Submitted:
08 April 2024
Posted:
09 April 2024
You are already at the latest version
Abstract
Tumor heterogeneity is a major obstacle in cancer therapy. We offer a novel perspective on tumor heterogeneity, informed by expanding upon the current view on tumor evolution. It is commonly known that in cancer, it is the advantageous genes and mutations that drive tumor capacities and the progression. We recognize an earlier, often overlooked, but necessary thus crucial step preceding the activation of advantageous genes functionality, namely, cellular response. We suggest that cancer is driven by an adaptive action taken by any organism when confronted with adversity - a cellular response to generate specific cancer capacities demanded to overcome survival threat - the responses that prompt the utilization of the functionality of advantageous genetic mutations and genes to attain these capacities and drive the disease. The ongoing development of cancer capabilities, as cells endeavor to obtain a competitive advantage, results in increased molecular chaos, observed as tumor heterogeneity. When the evolutionary drive for survival is impeded by therapeutic inhibition of critical genes, cell death occurs, a phenomenon thus termed oncogene addiction. We summarize the implications of this new framework for understanding tumor evolution and anti-cancer drug discovery, as well as understanding of basic cancer biology.
Keywords:
tumor evolution
; tumor heterogeneity
; oncogene addiction
; target therapy
Tumor Evolution and Heterogeneity Are Directed by Tumor Cell Response to the Need for Survival
Cancer is an umbrella term for a wide range of human disease that are all characterized by rapidly proliferating cells that have lost the ability to control some of the most basic cellular processes, such as division, migration, and death. In general, it is now well established that cancer cells are not uniform cellular population, but that they represent a highly heterogenous tumor cell populations, known as tumor heterogeneity [1,2]. Current understanding is tumor heterogeneity mainly originates from genomic instability [1,2]. During tumor evolution, beneficial mutations keep arising fueled from genomic instability that ultimately leads to heterogeneous cell populations. Our Perspective will add more insight into tumor heterogeneity by delving deeper into tumor evolution.
Tumor establishment and progression is a continuous process whereby cancer cells gradually acquire their hallmarks, typically progressing from proliferation to invasion to metastasis [3,4]. The continuous and sequential acquisition of these cancer hallmark capacities results in advantageous phenotypes that accumulate beneficial capacities need to support cancer cell survival and continue the disease under harsh conditions. Given their essentiality, common cancer hallmarks most likely represent necessary and specific advantageous characteristics in the process of adaptation. In turn, that adaptation forms the basis for tumor evolution, which follows a trajectory similar to the one encountered by any other organisms subjected to nature selection, promoting the survival of organisms with mutations that increase fitness and survival under given environmental conditions.
A basic concept is that organisms, when exposed to stressors, will react by developing specific capacities. There are three steps: step (i), exposure to stressors; step (ii), the organism’s response necessitates specific capacities to overcome the stressor; and step (iii), available resources are utilized to acquire these capacities. Human cells are no exception to this principle. Significant progress has been made in identifying cancer hallmark capacities and the critical gene mutations that lead to these hallmark tumor capacities. However, we contend that in most current cancer research, while the above step (iii) is widely recognized as clearly observed, what goes undiscerned and overlooked is steps (ii) and (i).
Cancer is known by its hallmark capacities. It is well established that the aggressive cancer behaviors confer survival advantage. The current view is that these hallmark tumor capacities arose as advantageous traits selected through natural selection, thereby supporting cell survival and enabling the disease to persist. The survival stress in advanced cancer is widely recognized and is believed to lead to more rigorous natural selection, consequently favoring the emergence of more aggressive traits. We contend what goes unrecognized is the survival benefit from these capacities – thus the motivation to generate these capacities is demanded by cellular response aiming to overcome the survival stress exposed (above step ii and i). It is the vigorous cellular responses in a bid to survive that prompt the cells to react with essential capacities before the cells are subjected to natural selection. We propose in cancer: step (i): cells encounter survival stress due to cellular damage, step (ii): cells respond to the stress and provoke necessary capacities that confer an advantage, and step (iii): cells harness essential genomic resources to develop hallmark capacities aiming to survive the natural selection. From this idea, we suggest the survival of tumor cells through the natural selection is not merely the underlying foundation to continue the disease as commonly understood, more critically, the term ‘cancer’ is defined by its hallmark capacities, thus, the disease continues for the same goal as its hallmark capacities: to sustain cell life, highlighting the intrinsic aim of cancer progress is to preserve cellular viability amidst stress.
