
Submitted:
08 April 2024
Posted:
10 April 2024
You are already at the latest version
Abstract
Antibody-drug conjugates (ADCs), a groundbreaking advancement in cancer therapy, synergistically combine the precision of monoclonal antibodies with the potent destruction capabilities of cytotoxic drugs, signifying a new era in the targeted oncological treatments. This comprehensive paper delves into the evolving field of antibody-drug conjugates (ADCs) and their pivotal role in cancer therapy. We have conducted an in-depth exploration of the structural components of ADCs, including target antigens, monoclonal antibodies, chemical linkers, and cytotoxic payloads, each playing a crucial role in determining the efficacy and safety profile of these novel therapies. A detailed market analysis reveals the leading pharmaceutical companies shaping the ADC landscape, reflecting the growing interest and potential in this therapeutic approach. We further dissect the engineering intricacies of ADCs, focusing on their ability to target specific cancer cell antigens while minimizing off-target effects, thus offering a more focused treatment with reduced systemic side effects compared to traditional chemotherapy. This approach holds promise in a heightened therapeutic index and decreased toxicity. The mechanisms and effector functions of ADCs are explored, highlight- ing their multifaceted strategy in combating cancer. We include a comprehensive look at how ADCs operate from initial blood circulation to the induction of apoptosis in cancer cells. We also review several key ADCs currently in use or under investigation, offering insights into their unique mechanisms of action, pharmacokinetics, and clinical implications. The discussion extends to drug creation modalities, focusing on innovative conjugation techniques that enhance ADC homogeneity and therapeutic efficacy. In conclusion, the paper underscores the transformative potential of ADCs in the realm of cancer treatment, marking a shift towards more precise and targeted therapeutic approaches. By offering a thorough analysis of ADC technology, this paper sheds light on the prospects of these groundbreaking therapies in the ongoing fight against cancer.
Keywords:
antibody-drug conjugates
; cancer
; monoclonal antibodies
1. Introduction
Bridging the gap between the destructive potential of conventional chemotherapeutics and highly specific monoclonal antibody technology for cancer therapeutic indications has been a recent area of interest for several large pharmaceutical companies. Antibody-drug conjugates (ADCs) are a significant advancement in cancer therapy, combining the specificity of antibodies with the potency of cytotoxic drugs. Cancer prevalence has become a global threat with the occurrence rising year over year, and the space for therapeutic capabilities has increased as well[1]. Cytotoxic agents or chemotherapies like 5-fluorouracil and 9-azaguanine have been primary options to initiate cell death in this patient population. Cisplatin and actinomycin D DNA intercalating agents have good efficacy but low specificity, causing healthy tissue degradation and high side effect outcomes[2]. It was previously found that mAb drugs were properly able to kill cancer cells but had some lower efficacies than standard chemotherapeutics[1]. However, with the advent of the novel technology of an ADC, there is greater potential to get similar specificity with greater impact and cancer cell death.[1,2]
Antibody-drug conjugates (ADCs) represent a significant breakthrough in the realm of targeted cancer therapy, merging the precision of monoclonal antibodies (mAbs) with the destructive power of cytotoxic drugs. This innovative approach emerged from the recognition that while mAbs excel in targeting specific tumor surface antigens, they often lack the lethal effectiveness of chemotherapy[4]. ADCs bridge this gap by effectively combining these two therapeutic approaches. They specifically target cancer cells, offering more focused treatment with reduced systemic side effects compared to traditional chemotherapy. Investigating the drug capabilities of an ADC allows for a higher level of control and potential for drug engineering when making functional manipulations to the general structure that exists for each of these drugs[3,4]. There are four components in consideration when working with an ADC, a highly specific antigen with the ability to internalize payloads, Monoclonal Antibodies, Chemical linkers, and Cytotoxic Drug attachments[4].
Target Antigen: The choice of target antigen is paramount. It should be an antigen predominantly or exclusively expressed on tumor cells to minimize off-target effects. The antigen ideally should be on the cell surface and capable of internalizing upon antibody binding, facilitating the delivery of the cytotoxic payload into the cancer cells[4,5].
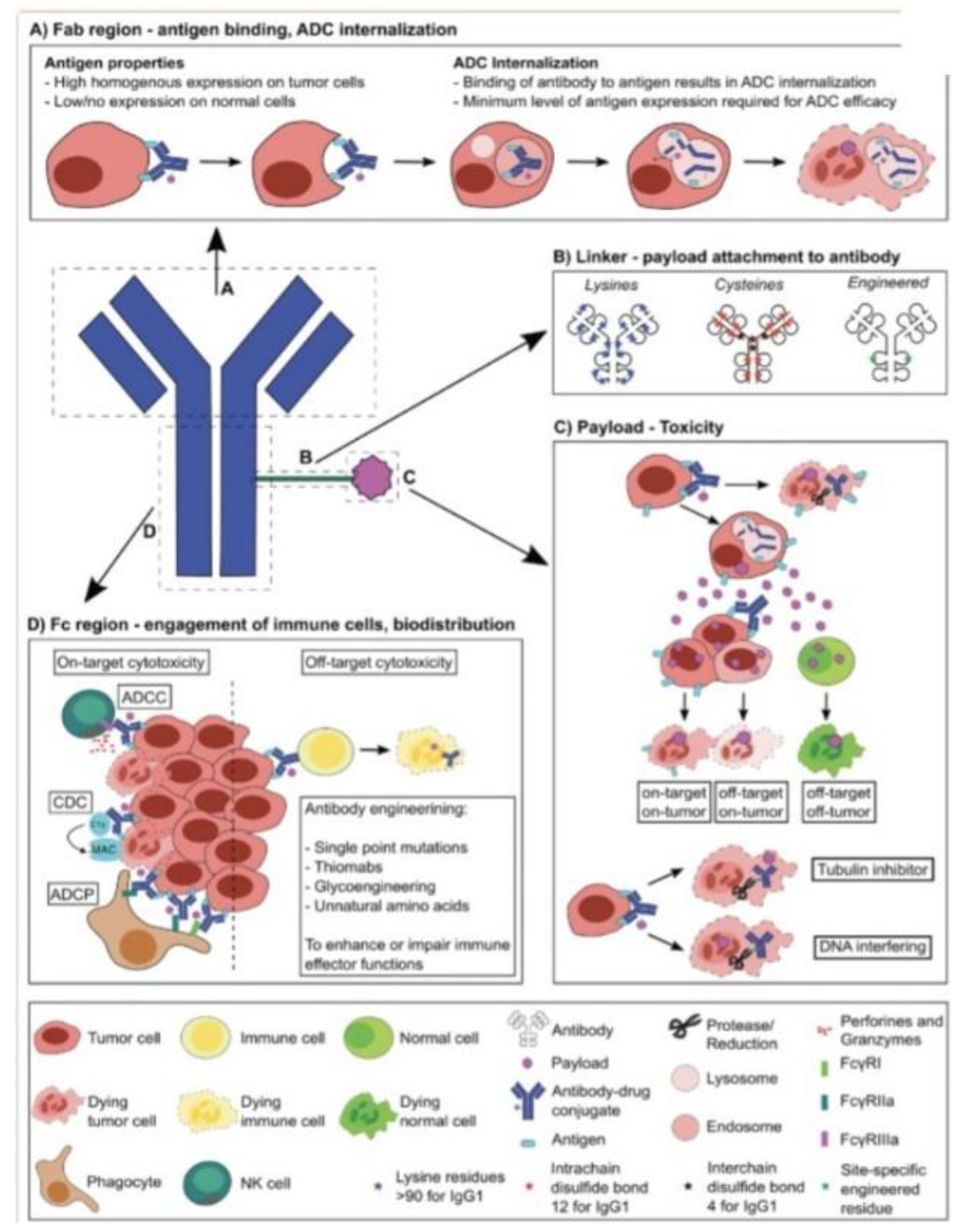
Figure 1.
Schematic of ADC structure and functionality with potential for high-scale engineering and specific cell targeting and destruction. A) Antibody binding to antigens with high specificity and internalization of the complex. B) Schematic overview of linker structure with binding of antibody to the cytotoxic payload. C) Revealing how toxicity occurs through payload internalization, linker cleavage, and molecular activities inducing cell death. D) Different immune effector functions induced by the Fc region engage cells and induce cellular responses based on the payload mechanism[3].
Figure 1.
Schematic of ADC structure and functionality with potential for high-scale engineering and specific cell targeting and destruction. A) Antibody binding to antigens with high specificity and internalization of the complex. B) Schematic overview of linker structure with binding of antibody to the cytotoxic payload. C) Revealing how toxicity occurs through payload internalization, linker cleavage, and molecular activities inducing cell death. D) Different immune effector functions induced by the Fc region engage cells and induce cellular responses based on the payload mechanism[3].
Monoclonal Antibody: This part of the ADC specifically binds to antigens overexpressed on the surface of cancer cells. The ideal antibody should have high binding affinity, efficient internalization, low immunogenicity, and a long plasma half-life. Most ADCs use humanized or fully human antibodies to reduce the risk of immune reactions. This component is designed to bind selectively to antigens overexpressed on cancer cells. The precision of mAbs in targeting tumor cells reduces the likelihood of affecting healthy cells, thereby enhancing the safety profile of the treatment[4].
Linker: This component connects the antibody to the cytotoxic drug. The linker is crucial because it must be stable enough to keep the payload attached during circulation in the bloodstream but also responsive to certain conditions within the tumor microenvironment to release the drug effectively. They can be cleavable, which releases the drug in response to certain conditions within the tumor environment, or non-cleavable, which relies on the degradation of the antibody for drug release. The choice of linker affects the efficacy and safety profile of the ADC[4,5].
Cytotoxic Drug (Payload): The role of the payload is to kill the cancer cells once the ADC has delivered it inside the target cells. This is a potent chemotherapeutic agent that, under normal circumstances, would be too toxic to administer systemically due to its non-specific nature. Once the ADC binds to its target antigen on the cancer cell surface, the complex is internalized, and the cytotoxic drug is released inside the cell, leading to cell death. Each one of these components will be investigated across several drug classifications, with an investigation of how engineering these molecules can lead to higher efficacy, binding capabilities, and decreased toxicity. The antibody-drug conjugates (ADCs) market has several leading companies that have been identified as key players in the industry. Antibody drugs, mAbs, bind receptors on cancer cells and linked cytotoxic compounds enter into the cells killing them with a smaller detriment to healthier cells. According to Mordor Intelligence, the top companies in the ADC market include Seagen Inc., F Hoffmann-La Roche Ltd, Pfizer Inc., AstraZeneca, Gilead Sciences, Inc., and others such as ImmunoGen Inc., Mersana Therapeutics Inc., Sorrento Therapeutics Inc., Oxford BioTherapeutics Ltd, AbbVie Inc., Takeda Pharmaceutical Company Ltd, ADC Therapeutics SA, GSK plc., and Daiichi Sankyo Company, Limited[5].
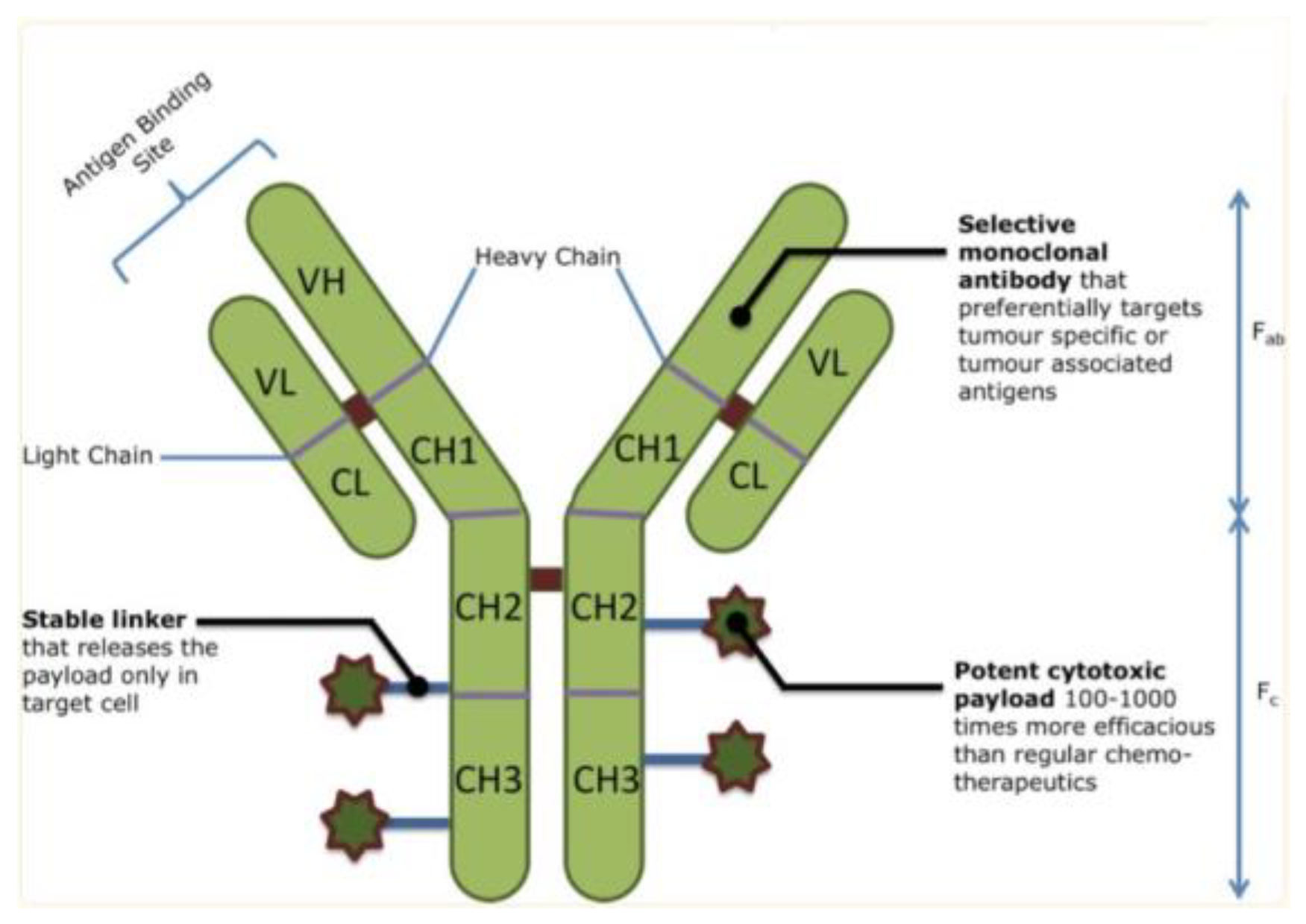
Figure 2.
