
Submitted:
09 April 2024
Posted:
10 April 2024
You are already at the latest version
Abstract
Chronic obstructive pulmonary disease (COPD) is a progressive lung disease that is commonly considered to be a potent driver of non-small cell lung cancer (NSCLC) development and related mortality. A growing body of evidence supports the role of the immune system, mainly played by alveolar macrophages (AMs), as key axes regulating the development of COPD or NSCLC phenotypes in response to harmful agents. MicroRNAs (miRNAs) are small non-coding RNAs that influence most biological processes and interfere with several regulatory pathways. The purpose of this study was to assess miRNA expression patterns in patients with COPD, NSCLC, and ever- or never-smoker controls to explore theirinvolvment in smoking-related diseases. Bronchoalveolar lavage (BAL) specimens were collected from a prospective cohort of 43 sex-matched subjects to determine expression of hsa-miR-223-5p, 16-5p, 20a-5p, -17-5p, 34a-5p and 106a-5p by RT-PCR. In addition, bioinformatic analysis of miRNA target genes linked to cancer was performed. Distinct and common miRNA expression levels were identified in each pathological group suggesting their possible role as an index of NSCLC or COPD microenvironment. Moreover, we identified miRNA targets linked to carcinogenesis using in silico analysis. In conclusion, this study identified miRNA signatures in AMs, allowing us to understand the molecular mechanisms underlying smoking-related conditions and potentially providing new insight for diagnosis or pharmacological treatment.
Keywords:
miRNA
; copd
; alveolar macrophages
; smoking-related lung diseases
; lung cancer
1. Introduction
Chronic obstructive pulmonary disease (COPD) is a fatal disease with high mortality worldwide placing a heavy economic burden on the healthcare systems [1,2]. It is an heterogenous condition characterized by progressive deterioration of lung function over time and is generally associated with lung inflammation triggered by harmful particles or gases (Pauwels et al., 2004; Kotlyarov, 2023; Urbanek et al., 2016; D’Agostino et al., 2002, 2005; Rouget et al., 2004; Gallelli et al., 2003). Besides that, COPD is commonly considered a potent driver of non-small cells lung cancer (NSCLC) development and related mortality, beyond a common etiology [10,11]. Indeed, lung cancer is up to five times more frequent in smokers with airflow obstruction than in those with normal lung function, and patients with COPD have twice the risk of cancer diagnosis [12]. Inflammation and tumorigenesis are closely linked, and chronic inflammation could lead to an increased rate of cell division, and to the chance of mutations and carcinogenesis [13,14,15,16]. Moreover, a growing body of evidence supports the role of the innate and adaptive immune systems, mainly played by alveolar macrophages (AMs), as key axes that regulate the development of COPD or NSCLC phenotypes in response to harmful agents [17,18,19,20,21,22,23] Despite this increased attention, the mechanistic nature of both diseases remains unclear, making novel biomarkers necessary to improve the prediction of cancer risk in subjects with COPD. Recently, epigenetic changes, including DNA methylation, covalent histone modifications, and microRNAs (miRNAs) expression, have been reported to play important roles in lung cancer and COPD onset [24,25]. Indeed, miRNAs are small non-coding, single stranded RNA molecules, capable of binding to mRNA sequences resulting in gene silencing via mRNA translational repression or target degradation. miRNAs expression can influence most biological processes and interfere with several regulatory pathways, playing a crucial role in the development of both innate and adaptive immune systems [26,27,28,29,30]. In addition, several authors identified serum miRNAs which could serve as biomarkers to help identify the risk of developing cancer in COPD patients [31,32]. In the present study, we focused on miRNAs that have recently been shown to influence immune system regulation or cancer hallmarks, such as cell proliferation and immunosuppression. Notably, hsa-miR-34a-5p has been reported to be dysregulated in various cancers and considered as a tumor suppressor because of its synergistic effect with p53 [33]. Hsa-miR-miR-223–5p was found down-regulated in the bronchial airway of lung cancer patients and regulated the malignant phenotypes of lung squamous cell carcinoma cells by pairing with OTX1 [34]. Similarly, hsa-miR-16-5p is a tumor inhibitor and a new biomarker for immune checkpoint inhibitors-dependent immunotherapy in lung adenocarcinoma by regulating the PD-L1 expression [35]. Furthermore, different authors showed that hsa-miR-106a-5p is overexpressed in the serum of patients with NSCLC [36] while hsa-miR-20a-5p and hsa-miR-17-5p have been reported as tumor suppressors [37,38,39]. However, miRNAs could be shared in COPD and cancer targeting candidate mRNA families implicated in the pathogenesis of both diseases [40]). Therefore, to obtain miRNA signatures between COPD and NSCLC that could be used to distinguish between smoking-related diseases, we evaluated miRNAs expression in AMs of bronchoalveolar lavage (BAL) from patients with COPD, NSCLC, and ever- or never- smoker controls.
