
Submitted:
04 March 2024
Posted:
09 April 2024
You are already at the latest version
Abstract
Cholangiocellular carcinoma (CCA) is the second most common primary liver cancer, with increasing incidence worldwide and inadequate therapeutic options. Intra- and extrahepatic bile ducts have distinctly different embryonic origins and developmental behavior, and accordingly intra- and extrahepatic CCAs (ICC vs. ECC) are molecularly different. A promising strategy in oncotherapy is targeted therapy, targeting proteins that regulate cell survival and proliferation; such as the MAPK/ERK and PI3K/AKT/mTOR signaling pathways. Inhibitors of these pathways have been tested previously in CCA cell lines. However, these cell lines could not be clearly assigned to ICC or ECC, and results indicated apoptosis induction by targeted therapeutics. We tested targeted therapeutics (selumetinib, MK2206) in three defined ICC cell lines (HuH28, RBE, SSP25). We observed cooperative effects of dual inhibition of the two pathways, in accordance with inhibition of phospho-AKT and phospho-ERK1/2 expression. Proliferation was blocked more effectively with dual inhibition than with each single inhibition, but cell numbers did not drop below baseline. Accordingly, we observed G1-phase arrest, but not apoptosis (cleaved caspase-3, AIFM1 regulation, subG1-phase) or necroptosis (phospho-MLKL). We conclude that dual inhibition of MAPK/ERK and PI3K/AKT/mTOR pathways is highly effective to block proliferation of ICC cell lines, but therapy must certainly be supplemented with standard therapies.
Keywords:
Cholangiocyte – primary liver cancer – selumetinib – AZD6244 – MK2206 – intrahepatic cholangiocarcinoma
Introduction
Cholangiocellular carcinoma (CCA) is a tumor arising from cholangiocytes in the bile ducts. It accounts for 3-5% of gastrointestinal cancers and is the second most common liver tumor after hepatocellular carcinoma (HCC) [1,2,3]. In CCA, a distinction must be made between intrahepatic (ICC) and extrahepatic (ECC) tumors depending on their anatomical localization [4]. ICC arises from bile ducts deeply within the liver, whereas ECC involves the larger perihilar and extrahepatic (distal) bile ducts [5,6]. Thereby, perihilar CCA is the most common form, accounting for 50-60% of CCAs, while 20-30% are distal CCAs. ICC accounts for 10% of all primary liver tumors [1]. Even though ICC remains a rare tumor, its incidence has been increasing worldwide in the last 30 years, e. g. in the USA by approximately 165% to currently 0.95 per 100,000 inhabitants [7]. The majority of CCAs, approximately 70%, develop for no apparent reason. However, there are a number of risk factors such as liver flukes Opisthorchis viverrini and Clonorchis sinensis, whose cercariae are transmitted through eating raw freshwater fish. Liver flukes occur primarily in East Asia and increase the risk of contracting CCA [4,8]. Primary sclerosing cholangitis (PSC), hepatitis B and C with associated cirrhosis, diabetes, or obesity are other risk factors [9]. Strikingly, in industrial countries, ICC often occurs in the absence of cirrhosis [5]. Diagnosis of ICC is difficult because symptoms appear late and tend to be nonspecific, such as weight loss, abdominal discomfort, jaundice, malaise, or hepatomegaly. A curative treatment option for ICC is surgical resection. However, this is only possible at early stages and can therefore be performed in no more than 20-40% of cases. In the majority of patients, the tumor is too advanced for resection [10]. The first-line chemotherapy for unresectable CCA consists of gemcitabine and cisplatin, and second-line chemotherapeutics can be effective in selected patients [11].
A promising strategy in cancer therapy is targeted therapy, which targets proteins that regulate cell survival and proliferation [12,13]. Although this concept has been proposed for CCA quite some time ago [14,15], it has not yet been firmly established in the clinic. In particular, the molecular heterogeneity of ECC and ICC must be taken into account [16], which is very likely due to the fact that extra- and intrahepatic bile ducts develop differently in the embryo. The embryonic development, differentiation and migration potency of the respective cell has a major influence on the pathophysiology. While the extrahepatic bile duct emerges directly by sprouting from the foregut endoderm, the intrahepatic bile ducts develop from bipotent progenitor cells, which transiently form a sheath of epithelial cells called ductal plate. Only later do the cells acquire a tubular morphology [17,18]. Therefore, ICC and ECC must be regarded as heterogenous tumor entities. For targeted therapy, this must be taken into account. For ICC patients, targeted therapy (with pemigatinib) has only been approved for a small cohort of patients with gene fusions affecting fibroblast growth factor receptor 2 (FGFR2) [19].
A frequently used strategy is targeted inhibition of the MAPK/ERK signaling pathway, which is hyperactive in at least 40% of cancers [20]. Furthermore, the PI3K/AKT/mTOR signaling pathway is highly active in numerous types of cancer [21]. Although the effects of inhibition of the two pathways were tested in CCA in vitro [15], the three human cell lines used in this study were either ECC (one cell line) or cannot be clearly assigned to ECC or ICC. Here, we focused on ICC and the cell lines HuH28, RBE and SSP25. We studied the AKT inhibitor MK2206 and the MEK inhibitor selumetinib, and determined their single and dual effects on cell cycle, proliferation, phosphorylation of signaling molecules and apoptosis. We observed cooperative effects of dual pathway inhibition on proliferation in conjunction with a G1 phase arrest, but no apoptosis. Therefore, cell counts never dropped below baseline. In sum, dual pathway inhibition is highly effective in blocking proliferation of ICC in vitro, but it must obviously be performed in conjunction with a standard therapy that kills the remaining tumor cells.
Material and Methods
Cell Culture
The human ICC cell lines (HuH28, RBE, SSP25) were purchased fresh from RIKEN BioResources Research Center (Tsukuba, Japan). They were incubated at 37°C with 5% CO2 under humidified atmosphere in RPMI medium (10% FCS, 1% penicillin/streptomycin). Cells were amplified, and used usually during passages 9-10, but not beyond passage 16.
Proliferation Assay and Drug Preparation
Proliferation assays were performed in 96-well plates as described previously [22]. Cells were seeded at a concentration of 5x103 cells/ml in 100 µl RPMI and incubated at 37°C over-night. Cells were treated with the inhibitors MK2206 and selumetinib (AZD-6244) (both from: Selleckchem, Munich, Germany) and DMSO (control). For single treatment 0.1 µM, 0.5 µM, 1 µM and 5 µM were used. Dual treatment: 0.5 µM (each) MK2206+selumetinib, and 1µM (each) MK2206+selumetinib were used. The stock solution was 10 mM of the respective drug dissolved in DMSO. Stock solutions were then diluted with cell medium to the respective final concentration. DMSO controls contained the same amount of DMSO as in the highest inhibitor concentration.
After 0 h, 24 h, 48 h and 72 h, cells were fixed with 5% glutaraldehyde and stained with crystal violet. After drying, 10% acetic acid was added and incubated for 15 minutes. Extinction was measured at 595 nm with iMark™ Microplate Absorbance Reader (Bio-Rad). Extinction was measured at various time points, normalized to the extinction at 0h, and converted to percent. Experiments were repeated at least 3 times with each 8 replicates.
Protein Extraction and Western Blot
Cells were seeded at concentrations of 3x106 cells/mL in RPMI in cell culture dishes and incubated for 12 h at 37°C. Then, cells were treated with inhibitors (1µM) for 12 h and washed with PBS containing 1 mM sodium orthovanadate on ice. Lysis was performed with a lysis buffer mixture containing RIPA lysis buffer (aqua dest., 140 mM NaCl, 10 mM TrisHcl pH 8, 1 mM EDTA, 1% Triton, 0,1% SDS, 0,1% Sodiumdeox) with 1 mM SOV and 1x Sample Complete Protease Inhibitor (Roche Diagnostics) for 30 minutes on ice. Lysates were centrifuged at 14,000 rpm for 15 minutes at 4°C and the supernatant was transferred. Protein concentration was measured by the Pierce BCA protein assay according to the manufacturer’s instructions (Thermo Fisher Scientific). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-Page) was performed. 20 µg protein were denatured at 70°C for 10 minutes. Proteins were transferred onto a PVDF membrane and incubated with a primary antibody (Table 1) over-night at 4°C followed by washing processes. Then, secondary horseradish peroxidase (HRP)-conjugated antibodies were incubated for 1h at RT. Visualization was performed with an enhanced chemiluminescence (ECL) solution (SignalFire™ ECL Reagent or Clarity Western ECL Substrate) in BioRad ChemiDoc (Bio-Rad). WB analyses were repeated at least three times, and luminescence signals were quantified using Image Lab Software (Bio-Rad).

Table 1.
