
Submitted:
08 April 2024
Posted:
09 April 2024
You are already at the latest version
Abstract
The clinical features of increasing muscle weakening are shared by a diverse set of illnesses known as muscular dystrophies. In early childhood, males with Duchenne muscular dystrophy (DMD) exhibit proximal muscle weakening and calf enlargement, making it the most frequent X-linked disease of muscular dystrophy in children. Development of motor skills is typically delayed, and wheelchair confinement eventually leads to early death from heart or lung problems. In terms of function, ambulation, quality of life, and life expectancy, treatment approaches like corticosteroid therapy and intermittent positive pressure breathing have improved. This is a case report of a 6-year-old kid who had early ambulatory stage Duchenne Muscular Dystrophy and complained to the neuro-physiotherapy department about frequent falls, walking difficulties, and dyspnea when walking. Other grievances included being unable to get off the ground and having trouble ascending stairs. He had physiotherapy and dynamic neuromuscular stabilization for his complaints. The main goals of treatment were educating the family, respiratory training, preventing ankle plantar flexors, maintaining strength, balance and gait training.
Keywords:
Duchenne muscular dystrophy
; Balance
; Gower’s sign
; Rehabilitation
; Physiotherapy
; Dynamic neuromuscular stabilization
1. Introduction
Duchenne muscular dystrophy (DMD) is a severe genetic illness defined by progressive muscle weakness. It is a multisystemic condition that results in degeneration of skeletal, heart and lungs [1]. The cause is the protein dystrophin, which helps to preserve the structural integrity of muscle cells. The initial symptoms appear in individuals between the ages of two and three. These include difficulty climbing stairs, a waddling gate, and frequent falls [1]. When the majority of individuals reach the age of 20, they end up wheelchair-dependent and need assisted breathing. Even with the finest care available, most DMD patients die from heart and respiratory illness around the ages of 20 and 40 [1]. Dystrophinopathies are X-linked recessive disorders that affect one in 5,000 to 6,000 live male neonates [2]. The estimated incidence of DMD is less than 10 cases per 100,000 males, and it seems to be constant throughout the world [2]. In contrast, for each 100,000 live male births, there are less than 8 cases of BMD [3]. With appropriate care, patients with DMD can survive well into their forties. This is largely due to advancements in the treatment of cardio-respiratory dysfunction and the development of care and management guidelines [2]. The prognosis for DMD patients has improved over time [3]. It is advised to look for DMD in young boys (between the ages of two and four) who show signs of weakness in muscles, hypertrophic calves, delayed motor milestones and the Gowers sign. It is important to suspect DMD because speech delay is common, and 30% of DMD patients have cognitive impairment at presentation [4].
Mutations in DMD, the gene encoding dystrophin, lead to DMD by inhibiting the synthesis of the muscle isoform of dystrophin (Dp427m) [4]. Mutation in DMD can also result in mild forms of Becker muscular dystrophy (BMD). Compared to DMD, BMD occurs later and advances less rapidly [4]. Dystrophin links cytoskeletal F-actin to the extracellular matrix, which is found in muscle via its N- and C-terminal domains [5]. In DMD, nonsense mutations and fragment shifters prematurely stop the translation of proteins, making dystrophin unstable and non-functional [6]. These dystrophin transcripts appear to be unaffected by nonsense-mediated destruction, whereas transcript production decreases due to epigenetic modifications [5].
A more precise indicator of muscle injury is CK levels. Young males with elevated plasma CK levels and characteristic DMD symptoms are likely to have DMD; to confirm the diagnosis, it is important to determine the DMD mutation in those people [7]. Notably, high plasma CK levels alone cannot diagnose DMD since they are an insensitive marker for skeletal muscle degeneration; genetic confirmation is essential [7]. Duchenne Muscular Dystrophy (DMD) has no known cure, but by treating its symptoms, a multidisciplinary strategy combining medicinal, surgical, and rehabilitative procedures can extend a patient’s life [8]. In addition to affecting the muscles, DMD also manifests in non-muscular areas, requiring coordinated therapy across the spectrum of the disease [8]. From diagnosis to end-of-life requirements, including the transfer from paediatric to adult care and advanced care planning, this care should be patient-centred [8]. When it comes to multidisciplinary symptom treatment, physical therapy is crucial. The main goal of physical therapy interventions is to slow down the progression of the disease. The objectives of physical therapy include preserving the child’s ability to prevent deformity, manage discomfort, and help support the family in order to prolong function [9].