An overview of ADC structure, with monoclonal antibody regions, a stable linker, and a potent cytotoxic payload that delivers specificity and toxicity to cancer cells[4].
Figure 2.
An overview of ADC structure, with monoclonal antibody regions, a stable linker, and a potent cytotoxic payload that delivers specificity and toxicity to cancer cells[4].
These companies have been recognized for their significant contributions and market share in the ADC industry. For instance, Seagen Inc. is known for its pioneering work in ADCs, particularly in cancer therapeutics. F Hoffmann-La Roche Ltd, another major player, has been active in developing ADCs with a focus on oncology and hematology, and is the leading patent filer for this cancer therapeutic space, with Gilead following in second. Pfizer and AstraZeneca are globally recognized pharmaceutical giants with a diverse portfolio that includes ADCs. Gilead Sciences has recently made headlines for its performance, posting a net income of $5.61 billion in FY 2023[5]. The ADC market is experiencing growth due to increasing incidences of cancer, technological advancements, and strategic partnerships among key players. The market is slightly fragmented and competitive with the presence of several global and local companies. North America, in particular, is expected to witness significant growth in this market due to the rising number of cancer cases and advancements in technology[5,37]. The very first ADC approved by the U.S. Food and Drug Administration (FDA) was Mylotarg (gemtuzumab ozogamicin) in 2000, used for treating acute myeloid leukemia (AML). This approval marked the advent of ADCs in cancer therapy[2]. As of December 2021, 14 ADC drugs have been approved for various cancers, including both hematological malignancies and solid tumors. Additionally, over 100 ADC candidates are currently undergoing various stages of clinical trials, and this data has come from 2021, there is a greater number present today in 2024, reflecting the growing interest and potential in this therapeutic approach[5].
2. Methods
The methodology for this comprehensive review on antibody-drug conjugates (ADCs) adheres to the PRISMA 2020 guidelines, aiming to systematically explore the literature related to the engineering, pharmacological characteristics, and clinical applications of ADCs in cancer therapy. An extensive literature search was conducted across several databases, including PubMed, Drug Bank, Medline, and Google Scholar, using a combination of terms related to ”antibody-drug conjugates,” ”cancer therapy,” ”monoclonal antibodies,” ”cytotoxic payloads,” and ”pharmacokinetics.” Additionally, manual searches of reference lists from key articles were performed to identify further relevant studies. The inclusion criteria for this review were strictly defined: studies had to be peer-reviewed articles focusing on ADCs in cancer treatment, with detailed information on the structure, engineering, and pharmacological properties of ADCs, including clinical trials and reviews discussing the application and efficacy of specific ADCs. The exclusion criteria were set to filter out studies with insufficient or irrelevant pharmacological data, articles not specifically addressing ADCs in the context of cancer therapy, and publications primarily focusing on non-cancerous applications of ADCs.
Figure 3.
A systematic approach to review with methods in accordance with PRISMA guidelines. This involves reports excluded due to irrelevance to our study, a lack of pharmacological data, or out of scope interventions. No Automation tools were used and databases utilized include Drugbank, Pubmed, Google Scholar, and Medline[38,39].
Figure 3.
A systematic approach to review with methods in accordance with PRISMA guidelines. This involves reports excluded due to irrelevance to our study, a lack of pharmacological data, or out of scope interventions. No Automation tools were used and databases utilized include Drugbank, Pubmed, Google Scholar, and Medline[38,39].
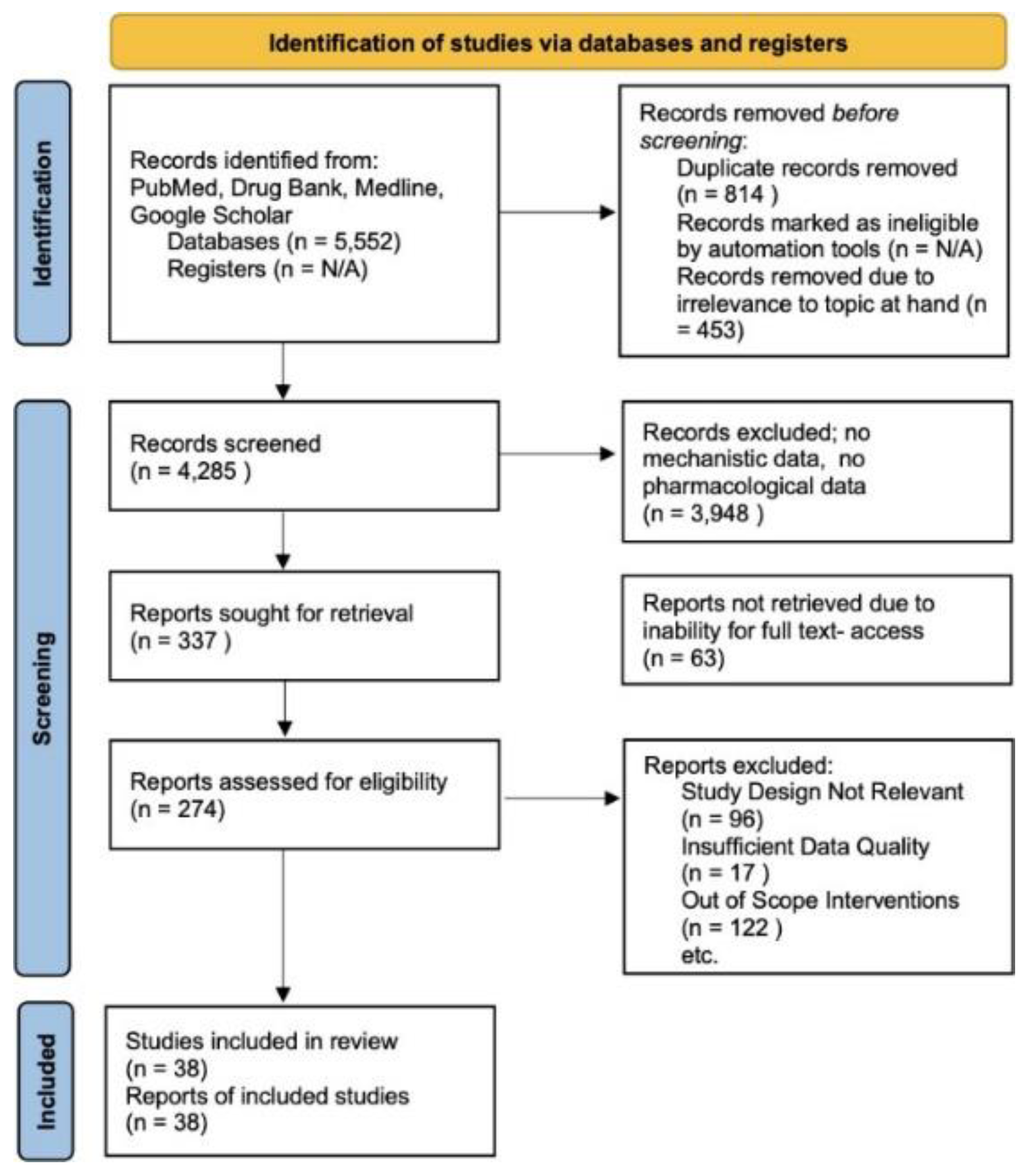
Initially, the search yielded 5,552 records. After removing duplicates (814 records) and irrelevant records (453 records), 4,285 records remained for screening. A significant number, 3,948, were excluded for lacking mechanistic or pharmacological data relevant to the review’s focus. Following this, 337 reports were subjected to a detailed evaluation. However, 63 were not retrievable in full text. Of the 274 assessed for eligibility, further exclusions were made based on irrelevant study design (96 reports), insufficient data quality (17 reports), and out-of-scope interventions (122 reports). Ultimately, 39 studies met all the inclusion criteria and were included in this review. Data from these studies were systematically extracted, focusing on the structural components, engineering techniques, pharmacokinetics, and clinical implications of ADCs. This included comprehensive information on target antigens, linkers, cytotoxic agents, therapeutic efficacy, and safety profiles. The extracted data were synthesized to provide an overarching view of the current ADC landscape in cancer therapy. The quality of the included studies was rigorously assessed using standardized tools, considering factors like study design, methodological rigor, and the relevance and reliability of the data. This review also adhered to ethical standards concerning data handling, ensuring that all analyzed studies complied with ethical research practices, and sensitive information was handled with confidentiality and respect.
3. ADC Engineering Approaches
Engineering a potent molecular and small molecule structure is essential when engineering ADCs. There are several components and a dynamic network of interactions that need to be taken into account with such a complex compound drug, including monoclonal antibody and antigen selection, as well as linker stability considerations in attachment to the cytotoxic payload. To ensure the successful development of an ADC, there must be increased and optimized target selection methodologies with engineered antibody-antigen binding selections. Moreover, the cytotoxic payload must be chosen carefully to have similar interference capabilities in the cell target whether it be microtubule destabilization or DNA intercalating mechanisms to induce cellular death. Innovative Linkers that maintain stability and proper binding metrics are essential to mitigate unintentional delivery of cytotoxic payload causing symptomatic responses to surrounding tissue and healthy cells. All of these factors ensure that drug resistance is not developed and maintains a highly specific outcome of cell death to primarily cancer cell types[4].
3.1. Antigen Binding
ADCs target specific antigens on cancer cells, including HER2, trop2, nectin4, and EGFR in solid tumors, and CD19, CD22, CD33, CD30, BCMA, and CD79b in hematological malignancies(blood cancers and malignancies)[6]. The targeting of these antigens ensures that the cytotoxic payload is delivered directly to cancer cells, minimizing damage to healthy cells. Additionally, some ADCs are designed to target components of the tumor microenvironment, such as the vasculature or stroma, which can also contribute to the efficacy of the treatment[7]. In the development of Antibody-Drug Conjugates (ADCs), the selection of the target antigen on tumor cells is a critical step, acting as the guiding beacon for the ADCs to accurately identify and target tumor cells[4]. This choice is pivotal because it dictates how the cytotoxic payloads are delivered into the cancer cells, commonly via mechanisms such as endocytosis. The ideal target antigen for ADC therapy should have a high expression specifically on tumor cells, with little to no presence in normal tissues, to minimize the risk of off-target effects and maximize the safety of the treatment. These antigens should be located on the surface or extracellular part of the tumor cells, making them accessible to the circulating ADCs[7]. Additionally, the target antigens should not be secretory to prevent undesirable ADC binding outside the tumor sites, which could reduce the efficacy of tumor targeting and raise safety concerns. Moreover, it is advantageous if these antigens can be internalized upon binding with the ADC, as this facilitates the entry of the ADC-antigen complex into the cancer cells, followed by the appropriate intracellular transport and release of the cytotoxic payload[4,6,7]. The ongoing research in oncology and immunology has broadened the horizons for ADC target antigens, extending beyond conventional tumor cell antigens to targets within the tumor microenvironment, such as the stroma and vasculature. This expansion in target selection is based on the emerging evidence that targeting these areas can significantly enhance the effectiveness of ADCs. For instance, targeting the matrix within the tumor environment could disrupt the concentration of growth factors vital for cancer cell survival, which often relies on angiogenesis and matrix factors. Targeting these more genetically stable cells in the tumor microenvironment also presents a potential strategy to mitigate the risk of mutation-induced drug resistance. This evolution in the approach to targeting reflects a more comprehensive strategy in ADC development, aiming to encompass a broader range of elements within the tumor microenvironment for more effective cancer therapy. With greater knowledge of Antigen targets and network interactions of downstream protein effects, these antigen-binding sites can be changed or manipulated to mitigate the downstream symptomatic effects of these highly cytotoxic drugs. Through a rigorous engineering approach of monoclonal antibody selection for highly specific interactions primarily with cancer tumor cells, a higher efficacy and safety profile can be reached for these drugs[4,7,8].