Primary antibodies and manufacturers.
| Primary Antibody | Manufacturer |
|---|---|
| β-actin - HRP conjugated | CellSignaling (Cambridge, UK) |
| phospho-AKT (Ser473) | CellSignaling (Cambridge, UK) |
| AKT | CellSignaling (Cambridge, UK) |
| anti-MLKL (Phospho S358) [ERP9514] | Abcam (Cambridge, UK) |
| anti-MLKL [ERP17514] | Abcam (Cambridge, UK) |
| cleaved caspase-3 (Asp175) | CellSignaling (Cambridge, UK) |
| phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) | CellSignaling (Cambridge, UK) |
| p44/42 MAPK (ERK1/2) | CellSignaling (Cambridge, UK) |
| α-tubulin - HRP conjugated | CellSignaling (Cambridge, UK) |
Semiquantitative Real-Time PCR (qPCR)
Cells were treated for 24h with 1 µM MK2206 or selumetinib, or with 1 µM of each drug.
For RNA isolation, medium was removed and cells were washed with PBS. NucleoZOL (Machery-Nagel) was used according to manufacturer’s instructions. 2 µg RNA were transcribed with Qiagen Omniscript reverse transcriptase (QIAGEN). The relative RNA expression was determined with the 2-ΔΔCT method [23]. Experiments were performed three times in duplicates. The following primers were used to detect Apoptosis inducing factor mitochondria associated-1 (AIFM1); Fwd: 5’-TGGGCTTATCAACAGTAGGAGC-3’; Rev: 5’-TTCTGGTGTCAGCCCTAACC-3’, Actin Fwd: 5’-GCATCCCCCAAAGTTCACAA-3’; Rev: 5’-AGGACTGGGCCATTCTCCTT-3’.
Flow Cytometry
Cells were seeded in 6-well plates in concentrations of 8x104 cells/mL and were treated with inhibitors or DMSO (control) after 24 h, and were further incubated for 24 h. Cells were washed with PBS and suspended in Nicoletti buffer with 50 µg/mL propidium iodide (PI). Flow cytometry was measured with the BD LSRFortessa™ X-20. The maximum number of detectable events was 5000. Experiments were repeated 5 times. Cell cycle analysis was performed using FlowJoTM Software.
Statistical Analysis
The data were analyzed with Graph Prism 5 software and Microsoft Excel 2019. The standard deviation (SD) was calculated for all experiments. Statistical significance was calculated with two-way-ANOVA (proliferation assays) or one-way-ANOVA (qPCR, flow cytometry) between treatment and DMSO-control group. Statistical level of significance is shown as: *p ≤0.05, **p ≤0.01, ***p ≤0.001, with 95% level of confidence.
Results
AKT Inhibitor MK2206 Effectively Reduces Proliferation in ICC cell lines
To determine the effects of MK2206, proliferation assays were performed with concentrations ranging from 0.1 µM – 5 µM; in addition to previous studies on 10 µM and 25 µM [24]. Results show that numbers of all three ICC cell lines (HuH28, RBE, SSP25) increased steadily over time in both the medium control group and the DMSO control group. The IC50 (95% CI) values after 72 h treatment were similar for all three cell lines: HuH28 5.92 µM (3.37-10.41); RBE 6.09 µM (2.9-12.69); SSP25 5.08 µM (2.8-9.2). MK2206 application induced a dose-dependent inhibition of cell proliferation in all cell lines (Figure 1). Highly significant inhibition in comparison with the DMSO control was observed at 0.5 µM after 24 h in HUH28 cells, resulting in 15% growth inhibition (Figure 1A and Suppl. Figure 1A). Cell counts after inhibition with 0.5 µM – 5 µM MK2206 reached the baseline (100%), but did not drop below this value (Figure 1A). MK2206 induced highly significant inhibition at 1 µM after 24h in RBE cells as well; again with 15% growth inhibition (Figure 1B and Suppl. Figure 1B). In SSP25, significant effects were noticed at 0.5 µM after 48 h (Figure 1C and Suppl. Figure 1C).
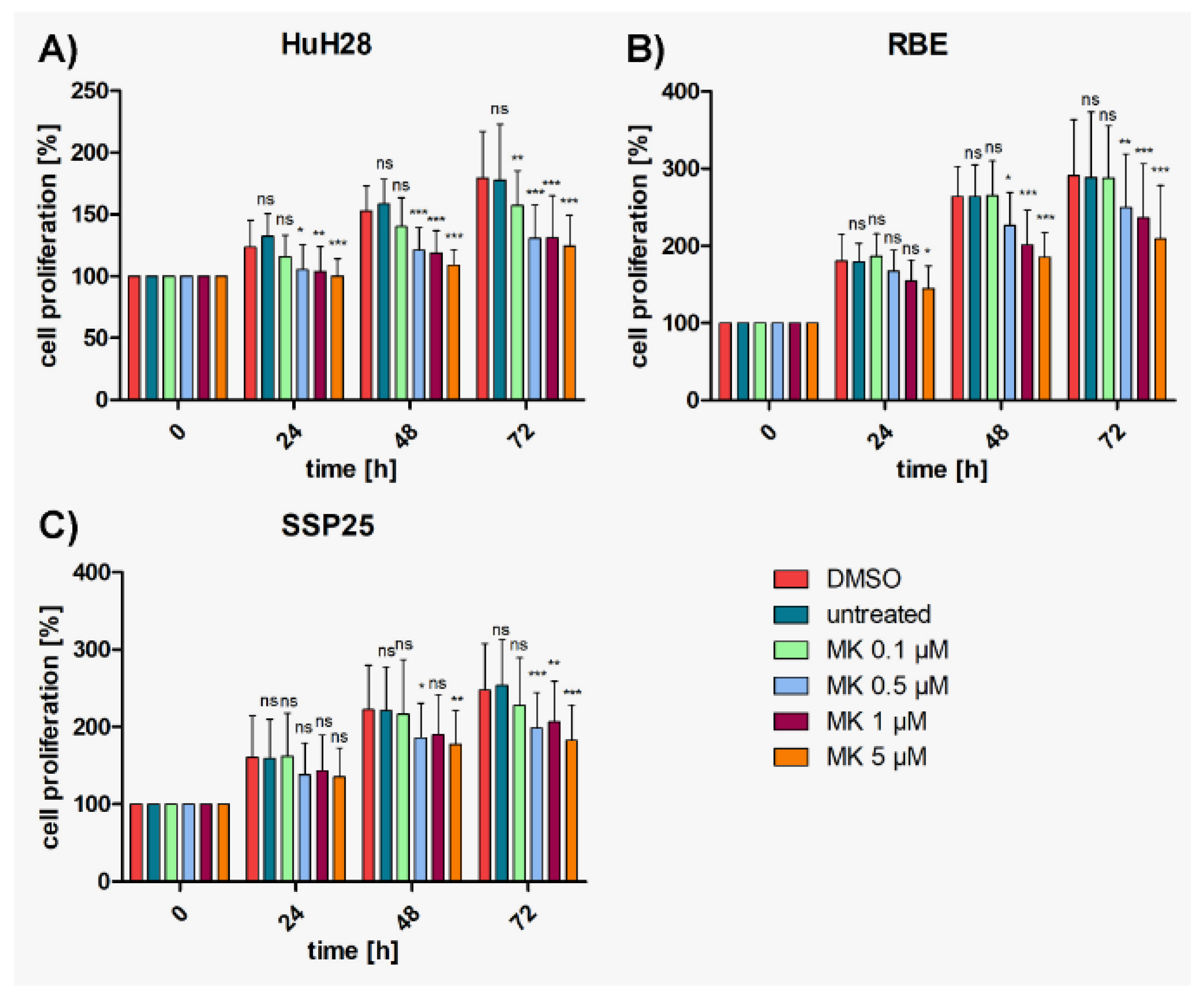
Figure 1.
Proliferation assays of ICC cell lines treated with MK2206. A) HuH28; B) RBE; C) SSP25. Cell numbers were calculated after 0, 24, 48 and 72 hours (h) with different concentrations of MK2206. Medium and DMSO controls are shown. Statistical analyses were performed compared to DMSO controls. Mean of n=3 independent experiments with each eight replicates are shown, as well as standard deviation and statistical significance. Note reduction of proliferation, but no drop below initial cell numbers. ns=not significant.
Figure 1.
Proliferation assays of ICC cell lines treated with MK2206. A) HuH28; B) RBE; C) SSP25. Cell numbers were calculated after 0, 24, 48 and 72 hours (h) with different concentrations of MK2206. Medium and DMSO controls are shown. Statistical analyses were performed compared to DMSO controls. Mean of n=3 independent experiments with each eight replicates are shown, as well as standard deviation and statistical significance. Note reduction of proliferation, but no drop below initial cell numbers. ns=not significant.