This idea is generated by inverting the sequence of the three steps mentioned above. We emphasize that the cells develop these hallmarks with specific purpose provoked by cellular response to overcome the survival stress. Specifically, while most current research is centered on the hallmark tumor capacities cells present as clearly observed, we expand the view to the original genesis of these capacities that adhere to the same basic rules observed in other organisms as mentioned above (step i, ii and iii). Several cancer progression models are well developed, such as clonal evolution, cancer stem cells model, and metabolic reprogramming. We contend the often neglected parts in these models are the trigger for the tumor hallmark capacities thus the development of the disease, namely, cellular response, and the trigger for the cellular response, namely, survival stressors, and the aim of the disease progression, namely, to survive.
Cancer is well known not a foreign pathogen invader, but what goes unrecognized is the core of cancer is directed by cellular response that perpetually strive for survival. The current belief is cancer is mainly driven by genetic mutations that activate genes that promote cancer growth, called oncogenes (e.g., Her2/neu, EGFR and BRAF, etc.) or inactivate genes that oppose tumor growth, called tumor suppressors (e.g., TP53, PTEN and RB1, etc.). We argue that tumor evolution is a directional and aimed process led by cellular response to survival stressors. Building on the basic three steps principle outlined above, we propose that cell response to survival threats needs to unfold prior to strategic harnessing of beneficial genes and random mutations, which are subsequently subjected to natural selection. It is the cellular response that compels cells to harness these critical genes’ functionalities to generate hallmark cancer capacities, thereby enhancing the cell’s ability to adapt. We contend the advantage is already built before the natural selection, and the subsequent natural selection sets the threshold to diminish the cells that cannot respond efficiently to gain the necessary capacities.
The concept can be applied to the context beyond cancer evolution. We propose the response of any alive organism to a stressor needs to unfold, before the organism starts to strategically harness the potential of random mutations and genes to generate specific capacities, which are subsequently subjected to the process of natural selection. This idea is reflected in an observation called mutation bias. Mutation bias influenced by stress response has been observed in recent studies in Arabidopsis and additional research on E. coli [5,6]. Our interpretation is the that the cellular response leads the directional evolution, where the genotype of the surviving cells may reflect how these cells leveraged the function of specific genes and mutations from their own genomic materials to develop necessary capacities aiming to overcome specific stressors. The same idea may also shed a light on how a protein’s function influences the shape of its fitness landscape, a fundamental question in evolution. Current studies usually is centered merely on the protein function and its fitness landscape (as in above step iii). We contend what goes undiscerned is the role of cellular responses to the specific stressors that exploit the protein function, thereby cellular response serves as an engineer, constructing the proteins fitness landscape and shaping the protein’s evolution (as above step i and ii).
In cancer, we understand that the stressor invoking the cellular response stems from the most immediate need for survival. This understanding is reinforced by a widely recognized observation known as oncogene addiction: when advantageous genes are eliminated, cells lose their malignant phenotypes and their ability to survive [7]. Our interpretation for oncogene addiction is: during therapeutic intervention, cells fail to respond effectively to survival stress when they cannot develop advantageous malignant hallmark capacities for survival - due to the loss of critical genes.
We suggest oncogene mutations typically become the catalyst for tumorigenesis only when cells begin to evolve; thus, without the survival stress to provoke the response, cells remain “normal” despite the presence of an oncogene. The concept may explain why, despite the presence of oncogene mutations, some cells, such as melanocytes with BRAF mutations [8], fibroblasts with HRAS mutations [9] and breast epithelial cells with PIK3CA mutations [10], exhibit normal phenotypes. There are also examples of cells with several inherited genetic defects in crucial genes (e.g., BRCA1/2, FAP, and RB1, among others) that remain dormant until being triggered into aggressive tumorigenesis [11,12,13].
We propose tumor progression is a continuous evolutionary process at each step of the capacities invoked by the cellular response, then acquired, and perpetuated. Specific beneficial mutations or cellular alternations keep exploited to attain each tumor hallmark and drive the disease. Inherent/inherited cellular diversity undoubtedly leads to varying levels of - susceptibility - to survival threats. However, it is the heterogeneous molecular responses to the survival threat - response based on the cellular diversity and which triggers the cells to exploit its own genomic and cellular machinery, that ultimately determine the cell fate for survivability. Increasing tumor heterogeneity is usually observed during tumor progression. Commonly, genomic instability has been viewed as the origin of tumor heterogeneity [1,2,14,15]. However, we argue, genomic instability is not the original cause for tumor heterogeneity; rather it is exploited to allow cells to rapidly respond to stressors by selecting variants with beneficial activity and function that enable them to successfully evolve advantageous capacities. As cells adapt to survive under stress, beneficial mutations and various cellular alternations keep exploited and emerge as the response, amidst genomic instability. The diversity generated at this juncture will be selectively favored by natural selection, and cells with more advantageous characteristics will proceed to the next beneficial capability demanded by the cellular response. In this idea, the essence of the disease lies not just in survival threat as the initial trigger, but more critically the cellular evolution under sustained survival threats, necessitates the continuous amplification of cancer hallmark capacities as cellular response amidst escalating cellular damages, ultimately leading to increasing molecular chaos within and among different tumors, namely, tumor heterogeneity (Figure 1).
