
Submitted:
08 April 2024
Posted:
09 April 2024
Read the latest preprint version here
Abstract
Receptor-interacting protein kinase 1 (RIPK1) plays a crucial role in controlling inflammation and cell death. Its function is tightly controlled through post-translational modifications, enabling its dynamic switch between promoting cell survival and triggering cell death. Phosphorylation of RIPK1 at various sites serves as a critical mechanism for regulating its activity, exerting either activating or inhibitory effects. Perturbations in RIPK1 phosphorylation status have profound implications for the development of severe inflammatory diseases in humans. This review explores the intricate regulation of RIPK1 phosphorylation and dephosphorylation and highlights the potential of targeting RIPK1 phosphorylation as a promising therapeutic strategy for mitigating human diseases.
Keywords:
RIPK1
; inflammation
; cell death
; phosphorylation
; dephosphorylation
; inflammatory diseases
Introduction
Receptor-interacting protein kinase 1 (RIPK1) plays a crucial role in regulating inflammation and cell death. Recent findings indicate that both genetic mutations and non-genetic factors influencing RIPK1 activity can lead to a range of inflammatory and degenerative diseases, highlighting the necessity for precise regulation of RIPK1 function in maintaining human health1,2.
The full-length human RIPK1 protein consists of 671 amino acids, with a molecular mass of approximately 76 kDa, sharing 68% identity with its mouse counterpart (Figure 1). Belonging to the RIP kinase family, RIPK1 is one of seven members, each featuring a homologous kinase domain (KD). Besides the common N-terminal kinase domain, RIPK1 possesses a C-terminal death domain (DD), facilitating its own dimerization or interaction with other death domain-containing proteins like TNFR1 (tumor necrosis factor receptor 1), TRADD (TNFR1-associated death domain protein) and FADD (Fas-associated death domain)3. Additionally, RIPK1 contains a bridging intermediate domain (ID) housing a RIP homotypic interaction motif (RHIM)4. The RHIM domain of RIPK1 facilitates its self-polymerization to form amyloid fibers5. It also allows interaction with other RHIM-containing proteins such as RIPK3, ZBP1 (Z-DNA binding protein 1, also known as DAI and DLM-1), and TRIF (TIR-domain-containing adapter-inducing interferon β)1,2.
RIPK1 primarily regulates inflammation through its scaffold function, while its involvement in cell death requires its kinase activity. The regulation of RIPK1 function involves various post-translational modifications, including ubiquitination, phosphorylation, and glycosylation. This review aims to summarize RIPK1’s impact on inflammation, cell survival, cell death, development and disease pathogenesis, with a focus on the role of phosphorylation and dephosphorylation.
RIPK1-Mediated Prosurvival and Inflammatory Signaling
RIPK1 was initially reported to strongly interact with the cell surface receptor Fas/APO-1 (CD95) and weakly with TNFR16. Subsequently, various members of the TNF superfamily, including TNFα, FasL, and TRAIL (TNF-related apoptosis-inducing ligand), were found to induce RIPK1-mediated NFκB activation3. Among these, the TNFα cascade has been extensively studied (Figure 2)7. Upon TNFα binding, RIPK1 and TRADD are rapidly recruited to TNFR1, initiating the assembly of complex I through mutual interactions between their death domains8-10. TRADD then recruits adaptor proteins TRAF2 and 5 (TNF receptor-associated factor protein 2/5), which in turn engage the E3 ubiquitin ligases cIAP1/2 (cellular inhibitor of apoptosis 1 and 2)11,12. cIAP1/2 catalyzes K63 ubiquitination of RIPK1, serving as a scaffold to recruit ubiquitin-binding proteins TAB2/3 (TAK1-binding protein 2/3) and TAK1 (transforming growth factor-β-activated kinase 1)13,14. TAK1 then activates the MAPK (mitogen-activated protein kinase) pathway, including p38 and MK2 (MAPK-activated kinase 2)15. Additionally, the K63 ubiquitin chain recruits another E3 complex LUBAC (the linear ubiquitin chain assembly complex), which catalyzes the M1 linear ubiquitin chains on RIPK1 and TNFR116,17. These linear ubiquitin chains recruit adaptor protein NEMO (NFκB essential modulator), which further engages IKKα/β (IκB kinase α/β) and TBK1/IKKε18−20. IKKα/β subsequently activates NFκB pathway21,22. Both the MAPK pathway and the NFκB pathway activate gene expression that promotes cell survival and inflammation to suppress cell death7,15.
RIPK1-Mediated Apoptosis
Apoptosis is regarded as a non-inflammatory form of programmed cell death, during which the contents of the dying cells are contained within apoptotic bodies. Caspases, a subfamily of cysteine proteases are both the initiators and executioners of apoptosis, as revealed by genetic and biochemical studies. Physiological and pathogen-related stimuli can trigger apoptosis through either extrinsic or intrinsic pathway. This process is vital for maintaining normal development and tissue homeostasis23,24.
RIPK1 primarily mediates extrinsic apoptosis induced by death receptor ligands, such as TNFα, FasL and TRAIL, dependent on cellular context1. Combining TNFα with a protein synthesis inhibitor cycloheximide (CHX), or an IAP antagonist Smac-mimetic can switch TNFα-induced inflammatory response to apoptosis (Figure 2)25. CHX promotes Caspase-8 activation by eliminating endogenous Caspase-8 inhibitor c-FLIP, leading to the formation of complex IIa including TRADD, FADD and Caspase-826. On the other hand, Smac-mimetic triggers cIAP1/2 autodegradation, releasing RIPK1 from the complex I to form complex IIb consisting of RIPK1, FADD, and Caspase-825,27,28. Activated Caspase-8 cleaves the preforms of caspasase-3/7 to execute apoptotic death. Activation of TNFR1 under various deficient conditions (TAK1, NEMO, TBK1, IKKα/β, A20 and ABIN1) also triggers RIPK1 activation and complex IIb formation, leading to RIPK1-dependent apoptosis29-35. Importantly, RIPK1 kinase activity is essential for TNF and Smac-mimetic-stimulated, but not for TNF and CHX-induced apoptosis, as demonstrated by RIPK1 knockdown, RIPK1 kinase inhibitor necrostatin, or kinase-dead mutants (K45A or D138N)10,25,36-38.
RIPK1-Mediated Necroptosis
Necroptosis, a proinflammatory form of programmed cell death, is characterized by cell membrane rupture and the release of damage-associated molecular patterns (DMAPs). While not essential for embryogenesis, necroptosis plays a vital role in immune defense against pathogens and is implicated in various human diseases, including inflammation, tissue damage and neurodegeneration39.
Necroptosis is initially suppressed by apoptosis, primarily through the cleavage of RIPK1 by activated Caspase-8 in complex II, for example, when induced by TNF and Smac-mimetic or CHX 40,41. However, when Caspase-8 is inactivated by specific inhibitors (such as Z-VAD-FMK) or genetic elimination, activated RIPK1 in complex IIb recruits RIPK3 through their respective RHIM domains, initiating the formation of another protein complex called the necrosome (Figure 2)5,42-46. Oligomerized RIPK3 then recruits the casein kinase 1 family proteins CK1α/δ/ε which phosphorylate Ser227 of human RIPK347. Phosphorylated RIPK3 subsequently recruits MLKL and phosphorylates human MLKL at Thr357 and Ser35848,49. Consequently, MLKL undergoes oligomerization into tetramers and amyloid-like polymers, which translocate to the plasma membrane, resulting in plasma membrane permeabilization50-56. In addition, activated MLKL translocates to the lysosomal membrane, where it forms amyloid-like polymers to facilitate lysosomal membrane permeabilization and the release of lysosomal proteases, thereby promoting cell death57.