MEK-Inhibitor Selumetinib Significantly Reduces Proliferation of HuH28 and RBE, but Less in SSP25
In controls, again, ICC cell lines (HuH28, RBE, SSP25) showed a steady increase of cell numbers over time. A dose-dependent inhibition of proliferation by selumetinib (AZD-6244) was observed in all three ICC cell lines, with HuH28 and RBE being more sensitive than SSP25. Again, cell numbers never dropped below baseline (Figure 2). Highly effective inhibition was observed in RBE cells with highest significance (p≤0.001) at 0.5 µM selumetinib after 48 h (Figure 2B); with 32% growth inhibition (Suppl. Figure 1B). In HuH28 cells, selumetinib inhibited cell proliferation highly significantly at 1 µM after 48 h; with 15% growth inhibition (Figure 2A and Suppl. Figure 1A). Effects of selumetinib were weakest in SSP25. However, significant effects were observed at 1 µM after 72 h; with 15% growth inhibition (Figure 1C and Suppl. Figure 1C).
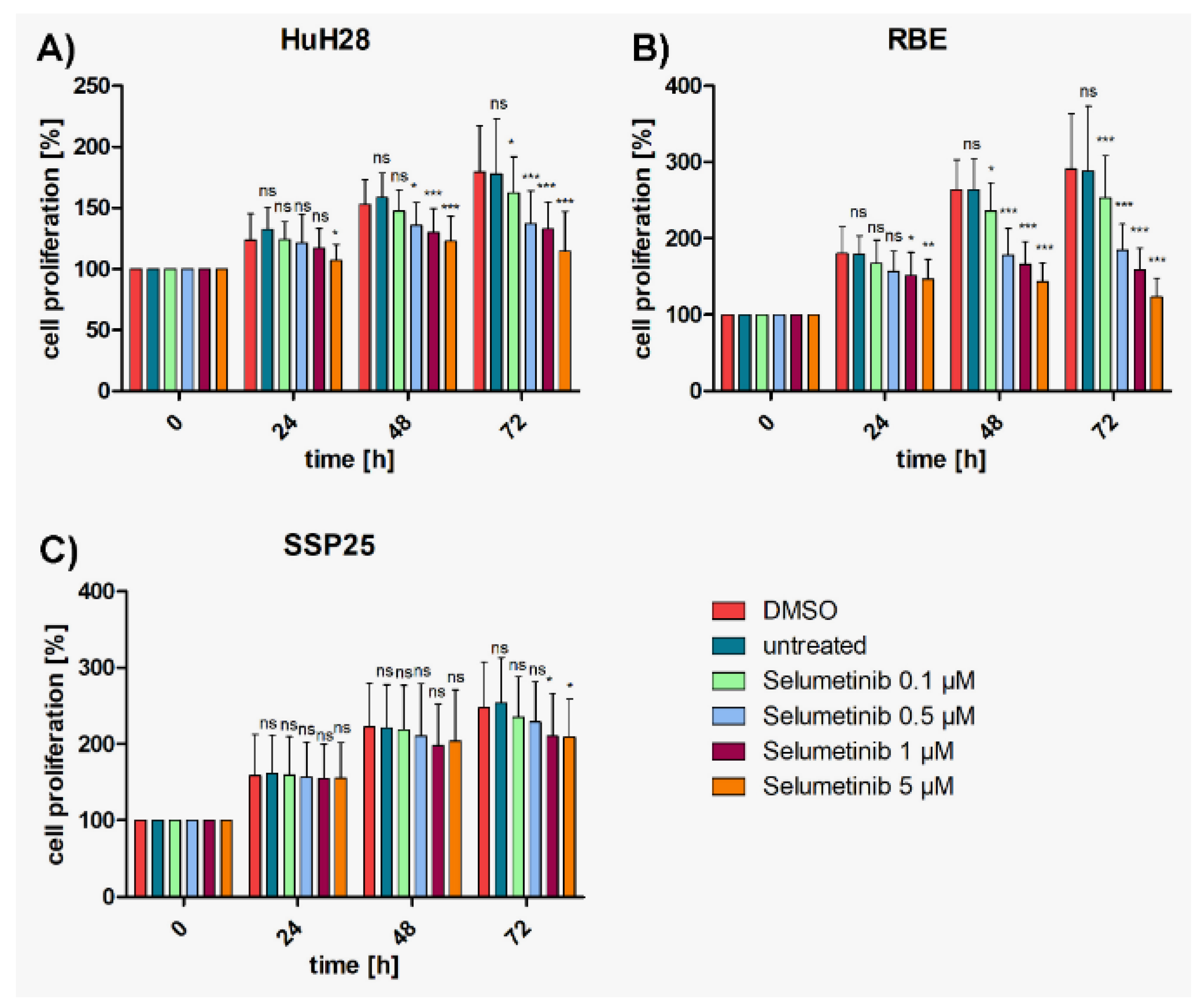
Figure 2.
Proliferation assays of ICC cell lines with selumetinib. A) HuH28; B) RBE; C) SSP25. Cell numbers were calculated after 0, 24, 48 and 72 hours (h) with different concentrations of selumetinib. Medium and DMSO controls are shown. Statistical analyses were performed compared to DMSO controls. Mean of n=3 independent experiments with each eight replicates are shown, as well as standard deviation and statistical significance. Note reduction of proliferation, but no drop below initial cell numbers. In SSP25, significant effects were seen only after 72h. ns=not significant.
Figure 2.
Proliferation assays of ICC cell lines with selumetinib. A) HuH28; B) RBE; C) SSP25. Cell numbers were calculated after 0, 24, 48 and 72 hours (h) with different concentrations of selumetinib. Medium and DMSO controls are shown. Statistical analyses were performed compared to DMSO controls. Mean of n=3 independent experiments with each eight replicates are shown, as well as standard deviation and statistical significance. Note reduction of proliferation, but no drop below initial cell numbers. In SSP25, significant effects were seen only after 72h. ns=not significant.
Dual Inhibition Shows Cooperative Effects as Compared with Single Treatments
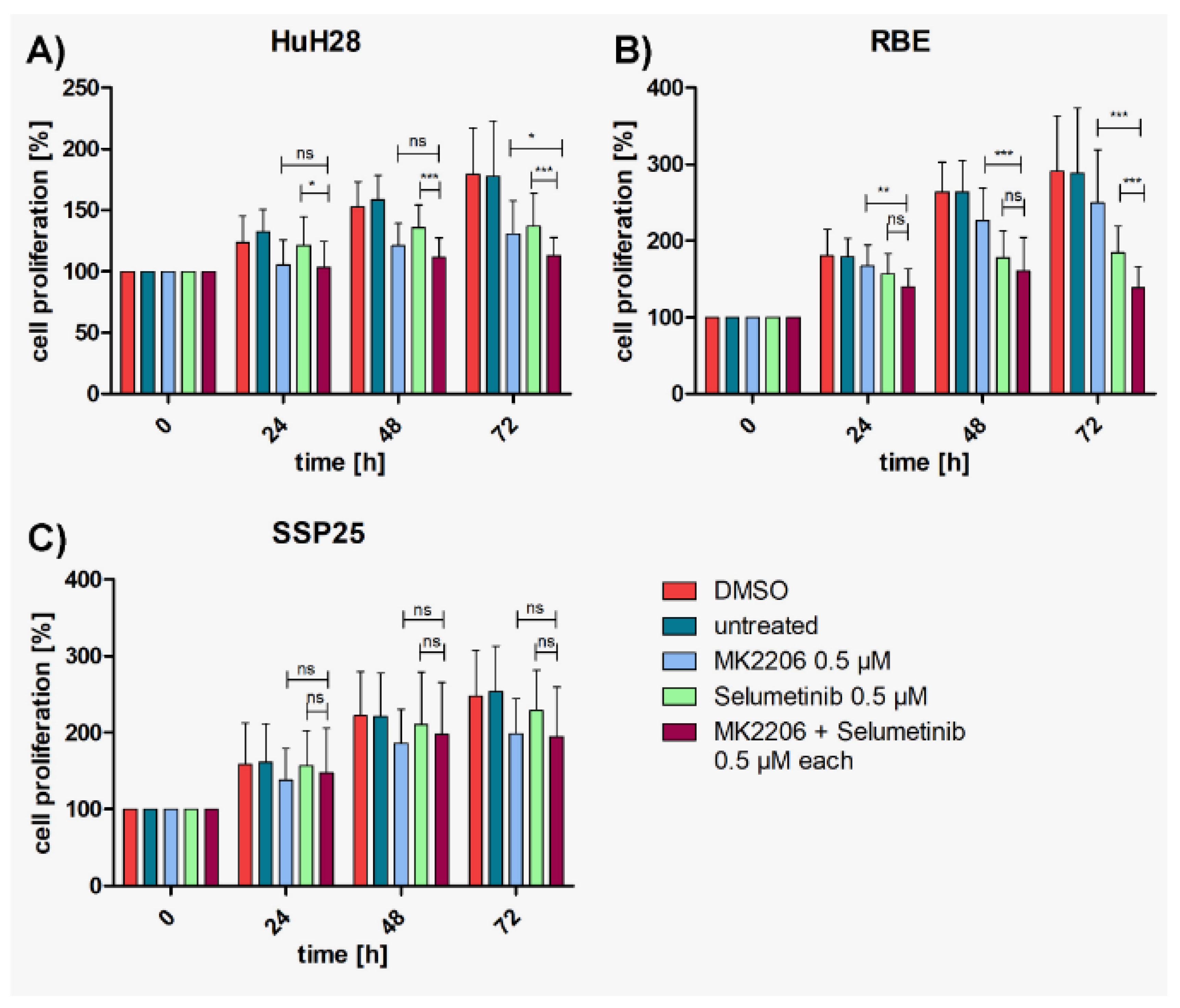
To investigate whether a cooperative effect could be achieved, suboptimal concentrations (each 0.5 µM or 1 µM) of MK2206 and selumetinib were tested in combination. Highly significant inhibition in comparison with DMSO controls was achieved with both concentrations already after 24 h in HuH28 and RBE, which is earlier than with each single application (Suppl. Figure 2). Cell numbers nearly reached baseline values but barely dropped below. In SSP25, the combined application of 0.5 µM of inhibitors achieved a significant cell number reduction only after 72 h. With each 1 µM of MK2206 and selumetinib robust effects were seen after 48 h (Suppl. Figure 2).
