Hsia, K.; Ho, Y.H.; Lin, Y.C.; Shen, T.L.; Lu, J.H.; Chen, S.K.; Yao, C.L.; Chen, B.P.C.; Lee, H. Lysophosphatidic acid receptors 2 and 3 regulate erythropoiesis at different hematopoietic stages. Biochim Biophys Acta Mol Cell Biol Lipids 2021, 1866, (1), 158818. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Hama, K.; Contos, J.J.; Anliker, B.; Inoue, A.; Skinner, M.K.; Suzuki, H.; Amano, T.; Kennedy, G.; Arai, H.; Aoki, J.; Chun, J. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature 2005, 435, (7038), 104–8. [Google Scholar] [CrossRef]
- Lai, S.L.; Yao, W.L.; Tsao, K.C.; Houben, A.J.; Albers, H.M.; Ovaa, H.; Moolenaar, W.H.; Lee, S.J. Autotaxin/Lpar3 signaling regulates Kupffer's vesicle formation and left-right asymmetry in zebrafish. Development 2012, 139, (23), 4439–48. [Google Scholar] [CrossRef]
- Yang, J.; Xu, J.; Han, X.; Wang, H.; Zhang, Y.; Dong, J.; Deng, Y.; Wang, J. Lysophosphatidic Acid Is Associated With Cardiac Dysfunction and Hypertrophy by Suppressing Autophagy via the LPA3/AKT/mTOR Pathway. Front Physiol 2018, 9, 1315. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Fan, G.; Wang, F.; Liu, S.; Li, T.; Cong, X.; Chun, J.; Chen, X. Protective Role for LPA(3) in Cardiac Hypertrophy Induced by Myocardial Infarction but Not by Isoproterenol. Front Physiol 2017, 8, 356. [Google Scholar] [CrossRef] [PubMed]
- Hama, K.; Aoki, J. LPA(3), a unique G protein-coupled receptor for lysophosphatidic acid. Prog. Lipid Res. 2010, 49, (4), 335–42. [Google Scholar] [CrossRef]
- Balijepalli, P.; Sitton, C.C.; Meier, K.E. Lysophosphatidic Acid Signaling in Cancer Cells: What Makes LPA So Special? Cells 2021, 10, (8). [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Okabe, K.; Kato, K.; Okumura, M.; Fukui, R.; Fukushima, N.; Tsujiuchi, T. Differential function of lysophosphatidic acid receptors in cell proliferation and migration of neuroblastoma cells. Cancer Lett. 2012, 316, (1), 91–6. [Google Scholar] [CrossRef]
- Okabe, K.; Hayashi, M.; Kato, K.; Okumura, M.; Fukui, R.; Honoki, K.; Fukushima, N.; Tsujiuchi, T. Lysophosphatidic acid receptor-3 increases tumorigenicity and aggressiveness of rat hepatoma RH7777 cells. Mol. Carcinog. 2013, 52, (4), 247–54. [Google Scholar] [CrossRef]
- Ueda, N.; Minami, K.; Ishimoto, K.; Tsujiuchi, T. Effects of lysophosphatidic acid (LPA) receptor-2 (LPA(2)) and LPA(3) on the regulation of chemoresistance to anticancer drug in lung cancer cells. Cell. Signal. 2020, 69, 109551. [Google Scholar] [CrossRef]
- Sun, K.; Cai, H.; Duan, X.; Yang, Y.; Li, M.; Qu, J.; Zhang, X.; Wang, J. Aberrant expression and potential therapeutic target of lysophosphatidic acid receptor 3 in triple-negative breast cancers. Clin. Exp. Med. 2015, 15, (3), 371–80. [Google Scholar] [CrossRef]
- Kato, K.; Yoshikawa, K.; Tanabe, E.; Kitayoshi, M.; Fukui, R.; Fukushima, N.; Tsujiuchi, T. Opposite roles of LPA1 and LPA3 on cell motile and invasive activities of pancreatic cancer cells. Tumour Biol 2012, 33, (5), 1739–44. [Google Scholar] [CrossRef]
- Zhao, P.; Yun, Q.; Li, A.; Li, R.; Yan, Y.; Wang, Y.; Sun, H.; Damirin, A. LPA3 is a precise therapeutic target and potential biomarker for ovarian cancer. Med Oncol 2022, 39, (2), 17. [Google Scholar] [CrossRef]
- Bahouth, S.W.; Nooh, M.M. Barcoding of GPCR trafficking and signaling through the various trafficking roadmaps by compartmentalized signaling networks. Cell. Signal. 2017, 36, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Alcántara-Hernández, R.; Hernández-Méndez, A.; Campos-Martínez, G.A.; Meizoso-Huesca, A.; García-Sáinz, J.A. Phosphorylation and Internalization of Lysophosphatidic Acid Receptors LPA1, LPA2, and LPA3. PLoS One 2015, 10, (10), e0140583. [Google Scholar] [CrossRef] [PubMed]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; Shenoy, S.K.; Gygi, S.P.; Lefkowitz, R.J. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal 2011, 4, (185), ra51. [Google Scholar] [CrossRef]
- Tobin, A.B. G-protein-coupled receptor phosphorylation: where, when and by whom. Br. J. Pharmacol. 2008, 153 Suppl. 1, S167–76. [Google Scholar] [CrossRef] [PubMed]
- Tobin, A.B.; Butcher, A.J.; Kong, K.C. Location, location, location. ..site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 2008, 29, (8), 413–20. [Google Scholar] [CrossRef]
- Hernández-Méndez, A.; Alcántara-Hernández, R.; García-Sáinz, J.A. Lysophosphatidic Acid LPA 1-3 receptors: signaling, regulation and in silico analysis of their putative phosphorylation sites. Recept Clin Invest 2014, 1, 236–252. [Google Scholar] [CrossRef]
- Ward, R.J.; Alvarez-Curto, E.; Milligan, G. Using the Flp-In T-Rex system to regulate GPCR expression. Methods Mol. Biol. 2011, 746, 21–37. [Google Scholar] [CrossRef]
- Thompson, A.K.; Mostafapour, S.P.; Denlinger, L.C.; Bleasdale, J.E.; Fisher, S.K. The aminosteroid U-73122 inhibits muscarinic receptor sequestration and phosphoinositide hydrolysis in SK-N-SH neuroblastoma cells. A role for Gp in receptor compartmentation. J. Biol. Chem. 1991, 266, (35), 23856–23862. [Google Scholar] [CrossRef]
- Smith, R.J.; Sam, L.M.; Justen, J.M.; Bundy, G.L.; Bala, G.A.; Bleasdale, J.E. Receptor-coupled signal transduction in human polymorphonuclear neutrophils: effects of a novel inhibitor of phospholipase C-dependent processes on cell responsiveness. J. Pharmacol. Exp. Ther. 1990, 253, (2), 688–97. [Google Scholar]
- Takasaki, J.; Saito, T.; Taniguchi, M.; Kawasaki, T.; Moritani, Y.; Hayashi, K.; Kobori, M. A novel Galphaq/11-selective inhibitor. J. Biol. Chem. 2004, 279, (46), 47438–45. [Google Scholar] [CrossRef]
- Uemura, T.; Kawasaki, T.; Taniguchi, M.; Moritani, Y.; Hayashi, K.; Saito, T.; Takasaki, J.; Uchida, W.; Miyata, K. Biological properties of a specific Galpha q/11 inhibitor, YM-254890, on platelet functions and thrombus formation under high-shear stress. Br. J. Pharmacol. 2006, 148, (1), 61–9. [Google Scholar] [CrossRef]