RIPK1-mediated necroptosis requires its kinase activity, similar to its involvement in RIPK1-dependent apoptosis. For example, the RIPK1 inhibitor Nec-1 effectively prevents necroptosis induced by TNF, TLR-ligands and interferons43,58,59. Moreover, mice with kinase-dead RIPK1 knock-in mutations are resistant to TNF-induced necroptosis and systemic inflammatory response syndrome (SIRS), similar to RIPK3 knockout mice, and demonstrate superior resistance compared to MLKL knockout mice38,60,61.
Multiple innate immune signaling molecules, including death receptors (such as TNFR1), pathogen recognition receptors (such as TLR3 and TLR4), and cytosolic RNA sensor ZBP1, can induce necroptosis. Activation of these pathways all lead to the interaction of RHIM-containing proteins, such as RIPK1, TRIF or ZBP1 with the RHIM domain of RIPK3, activating RIPK3 and MLKL to promote necroptosis58,62-64.
The role of apoptosis in suppressing necroptosis is crucial for embryonic development. Deficiency in apoptosis components, such as the knockout of Caspase-8 or FADD, often results in late gestation embryonic lethality, primarily due to hyperactivation of necroptosis65,66. Simultaneous deletion of RIPK3 or MLKL can rescue the embryonic lethality of these mice, albeit with immune deficiencies in adulthood67,68 69.
Phosphorylation of RIPK1
- (1)
- Auto-activating phosphorylation
The serine-threonine kinase activity of RIPK1 is crucial for both complex IIb-dependent apoptosis and necroptosis70. Typically, kinases adopt a closed conformation and require phosphorylation in the activation loop, also known as the T-loop, to activate their kinase activity71. These activating phosphorylation events can be catalyzed by upstream kinases or achieved through autophosphorylation. Currently, autophosphorylation is the only known mechanism for activating RIPK1 (Figure 3). For instance, autophosphorylation of S161 stabilizes the open conformation of the T-loop and promotes human RIPK1 kinase activation to induce necroptosis43. Furthermore, mitochondrial reactive oxygen species (ROS) modify three essential cysteine residues of RIPK1, leading to cysteine-mediated aggregation of RIPK1 and subsequent autophosphorylation on S161, which is critical for RIPK1 to effectively promote necrosome formation and cell death72. Moreover, S166 autophosphorylation of RIPK1 is indispensable for MLKL activation and necrosome formation. Mutation of S166 effectively prevents multiple RIPK1 kinase-dependent inflammatory lesions in vivo, such as intestinal colitis, hepatitis, liver tumorigenesis, skin inflammation, and TNF-induced SIRS. Interestingly, while autophosphorylation of Ser166 is essential, it alone is not adequate to initiate RIPK1-mediated cell death73. Multiple autophosphorylation sites, including serine residues 14/15, 20, 161, and 166, cooperate to induce conformational changes in RIPK143,74. These changes facilitate its association with cell death effectors such as FADD and RIPK3, promoting the assembly of cell death-inducing signaling complexes, such as complex II and the necrosome. It is noteworthy that recombinant RHIM domain of RIPK1 exhibits a significantly higher affinity toward itself than the RHIM domain of RIPK35. Autophosphorylation of RIPK1 is thought to change its conformation, favoring the interaction between the RIPK1 and RIPK3 RHIM domains over the interactions between RIPK1 RHIM domains, thereby promoting necrosome formation47.
- (2)
- Inhibitory phosphorylation
The kinase activity of RIPK1 is tightly controlled at multiple levels to prevent spontaneous activation. Various post-translational modifications on RIPK1, such as ubiquitination and inhibitory phosphorylation, are intricately connected to keep RIPK1 kinase activity in check. For instance, in complex I, RIPK1 undergoes K63 ubiquitination by cIAP1/2 and M1 linear ubiquitination by LUBAC. These modifications stabilize Complex I, inhibiting its dissociation and formation of cell death-promoting complex II. In addition to activating the MAPK pathway and the NFκB pathway to activate gene expression that promotes cell survival and inflammation, TAK1 and IKK kinases further suppress cell death by performing inhibitory phosphorylations on RIPK1 to block its kinase activity (Figure 3). For instance, TAK1 activates MK2, which directly phosphorylates S320 and S335 of human RIPK1, or S321 and S336 of mouse RIPK1, to inhibit RIPK1 kinase activity and subsequent apoptosis or necroptosis75-77. Interestingly, TAK1 is also reported to directly phosphorylate mouse RIPK1 at S32129. In addition, TAK1 activates IKKα/β which in turn phosphorylates S25 in the kinase domain of RIPK1. Phosphorylation of S25 prevents ATP binding and inhibits RIPK1 kinase activation78. Furthermore, TBK1/IKKε phosphorylates T189 in the kinase domain to inhibit RIPK1 kinase activity32,79. It is important to note that MK2 phosphorylates cytosolic RIPK1, while IKKα/β, TBK1/IKKε and TAK1 phosphorylate ubiquitinated RIPK1 in complex I.
Recently, kinases outside of TNF pathway are also found to directly phosphorylate RIPK1 to inhibit its kinase activity and cell death. For example, glucose starvation activates AMPK (adenosine monophosphate-activated protein kinase) which phosphorylates S416 of human RIPK1 (or S415 of mouse RIPK1) to inhibit RIPK1 kinase activity and cell death80.
In addition to serine/threonine-phosphorylation, tyrosine phosphorylation has also been found to inhibit RIPK1 activity. Studies have shown that JAK1 (Janus Kinase 1) and Src kinases phosphorylate Y384 of human RIPK1 (or Y383 of mouse RIPK1) to inhibit RIPK1 kinase activity and subsequent cell death81.
Inhibitory phosphorylation of RIPK1 plays a pivotal role in host defense against pathogens and modulates inflammatory responses. For example, the gram-negative bacterial pathogen Yersinia counters host defense by inhibiting NFκB- and MAPK-mediated pro-inflammatory cytokines expression while promoting RIPK1 activation-dependent cell death82. Its effector protein acetyltransferase YopP/J elicits multiple functions in the process. First, it inactivates IKKα/β and ΤAΚ1 to block NF-κB and MAPK activation83. Second, it blocks the inhibitory phosphorylation of S25 of RIPK1 by IKKα/β to promote RIPK1-dependent macrophage cell death78. Lastly, it inactivates MK2, preventing inhibitory phosphorylation of S321 and S335 on RIPK1, thereby activating RIPK1-dependent cell death75,76,84. As a consequence, mice expressing the S25D-RIPK1 mutant fail to activate RIPK1-dependent cell death and are defective in defending against Yersinia infection, similar to the mice expressing the RIPK1 kinase-dead mutant K45A78. In addition, inhibitors of TAK1, IKKα/β, IKKε and MK2, the kinases responsible for the inhibitory phosphorylation of RIPK1, all exacerbate TNF-induced necroptosis and SIRS31,33,76,84,85.
Together, the inhibitory phosphorylations by these kinases function as crucial checkpoints to prevent RIPK1 kinase activation and subsequent cell death. Dysregulation of any of these inhibitory phosphorylation events leads to elevated cell death and is frequently associated with inflammatory diseases.
Dephosphorylation of RIPK1
Protein phosphatases play a complementary role in regulating phosphorylation homeostasis. These enzymes are classified into three main families based on the sequence similarity of the catalytic domain and substrate specificity: PTPs (protein tyrosine phosphatases), PPPs (phosphoprotein phosphatases), and PPMs (protein phosphatase metal-dependent). PTPs specifically dephosphorylate phosphotyrosine residues, while PPPs and PPMs dephosphorylate phosphoserine and phosphothreonine residues. In addition, a subfamily of PTPs, called the dual specificity phosphatases, dephosphorylate all three phospho-amino acids. PPPs and PPMs differ in that PPMs require metal ions, such as magnesium or manganese, for their activity and function as single subunit enzymes, while PPPs require regulatory subunits86.