In order to attain ideal, comprehensive bodily function, dynamic neuromuscular stabilization (DNS) combines brain stimulation with manipulation, mobilization, postural awareness, breathing training, and teaching. It is a neuromuscular technique that diagnoses and treats motor abnormalities by utilizing the developing process of infants’ movements [10]. It's a rehabilitation approach where deep abdominal muscles are employed to activate the diaphragm before movement [10]. With a focus on diagnosis, DNS is founded on neurophysiology, neuroanatomy, muscular physiology, and kinesiology [11]. The Integrated Spinal Stabilisation System (ISSS), which is made up of the diaphragm, transvers-abdominis, multifidus, pelvic floor, cervical flexors and extensors, and other intrinsic muscles of the spine, is precisely co-activated in this process. By using developmental positions to establish appropriate neural pathways and support the infant’s progress, DNS is used with paediatric patients to resolve notable deficits in developmental milestones.
Attaining optimal trunk stabilization serves as a cornerstone in all training regimens. In order to cure respiratory issues, postural errors must be addressed, and the costovertebral joints must be mobilized and interact with intercostal tissue and trunk fascia. Notably, stabilization and breathing patterns are interlinked, as respiratory muscles function as stabilizers and vice versa. Thus, integrating exercises that target respiratory-postural function in diverse positions is advantageous.
2. Case Presentation
As told by his mother (primary caregiver), a 6-year-old kid complained of difficulty in standing from sitting position, frequent stumbles, walking difficulties and dyspnea when walking over the past year. He was seen at the neuro-physiotherapy department. Having trouble mounting stairs and being unable to get off the ground were two more related problems. It was stated that he had a normal cognitive quotient. A serological examination revealed an increased level of creatine kinase (CK) at 11975 U/L. His maternal uncle passed away from the same disease at a young age thus the family history was important. At the age of two, the patient reports having toe-walked. Timeline shown in Table 1.
2.1. Clinical Examination
After gaining oral consent from the caregiver, a physiotherapy assessment was conducted. His speech, hearing and vision were intact. He was assessed in supine lying position. The patient was afebrile, i.e. 370 C, his respiratory rate was 24 breaths/ minute, his pulse rate was 98 beats/minute, and he was able to sustain oxygen saturation in room air. Upon assessment, the infant appeared obese, and both of his feet had Pes planus. There was Gower’s sign, proximal weakness, and hypertrophy of the calf muscles. Muscle tone, cranial nerve testing, and thinning and twitching of the muscles were all normal. Upon neurological assessment, all of the sensations were intact. Muscle tone was normal (2+). The superficial and deep tendon reflexes were normal (2+). Bilateral upper and lower limb manual muscle testing and Range of motion, as seen in Table 2. The girth measurement showed hypertrophied calves. The patient was experiencing the disease’s ambulatory phase. Chest expansion was found to be reduced at the axillary, nipple, and xiphisternum levels, respectively, during the respiratory examination. With the use of a spirometer, the vital lung capacity was determined to be 600 CC.
2.2. Posture and Gait Assessment
A posture assessment in the standing position indicated a hyper-lordotic curve in the lumbar spine, an anterior pelvic tilt, forward shifting of the weight, and bilateral thick calf muscles. The examination in the sitting position revealed a disappearance of the hyper-lordotic curve and an increase in thoracic kyphosis. The results of the gait study showed a broad base of support, a reduced heel strike, a shorter stride length, a waddling gait, and an enhanced arm swing. Figure 2(a) shows that lower limb is externally rotated and ankle plantarflexed and Figure 2(b) shows clinical photograph of patient.