- Schonwasser, D.C.; Marais, R.M.; Marshall, C.J.; Parker, P.J. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 1998, 18, (2), 790–8. [Google Scholar] [CrossRef]
- von Kleist, L.; Stahlschmidt, W.; Bulut, H.; Gromova, K.; Puchkov, D.; Robertson, M.J.; MacGregor, K.A.; Tomilin, N.; Pechstein, A.; Chau, N.; Chircop, M.; Sakoff, J.; von Kries, J.P.; Saenger, W.; Krausslich, H.G.; Shupliakov, O.; Robinson, P.J.; McCluskey, A.; Haucke, V. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 2011, 146, (3), 471–84. [Google Scholar] [CrossRef]
- Shiraki, A.; Shimizu, S. The molecular associations in clathrin-coated pit regulate beta-arrestin-mediated MAPK signaling downstream of mu-opioid receptor. Biochem. Biophys. Res. Commun. 2023, 640, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Avendaño-Vázquez, S.E.; García-Caballero, A.; García-Sáinz, J.A. Phosphorylation and desensitization of the lysophosphatidic acid receptor LPA1. Biochem. J. 2005, 385, (Pt 3), 677–84. [Google Scholar] [CrossRef]
- Ohta, H.; Sato, K.; Murata, N.; Damirin, A.; Malchinkhuu, E.; Kon, J.; Kimura, T.; Tobo, M.; Yamazaki, Y.; Watanabe, T.; Yagi, M.; Sato, M.; Suzuki, R.; Murooka, H.; Sakai, T.; Nishitoba, T.; Im, D.S.; Nochi, H.; Tamoto, K.; Tomura, H.; Okajima, F. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol. Pharmacol. 2003, 64, (4), 994–1005. [Google Scholar] [CrossRef]
- Uemura, T.; Takamatsu, H.; Kawasaki, T.; Taniguchi, M.; Yamamoto, E.; Tomura, Y.; Uchida, W.; Miyata, K. Effect of YM-254890, a specific Galphaq/11 inhibitor, on experimental peripheral arterial disease in rats. Eur. J. Pharmacol. 2006, 536, (1–2), 154. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Alqahtani, S.; Nasrullah, M.Z.A.; Shen, J. Functional evidence for biased inhibition of G protein signaling by YM-254890 in human coronary artery endothelial cells. Eur. J. Pharmacol. 2021, 891, 173706. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Caron, M.G.; Barak, L.S. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 2000, 275, (22), 17201–10. [Google Scholar] [CrossRef]
- Tohgo, A.; Choy, E.W.; Gesty-Palmer, D.; Pierce, K.L.; Laporte, S.; Oakley, R.H.; Caron, M.G.; Lefkowitz, R.J.; Luttrell, L.M. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J. Biol. Chem. 2003, 278, (8), 6258–67. [Google Scholar] [CrossRef]
- Daaka, Y.; Luttrell, L.M.; Ahn, S.; Della Rocca, G.J.; Ferguson, S.S.; Caron, M.G.; Lefkowitz, R.J. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, (2), 685–8. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, (10), 1285–92. [Google Scholar] [CrossRef]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; Barty, A.; Latorraca, N.R.; Chapman, H.N.; Hubbell, W.L.; Dror, R.O.; Stevens, R.C.; Cherezov, V.; Gurevich, V.V.; Griffin, P.R.; Ernst, O.P.; Melcher, K.; Xu, H.E. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 2017, 170, (3), 457–469 e13. [Google Scholar] [CrossRef]
- Alcántara-Hernández, R.; Hernández-Méndez, A.; Romero-Ávila, M.T.; Alfonzo-Méndez, M.A.; Pupo, A.S.; García-Sáinz, J.A. Noradrenaline, oxymetazoline and phorbol myristate acetate induce distinct functional actions and phosphorylation patterns of alpha1A-adrenergic receptors. Biochim. Biophys. Acta 2017, 1864, (12), 2378–2388. [Google Scholar] [CrossRef]
- Alfonzo-Méndez, M.A.; Carmona-Rosas, G.; Hernández-Espinosa, D.A.; Romero-Ávila, M.T.; García-Sáinz, J.A. Different phosphorylation patterns regulate alpha1D-adrenoceptor signaling and desensitization. Biochim. Biophys. Acta 2018, 1865, (6), 842–854. [Google Scholar] [CrossRef]
- Hernández-Espinosa, D.A.; Carmona-Rosas, G.; Alfonzo-Méndez, M.A.; Alcántara-Hernández, R.; García-Sáinz, J.A. Sites phosphorylated in human alpha1B-adrenoceptors in response to noradrenaline and phorbol myristate acetate. Biochim Biophys Acta Mol Cell Res 2019, 1866, (10), 1509–1519. [Google Scholar] [CrossRef]
- Brose, N.; Rosenmund, C. Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J. Cell Sci. 2002, 115, (Pt 23), 4399–411. [Google Scholar] [CrossRef]
- Colon-Gonzalez, F.; Kazanietz, M.G. C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochim. Biophys. Acta 2006, 1761, (8), 827–37. [Google Scholar] [CrossRef]
- García-Sáinz, J.A.; Romero-Ávila, M.T.; Ruiz-Arriaga, A.; Ruiz-Puente, J.; Agundis, C.; Ortiz, V.; Isibasi, A. Characterization and detoxification of an easily prepared acellular pertussis vaccine. Antigenic role of the A protomer of pertussis toxin. Vaccine 1992, 10, (5), 341–4. [Google Scholar]
- Bouzo-Lorenzo, M.; Santo-Zas, I.; Lodeiro, M.; Nogueiras, R.; Casanueva, F.F.; Castro, M.; Pazos, Y.; Tobin, A.B.; Butcher, A.J.; Camina, J.P. Distinct phosphorylation sites on the ghrelin receptor, GHSR1a, establish a code that determines the functions of beta-arrestins. Sci Rep 2016, 6, 22495. [Google Scholar] [CrossRef] [PubMed]
- Lazari, M.F.; Liu, X.; Nakamura, K.; Benovic, J.L.; Ascoli, M. Role of G protein-coupled receptor kinases on the agonist-induced phosphorylation and internalization of the follitropin receptor. Mol. Endocrinol. 1999, 13, (6), 866–78. [Google Scholar] [CrossRef]
- Martínez-Morales, J.C.; González-Ruiz, K.D.; Romero-Ávila, M.T.; Rincón-Heredia, R.; Reyes-Cruz, G.; García-Sáinz, J.A. Lysophosphatidic acid receptor LPA(1) trafficking and interaction with Rab proteins, as evidenced by Forster resonance energy transfer. Mol. Cell. Endocrinol. 2023, 570, 111930. [Google Scholar] [CrossRef] [PubMed]
- García-Sáinz, J.A.; Romero-Ávila, M.T.; Medina, L.C. alpha(1D)-Adrenergic receptors constitutive activity and reduced expression at the plasma membrane. Methods Enzymol. 2010, 484, 109–25. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, (6), 3440–50. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, (5259), 680–5. [Google Scholar] [CrossRef]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, (5), 629–33. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ. In National Institutes of Health http://rsb.info.nih.gov/ij/, 1997-2004.