Compared with single MK2206 (0.5 µM) treatment, dual inhibition with each 0.5 µM MK2206 and selumetinib significantly decreased proliferation of HuH28 and RBE cells after 72h (Figure 3A,B); with 37% growth inhibition in HuH28, and 52% in RBE (Suppl. Figure 1A,B). No cooperative effect was observed in SSP25 with each 0.5 µM (Figure 3C). However, dual treatment of SSP25 with 1 µM induced a significant decrease of cell proliferation after 72 h (Figure 4C); with 32% growth inhibition (Suppl. Figure 1C).
Figure 3.
Figure 3. Proliferation assays of ICC cell lines with MK2206 and selumetinib, dual compared with single treatments. A) HuH28; B) RBE; C) SSP25. Cell numbers were calculated after 0, 24, 48 and 72 hours (h). Application of each 0.5 µM MK2206 and selumetinib. Mean of n=3 independent experiments with each eight replicates are shown, as well as standard deviation and statistical significance calculated with Two-Way ANOVA. Note highly significant effects of dual treatment in RBE after 72h, significant effects in HuH28, but no effects in SSP25. ns=not significant.
Figure 3.
Figure 3. Proliferation assays of ICC cell lines with MK2206 and selumetinib, dual compared with single treatments. A) HuH28; B) RBE; C) SSP25. Cell numbers were calculated after 0, 24, 48 and 72 hours (h). Application of each 0.5 µM MK2206 and selumetinib. Mean of n=3 independent experiments with each eight replicates are shown, as well as standard deviation and statistical significance calculated with Two-Way ANOVA. Note highly significant effects of dual treatment in RBE after 72h, significant effects in HuH28, but no effects in SSP25. ns=not significant.
The cell line HuH28 showed a strong significant decrease in cell number after 48 h of dual treatment with 1 µM compared with each single treatment with MK2206 or selumetinib (Figure 4A); with 39% growth inhibition (Suppl. Figure 1A).
Figure 4.
Proliferation assays of ICC cell lines with MK2206 and selumetinib, dual compared with single treatments. A) HuH28; B) RBE; C) SSP25. Cell numbers were calculated after 0, 24, 48 and 72 hours (h). Application of each 1 µM MK2206 and selumetinib. Mean of n=3 independent experiments with each eight replicates are shown, as well as standard deviation and statistical significance calculated with Two-Way ANOVA. Note highly significant effects of dual treatment in RBE starting after 24h, in HuH28 starting after 48h, and in SSP25 after 72h. ns=not significant.
Figure 4.
Proliferation assays of ICC cell lines with MK2206 and selumetinib, dual compared with single treatments. A) HuH28; B) RBE; C) SSP25. Cell numbers were calculated after 0, 24, 48 and 72 hours (h). Application of each 1 µM MK2206 and selumetinib. Mean of n=3 independent experiments with each eight replicates are shown, as well as standard deviation and statistical significance calculated with Two-Way ANOVA. Note highly significant effects of dual treatment in RBE starting after 24h, in HuH28 starting after 48h, and in SSP25 after 72h. ns=not significant.
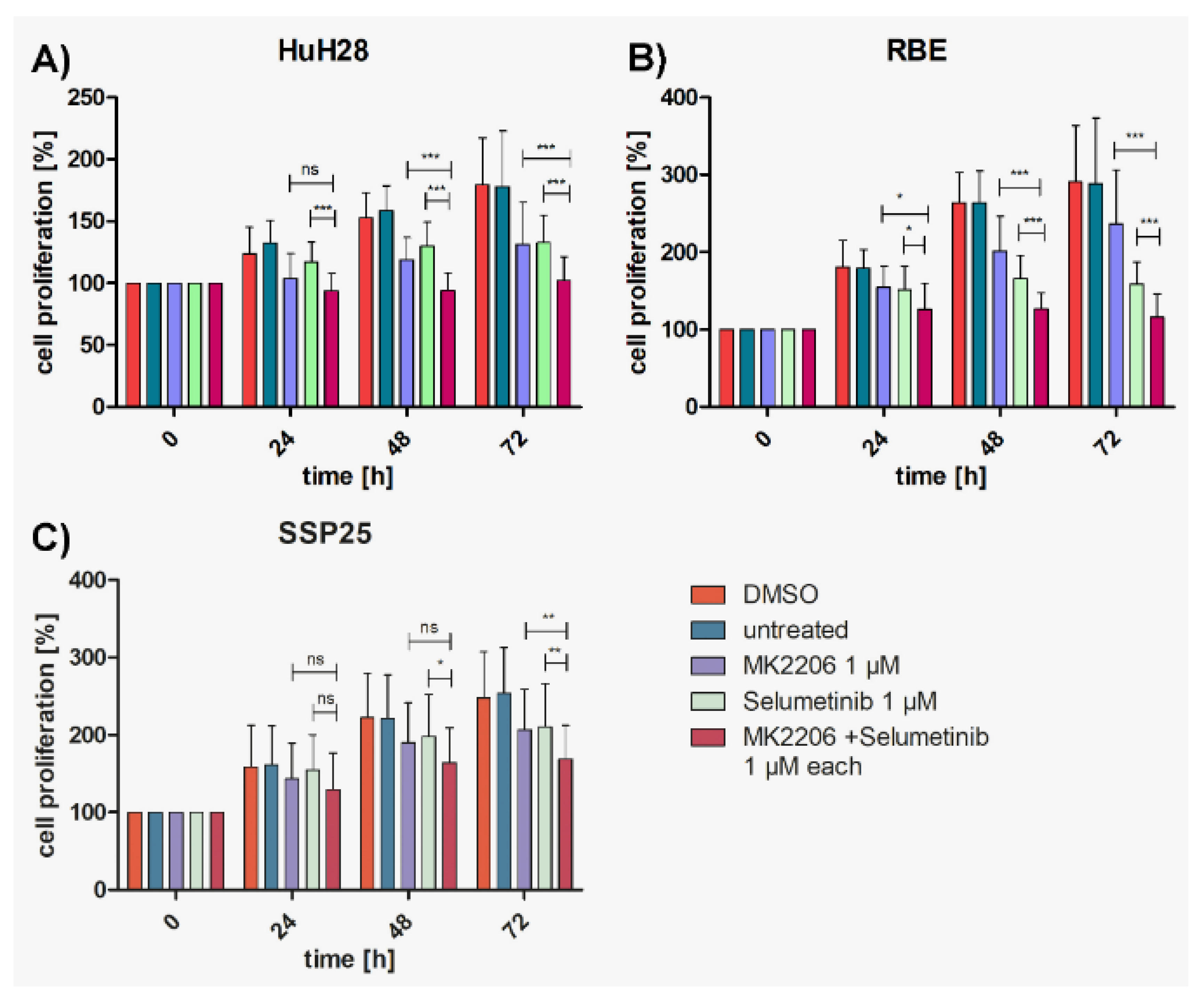
In RBE, dual treatment with 1 µM induced a significant decrease in cell proliferation compared to the respective single treatments already after 24 h (reaching 30% growth inhibition; see: Suppl. Figure 1B), which was highly significant (p≤0.001) thereafter (Figure 4B); with 60% growth inhibition after 72 h; see: Suppl. Figure 1B).