Taken together, we propose the presence of survival-driven responses is required to trigger tumorigenesis. We propose a model of cancer initiation, progression, and heterogeneity based on the cell response to stressors that rewire normal cell genomic machine into one that favors cancer hallmarks as necessary for survival. Prolonged stress conditions further drive evolution along the accepted trajectories of natural selection and survival of the fittest. In its core, cancer is cell’s persistent seeking for survival.
Survival-Driven Cell Response, Oncogene Addiction, and Implications for Targeted Therapy Development
Oncogene addiction refers to the reliance of cancer cells on one or more genes for their survival [7]. As a result, inhibiting the proteins corresponding to these oncogenes can lead to a complete halt or regression in tumor growth [7]. This concept of “addiction” explains the selectivity and effectiveness seen in certain molecularly targeted therapies currently used in clinical practice despite the common occurrence of resistance at later stages of treatment [16,17,18].
The concept outlined here suggests that cells continuously develop advantageous capacities in response to environmental pressures. Consequently, beneficial gene mutations keep exploited, including but not limited to oncogenes. This process contributes to the evolution of adaptive capabilities, with survival being a primary objective of this evolution. When treatment agents inhibit these advantageous genes, the evolutionary process is disrupted, leading to cell death, a phenomenon thus observed as oncogene addiction. Moreover, the occurrence of oncogene addiction further reinforces the notion that survival is a key objective driving the evolution. It is crucial to understand that any genes that contribute to tumor hallmark capacities are essential for the cells to respond to and overcome environmental pressures, thereby facilitating survival. As such, when oncogenes are inhibited, or tumor suppressors are activated, the initial observation post-treatment is a decrease in the cells’ aggressiveness, and this is almost immediately followed by cell death when the cells are removed from their evolutionary trajectory. Intriguingly, some studies [19,20,21] observed the cells may revert to a normal state in the presence or absence of the oncogene. Our interpreation is this is when survival condition is improved, cellular response to adapt becomes unnecessary, the cells revert to a normal state by ceasing the generation of aggressive traits - regardless the critical genes’ presence or absence.
In this idea, each tumor capability represents a step in the evolutionary process; when the oncogene fosters tumor capacities, the more reliant cells are on this particular gene for tumor capacities and continued evolution, the more significant the evolutionary loss will be when the gene is inhibited. The cessation of the evolutionary process, in turn, leads to cell death (Figure 2). Activating more tumor capabilities thus displaying increased aggressiveness are the primary factors determining the degree of cell addiction to the genes that drive this aggressiveness. We suggest any cells in the tumor population are addicted at any time of the disease when the aggressive capacities are provoked by cellular response. We suggest being aggressive is the merely a phenotype cell presents, the critical part is what is behind it and the more important part is the critical genes and the networks the cell exploited to support the aggressive tumor capacities and these are the genes to be “addicted” to. It is well known advanced tumor usually needs additional treatment. We suggest what goes unrecognized is in the advanced stage of the disease, when it is more likely that cells construct intricate networks to support their continued evolving tumor hallmarks, the target treatment usually will becomes more complicated than the initial stage of the disease.
ur ideas are also reflected in a concept developed recently – non-oncogene addiction. A study identified the pivotal role of CK2 in various survival pathways and CK2 arised as a potential target that could provide a therapeutic approach against multiple myeloma [32]. Additional study on the enzyme Taspase1 has identified it as a crucial non-oncogene addiction protease coordinating cancer cell survival [33]. The critical genes in “non-oncogene addiction” are well known typically involved in cell survival – we suggest the concept is akin to “oncogene addiction” - the dependency on crucial genes to foster hallmark capacities under stress provoked by celluar response. We suggest any other tactics cells use to ensure and sustain cell survival can fall under the term “addiction”. Future studies could be conducted.