PP1(protein phosphatase 1) is an important subfamily of PPPs. Its catalytic subunits (PP1c), including PP1α, PP1β, and PP1γ, are responsible for dephosphorylation of the majority phospho-serine and phospho-threonine sites in mammalian cells, regulating a broad range of cellular processes. Each PP1 catalytic subunit is obligatorily complexed with one or two regulatory subunits to form distinct PP1 holoenzymes. The regulatory subunits, also known as PP1-interacting proteins (PIPs) or regulatory interactors of protein phosphatase one (RIPPOs), determine substrate specificity by directing PP1c to the subcellular locations of its substrates and modulating its activity towards different substrates. There are approximately 200 validated PIPs, which assemble into more than 650 different PP1 holoenzymes in mammalian cells, enabling the dephosphorylation of diverse substrates87.
While numerous kinases have been identified to phosphorylate RIPK1, only a limited number of phosphatases are found to dephosphorylate RIPK1 or RIPK3. For example, Ppm1b, a metal ion-dependent phosphatases, dephosphorylates and inactivates RIPK3 to prevent the recruitment of MLKL into the necrosome, thus inhibiting subsequent necroptosis. Moreover, Ppm1b-/- mice exhibited heightened sensitivity to TNF-induced SIRS compared to WT mice, confirming its role in inhibiting necroptosis in vivo88.
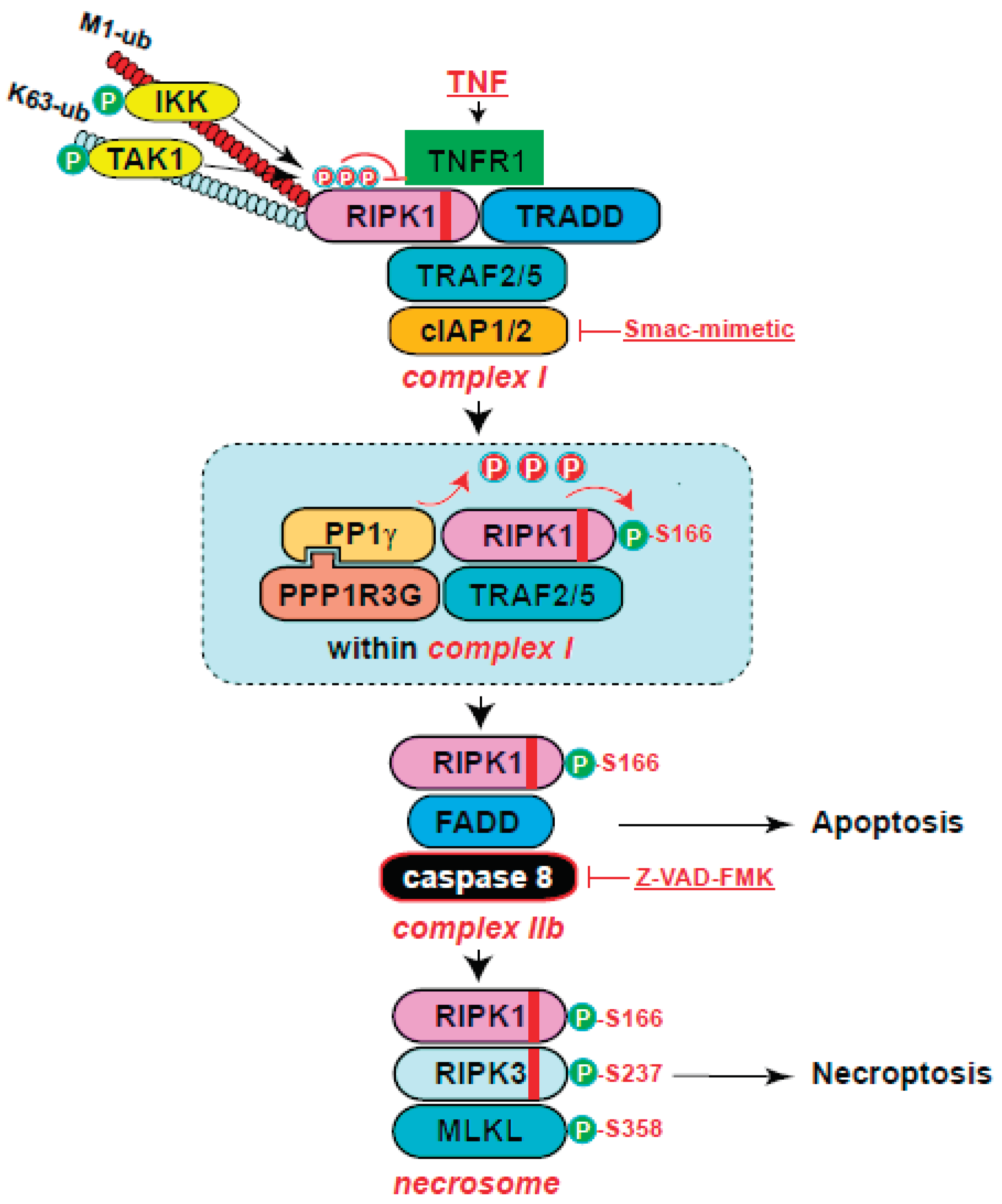
A sensitized CRISPR whole genome knockout screen revealed that PPP1R3G (protein phosphatase 1 regulator subunit 3G) is essential for necroptosis89. Specifically, PPP1R3G forms a holoenzyme with PP1γ to directly dephosphorylate the inhibitory phosphorylates sites of RIPK1, including S25, S320 and S335, thereby activating RIPK1-dependent apoptosis and necroptosis (Figure 4). An interesting note is that the holoenzyme does not remove the activating phosphorylation of S166 in vitro. In this context, upon treatment with TNF/Smac-mimetic/Z-VAD-FMK (T/S/Z), TRAF2 interacts with PPP1R3G to recruit the PPP1R3G/ PP1γ holoenzyme to complex I, where PP1γ dephosphorylates the inhibitory phosphorylation sites of RIPK1, activating RIPK1 kinase. Loss of PPP1R3G leads to loss of RIPK1 autophosphorylation at S166 and subsequent failure to form complex IIb to induce cell death. Like many other PP1 regulatory subunits, PPP1R3G interacts with PP1γ through a RVXF motif (X stands for any amino acids)90. Mutation of RVQF in PPP1R3G to RAQA disrupts the interaction with PP1γ. Importantly, the RAQA mutant fails to rescue RIPK1 activation and cell death in PPP1R3G knockout cells. Furthermore, prevention of RIPK1 inhibitory phosphorylations with p38 or IKK inhibitors or mutation of serine 25 of RIPK1 to alanine largely restores cell death in PPP1R3G-knockout cells. Finally, Ppp1r3g-/- mice are protected from TNF-induced SIRS, confirming the important role of PPP1R3G in regulating apoptosis and necroptosis in vivo. Due to experimental sensitivity limitations, the authors were unable to determine if PPP1R3G/PP1γ removes the inhibitory phosphorylation of T189. This warrants further analysis in the future. Additionally, it will be interesting to investigate if the PPP1R3G/PP1γ holoenzyme removes inhibitory phosphorylation of S415.
A recent study revealed that the PPP6C (protein phosphatase 6 catalytic subunit) is essential for necroptosis, identified through a CRISPR whole-genome knockout screen91. As previously reported92, PPP6C is recruited to complex I through TAB2, and dephosphorylates TAK1 to prevent inhibitory phosphorylations of RIPK1, thus activating TNF-induced necroptosis. Gastrointestinal tract-specific deletion of one alle of Ppp6c in mice could partially alleviate cecum damage caused by TNF-induced SIRS, confirming its role in necroptosis activation.