MK2206 Alone or in Combination Effectively Inhibits Phosphorylation of AKT (Ser473)
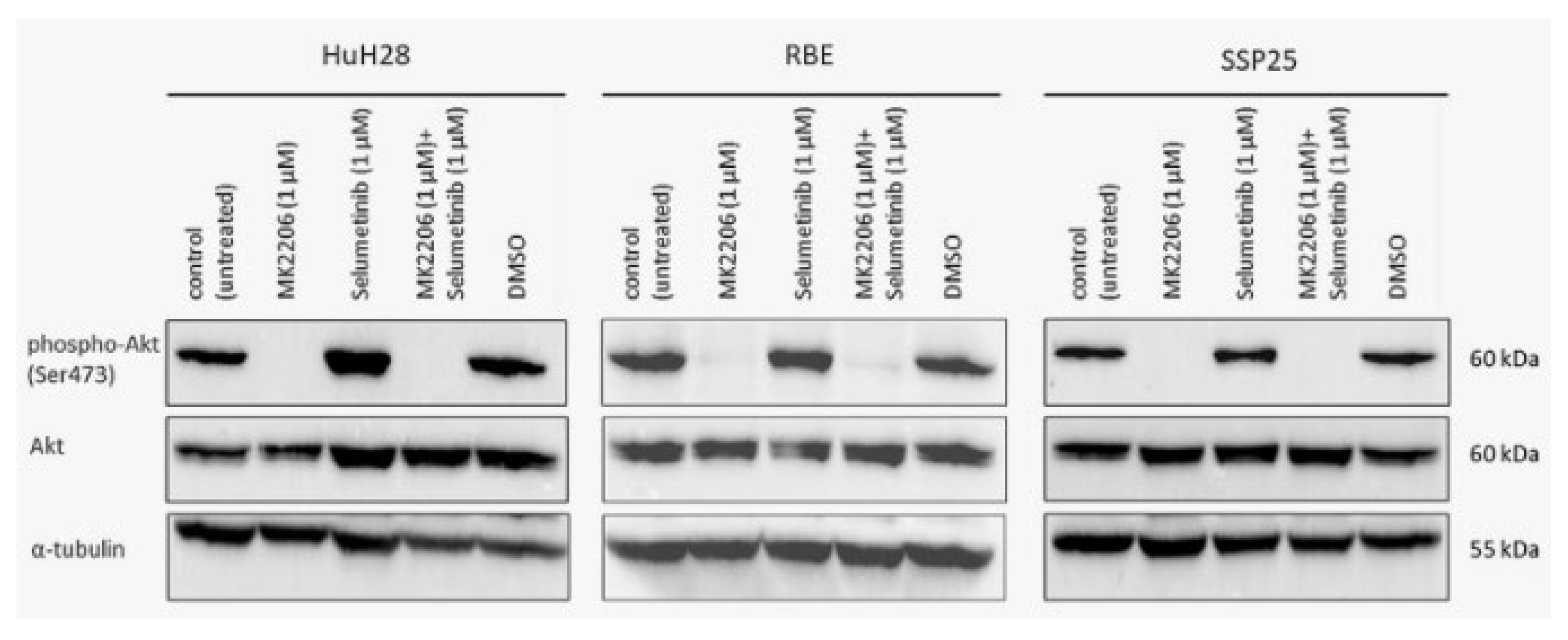
In all three ICC cell lines, single inhibition with MK2206 as well as dual inhibition with MK2206 and selumetinib caused significant down-regulation of phospho (p)-AKT at Ser473 after 12 h. Total AKT protein and loading control α-tubulin remained unchanged throughout the treatment (Figure 5).
Figure 5.
Western blot analysis of phospho-AKT (Ser473) and total AKT in ICC cell lines. A representative blot for HuH28, RBE and SSP25 of n=3 independent experiments is shown. Loading control was performed against α-tubulin. Note the distinct downregulation of pAKT by MK2206 alone or in combination with selumetinib.
Figure 5.
Western blot analysis of phospho-AKT (Ser473) and total AKT in ICC cell lines. A representative blot for HuH28, RBE and SSP25 of n=3 independent experiments is shown. Loading control was performed against α-tubulin. Note the distinct downregulation of pAKT by MK2206 alone or in combination with selumetinib.
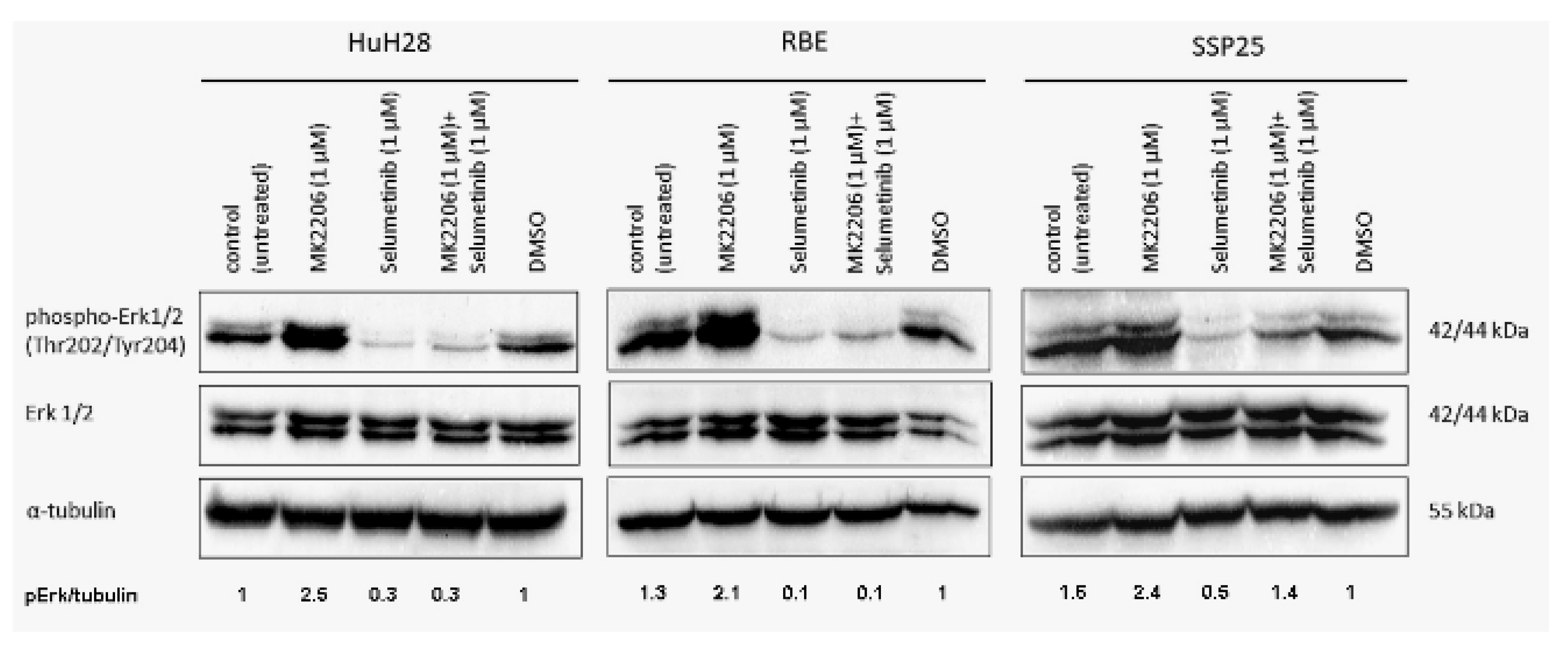
Selumetinib Alone or in Combination Effectively Inhibits ERK1/2 Phosphorylation (Thr202/Tyr204)
In all three ICC cell lines, the signal of phospho (p)-ERK1/2 (Thr202/Tyr204) was down-regulated after 12 h through selumetinib, both in single or in dual inhibition with MK2206. Interestingly, selumetinib inhibits ERK1/2 phosphorylation in SSP25, but in combination with MK2206 the inhibitory effect is neutralized when compared to the DMSO controls (Figure 6). However, our proliferation studies show a cooperative effect after 72 h (Figure 4C). Upregulation of pERK1/2 by single MK2206 was observed in all 3 cell lines, indicating interactions between the two pathways (Figure 6). Total ERK1/2 protein and α-tubulin remained unchanged.
Figure 6.
Western blot analysis with phospho-ERK1/2 in ICC cell lines. A representative blot of HuH28, RBE and SSP25 of n=3 independent experiments is shown. Loading control was performed against α-tubulin. Quantification of pERK1/2 was normalized to α-tubulin and the mean value of n=3 experiments is shown. Note downregulation of pERK1/2 by selumetinib and upregulation by MK2206. In HuH28 and RBE, dual application also downregulates pERK1/2, but in SSP25 the effect becomes neutralized.
Figure 6.
Western blot analysis with phospho-ERK1/2 in ICC cell lines. A representative blot of HuH28, RBE and SSP25 of n=3 independent experiments is shown. Loading control was performed against α-tubulin. Quantification of pERK1/2 was normalized to α-tubulin and the mean value of n=3 experiments is shown. Note downregulation of pERK1/2 by selumetinib and upregulation by MK2206. In HuH28 and RBE, dual application also downregulates pERK1/2, but in SSP25 the effect becomes neutralized.
Dual Inhibition with MK2206 and Selumetinib Causes Cell Cycle Arrest in ICC Cell Lines
In order to determine which mechanisms may be responsible for the decrease in cell numbers, we studied apoptosis by WB analysis against cleaved caspase-3, and qPCR against Apoptosis inducing factor mitochondria associated-1 (AIFM1). Additionally, we studied necroptosis with antibodies against (phospho) Mixed lineage kinase domain like (p-MLKL), and we used flow cytometry to determine the cell cycle.
