
Submitted:
08 April 2024
Posted:
09 April 2024
You are already at the latest version
Abstract
To develop peptide drugs targeting integrin receptors, synthetic peptide ligands endowed with well-defined selective binding motifs are necessary. The snake venom KTS-containing disintegrins, that selectively block collagen α1β1 integrin, were used as lead compounds for the synthesis and structure-activity relationship of a series of linear peptides containing the KTS-pharmacophore and alternating natural amino acids and 3-aminobenzoic acid (MABA). To ensure a better stiffness and metabolic stability, one, two and three MABA residues, were introduced around the KTS pharmacophore motif. Molecular dynamics simulations have determined that the solution conformation of MABA peptide 4 is more compact, underwent larger conformational changes until convergence, and spent most of the time in a single cluster. The peptides’ binding affinity has been characterized by enzyme linked immunosorbent assay in which the most potent peptide 4 inhibited with IC50 of 324±8 µM and 550±45 µM the binding of GST-α1-A domain to collagen IV fragment CB3, and the cell adhesion to collagen IV using α1-overexpressor cells, respectively. Docking studies and MM-GBSA calculations confirmed that peptide 4 binds a smaller region of the integrin near the collagen- binding site and penetrated deeper into the binding site near Trp1. Peptide 4, inhibited tube formation by endothelial cell migration in the Matrigel angiogenesis in vitro assay. Peptide 4 was acutely tolerated by mice, showed stability in human serum, decreased tumor volume and angiogenesis, and significantly increased the survival of mice injected with B16 melanoma cells. These findings propose that MABA-peptide 4 can further serve as an α1β1-integrin antagonist lead compound for further drug-optimization in angiogenesis and cancer therapy.
Keywords:
MABA
; KTS
; peptide synthesis
; modeling
; conformation
; stability
; integrin α1
; cell adhesion
; angiogenesis
; melanoma tumor
1. Introduction
Integrins are considered the major cell-adhesion transmembrane receptors that play multiple roles as extracellular matrix-cytoskeletal linkers and transducers in biochemical and mechanical signals between cells and their environment in a wide range of physiological and pathological conditions. Every integrin heterodimer comprises an α-subunit and a β-subunit in a noncovalent complex, and 18 α- and 8 β-subunits create 24 functionally distinct heterodimeric transmembrane receptors. Their functions are regulated by a delicate balance between active and inactive conformations, which are converted between each other via multiple mechanisms, including protein-protein phosphorylation and interactions with adapter proteins (e.g., kindlins), and trafficking [1]. Due to their exposure on the cell surface and sensitivity to the molecular blockade, integrins have been intensively investigated as pharmacological targets [2]. However, given the complexity and sometimes opposite characteristics, integrin-targeting therapeutics has been a challenge [3] with ongoing research efforts towards drug development for different unmet clinical needs [4,5]. Upon ligand binding, integrins cluster and recruit via their cytoplasmic domains cytoskeletal, adaptor and signaling proteins, thus eventually forming focal adhesions that anchor the cell to the bound extracellular matrix ligands, conveying bidirectional signals into the cell [6]. From the focal adhesion sites signal pathways diverge and regulate adhesion, migration, proliferation, and survival [7]. Integrins play an important role in the angiogenic process, the generation of new capillaries, in physiological as well as in pathological blood vessel formation [8]. The vascularization of tumors implanted into integrin α1-knock out mice was clearly decreased, with a reduction in capillary size and number [9]. Both integrins, α1β1 and α2β1 integrin, are collagen receptors, and contribute to the metastatic process as evidenced by the observation that inhibitors or genetic ablation of these integrins [10], such as in integrin α1-knock out mice[11], markedly reduced tumor volume in comparison with wild-type mice. These preclinical findings indicate the important role of α1β1 in providing critical support for endothelial cell migration, and tumor angiogenesis [12,13], prompting peptidomimetic antagonists development towards this collagen binding integrin [14] that could be instrumental as anti-angiogenic and anti-cancer drugs [4].
Snake venom disintegrins are the most potent natural and selective inhibitors of integrins. KTS-disintegrins such as obtustatin [15,16] and viperistatin [17,18] uniquely contain the Lys-Thr-Ser tripeptide motif and bind selectively to the α1β1 integrin [19,20]. This KTS sequence motif has been used by us as a pharmacophore for developing different linear and cyclic peptides antagonists of α1β1 integrins with antiangiogenic and antitumor activity [14,21]. In an attempt to overcome the proteolytic susceptibility of KTS-peptides, we sought to investigate in the present study the structure-function activities of KTS-linear peptides incorporating 3-aminobenzoic acid.
3-Aminobenzoic acid (also known as meta-aminobenzoic acid or MABA) consists of a benzene ring substituted with an amino group and a carboxylic acid with the molecular formula H2NC6H4CO2H. Because unnatural amino acids such as MABA are not found in proteins, most enzymes do not recognize them as cleavage sites for proteolytic degradation. In synthetic peptides, however, these building blocks can be incorporated into peptides for a variety of pharmaceutical applications, including conformational constraints, pharmacologically active ingredients, construction of diverse combinatorial libraries, and in robust molecular scaffolds. The introduction of MABA into acyclic tripeptides [22], leads to reduced conformational flexibility, and inhibits the formation of intramolecular hydrogen bonds that could affect binding pockets by folding-induced selectivity [23,24]. The aromatic subunits also allow the introduction of further substituents, and can mediate interactions with complementary binding receptor partners [25]. Considering oral delivery route in MABA peptide- drug development it is important to notice that MABA is transported through the intestine by a carrier-mediated transport system, important for its absorption and metabolic characteristics [25]. Active ion transporters have been developed from acyclic octapeptides comprising MABA [26] and MABA monopeptide esters showed promising antimicrobial activity towards gram-positive bacteria [27]. Different MABA analogs showed anticoagulant and antiplatelet activities [28], hexapeptides with MABA were evaluated as potential isosteres of the Val-Thr dipeptide unit and a peptidomimetic incorporating MABA proved to effectively inhibit oligosaccharyl transferase, the enzyme responsible for asparagine-linked glycosylation in the lumen of the endoplasmic reticulum [29].
With this background, KTS-linear peptides targeting α1β1- integrin, with alternating natural and unnatural amino acid sequence, the latter of which were MABA, were designed, synthesized, modeled and characterized in vitro and in vivo, hypothesizing that MABA will make the peptide metabolically stable, with reduced conformational flexibility, while preserving its integrin binding properties and activities.
2. Results and Discussion
2.1. Design and Synthesis of MABA-Peptides
To investigate the effect of number and position of MABA units, eight peptide analogs containing the KTS binding motif to α1β1 integrin were prepared according to standard Fmoc solid phase peptide synthesis (SPPS) conditions employing rink amide resin and HBTU/HOBt as coupling reagents (Table S1 and Figures S1–S9). All the MABA-peptide sequences were derived with some modifications from the disintegrin Viperistatin sequence at positions 20-36 (20W-KTS-R-TSHYCTGKSCDG36) [1,4,17].
In all cases, the KTS- pharmacophore responsible for the selective binding to the α1β1 integrin was preserved, while cysteine at position 34 was replaced with serine, and the cysteine at position 29 was omitted (peptide 8), or replaced by MABA unit, or glycine. Peptide 1 indeed includes these two changes. One to three MABA units at different positions around the KTS motif were introduced in order to form a bent in the peptide chain and to restrict conformational diversity. In peptide 2, the MABA element was inserted between the KTS and the RTS motifs, while the amino acids threonine and glycine at positions 30 and 31 were omitted. The sequence of peptide 3 is similar to that of peptide 2 with one distinction: an additional KTS unit was attached to the amino terminal end of the sequence. Peptide 4 includes two MABA units. One is located as is in peptide 2 and the second was inserted between positions 24-25 of Viperistatin-derived peptide 8. The amino acid sequence is the same as in peptide 2. Peptide 5 also includes two MABA elements at the same positions as in peptides 1 and 2, however, the amino acid sequence corresponds to the sequence of peptide 1. Peptide 6 is like peptide 4 and incorporates two changes, one is the exchange of the serine at position 23 for arginine, and the second is in the amino acid sequence that is similar to that of peptide 1. Peptide 7 is composed of three MABA units. Two are in the same positions as in peptide 6, and the third is as in peptide 1. Peptide 8 is the original sequence motif of Viperistatin lacking the cysteine at position 29 (Table S1). All peptides were synthesized by SPPS applying Fmoc chemistry and purified by preparative HPLC to > 95 % homogeneity (Table S1 and Figures S1-S8) and their structure confirmed by analytical LC/MS (Table S1 and Figures S1-S8).
2.2. MABA-Peptides Inhibit Integrin Binding and Cell Adhesion
In a previous study linear obtustatin and viperistatin peptide analogs containing the KTS motif were synthesized and their biologic activity was investigated [14], representing the starting point for the synthesis of small peptides lead compounds toward the collagen-binding integrins (α1β1, α2β1). As shown earlier, Thr22 appeared to be the most critical residue for integrin binding, whereas substitution of the flanking lysine or serine residues for alanine resulted in a less pronounced decrease in the anti-α1β1 integrin activity of the disintegrin [20]. Moreover, the presence of Arg24 next to the K21T22S23 motif strongly increases the binding to the α1β1integrin, suggesting that the positively charged side chain of Arg forms a better interaction than the alkyl chain of Leu within the ligand-binding pocket of the α1β1integrin [20]. To investigate the influence of MABA on the α1β1integrin receptor binding and cell adhesion, a library of eight peptides has been examined by competition to collagen IV fragment CB3 binding to the GST-α1-A domain using ELISA, and by cell adhesion to immobilized collagen IV using α1-overexpressor cells. Peptides 1-8 were incubated together with the GST-linked α1-A domain, and allowed to bind to the immobilized CB3 (collagen IV fragment). The amount of the bound recombinant α1-A domain provided information on the inhibitory potential and the recognition ability of these peptides (Table 1). Peptides 1-8 dose dependently inhibited binding of GST-α1-A domain, with modest IC50 values, in the hundreds micromolar range (Table 1). Peptides 3 and 4 were the most potent, albeit with low avidity of IC50 442±12 and 324±8 µM, respectively. These findings suggest that peptides 1-8 recognize with low potency a sequence motif in the α1-A domain. The ability of peptides 1-8 to inhibit the adhesion of α1-K562 (human erythroleukemic cells, overexpressing α1β1 integrin) to immobilized collagen IV was also evaluated. This cellular model was widely used by us to characterize potential antagonists of α1β1 integrin [14,17,30]. In these experiments, the cells were applied on plates coated with different extracellular matrix proteins and allowed to adhere before detection of the adhered cells number. Under these conditions, no significant cell adhesion to BSA-coated plates (negative control) or to non-specific substrate-coated plates was observed. The results are summarized in Table 1. Peptide 3 retained a notable inhibition of the adhesion of the cells to immobilized collagen IV, with an IC50 of >900 μM. The rest of the compounds poorly affected the adhesion of the cells to immobilized collagen IV (Table 1). Peptide 4, exhibited the highest potency as an inhibitor of the cell adhesion mediated by α1β1 integrin with an IC50 of 550±45 µM, similar to the value obtained by the competitive ELISA binding assay. The selectivity of the inhibitory effects of MABA-peptide 4, compared to Viperistatin, on various integrins was confirmed in cell adhesion assays using cell lines expressing different major integrin subunits (Table S2). Noteworthy, peptide 4, like Viperistatin, showed an outstanding selectivity towards the α1β1 integrin, about five fold lower inhibitory activity toward α2β1, and did not show any activity toward other integrins such as α6β1, α5β1, α4β1, α9β1, αvb3 and αIIbb3. These findings demonstrate that target integrin selectivity can be preserved by switching from proteins to small MABA- peptidomimetics, but further optimization is required to improve their potency.