Adaptive drug resistance is already well-understood as the result of survival response. For instance, the re-activation of p-ERK independent of BRAFV600E can occur through the activation or overexpression of CRAF. Also, BRAFT1799A’s amplification or the upregulation of BRAF protein levels can mediate vemurafenib resistance [25,26,27]. Many of remaining cells are found to be aggressive. The current view is being aggressive and being resistant are two separate processes. The unrecognized part is the added pressure from drug treatment prompts cells capable of responding to seek mutations that further enhance their aggressive hallmark tumor capacities for survival, leading to a survival-of-the-fittest scenario. Being aggressive is a response of the cells to resist the heightened survival stress from the drug, observed as being resistant. Suppose the gene is not the primary or sole contributor to the cells’ aggressiveness, and several other contributing factors could emerge. In that case, inhibiting this oncogene is unlikely to result in oncogene addiction unless multiple targets are inhibited. This logic applies to strategies involving multiple critical genes, such as simultaneously targeting KRAS, CDKN2A, TP53, and SMAD4 in pancreatic cancer [28] or targeting MEK, NRAS, or other components of the MAPK pathway in melanoma, besides BRAF [29]. An often unnoticed aspect is that these multiple targets often need to collaborate to achieve essential tumor hallmarks. Hence, they all need to be decreased to disrupt the cell evolution process.
We suggest the “addiction” state is an outcome provoked by cellular response to survival stress. When cells are more addicted, more cellular response triggers the intense exploitation on critical gene or its network to generate hallmark capacities, suggesting cells are in more struggling state under more escalating survival conditions. Under such conditions, it is usually observed a heightened compulsion in cells to develop stemness, and these cells exhibit a stronger predisposition towards tumorigenesis, a trend substantiated by studies [22,23,24]. In our idea, the possible enhanced survivability associated with later phased stem cells is offset by persistent survival threats. Any cells that are compelled to respond will become tumorigenic and once cancer stem cells are compelled to respond, their tumorigenic potential, coupled with the molecular alterations derived and accumulated in the later phase of the disease when they are derived, is further amplified by their capacity to display one of the primary characteristics of tumors – uncontrolled proliferation. This proliferation then sets off a cascade of subsequent steps more attainable and driven by heightened survival threats; thus cells presents more tumorigenic hallmarks, surely, in our idea, these stem cells will also become “addicted” to the genes networks that support he aggressive capacities. Furture research could be conducted.
Overall, our model collectively interprets oncogene addiction as a result of cancer cells’ unsuccessful response to survival threats. The essence of targeted therapy, within this framework, is to strategically target orthogonal cancer dependencies, aiming to simultaneously eliminate multiple cancer hallmarks, rendering the cells compromised and not able to recover effectively.
3). Deepening Insights into Oncogenes and Tumor Suppressors
During the early stages of the disease, it is widely observed that invasive cells constitute only a minority of the cell population within the primary tumor. The limited growth potential observed in these invasive cells suggests that they acquire their proliferative capacity at a distant metastatic site. This underscores that a disease cannot be deemed aggressive if a cell can only proliferate without the ability to invade, or conversely, if a cell can only invade without the ability to proliferate.
It is reasonable to assume that different cellular behaviors, such as proliferation and invasion, necessitate distinct oncogenic networks, signaling pathways, and oncogenic mutations for optimal activation, as observed [30,31]. The apparent inhibited growth ability in invasive cells as mentioned above may exemplify how one oncogenic network supporting invasion reduces the intensity of another oncogenic network that supports growth. This concept can be extended to other cancer hallmark behaviors, as each of them is regulated by various oncogenic networks, mutations and different aboundance of the oncogenes. Consequently, cells must adjust these driving factors for each behavior to achieve the ideal level needed for disease progression. At the population level, this modulation manifests as heterogeneity. At the single-cell level, it is seen as cell plasticity.
We propose a focus on modulating target oncogene heterogeneity as an alternative method to implementing a strict inhibition on the advantageous genes. In our paper, treatment-induced pressure often leads the cells to activate a wider spectrum of tumorigenic capacities to cope with the added stress due to the treatment. In clinic, these cells are observed to be resistant. We propose when the complete eradication of all residual cancer cells proves challenging, causing a shift to an aggressive state, it becomes more necessary to develop treatment strategies that extend beyond merely aiming at the strenuous inhibition of the oncogenes. The aim should be to achieve homogeneity of the oncogene across cells. This strategy can be coupled with other debulking methods, such as surgery or radiation, for effective disease control. On the other side, in tumors with greater homogeneity, we propose to emphasize the identification of the precise total oncogene levels within the tumor. The goal is to adjust these levels such that individual tumorigenic behaviors experience a significant decrease from their peak, suggesting that the cells have shifted away from their utmost aggressiveness. In contrast to most current treatment paradigms, these approaches give precedence to halting disease progression rather than attempting to eliminate cells outright. The critical part is once the cells are on hold of the progression, other means of debulking should be applied for possible eradication. We suggest this may reduce the risk of provoking an aggressive cell state in the residual cells as challenged in most current approaches.