2. Materials and Methods
2.1. Study Population
We performed an observational clinical study on 43 age- and sex-matched subjects, aged 18 years or older, referred to the “Mater Domini” Hospital in Catanzaro, Italy, with a suspected diagnosis of pulmonary neoplasia, undergoing routine bronchoscopy and BAL. This study is a part of the clinical trial recorded at clinicaltrials.gov (NCT04654104) and approved by the local Ethics Committee “Calabria Centro” (n. 361 of 22 October 2020). This study was conducted in compliance with the Institutional Review Board/Human Subjects Research Committee, the Declaration of Helsinki, and the Guidelines for Good Clinical Practice criteria. Before the beginning of the study, all the enrolled patients or legal guardians signed the informed consent and underwent spirometry according to international guidelines [41]. At enrollment patients’ demographics and clinical and social history were obtained by physicians: age, gender, smoking history (pack/years), spirometry data, comorbidities, and pharmacological treatments. The exclusion criteria were active pulmonary infections, autoimmune diseases, extrapulmonary neoplasia, or other airflow obstruction, such as asthma or bronchiectasis, or those who did not sign the informed consent. The main clinical and pathological characteristics of cohorts are summarized in Table 1. The enrolled subjects were classified into four groups according to their clinical and pathological (bronchoscopy-guided) diagnoses:1) healthy never-smokers control (“HNS”; n = 9); 2) healthy ever-smokers control (“HS”; n = 11); 3) smokers with Global Initiative for Obstructive Lung Diseases (GOLD) stage 1–4 (“COPD,” n =11); 4) non-small cell lung cancer (“NSCLC”; n=12). Within each group, subjects were comparable with respect to COPD severity and/or lung cancer histology. Indeed, only those with NSCLC were enrolled among the subject’s presenting cancer. The most frequent comorbidities were hypertension and diabetes mellitus; bronchodilators were the most common medications used.
2.2. Bronchoalveolar Lavage
All subjects underwent bronchoscopy for clinical indications, and BAL was obtained with a bronchoscope according to international guidelines (41 The procedure involved premedication (20 mg codeine per os) and local anesthesia of the larynx and lower airways (0.5% tetracaine in the oropharynx, 8 cc 0.5% tetracaine in lower airways). Transcutaneous oxygen saturation was monitored continuously through an oximeter with a finger probe. BAL was obtained by instilling a total volume of 200 ml of sterile isotonic saline solution (37 °C) in the right middle lobe. Specifically, fraction I, returned after instilling 50 ml of saline, and fraction II, returned after instilling 3 × 50 ml of saline, was collected in a siliconized specimen trap and immediately kept on ice. BAL was filtered through nylon gauze and centrifuged (10 min at 400 g at 4 °C). The cell pellet was washed twice, counted, and resuspended in PBS. Cells were counted in a Bürker chamber and cell yield was determined by total cell number per fraction/total recovered volume per fraction. Cell viability was determined by Trypan blue and differential cell count was performed by microscopy on the cytospin slide after staining with QUICK-DIFF KIT (Reastain®, Reagena Toivala, Finland); at least 100 cells were counted. AMs were identified as greater than 95% by morphology.
2.3. Clinical Biochemistry Assays and Real Time PCR (RT-PCR)
To extract miRNAs (total RNA) from AMs from BAL, we adopted the best method available, the miRNeasy Mini kit (QIAGEN, Hilden Germany) according to manufacturer instructions. RNA was eluted at a volume of 15 µL with RNAse free water. The qubit RNA Integrity and Quality (IQ) assay (catalog number Q33222) was performed to check for RNA degradation, then microRNAs were quantified with Qubit™ microRNA assay kit (catalog number Q 32881). For both kits, Qubit 4 fluorometer (Thermo Fisher Scientific,Waltham, MA, USA) (serial number 2322618032114) was used.