2.3. Prognosis
DMD is an incurable condition that worsens over time. Although respiratory infections and cardiomyopathy are typically the cause of death for DMD patients, advancements in cardiac and respiratory treatment can increase survival rates. The loss of muscle mass can be slowed down with the usage of steroids. Table 3 lists medications along with their dosages. Table 4 presents the management of physiotherapy. Table 3 lists medications along with their dosages. Table 4 presents the management of physiotherapy.
3. Discussion
This case study of dynamic neuromuscular stabilization and physiotherapy for a 6-year-oldmale with Duchenne Muscular Dystrophy exemplifies the profound impact of comprehensive rehabilitation strategies on improving functional outcomes and enhancing quality of life in children with neuromuscular disorders. This discussion aims to explore the implication of the reported findings, emphasizing the multifaceted approach to paediatric rehabilitation and the pivotal role of interdisciplinary and family involvement.
The comprehensive rehabilitation program described in the case study integrates DNS principles with physiotherapy techniques, such as strengthening exercises, stretching, and manual therapy. This multidisciplinary approach targets not only the physical aspects of the condition but also addresses functional limitations, respiratory function, and psychosocial well-being [12]. The observed improvements in motor function, postural control, and respiratory capacity underscore the transformative impact of rehabilitation interventions on the child's functional abilities and overall quality of life. By incorporating DNS principles, which emphasize the activation of core stabilizing muscles and optimal movement patterns, the child gains better control over movement and posture, leading to enhanced mobility and participation in daily activities [13]. Furthermore, physiotherapy interventions play a crucial role in addressing muscle weakness, contractures, and respiratory complications commonly associated with DMD. Through targeted exercises and respiratory support, the child experiences improved strength, endurance, and respiratory function, thereby enhancing overall functional capacity and reducing the risk of secondary complications [14,15].
Family involvement is paramount in paediatric rehabilitation, particularly in the case of chronic and progressive conditions like DMD. Caregivers play a central role in supporting the child’s rehabilitation journey, implementing home exercise programs, and advocating for their needs within the healthcare system. Educating and empowering families equips them with the knowledge and skills necessary to provide ongoing support and ensure continuity of care beyond clinical settings [16,17]. Despite the promising outcomes observed in this case study, paediatric rehabilitation for DMD poses significant challenges and considerations. Disease progression, comorbidities, and access to specialized care and resources can impact the efficacy of interventions and the trajectory of the child’s condition. Moreover, the transition from paediatric to adult services requires careful planning and coordination to ensure continuity of care and optimize long-term outcomes [18].
Advancements in rehabilitation technologies, such as assistive devices, orthotics, and respiratory support aids, offer promising avenues for enhancing outcomes in children with DMD. Integrating these technologies into rehabilitation protocols can improve functional independence, facilitate participation in daily activities, and enhance overall quality of life. However, ensuring equitable access to these technologies and addressing financial barriers remains a critical concern [19,20].
4. Conclusions
This case study showcases how Dynamic Neuromuscular Stabilization and physiotherapy significantly improved functional outcomes and quality of life for a 6-year-old boy with Duchenne muscular dystrophy. Integrated DNS principles and traditional physiotherapy techniques led to enhancements in motor function, stability, and overall well-being. These improvements supported increased independence, eased family burdens, and fostered positive familial dynamics. The study demonstrates the effectiveness of holistic rehabilitation methods for meeting the complex demands of children with neuromuscular disorders. Emphasizing evidence-based, multidisciplinary interventions tailored to individuals' specific challenges and goals, the research calls for continued exploration of rehabilitation strategies that prioritize functional gains, autonomy, and high-quality living experiences for paediatric patients and their families.