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; Tinevez, J.Y.; White, D.J.; Hartenstein, V.; Eliceiri, K.; Tomancak, P.; Cardona, A. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012, 9, (7), 676–82. [Google Scholar] [CrossRef]
- Carmona-Rosas, G.; Alfonzo-Méndez, M.A.; Hernández-Espinosa, D.A.; Romero-Ávila, M.T.; García-Sáinz, J.A. A549 cells as a model to study endogenous LPA1 receptor signaling and regulation. Eur. J. Pharmacol. 2017, 815, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Zhou, J.; Zhang, Y.; Shi, Y.; Peng, D.; Xue, Y. GPS 5. 0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genomics Proteomics Bioinformatics 2020, 18, (1), 72–80. [Google Scholar] [CrossRef]
- Dou, Y.; Yao, B.; Zhang, C. PhosphoSVM: prediction of phosphorylation sites by integrating various protein sequence attributes with a support vector machine. Amino Acids 2014, 46, (6), 1459–69. [Google Scholar] [CrossRef]
- Dou, Y.; Yao, B.; Zhang, C. Prediction of Protein Phosphorylation Sites by Integrating Secondary Structure Information and Other One-Dimensional Structural Properties. Methods Mol. Biol. 2017, 1484, 265–274. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
LPA3 receptors expressed are functional and markedly sensitive to heterologous but not to homologous desensitization. Panel A, Representative calcium tracings in response to 1 µM LPA (arrow) in HEK 293 cells (green line), and HEK293 TREx TM transfected to express LPA3 receptors (induced, black continuous line, and not induced, black dotted line). Panel B, calcium response to 1 µM LPA (arrow) of LPA3-expressing cells incubated in buffer with (black line) or without (red dotted line) calcium. Panel C, intracellular calcium tracings of LPA3-expressing cells preincubated for 30 min in the absence of any agent (black line) or presence of 10 µM U73122 (phospholipase C inhibitor, orange line) or 10 µM U73343 (inactive analog, red line) and challenged with 1 µM LPA (arrow). Thapsigargin 1 µM (Thap, second arrow). Panel D, calcium tracings of induced cells preincubated overnight without (black line) and with pertussis toxin (purple line); arrow indicated the addition of 1 µM LPA. Panel E, calcium tracings in response to 1 µM LPA of cells preincubated without any agent (black line) or with 1 µM LPA for 5 min and washed before re-challenging (black dotted line) with 1 µM PMA (PMA, red line). Panel D, calcium tracings in response to LPA of cells preincubated without any agent (None, open bar), with 1 µM LPA for 5 min and washed before re-challenging (LPA, dashed bar) or with 1 µM PMA (PMA, red dashed bar). The means are plotted, and vertical lines indicate the SEM of 4-5 experiments performed on different days and distinct cell cultures. ***p< 0.001 vs. baseline.
Figure 1.
LPA3 receptors expressed are functional and markedly sensitive to heterologous but not to homologous desensitization. Panel A, Representative calcium tracings in response to 1 µM LPA (arrow) in HEK 293 cells (green line), and HEK293 TREx TM transfected to express LPA3 receptors (induced, black continuous line, and not induced, black dotted line). Panel B, calcium response to 1 µM LPA (arrow) of LPA3-expressing cells incubated in buffer with (black line) or without (red dotted line) calcium. Panel C, intracellular calcium tracings of LPA3-expressing cells preincubated for 30 min in the absence of any agent (black line) or presence of 10 µM U73122 (phospholipase C inhibitor, orange line) or 10 µM U73343 (inactive analog, red line) and challenged with 1 µM LPA (arrow). Thapsigargin 1 µM (Thap, second arrow). Panel D, calcium tracings of induced cells preincubated overnight without (black line) and with pertussis toxin (purple line); arrow indicated the addition of 1 µM LPA. Panel E, calcium tracings in response to 1 µM LPA of cells preincubated without any agent (black line) or with 1 µM LPA for 5 min and washed before re-challenging (black dotted line) with 1 µM PMA (PMA, red line). Panel D, calcium tracings in response to LPA of cells preincubated without any agent (None, open bar), with 1 µM LPA for 5 min and washed before re-challenging (LPA, dashed bar) or with 1 µM PMA (PMA, red dashed bar). The means are plotted, and vertical lines indicate the SEM of 4-5 experiments performed on different days and distinct cell cultures. ***p< 0.001 vs. baseline.