RIPK1 in Development
The scaffolding function, rather than kinase activity, of RIPK1 plays an important prosurvival role in regulating early postnatal lethality and inflammatory response by preventing apoptosis and necroptosis. Specifically, the death domain of RIPK1 binds the death domain of FADD to prevent FADD and Caspase-8-dependent apoptosis, while the RHIM domain of RIPK1 binds RHIM domains of RIPK3 and ZBP1, prevent their hyperactivation-induced necroptosis. For example, genetic deletion of RIPK1 in mice causes postnatal lethality93. While double knockout of RIPK3, Caspase-8 or FADD along with RIPK1 only marginally prolonged survival94-96, triple knockout of RIPK1, RIPK3 and either Caspase-8 or FADD rescued RIPK1-deficient mice, allowing them to survive weaning and mature normally97,98. The RHIM domain of RIPK1 inhibits ZBP1-RIPK3-MLKL-mediated necroptosis, crucial for preventing late embryonic lethality and adult skin inflammation99,100. Moreover, RIPK1 is essential for maintaining the survival of intestinal epithelial cells (IECs) by blocking apoptosis and necroptosis101. Additionally, mice harboring RIPK1 kinase-dead knock-in mutants, including D138N and K45A, survive to adulthood with no gross or histological abnormalities, indicating that RIPK1 kinase activity is dispensable for survival37,38.
RIPK1-Mediated Inflammatory Diseases
Many human inflammatory and neurodegenerative diseases have been found to be associated with abnormal RIPK1 expression or activity. Reports of genes mutations or non-genetic factors that affect RIPK1 activity are accumulating, highlighting the importance of RIPK1 regulation in human diseases.
Reduced RIPK1 expression can lead to various human diseases, largely due to the hyperactivation of RIPK3, ZBP1 and Caspase-8. As discussed previously, RIPK1 neutralizes RIPK3 and ZBP1 through RHIM domain interaction under normal conditions, and loss of RIPK1 leads to overactivation of ZBP1 and RIPK3, resulting in excessive necroptosis and systemic inflammation. At the meantime, RIPK1 inhibits FADD/Caspase-8-mediated apoptosis through death domain interaction during development, and loss of RIPK1 leads to excessive apoptosis. In humans, rare homozygous loss of function (LoF) mutations in RIPK1, including missense, nonsense and frameshift mutations, cause combined immunodeficiency and inflammatory bowel disease (IBD). Many of these patients also suffer from lymphopenia, recurrent infections, and arthritis102-105.
Conversely, elevated RIPK1 activity is also implicated in various human diseases due to heightened inflammation and cell death. For instance, rare mutations in RIPK1, such as D324N, D324H and D324Y at the Caspase-8 recognition site LQLD, block Caspase-8-mediated cleavage of RIPK1, resulting in an autosomal dominant autoinflammatory disease, characterized by recurrent fevers and lymphadenopathy106-108. Patients with these variants often have increased pro-inflammatory cytokines and chemokines, such as IL-6, TNF and CXCL2/3, and their peripheral blood mononuclear cells are hypersensitive to RIPK1 activation-dependent apoptosis and necroptosis induced by TNFα.
Furthermore, mutations in other genes that result in hyperactivation of RIPK1 kinase activity also lead to human diseases. For instance, monogenic mutations in genes like IKBKG (encoding NEMO), TNIP1 (encoding ABIN1), TNFAIP3 (encoding A20), and members of the LUBAC complex, have been linked to auto-immune and inflammatory disorders, such as inflammatory bowel disease, psoriasis, rheumatoid arthritis and multiple sclerosis109-112. Interestingly, these genes are also involved in regulating NFκB signaling113. Animal model studies demonstrate that genetic or pharmacological inhibition of RIPK1 kinase activity can alleviate pathological symptoms, indicating that the pathogenesis resulting from these mutations may be driven more by dysregulated RIPK1-dependent cell death and inflammatory mechanisms rather than failure to activate NFκB35,60.
Several chronic neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS), Alzheimer disease (AD) and Parkinson disease (PD), are also linked to increased activation of RIPK1114-116. For example, mutations in the optineurin (OPTN) gene have been implicated in human ALS. In mouse models, loss of OPTN leads to elevated RIPK1 activity, as well as downstream RIPK3 and MLKL activation, resulting in axon degeneration, which is partially rescued by Ripk3 loss-of-function or treatment with a RIPK1 inhibitor necrostatin114. Furthermore, aging-induced reduction of TAK1 expression combined with TBK1 mutations promotes the onset of neurodegenerative diseases, including ALS and frontotemporal dementia (FTD). This is mainly attributed to the hyperactivation of RIPK1, due to decreased inhibitory phosphorylations resulting from reduced activity of TAK1 and TBK1. Importantly, the ALS/FTD phenotype is partially rescued by a single alle of kinase-dead RIPK132.
Therapeutic Perspectives
Elevated RIPK1 activity is associated with numerous human diseases, making it a crucial target for therapeutic interventions. In theory, RIPK1 kinase inhibitors will prevent RIPK1 hyperactivation-induced inflammatory diseases, while preserving RIPK1 scaffold function to maintain its basal inhibition on RIPK3, ZBP1 and FADD/Caspase-8, thus averting unwanted necroptosis or apoptosis. Indeed, in mouse models, RIPK1 inhibitors have been shown to prevent or alleviate clinical symptoms of various diseases, including SIRS, ischemia-induced tissue injury, neurodegeneration, bacterial and viral infection2. Currently, numerous RIPK1 inhibitors are in different phases of clinical trials for a spectrum of human inflammatory diseases, ranging from rheumatoid arthritis, cutaneous lupus erythematosus, ulcerative colitis, SARS-CoV-2 infection, to Alzheimer’s disease and ALS117-119 (ClinicalTrials.gov). While many of these inhibitors have passed the phase I safety test, none has progressed to phase III trial yet.
Considering the diverse functions of RIPK1 and the uncertain outcomes of the clinical trials involving RIPK1 inhibitors, there is a pressing need to identify novel targets for specifically inhibiting its cell death-promoting activity. Considering the pivotal role of PPP1R3G/PP1γ in removing inhibitory phosphorylation sites on RIPK1, it emerges as a promising alternative therapeutic target. Notably, PPP1R3G interacts with PP1γ through a short RVQF motif, presenting a unique opportunity to develop short peptide-mimetics that disrupt PPP1R3G and PP1γ interaction, blocking RIPK1-dependent apoptosis and necroptosis. The same approach has been successfully employed in designing the Smac-mimetics, which mimics the four-residue AVPI sequence in the SMAC (Second Mitochonria-derived Activator of Caspases) protein. These mimetics specifically block the interaction between SMAC and IAPs (Inhibitor of Apoptosis Proteins), thereby inducing apoptosis27,28,120. Unlike RIPK1 inhibitors, these PPP1R3G/PP1γ interaction inhibitors will maintain the inhibitory phosphorylation sites on RIPK1, preventing its hyperactivation, while not altering RIPK1 scaffold function, thereby preserving its other functions. At the same time, these inhibitors would have minimal impact on the phosphatase activity of PP1γ, thus maintaining its other vital functions. This innovative approach holds potential for therapeutic interventions targeting inflammatory diseases associated with heightened RIPK1 activity, while minimizing any adverse effects on cellular homeostasis.
Conclusion and Future Perspectives
The critical role of RIPK1 in modulating both cell survival and cell death underscores the necessity for tight regulation of its activity. In addition to phosphorylation, other post-translational modifications like ubiquitination and glycosylation also significantly impact RIPK1's scaffolding function and kinase activity. Yet, the intricate interplay among these modifications and how they collectively regulate RIPK1 function still require further elucidation. A comprehensive understanding of this complex regulatory network holds promise for the development of more effective treatments targeting RIPK1-related conditions in the future.