2.3. In Vitro Metabolic Stability of MABA-Peptides
To investigate the proteolytic susceptibility of the synthetic peptides, the degradation of intact peptides 2 (one MABA unit), 4 (two MABA units) and 8 (linear peptide) (Table 1) incubated in rat serum at 37°C for different periods of time, was followed by LC-MS. The results are presented in Figure 1. Peptide 8, lacking MABA was rapidly degraded, with a half-life of about 20 min. In contrast, peptides 4 and 2 containing two, and one MABA respectively, were stable up to 60 min. Similar findings were obtained by incubating the MABA-peptides in human serum (data not shown). Therefore, the water solubility and metabolic stability enable the MABA-peptides to circumvent some challenges of peptides' usage as candidates in drug development [31].
2.4. In Vitro Anti-Angiogenic Effect of the MABA-Peptides
Angiogenesis is a cellular process characterized by a number of events, including endothelial cell migration, invasion, and assembly into capillaries. In vitro endothelial tube formation assays are performed using primary human umbilical vein endothelial cells that upon applying onto Matrigel matrix, reorganize to create tube-like structure, which may be used as a model for studying some aspects of in vitro angiogenesis [32]. Number of nodes, junctions, meshes and total tube length were the parameters chosen for quantification of the antiangiogenic effects of the MABA-peptides. Nodes function quantifies points where branches of the network converge, a junction is a collection of nodes, a mesh is an area entirely closed by surrounding branches and tubes length is the total area of skeletonized network branches [32,33] . HUVEC cells plated on Matrigel supplemented with the angiogenic vascular endothelial growth factor VEGF (30 ng/mL), formed a capillary-like network within 16 h (Figure 2). In the presence of peptides 3 and 4 (500 μM), the extent of tube formation evaluated by all parameters was significantly reduced in comparison to that in the cells treated with the vehicle alone (containing up to 1% DMSO dissolved in cell culture medium). Interestingly, peptide 4 exhibited the greatest inhibitory effect in blocking VEGF-induced angiogenesis. The number of junctions, nodes, meshes and tube length were reduced by 70-80 % (p≤ 0.01 and 0.001 vs control). The minimal concentration of peptides 3 and 4 yielding a complete inhibition of endothelial morphogenesis on Matrigel was about 200 μM, whereas the other peptides 1, 2, 6, 7 and 8 in milimolar concentrations did not cause any significant reduction in the level of angiogenesis (Figure S10).
2.5. Anti-Tumor Effect of the MABA-Peptides in a Murine Melanoma Model
To evaluate their antitumor efficacy, the MABA-peptides were administered systemically by intraperitoneal injection, three times a week, for three weeks with a cumulative dose of 90 mg/kg in C57BL/6 mice bearing B16-F10 melanoma tumors. Figure 3A shows the mean tumor volume of mice treated with MABA peptide-4 as compared to saline-treated mice. A significant (p < 0.01 vs. untreated sick mice) delay of tumor growth was observed in the peptide 4 treated mice at 14 and 16 days after tumor cells injection. Body weights were monitored twice a week throughout the study and were not statistically different between the groups at any time (25-30 gram), indicating lack of toxicity. Figure 3B shows the Kaplan-Meier survival plots in healthy, sick tumor-bearing mice group untreated or treated with saline or peptides 3 to 6. The survival curves of peptides 3, 4-treated mice were significantly different, from the curves of controls, and peptides 5 and 6 treated mice. Peptides 4 and 3 significantly increased the survival of the mice with melanoma tumors from median survival time of 32-35 days (control) to 73 and 50 days (p ≤ 0.0001, Log-rank Mantel-Cox test) , respectively (Table S3). All mice treated with these peptides did not show any disease clinical signs at the end of 80 days, and showed no metastases in the lung at necropsy. Overall, these data indicate that MABA-peptide 4, and to a lower efficacy, MABA-peptide 3 treatment induced a significant increase in the survival of tumor bearing mice. To verify the antiangiogenic effect of peptide 4, melanoma tumor tissue sections were immunostained with a CD31 (an endothelial cell maker of blood vessels) antibody. As shown in Figure 3C, fewer CD31-positive microvessels were observed in the tumors of mice treated with peptide 4 compared to control. These data indicate that MABA-peptide 4 significantly inhibited melanoma growth and angiogenesis in mice. Because tumor growth and angiogenesis affect each other, the question arises as to whether inhibition of angiogenesis by MABA-peptide 4, as also found by tube formation assay in vitro (Figure 2), directly contributes to the anti-melanoma effects of peptide 4. The answer to this question requires further experimentation. These results also indicate that in spite of the low affinity of MABA-peptide 3 and 4 to the α1β1integrin receptor binding and cell adhesion (Table 1), using a high dose of these peptides, positive antiangiogenic and anti-tumor effects can be generated, encouraging further optimization of these lead peptides.
2.6. Safety of MABA-Peptide 4
To verify that the antiangiogenic effect of peptide 4 is not due to cytotoxicity to endothelial cells, HUVECs cell cultures were treated for 7 days with 25 mM peptide 4, and the amount of LDH release in the medium was estimated. No significant release of LDH over the control (less than 4 %) was measured, indicating absence of necrotic cell death. Considering that the IC50 value for inhibition of cell adhesion is 0.55 mM, we trust that the therapeutic index of peptide 4 in vitro is higher than 50, and that it has high safety if endothelial cells are exposed to it.
To investigate the safety of peptide 4 in mice, male mice were intraperitoneal injected three times a week, with a cumulative dose of 90 mg/kg for three consecutive weeks (9 injections). Acute tolerability was observed since the mice did not suffer from weakness, paralysis or altered motor activity, and no irregular behavior was observed. Cutaneous hematomas around the injection or at distant locations site were not observed within 24 hours after injection. Furthermore, no mice sudden deaths or infections occurred during the 3 weeks of observation. Body weight increased significantly from 18 to 21 gram as in the untreated mice. After 10 hours, the blood of mice injected with peptide 4 was submitted for hematologic and biochemical analysis. The values for white blood cells, red blood cells, and platelet counts were in the normal range. Alkaline phosphatase and LDH values were in the range of 100–214 units/liter and 1000–2400 units/liter, respectively, in blood samples of mice injected with peptide 4 and compared with control mice, ruling out any acute toxic effects on liver and other tissues. Blood urea and creatinine were in the range of 35mg/dl and 1.2 mg/dl respectively, and did not differ between mice injected with peptide 4 and control mice, suggesting no toxic effects to the kidney. These findings support the safety of peptide 4.
2.7. Molecular Modeling
To get atomic level insight into the solution structures, and bound structures of some of the peptides considered in this work, we subjected two of them, peptide 4 (the most active one containing two MABA units), and peptide 2 (the third most active one containing a single MABA unit yet with a sequence highly similar to the one of peptide 4), to microsecond long molecular dynamics (MD) simulations, followed by molecular docking, and MM-GBSA calculations. The MD simulations of both peptides were initiated from an extended conformation and were carried out for three microseconds each in an explicit solvent model. RMSD plots (Figure S11) suggest that both simulations have reached convergence about half time through the simulations. However, surprisingly, the simulation of peptide 4 bearing the two supposedly rigidifying MABA units took longer to converge and demonstrated larger conformational changes until convergence. On the other hand, the radius of gyration (Rg) plots suggested that the solution conformation of peptide 4 is more compact than that of peptide 2 (Figure 4).
Next, the resulting trajectories were clustered giving rise to seven clusters for peptide 4 (with 14282, 508, 125, 66, 16, 3, and 1 members) and six clusters for peptide 2 (with 14212, 495, 200, 77, 12, and 5 members). These numbers clearly indicate that both peptides in solution spend most of the time in a single cluster. The central structures from the largest cluster for peptides 2 and 4 are presented in Figure S12 and demonstrate the compact conformation in solution of these peptides.
Next, the central structure of each cluster was docked into the crystal structure of the alpha-1 integrin I-domain which was crystalized in complex with a collagen-mimetic peptide (PDB code 2M32) using Schrodinger’s protein-protein docking tool (PIPER). For each input structure, PIPER produces multiple clusters. The centroids of these clusters were ranked based on their PIPER scores, and the best scoring pose was subjected to MM-GBSA calculations, in order to compare the predicted binding free energies between peptide 2 and 4 (PIPER scores can only be used to compare between different poses of the same peptide whereas MM/GBSA energies could be used to compare the binding affinities of different peptides). The results presented in Table 2 suggest that, in agreement with the experimental findings, peptide 4 is a better binder to the integrin than peptide 2.
Figure 5 presents the binding modes of the two lowest energy poses of peptides 2 and 4 at their predicted integrin binding sites as obtained from the MM/GBSA calculations, and Table 3 provides a summary of their interactions with binding site residues. Both peptides bind the integrin in the vicinity of the collagen-mimetic peptide binding site at two different regions, albeit with some overlap. Thus, this overlap region (around Asp177) might serve as a hotspot for the design of additional integrin KTS-ligands. Peptide 2 makes more interactions with the surface of the integrin, covering a larger region, whereas peptide 4 interacts with a smaller region of the integrin, but “penetrates” deeper into the binding site, in particular in the vicinity of Trp1. The rather small overlap between the binding sites of the two peptides might seem at first glance surprising, in light of the high sequence similarity between them. We note however that the presence of a single MABA group in peptide-2 vs. the two MABA groups in peptide-4, lead to substantial conformational differences between the two peptides both in their unbound and bound conformations. Thus, the RMSD difference between the centroids of the two largest clusters obtained from the solution simulations of the peptide (based on the backbone of their common sequences) is 6.7 Å, whereas the RMSD difference between their bound conformations (calculated in the same way) is 7.9 Å.