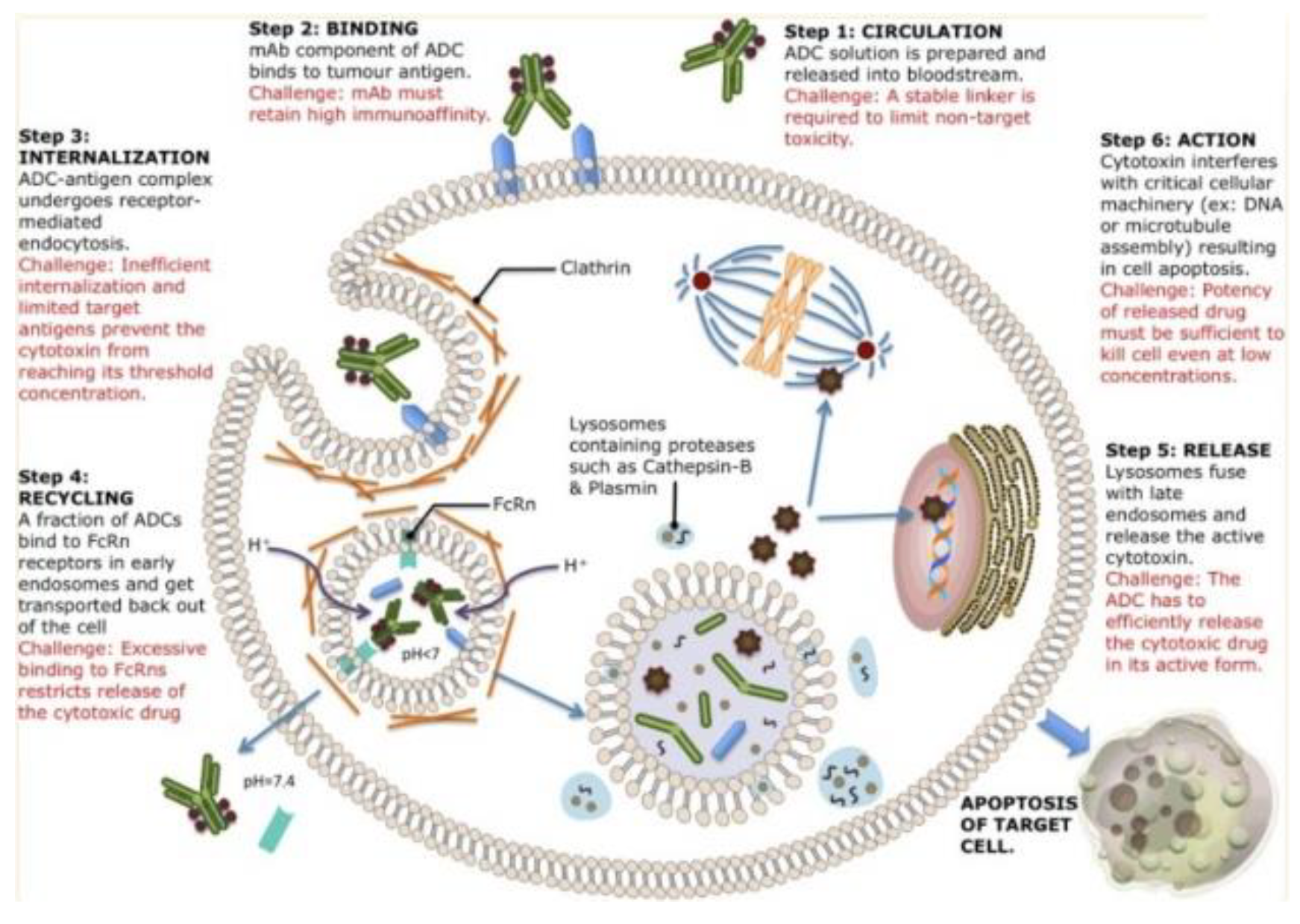
Figure 4.
An overview of mechanistic action of ADCs from blood circulation to apoptotic activities. Each ADC will bind to different receptors on the cell surface and be internalized with different mechanisms. After internalization, there are several pathways of cell destruction including DNA or microtubule interference causing cell destruction potential[4].
Figure 4.
An overview of mechanistic action of ADCs from blood circulation to apoptotic activities. Each ADC will bind to different receptors on the cell surface and be internalized with different mechanisms. After internalization, there are several pathways of cell destruction including DNA or microtubule interference causing cell destruction potential[4].
3.2. Antibody Specifications
The choice of antibody for tumor-antigen targeting capabilities is essential for a properly working ADC mechanism of action. The antibodies allow for a highly specific, stable, and strong linkage between the cytotoxic payload and the cell through antibodyantigen interactions. Antibodies chosen for these drugs must have high binding affinity to the target antigen of choice, facilitate efficient internalizations, and have a high biocompatibility profile[7]. Along with low immunogenicity, these drugs must be longlasting in the body to allow for efficiently dispersed cell death with a long plasma half-life. Because of the necessity to have low immunogenicity, mouse-derived antibodies have been replaced with humanized antibodies to mitigate their high failure rates and low biocompatibility. Chimeric and humanized antibodies have been highly effective and common with the advent of recombinant technology allowing for greater biocompatibility profiles. Immunoglobulin G (IgG) antibodies are predominantly used in ADCs specifically IgG1 subtypes due to their strong effector functions like antibody-dependent cell-mediated cytotoxicity(ADCC), antibody-dependent phagocytosis(ADCP), and complement-dependent cytotoxicity(CDC)[7]. IgG1 is highly favored for ADCs due to their strong effector functions and a high abundance in serum. IgG3 has a much faster clearance rate and is rarely used. IgG2 dimerizes and aggregates causing decreased concentration of the drug and IgG4 has a low efficacy as well. The binding affinity between antibody and surface antigen on tumor cells determines the efficiency of internalization of the antibody-antigen complex. Higher affinity antibodies allow for greater internalizations but could reduce the penetration into solid tumors. Moreover, the size of the antibody also affects tumor penetration with larger antibodies presenting barriers to penetration through blood capillaries and tumor microenvironment matrices. Miniaturized antibodies without Fc segment retain higher affinities and specificity, penetrate solid tumors more easily, and have reduced halflives. Designing and optimizing these miniaturized antibodies requires consideration of various factors to optimize efficacy and pharmacokinetic profiles[6,7].
3.3. Chemical Linker Specifications
Chemical Linkers contained within ADCs bridge the antibody and cytotoxic drugs, playing a critical role in the stability of ADC and release profiles of payloadtherefore impacting the therapeutic index of ADCs. Ideal linkers should not induce ADC aggregations and should limit the premature release of payloads in plasma while promoting the release of the drug at target antigen and tumor sites for internalization. Cleavable linkers exploit the microenvironmental differences in systemic circulations and tumor cells to get higher levels of accurate payload release[7]. They can be chemical cleavage linkers like hydrazone or disulfide bonds or enzyme cleavage linkers like glucuronide and peptide bonds. Hydrazone linkers are pH sensitive and stable in blood circulation and release the chemotherapeutic agent payload in lysosomes upon internalization to cancer cells. Disulfide linkers are sensitive to intracellular reductive glutathione, releasing payloads in cancer cells. Enzymesensitive linkers such as peptide-based and beta glucuronide linkers are cleaved by lysosomal proteases or beta-glucuronidase, respectively, in tumor cells, minimizing off-target effects. Non-cleavable linkers like thioethers and maleimidocaproyl groups are inert to chemical and enzymatic environments in vivo leading to low off-target toxicity and increased plasma stability[4]. These linkers rely on enzymatic hydrolysis of the antibody component to release the payload. T-Dm1 is an ADC using a thioether linker with the antibody linked with DM1 via a thioether linker, providing stability in blood and releasing active metabolite after antibody degradation inside cancer cells[7].
3.4. Cytotoxic Payload Specifications
In the realm of cancer therapy, antibody-drug conjugates (ADCs) represent a significant advancement, with the cytotoxic payload being the critical component responsible for the destruction of cancer cells upon internalization. These payloads, due to the limited efficiency of ADC delivery to tumor sites, demand high potency, often reflected in IC50 values ranging in the nanomolar to picomolar scale. ADCs typically utilize various cytotoxic agents, broadly categorized into tubulin inhibitors, DNA damaging agents, and the emerging class of immunomodulators. Tubulin inhibitors, like auristatin and maytansinoid derivatives, disrupt cell division by targeting microtubule functions. DNA damaging agents, including calicheamicin and pyrrolobenzodiazepines, induce lethal DNA damage in cancer cells. The newest category, immunomodulators, aims to stimulate the immune system, thereby enhancing tumor regression and sustained anti-tumor immunity, with TLR and STING agonists being notable examples. A promising development in this area is ISACs (immunestimulating antibody conjugates), which merge precise antibody targeting with the immunomodulatory impact of small molecule payloads, exemplified by BDC-1001 and CRD5500[7–9].
The incorporation of novel payloads with unique mechanisms is expanding ADC applications, potentially offering enhanced efficacy and reduced side effects in cancer treatment. Notably, ADCs have a significantly larger molecular weight than conventional cytotoxic drugs, which may restrict their tumor penetration efficiency. The research underscores that only a fraction of administered ADCs reach tumor cells, thus emphasizing the necessity for potent payloads in ADC design. Delivery of these payloads hinges on the internalization of the ADC-antigen complex, either via antigen-dependent endocytosis or antigenindependent pinocytosis. Following internalization, the complex is transported to endosomes or lysosomes, where payloads are released, depending on the nature of their linkers and the specific intracellular environment. Importantly, some ADCs exhibit a ”bystander effect,” impacting neighboring cancer cells that lack the target antigens. This effect is particularly vital for ADCs aimed at tumors with varied antigen expression and relies on the ability of the released payload to permeate cell membranes, favoring nonpolar over polar molecules due to their higher likelihood of cell membrane crossing[6,8,9].
Traditional payloads are primarily chemotherapeutic drugs, microtubule inhibitors, and DNAdamaging elements. However, with relatively low therapeutic indexes, these drugs can be optimized or other investigational pathways could be investigated to mitigate these side effects, therefore optimizing payload capabilities. Currently, payloads consist of mechanisms involving microtubule targeting, Maytansinoids, Auristatin, Eribulin, Tubulysins, Cryptophycins, EG5 inhibitors, DNA targeting payloads, Enediyne, Topoisomerase I inhibitors, PBD, Duocarmycins, RNA targeting payloads, Thailanstatin, Amatoxins, immune ADC payloads, toll-like receptor agonists, STING agonists, and Glucocorticoid receptor modulators all with differing therapeutic indices. Several new alternative payloads are being investigated across different classes with highly prominent pathways being bcl-xL inhibitors, NAMPT inhibitors, Carmaphycins, PROTAC payloads, NIRPIT drug payloads, and dual payload combination therapies. Moreover, the development of a PDC or peptide drug conjugate has greater advantages like lower molecular weight, higher tumor penetration, high biocompatibility, lower manufacturing costs, and greater uniformity across synthesization[8].