Acknowledgments
This study is supported by the National Institute of General Medical Sciences NIGMS (R01GM147474 to Z.W).
References
- He, S. & Wang, X. RIP kinases as modulators of inflammation and immunity. Nat Immunol 2018, 19, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, L. , Ofengeim, D. & Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov 2020, 19, 553–571. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A. & Dixit, V. M. Death receptors: signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Sun, X. , Yin, J., Starovasnik, M. A., Fairbrother, W. J. & Dixit, V. M. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem 2002, 277, 9505–9511. [Google Scholar] [CrossRef] [PubMed]
- Li, J. et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Stanger, B. Z. , Leder, P., Lee, T. H., Kim, E. & Seed, B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 1995, 81, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Chen, G. & Goeddel, D. V. TNF-R1 signaling: a beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H. , Xiong, J. & Goeddel, D. V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H. , Huang, J., Shu, H. B., Baichwal, V. & Goeddel, D. V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef]
- Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Rothe, M. , Pan, M. G., Henzel, W. J., Ayres, T. M. & Goeddel, D. V. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 1995, 83, 1243–1252. [Google Scholar] [CrossRef]
- Mahoney, D. J. et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. P Natl Acad Sci USA 2008, 105, 11778–11783. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M. J. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008, 30, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, A. et al. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Molecular cell 2004, 15, 535–548. [Google Scholar] [CrossRef]
- Wang, C. et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T. et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J 2006, 25, 4877–4887. [Google Scholar] [CrossRef]
- Haas, T. L. et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Ea, C.-K. , Deng, L., Xia, Z.-P., Pineda, G. & Chen, Z. J. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Molecular cell 2006, 22, 245–257. [Google Scholar]
- Tokunaga, F. et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nature cell biology 2009, 11, 123–132. [Google Scholar] [CrossRef]
- Rahighi, S. et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef]
- Mercurio, F. et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J. A. , Hayakawa, M., Rothwarf, D. M., Zandi, E. & Karin, M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388, 548–554. [Google Scholar] [PubMed]
- Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J. & Ofengeim, D. A guide to cell death pathways. Nat Rev Mol Cell Biol, 2023. [Google Scholar] [CrossRef]
- Wang, L. , Du, F. & Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Kreuz, S. , Siegmund, D., Scheurich, P. & Wajant, H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol 2001, 21, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Li, L. et al. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science 2004, 305, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Oost, T. K. et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem 2004, 47, 4417–4426. [Google Scholar] [CrossRef] [PubMed]
- Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nature communications 2017, 8, 359–359. [Google Scholar] [CrossRef] [PubMed]
- Vlantis, K. et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-κB-Dependent and -Independent Functions. Immunity 2016, 44, 553–567. [Google Scholar] [CrossRef]
- Lafont, E. et al. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nature cell biology 2018, 20, 1389–1399. [Google Scholar] [CrossRef]
- Xu, D. et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell, 2018; 174, 1477-1491.e1419. [Google Scholar] [CrossRef]
- Dondelinger, Y. et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell 2015, 60, 63–76. [Google Scholar] [CrossRef]
- Priem, D. et al. A20 protects cells from TNF-induced apoptosis through linear ubiquitin-dependent and -independent mechanisms. Cell death & disease 2019, 10, 692–692. [Google Scholar] [CrossRef]
- Dziedzic, S. A. et al. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nature cell biology 2018, 20, 58–68. [Google Scholar] [CrossRef]
- Degterev, A. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Berger, S. B. et al. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. Journal of Immunology 2014, 192, 5476–5480. [Google Scholar] [CrossRef]
- Polykratis, A. et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. Journal of immunology (Baltimore, Md. : 1950) 2014, 193, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Shan, B. , Pan, H., Najafov, A. & Yuan, J. Necroptosis in development and diseases. Genes Dev 2018, 32, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y. , Devin, A., Rodriguez, Y. & Liu, Z. G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes & development 1999, 13, 2514–2526. [Google Scholar] [CrossRef]
- Newton, K. et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef]
- Holler, N. et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature chemical biology 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D. W. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Hanna-Addams, S. , Liu, S., Liu, H., Chen, S. & Wang, Z. CK1alpha, CK1delta, and CK1epsilon are necrosome components which phosphorylate serine 227 of human RIPK3 to activate necroptosis. Proc Natl Acad Sci U S A 2020, 117, 1962–1970. [Google Scholar]
- Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A 2012, 109, 5322–5327. [Google Scholar] [CrossRef]
- Wang, H. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Su, L. et al. A plug release mechanism for membrane permeation by MLKL. Structure 2014, 22, 1489–1500. [Google Scholar] [CrossRef]
- Cai, Z. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Chen, X. et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 2014, 24, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J. M. et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A 2014, 111, 15072–15077. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y. et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Reports 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. et al. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc Natl Acad Sci U S A, 2017; 114, E7450–E7459. [Google Scholar] [CrossRef]
- Liu, S. et al. MLKL polymerization-induced lysosomal membrane permeabilization promotes necroptosis. Cell Death Differ 2024, 31, 40–52. [Google Scholar] [CrossRef] [PubMed]
- He, S. , Liang, Y., Shao, F. & Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. P Natl Acad Sci USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef]
- Tanzer, M. C. et al. Combination of IAP antagonist and IFNγ activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell death and differentiation 2017, 24, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Duprez, L. et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 2011, 35, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Newton, K. et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell death and differentiation 2016, 23, 1565–1576. [Google Scholar] [CrossRef]
- Kaiser, W. J. et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. The Journal of biological chemistry 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Upton, J. W. , Kaiser, W. J. & Mocarski, E. S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar]
- Thapa, R. J. et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E. E. et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W. C. et al. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 1998, 279, 1954–1958. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W. J. et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A. et al. Catalytic activity of the caspase-8-FLIP L complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Diaz, S. et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Delanghe, T. , Dondelinger, Y. & Bertrand, M. J. M. RIPK1 Kinase-Dependent Death: A Symphony of Phosphorylation Events. Trends Cell Biol 2020, 30, 189–200. [Google Scholar]
- Taylor, S. S. & Kornev, A. P. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci 2011, 36, 65–77. [Google Scholar]