A comparison between the solution conformations and the bound (i.e., bioactive) conformations of the two peptides is informative. For peptide 2, the RMSD values between the docked conformation and the six cluster centers are 6.2Å, 3.7Å, 4.3Å, 5.8Å, 5.6Å, and 7.2Å. For peptide 4, the RMSD values between the docked conformation and the seven cluster centers are 8.2Å, 7.3Å, 6.1Å, 11.3Å, 5.7Å, 8.8Å, and 7.7Å. Moreover, for both peptides, the lowest energy-binding pose (based on the MM/GBSA energies) came from one of the least populated solution clusters (clusters 2 and 3 for peptide 2 and 4, respectively). Taken together these data suggest that the two peptides had to undergo a substantial conformational change to bind the integrin, a change accompanied by an enthalpy “penalty”. This in turn may indicate that yet more peptides that are active could be designed by further constraining their unbound conformations to the respective docked conformations. Finally, we calculated the RMSD differences between the solution conformations of peptide-2 and peptide-4 (represented by the centroid of their largest cluster) and a representative conformation of Viperistatin taken from the MD simulation reported in reference [14]. Viperistatin is a 41-resudues peptide, snake venom toxin known to strongly, and selectively inhibit α1β1 integrins. While a direct comparison between the peptides considered in this work and Viperistatin is hampered by the large differences in their sizes, the results (RMSD values of 7.0 Å and 6.6 Å for peptide-2 and peptide-4 respectively, based on the backbone sequence common to Viperistatin and the two peptides) suggest that both peptides share some structural similarity with the more potent compounds. Interestingly, the more active peptide (peptide-4) is slightly more similar to Viperistatin than the less potent compound (peptide-2) although the difference is not significant.
3. Conclusion
Herein we described the synthesis, structure-activity relationship and molecular modeling of synthetic linear KTS-peptides containing meta-aminobenzoic acid (MABA) as antagonists of α1β1 integrin. The impact of MABA location, and the numbers of MABA in the KTS-pharmacophore, on the binding of GST-α1-A domain to collagen IV fragment CB3, and α1-overexpressor cells adhesion to collagen IV indicated that peptide 4 containing two MABA units was the most potent, albeit with low avidity and suggesting recognition of a binding motif in the α1-A domain. Peptide 4, showed an outstanding selectivity towards the α1β1 integrin, about five folds lower activity towards α2β1, and did not show any inhibitory activity toward α6β1, α5β1, α4β1, α9β1, αvb3 and αIIbb3 integrins. These findings demonstrate that integrin target selectivity can be preserved by switching from proteins to small MABA- peptidomimetics, but further optimization is obligatory to improve their binding avidity for pharmaceutical purposes. MABA-peptides 4 and 2 containing two and one MABA unit respectively, demonstrated metabolic stability in rat and human serum. Peptide 4 and to a lower extent peptide 3 containing one MABA unit, exhibited the greatest inhibitory effect in blocking VEGF-induced angiogenesis in the Matrigel-tube formation in vitro assay, and significantly increased the survival of the mice with melanoma tumors. Peptide 4 was found safe in mice upon intraperitoneal injection. Molecular dynamics simulations followed by molecular docking and MM-GBSA calculations, indicated that peptide 4 bearing the two rigidifying MABA units took longer to converge, demonstrated larger conformational changes until convergence, and spend most of the time in water as a single cluster. In accordance with the beta-turn inducing properties of the MABA unit, the radius of gyration plots suggested that the solution conformation of peptide 4 is more compact than that of peptide 2, property that may affect the metabolic stability and binding to the integrin. Peptides 4 and 2 cluster conformation was docked into the crystal structure of the α1 integrin I-domain which was crystalized in complex with a collagen peptide using Schrodinger’s protein-protein docking tool (PIPER). The centroids of these clusters were ranked based on their PIPER scores, and the best scoring pose was subjected to MM-GBSA calculations in order to compare the predicted binding free energies. The results indicated that peptide 4 is a better binder to the integrin than peptide 2 explaining the differences between these two peptides in their biological activities. According to the model, both peptides bind the α1 integrin I-domain in the vicinity of the collagen-mimetic peptide binding site at two different regions, albeit with some overlap. Thus, this overlap region (around Asp177) might serve as a hotspot for the design of additional integrin KTS-ligands. Peptide 2 makes more interactions with the surface of the integrin, covering a larger region, whereas peptide 4 interacts with a smaller region of the integrin, but penetrates deeper into the binding site, in particular in the vicinity of Trp1. The docking studies suggest that the number of MABA units and their location in the KTS-peptide result in slightly diverse interaction patterns with the α1 integrin I-domain, thus leading to different binding potencies and affecting the antiangiogenic and antitumor activities of MABA- peptides. Although additional preclinical characterizations are required, MABA peptide 4 lead compound that is slightly more similar to Viperistatin, can pave the way for the rational design of an improved α1β1-selective peptidomimetic antagonist drug.
4. Materials and Methods
4.1. Materials
Collagen IV (from bovine placenta villi) and vitronectin were purchased from Chemicon (Temecula, CA), and collagen I (from rat tail) and Matrigel from BD Biosciences (Bedfor, MA). Human fibronectin and laminin were purchased from Sigma-Aldrich (St. Louis, MO). 96-well polystyrene EIA/RIA plates were obtained from Nunc (Roskilde, Denmark Bovine serum albumin (BSA) and Hank’s Balanced Salt Solution (HBSS) sulfate was purchased from Sigma-Aldrich (St. Louis, MO). The celltracker™ green 5-chloromethylfluorescein diacetate (CMFDA), was purchased from Invitrogen-Molecular Probes (Eugene, Oregon, USA). Rabbit polyclonal antibodies against GST were purchased from Molecular Probes (Nijmegen, The Netherlands). Alkaline phosphatase-conjugated anti-rabbit antibody and p-nitrophenyl phosphate were from Sigma-Aldrich (St. Louis, MO). Viperistatin was isolated from the venom of Vipera xantina palestinae as previously described [10].
4.2. Cell Lines
Human umbilical vein endothelial cells (HUVECs) were obtained from Lonza Bioscience (Haifa, Israel) and B16 melanoma was obtained from the American Type Culture Collection (Manassas, VA, USA).
4.3. Peptide Synthesis Reagents and General Procedure for Peptides Preparation and Characterization
4.3.1. General
All protected amino acids and Rink Amide resin were purchased from GL Biochem Ltd. (Shanghai, People’s Republic of China). N,N-diisopropylethylamine (DIPEA), 2-(1H-benzotriazole-1yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and 1-hydroxybenzo-triazole (HOBt) were purchased from BioLab Ltd (Jerusalem, Israel). All coupling reagents, chemicals, and solvents were purchased from Sigma-Aldrich. The peptidomimetics were synthesized on a solid support by the standard Solid Phase Peptide Synthesis (SPPS) fluorenylmethyloxycarbonyl (Fmoc) methodology. The synthesis was carried out manually on a Rink Amide resin using Fmoc-protected amino acids. Meta-aminobenzoic acid (MABA) was also Fmoc protected. Coupling was performed for 1 hour with four equivalents of TBTU and one equivalent of amino acid in the presence of eight equivalents of DIEA. Fmoc groups were removed with 20% piperidine in DMF. Washings after Fmoc removal and couplings were performed with DMF three times. After the final assembly, the peptidyl-resin was washed three times with DMF, three times with DCM, and three times with MeOH. The resin was then dried 3h in oven at 60 0C. Cleavage from the resin and full deprotection of peptides was carried out using a mixture of trifluoroacetic acid (TFA)/phenol/water/triisopropylsilane [88:5:5:2 (v/v/v/v)] for 3 hours at room temperature [33]. The resin was removed by filtration and the residual peptidomimetics were precipitated by addition of cold diethyl ether to the filtrate. The precipitate was separated by centrifugation at 3500 rpm for 10 minutes, dried under vacuum, solubilized in water, and lyophilized. The synthesized peptidomimetics were purified by preparative reverse-phase (RP)-HPLC using either Kromasil and NanoChrom, ChromCore 120 C18 HPLC columns (5 µm particle size, pore size 100 A, 250 mm × 4.6 mm, Sigma Aldrich), with an elution gradient of 0–100% acetonitrile with 0.1% trifluoroacetic in 100% water, at a flow rate of 1.0 ml/min using a Dionex UltiMate 3000 analytical HPLC (Thermo Scientific, Waltham, MA) with detection at a wavelength of 220 nm. The solid phase peptide synthesis (SPPS) was carried out according to the Fmoc method applying Wang resin (1.5mmole/g for linear peptides and 0.5mmole/g for cyclic peptides) as a solid support.
4.3.2. Loading
0.1 mmol of the Wang resin was swelled for 1h in NMP, and drained by filtration. Then, 0.5 mmol (5eq) of FmocAa and 0.5mmol (5eq) of HBTU were dissolved in 2ml of NMP+1mmol (10eq) of DIEA (10eq=1.74ml) and stirred for 3min. Subsequently the solution was added to the resin. The resin was then vortex with for 3h at RT, filtrated and washed with NMPx4 and DCMx4.
4.3.3. Fmoc Deprotection
Removal of the Fmoc protecting group was implemented by treating the pre-swollen resin with 2ml of 20% piperidine in NMP for 15min. The solution was then filtered and the resin washed with NMP (2x2ml). The piperidine step is repeated once again and the resin washed with NMP(4x2ml) and DCM (4x2ml).
4.3.4. Elongation with the FmocAa
The reaction progress is checked by the Kaiser test. Finally, after the last deprotection step, the resin is washed with NMP (4 × 2 ml), DCM (4 × 2 ml), MeOH (1 × 2 mL), DCM (4 × 2 mL), then dried under vacuum.
4.3.5. Cleavage from Resin
TFA /Et3SiH (90/10), 1.5 ml, is added to the resin and vortex for 3.5 h. Then the solvent was removed by filtration and the resin was washed again with TFA/H2O (95/5, 1 ml).The TFA solutions were collected and the peptide was precipitate from the solution by addition of cold Et2O. The solid precipitation was washed with cold Et2O and dried. The synthetic peptide was dissolved in distilled water, lyophilize, and applied to HPLC purification. The collected fractions were further analyzed by MS.
4.3.6. Mass Spectroscopy
The single-quad liquid chromatograph ultra-fast mass spectrometer LC MS- Model 2020 (Shimadzu, Kyoto, Japan) comprised a binary pump (20AD), vacuum degasser, thermostatic auto sampler (SIL 20ACHT), thermostatic column compartment (CTO 20A), photodiode detector (SPD M 20A) and mass analyzer (MS 2020) with both electrospray ionization (ESI) and atmospheric-pressure chemical ionization (APCI) systems. Optimized mass spectra were acquired with an interface voltage of 4.5 kV, a detector voltage of 1.2 kV, a heat block temperature of 300 °C and a desolvation gas temperature of 200 °C. Nitrogen was used as the nebulizer gas at a flow rate of 1.5 L/min and dry gas flow of 10 L/min. LC MS data acquisition and processing was performed by a single method that quickly review the ESI and DUIS results in the data browser window (an/products/liquid-chromatograph-mass-spectrometry/single-quadrupole-lc-ms/lcms-2020/spec.html ).