Overall, this perspective suggests that human cancer treatment strategies could go beyond methods that only based on the concept of oncogene addiction. They could also encompass approaches to stabilize the expression state and to achieve precise levels of the addicted oncogenes, leading to new drug designs.
Final Remark
Here, we proposed a model of survival – driven cancer cell response as the central concept. The cell response determines different aspects of tumor emergence, progression, evolution, heterogeneity, and oncogene addiction. In general, being able to respond to stressors and initiate capacities to overcome the stressor is a defining characteristic of living organisms. The human cells are no different, and we take this as a starting point for rationalizing: (1) tumor emergence, which occurs when cells with accumulated damage respond by upregulating survival, as opposed to death; (2) tumor heterogeneity, which arises from molecular responses as cells evolve to develop advantageous hallmark tumor capabilities under survival pressure; (3) oncogene addiction, which emerges when cell failed to respond by losing critical genes to generate tumor capacities facilitating survival; (4) tumor evolution, whereby cellular response generates heterogeneity from which fittest clones emerge; Taken together, our model takes a series of complex processes and attempts to specify survival-driven human cell inherent responses as the earliest events that lead to transformation, and, via evolution, give rise to the complexity and increasing aggressiveness seen in late stage cancers. The ideas explored herein pave the way for novel cancer therapies and the betterment of human health. Nevertheless, we acknowledge some of the content has not been tested and that outlines possible testable hypothesis for future studies.
Contributions
HZ generated the concepts and wrote the paper.
Acknowledgment
This paper was supported by the Elsa U. Pardee Foundation (CA-0122861 to YZ). I extend my gratitude to Robert Judson-Torres (University of Utah) and William Weiss (University of California, San Francisco) for their comments that encouraged the inception of this paper. The current version was only made possible by the assistance with content and English editing provided by Life Science Editors, American Manuscript Editors and AJE (part of Springer Nature).
Author information
This paper was generated and written by HZ.
Footnote
English editing on the the very earliest version was assisted by ChatGPT.
References
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [CrossRef]
- Proietto, M.; Crippa, M.; Damiani, C.; Pasquale, V.; Sacco, E.; Vanoni, M.; Gilardi, M. Tumor heterogeneity: preclinical models, emerging technologies, and future applications. Front. Oncol. 2023, 13. [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [CrossRef]
- Hanahan D, Weinberg RA. 2000. The Hallmarks of Cancer. Cell. 100(1):57–70.
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Becker, C.; Lensink, M.; Exposito-Alonso, M.; Klein, M.; Hildebrandt, J.; Neumann, M.; Kliebenstein, D.; et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022, 602, 101–105. [CrossRef]
- Marciano, D.C.; Wang, C.; Hsu, T.-K.; Bourquard, T.; Atri, B.; Nehring, R.B.; Abel, N.S.; Bowling, E.A.; Chen, T.J.; Lurie, P.D.; et al. Evolutionary action of mutations reveals antimicrobial resistance genes in Escherichia coli. Nat. Commun. 2022, 13, 1–13. [CrossRef]
- Weinstein IB, Joe A. 2008. Oncogene Addiction. Cancer Research. 68(9):3077–80.
- Kumar, R.; Angelini, S.; Snellman, E.; Hemminki, K. BRAF Mutations Are Common Somatic Events in Melanocytic Nevi11Tables 2 and 3 can be found at http://www.blackwellpublishing.com/products/journals/suppmat/jid/jid22225/jid22225sm.htm. J. Investig. Dermatol. 2004, 122, 342–348. [CrossRef]
- Benanti, J.A.; Galloway, D.A. Normal Human Fibroblasts Are Resistant to RAS-Induced Senescence. Mol. Cell. Biol. 2004, 24, 2842–52. [CrossRef]
- Isakoff, S.J.; Engelman, J.A.; Irie, H.Y.; Luo, J.; Brachmann, S.M.; Pearline, R.V.; Cantley, L.C.; Brugge, J.S. Breast Cancer–Associated PIK3CA Mutations Are Oncogenic in Mammary Epithelial Cells. Cancer Res. 2005, 65, 10992–11000. [CrossRef]
- Foulkes, W.D. Inherited Susceptibility to Common Cancers. New Engl. J. Med. 2008, 359, 2143–2153. [CrossRef]
- Fearon, E.R. Human Cancer Syndromes: Clues to the Origin and Nature of Cancer. Science 1997, 278, 1043–1050. [CrossRef]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.-C. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [CrossRef]
- Atkins, K. Whole-Genome Doubling and Aneuploidy in Human Cancer. Oncotarget https://www.oncotarget.org/2023/08/10/whole-genome-doubling-and-aneuploidy-in-human-cancer/ (2023).