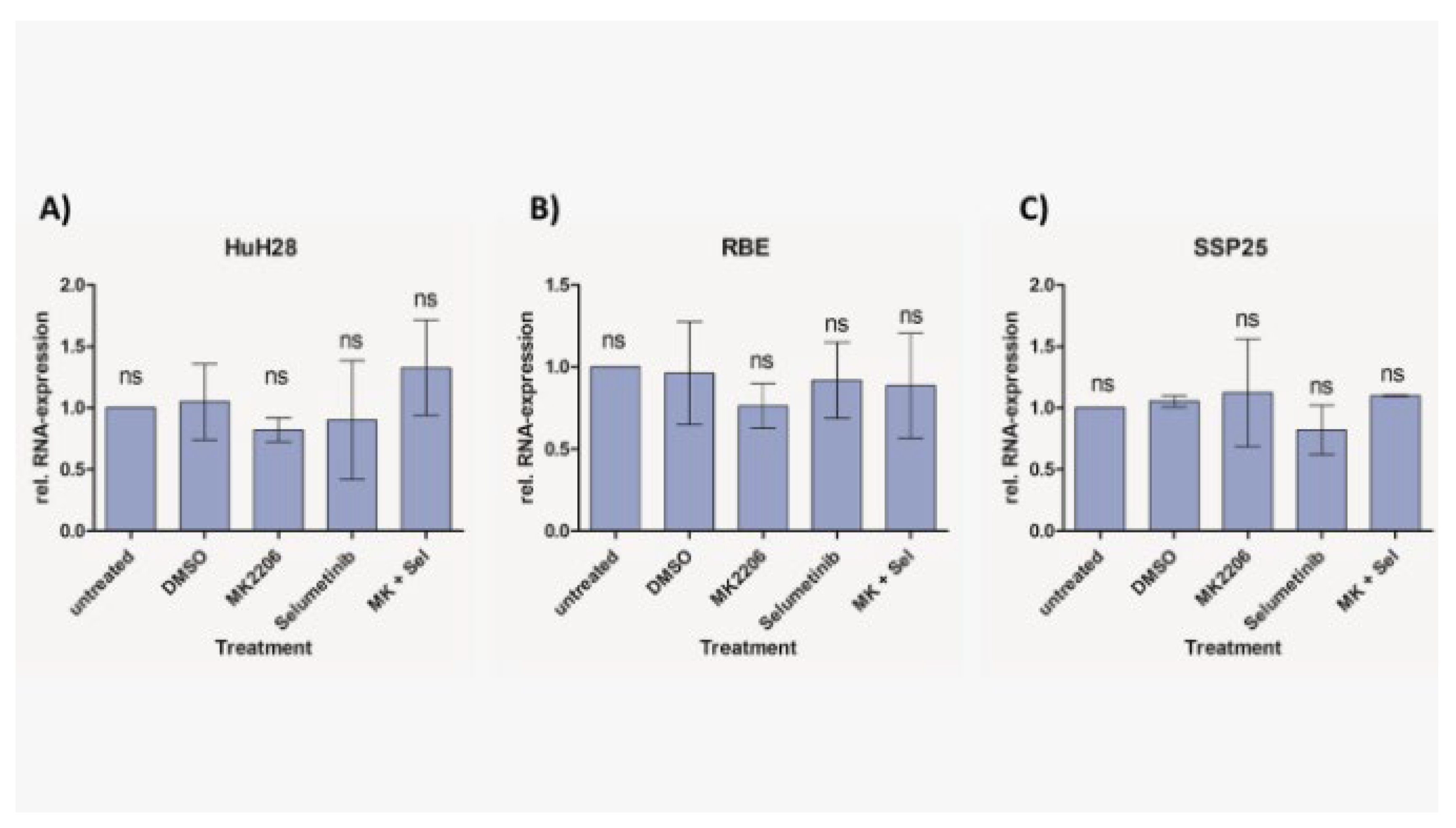
We did not observe cleaved caspase-3 after inhibitor treatment in all three ICC cell lines (Figure 7). As a positive control we incubated cells with 1 µM staurosporine, which induces caspase-dependent and -independent apoptosis [25]. Especially, in SSP25, staurosporine was highly effective and dramatically reduced cell numbers. As a result, only very weak signals could be found after 12 h staurosporine incubation (Figure 7). Transcriptional regulation of AIFM1 during apoptosis has been described [26,27]. Our qPCR studies did not show any AIFM1 regulation by the two inhibitors (Figure 8), which again excludes involvement of apoptosis as a contributor to cell number reduction in the experiments. Additionally, we did not notice phosphorylation of MLKL at Ser358 in inhibitor-treated ICC cells, which excludes necroptosis (data not shown).
Figure 7.
Western blot analysis of cleaved caspase-3 in ICC cell lines. A representative blot of HuH28, RBE and SSP25 of n=3 independent experiments is shown. Loading control was performed against β-actin. For positive control, cells were treated with 1 µM staurosporine for 12h, which reduced cell numbers, especially in SSP25.
Figure 7.
Western blot analysis of cleaved caspase-3 in ICC cell lines. A representative blot of HuH28, RBE and SSP25 of n=3 independent experiments is shown. Loading control was performed against β-actin. For positive control, cells were treated with 1 µM staurosporine for 12h, which reduced cell numbers, especially in SSP25.
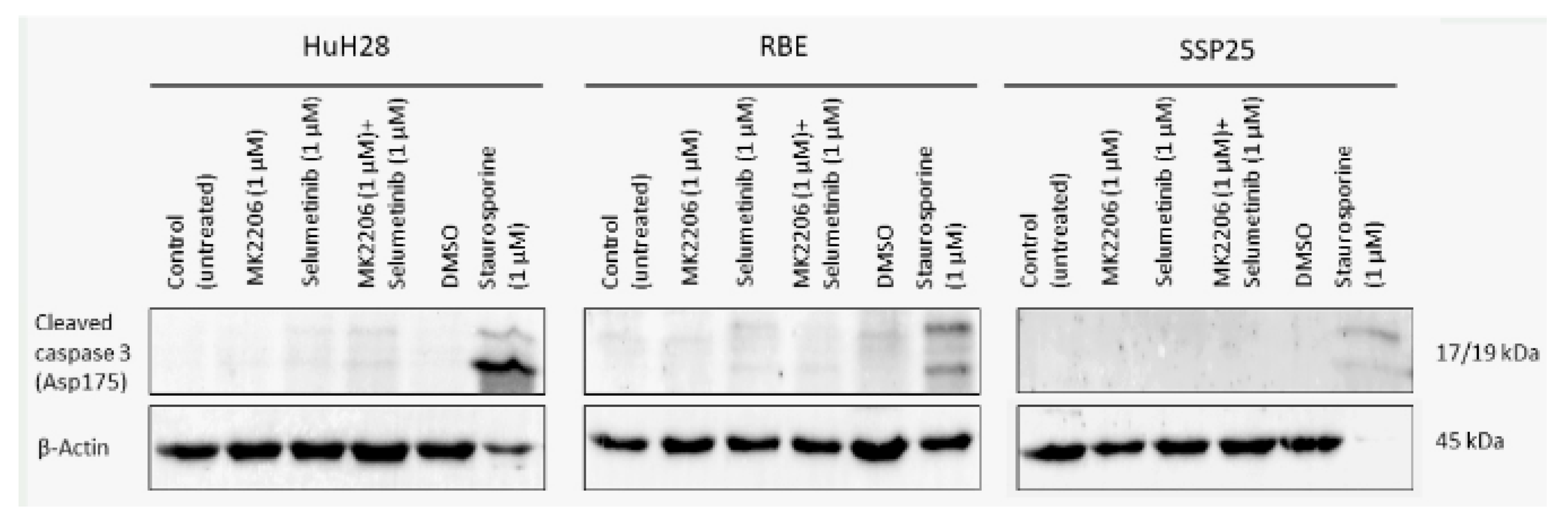
Figure 8.
Relative mRNA expression of AIFM1 in ICC cell lines measured with qPCR. Treatment of HuH28 (A), RBE (B), and SSP25 (C) with each 1µM inhibitor as indicated. Mean and standard deviation of n=3 independent experiments are shown. Untreated control probe was used as reference. No regulation of AIFM1 was noted.
Figure 8.
Relative mRNA expression of AIFM1 in ICC cell lines measured with qPCR. Treatment of HuH28 (A), RBE (B), and SSP25 (C) with each 1µM inhibitor as indicated. Mean and standard deviation of n=3 independent experiments are shown. Untreated control probe was used as reference. No regulation of AIFM1 was noted.