4.4. ELISA to Test GST-α1A Binding to Collagen IV Fragment CB3
The inhibition enzyme-linked immunosorbent assay (ELISA) was performed as previously described [35], with the following modifications: CB3 (collagen IV fragment) was immobilized overnight at 4°C on a microtiter plate at 10 mg/ml in Tris-buffered saline (TBS)/MgCl2 (50 mM Tris-HCl, 150 mM NaCl, and 2 mM MgCl2, pH 7.4) and 0.1 M acetic acid, respectively. After blocking the plate with 1% BSA in TBS/MgCl2, the GST-tagged α1A-domain was allowed to bind to collagen IV-fragment CB3 in the presence or absence of different peptides for 2 hours at room temperature. The bound GST-α1A domain was fixed for 10 minutes with 2.5% glutaraldehyde in HEPES buffer (50 mM HEPES, 150 mM NaCl, and 2 mM MgCl2, and 1mM MnCl2, pH 7.4). The bound GST-α1A was quantified with rabbit polyclonal antibodies against GST, followed by alkaline phosphatase–conjugated anti-rabbit antibody, which served as the primary and secondary antibodies, respectively, each diluted in 1% BSA in TBS/MgCl2. The conversion of p-nitrophenyl phosphate was measured at 405 nm using an enzyme-linked immunosorbent assay reader (BioTek, Bad Friedrichshall, Germany). Nonspecific binding was assessed by binding of GST-α1A to BSA in the presence of 10 mM EDTA.
4.5. Cell Adhesion Assay
The assay was carried out as described previously [30]. The day before the experiment, each well of a 96-well plate was coated with 10 µg/mL collagen IV or 1 µg/mL collagen I in 0.02 M acetic acid and incubated overnight at 4°C. Thereafter, nonspecific binding was blocked by incu bating the wells with 1% (w/v) BSA in HBSS containing 5 mM magnesium chloride (MgCl2), at room temperature for 1 hour before use. The cells were labeled by incubation with 12.5 µM CMFDA in HBSS without 1% BSA at 37°C for 30 minutes. The labeled cells were then centrifuged at 1000 rpm, and washed twice with HBSS containing 1% BSA, to remove excess CMFDA. Labeled cells (1×105 cells/well) were added to each well, in the presence or absence of peptides, and incubated at 37°C for 60 minutes. In the presence of peptide, cells were added to the well after prior incubation with peptide for 30 minutes at 37°C. Unbound cells were removed by washing the wells three times with 1% (w/v) BSA in HBSS, and bound cells were lysed by the addition of 0.5% Triton X-100 (diluted in double-distilled water). The fluo rescence in each well was quantified with a SPECTRAFluor Plus plate reader (Tecan, San Jose, CA, USA) at excitation wavelength 485 nm and emission wavelength 530 nm. To determine the number of adhered cells from the fluorescence values, a standard curve was generated using serial known numbers of CMFDA-labeled cells.
4.6. Stability of the Peptides in Rat Serum
1.5 milliliter of fresh plasma obtained from male Wistar rats (Harlan, Israel) was incubated at 37°C with peptide stock solution to make a final peptide concentration of 10 µg/mL. Metoprolol (1 µg/mL) was used as an internal standard. Triplicate samples were taken at time 0 and after 5, 10, 20, 30 and 60 min. 50 µL of the reaction solution was removed and added to 100 µL of ice-cold acetonitrile to precipitate serum proteins. The samples were vortex-mixed for 1 minute and then centrifuged at 14 600 x g for 10 min to precipi tate serum proteins. The supernatant was then transferred to fresh glass tubes and evaporated to dryness (Vacuum Evaporation System, Labcon-co, Kansas City, MO, USA). Then, the glass tubes were reconstituted in 80 μL of a solution of 50 % water and 50 % acetonitrile supplemented with 0.1 % formic acid, and centrifuged second time (14 600 g, 10 min). The resulting solution was injected (10 μL) into the HPLC-MS device (HPLC−MS Waters 2695 Separation Module, equipped with a Micromass ZQ detector). The column used was Kinetex® (Phenomenex) 2.6 μm EVO C18 100 Å, LC column 100 mm x 2.1 mm. A linear gradient from 80 % buffer A (0.1 % formic acid in water), to 10–90 % buffer A and buffer B (0.1 % formic acid in acetonitrile), was applied for 15 min. The flow rate used was 0.2 mL/min at 25°C [36].
4.7. Tube Formation-Angiogenesis Assay
Tube formation assay was performed as previously described [37]. Briefly, human umbilical vein endothelial cells (HUVEC) were serum-starved in medium (PeproGrow endothelial cell media, PeproThec Co., Rocky Hill, NJ, USA) for 3 hours before applying to Matrigel® polymerised in Ibidi angiogenesis μ-slides and 96-well plates at 37 °C for 1 h. 2 x 104 cells per well were treated with 100 µg/mL of the tested peptide or left without treatment (control). The cells were cultured at 37 °C, with 5% CO2 for 6 hours, in the presence of a complete, chemically defined formulation medium for the in vitro cultivation of endothelial cells from large blood vessels and angiogenesis VEGF inducer (PeproGrow MacroV, PeproThec Co., Rocky Hill, NJ, USA), and then analyzed by light microscopy (IX-73, OLYMPUS, Japan). The quantitation of tube length, nodes, branches, mesh was performed with the angiogenesis analyzer for ImageJ [33].
4.8. In Vivo Characterization of Anti-Tumor Activity
C57BL/6 young male mice ( aged 4–6 weeks, weighing 25 g) , were purchased from Envigo RMS Ltd. (Rehovot, Israel), and served as subjects in these experiments, after acclimation for four days to laboratory conditions. During the entire study duration, animals were housed within a limited access in a specific pathogen free rodent facility, and kept in groups with a maximum of seven mice per cage made of polypropylene (Euro standard type IV, floor area of the cage: 425 × 266 × 185 mm (800 cm2), fitted with solid bottoms, and a static cage filter top (Tecniplast Co., Buguggiate, Italy), filled with 7090 Teklad sani-chips animal bedding (Envigo RMS Ltd., Rehovot, Israel), and having two plastic tubes in each cage as enrichment material. Three days prior to implantation of tumor cells, the mice were provided ad libitum with a commercial Teklad 2018S global 18% protein rodent diet (Envigo RMS Ltd., Rehovot, Israel), had free access to drinking water, that was monitored periodically, and supplied to each cage via polyethylene bottles with stainless steel sipper tubes. Mice were housed in automatically controlled environmental conditions, and the temperature was maintained at 17–23◦C, with a relative humidity of 30-70 % on a 12-h light/dark cycle (light cycle: 7 a.m.–7 p.m; dark cycle: 7 p.m.–7 a.m.) and 15–30 air changes/h in the study room. During the acclimation period, the mice were assigned to experimental groups using an online random number generator (https://www.graphpad.com/quickcalcs/randomize1/(accessed on 10 June 2021) assigning 7 mice subjects to a group. The experiments included seven groups (a. Healthy-Saline, Control; b. Sick-untreated, Control; c. Sick-Saline, Control; d-h. MABA-peptide 3 to 6- treated Sick mice), and was repeated twice (two blocks) during a 12-month period. Cages were likewise allocated to treatment groups with randomly generated numbers. Body weights were measured throughout the study. Two female experimenters were blinded to the allocation of the mice and conduct of the experiments, and another female investigator was blinded for the outcome assessments and data analysis. All procedures were carried out in compliance with the ARRIVE guidelines and adhered to the guidelines of the joint ethics committee (IACUC) of the Hebrew University (an AAALAC International accredited institute), and Hadassah Medical Center in Jerusalem, Israel that approved the study protocol for animal welfare (MD-09-11910-5).
The B16 murine melanoma cell line was maintained in RPMI 1640 (Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Sigma, St Louis, MO), 20 mM HEPES (Gibco BRL), penicillin and streptomycin 100 IU/ml and 100 µg/ml, respectively. Cells in log-phase growth (24–48 h after plating) were harvested by brief treatment with trypsin, and washed with phosphate-buffered saline (PBS). The cells were first incubated with the peptides in PBS for 2 hours at 37ºC or left untreated (control), and thereafter 7 x 105 cells were injected subcutaneously in 0.15 ml PBS in the right flank of the mice. Eight mice were used for each group examined. The peptides were administered three times a week, for three weeks after cell inoculation, by intraperitoneal injection of a 10 mg/kg dose (cumulative dose of 90 mg/kg/mouse) in 0.2 ml PBS. Mice survival was followed-up every day for 80 days after cells inoculation and the death was recorded. Toxicity was evaluated by mice examination and body weight assessment. In a case, that a mouse showed clinical signs of discomfort or pain, it was euthanized and necropsy was performed. Mice survival was analyzed by the Kaplan-Meier method. The median survival of groups of B16 melanoma mice were compared to control group of sick tumor-implanted but non-peptide-treated mice with GraphPad Prism 5, performing both the log-rank (Mantel-Cox) test and the Gehan-Breslow-Wilcoxon test (Table S2). Experiments were performed under double blind testing. Palpable tumors under the skin were measured in two perpendicular diameters using a digital caliper. Tumor volume (V), which correlates well with tumor weight, was calculated as V = 0.5LW2 (L is the largest length diameter and W is the smallest width diameter). Sixteen days after implantation of the cells, mice were killed under pentobarbital anesthesia (120 mg/kg) and tumors were excised and weighed. Some mice were killed 3 days after implantation of the cells to determine tumor size, and at this time tumors were about 2 mm diameter and therefore were not yet dependent on angiogenesis. Sections of tumors were formalin fixed for further study. Immunohistochemistal analyses of blood vessel formation were performed with goat anti-mouse CD31 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) using the labeled streptavidin-biotin method. Briefly, sections were deparafinized in xylol and rehydrated in a graded alcohol series. Antigen retrieval was carried out by autoclaving sections in retrieval buffer (10 mM EDTA citrate buffer, pH 6.0) for 3 min. Endogenous peroxidase activity was blocked by incubation in 3% hydrogen peroxide at room temperature in the dark for 20 min. Non-specific binding of reagents was reduced by incubation of sections for 30 min in 5% normal rabbit serum. Sections were then incubated with goat anti-mouse CD31 (dilution 1/200) antibody overnight at 4°C, followed by incubation with biotinylated rabbit anti-goat IgG, and then streptavidin-biotin-horseradish peroxidase complex at 37°C for 1 h. A negative control was included with each run by replacing the primary antibody for non-immune rabbit serum. The nuclei were counter-stained with hematoxylin. Representative images were taken under a light microscope (×400) in randomly selected fields and the number of blood vessels stained brown were counted. Statistical analysis of the differences in tumor volume and blood vessel density were performed using one-way analysis of variance (ANOVA) and a value of p < 0.05 was considered statistically significant.