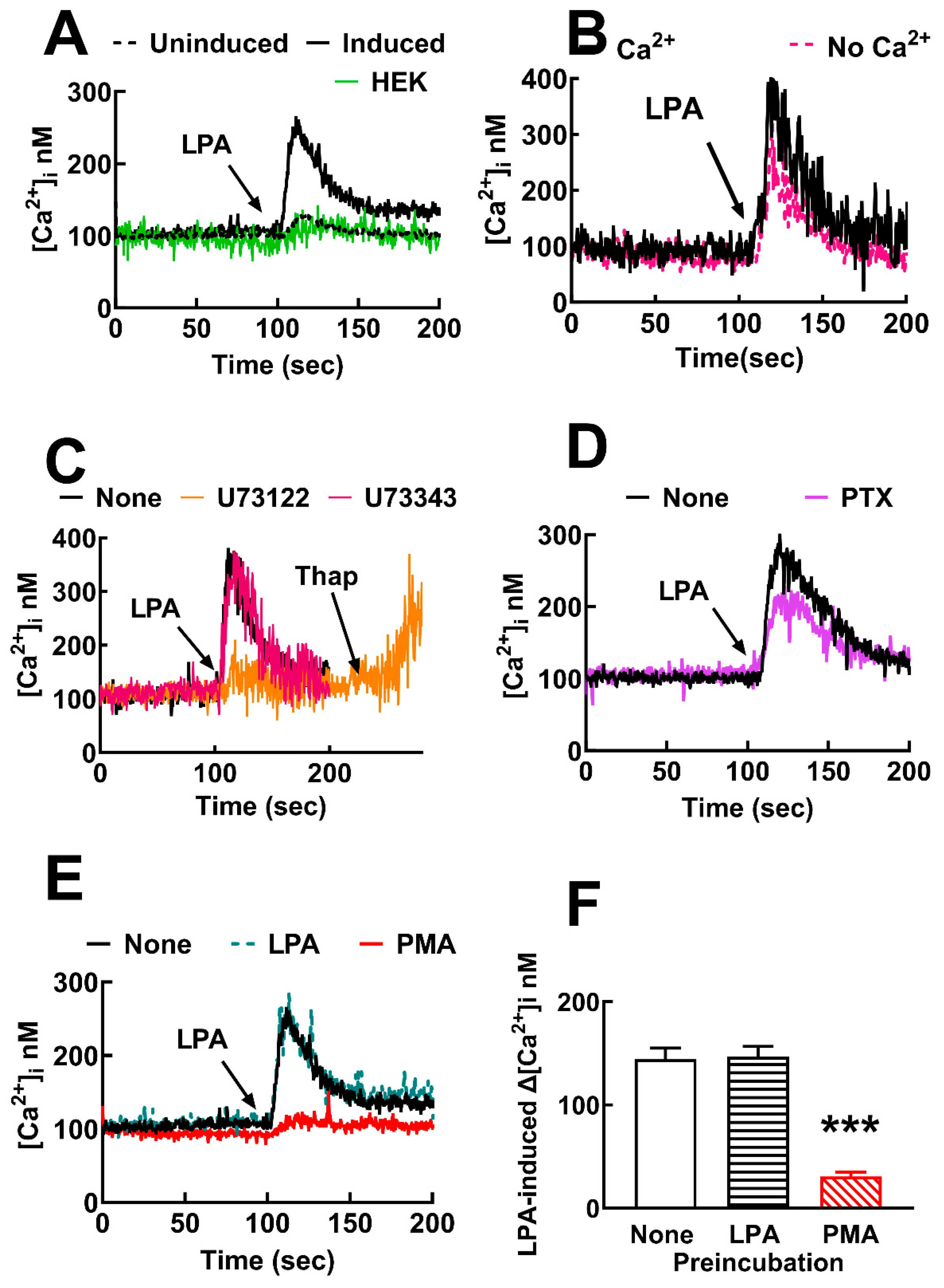
Figure 2.
Time-course of LPA- and PMA-induced ERK 1/2 phosphorylation in LPA3 receptor-expressing cells. Cells were stimulated with 1 µM LPA (black symbols and line) or 1 µM PMA (graphical black). The mean is plotted, and vertical lines indicate the SEM of 7-8 experiments performed on different days and distinct cell cultures. ***p < 0.0001, **p < 0.001, and *p< 0.01 vs. baseline value. Representative Western blots are presented above the Figure.
Figure 2.
Time-course of LPA- and PMA-induced ERK 1/2 phosphorylation in LPA3 receptor-expressing cells. Cells were stimulated with 1 µM LPA (black symbols and line) or 1 µM PMA (graphical black). The mean is plotted, and vertical lines indicate the SEM of 7-8 experiments performed on different days and distinct cell cultures. ***p < 0.0001, **p < 0.001, and *p< 0.01 vs. baseline value. Representative Western blots are presented above the Figure.
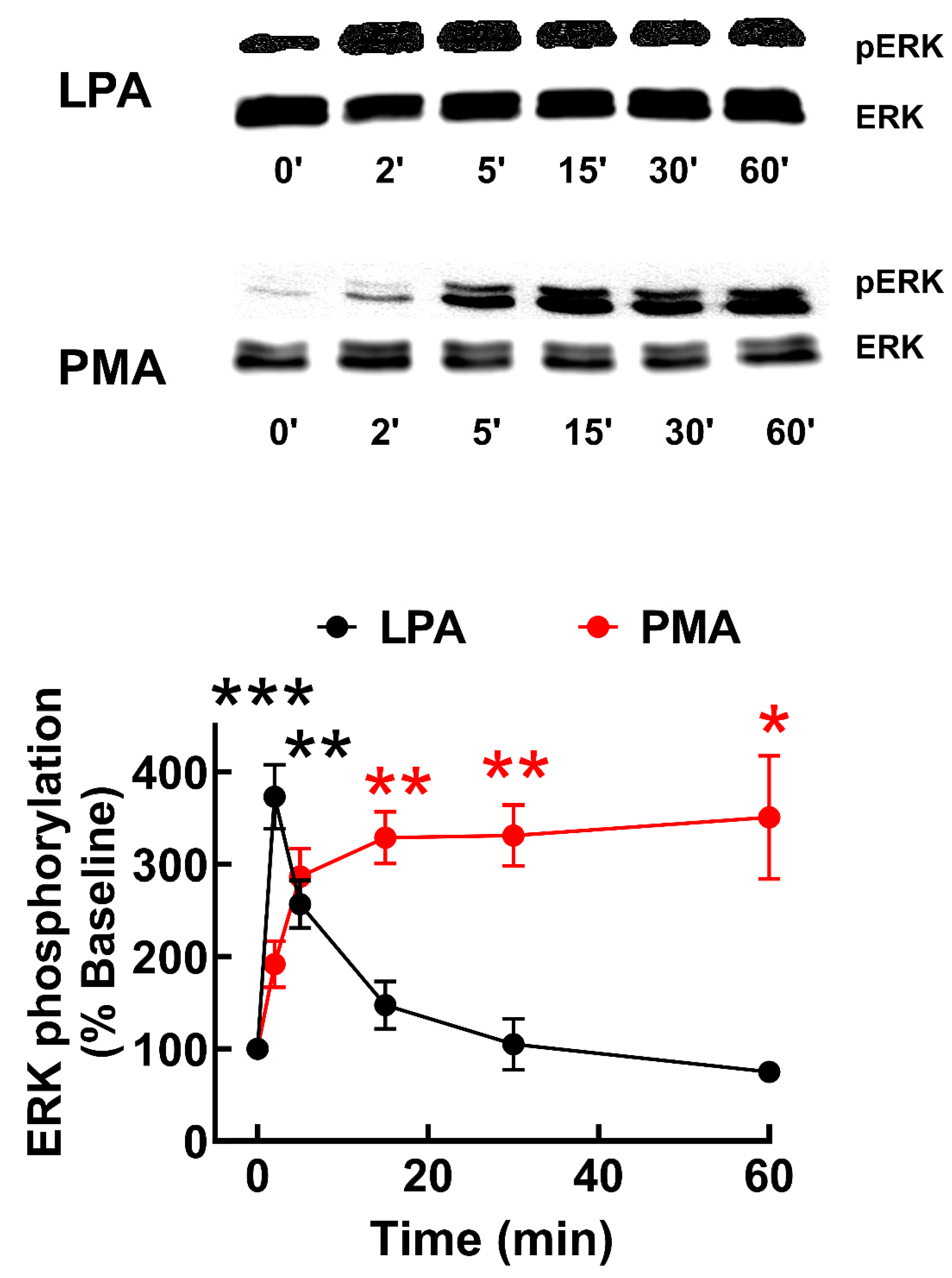
Figure 3.