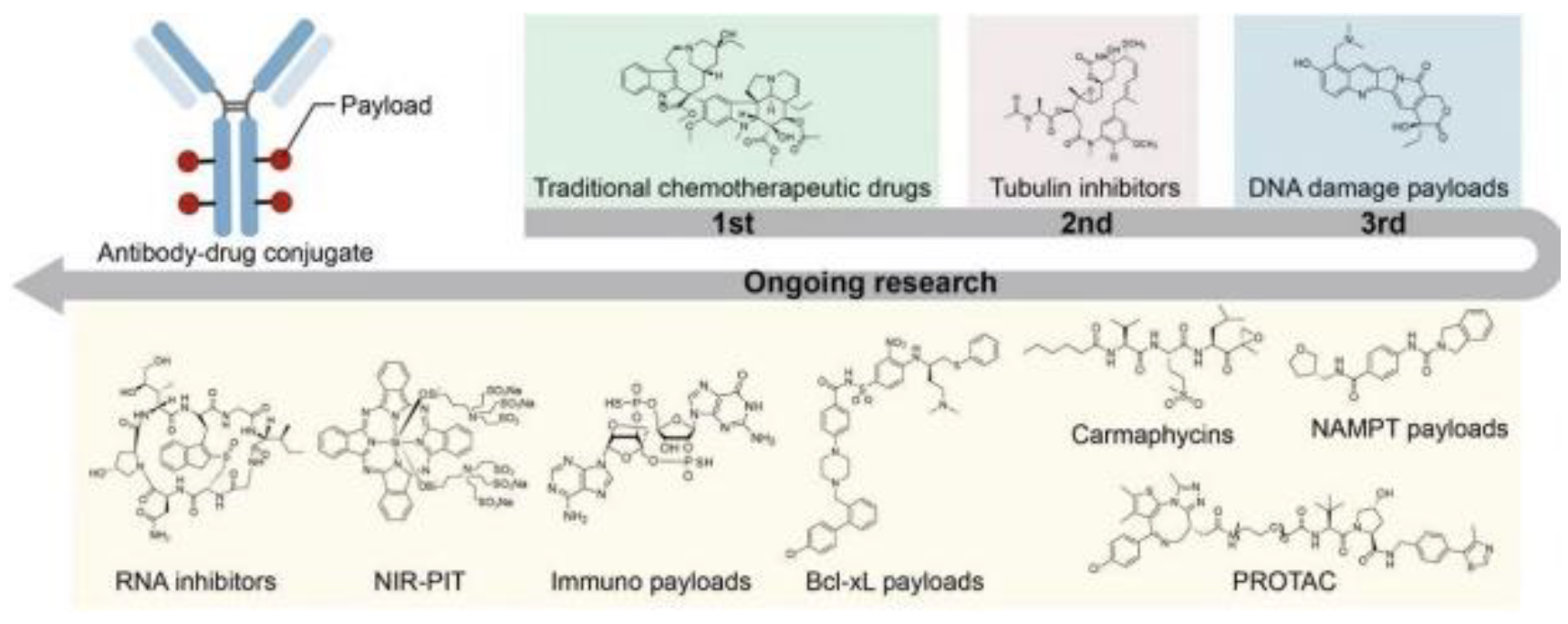
Figure 5.
This figure outlines current cytotoxic payload capabilities, with potential expansionary investigations into RNA inhibition, and immunologicaloriented payloads, along with several other potential advancements. Optimizing the payload can lead to future mitigations of side effects and greater apoptotic potential within cancer cells[8].
Figure 5.
This figure outlines current cytotoxic payload capabilities, with potential expansionary investigations into RNA inhibition, and immunologicaloriented payloads, along with several other potential advancements. Optimizing the payload can lead to future mitigations of side effects and greater apoptotic potential within cancer cells[8].
3.5. Mechanisms and Effector Functions
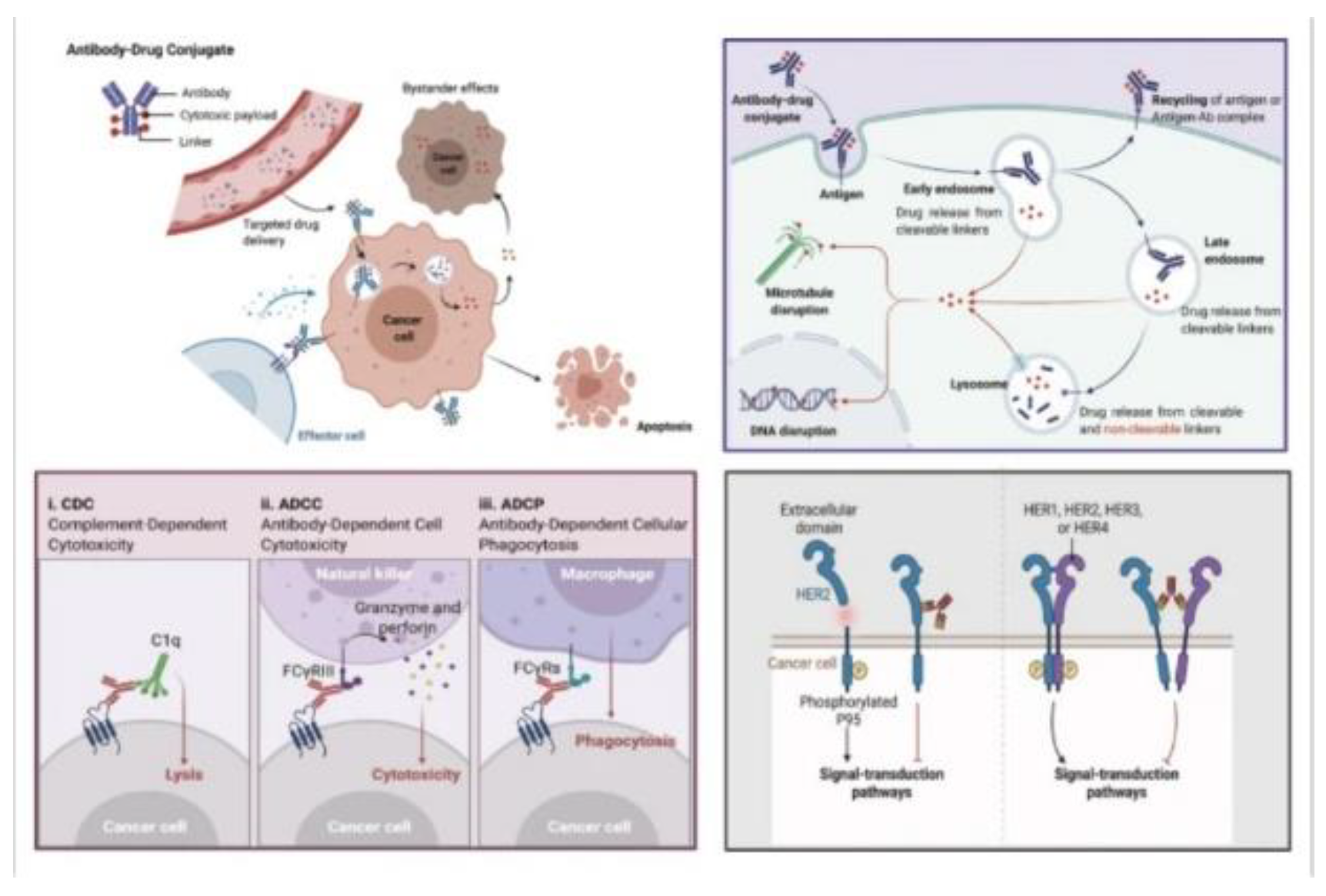
Antibody-drug conjugates (ADCs) play a dual role in cancer therapy, serving as precision-guided ”biological missiles”[2] that offer specific targeting capabilities coupled with potent effects in killing cancer cells. This approach notably improves the therapeutic window and diminishes off-target side effects. The primary mechanism of ADCs involves several crucial steps. Initially, the monoclonal antibody (mAb) component binds to antigens specifically expressed on cancer cells, followed by the internalization of the ADC through endocytosis. This process transitions from early endosomes to late endosomes, and ultimately to fusion with lysosomes.
Within the lysosomes, cytotoxic payloads are released via chemical or enzyme-mediated mechanisms, targeting vital cellular components such as DNA or microtubules and leading to cell death or apoptosis. If the payload can cross cell membranes, it may also induce a bystander effect, thereby enhancing the ADC’s efficacy by affecting neighboring cancer cells. In addition to direct cytotoxicity, ADCs exert anticancer effects through various mechanisms. These include Antibody-Dependent Cellular Cytotoxicity (ADCC), where the antibody’s Fab segment binds to cancer cell antigens, and the Fc segment interacts with immune effector cells, resulting in direct cell killing. Antibody-dependent cellular Phagocytosis (ADCP) involves immune cells, particularly macrophages, engulfing cancer cells coated with ADCs, aiding in tumor clearance. Complementdependent cytotoxicity (CDC) is triggered when antibodies on cancer cell surfaces activate the complement system, leading to cell lysis. Furthermore, ADCs can inhibit downstream signaling pathways essential for cell survival and proliferation, such as the inhibition of signal transduction pathways like PI3K or MAPK by the antibody component in T-DM1, which targets HER2 receptors, ultimately inducing apoptosis. Overall, ADCs utilize a multifaceted strategy in combating cancer, combining the targeted delivery of cytotoxic payloads with immune-mediated mechanisms and inhibition of critical signaling pathways in cancer cells. This comprehensive approach underscores the versatility and effectiveness of ADCs in cancer therapy[2,6,7].
Figure 6.
A general overview of mechanisms of ADCs for killing cancer cells(Upper-Right); CDC, ADCC, ADCC effects of eliciting antitumor immunity(Lower Left); Antibody component retains activity profile and can interfere with target functionality, dampening downstream signaling, inhibiting tumor growth(Lower Right)[2].
Figure 6.
A general overview of mechanisms of ADCs for killing cancer cells(Upper-Right); CDC, ADCC, ADCC effects of eliciting antitumor immunity(Lower Left); Antibody component retains activity profile and can interfere with target functionality, dampening downstream signaling, inhibiting tumor growth(Lower Right)[2].
3.6. Drug Synthesis and Conjugation Modalities
The construction of antibody-drug conjugates (ADCs) critically hinges on the method used to attach the small molecule moiety (comprising the linker and payload) to the antibody. Traditional stochastic conjugation techniques, which typically attach payloads to naturally occurring lysine or cysteine residues on antibodies, face challenges like heterogeneous drug-antibody ratios (DAR) and the risk of offtarget toxicity due to premature payload release. To overcome these issues, site-specific conjugation methods have been devised, greatly enhancing ADC homogeneity and therapeutic efficacy[3,8].
One such technique involves engineered reactive cysteine residues, exemplified by ThioMab technology, which has demonstrated remarkable consistency in DAR. Disulfide re-bridging conjugation is another innovative approach, where cysteine-selective crosslinking agents are used to reattach polypeptide chains and append payloads, achieving DAR values of 4, 8, or 16. The introduction of unnatural amino acids for site-specific conjugation introduces unique functional groups, although this method can complicate antibody production and potentially increase immunogenicity. Enzyme-assisted ligation represents another strategy, altering specific amino acid sequences in the antibody for precise conjugation, yet it too may raise immunogenic concerns[7].
Additionally, glycan remodeling and glycoconjugation leverage reactive sites on N-glycans within the antibody’s Fc fragment for site-specific attachment. Recent advancements include technologies like pClick, which facilitate efficient site-specific conjugation without necessitating antibody engineering, offering a streamlined and effective route for ADC development. Each of these methods reflects the evolving landscape of ADC technology, striving to optimize therapeutic effectiveness while minimizing adverse effects[2,3].
4. ADC Drug Classifications and Review
Outlining key players within the development of ADCs based on competent market capitalization and drug safety and efficacy, the preceding reviews are outlining certain mechanistic functions, pharmacological profiles, and disease indications of 10 Antibody Drug Conjugates. These drugs outline the potential for these potent therapeutic compounds to be a breakthrough within cancer treatment. There are still drawbacks currently including some drugs with a low therapeutic index or drugs like Lumoxiti being withdrawn from the market due to low clinical adoption. Currently many ADCs have barriers to market entry due to alternatives with higher safety and efficacy profiles with reduced symptomatic effects. With greater investigations and optimized drug engineering, there is potential for these drugs to reduce symptoms and neighboring healthy tissue damage while still delivering potent payloads, inducing cancer cell apoptosis.
4.1. Adcetris(Brentuximab Vedotin)
Brentuximab vedotin, marketed under the brand name ADCETRIS, stands as a pivotal intravenous antibody-drug conjugate (ADC) particularly effective against CD30-positive cancer cells, with a significant impact on classical Hodgkin lymphoma (HL). Brentuximab vedotin is a chimeric immunoglobulin G1 antibody-drug conjugate targeting CD30, linked to monomethyl auristatin E (MMAE) through a protease-cleavable linker. It effectively induces apoptotic death in CD30-expressing tumor cells by binding to their surface[9]. This ADC has shown high objective response rates in patients with relapsed or refractory CD30-positive HL, evidenced by clinical trials and real-world applications, including scenarios where retreatment is necessary[9]. From a pharmacokinetic perspective, data derived from phase 1 trials and population analysis involving 314 patients revealed that brentuximab vedotin and its antibody component share similar profiles. Its maximum serum concentrations are usually reached near the end of a 30-minute infusion, while MMAE peaks between 1 to 3 days later[10]. Dose-proportional exposure and minimal accumulation over multiple doses were noted, with steady-state achieved within 21 days of each 3-week dose. MMAE exposure declines to about 50-80 percent with continued administration. Notably, as a consolidation therapy following autologous hematopoietic stem cell transplant (ASCT), brentuximab vedotin has substantially extended progression-free survival compared to a placebo, with these benefits persisting during extended follow-up periods[9,11,12].