The expression levels of hsa-miR-223-5p, 16-5p, 20a-5p, -17-5p, 34a-5p and 106a-5p was performed through TaqMan™ Advanced miRNA Assay RT-PCR, following the Thermo Fisher Scientific procedures (Waltham, MA, USA) selecting U6 snRNA as housekeeping miRNA (Gallelli et al., 2019). The sequences used were:
- hsa-miR-223-5p: CGUGUAUUUGACAAGCUGAGUU; (Assay ID 477984_mir)
- hsa-miR-16-5p: UAGCAGCACGUAAAUAUUGGCG; (Assay ID 477860_mir)
- hsa-miR-20a-5p: UAAAGUGCUUAUAGUGCAGGUAG; (Assay ID 478317_mir)
- hsa-miR-17-5p: CAAAGUGCUUACAGUGCAGGUAG; (Assay ID 478447_mir)
- hsa-miR-34a-5p: UGGCAGUGUCUUAGCUGGUUGU; (Assay ID 478048_mir)
- hsa-miR-106a-5p: AAAAGUGCUUACAGUGCAGGUAG; (Assay ID 478225_mir)
- U6 snRNA: GTGCTCGCTTCGGCAGCACATATACTAAAATTGGAACGATACAGAGAAGATTAGCATGGCCCCTGCGCAAGGATGACACGCAAATTCGTGAAGCGTTCCATATTTT; (AssayID: 001973).
Briefly, 5–10 ng of total RNA was subjected to reverse transcription polymerase chain reaction using the TaqMan MicroRNA Reverse Transcription kit for all the miRNA targets chosen, according to manufacturer’s instructions. The thermocycling conditions were as follows:42 °C for 30 min, 85 °C for 5 min 85 °C and 4 °C for 5 min. RT-PCR was performed using the TaqMan™ Fast Advanced Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol and QuantumStudio5TM Real-Time PCR Systems. The thermocycling conditions were as follows:95°C for 10 min and 40 cycles of 15 s at 95 °C, followed by 1 min at 60 °C. Nine biological replicates for the HNS group, eleven for HS, eleven for NSCLC, and twelve for COPD were analyzed, and all samples were run in triplicate; after the achievement of the RT-PCR, the cycle threshold (Ct) of the reactions was determined. Gene expression stability was evaluated using RefFinder, a web tool integrating geNorm, Normfinder, BestKeeper and the comparative ΔΔCt method. Data from all RT–PCR experiments and miRNA expression was analyzed by normalizing to the endogenous miRNA control applying the comparative 2ˆ-ΔΔCt method, where ΔCt = CtmiRNA – Ct housekeeping miRNA, whereas the relative quantification of differences in expression was conducted with ΔΔCt = ΔCtHS/COPD/NSCLC − ΔCtHNS.
2.4. In Silico Prediction of hsa-miRs Target Genes
To identify genes as targets of hsa-miR-223-5p, 16-5p, 20a-5p, -17-5p, 34a-5p and 106a-5p linked to NSCLC and/or COPD, we performed in silico analysis. The in-silico identification of the target genes was performed using DIANA Tools (http://diana.imis.athena-innovation.gr/DianaTools/index.php, accessed on 8 January 2024) and miRTargetLink 2.0 (https://ccb-compute.cs.uni-saarland.de/mirtargetlink2 accessed on 10 January 2024) databases. These databases were used to check which miRNA target genes were already validated experimentally. Possible biochemical pathways were checked using the GeneCard database (https://www.genecards.org/ accessed on18 January 2024), allowing genomic, proteomic, transcriptomic, genetic, and functional information on all known and predicted human genes.
2.5. Statistical Analysis
Unless specified, all data are expressed as mean ± standard deviation (SD). The one-way ANOVA test followed by Tukey Multiple comparison test was used to evaluate the differences between the groups. We used nominal (sex, comorbidity, and treatment) and categorical variables and the correlation between clinical characteristics was calculated using one-way ANOVA followed by Tukey Multiple Comparison Test. GraphPad software (version 9.1.0) was used for statistical analyses (GraphPad Software, San Diego, CA, USA). Differences were considered statistically significant at p < 0.05.
3. Results
3.1. miRNA Expression Levels
hsa-miR-223-5p
We identified common and discriminating miRNA signatures for each pathological entity (NSCLC and COPD) or smoking habit (HS) compared to the control group (HNS). RT-PCR analysis indicated that HS, COPD, and NSCLC shared a significant negative modulation of hsa-miR-223-5p expression levels compared to HNS (p <0.0001, respectively) (Figure 1). Specifically, among the three groups, NSCLC showed the lowest values (p <0.0001, respectively) (Figure 1).
hsa-miR-16-5p and 20a-5p
hsa-miR-17-5p
HS and COPD shared a significantly positive modulation of hsa-miR-17-5p expression levels compared to HNS (p <0.05 and p<0.0001, respectively) and particularly to NSCLC (p <0.0001, respectively), in which a significant downregulation was observed with respect to healthy subjects (p <0.05) (Figure 4).