Time-course of LPA- and PMA-mediated LPA3 internalization. Cells were incubated for the times indicated with 1µM LPA (black symbols and line) or 1µM PMA (red symbols and line). Internalization is presented as the percentage of baseline intracellular fluorescence. The mean is plotted, and vertical lines indicate the SEM of 14 images obtained from 5 experiments performed on different days and cell cultures. ***p < 0.001 vs. baseline value. Representative images are presented above the graph. Bars 10 µm.
Figure 3.
Time-course of LPA- and PMA-mediated LPA3 internalization. Cells were incubated for the times indicated with 1µM LPA (black symbols and line) or 1µM PMA (red symbols and line). Internalization is presented as the percentage of baseline intracellular fluorescence. The mean is plotted, and vertical lines indicate the SEM of 14 images obtained from 5 experiments performed on different days and cell cultures. ***p < 0.001 vs. baseline value. Representative images are presented above the graph. Bars 10 µm.
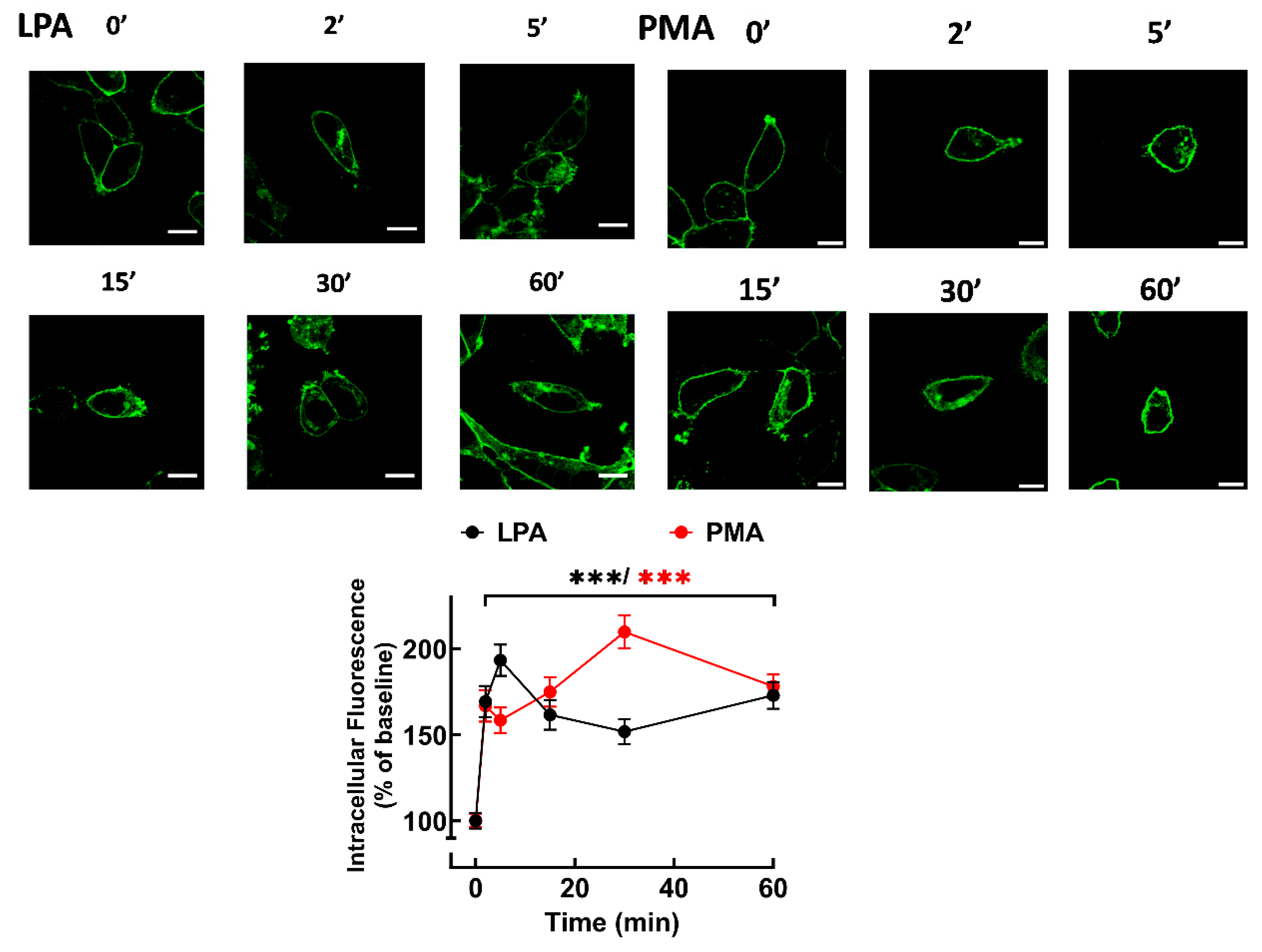
Figure 4.
LPA-induced LPA3 receptor-β-arrestin interaction. Panel A, Time-course of LPA3 receptor-β-arrestin interaction as reflected by FRET. Cells were challenged with 1µM LPA at time 0. The means are plotted, and vertical lines indicate the SEM of 5-6 experiments performed on different days and distinct cell cultures. ***p < 0.0001 and *p < 0.05 vs. baseline value. Panel B, Representative FRET images. Panel C, Representative images showing colocalization (white) of LPA3-eGFP receptors (green) and β-arrestin-mCherry (red). Bars 10 µm. .
Figure 4.
LPA-induced LPA3 receptor-β-arrestin interaction. Panel A, Time-course of LPA3 receptor-β-arrestin interaction as reflected by FRET. Cells were challenged with 1µM LPA at time 0. The means are plotted, and vertical lines indicate the SEM of 5-6 experiments performed on different days and distinct cell cultures. ***p < 0.0001 and *p < 0.05 vs. baseline value. Panel B, Representative FRET images. Panel C, Representative images showing colocalization (white) of LPA3-eGFP receptors (green) and β-arrestin-mCherry (red). Bars 10 µm. .