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, NC, SH, AS and RR.; methodology, NC, SH, software, NC, SH, validation, SH, RR formal analysis, NC, SH,.; investigation, NC, SH, AS.; resources, NC, SH,.; data curation, SH,RR.; writing—original draft preparation, NC, SH,.; writing—review and editing NC, SH,.; visualization, AS, RR.; supervision, NC, SH,.; project administration, NC, SH,.; funding acquisition, NC, SH.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mercuri E, Bönnemann CG, Muntoni F: Muscular dystrophies. Lancet. 2019, 30, 2025–2038. [CrossRef]
- Ryder S, Leadley RM, Armstrong N, et al. : The burden, epidemiology, costs and treatment for duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. 2017, 26, 79. [Google Scholar] [CrossRef]
- Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N: A systematic review and meta-analysis on the epidemiology of duchenne and becker muscular dystrophy. Neuromuscul Disord. 2014, 24, 482–491. [CrossRef] [PubMed]
- Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT: Entries in the leiden duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006, 34, 135–144. [CrossRef] [PubMed]
- García-Rodríguez R, Hiller M, Jiménez-Gracia L, et al. : Premature termination codons in the DMD gene cause reduced local mRNA synthesis. Proc Natl Acad Sci U S A. 2020, 14, 16456–16464. [Google Scholar] [CrossRef]
- Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM: An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988, 2, 90–95. [CrossRef] [PubMed]
- Aartsma-Rus A, Hegde M, Ben-Omran T, et al. : Evidence-based consensus and systematic review on reducing the time to diagnosis of duchenne muscular dystrophy. J Pediatr. 2019, 204, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Moxley RT, Pandya S, Ciafaloni E, Fox DJ, Campbell K: Change in natural history of duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. J Child Neurol. 2010, 25, 1116–1129. [CrossRef] [PubMed]
- Duan D, Goemans N, Takeda SI, Mercuri E, Aartsma-Rus A: Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021, 18, 13. [CrossRef]
- Mahdieh L, Zolaktaf V, Karimi MT: Effects of dynamic neuromuscular stabilization (DNS) training on functional movements. Hum Mov Sci. 2020, 70, 102568. [CrossRef] [PubMed]
- Kim DH, An DH, Yoo WG: Effects of 4 weeks of dynamic neuromuscular stabilization training on balance and gait performance in an adolescent with spastic hemiparetic cerebral palsy. J Phys Ther Sci. 2017, 29, 1881–1882. [CrossRef] [PubMed]
- Sharma K, Yadav A: Dynamic neuromuscular stabilization- a narrative review. Int J Health Sci Res. 2020, 10, 221–231.
- Frank C, Kobesova A, Kolar P: Dynamic neuromuscular stabilization & sports rehabilitation. Int J Sports Phys Ther. 2013, 8, 62–73.
- Shahade PS, Mundada PH, Samal SS: Perks of rehabilitation in improving motor function in a nine-year-old male with duchenne muscular dystrophy: a case report. Cureus. 2022, 11, 30162. [CrossRef]
- Hammer S, Toussaint M, Vollsæter M, et al. : Exercise training in duchenne muscular dystrophy: a systematic review and meta-analysis. J Rehabil Med. 2022, 11, 00250. [Google Scholar] [CrossRef]
- Magliano L, D'Angelo MG, Vita G, et al. : Psychological and practical difficulties among parents and healthy siblings of children with duchenne vs. becker muscular dystrophy: an Italian comparative study. Acta Myol. 2014, 33, 136–143. [Google Scholar]
- Magliano L, Politano L: Family context in muscular dystrophies: psychosocial aspects and social integration. Acta Myol. 2016, 35, 96–99.
- Case LE, Apkon SD, Eagle M, et al. : Rehabilitation management of the patient with duchenne muscular dystrophy. Pediatrics. 2018, 142, 17–33. [Google Scholar] [CrossRef] [PubMed]
- e Souza MA, Figueiredo MM, de Baptista CR, Aldaves RD, Mattiello-Sverzut AC: Beneficial effects of ankle-foot orthosis daytime use on the gait of duchenne muscular dystrophy patients. Clin Biomech. 2016, 35, 102–110. [CrossRef] [PubMed]
- Gupta A, Nalini A, Arya SP, Vengalil S, Khanna M, Krishnan R, Taly AB: Ankle-foot orthosis in duchenne muscular dystrophy: a 4 year experience in a multidisciplinary neuromuscular disorders clinic. Indian J Pediatr. 2017, 84, 211–215. [CrossRef] [PubMed]
Figure 2.
(a) Lower limb externally rotated and ankle plantarflexed; (b) Clinical photograph of patient.