The safety profile of brentuximab vedotin has been deemed acceptable in both controlled trials and realworld settings. In March 2018, the FDA approved Adcetris (brentuximab vedotin) for treating adult patients with previously untreated stage III or IV classical Hodgkin lymphoma in combination with chemotherapy. This approval broadened its therapeutic use, adding to its existing approvals for Hodgkin’s lymphoma post-relapse or post-stem cell transplantation, systemic anaplastic large cell lymphoma after other treatments failed, and primary cutaneous ALCL following unsuccessful treatments. Lymphoma originates in the lymphatic system, with Hodgkin lymphoma having two main types, most commonly the classical type characterized by ReedSternberg cells. The ECHELON-1 study showed Adcetris combined with chemotherapy improved efficacy compared to previous standards, notably eliminating the highly toxic agent bleomycin from the regimen, signifying significant advancement in treatment. The most common manageable adverse events include peripheral sensory neuropathy and neutropenia. Given the limited range of treatment options available for relapsed or refractory HL, brentuximab vedotin emerges as a vital therapeutic choice. It is especially relevant for patients who have not responded to high-dose chemotherapy/ASCT or those who have undergone multiple prior chemotherapy treatments. Furthermore, its use as post-ASCT consolidation therapy offers a promising option for highrisk patients, underscoring its significance in the landscape of HL treatment[10–12].
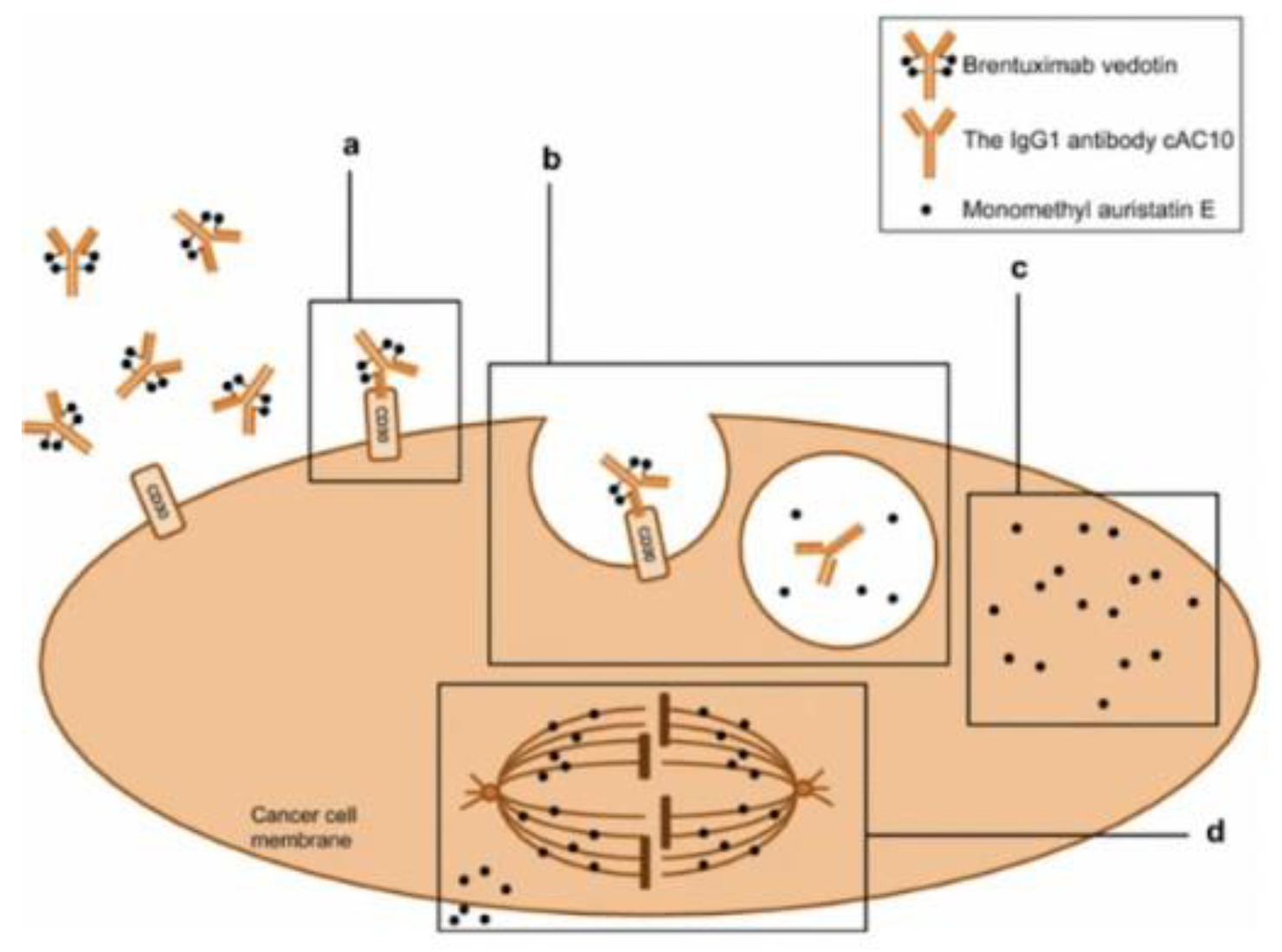
Figure 7.
Mechanism of action of Brentuximab Vedotin in CD-30 positive tumor cell. 1)Adcetris binds to the CD30 membrane target receptor. 2) drug complex internalized by a lysosome and enzymes cleave the linker between monoclonal antibody and cytotoxic payload MMAE. 3) MMAE is released within the cell where it binds to tubulin inducing apoptosis[11].
Figure 7.
Mechanism of action of Brentuximab Vedotin in CD-30 positive tumor cell. 1)Adcetris binds to the CD30 membrane target receptor. 2) drug complex internalized by a lysosome and enzymes cleave the linker between monoclonal antibody and cytotoxic payload MMAE. 3) MMAE is released within the cell where it binds to tubulin inducing apoptosis[11].
4.2. Kadcyla(Trastuzumab Emtansine)
Breast cancer (BC) is a common disease globally and presents substantial public health challenges. Recent advances in molecular biology and immunotherapy have led to targeted treatments based on specific patient and disease profiles[10]. A key element in BC pathophysiology is the overexpression of Human Epidermal Growth Factor Receptor 2 (HER2), which is also a critical biomarker for therapy selection. Trastuzumab, a recombinant igG1 humanized monoclonal antibody that targets HER2, was the first biologic drug approved for treating HER2-positive BC and continues to be a cornerstone of therapy, even with the emergence of other anti-HER2 agents like pertuzumab and lapatinib. However, issues such as cardiotoxicity and resistance to treatment have been noted, and there remains a lack of consistent resolution on various therapeutic aspects in existing literature. Therefore, a thorough review of trastuzumab is necessary to equip clinicians with vital information regarding its pharmacokinetics, pharmacodynamics, clinical usage, and ongoing setbacks/ symptomatic responses[10,13].
HER2, part of the tyrosine kinase receptor family, is essential in both normal and malignant breast tissues, activated through dimerization with other HER family members like HER1 (EGFR), HER3, and HER4, rather than ligand-binding. In breast cancer (BC), 15-30 percent of cases exhibit HER2 overexpression, categorizing them as HER2-positive, a subtype linked to increased tumorigenicity due to constant activation of downstream signaling pathways. This overexpression is associated with a more aggressive BC phenotype, marked by rapid tumor growth, early metastasis, and reduced survival rates. Trastuzumab, a monoclonal antibody targeting HER2, has revolutionized HER2-positive BC treatment but is contingent on HER2 overexpression. Determining HER2 status, crucial in BC management, is conducted through immunohistochemical analysis or in situ hybridization, with borderline cases retested to guide treatment decisions, highlighting the importance of understanding and accurately assessing HER2 in tailoring effective breast cancer therapies[13,14].
Figure 8.
Mechanism of action of Trastuzumab Emtansine. This drug has an ADCC induction profile undergoing antibody-dependent cellular cytotoxicity. It binds to subdomain IV of the HER2 receptor, leading to receptor-mediated internalization with concurrent endosomal/lysosomal intake and linker degradation releasing DM1-containing cytotoxic catabolites[13].
Figure 8.
Mechanism of action of Trastuzumab Emtansine. This drug has an ADCC induction profile undergoing antibody-dependent cellular cytotoxicity. It binds to subdomain IV of the HER2 receptor, leading to receptor-mediated internalization with concurrent endosomal/lysosomal intake and linker degradation releasing DM1-containing cytotoxic catabolites[13].
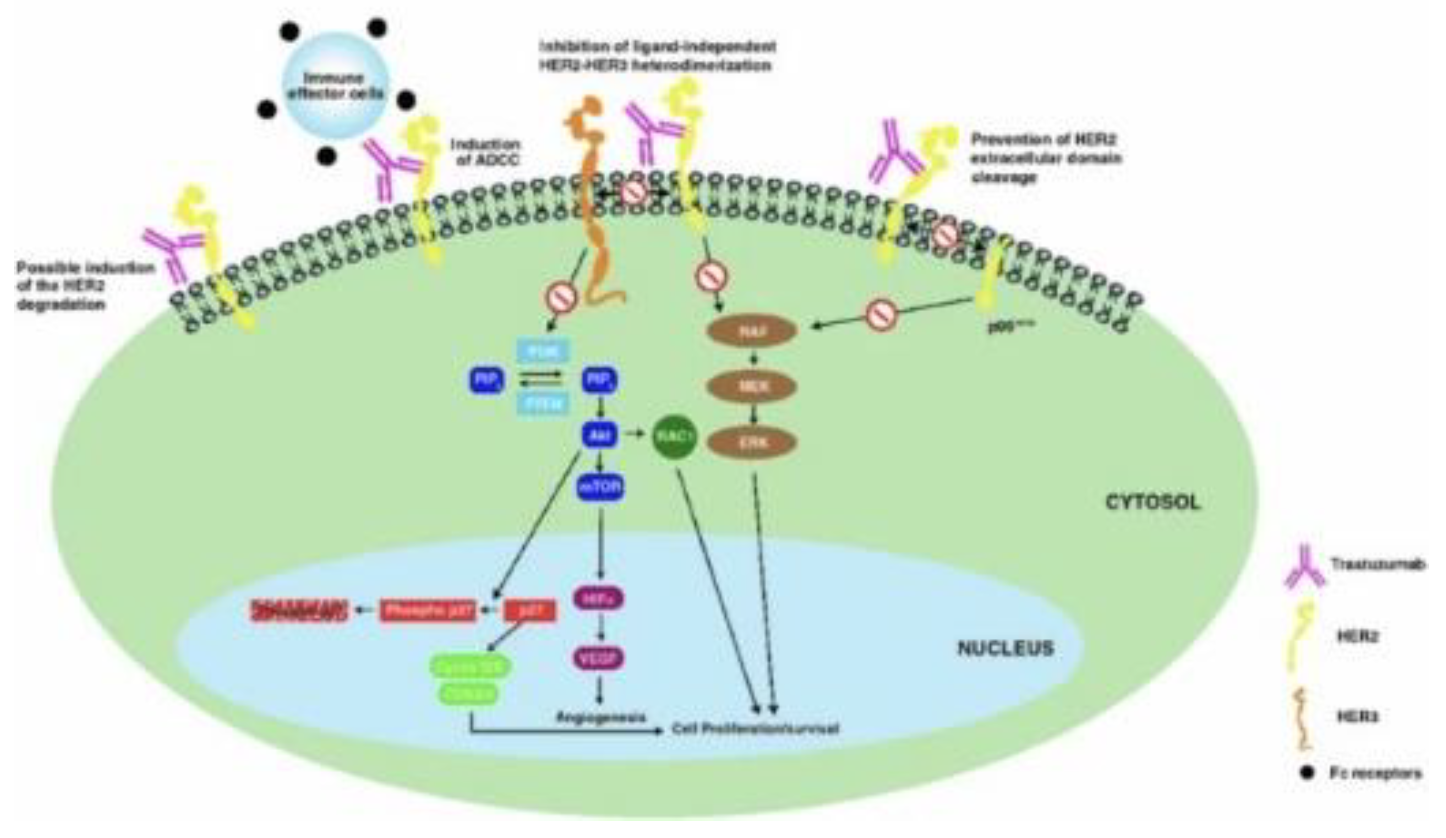
Trastuzumab emtansine, a hydrophilic molecule, operates by binding to the HER2 receptor’s subdomain IV and undergoing receptor-mediated internalization via endocytosis[15]. It follows the endosomallysosomal pathway, where it degrades to release DM1-containing cytotoxic catabolites. DM1, a positively charged molecule linked to lysine, disrupts microtubule assembly, leading to cell cycle arrest and apoptotic death. This drug maintains the antitumor efficacy of trastuzumab, with a similar affinity for HER2-expressing cells. It not only mediates antibody-dependent cell-mediated cytotoxicity but also hinders HER2 extracellular domain shedding and blocks HER3 phosphorylation, thereby impeding the PI3K pathway. Targeting HER2-positive cells selectively, trastuzumab emtansine is more potent than trastuzumab alone. Additionally, it disrupts epithelial-to-mesenchymal transition and reduces the clonogenic potential of cancer stem cells. Resistance to trastuzumab emtansine can develop, potentially due to the upregulation of multi-drug resistance transporters or increased expression of EGFR and other receptors[13–15].