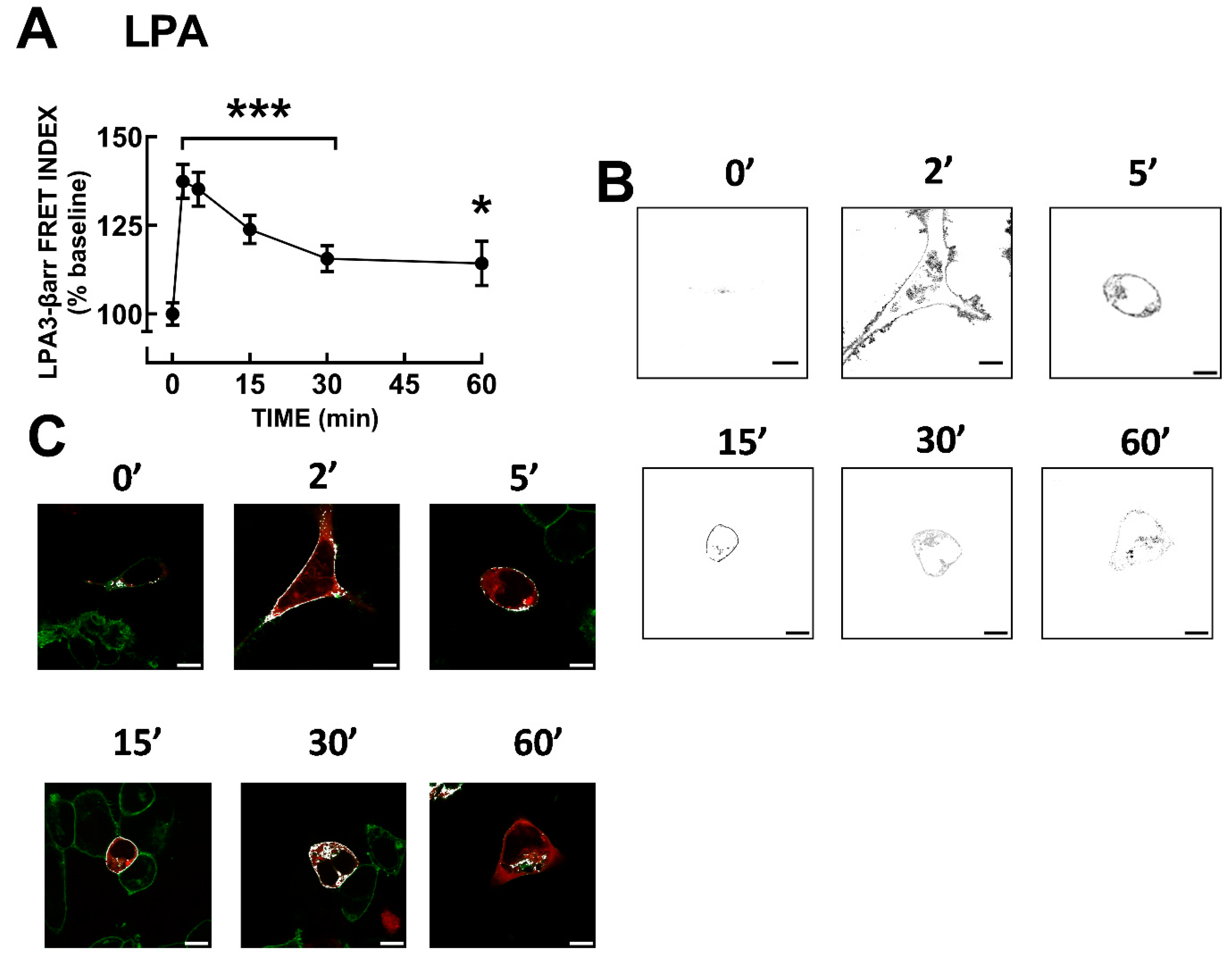
Figure 5.
PMA-induced LPA3 receptor-β-arrestin interaction. Panel A, Time-course of LPA3 receptor-β-arrestin interaction as reflected by FRET. Cells were challenged with 1µM PMA at time 0. The means are plotted, and vertical lines indicate the SEM of 5-6 experiments performed on different days and distinct cell cultures. ***p < 0.0001 and *p < 0.05 vs. baseline value. Panel B, Representative FRET images. Panel C, Representative images showing colocalization (white) of LPA3-eGFP receptors (green) and β-arrestin-mCherry (red). Bars 10 µm.
Figure 5.
PMA-induced LPA3 receptor-β-arrestin interaction. Panel A, Time-course of LPA3 receptor-β-arrestin interaction as reflected by FRET. Cells were challenged with 1µM PMA at time 0. The means are plotted, and vertical lines indicate the SEM of 5-6 experiments performed on different days and distinct cell cultures. ***p < 0.0001 and *p < 0.05 vs. baseline value. Panel B, Representative FRET images. Panel C, Representative images showing colocalization (white) of LPA3-eGFP receptors (green) and β-arrestin-mCherry (red). Bars 10 µm.
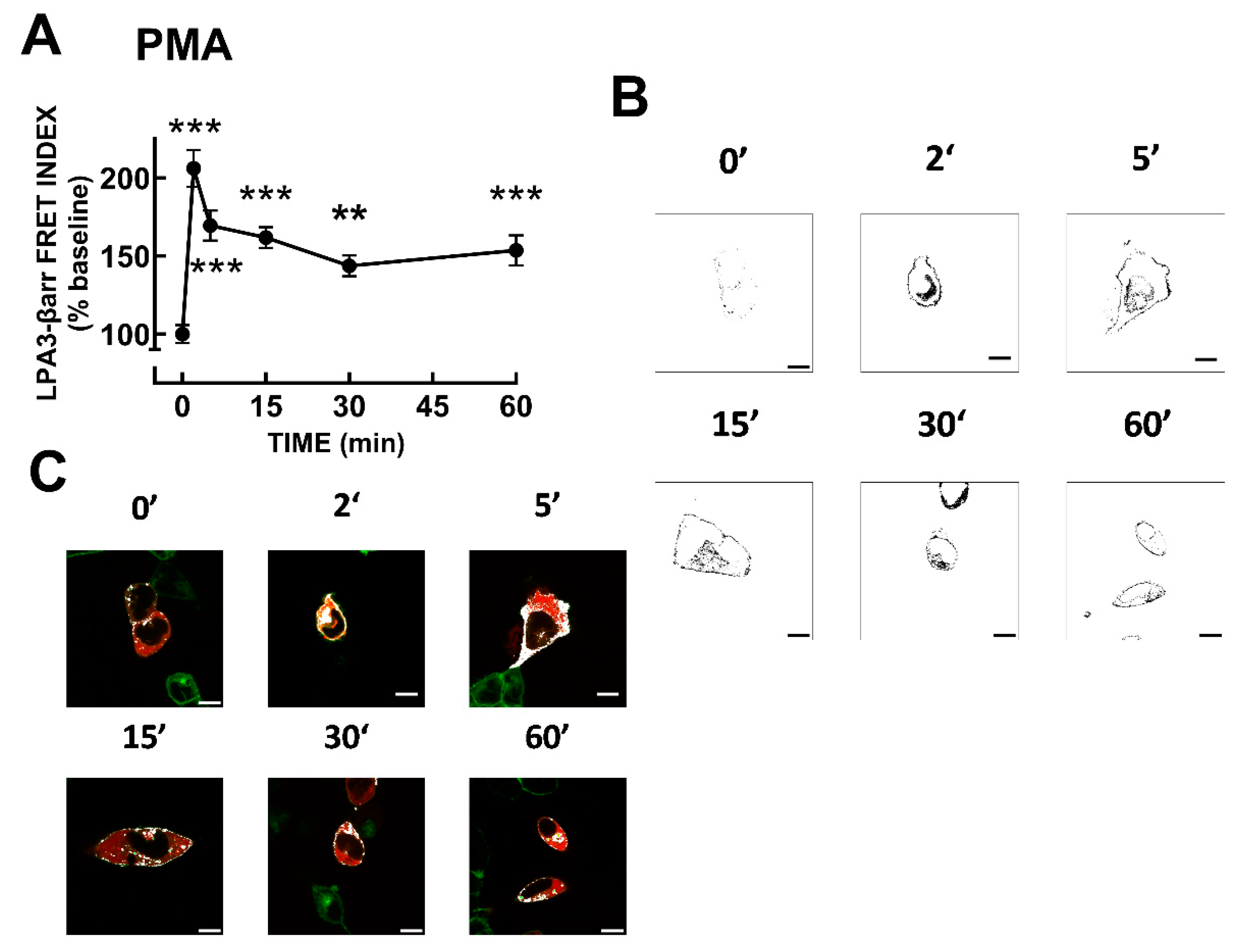
Figure 6.