4.9. Cell Death
Cell death was assessed by measuring the release of lactate dehydrogenase ( LDH) into the medium using the Lactate Dehydrogenase necrotic assay [38]. LDH activity was determined spectrophotometrically at 340 nm by following the rate of conversion of oxidized nicotinamide adenine dinucleotide (NAD) to the reduced form of (NADH). LDH release was expressed as the optical density (OD) units.
4.10. Toxicity to Mice
Experiments with animals and animal care were approved by the Hebrew University Committee of Ethics and were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Adult male C57BL/6 mice (n = 10) weighing 18 gram, and aged 4 weeks, were housed in temperature-controlled rooms (22–25°C), with access to water and food ad libitum. Five mice were intraperitoneal injected twice a week, with a dose of 50 mg/kg of peptide 4 for four consecutive weeks (8 injections) and the control group of five mice were injected with saline. The animals were daily examined for autonomic symptoms, paralysis, motor activity, and regular behavior. Blood samples were taken from control and peptide-injected mice after 10 hours from injection and were analyzed for hematocrit and biochemical analysis at Koret School of Veterinary Medicine, Faculty of Agricultural, Food and Environment, The Hebrew University of Jerusalem.
4.11. Molecular Dynamic Simulations
The starting structures for the simulations of compounds 2 and 4 were built in Maestro 3D Builder as extended peptides and the two termini were kept charged. The structures were minimized using the protein preparation protocol [39] as implemented in Maestro (Schrödinger Release 2022-2) and proper protonation states of residues were determined. Quantum mechanical calculations were performed for the minimized conformation of the compounds to calculate atomic charges to be used in molecular dynamics (MD) simulations. Geometry optimization was performed for each compound, and the electrostatic potentials were calculated at the HF/6-31G* level using the Gaussian 09 software. The partial atom charges were determined using the RESP method [40] with the AnteChamber software [41,42]. MD simulations were carried out using the AMBER99SB-ILDN force field [43] as implemented in the Gromacs program (v. 2021.1) using GPU accelerators [44,45,46]. The compounds were first minimized in a cubic box of TIP3P water molecules with an extra extension along each axis of the peptide of 10 Å under periodic boundary conditions. Ions were added to the solution to make the system electrically neutral. After the minimization, compounds were equilibrated first under NVT conditions (at T = 300°K for 10 ns) employing the V-Rescale thermostat [47] and subsequently under NPT conditions (at T = 300°K and P = 1 bar for 10 ns) employing the V-Rescale thermostat [47] and the Parrinello-Rahman barostat [48]. The simulations were run with a time step of 2 fs using the leap-frog algorithm. Long-range electrostatic interactions were computed using Particle Mesh Ewald Summation [49,50]. All simulations employed a 10 Å cutoff for van der Waals and Coulomb interactions using the Verlet cutoff scheme [51]. The LINCS algorithm was used to constrain bond lengths [52]. Production runs were performed under NPT conditions as described above and were run for 3 μs each [53]. Root Mean square deviation (RMSD) calculations indicated that both simulations are converged (Supporting Information, Figure S1).
To explore the conformational space of the compounds, we performed clustering of 15000 frames retrieved from the MD production run using the Gromos method [54] as implemented in the Gromacs software. The algorithm counts the number of neighbors of each structure using a cut-off (0.5 nm), forms a cluster from the structure with the largest number of neighbors with all its neighbors and eliminates them from the pool of structures. The process was iterated until all structures have been considered. This procedure resulted in a total of 6 and 7 clusters for compound 2 and 4, respectively. The most populated cluster for all compounds was the first one, including 14212 conformers in case of compound 2, and 14282 conformers in case of compound 4. The central structure (i.e., the structure with the smallest average RMSD from all other structures of the cluster) of the biggest cluster of compounds 2 and 4 are presented in Figure S2 (Supporting Information)
4.12. Molecular Docking
To gain atomic-level insight into potential binding modes of peptides 2 and 4 to the integrin I-domain and to obtain approximated binding free energies, the central structure of each cluster, was docked into the crystal structure of the alpha-1 integrin I-domain which had crystalized in complex with a collagen-mimetic peptide (PDB code 2M32 [55]). Prior to docking, the protein structure was prepared with the Protein Preparation Wizard [39,56] to assign correct protonation states for all residues at physiological pH and to add missing hydrogens. Docking was performed by means of the PIPER algorithm, a protein-protein docking tool [56,57] which is implemented in the Schrödinger software. The algorithm rotates the ligand into a large number of different orientations with respect to the receptor, and each of these orientations is scored by the PIPER’s scoring function. Finally, PIPER clusters the resulting poses based on their RMSD values and outputs the centroids of the clusters with the respective PIPER scores.
4.13. MM/GBSA Calculations
MM/GBSA calculations were performed using Schrodinger’s Prime module with default parameters. Protein flexibility was considered using Hierarchical Sampling method in radius 3A from the peptide.
4.14. Statistical Analysis
Statistical analyses were performed using the software GraphPad Prism version 8.0.2 (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was determined by the one-way analysis of variance (ANOVA) or Student t-test. Median survival of mice was analyzed by Log-rank Mantel-Cox test. Normal distribution was confirmed using Shapiro-Wilk test (α = 0.05). The experiments were performed three times, and results are expressed as mean ± SEM. The differences were considered significant at p ≤ 0.05.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
CRediT authorship contribution statement
Conception and design, analysis and interpretation of data: P. Lazarovici, C. Gilon, A. Hoffman, J. A, Eble & H. Senderowitz; Synthesis, chemical purification, characterization, and quality control: M. S. Naamneh, T. Momic, C. Gilon & J. Katzhendler; Biochemical and cellular assays: J. Grosche, J. A, Eble , C. Marcinkiewicz; Metabolic stability assay: M. Klazas, A. Hoffman; Therapy study, animal handling and tumor measurements: T. Momic, P. Lazarovici; Molecular modelling and docking: N. Khazanov, H. Senderowitz; Review & revision of the manuscript: P. Lazarovici, C. Gilon, A. Hoffman , H. Senderowitz and J. A, Eble; The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Data availability
Data will be made available on request.
Acknowledgments
PL holds the Jacob Gitlin Chair in Physiology and is affiliated and supported in part by David R. Bloom Center for Pharmacy, Dr. Adolf and Klara Brettler Center for Research in Molecular Pharmacology and Therapeutics, and the Grass Center for Drug Design and Synthesis of Novel Therapeutics at the Hebrew University of Jerusalem, Israel. We would like to thank Mrs Zehava Cohen for help with artwork. PL would like to thank the financial support by the Alex Grass Center for Drug Design and Synthesis of Novel Therapeutics at The Hebrew University. The Deutsche Forschungsgemeinschaft (DFG) within the CRC1009 financially supported J.A.E. (Grant no. 446302350; SFB1009-A09).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Abbreviations
MABA, meta-aminobenzoic acid; SPPS, solid-phase peptide synthesis; TFA, trifluoroacetic acid; KTS, Lys-Thr-Ser tripeptide pharmacophore selective motif for binding collagen-integrin receptors; ELISA, enzyme-linked immunosorbent assay; HPLC, high pressure liquid chromatography; LC-MS, liquid chromatography-mass spectrometry; HUVEC, Human umbilical vein endothelial cells; VEGF, vascular endothelial growth factor; B16, a melanoma tumor cell line; CD31, platelet endothelial cell adhesion molecule 1 (PECAM-1) is a blood vessel marker; HBSS, Hank’s balanced salt solution; DIPEA, N,N-diisopropylethylamine; HBTU, 2-(1H-benzotriazole-1yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HOBt, 1-hydroxybenzo-triazole; FmocAa, fluorenylmethoxycarbonyl-conjugated amino acid; CB3, a collagen IV fragment; CMFDA, 5-chloromethylfluorescein diacetate; MD, molecular docking; MM-GBSA, molecular mechanics with generalized Born and surface area solvation; RMSD, root mean square deviation; PIPER, Schrodinger’s protein-protein docking tool.
Appendix A. Supplementary Material
Supplementary material related to this article (Synthesis of a peptide with MABA units, HPLC and MS analyzes of all MABA-peptides, tube formation-angiogenesis assay, ELISA assay’s selectivity for integrins, statistics of the median survival of groups of B16 melanoma bearing mice, RMSD plots and central structures of the biggest cluster from the Molecular Dynamic simulation) can be found at http://.....