Figure 2.
(a) Lower limb externally rotated and ankle plantarflexed; (b) Clinical photograph of patient.
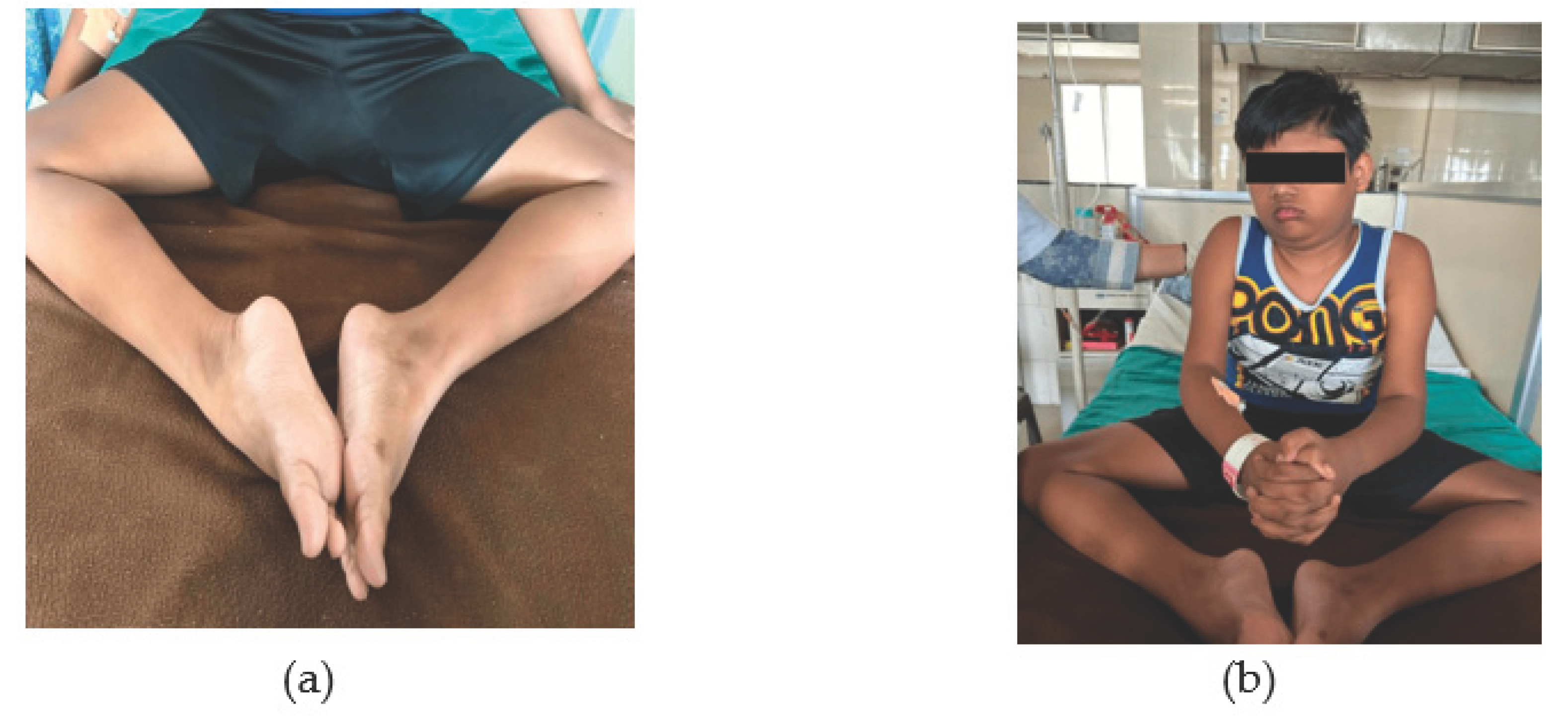

Table 1.
Timeline of events.
| Dates | Events |
|---|---|
| July 2023 | Diagnosed with Duchenne Muscular Dystrophy |
| 08/02/2024 | Got admitted with above complains |
| 09/02/2024 | Physiotherapy started |
Table 2.