- Zhang, Y. et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nature Communications, 2017; 8. [Google Scholar]
- Laurien, L. et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nature communications 2020, 11, 1747–1747. [Google Scholar] [CrossRef]
- McQuade, T. , Cho, Y. & Chan, F. K. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem J 2013, 456, 409–415. [Google Scholar]
- Menon, M. B. et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat Cell Biol 2017, 19, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y. et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol 2017, 19, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Jaco, I. et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Molecular cell, 2017; 66, 698-710.e695. [Google Scholar]
- Dondelinger, Y. et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat Commun, 2019; 10, 1729. [Google Scholar]
- Lafont, E. et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol 2018, 20, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T. et al. Metabolic orchestration of cell death by AMPK-mediated phosphorylation of RIPK1. Science 2023, 380, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Tu, H. , Xiong, W., Zhang, J., Zhao, X. & Lin, X. Tyrosine phosphorylation regulates RIPK1 activity to limit cell death and inflammation. Nat Commun, 2022; 13, 6603. [Google Scholar]
- Peterson, L. W. et al. RIPK1-dependent apoptosis bypasses pathogen blockade of innate signaling to promote immune defense. The Journal of experimental medicine 2017, 214, 3171–3182. [Google Scholar] [CrossRef] [PubMed]
- Orth, K. et al. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science 2000, 290, 1594–1597. [Google Scholar] [CrossRef] [PubMed]
- Jaco, I. et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell, 2017; 698-710, e695. [Google Scholar]
- Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun. 2017; 8, 359. [Google Scholar]
- Barford, D. , Das, A. K. & Egloff, M. P. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct 1998, 27, 133–164. [Google Scholar] [PubMed]
- Cohen, P. T. Protein phosphatase 1--targeted in many directions. J Cell Sci 2002, 115, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Chen, W. et al. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat Cell Biol 2015, 17, 434–444. [Google Scholar] [CrossRef]
- Du, J. et al. RIPK1 dephosphorylation and kinase activation by PPP1R3G/PP1γ promote apoptosis and necroptosis. Nature communications 2021, 12, 7067–7067. [Google Scholar] [CrossRef]
- Egloff, M. P. et al. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J 1997, 16, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y. et al. Deficiency of PPP6C protects TNF-induced necroptosis through activation of TAK1. Cell death & disease 2022, 13, 618–618. [Google Scholar]
- Broglie, P. , Matsumoto, K., Akira, S., Brautigan, D. L. & Ninomiya-Tsuji, J. Transforming growth factor beta-activated kinase 1 (TAK1) kinase adaptor, TAK1-binding protein 2, plays dual roles in TAK1 signaling by recruiting both an activator and an inhibitor of TAK1 kinase in tumor necrosis factor signaling pathway. J Biol Chem 2010, 285, 2333–2339. [Google Scholar]
- Kelliher, M. A. et al. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 1998, 8, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. et al. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011, 471, 373–376. [Google Scholar] [CrossRef]
- Dillon, C. P. et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep 2012, 1, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. , Dowling, J. P. & Zhang, J. RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell death & disease 2019, 10, 245–245. [Google Scholar]
- Dillon, C. P. et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 2014, 157, 1189–1202. [Google Scholar] [CrossRef]
- Kaiser, W. J. et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. P Natl Acad Sci USA 2014, 111, 7753–7758. [Google Scholar] [CrossRef]
- Lin, J. et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 2016, 540, 124–128. [Google Scholar] [CrossRef]
- Newton, K. et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 2016, 540, 129–133. [Google Scholar] [CrossRef]
- Dannappel, M. et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 2014, 513, 90–94. [Google Scholar] [CrossRef]
- Cuchet-Lourenço, D. et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 2018, 361, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. P Natl Acad Sci USA 2019, 116, 970–975. [Google Scholar] [CrossRef]
- Uchiyama, Y. et al. Primary immunodeficiency with chronic enteropathy and developmental delay in a boy arising from a novel homozygous RIPK1 variant. J Hum Genet 2019, 64, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Lin, L. et al. Clinical phenotype of a Chinese patient with RIPK1 deficiency due to novel mutation. Genes Dis 2020, 7, 122–127. [Google Scholar] [CrossRef]
- Lalaoui, N. et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 2020, 577, 103–108. [Google Scholar] [CrossRef]
- Tao, P. et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature 2020, 577, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Tapiz, I. R. A. J. et al. Characterization of Novel Pathogenic Variants Leading to Caspase-8 Cleavage-Resistant RIPK1-Induced Autoinflammatory Syndrome. J Clin Immunol 2022, 42, 1421–1432. [Google Scholar] [CrossRef]
- Smahi, A. et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 2000, 405, 466–472. [Google Scholar]
- Rizk, M. M. , Elsayed, E. T., ElKeraie, A. F. & Ramzy, I. Association of Tumor Necrosis Factor Alpha-Induced Protein 3 Interacting Protein 1 (TNIP1) Gene Polymorphism (rs7708392) with Lupus Nephritis in Egyptian Patients. Biochem Genet 2018, 56, 478–488. [Google Scholar] [PubMed]
- Sato, S. et al. Juvenile onset autoinflammatory disease due to a novel mutation in TNFAIP3 (A20). Arthritis Res Ther, 2018; 20, 274. [Google Scholar]
- Boisson, B. et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. The Journal of experimental medicine 2015, 212, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Aksentijevich, I. & Zhou, Q. NF-κB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases. Front Immunol 2017, 8, 399–399. [Google Scholar] [PubMed]
- Ito, Y. et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science (New York, N.Y.) 2016, 353, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Ofengeim, D. et al. RIPK1 mediates a disease-associated microglial response in Alzheimer's disease. P Natl Acad Sci USA, 2017; 114, E8788–E8797. [Google Scholar]
- Iannielli, A. et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson's Disease Models. Cell reports 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Grievink, H. W. et al. DNL104, a Centrally Penetrant RIPK1 Inhibitor, Inhibits RIP1 Kinase Phosphorylation in a Randomized Phase I Ascending Dose Study in Healthy Volunteers. Clin Pharmacol Ther 2020, 107, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Weisel, K. et al. A randomised, placebo-controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol, 2021; 8. [Google Scholar]
- Vissers, M. et al. Safety, pharmacokinetics and target engagement of novel RIPK1 inhibitor SAR443060 (DNL747) for neurodegenerative disorders: Randomized, placebo-controlled, double-blind phase I/Ib studies in healthy subjects and patients. Clin Transl Sci 2022, 15, 2010–2023. [Google Scholar] [CrossRef]
- Bai, L. , Smith, D. C. & Wang, S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol Ther 2014, 144, 82–95. [Google Scholar]
Figure 1.
Domain structure of RIPK1. KD, kinase domain. ID, intermediate domain. RHIM, RIP homotypic interaction motif. DD, death domain. RIPK1, receptor-interacting protein kinase 1. RIPK3, receptor-interacting protein kinase 3. ZBP1, Z-DNA binding protein 1, also known as DAI (DNA-dependent activator of interferon regulatory factors) and DLM-1. TRIF, TIR-domain-containing adapter-inducing interferon β.
Figure 1.
Domain structure of RIPK1. KD, kinase domain. ID, intermediate domain. RHIM, RIP homotypic interaction motif. DD, death domain. RIPK1, receptor-interacting protein kinase 1. RIPK3, receptor-interacting protein kinase 3. ZBP1, Z-DNA binding protein 1, also known as DAI (DNA-dependent activator of interferon regulatory factors) and DLM-1. TRIF, TIR-domain-containing adapter-inducing interferon β.