References
- Moreno-layseca, P.; Icha, J.; Hamidi, H.; Ivaska, J. Europe PMC Funders Group Integrin trafficking in cells and tissues. Nat Cell Biol. 2019, 21, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2021, 21, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef]
- Cox, D. How not to discover a drug - integrins. Expert Opin. Drug Discov. 2020, 16, 197–211. [Google Scholar] [CrossRef]
- Kadry, Y.A.; Calderwood, D.A. Chapter 22: Structural and signaling functions of integrins. Biochim. et Biophys. Acta (BBA) - Biomembr. 2020, 1862, 183206–183206. [Google Scholar] [CrossRef] [PubMed]
- Topple, A.; Fifkova, E.; Baumgardner, D.; Cullen-Dockstader, K. Effect of age on blood vessels and neurovascular appositions in the CA1 region of the rat hippocampus. Neurobiol. Aging 1991, 12, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Pain, B.; Woods, C.; Saez, J.; Flickinger, T.; Raines, M.; Peyroll, S.; Moscovici, C.; Moscovici, M.; Kung, H.-J.; Jurdic, P.; et al. EGF-R as a hemopoietic growth factor receptor: The c-erbB product is present in chicken erythrocytic progenitors and controls their self-renewal. Cell 1991, 65, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Moberg, P.E.; Miles, L.A.; Wagner, S.; Soloway, P.; Gardner, H.A. Elevated matrix metalloprotease and angiostatin levels in integrin α1 knockout mice cause reduced tumor vascularization. Proc. Natl. Acad. Sci. 2000, 97, 2202–2207. [Google Scholar] [CrossRef]
- Staniszewska, I.; Walsh, E.M.; Rothman, V.L.; Gaathon, A.; Tuszynski, G.P.; Calvete, J.J.; Lazarovici, P.; Marcinkiewicz, C. Effect of VP12 and viperistatin on inhibition of collagen-receptor-dependent melanoma metastasis. Cancer Biol. Ther. 2009, 8, 1507–1516. [Google Scholar] [CrossRef]
- Chen, X.; Su, Y.; Fingleton, B.; Acuff, H.; Matrisian, L.M.; Zent, R.; Pozzi, A. Increased plasma MMP9 in integrin α1-null mice enhances lung metastasis of colon carcinoma cells. Int. J. Cancer 2005, 116, 52–61. [Google Scholar] [CrossRef]
- Senger, D.R.; Perruzzi, C.A.; Streit, M.; Koteliansky, V.E.; de Fougerolles, A.R.; Detmar, M. The α1β1 and α2β1 Integrins Provide Critical Support for Vascular Endothelial Growth Factor Signaling, Endothelial Cell Migration, and Tumor Angiogenesis. Am. J. Pathol. 2002, 160, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Niland, S.; Schulz, J.-N.; Wang, F.; Eble, J.A.; Leitges, M.; Mauch, C.; Krieg, T.; Zigrino, P.; Eckes, B. Role of Integrins α1β1 and α2β1 in Wound and Tumor Angiogenesis in Mice. Am. J. Pathol. 2016, 186, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Momic, T.; Katzehendler, J.; Benny, O.; Lahiani, A.; Cohen, G.; Noy, E.; Senderowitz, H.; Eble, J.A.; Marcinkiewicz, C.; Lazarovici, P. Vimocin and Vidapin, Cyclic KTS Peptides, Are Dual Antagonists ofα1β1/α2β1Integrins with Antiangiogenic Activity. J. Pharmacol. Exp. Ther. 2014, 350, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Murciano, M.P.; Monleón, D.; Calvete, J.J.; Celda, B.; Marcinkiewicz, C. Amino acid sequence and homology modeling of obtustatin, a novel non-RGD-containing short disintegrin isolated from the venom ofVipera lebetina obtusa. Protein Sci. 2003, 12, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Murciano, M.P.; Monleón, D.; Marcinkiewicz, C.; Calvete, J.J.; Celda, B. NMR Solution Structure of the Non-RGD Disintegrin Obtustatin. J. Mol. Biol. 2003, 329, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Kisiel, D.G.; Calvete, J.J.; Katzhendler, J.; Fertala, A.; Lazarovici, P.; Marcinkiewicz, C. Structural determinants of the selectivity of KTS-disintegrins for the α1β1 integrin. FEBS Lett. 2004, 577, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Momic, T.; Arlinghaus, F.T.; Arien-Zakay, H.; Katzhendler, J.; Eble, J.A.; Marcinkiewicz, C.; Lazarovici, P. Pharmacological Aspects of Vipera xantina palestinae Venom. Toxins 2011, 3, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.M.; Marcinkiewicz, C. Non-RGD-containing snake venom disintegrins, functional and structural relations. Toxicon 2011, 58, 355–362. [Google Scholar] [CrossRef]
- Brown, M.C.; Eble, J.A.; Calvete, J.J.; Marcinkiewicz, C. Structural requirements of KTS-disintegrins for inhibition of α1β1 integrin. Biochem. J. 2008, 417, 95–101. [Google Scholar] [CrossRef]
- T. Momic, J. Katzhendler, C. Marcinkiewicz, A.J. Eble, P. Lazarovici, Medicinal Chemistry Approach for Solid Phase Synthesis of Peptide Mimetics of Viperistatin Disintegrin as Lead Compounds for α1/α2 Integrin Receptors. In: G. Kokotos, V. Constantionou-Kokotou, J. Matsoukas (Eds.), Proceedings of the 32nd European Peptide Symposium, Athens, Greece, 2012, 304-5.
- Zhang, D.-W.; Zhao, X.; Hou, J.-L.; Li, Z.-T. Aromatic Amide Foldamers: Structures, Properties, and Functions. Chem. Rev. 2012, 112, 5271–5316. [Google Scholar] [CrossRef] [PubMed]
- Kubik, S. Synthetic Receptors Based on Abiotic Cyclo(pseudo)peptides. Molecules 2022, 27, 2821. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-M.; Chen, S.-G.; Zhao, X.; Jiang, X.-K.; Li, Z.-T. meta-Substituted benzamide oligomers that complex mono-, di- and tricarboxylates: folding-induced selectivity and chirality. Org. Biomol. Chem. 2011, 9, 8122–8129. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Sakane, T.; Shibukawa, M.; Hashida, M.; Sezaki, H. Absorption and Metabolic Characteristics of p-Aminobenzoic Acid and Its Isomer, m-Aminobenzoic Acid, from the Rat Small Intestine. J. Pharm. Sci. 1991, 80, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Benke, B.P.; Madhavan, N. Active ion transporters from readily accessible acyclic octapeptides containing 3-aminobenzoic acid and alanine. Chem. Commun. 2013, 49, 7340–7342. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, N.; Poojary, B.; Rai, V.; Vasantha, S.P. A preliminary study on quinazolinylaminobenzoyl monopeptide esters as effective Gram-positive bacteriostatic agents. Futur. Med. Chem. 2019, 11, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, W.; Bae, J.-S.; Ma, E. Synthesis and in Vitro and in Vivo Anticoagulant and Antiplatelet Activities of Amidino- and Non-Amidinobenzamides. Molecules 2016, 21, 676. [Google Scholar] [CrossRef] [PubMed]
- Weerapana, E.; Imperiali, B. Peptides to peptidomimetics: towards the design and synthesis of bioavailable inhibitors of oligosaccharyl transferase. Org. Biomol. Chem. 2002, 1, 93–99. [Google Scholar] [CrossRef]
- P. Lazarovici, C. P. Lazarovici, C. Marcinkiewicz, P.I. Lelkes, Cell-based adhesion assays for isolation of snake venom’s integrin antagonists, in: Methods Mol. Biol., Humana Press Inc., (2020) pp. 205–223. [CrossRef]
- La Manna, S.; Di Natale, C.; Florio, D.; Marasco, D. Peptides as Therapeutic Agents for Inflammatory-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2714. [Google Scholar] [CrossRef]
- P. Lazarovici, A. P. Lazarovici, A. Lahiani, G. Gincberg, D. Haham, A. Fluksman, O. Benny, C. Marcinkiewicz, P.I. Lelkes, Nerve growth factor-induced angiogenesis: 1. Endothelial cell tube formation assay, Methods Mol. Biol. 1727 (2018) 239–250. [CrossRef]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ — A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Fields, C.G.; Lovdahl, C.M.; Miles, A.J.; Hageini, V.L.M.; Fields, G.B. Solid-Phase synthesis and stability of triple-helical peptides incorporating native collagen sequences. Biopolym. 1993, 33, 1695–1707. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.A.; Tuckwell, D.S. The α2β1 integrin inhibitor rhodocetin binds to the A-domain of the integrin α2 subunit proximal to the collagen-binding site. Biochem. J. 2003, 376, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Schumacher-Klinger, A.; Fanous, J.; Merzbach, S.; Weinmueller, M.; Reichart, F.; Räder, A.F.B.; Gitlin-Domagalska, A.; Gilon, C.; Kessler, H.; Hoffman, A. Enhancing Oral Bioavailability of Cyclic RGD Hexa-peptides by the Lipophilic Prodrug Charge Masking Approach: Redirection of Peptide Intestinal Permeability from a Paracellular to Transcellular Pathway. Mol. Pharm. 2018, 15, 3468–3477. [Google Scholar] [CrossRef]
- Fluksman, A.; Steinberg, E.; Orehov, N.; Shai, E.; Lahiani, A.; Katzhendler, J.; Marcinkiewicz, C.; Lazarovici, P.; Benny, O. Integrin α2β1-Targeted Self-Assembled Nanocarriers for Tumor Bioimaging. ACS Appl. Bio Mater. 2020, 3, 6059–6070. [Google Scholar] [CrossRef]
- Y. Maatuf, A. Y. Maatuf, A. Priel, P. Lazarovici, Measurements of Cell Death Induced by Snake and Spider's Venoms and Derived Toxins. Methods Mol Biol. (2020) 2068:239-268. [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Woods, R.; Chappelle, R. Restrained electrostatic potential atomic partial charges for condensed-phase simulations of carbohydrates. J. Mol. Struct. THEOCHEM 2000, 527, 149–156. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. Best bang for your buck: GPU nodes for GROMACS biomolecular simulations. J. Comput. Chem. 2015, 36, 1990–2008. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2007, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101–074101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef]
- B. Hess, H. B. Hess, H. Bekker, H.J.C. Berendsen, J.G.E.M. Fraaije, LINCS: A Linear Constraint Solver for molecular simulations, J. Comput. Chem. 18 (1997) 1463–1472. [CrossRef]
- Hockney, R.; Goel, S.; Eastwood, J. Quiet high-resolution computer models of a plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- X. Daura, K. X. Daura, K. Gademann, B. Jaun, D. Seebach, W.F. Van Gunsteren, A.E. Mark, Peptide folding: When simulation meets experiment, Angew. Chemie - Int. Ed. 38 (1999) 236–240. [CrossRef]
- Chin, Y.K.-Y.; Headey, S.J.; Mohanty, B.; Patil, R.; McEwan, P.A.; Swarbrick, J.D.; Mulhern, T.D.; Emsley, J.; Simpson, J.S.; Scanlon, M.J. The Structure of Integrin α1I Domain in Complex with a Collagen-mimetic Peptide. J. Biol. Chem. 2013, 288, 36796–36809. [Google Scholar] [CrossRef] [PubMed]
- Chuang, G.-Y.; Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. DARS (Decoys As the Reference State) Potentials for Protein-Protein Docking. Biophys. J. 2008, 95, 4217–4227. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Metabolic stability of the MABA-peptides in rat plasma (n=3; p≤ 0.05). The amount of the peptides is presented as a percentage of the sample at time 0. .
Figure 1.
Metabolic stability of the MABA-peptides in rat plasma (n=3; p≤ 0.05). The amount of the peptides is presented as a percentage of the sample at time 0. .