4.3. Blenrep (Belantamab Mafodotin)
Blenrep is a first-in-class ADC targeting relapsed and/or refractory multiple myeloma in adult patients. This drug approach combines a BCMA targeting antibody for highly specific interactions and cytotoxic payload of monomethyl auristatin F(MMAF) which is a microtubule inhibitor, causing cell destabilizations and eventually death. The drug comprises a humanized igG1 monoclonal antibody, an MMAF payload to disrupt microtubules, and a protease-resistant linker connecting the two ensuring stability within the circulation. The antibody primarily binds to the BCMA protein that’s specifically and highly expressed in multiple myeloma, obtaining a highly specific targeted approach to minimize cell death in surrounding tissue. Upon binding, the ADC-receptor complex is internalized to the cell, the linker cleaves, and MMAF is released to induce cell death. MMAF can bind and disrupt the polymerization of microtubules which is essential for cell division and survival, ultimately leading to a cell cycle arrest at the G2/M stage, eventually leading to apoptosis[16].
Belantamab mafodotin is administered as an intravenous infusion every three weeks. Once administered, it reaches its peak concentration (Cmax) in approximately 0.78 hours, with a mean distribution volume of about 11 liters. The drug is cleared from the body over a period, having a mean half-life (t 1/2) of 12-14 days, with steady-state typically achieved around the first treatment cycle. Eventually, the drug is broken down into smaller peptides and amino acids like other proteins, with oxidation and demethylation breaking down the payload. In terms of binding, belantamab mafodotin demonstrates a high affinity for BCMA (B-cell maturation antigen), with a Kd (dissociation constant) of approximately 1 nM, ensuring precise targeting and efficient drug delivery. This antibody is uniquely afucosylated, meaning it lacks fucose sugar residues. This fucosylation enhances its binding affinity to the FcRIIIa receptor found on immune cells, which significantly boosts its ability to promote antibody-dependent cell-mediated cytotoxicity (ADCC), a crucial mechanism in its therapeutic action. This drug is given every three weeks through IV with a long duration of action, though some minor setbacks exist with a narrow therapeutic index needing consistent monitoring and keratopathy risk or severe eye damage[16,17].
4.4. Enhertu (Trastuzumb Deruxtecan)
Trastuzumab Deruxtecan (T-DXd, also known as DS-8201) marks a notable advancement in treating metastatic breast cancer, especially in patients with HER2-positive (HER2+) tumors. As an antibodydrug conjugate (ADC), T-DXd uniquely combines a HER2-targeted antibody with a potent cytotoxic drug, ensuring precise chemotherapy delivery to HER2-overexpressing cancer cells while minimizing impact on healthy tissues. Its mechanism involves the anti-HER2 antibody’s specificity in binding to HER2expressing cancer cells, followed by internalization and release of the cytotoxic drug within the cell, leading to focused cell death. This method boosts antitumor activity and lessens exposure of non-target cells to the toxic agent, thereby potentially reducing side effects. Trastuzumab deruxtecan employs a dual-action approach in targeting HER2-positive cancer cells, blending targeted delivery with a potent payload release. The targeted delivery aspect involves a humanized anti-HER2 IgG1 antibody that specifically binds to the HER2 protein on cancer cells’ surfaces, thereby minimizing damage to healthy tissues. This antibody fragment’s humanized nature closely mimics human antibodies, further lowering the risk of adverse reactions. In terms of payload release, the deruxtecan (DXd) molecule, a powerful topoisomerase I inhibitor, is linked to the antibody with a cleavable peptide linker. Once the antibody binds to HER2, the ADC is internalized by the cancer cell, where lysosomal enzymes then cleave the linker, releasing DXd inside the cancer cell. The released DXd impedes DNA replication by inhibiting topoisomerase I, leading to DNA damage and triggering apoptosis in cancer cells[3,4,18].
Clinically, T-DXd has demonstrated significant efficacy, as seen in the phase 2 DESTINY-Breast01 trial, particularly for HER2+ metastatic breast cancer patients who had undergone multiple previous anti-HER2 therapies. It showed an impressive objective response rate of 60.9 percent and a median progression-free survival of 16.4 months, underpinning its accelerated approval for this group. Moreover, T-DXd’s effectiveness extends beyond strictly HER2+ breast cancer, showing potential in treating tumors with low HER2 expression, thereby broadening the spectrum of patients who could benefit from this therapy. However, there do exist some safety concerns including pneumonitis, interstitial lung disease, and embryo-fetal toxicity. Indications for this drug include unresectable HER2-positive breast cancer after previous anti-HER2 therapy administration. The DXd payload has a high plasma protein binding of 97 percent showing some potential interactions with target cells, with a drug half-life of 5.8 days and a dosing interval of every 3 weeks[18,19].
4.5. Trodelvy (Sacituzumab Govitecan)
Sacituzumab govitecan, branded as Trodelvy, is a groundbreaking antibody-drug conjugate (ADC) in the treatment of solid tumors, notably metastatic triple-negative breast cancer (mTNBC) and urothelial cancers. Gaining accelerated U.S. approval in April 2020 for mTNBC patients with prior treatments, marks a significant milestone in oncology, particularly for challenging cancer subtypes. Currently under phase III development for broader applications in breast cancer and phase II trials for urothelial cancer, its scope extends to various malignancies, indicating its versatility[20]. Sacituzumab govitecan targets Trop-2, a cell surface antigen prevalent in these cancers, and employs SN-38, a potent topoisomerase I inhibitor, delivering the cytotoxic agent precisely within tumor cells. SN-38 is an active metabolite in Irinotecan and this covalently links to the antibody with a hydrolyzable CL2A linker to ensure proper cytotoxic activity at the tumor site. The potent SN-38 internalizes Trop-2 expressing cells, halting DNA replication through inhibition of topoisomerase I, leading to cancer cell death. Recent studies indicate that SN-38 may also have additional celldisrupting mechanisms with specific interference of FUBP1 protein halting cancer gene expression. This delivery mechanism provides up to 136-fold more SN-38 than pure irinotecan delivery, potentially overcoming cancer cell resistance mechanisms like Rad51mediated DNA repair in TNBC and other solid tumors. There are also opportunities for combination therapies with resistance linked to ABCG2 efflux transporter expression in which combination with ABCG2 inhibitors may improve efficacy and overcome resistance[7,20,21].
This targeted mechanism, coupled with its high drug-to-antibody ratio, leads to effective tumor cell destruction while minimizing systemic exposure, showcasing the shift toward precision medicine in oncology. Sacituzumab govitecan, at a 10 mg/kg dose, shows a distribution volume of 0.045 L/kg and a clearance rate of 0.002 L/h/kg, with a mean halflife of 16 hours, extending to 18 hours for the free SN-38 component. Metabolism by UGT1A1, particularly influenced by the UGT1A1*28 allele, can affect the risk of neutropenia. While mild hepatic impairment doesn’t significantly impact its pharmacokinetics, the effects in moderate to severe cases are less clear. With minimal renal elimination and potential interactions with UGT1A1 modifiers, careful monitoring is advised, especially in patients with hepatic or renal concerns. Despite its significant therapeutic benefits, its safety profile, including a black box warning for neutropenia and diarrhea, necessitates careful patient monitoring[20,21].
4.6. Polivy (Polatuzumab Vedotin)
Diffuse large B-cell lymphoma (DLBCL), the most common non-Hodgkin lymphoma subtype accounts for 25 percent to 40 percent of cases worldwide. This high-grade B-cell lymphoma, characterized by rapidly expanding nodal or extranodal tumors, has seen significant therapeutic advancements. The addition of rituximab to CHOP chemotherapy has notably improved remission rates in nearly 60% of DLBCL patients. Despite efforts with agents like lenalidomide and ibrutinib, overall survival (OS) and progression-free survival (PFS) enhancements have been limited. However, a 2021 study demonstrated improved PFS with polatuzumab vedotin (pola-RCHP) in intermediate/high-risk DLBCL patients. DLBCL treatment still faces challenges, with a 5-year relapse rate post-treatment between 20% and 30% and OS rates of 55% to 65%. For relapsed/refractory (R/R) DLBCL, salvage chemotherapy followed by autologous hematopoietic stem cell transplantation (auto-HSCT) has been standard since 1995, offering remission in 30% to 40% of patients. Polatuzumab vedotin (POLIVY) and polatuzumab vedotin-piiq, both anti-CD79b ADCs combining a monoclonal antibody with monomethyl auristatin E (MMAE), have emerged as prominent treatments, especially for R/R DLBCL[22,23].
These therapies, targeting CD79B-expressing Bcells and inducing cell death via cytotoxic payload MMAE, have been further developed and approved for use in combination with bendamustine and rituximab (BR), reflecting the shift towards precision medicine and targeted therapies in DLBCL. POLIVY is designed to target dividing B cells especially cancerous, CD79 b-expressing cells. The antibody binds to this cell surface marker present on these cells, and POLIVY is internalized where it releases the payload MMAE, a potent anti-mitotic agent that disrupts microtubule functionality leading to cell death. The linker involved is a protease cleavable linker (mcvc-PAB) that releases the payload once internalized to the cell. Despite over 50 percent of DLBCL patients achieving a cure, treatment efficacy varies among subtypes and sites, with some responding better to Burkitt’s lymphoma regimens than CHOP-R. Relapsed or refractory disease typically surfaces within two years of diagnosis, necessitating intensive treatments. Patients with doubleor triple-hit lymphomas have poorer prognosis, and while R-CHOP remains foundational, a complete cure remains elusive. Second-line regimens like DHAP, ESHAP, mini-BEAM, and ICE, particularly when combined with rituximab, have shown improved outcomes, highlighting the ongoing evolution in DLBCL management strategies. After the initial 1.8 mg/kg dose of polatuzumab vedotin, the mean maximum concentrations (Cmax) of antibodyconjugated MMAE and unconjugated MMAE are 803 ± 233 ng/mL and 6.82 ± 4.73 ng/mL, respectively, with their respective area under the concentrationtime curve (AUCinf) values being 1860 ± 966 day x ng/mL and 52.3 ± 18.0 day x ng/mL. MMAE, the drug’s active component, shows a plasma protein binding of 71 percent to 77 percent and is primarily metabolized by CYP3A4/5, leading to its excretion largely in feces and partially in urine; polatuzumab vedotin itself undergoes catabolism into small peptides, amino acids, and related catabolites. The terminal half-life of polatuzumab vedotin is approximately 12 days at Cycle 6, with a predicted clearance of 0.9 L/day, while unconjugated MMAE has a shorter half-life of around four days[22–24].
4.7. Mylotarg (Gemtuzumab Ozogamicin)
Gemtuzumab ozogamicin (GO), an antibody-drug conjugate targeting CD33 linked to the toxin calicheamicin, was initially FDA-approved in 2000 for CD33+ acute myeloid leukemia (AML). However, it was withdrawn due to efficacy concerns and increased hepatotoxicities, including hepatic venoocclusive disease (VOD), as observed in the SWOG0106 study. Subsequent research, including the ALFA-0701 study, showed that lower, fractionated doses of GO combined with standard chemotherapy (SC) led to improved survival and reduced toxicities. A meta-analysis across five phase 3 studies confirmed these benefits, with a 3 mg/m2 dose associated with lower hepatoxicity than the 6 mg/m2 dose, culminating in GO’s 2017 reapproval for treating CD33+ AML[13]. Gemtuzumab ozogamicin, a recombinant humanized IgG4 kappa antibody conjugated with calicheamicin, a cytotoxic antitumor antibiotic derived from Micromonospora echinospora ssp. calichensis targets the CD33 antigen found on leukemic myeloblasts in most acute myeloid leukemia (AML) patients. About 50 percent of the antibody carries 4-6 moles of calicheamicin per mole of antibody, focusing on inhibiting growth and inducing cell death in cancerous cells. Initially approved by the FDA in 2000 for CD33-positive AML in the first relapse in patients 60 years or older not suited for cytotoxic chemotherapy, it was withdrawn in 2010 due to safety concerns and insufficient clinical benefit. However, following reassessment and adaptation of dosing regimens, it was reapproved in 2017 for adults with newly diagnosed CD33-positive AML, in combination with chemotherapy or as monotherapy, and for patients aged 2 years and older with relapsed or refractory CD33-positive AML[13,26].