LPA- and PMA-induced LPA3 receptor phosphorylation. Cells were challenged with 1 µM LPA or 1 µM PMA at time 0. Receptor phosphorylation is expressed as the percentage of the baseline value. The means are plotted, and vertical lines indicate the SEM of 9 experiments performed on different days using different cell cultures. **p < 0.01, *p < 0.05 vs. baseline value. Representative autoradiographs (32P) and Western blots (WB) are presented above the graph.
Figure 6.
LPA- and PMA-induced LPA3 receptor phosphorylation. Cells were challenged with 1 µM LPA or 1 µM PMA at time 0. Receptor phosphorylation is expressed as the percentage of the baseline value. The means are plotted, and vertical lines indicate the SEM of 9 experiments performed on different days using different cell cultures. **p < 0.01, *p < 0.05 vs. baseline value. Representative autoradiographs (32P) and Western blots (WB) are presented above the graph.
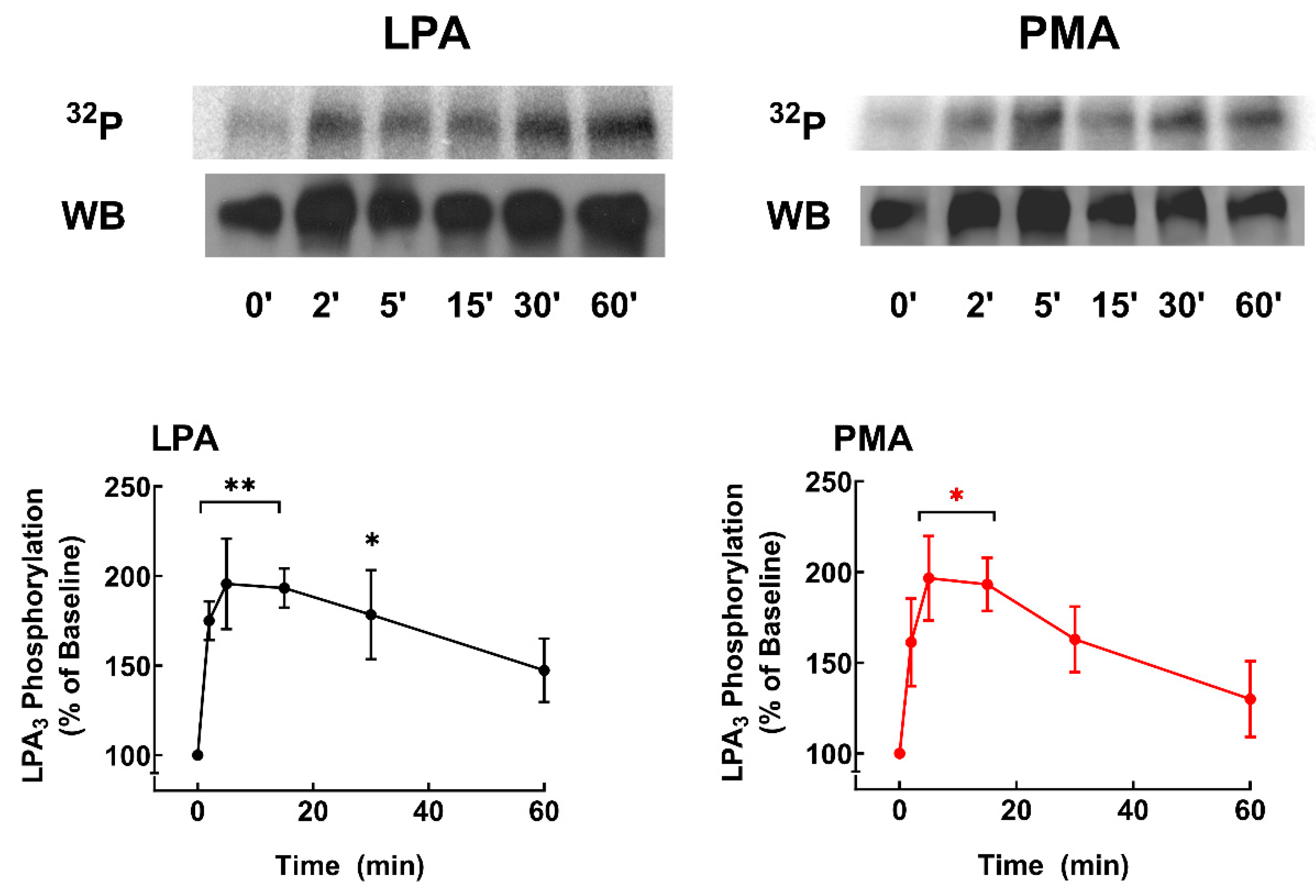
Figure 7.
Models showing the LPA3 receptor phosphorylation sites predicted in silico (panel A) and obtained experimentally (mass spectrometry, panel B). Phosphorylation sites are indicated in red.
Figure 7.
Models showing the LPA3 receptor phosphorylation sites predicted in silico (panel A) and obtained experimentally (mass spectrometry, panel B). Phosphorylation sites are indicated in red.