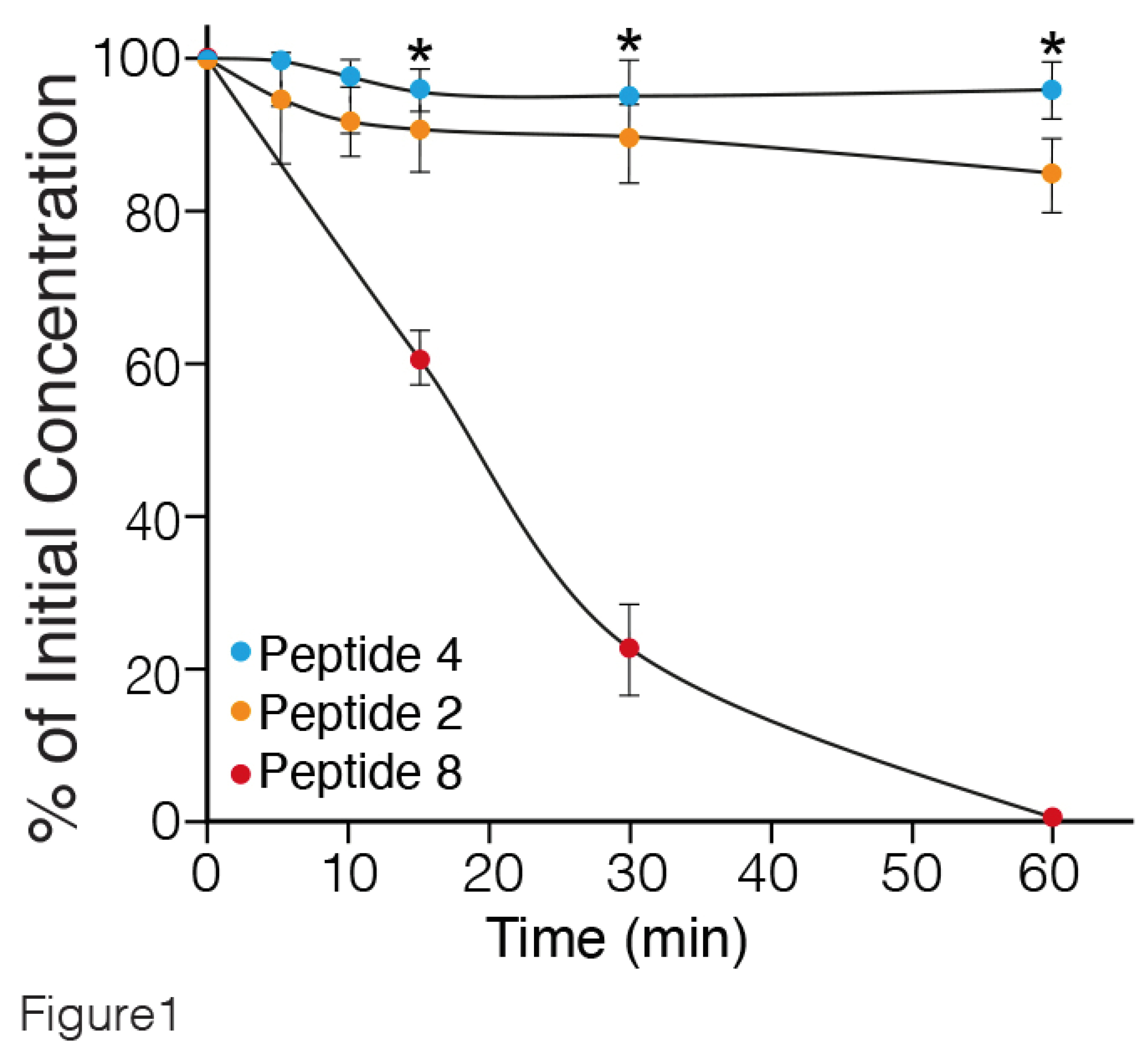
Figure 2.
Quantification of tube formation after treatment of HUVEC cells with MABA-peptides in the Matrigel assay. (a) Phase contrast images taken after 6 h of the treatment. The left column of the images indicates phase contrast images, the right column indicates the angiogenesis analyzer of quantification of tube formation as the number of junctions, nodes, meshes or total tube length per field of view. Error bars indicate SEM calculated from three different experiments performed in triplicates (*p≤ 0.05; **p≤ 0.01;***p≤ 0.001).
Figure 2.
Quantification of tube formation after treatment of HUVEC cells with MABA-peptides in the Matrigel assay. (a) Phase contrast images taken after 6 h of the treatment. The left column of the images indicates phase contrast images, the right column indicates the angiogenesis analyzer of quantification of tube formation as the number of junctions, nodes, meshes or total tube length per field of view. Error bars indicate SEM calculated from three different experiments performed in triplicates (*p≤ 0.05; **p≤ 0.01;***p≤ 0.001).
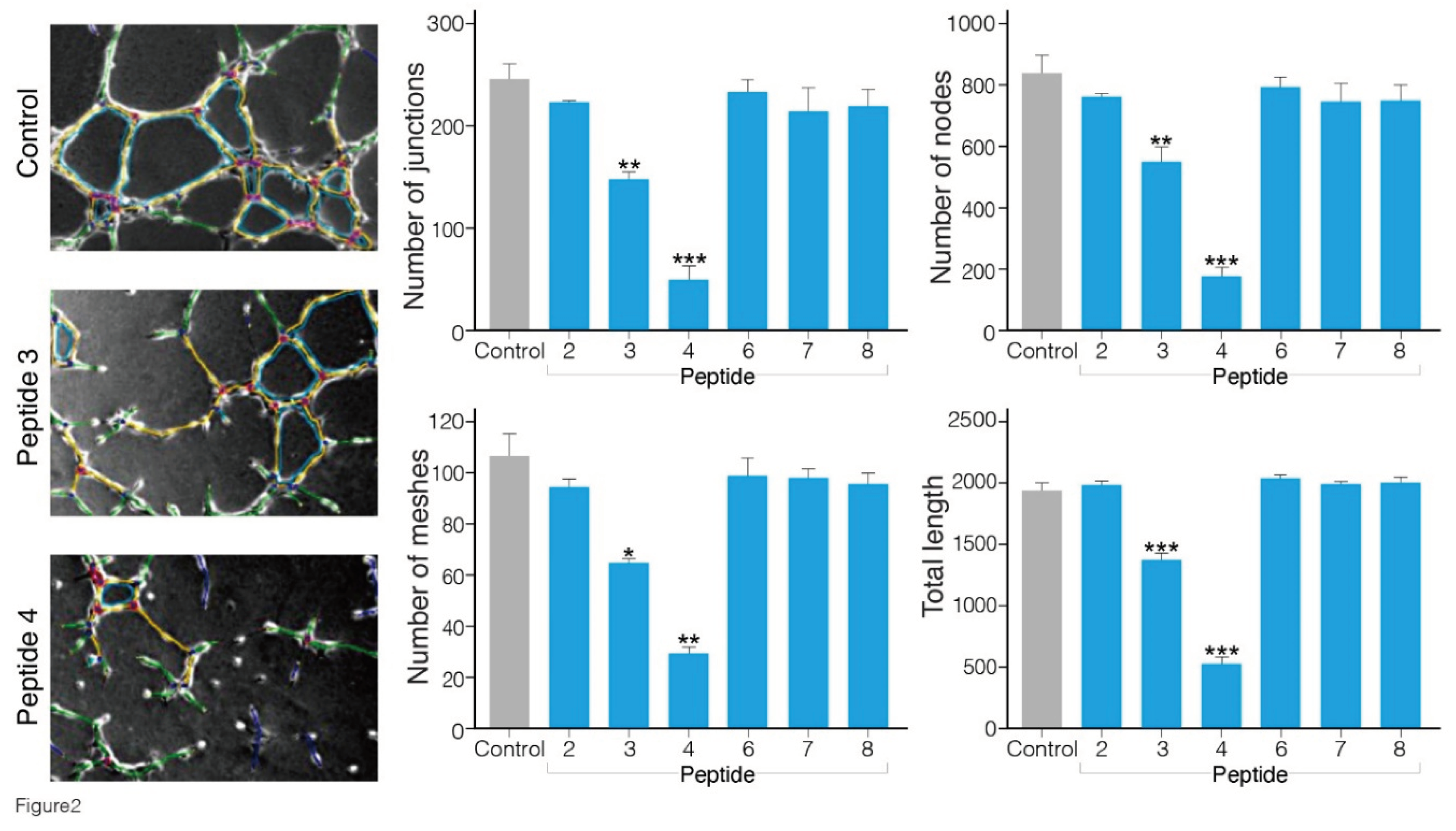
Figure 3.
Anti-melanoma effects of the MABA-peptides in C57BL/6 mice bearing B16 melanomas. The cells (7 x 105) were subcutaneously inoculated at the right flank of individual mice. Immediately after cell injection, mice were randomly divided into different groups (n = 8) and intraperitoneal administered with vehicle control (PBS solution) or 10 mg/kg of MABA-peptide three times a week. Tumor volume was measured at the indicated days. At day 16, mice were sacrificed and tumors were dissected; (A) Tumor volume of MABA-peptide 4 treated mice (light blue) compared to untreated sick group (blue); *p < 0.01 versus sick group (B); Kaplan-Meier survival plots of saline- (dashed line) or the MABA-peptide-treated (solid lines) tumor-bearing mice; (C) Inhibition of intra-tumoral angiogenesis. Representative images (left panel; X400) of vascular growth, detected by immune staining of CD31 in melanoma sections from mice with different treatments. Number of brown stained, CD31-exposing vessels (encircled in yellow) of each group was calculated using Image J software (right panel). Data are presented as mean ± SEM of 7 mice. **p < 0.01 versus control group.
Figure 3.
Anti-melanoma effects of the MABA-peptides in C57BL/6 mice bearing B16 melanomas. The cells (7 x 105) were subcutaneously inoculated at the right flank of individual mice. Immediately after cell injection, mice were randomly divided into different groups (n = 8) and intraperitoneal administered with vehicle control (PBS solution) or 10 mg/kg of MABA-peptide three times a week. Tumor volume was measured at the indicated days. At day 16, mice were sacrificed and tumors were dissected; (A) Tumor volume of MABA-peptide 4 treated mice (light blue) compared to untreated sick group (blue); *p < 0.01 versus sick group (B); Kaplan-Meier survival plots of saline- (dashed line) or the MABA-peptide-treated (solid lines) tumor-bearing mice; (C) Inhibition of intra-tumoral angiogenesis. Representative images (left panel; X400) of vascular growth, detected by immune staining of CD31 in melanoma sections from mice with different treatments. Number of brown stained, CD31-exposing vessels (encircled in yellow) of each group was calculated using Image J software (right panel). Data are presented as mean ± SEM of 7 mice. **p < 0.01 versus control group.