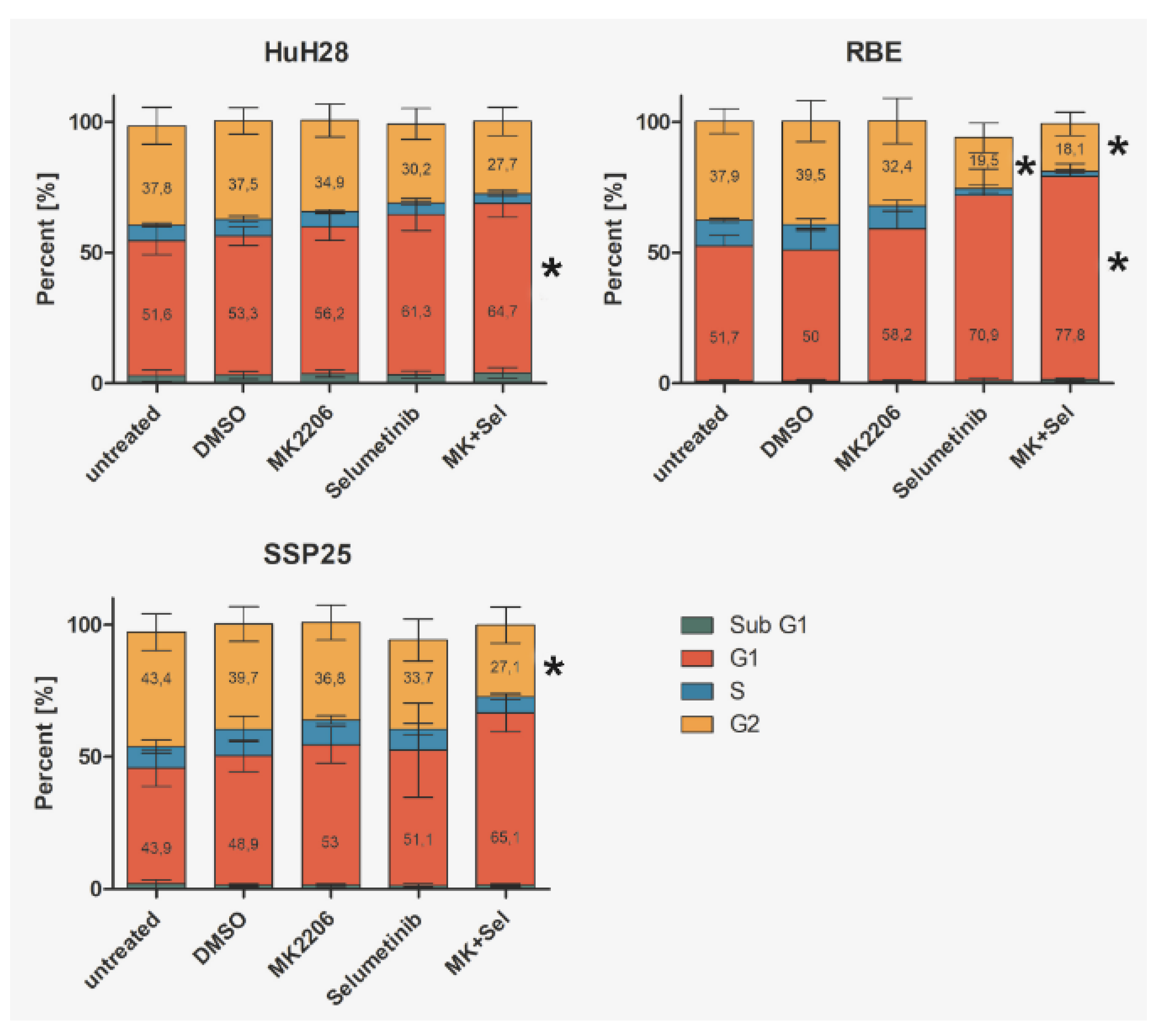
Flow cytometry showed a slight increase of cells in the G1 phase after single treatment of HuH28 with 1µM MK2206 or selumetinib. Percentage of cells in G2 phase slightly decreased. Dual inhibition induced a significant increase of cells in G1 phase as compared to DMSO control (Figure 9A). Similar results were obtained in RBE cells. Here, selumetinib induced a significant decrease of cells in G2, while dual inhibition caused both a significant decrease of G2 phase and a significant increase of G1 phase cells (Figure 9B). In SSP25, we observed a tendency for an increase in G1 and a significant decrease of cells in G2 phase after dual inhibition (Figure 9C). We did not notice any changes in the Sub-G1 phase (Figure 9), which is characteristic for apoptotic and fragmented cells [28].
Figure 9.
Flow cytometry analysis of ICC cell lines. Treatment of HuH28 (A), RBE (B), and SSP25 (C) with each 1µM inhibitor as indicated. Mean and standard deviation of the percentage of each cell cycle phase of n=5 independent experiments are shown. Significance was calculated in comparison to DMSO controls with One Way ANOVA and *p <0.05. Note significant effects of dual inhibitor treatment.
Figure 9.
Flow cytometry analysis of ICC cell lines. Treatment of HuH28 (A), RBE (B), and SSP25 (C) with each 1µM inhibitor as indicated. Mean and standard deviation of the percentage of each cell cycle phase of n=5 independent experiments are shown. Significance was calculated in comparison to DMSO controls with One Way ANOVA and *p <0.05. Note significant effects of dual inhibitor treatment.
Discussion
Dual Inhibition of MAPK/ERK and PI3K/AKT/mTOR is Highly Effective in ICC
Because of the increasing incidence of CCA and the limited therapeutic options, new drugs are urgently needed. A more specific combination of targeted drugs could be a solution to the problem. It has become increasingly clear that from both the molecular and the surgical point of view, CCA is not a uniform tumor entity. Topographically, CCA is subdivided into intrahepatic (ICC) and extrahepatic (ECC) tumors, and ECC can be further divided into perihilar and distal [4,5,6]. Based, most likely, on the differential embryonic origin of intra- and extrahepatic cholangiocytes [17,18] distinct molecular differences between ICC and ECC have been observed. ICC patients are more likely to have FGFR2 fusions, FGFR mutations and IDH1 mutations than ECC patients. ECC patients, however, show more commonly KRAS, TP53, SMAD4 and APC mutations [16]. Transcriptomic analysis of ICC and ECC cells identified subgroups which revealed different tumor biology. Thereby, gene expression profiles from 340 ICC and 203 ECC patients were compared. ICC were enriched in molecular pathways such as EMT, IL6, RTK-RAS-PIK3K, and DNA repair. ECC were enriched in EGFR, VEGF signaling, and integrin-pathways [29]. This suggests that differential treatment for the subtypes of CCA will be necessary to achieve promising therapeutic result.
Here, we focused on ICC and studied three human ICC cell lines in vitro (HuH28, RBE and SSP25) in order to determine the effects of inhibitors of two common tumor signaling pathways. We tested the AKT-inhibitor MK2206 and the MEK-inhibitor selumetinib (AZD6244) and studied proliferation, phosphorylation of signaling molecules of the PI3K/AKT/mTOR and the MAPK/ERK pathways, as well as cell cycle. We used concentrations of MK2206 well below the IC50 values, assuming that cooperative effects should be seen at a significantly reduced dose. Basically, this type of research is not new. However, it has so far been performed on uncharacterized CCA cell lines and on an ECC lines [15]. We show that ICC cell lines behave differently in their response to the inhibitors. Thereby, single treatment with MK2206 or selumetinib induced inhibition of proliferation, and the effect was clearly enhanced by dual application of the inhibitors. Thereby, HuH28 and RBE were more sensitive, but at 1 µM dosage of inhibitors SSP25 responded significantly, too. Western blot analyses with antibodies against p-AKT (Ser473) and p-ERK confirmed the effectiveness of the two inhibitors. Thereby, single as well as dual inhibition showed downregulation of phosphorylation of the respective proteins in all three ICC cell lines. A potential direct interaction between the two pathways was well visible. The WB pERK quantification showed that selumetinib significantly downregulates pERK, while MK2206 upregulates pERK both alone and, in the SSP25, also in the combination with selumetinib. We have previously observed upregulation of pERK by MK2206 in various liver cancer cell lines [24]. Such interactions have been observed in other tumor types including melanoma, colorectal, pancreatic and breast carcinoma [30,31], and were also reported for CCA [15]. For review on the crosstalk of the pathways see: [32,33].
Dual Inhibition of MAPK/ERK and PI3K/AKT/mTOR Does not Induce Apoptosis in ICC
In contrast to previous studies on CCA cell lines [15], we did not observe any signs for apoptosis induction by dual inhibition of MAPK/ERK and PI3K/AKT/mTOR signaling pathways in ICC. We did not observe cleaved caspase-3, which is the dominant effector caspase [34,35]. We think that this has clear consequences for the potential treatment of patients. Neither single treatment with MK2206 or selumetinib nor dual treatment with both inhibitors reduced cell numbers below baseline. The inhibitors effectively induced a blockade of the proliferation, but not apoptosis or necroptosis. In line with this, we observed cell cycle arrest, which was evident from a significant increase in cells in the G1 phase. Signs of apoptosis could not be detected, such as increased appearance of cells in the sub-G1 phase, cleavage of caspase-3, or expression of AIFM1. We also did not observe phosphorylation of MLKL, which is would have been a sign for necroptosis [36].