- Comaills, V.; Castellano-Pozo, M. Chromosomal Instability in Genome Evolution: From Cancer to Macroevolution. Biology 2023, 12, 671. [CrossRef]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2018, 1866, 165339. [CrossRef]
- Shiokawa D, Sakai H, Ohata H, Miyazaki T, Kanda Y, et al. 2020. Slow-Cycling Cancer Stem Cells Regulate Progression and Chemoresistance in Colon Cancer. Cancer Res. 80(20):4451–64.
- Robinson, N.; Taylor, D.J.; Schiemann, W.P. Stem cells, immortality, and the evolution of metastatic properties in breast cancer: telomere maintenance mechanisms and metastatic evolution. J. Cancer Metastasis Treat. 2019, 5, 62. [CrossRef]
- Felsher, D.W.; Bishop, J. Reversible Tumorigenesis by MYC in Hematopoietic Lineages. Mol. Cell 1999, 4, 199–207. [CrossRef]
- Schmidt, A.V.; Monga, S.P.; Prochownik, E.V.; Goetzman, E.S. A Novel Transgenic Mouse Model Implicates Sirt2 as a Promoter of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 12618. [CrossRef]
- McNeal, A.S., Belote,R.L.,Zeng,H.,Urquijo,M.,Barker,K.,Torres,R.,Curtin,M.,Shain,A.H.,Andtbacka, R. H., Holmen, S., Lum, D. H., McCalmont, T. H., VanBrocklin, M. W., Grossman, D., Wei, M. L., Lang, U. E., & Judson-Torres, R. L. (2021). BRAFV600E induces reversible mitotic arrest in human melanocytes via microrna-mediated suppression of AURKB. ELife, 10, e70385.
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [CrossRef]
- He, H.; Wang, S.; Zhang, W.; Gao, S.; Guan, H.; Zhou, P. Downregulation of TAB182 promotes cancer stem-like cell properties and therapeutic resistance in triple-negative breast cancer cells. BMC Cancer 2023, 23, 1–17. [CrossRef]
- Lei, M.M.L.; Lee, T.K.W. Cancer Stem Cells: Emerging Key Players in Immune Evasion of Cancers. Front. Cell Dev. Biol. 2021, 9. [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired Resistance to BRAF Inhibitors Mediated by a RAF Kinase Switch in Melanoma Can Be Overcome by Cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [CrossRef]
- Shi, H. et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun 3, 724 (2012).
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [CrossRef]
- Hu, H.-F.; Ye, Z.; Qin, Y.; Xu, X.-W.; Yu, X.-J.; Zhuo, Q.-F.; Ji, S.-R. Mutations in key driver genes of pancreatic cancer: molecularly targeted therapies and other clinical implications. Acta Pharmacol. Sin. 2021, 42, 1725–1741. [CrossRef]
- Wang, B.; Zhang, W.; Zhang, G.; Kwong, L.; Lu, H.; Tan, J.; Sadek, N.; Xiao, M.; Zhang, J.; Labrie, M.; et al. Targeting mTOR signaling overcomes acquired resistance to combined BRAF and MEK inhibition in BRAF-mutant melanoma. Oncogene 2021, 40, 5590–5599. [CrossRef]
- Endo, Y.; Lyon, S.; Shen, Y.; Mohan, N.; Wu, W.J. Cell proliferation and invasion are regulated differently by EGFR and MRP1 in T-DM1-resistant breast cancer cells. Sci. Rep. 2019, 9, 16383. [CrossRef]
- Swami, P.; Thiyagarajan, S.; Vidger, A.; Indurthi, V.S.K.; Vetter, S.W.; Leclerc, E. RAGE Up-Regulation Differently Affects Cell Proliferation and Migration in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2020, 21, 7723. [CrossRef]
- Chen, D.Y.; Liu, H.; Takeda, S.; Tu, H.-C.; Sasagawa, S.; Van Tine, B.A.; Lu, D.; Cheng, E.H.-Y.; Hsieh, J.J.-D. Taspase1 Functions as a Non-Oncogene Addiction Protease that Coordinates Cancer Cell Proliferation and Apoptosis. Cancer Res 2010, 70, 5358–5367. [CrossRef]
- Mandato, E.; Manni, S.; Zaffino, F.; Semenzato, G.; Piazza, F. Targeting CK2-driven non-oncogene addiction in B-cell tumors. Oncogene 2016, 35, 6045–6052. [CrossRef]
Figure 1.
Tumor evolution and tumor heterogeneity. Tumor evolution is led by cellular response to survival threats, specifically, the response activates the cells genomic machine, utilizing advantageous genetic mutations and other cellular content to develop capabilities that enhance survival. The goal of tumor evolution is to sustain cells under stress – the initiative to develop necessary tumor hallmark capacities. During the disease progression, continual stress leads to interconnected hallmarks of cancer become progressively engaged provoked by escalating cellular damages, ultimately resulting in molecular chaos, within and among tumors, namely, tumor heterogeneity.