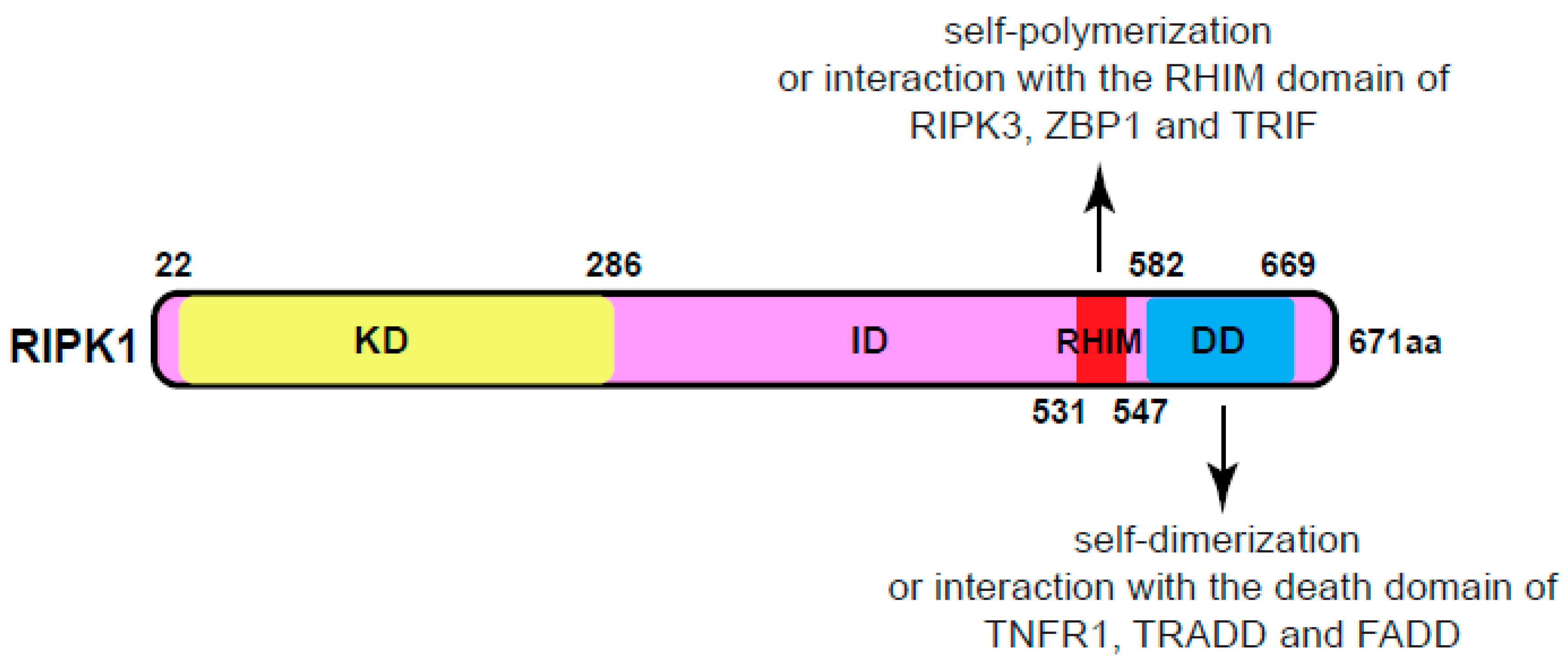
Figure 2.
Pleiotropic TNF signaling pathways. (1) Inflammation and cell survival. Engaging of TNF with its receptor TNFR1 leads to the recruitment of RIPK1 and TRADD through death domain interactions to initiate complex I formation. TRADD then recruits adaptor protein TRAF2/5, which binds E3 ligase cIAP1/2. cIAP1/2 catalyzes K63 ubiquitination of RIPK1, serving as a scaffold to recruit ubiquitin-binding proteins TAB2/3 and associated TAK1, activating downstream MAPK pathway. Additionally, the K63 ubiquitin chain recruits another E3 complex LUBAC, which catalyzes the M1 linear ubiquitin chains on RIPK1 and TNFR1. These linear ubiquitin chains recruit adaptor protein NEMO and associated IKKa/b, phosphorylating IkBa to promote its degradation and subsequent NFκB activation. Both the MAPK pathway and the NFκB pathway activate gene expression that promotes cell survival and inflammation. (2) Apoptosis. Under TNF treatment with protein synthesis inhibition by cycloheximide (CHX), complex I is converted to complex IIa, containing TRADD, FADD and Caspase-8, leading to oligomerization and activation of Caspase-8 and subsequent apoptosis. Alternatively, co-treatment of TNF with a cIAP1/2 inhibitor Smac-mimetic converts complex I to complex IIb, containing RIPK1, FADD and Caspase-8, which also activates Caspase-8 and apoptosis. (3) Necroptosis. Inhibition of apoptosis with Z-VAD-FMK, along with the presence of RIPK3, leads to the conversion of complex II into the necrosome. The core components of necrosome includes RIPK1, RIPK3 and MLKL, resulting in polymerization and membrane translocation of MLKL and subsequent cell death. In general, the scaffold function of RIPK1 is important for inflammation and cell survival, while the kinase activity is important for complex IIb-dependent apoptosis as well as necroptosis. TNF, tumor necrosis factor. TNFR1, tumor necrosis factor receptor 1. TRADD, TNFR1-associated death domain protein. TRAF2 and 5, TNF receptor-associated factor protein 2/5. cIAP1/2, cellular inhibitor of apoptosis 1 and 2. TAB2/3, TAK1-binding protein 2/3. TAK1, transforming growth factor-β-activated kinase 1. LUBAC, the linear ubiquitin chain assembly complex. NEMO, NFκB essential modulator. IKKa/b, IkB kinase a/b. IkBa, inhibitor of kB alpha. NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells. MAPK, mitogen-activated protein kinase. FADD, Fas-associated death domain. MLKL, mixed-lineage kinase like protein.
Figure 2.
Pleiotropic TNF signaling pathways. (1) Inflammation and cell survival. Engaging of TNF with its receptor TNFR1 leads to the recruitment of RIPK1 and TRADD through death domain interactions to initiate complex I formation. TRADD then recruits adaptor protein TRAF2/5, which binds E3 ligase cIAP1/2. cIAP1/2 catalyzes K63 ubiquitination of RIPK1, serving as a scaffold to recruit ubiquitin-binding proteins TAB2/3 and associated TAK1, activating downstream MAPK pathway. Additionally, the K63 ubiquitin chain recruits another E3 complex LUBAC, which catalyzes the M1 linear ubiquitin chains on RIPK1 and TNFR1. These linear ubiquitin chains recruit adaptor protein NEMO and associated IKKa/b, phosphorylating IkBa to promote its degradation and subsequent NFκB activation. Both the MAPK pathway and the NFκB pathway activate gene expression that promotes cell survival and inflammation. (2) Apoptosis. Under TNF treatment with protein synthesis inhibition by cycloheximide (CHX), complex I is converted to complex IIa, containing TRADD, FADD and Caspase-8, leading to oligomerization and activation of Caspase-8 and subsequent apoptosis. Alternatively, co-treatment of TNF with a cIAP1/2 inhibitor Smac-mimetic converts complex I to complex IIb, containing RIPK1, FADD and Caspase-8, which also activates Caspase-8 and apoptosis. (3) Necroptosis. Inhibition of apoptosis with Z-VAD-FMK, along with the presence of RIPK3, leads to the conversion of complex II into the necrosome. The core components of necrosome includes RIPK1, RIPK3 and MLKL, resulting in polymerization and membrane translocation of MLKL and subsequent cell death. In general, the scaffold function of RIPK1 is important for inflammation and cell survival, while the kinase activity is important for complex IIb-dependent apoptosis as well as necroptosis. TNF, tumor necrosis factor. TNFR1, tumor necrosis factor receptor 1. TRADD, TNFR1-associated death domain protein. TRAF2 and 5, TNF receptor-associated factor protein 2/5. cIAP1/2, cellular inhibitor of apoptosis 1 and 2. TAB2/3, TAK1-binding protein 2/3. TAK1, transforming growth factor-β-activated kinase 1. LUBAC, the linear ubiquitin chain assembly complex. NEMO, NFκB essential modulator. IKKa/b, IkB kinase a/b. IkBa, inhibitor of kB alpha. NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells. MAPK, mitogen-activated protein kinase. FADD, Fas-associated death domain. MLKL, mixed-lineage kinase like protein.