The results suggest that dual application of MK2206 and selumetinib in ICC could stop tumor growth but does not eliminate residual tumors, and therapy must certainly be supplemented with standard therapies. A phase I dose-escalation study of MK2206 and selumetinib in therapy-refractory solid tumors has shown clinical effects in patients with different tumors with K-RAS mutations. However, the study also shows that not all patients responded well, and it was assumed that this was due to heterogeneity of tumor biology. However, the study did not include patients with biliary or hepatic carcinoma [37]. A biomarker-driven phase II trial in patients with colorectal cancer has also been conducted for MK2206 and selumetinib, in which no clinical efficacy was observed [38]. Nevertheless, dual inhibition with MK2206 and selumetinib could be an option for patients with ICC in combination with standard therapies.
Abbreviations:
| AIFM1 | Apoptosis Inducing Factor Mitochondria Associated 1 |
| AKT | Protein kinase B |
| CCA | Cholangiocellular carcinoma |
| DMSO | Dimethyl sulfoxide |
| ECC | Extrahepatic cholangiocellular carcinoma |
| ERK | Extracellular signal-regulated kinase |
| FGFR2 | Fibroblast growth factor receptor 2 |
| ICC | Intrahepatic cholangiocellular carcinoma |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen-activated protein kinase kinase |
| MLKL | Mixed Lineage Kinase Domain Like |
| mTOR | Mammalian target of rapamycin |
| PBS | Phosphate-buffered saline |
| PI3K | Phosphatidylinositol-3-kinase |
Author Contributions
J.W.: conceptualization, data curation, writing - review and editing; J.S.: investigation, data curation, writing – draft; M.V., N.S. and J.B.: investigation and data curation.
Funding
Not applicable.
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Data Availability Statement
All data are included in the manuscript and the supplementary material data files. Further enquiries can be directed to the corresponding author.
Acknowledgments
We are grateful to Ch. Zelent and B. Manshausen for their expert technical assistance. We acknowledge support by the Open Access Publication Funds of the Georg-August University Göttingen.
Conflicts of Interest Statement
The authors declare no competing interests.
References
- Turati, F.; Bertuccio, P.; Negri, E.; La Vecchia, C. Epidemiology of cholangiocarcinoma. Hepatoma Res. 2022, 8, 19. [CrossRef]
- Poultsides GA, Zhu AX, Choti MA, Pawlik TM. Intrahepatic cholangiocarcinoma. Surg Clin North Am. 2010 Aug;90(4):817–37.
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [CrossRef]
- Vogel, A.; Wege, H.; Caca, K.; Nashan, B.; Neumann, U. The Diagnosis and Treatment of Cholangiocarcinoma. Dtsch. Aerzteblatt Online 2014, 111, 748–54. [CrossRef]
- Sia, D.; Tovar, V.; Moeini, A.; Llovet, J.M. Intrahepatic cholangiocarcinoma: pathogenesis and rationale for molecular therapies. Oncogene 2013, 32, 4861–4870. [CrossRef]
- Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013 Dec;145(6):1215–29.
- Gupta, A.; Dixon, E. Epidemiology and risk factors: intrahepatic cholangiocarcinoma. HepatoBiliary Surg. Nutr. 2017, 6, 101–104. [CrossRef]
- Kaewpitoon N, Kaewpitoon SJ, Pengsaa P, Sripa B. Opisthorchis viverrini: The carcinogenic human liver fluke. World J Gastroenterol. 2008 Feb 7;14(5):666–74.
- Vogel A, Saborowski A. Cholangiocellular Carcinoma. Digestion. 2017;95(3):181–5.
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.-W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [CrossRef]
- Wang, M.; Chen, Z.; Guo, P.; Wang, Y.; Chen, G. Therapy for advanced cholangiocarcinoma: Current knowledge and future potential. J. Cell. Mol. Med. 2020, 25, 618–628. [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 1–37. [CrossRef]
- Schmitz KJ, Lang H, Wohlschlaeger J, Sotiropoulos GC, Reis H, Schmid KW, et al. AKT and ERK1/2 signaling in intrahepatic cholangiocarcinoma. World J Gastroenterol. 2007 Dec 28;13(48):6470–7.
- Ewald F, Nörz D, Grottke A, Hofmann BT, Nashan B, Jücker M. Dual Inhibition of PI3K-AKT-mTOR- and RAF-MEK-ERK-signaling is synergistic in cholangiocarcinoma and reverses acquired resistance to MEK-inhibitors. Invest New Drugs. 2014 Dec;32(6):1144–54.
- Spencer, K.; Pappas, L.; Baiev, I.; Maurer, J.; Bocobo, A.G.; Zhang, K.; Jain, A.; De Armas, A.D.; Reyes, S.; Le, T.M.; et al. Molecular profiling and treatment pattern differences between intrahepatic and extrahepatic cholangiocarcinoma. JNCI J. Natl. Cancer Inst. 2023, 115, 870–880. [CrossRef]
- Strazzabosco, M.; Fabris, L. Development of the bile ducts: Essentials for the clinical hepatologist. J. Hepatol. 2012, 56, 1159–1170. [CrossRef]
- Muntean, A.; Davenport, M. Biliary atresia & choledochal malformation–—Embryological and anatomical considerations. Semin. Pediatr. Surg. 2022, 31, 151235. [CrossRef]
- EMA. Pemazyre [Internet]. European Medicines Agency. 2021 [cited 2023 Nov 22]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/pemazyre.
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [CrossRef]
- Rychahou PG, Jackson LN, Silva SR, Rajaraman S, Evers BM. Targeted Molecular Therapy of the PI3K Pathway: Therapeutic Significance of PI3K Subunit Targeting in Colorectal Carcinoma. Ann Surg. 2006 Jun;243(6):833.
- Becker, J.; Erdlenbruch, B.; Noskova, I.; Schramm, A.; Aumailley, M.; Schorderet, D.F.; Schweigerer, L. Keratoepithelin Suppresses the Progression of Experimental Human Neuroblastomas. Cancer Res 2006, 66, 5314–5321. [CrossRef]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods San Diego Calif. 2001 Dec;25(4):402–8.
- Malik, I.A.; Rajput, M.; Werner, R.; Fey, D.; Salehzadeh, N.; von Arnim, C.A.F.; Wilting, J. Differential in vitro effects of targeted therapeutics in primary human liver cancer: importance for combined liver cancer. BMC Cancer 2022, 22, 1–11. [CrossRef]
- Belmokhtar, C.A.; Hillion, J.; Ségal-Bendirdjian, E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 2001, 20, 3354–3362. [CrossRef]
- Liu H, Liu C, Wang M, Sun D, Zhu P, Zhang P, et al. Tanshinone IIA affects the malignant growth of Cholangiocarcinoma cells by inhibiting the PI3K-Akt-mTOR pathway. Sci Rep. 2021 Sep 29;11(1):19268.
- Zhang, Y.; Yang, Y.; Liu, R.; Meng, Y.; Tian, G.; Cao, Q. Downregulation of microRNA-425-5p suppresses cervical cancer tumorigenesis by targeting AIFM1. Exp. Ther. Med. 2019, 17, 4032–4038. [CrossRef]
- Wenzel, U.; Kuntz, S.; Brendel, M.D.; Daniel, H. Dietary flavone is a potent apoptosis inducer in human colon carcinoma cells.. 2000, 60, 3823–31.
- Silvestri, M.; Vu, T.N.; Nichetti, F.; Niger, M.; Di Cosimo, S.; De Braud, F.; Pruneri, G.; Pawitan, Y.; Calza, S.; Cappelletti, V. Comprehensive transcriptomic analysis to identify biological and clinical differences in cholangiocarcinoma. Cancer Med. 2023, 12, 10156–10168. [CrossRef]
- Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011 Jun;36(6):320–8.
- Britten, C.D. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother. Pharmacol. 2013, 71, 1395–1409. [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [CrossRef]
- Cao Z, Liao Q, Su M, Huang K, Jin J, Cao D. AKT and ERK dual inhibitors: The way forward? Cancer Lett. 2019 Sep 10;459:30–40.
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [CrossRef]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner Caspase-3, -6, and -7 Perform Distinct, Non-redundant Roles during the Demolition Phase of Apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [CrossRef]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65.e7. [CrossRef]
- Tolcher AW, Baird RD, Patnaik A, Moreno Garcia V, Papadopoulos KP, Garrett CR, et al. A phase I dose-escalation study of oral MK-2206 (allosteric AKT inhibitor) with oral selumetinib (AZD6244; MEK inhibitor) in patients with advanced or metastatic solid tumors. J Clin Oncol. 2011 May 20;29(15_suppl):3004–3004.
- Do, K.; Speranza, G.; Bishop, R.; Khin, S.; Rubinstein, L.; Kinders, R.J.; Datiles, M.; Eugeni, M.; Lam, M.H.; Doyle, L.A.; et al. Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Investig. New Drugs 2015, 33, 720–728. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