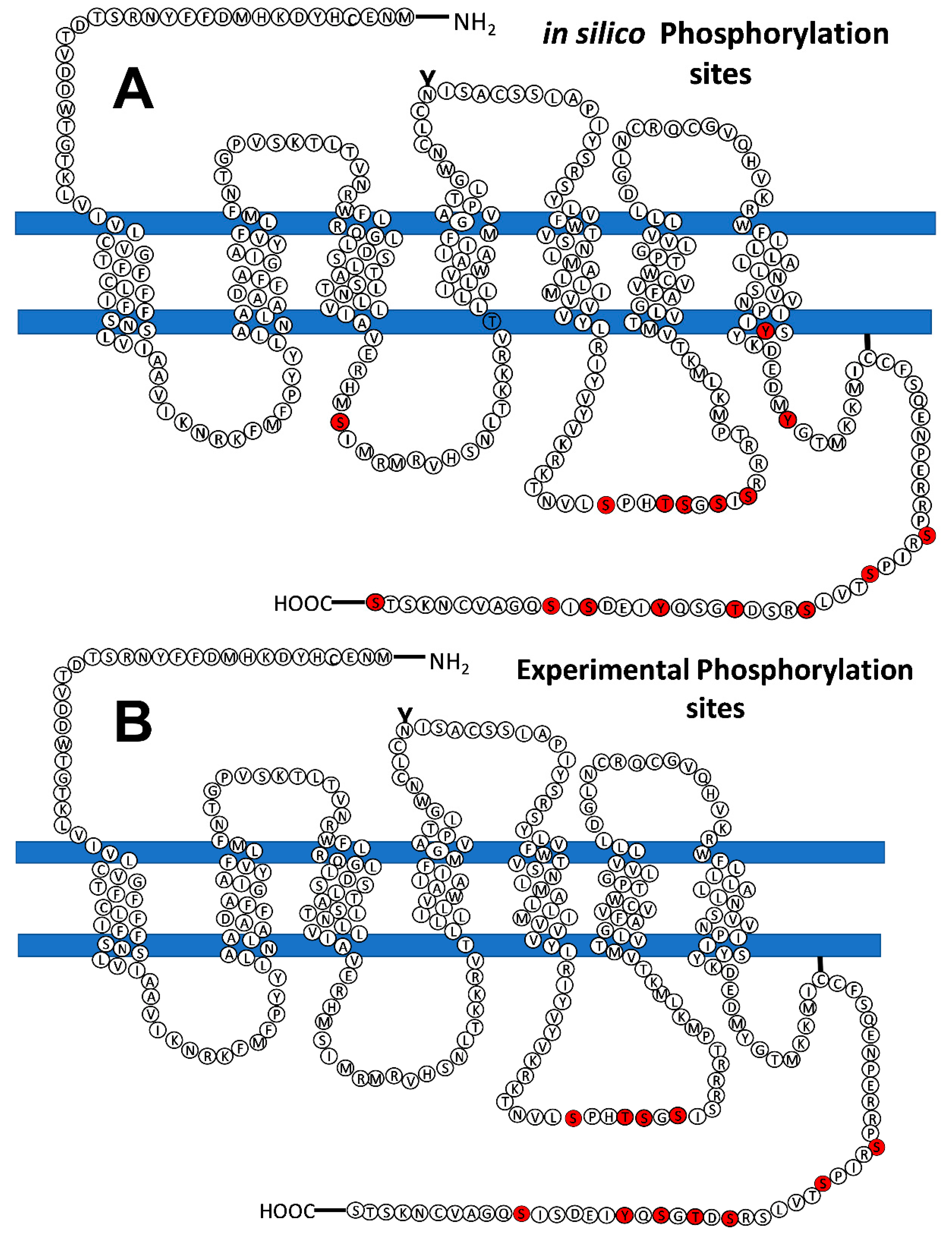
Figure 8.
Sequence alignment of the intracellular loop 3 and the carboxyl terminus of the three human LPA receptors belonging to the lysophosphatidic acid family. Amino acids detected phosphorylated in mass spectrometry in the LPA3 receptors or conserved in the other subtypes are marked in red.
Figure 8.
Sequence alignment of the intracellular loop 3 and the carboxyl terminus of the three human LPA receptors belonging to the lysophosphatidic acid family. Amino acids detected phosphorylated in mass spectrometry in the LPA3 receptors or conserved in the other subtypes are marked in red.

Figure 9.
Sequence alignment of the intracellular loop 3 and the carboxyl terminus of the three human LPA receptors belonging to the lysophosphatidic acid family. Conserved amino acids detected phosphorylated (human) among LPA3 receptors orthologs. The intracellular loop 3 of the distinct orthologs is indicated in green, and the conserved (compared to the human ortholog) phosphorylated amino acids are marked in red. The carboxyl terminus is indicated in yellow, and the conserved phosphorylated amino acids are marked in blue.
Figure 9.
Sequence alignment of the intracellular loop 3 and the carboxyl terminus of the three human LPA receptors belonging to the lysophosphatidic acid family. Conserved amino acids detected phosphorylated (human) among LPA3 receptors orthologs. The intracellular loop 3 of the distinct orthologs is indicated in green, and the conserved (compared to the human ortholog) phosphorylated amino acids are marked in red. The carboxyl terminus is indicated in yellow, and the conserved phosphorylated amino acids are marked in blue.


Table 1.
Receptor phosphorylation sites detected in silico. Protein kinases are abbreviated as suggested by Uniprot (https://www.uniprot.org/uniprotkb/P80885/entry). IL3, intracellular loop 3; Ctail, carboxyl terminus. .
Table 1.
Receptor phosphorylation sites detected in silico. Protein kinases are abbreviated as suggested by Uniprot (https://www.uniprot.org/uniprotkb/P80885/entry). IL3, intracellular loop 3; Ctail, carboxyl terminus. .
| Kinases | Site | Amino acid | Sequence | Domain |
|---|---|---|---|---|
| CAMK, CAMKL CK1/VRK Pyk 2, JAK GRK 4 CAMK, MAPK PKC α, β, γ, δ TK TK PKA, PKC α, ζ, GRK 3 PKA, PKC α, ε, GRK 3, 5 Akt PKA GRK 2 AGC/GRK AGC/GRK PKC γ, δ, ζ, GRK 1, 2, 4, |
130 221 224 225 227 229 293 301 321 325 329 333 337 341 343 353 |
S S T S S S Y Y S S S T Y S S S |
IAVERHMSIMRMRVH KRKTNVLSPHTSGS TNVLSPHTSGSISRR NVLSPHTSGSISRRR LSPHTSGSISRRRTP PHTSGSISRRRTPMK SVVNPIIYSYKDEDM SYKDEDMYGTMKKMI ENPERRPSRIPSTVL RRPSRIPSTVLSRSD RIPSTVLSRSDTGSQ TVLSRSDTGSQYIED RSDTGSQYIEDSISQ GSQYIEDSISQGAVC QYIEDSISQGAVCNK AVCNKSTS |
IL 2 IL 3 IL 3 IL 3 IL 3 IL 3 Ctail Ctail Ctail Ctail Ctail Ctail Ctail Ctail Ctail Ctail |
Table 2.
Receptor sites detected phosphorylated by mass spectrometry. Conditions: baseline (B, no color background), and stimulations by 1 µM LPA (grey) or 1 µM PMA (red); the numbers inside the boxes indicate the times peptides were detected phosphorylated in the experiments.
Table 2.
Receptor sites detected phosphorylated by mass spectrometry. Conditions: baseline (B, no color background), and stimulations by 1 µM LPA (grey) or 1 µM PMA (red); the numbers inside the boxes indicate the times peptides were detected phosphorylated in the experiments.
| Location | Amino acid | B | LPA | PMA |
|---|---|---|---|---|
|
221* 224* 225* 229* 321* 325* 331 333* 335 337* 343* |
S T S S S S S T S Y S |
33 13 59 31 27 23 21 20 |
26 18 12 18 40 11 15 19 |
49 18 14 90 31 22 20 13 20 |
* Suggested in the in silico analyses.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim respon-sibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