hsa-miR-34a-5p
Interestingly, RT-PCR results showed that hsa-miR-34a-5p was exclusively upregulated in patients with COPD (p <0.0001), while HS and NSCLC shared a significant negative modulation of this miRNA compared to HNS (p <0.01, respectively) (Figure 5).
has-miR-106a-5p
The three pathological groups shared upregulated hashsa-miR-106a-5p expression levels as compared with the control group (p <0.0001 for HS and COPD group, p <0.01 for NSCLC group), with the highest levels observed in HS subjects versus COPD and NSCLC groups (p <0.0001, respectively) (Figure 6).
3.2. In Silico Identification of Target mRNAs
To assess the potential interconnection of miRNAs discriminating each pathological entity studied, we performed in silico prediction analysis based on sequence similarity between miRNAs and mRNAs. Two different databases were used for the analysis and data were compared with respect to the number of target genes experimentally validated. The results are reported in Table 2. The numbers of validated target genes were higher in miR target Link 2.0; therefore, it was used for bioinformatics analysis. We analyzed all selected miRNA targets, focusing on those that are common to several miRNA and implicated in the cellular pathways responsible for carcinogenesis. Abbreviations, names of the selected genes, methods, tissues of validation and references are reported in Table 3. The results showed that the miRNA may be involved in regulation of several lung cancer driver genes, such as BCL2, CYCS, E2F2, MCL1 or MYC, among others. However, other target genes linked to tumor invasion and metastasis were found, as shown in Table 4. Given that several authors have experimentally confirmed all genes, miRNA might modulate these targets in a coordinated or individual manner, affecting several hallmarks of lung carcinogenesis.
4. Discussion
In this study, we assessed the role of miRNAs in two smoking-related diseases, COPD and NSCLC, by discriminating between common and selectively dysregulated miRNAs associated with each condition or smoking habit. NSCLC and COPD are lung diseases that share etiological and pathogenic factors [10,11,12]. Indeed, inflammation and tumorigenesis are closely linked, and chronic inflammation in COPD can lead to an increased rate of cell division and the chance of mutations and carcinogenesis [13,14,15,16]. Given the high prevalence of lung cancer in COPD patients, the search for new biomarkers has prompted in-depth epidemiological studies [42,43]. Here, we analyzed a miRNA panel identifying miRNAs that may be potentially useful as biomarkers or for therapeutic purposes. This analysis was carried out in AMs recovered from BAL, a precious biological sample that is highly representative of the pulmonary microenvironment [44]. First, we identified two similarly dysregulated miRNAs in HS, COPD patients, and NSCLC patients, suggesting common pathological mechanisms underlying these conditions. Specifically, hsa-miR-223-5p levels were decreased and hsa-miR-106a-5p levels were increased, in these groups compared with those in healthy never-smokers. hsa-miR-223-5p was often reported to be downregulated in NSCLC tissue and cells leading in turn to the upregulation of many target genes that contribute to tumor proliferation, migration, and invasion [45,46]. In addition, our findings in the ever-smokers and COPD group suggested that hsa-miR-223-5p expression could indicate a common pathway between COPD and NSCLC, probably sustained by cigarette smoke exposure. Several authors have described hsa-miR-223-5p role in macrophage differentiation, neutrophil recruitment, and pro-inflammatory responses, which are the key features of lung inflammation and remodeling [47]. For instance, Schembri et al. reported lower hsa-miR-223-5p expression levels in bronchial epithelial cells from current smokers than in those from never-smokers [48] et al. ). BTG3, a member of the B-cell translocation gene/transducer of the ErbB2 antiproliferative protein family, which regulates cell cycle progression and differentiation in various cell types, has been reported to be downregulated in NSCLC specimens and cell lines, with the upregulation of hsa-miR-106a-5p [49]. Furthermore, Ye et al. showed that hsa-miR-106a-5p is overexpressed in the serum of patients with NSCLC compared to healthy subjects, thus playing a pivotal role in the onset and development of the disease [36]. Consistent with these data, we observed a significant positive modulation of hsa-miR-106a-5p in the NSCLC, COPD, and HS groups compared to healthy individuals, thus providing evidence of cancer-like dysregulation in COPD and smoking habits. Although a few studies have investigated hsa-miR-106a-5p expression patterns associated with cigarette smoke and chronic lung diseases, Sharma et al. reported that it negatively regulates IL-10 expression in an in vitro and in vivo model of airway inflammation, which is in accordance