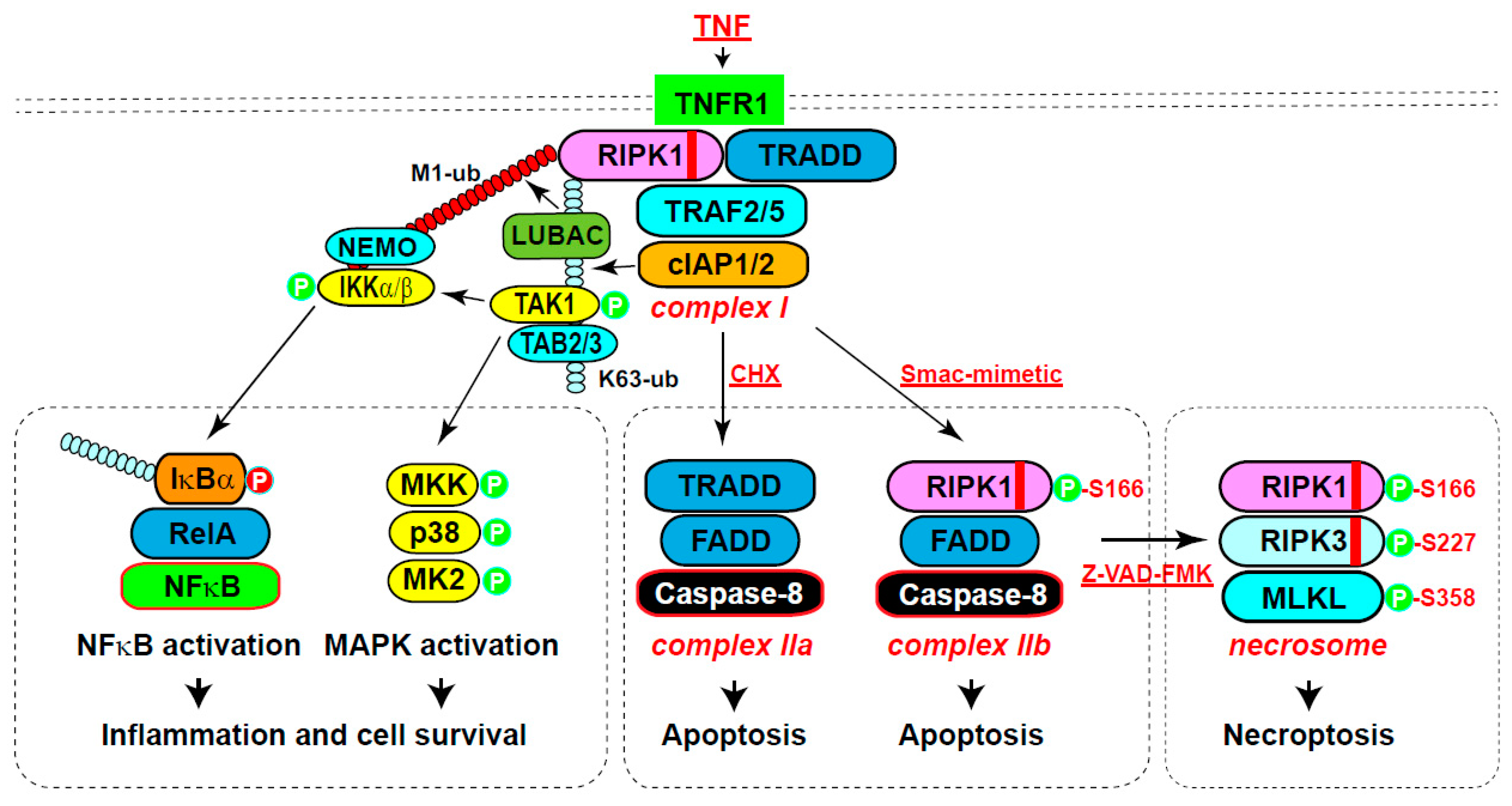
Figure 3.
Phosphorylation events impacting RIPK1 kinase activity. In this context, green character denotes auto-activating phosphorylation, while red character denotes inhibitory phosphorylation. Notably, the kinase responsible for S89 phosphorylation has not yet been reported. TBK1, TANK-binding kinase 1. IKKe, IkB kinase e. MK2, MAPK-activated protein kinae 2. JAK1, Janus kinase 1. AMPK, AMP-activated protein kinase.
Figure 3.
Phosphorylation events impacting RIPK1 kinase activity. In this context, green character denotes auto-activating phosphorylation, while red character denotes inhibitory phosphorylation. Notably, the kinase responsible for S89 phosphorylation has not yet been reported. TBK1, TANK-binding kinase 1. IKKe, IkB kinase e. MK2, MAPK-activated protein kinae 2. JAK1, Janus kinase 1. AMPK, AMP-activated protein kinase.
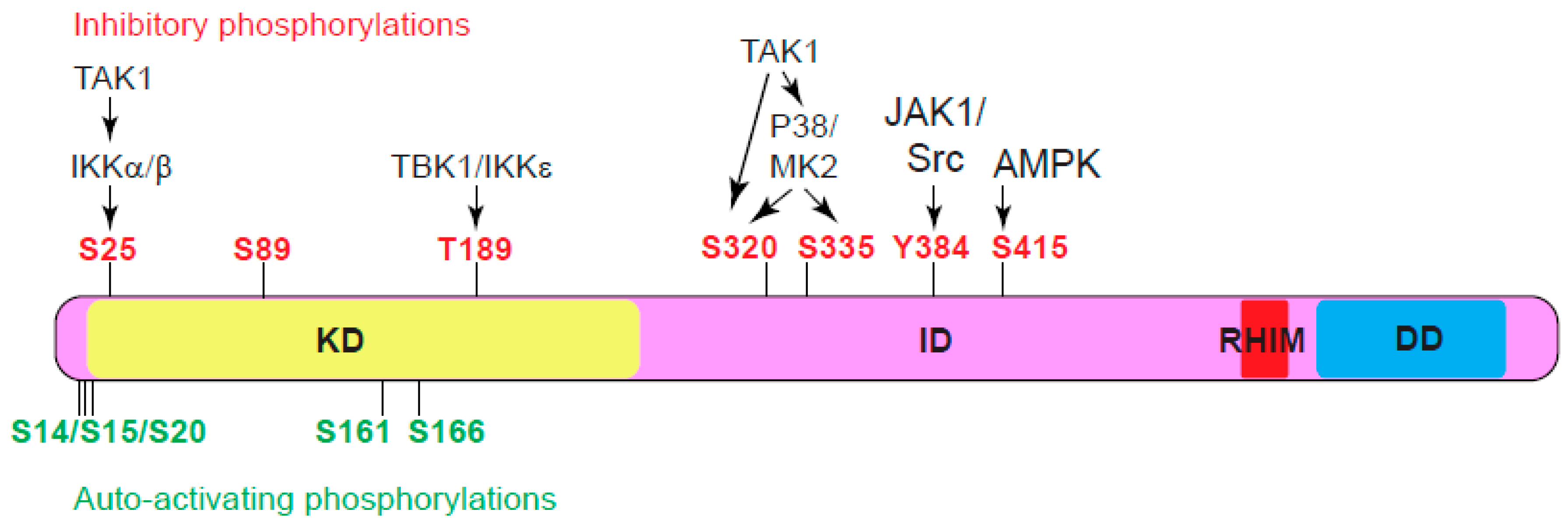
Figure 4.
The PPP1R3G/PP1g holoenzyme removes the inhibitory phosphorylations of RIPK1 to activate RIPK1-dependent apoptosis and necroptosis. Inhibitory phosphorylations on RIPK1, catalyzed by multiple kinases, serve as important checkpoints for cell death activation. Following cell death induction, the PPP1R3G/PP1g holoenzyme is recruited to complex I to remove the inhibitory phosphorylations on RIPK1. This process enables RIPK1 autophosphorylation to activate its kinase activity and downstream cell death.
Figure 4.
The PPP1R3G/PP1g holoenzyme removes the inhibitory phosphorylations of RIPK1 to activate RIPK1-dependent apoptosis and necroptosis. Inhibitory phosphorylations on RIPK1, catalyzed by multiple kinases, serve as important checkpoints for cell death activation. Following cell death induction, the PPP1R3G/PP1g holoenzyme is recruited to complex I to remove the inhibitory phosphorylations on RIPK1. This process enables RIPK1 autophosphorylation to activate its kinase activity and downstream cell death.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