Figure 1.
Tumor evolution and tumor heterogeneity. Tumor evolution is led by cellular response to survival threats, specifically, the response activates the cells genomic machine, utilizing advantageous genetic mutations and other cellular content to develop capabilities that enhance survival. The goal of tumor evolution is to sustain cells under stress – the initiative to develop necessary tumor hallmark capacities. During the disease progression, continual stress leads to interconnected hallmarks of cancer become progressively engaged provoked by escalating cellular damages, ultimately resulting in molecular chaos, within and among tumors, namely, tumor heterogeneity.
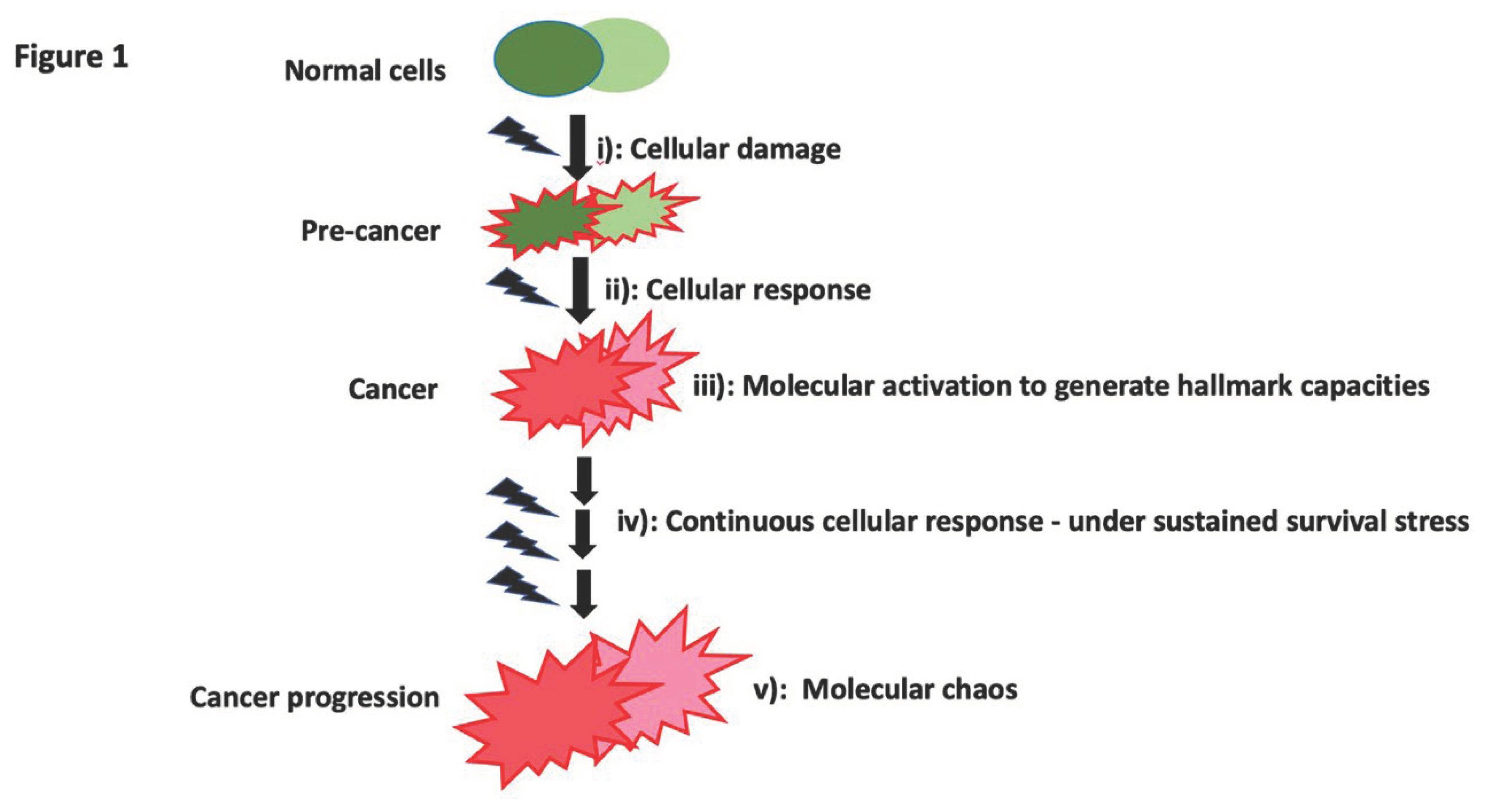
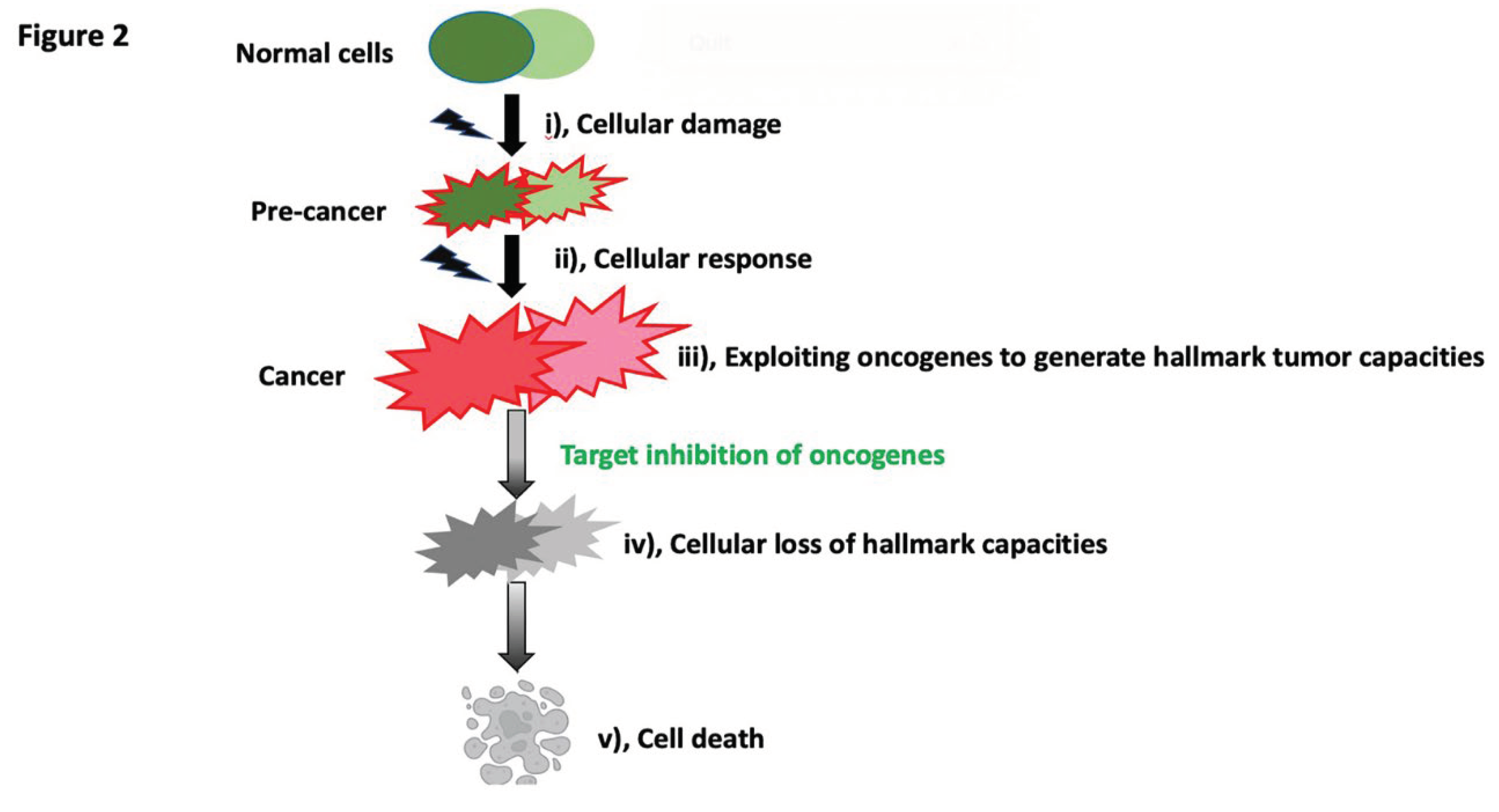
Figure 2.
Oncogene Addiction. During tumor evolution, advantageous genes are selected and activated in response to the cellular survival threats. When advantageous oncogenes are inhibited, cells lose their survival response mechanism to generate essential hallmark capacities. Subsequently, this evolutionary disruption results in cell death, a phenomenon termed “oncogene addiction”.
Figure 2.
Oncogene Addiction. During tumor evolution, advantageous genes are selected and activated in response to the cellular survival threats. When advantageous oncogenes are inhibited, cells lose their survival response mechanism to generate essential hallmark capacities. Subsequently, this evolutionary disruption results in cell death, a phenomenon termed “oncogene addiction”.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