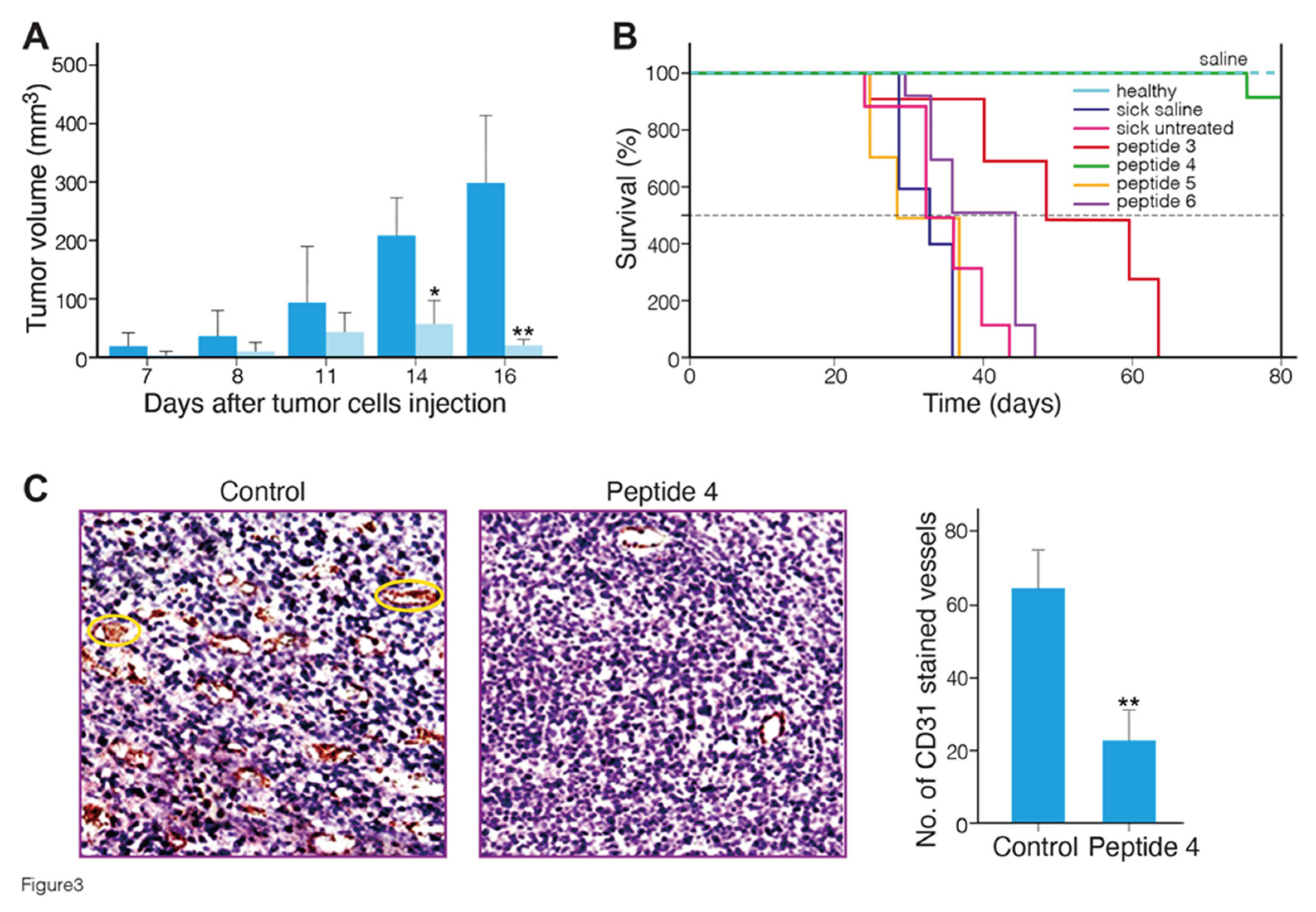
Figure 4.
Radius of gyration (Rg) plots for peptides 2 and 4 as a function of simulations time.

Figure 5.
The binding modes of the lowest energy poses (based on MM/GBSA energies) of peptides 2 and 4 in their predicted integrin binding sites, in comparison with the binding mode of the crystallized collagen-mimetic peptide. Peptides 2 and 4 are colored blue and orange, respectively. The α1 integrin I-domain protein is depicted in ribbon form, highlighted in green, while three crystallized collagen-mimetic peptides (PDB code 2M32) are distinguished by their coloring in yellow, cyan, and violet.
Figure 5.
The binding modes of the lowest energy poses (based on MM/GBSA energies) of peptides 2 and 4 in their predicted integrin binding sites, in comparison with the binding mode of the crystallized collagen-mimetic peptide. Peptides 2 and 4 are colored blue and orange, respectively. The α1 integrin I-domain protein is depicted in ribbon form, highlighted in green, while three crystallized collagen-mimetic peptides (PDB code 2M32) are distinguished by their coloring in yellow, cyan, and violet.
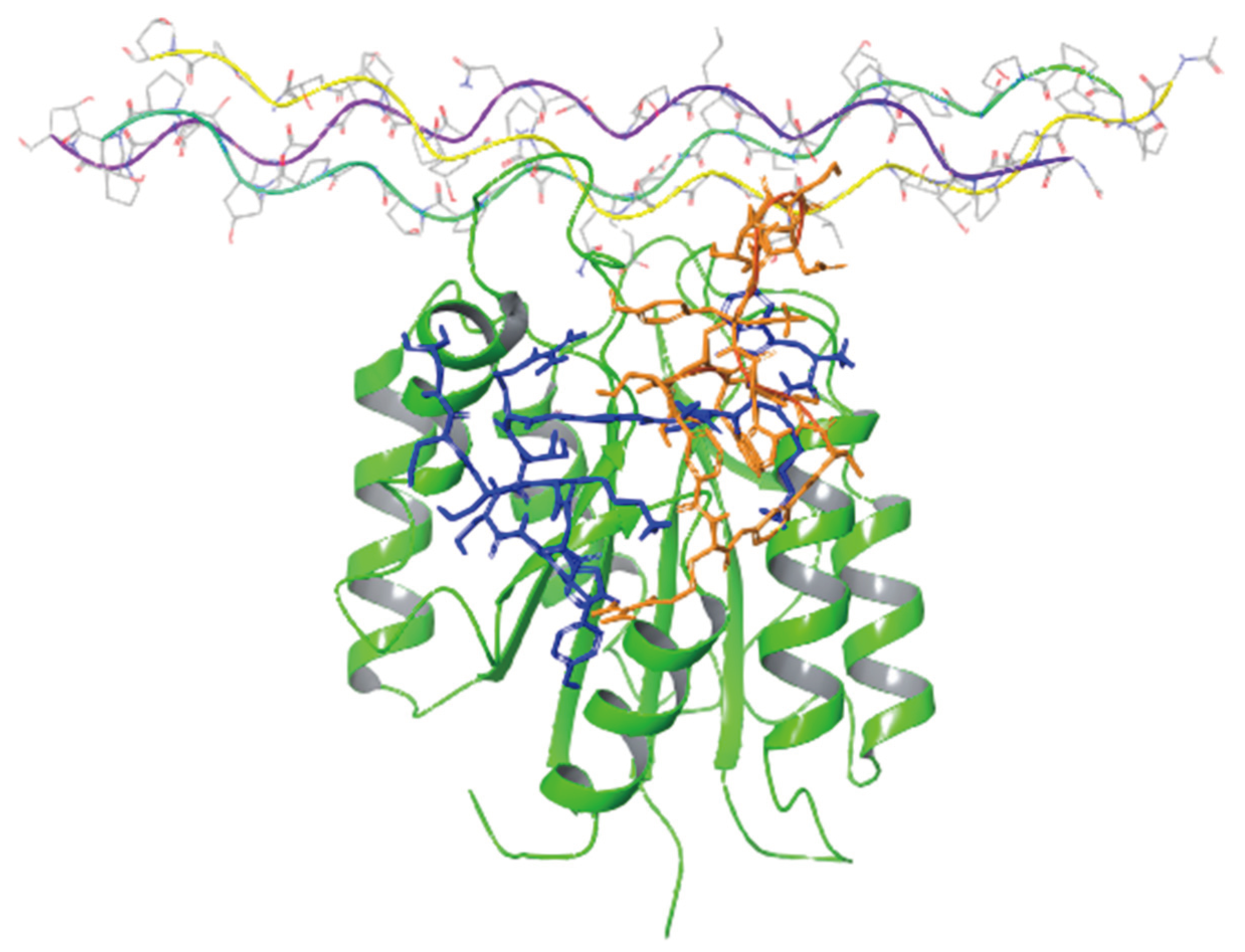

Table 1.
Inhibitory effect of the synthetic KTS(R)-peptides containing meta-aminobenzoic acid on binding of GST-α1 A domain to collagen IV fragment CB3 and on α1- overexpressor cells.
Table 1.
Inhibitory effect of the synthetic KTS(R)-peptides containing meta-aminobenzoic acid on binding of GST-α1 A domain to collagen IV fragment CB3 and on α1- overexpressor cells.
| Peptide Number |
Sequence | α1β1 binding in vitro assay IC50 (µM) |
α1β1adhesion in cell assay IC50 (µM) |
|---|---|---|---|
| 1 | H2N-W-KTS-R-TSHY-MABA-TGKSDG-COOH | 828 | ≥3000 |
| 2 | H2N-W-KTS- MABA-R-TSHY-GKSDG-COOH | 600 | ≥3000 |
| 3 | H2N-KTS-W-KTS-MABA-R-TSHY-GKSDG-COOH | 4 42 | ≥900 |
| 4 | H2N-W-KTS-MABA-R-MABA-TSHY-GKSDG-COOH | 32 | 550 |
| 5 | H2N-W-KTS-MABA-RTSHY-MABA-TGKSDG-COOH | 1000 | ≥3000 |
| 6 | H2N-W-KTR-MABA-R-MABA-TSHY-TGKSDG-COOH | 2142 | 200696 |
| 7 | H2N-W-KTR-MABA-R-MABA-TSHY-MABATGKSDG-COOH | 2481 | ≥5000 |
| 8 | H2N-20W-KTS-R-TSHY-TGKSDG36-COOH | 970 | ≥3000 |
Table 2.
MM/GBSA binding free energies (in kcal/mol) for the centroid poses of peptides 2 and 4 from the corresponding clusters.
Table 2.
MM/GBSA binding free energies (in kcal/mol) for the centroid poses of peptides 2 and 4 from the corresponding clusters.
| Peptide 2 | Peptide 4 | |
|---|---|---|
| Cluster No. | MM/GBSA energy | MM/GBSA energy |
| 1 | -35.8 | -55.7 |
| 2 | -88.1 | -47.2 |
| 3 | -39.7 | -91.1 |
| 4 | -70.0 | -80.3 |
| 5 | -68.9 | -73.8 |
| 6 | -41.5 | -77.3 |
| 7 | -52.8 |
Table 3.
Interactions formed between the lowest energy poses (based on MM/GBSA calculations) of peptides 2 and 4 at their predicted binding sites on the integrin structure. *.
Table 3.
Interactions formed between the lowest energy poses (based on MM/GBSA calculations) of peptides 2 and 4 at their predicted binding sites on the integrin structure. *.
| Peptide 4 | Interactions between peptide and protein | Peptide 2 | Interactions between peptide and protein |
|---|---|---|---|
| Trp1 | HB with BB of Tyr 17 | Trp1 | |
| Lys2 | Lys2 | BB HB with Val175 and HB with Asp28 | |
| Thr3 | HB with Ser176 | Thr3 | HB with BB of Leu143 |
| Ser4 | Ser4 | HB BB of Ala174 | |
| MABA 5 | MABA 5 | ||
| Arg6 | 2 HB with Asp177 | Arg6 | HB with Thr153 |
| MABA 7 | Thr7 | HB with Asn174 | |
| Thr8 | Ser8 | HB with Thr168 | |
| Ser9 | HB with Ser21 | His9 | |
| His10 | Tyr10 | HB with Thr183 | |
| Tyr11 | HB with BB of Tyr146 | Gly11 | |
| Gly12 | BB HB with Tyr146 | Lys12 | HB with Asp177 |
| Lys13 | Ser13 | ||
| Ser14 | Asp14 | ||
| Asp15 | Gly15 | BB HB with Glu154 | |
| Gly16 |
*HB and BB stand for hydrogen bond and backbone, respectively. Interactions common to both peptides are highlighted. .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