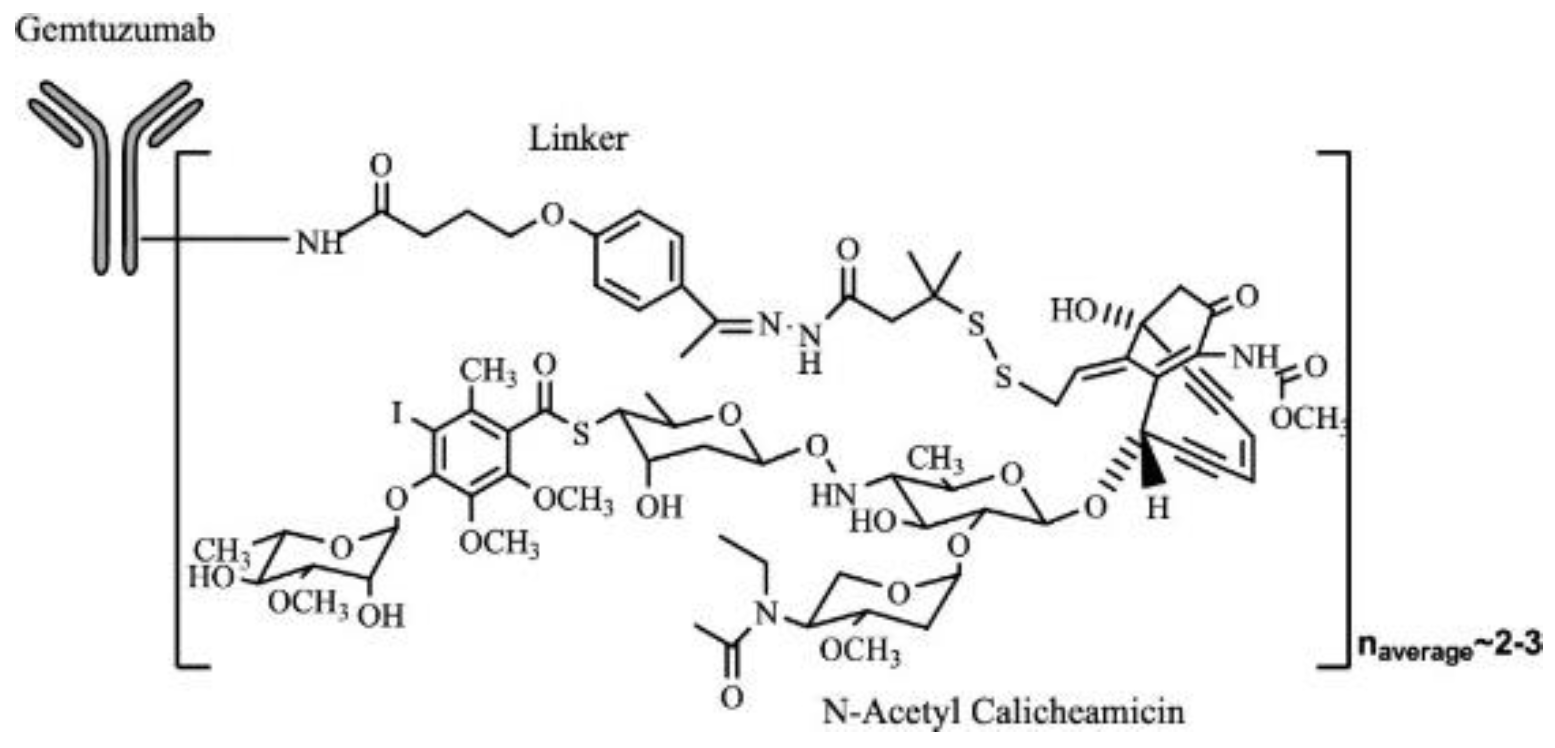
Figure 9.
A representative example of Mylotarg molecularly with Gemtuzumab monoclonal antibody bound with a linker to small molecule N-acetyl calicheamicin. This follows a similar pattern of ADC structures represented through a monoclonal antibody bound to a linker and cytotoxic component[25].
Figure 9.
A representative example of Mylotarg molecularly with Gemtuzumab monoclonal antibody bound with a linker to small molecule N-acetyl calicheamicin. This follows a similar pattern of ADC structures represented through a monoclonal antibody bound to a linker and cytotoxic component[25].
4.8. Besponsa (Inotuzumab Ozogamicin)
Acute lymphoblastic leukemia (ALL) impacts both children and adults, with B-cell lineage ALL notably prevalent in adult cases. This drug targets leukemic blasts expressing CD22 antigen, present in over 90% of patients. Inotuzumab is a humanized igG4 monoclonal antibody and ozogamicin is derived from calicheamicins, a potent antitumor antibiotic targeting DNA in the minor groove, inducing strand scission. After internalization to leukemic cells, it undergoes hydrolysis by endosomes and lysosomes, releasing the active ozogamicin causing apoptosis in the nucleus. Although high complete remission rates are observed in adults newly diagnosed with B-cell ALL, the prognosis is often poor due to frequent relapses, underscoring the need for new treatments. CD22, a B-cell-specific antigen present in most B-cell ALL cases, has emerged as a vital therapeutic target. Inotuzumab ozogamicin (Besponsa) is an antibody-drug conjugate (ADC) targeting CD22, designed to deliver the cytotoxic agent Calich-DMH to CD22-expressing B-cells. This ADC has demonstrated dose-dependent inhibition of B-cell ALL cell lines and anti-tumor efficacy in preclinical models, with continuous exposure being more effective than pulse exposure, influencing the fractionated dosing approach in clinical trials[27,28].
In patients with relapsed/refractory B-cell ALL, inotuzumab ozogamicin administered at 1.8 mg/m2/cycle has shown promising results.ALL is a rapidly progressing cancer of the bone marrow characterized by high mortality rates and poor response to standard chemotherapies in relapsed conditions. In a randomized trial, inotuzumab ozogamicin demonstrated higher percentages of patients achieving longer periods of complete remission with no evidence of disease compared to patients receiving alternative chemotherapy. However, it carries risks like veno-occlusive liver disease (VOD) or sinusoidal obstruction syndrome (SOS) and potential QT interval prolongation, especially in patients with a predisposition to cardiac issues, concurrent medications prolonging QT interval, or electrolyte imbalances. The development of anti-drug antibodies (ADAs) during treatment has been noted but appears to have minimal impact on drug clearance. Inotuzumab ozogamicin’s pharmacokinetics involves a 12.3-day half-life and follows a two-compartment model with both linear and time-dependent clearance components, illustrating its complex interaction within the body[27–29].
4.9. Lumoxiti (Moxetumomab Pasudotox)
Moxetumomab pasudotox-tdfk (LUMOXITI), developed by MedImmune and AstraZeneca, is an anti-CD22 recombinant immunotoxin for hairy cell leukemia (HCL). Consisting of an anti-CD22 monoclonal antibody fragment fused with a fragment of Pseudomonas exotoxin A (PE38), it binds to CD22 on malignant B-cells, inducing apoptosis through the ADP ribosylation of elongation factor-2. After being engulfed into the cell, the toxin takes on conformational changes, inactivating the eukaryotic translation elongation factor(eEF-2) by transferring ADP from NAD to a modified histidine at position 715 on eEF-2, leading to inhibited protein synthesis in the cell and eventually cell death. FDAapproved for adult patients with relapsed or refractory HCL after at least two prior therapies, moxetumomab pasudotox is administered intravenously and has demonstrated efficacy in rapidly depleting circulating CD19+ B cells and tumor reduction. Despite its success, the drug has boxed warnings for capillary leak syndrome and hemolytic uremic syndrome. Optimized from CAT-3888, it showcases improved affinity for CD22 and is a testament to targeted therapy advancements in addressing relapsed/refractory HCL, meeting a critical unmet need[30,31].
Its pharmacokinetic properties show doseproportional concentration, steady-state peak plasma concentration, and elimination through proteolytic degradation, unaffected by factors like age, gender, and mild organ impairments. It is particularly targeted towards adults with HCL characterized by an accumulation of abnormal B lymphocytes exhibiting “hairy projections” on their surface. MxP is administered intravenously over 30 minutes on days 1, 3, and 5 of a 28-day cycle, with a recommended dosage of 0.04 mg/kg. Treatment may consist of up to 6 cycles, depending on the patient’s response and tolerance. The drug’s metabolism in patients with moderate to severe hepatic or renal impairment remains unclear and the safety profile includes warnings for adverse renal reactions. This drug was discontinued by AstraZeneca in 2023 for precursor cell lymphoblastic leukemia/lymphoma, non-Hodgkin’s lymphoma, and chronic lymphocytic leukemia due to low clinical uptake since 2018 when it was approved by the FDA. This was due to the complexity of administration, the need for additional therapies to reduce toxicity, and the required safety monitoring consistently not due to safety or efficacy concerns. This drug target, if potentially enhanced by data analytical models and drug design could potentially pose great therapeutic capacities if re-investigated and optimized in the future[30–33].
4.10. Zynlonta(Loncastuximab Tesirine)
CD19, a key antigen in B-cell malignancies, presents an ideal target for antibody-drug conjugates (ADCs), particularly in treating relapsed or refractory diffuse large B-cell lymphoma (DLBCL), the most common subtype of B-cell non-Hodgkin lymphoma. Standard front-line treatment is R-CHOP resulting in a cure 60-65% of the time. Many patients that are r/r have poor prognoses, necessitating alternative therapies as approximately 40% of frontline treatments fail. Loncastuximab tesirine, a novel ADC comprising a humanized igG1, anti-CD19 monoclonal antibody conjugated to a pyrrolobenzodiazepine dimer warhead, was approved by the FDA in 2021 for r/r DLBCL after two or more systemic therapies[34]. It works by binding to CD19 on B-cells, facilitating internalization and release of the cytotoxic payload SG3199, which forms DNA crosslinks, leading to cell death. This drug has exhibited the bystander-killing effect meaning it also induces cell death in neighboring CD19Cells showing potentially high therapeutic efficacy. This therapy addresses the critical need for effective treatments in r/r DLBCL, where standard therapies and CAR T-cell options have limitations in efficacy, safety, and accessibility[34].
Loncastuximab Tesirine shows antitumor activity against malignant B-cells, but higher exposure during the first therapy cycle is associated with an increased occurrence of Grade 2 adverse reactions. Administered intravenously, loncastuximab tesirine has a Cmax of 2995 g/L in Cycle 1 and 3155 g/L in Cycle 2, with an AUC ranging from 15,245 to 22,823 g*day/L. Its average distribution volume is around 7.11 liters, and while protein binding data are lacking, the antibody part is broken down into peptides, and SG3199 is metabolized by CYP3A4/5. SG3199’s excretion pathways in humans are not fully understood but are believed to be minimally renal. The halflife of loncastuximab tesirine-lpyl ranges from 7.06 to 12.5 days, with its clearance decreasing over time. However, higher doses can lead to increased toxicity and potential discontinuation of the drug[34–36].
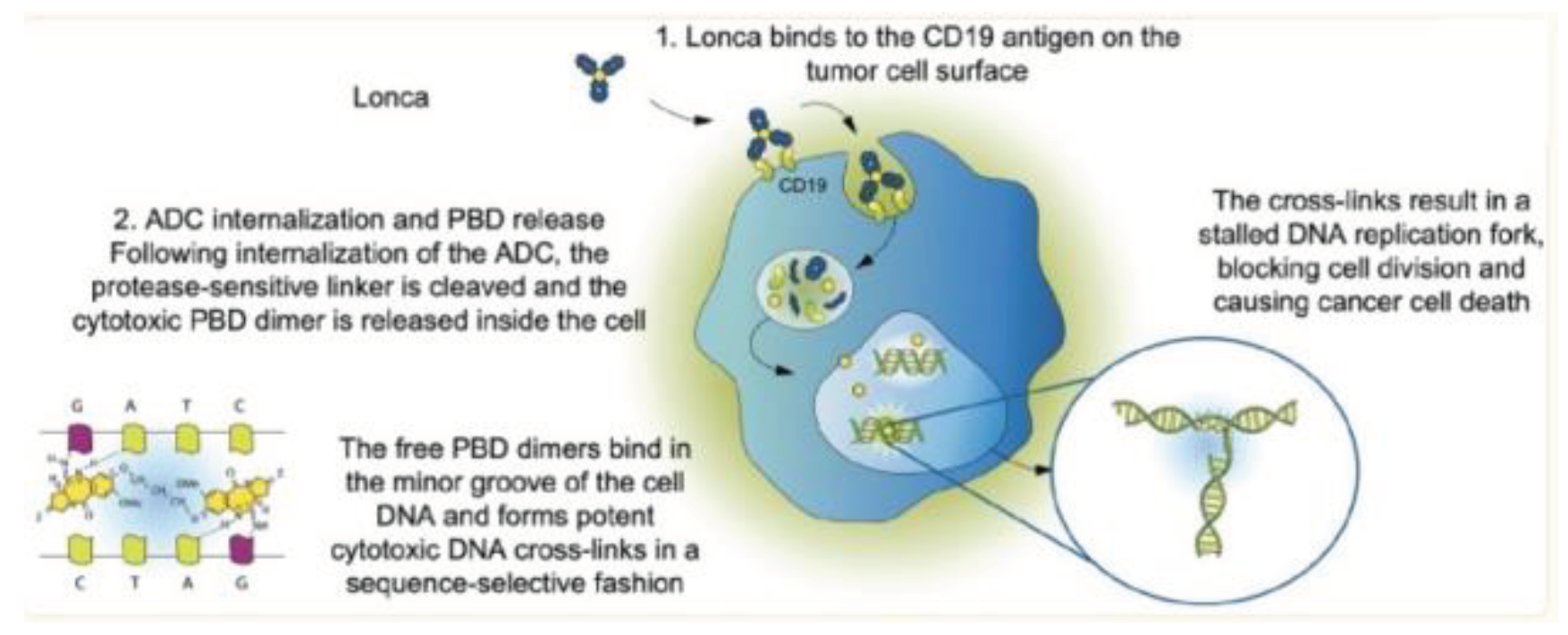
Figure 10.