Manual muscle testing and Range of motion.
| Joints | Movements | Range of motion | Manual muscle testing | ||
|---|---|---|---|---|---|
| Right | Left | Right | Left | ||
| Shoulder joint | Flexion | 0-1800 | 0-1800 | 4/5 | 4/5 |
| Extension | 0-350 | 0-390 | 4/5 | 4/5 | |
| Abduction | 0-1600 | 0-1630 | 4/5 | 4/5 | |
| Elbow joint | Flexion | 0-1450 | 0-1470 | 4/5 | 4/5 |
| Hip joint | Flexion | 0-740 | 0-720 | 3/5 | 3/5 |
| Extension | 0-160 | 0-190 | 3/5 | 3/5 | |
| Abduction | 0-300 | 0-350 | 3/5 | 3/5 | |
| Adduction | 0-200 | 0-240 | 3/5 | 3/5 | |
| Knee joint | Flexion | 0-1200 | 0-1200 | 3/5 | 3/5 |
3: Full range of motion against gravity; 4: Full range of motion against gravity with moderate resistance; 5: Full range of motion against gravity with maximum resistance.
Table 3.
Medications and its dosage.
| Medication | Dosage | Duration |
|---|---|---|
| Syrup Zincovit | 5 ml | BD |
| Syrup TusQ | 3.5 ml | BD |
| Tablet Folic acid | 5 mg | OD |
| Tablet iron | 30 mg | OD |
| Tablet deflazacort | 6 mg | OD |
BD: Bis in die (twice a day); OD: Once daily.
Table 4.
Physiotherapy Intervention.
| Goals | Therapeutic exercise |
|---|---|
| Patient education | Patient education is of paramount importance in Duchenne Muscular Dystrophy (DMD) due to the progressive nature of the condition and its impact on multiple aspects of an individual’s life. Here are several key reasons why patient education is crucial in DMD. The importance of treatment adherence is to be done. |
| To prevent plantar flexion of bilateral ankles | The splinting technique was applied using Ankle-foot orthosis |
| To promote healthy weight | Ensure that individuals with DMD receive adequate nutrition to support their energy needs while managing calorie intake to achieve or maintain a healthy weight. A balanced diet rich in nutrients such as protein, vitamins, and minerals is essential for supporting muscle function, growth, and overall health. |
| To promote and maintain strength of the upper and lower limb |
Proprioceptive neuromuscular facilitation technique for bilateral upper and lower limb (D1-D2 pattern) for bilateral upper and lower limbs, (Rhythmic initiation), (10 reps, 3 sets) |
| To promote Respiratory functions | Exercises in the Supine position • Position: Lie on your back. Set 1: Perform 10 repetitions with a 1-second inhale followed by a 2-second exhale. Rest for 60-90 seconds between sets. Set 2: Increase to 15 repetitions with a 2-second inhale and a 4-second exhale. Maintain a rest period of 60-90 seconds. Set 3: Progress to 20 repetitions with a 3-second inhale and a 6-second exhale. Allow a longer rest period of 120-150 seconds. Supine Position 90/90: Lie on your back with your arms positioned outside your body, mimicking the posture of a 3-month-old. Supine Position 90/90: Place your hands on your abdomen, replicating the position of a 4-month-old. Exercises in the Prone position • Prone Position: Support yourself on your elbows, resembling a position typically seen in infants at around 3 months old. • Creeping Position: With one hip and knee flexed, support yourself on your elbows while ensuring the opposite ASIS (anterior superior iliac spine) and medial epicondyle of the knee are in contact with the surface, resembling the posture of a 4.5-month-old. |
| To increase lung vital capacity | Devices like spirometers was used to increase lung vital capacity. |
| Balance and gait training | Tall kneeling • Tall knee walking • Tandem standing: one foot is placed in front of the other; this will work on balance when both feet are not next to each other • Kicking a ball: kicking a ball can also strengthen a child’s balance skills. • Standing on one leg: helps to control and stability when stopping and starting to step • Zig-zag walking • Walking over the obstacles • Treadmill training (5 minutes) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