Mechanism of action of Loncastuximab Tesirine. Lonca binds to the CD19 antigen on the tumor cell surface the ADC is internalized and the cytotoxic payload is released with free PBD dimers binding the minor groove of cell DNA causing apoptosis[35].
Figure 10.
Mechanism of action of Loncastuximab Tesirine. Lonca binds to the CD19 antigen on the tumor cell surface the ADC is internalized and the cytotoxic payload is released with free PBD dimers binding the minor groove of cell DNA causing apoptosis[35].
5. Discussion and Further Analysis
The rapid advancement in cancer therapeutics, particularly the development of antibody-drug conjugates (ADCs), marks a significant stride in oncology. These sophisticated drugs utilize monoclonal antibodies to deliver cytotoxic payloads directly to cancer cells, offering a targeted approach to cancer treatment. This specificity in targeting cancer cells reduces collateral damage to healthy tissues, thus optimizing therapeutic indexes and potentially improving disease outcomes. ADCs have demonstrated high therapeutic capabilities, especially in hematological disorders, including various leukemia derivatives. Their potential for curative treatment in these cancers is increasingly evident. However, expanding their application to solid tumors presents additional challenges. Solid tumors’ complex three-dimensional structure complicates drug penetration and binding, necessitating higher drug concentrations, which may lead to increased side effects, similar to those observed in traditional chemotherapeutics. The engineering of ADCs plays a crucial role in overcoming these challenges. Advances in linker technology are vital, balancing the need for stability in the bloodstream with effective payload release within the cancer cell. Additionally, the development of novel cytotoxic payloads, distinct from traditional chemotherapy agents, may offer enhanced efficacy and fewer side effects. Future research and development in ADC technology are geared towards expanding their scope and refining their efficacy. Efforts to enhance antibody selectivity and binding efficiency, coupled with innovative payload delivery methods, promise more personalized and targeted cancer treatments. The goal is to maximize cancer cell destruction while mitigating damage to healthy cells and reducing systemic side effects. In conclusion, the evolution of ADCs signifies a shift towards more precise and targeted cancer treatments. While treating solid tumors remains a challenge, ongoing innovations in ADC design and functionality are paving the way for more effective and safer therapies. This progress in ADC technology holds the potential to transform cancer treatment paradigms, offering hope for more effective curative options across a broader range of cancers.
Contributions
C.K. conceived the review article, C.K., O.F., A.K. collected data, organized figures, and performed all meta-analyses of the literature provided in the paper. C.C. contributed to the oversight of this article as the principal investigator along with C.K., provided edits, and helped write portions of this article.
Acknowledgments
The authors are grateful to the various research studies, clinical trials, and individuals behind the work of tackling antibody drug conjugates and their respective disease conditions. The authors would like to extend their gratitude to the entire Research and Development team at Nexabio Venture Solutions for their guidance, support, and contributions to the overall team dynamics.
Competing Interests
For C.K., O.F., A.K., C.C., and authors that are affiliated with Nexabio Venture Solutions LLC: Nexabio Venture Solutions LLC is a corporation focused on AI-driven drug discovery. We specialize in repurposing abandoned therapeutics with an emphasis on developing computational and predictive modeling for enhanced understanding of diseases and drugs. Nexabio Venture Solutions LLC holds proprietary algorithms in the field of drug repurposing for several disease states and drug targets. The authors are engaged in creating and applying AI models to facilitate drug discovery and to provide greater insights into a broad array of conditions, including metabolic diseases. O.F., C.K., C.C., and A.K. are affiliated with Nexabio Venture Solutions LLC, and have contributed to the research and development of the disease/drug models discussed in this review. No other conflicts are reported.
References
- Khongorzul, P., Ling, C. J., Khan, F. U., Ihsan, A. U., & Zhang, J. (2020). Antibody–Drug Conjugates: A Comprehensive Review. Molecular Cancer Research, 18(1), 3–19. [CrossRef]
- Fu, Z., Li, S., Han, S. et al. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Sig Transduct Target Ther 7, 93 (2022). [CrossRef]
- Hoffmann, R. M., Coumbe, B. G. T., Josephs, D. H., et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology, 2017 Nov 20;7(3):e1395127. [CrossRef]
- Peters, C., & Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep, 2015 Jun 12;35(4):e00225. [CrossRef]
- Pharmaceutical Technology. (2023, January 6). Who are the leading innovators in antibody drug conjugates (ADC) cancer therapy for the pharmaceutical industry? https://www.pharmaceutical-technology.com/data-insights/.
- Scott, L. J. (2017). Brentuximab Vedotin: A Review in CD30-Positive Hodgkin Lymphoma. Drugs, 77(4), 435–445. [CrossRef]
- DrugBank. (n.d.). Brentuximab vedotin: Uses, Interactions, Mechanism of Action. Retrieved from https://go.drugbank.com/drugs/DB08870.
- Maximiano, S., Magalh˜aes, P., Guerreiro, M. P., & Morgado, M. (2016). Trastuzumab in the Treatment of Breast Cancer. BioDrugs, 30(2), 75–86. [CrossRef]
- Dhillon, S. (2014). Trastuzumab emtansine: a review of its use in patients with HER2positive advanced breast cancer previously treated with trastuzumab-based therapy. Drugs, 74(6), 675–686. https://doi.org/10.1007/innovators-antibody-drug-conjugates-adc-cancers-4t0h2e6r5a-p0y1-4p-h0a2r0m1a-c0eutical.
- Riccardi, F., Dal Bo, M., Macor, P., Toffoli, G. A comprehensive overview on antibodydrug conjugates: from the conceptualization to cancer therapy. Front Pharmacol, 2023 Sep 18;14:1274088. [CrossRef]
- Shastry, M. et al. Rise of Antibody-Drug Conjugates: The Present and Future. Am Soc Clin Oncol Educ Book 43, e390094 (2023). [CrossRef]
- Wang, Z., Li, H., Gou, L., Li, W., Wang, Y. Antibody–drug conjugates: Recent advances in payloads. Acta Pharmaceutica Sinica B, Volume 13, Issue 10, 2023, Pages 4025-4059. [CrossRef]
- Zhao, M. K. (2017). Monoclonal Antibodies. In Reference Module in Life Sciences, Book chapter: Antibody drug Conjugates beyond cytotoxic payloads. Elsevier. https://www.sciencedirect.com/topics/neuroscience/antibody-drug-conjugate.
- DrugBank. (n.d.). Trastuzumab: Uses, Interactions, Mechanism of Action. Retrieved from https://go.drugbank.com/drugs/DB00072.
- Markham, A. Belantamab Mafodotin: First Approval. Drugs. 2020 Oct;80(15):16071613. [CrossRef]
- DrugBank. (n.d.). Belantamab mafodotin: Uses, Interactions, Mechanism of Action. Retrieved from https://go.drugbank.com/drugs/DB15719.
- Lee, J., & Park, Y. H. Trastuzumab deruxtecan for HER2+ advanced breast cancer. Future Oncol. 2022 Jan;18(1):7-19. [CrossRef]
- DrugBank. (n.d.). Trastuzumab deruxtecan: Uses, Interactions, Mechanism of Action. Retrieved from https://go.drugbank.com/drugs/DB14962.
- Syed, Y. Y. Sacituzumab Govitecan: First Approval. Drugs. 2020 Jul;80(10):10191025. [CrossRef]
- DrugBank. (n.d.). Sacituzumab Govitecan: Uses, Interactions, Mechanism of Action. Retrieved from https://go.drugbank.com/drugs/DB12893.
- Sehn, L. H., Herrera, A. F., Flowers, C. R., et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2020 Jan 10;38(2):155-165. [CrossRef]
- Raqib, M. A., Haseeb, A., Shafique, M. A., et al. (2023) Advances in Polatuzumab Vedotin-PIIQ Therapy: A Review of Treatment Efficacy in Diffuse Large B Cell Lymphoma and High-Grade B Cell Lymphoma, Pediatric Health, Medicine and Therapeutics, 14:, 323-331. [CrossRef]
- Polivy. Available online: www.polivy.com/content/dam/gene/polivy/patient/en_us/rrdlbcl/pdfs/polivy-3l-patient-brochure.pdf Accessed 1 Apr. 2024.
- Swaminathan, M., & Cortes, J. E. Update on the role of gemtuzumab-ozogamicin in the treatment of acute myeloid leukemia. Ther Adv Hematol. 2023 Feb 9;14:20406207231154708. [CrossRef]
- DrugBank. (n.d.). Gemtuzumab ozogamicin: Uses, Interactions, Mechanism of Action. Retrieved from https://go.drugbank.com/drugs/DB00056.
- Al-Salama, Z. T. Inotuzumab Ozogamicin: A Review in Relapsed/Refractory B-Cell Acute Lymphoblastic Leukaemia. Targ Oncol, 13, 525–532 (2018). [CrossRef]
- Yurkiewicz, I. R., Muffly, L., & Liedtke, M. Inotuzumab ozogamicin: a CD22 mAb-drug conjugate for adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Drug Des Devel Ther. 2018 Jul 24;12:2293-2300. [CrossRef]
- DrugBank. (n.d.). Inotuzumab ozogamicin: Uses, Interactions, Mechanism of Action. Retrieved from https://go.drugbank.com/drugs/DB05889.
- Dhillon, S. Moxetumomab Pasudotox: First Global Approval. Drugs. 2018 Nov;78(16):1763-1767. [CrossRef]
- DrugBank. (n.d.). Moxetumomab pasudotox: Uses, Interactions, Mechanism of Action. Retrieved from https://go.drugbank.com/drugs/DB12688.
- Ryan, C. (2023, January 13). AstraZeneca to Discontinue Moxetumomab Pasudotox in US for Hairy Cell Leukemia. OncLive. https://www.onclive.com/view/astrazeneca-to-discontinue-moxetumomab-pasudotox-in-u.
- Medthority. (2023, January 14). AstraZeneca to discontinue selling Lumoxiti in the US due to low clinical uptake. Retrieved from https://www.medthority.com/news/2023/1/astrazeneca-to-discontinue-selling-lumoxiti-in-the-us.
- Xu, B. Loncastuximab tesirine: an effective therapy for relapsed or refractory diffuse large B-cell lymphoma. Eur J Clin Pharmacol. 2022 May;78(5):707-719. [CrossRef]
- Furqan, F., & Hamadani, M. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma: a review of clinical data. Ther Adv Hematol. 2022 Mar 22;13:20406207221087511. [CrossRef]
- DrugBank. (n.d.). Loncastuximab tesirine: Uses, Interactions, Mechanism of Action. Retrieved from https://go.drugbank.com/drugs/DB16222.
- MarketsandMarkets. (2023). Antibody Drug Conjugates (ADC) Market by Product, Linker Type, Payload Type, Target, Disease, Region Global Forecast to 2028 (Report No. BT 8808). https://www.marketsandmarkets.com/Market-Reports/antibody-drug-conjugates-market-122857391.html.
- Haddaway, N. R., Page, M. J., Pritchard, C. C., & McGuinness, L. A. (2022). PRISMA2020: An R package and Shiny app for producing PRISMA 2020-compliant flow diagrams, with interactivity for optimised digital transparency and Open Synthesis Campbell Systematic Reviews, 18, e1230. [CrossRef]
- Page, M. J., McKenzie, J. E., Bossuyt, P. M., Boutron, I., Hoffmann, T. C., Mulrow, C. D., et al. (2021). The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ, 372:n71. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