with our data [50]. Interestingly, COPD exhibited different trends in hsa-miR-16-5p, 20a-5p, and 34a-5p expression compared to HS and NSCLC. This evidence suggests a crucial role for cigarette smoking in the mechanisms responsible for the development of a cancer-like milieu and that the dysregulation of these miRNAs in COPD subjects towards the levels found in NSCLC could be indicative of an overlap between the two phenotypes. Programmed cell death ligand-1 (PDL1) is a well-known immune checkpoint that is upregulated in patients with lung cancer, allowing tumors to escape immune attack, cell growth, and metastasis [51]. Chen et al. reported that overexpression of PD-L1 accelerated tumor growth and decreased exosomal hsa-miR-16-5p content in cell culture media, while exosomal miR-16-5p overexpression in cell culture media inhibited tumor development by decreasing PD-L1 expression [35]. Wei et al. reported a 5.5-fold suppressed expression of hsa-miR-16-5p in lung cancer tissues in comparison to the normal adjacent tissues [52]. Consistent with these data, we observed a significant decrease in hsa-miR-16-5p in both HS and NSCLC patients with respect to subjects with COPD. Similarly, hsa-miR-20a-5p showed a different trend between ever-smokers and NSCLC patients compared with COPD subjects. Angiogenesis is an essential feature of carcinogenesis involving molecules in the lung microenvironment that recruit endothelial cells to promote the formation of intra-tumoral capillaries and primary tumor growth, proliferation, and metastasis [53]. Among these molecules, RRM2 plays a crucial regulatory role in controlling intratumoral capillary formation through the PI3K/Akt axis [54]. Han et al. found that the expression of RRM2 was upregulated, whereas the expression of hsa-miR-20a-5p was downregulated in cancer tissues compared to adjacent tissues in NSCLC patients, identifying hsa-miR-20a-5p as a potential tumor suppressor [37]. Accordingly, our findings showed a negative modulation of hsa-miR-20a-5p not only in the NSCLC group but also in ever-smokers, suggesting the ability of cigarette smoke to modulate its expression in a cancer-like manner.
hsa-miR-34a-5p has been extensively evaluated in NSCLC and is commonly considered a tumor suppressor because of its modulation of cellular proliferation, apoptosis, and immune evasion [33]. In a previous study, we reported that NSCLC and COPD patients differed in the hsa-miR-34a-5p lung tissue levels, showing an increase and decrease, respectively, compared to healthy controls [55]. In line with these observations, AMs recovered from the BAL of lung cancer and COPD subjects showed an opposite trend in hsa-miR-34a-5p levels. Moreover, there were no statistical differences between ever-smokers and patients with lung cancer, suggesting a similar hsa-miR-34a-5p modulation. Cigarette smoking directly damages bronchial epithelial cells, the first barrier for the respiratory tract, and the infiltration of immune cells in the lung tissue, including macrophages and neutrophils [56]. SIRPα, a member of the immunoglobulin superfamily, modulates many aspects of the inflammatory response including activation, chemotaxis, and phagocytosis [57]. In macrophages, the upregulation of hsa-miR-17-5p by lipopolysaccharides (LPS) serves as the mechanism underlying LPS-induced SIRPα reduction and macrophage activation [58]. This was consistent with our finding of higher hsa-miR-17-5p levels in both COPD patients and ever-smokers, supporting the importance of smoking in the mechanisms underlying COPD. Intriguingly, NSCLC showed the lowest levels of hsa-miR-17-5p, in accordance with several studies that reported it as a tumor suppressor [38,39] However, hsa-miR-17-5p regulatory effects on tumor progression are different and cannot be generalized, making its role controversial [59]. It is important to point out that although all subjects with COPD were current smokers, our data revealed that hsa-miR-16-5p, 20-5p, and 34a-p expression levels differed from those in ever-smokers. Generally, the intensity of the reaction against immunogenic antigens produced in response to cigarette smoke varies across a wide range of disease manifestations, such as COPD or lung cancer. Moreover, recent studies have highlighted the crucial role of immune responses in the regulation of the development of COPD or NSCLC phenotypes in response to smoke, assuming that COPD may be associated with a shift from an innate response to an adaptive immune response with hallmarks typical of autoimmune processes [60,61]. In this context, dysregulation of these specific miRNAs could reflect phenotype switching or the onset of different lung manifestations, underlining the prominent but not exclusive role of cigarette smoking. Indeed, miRNA expression profiles could be confounded by other environmental conditions, which could further modulate the correlation between the expression of miRNAs and their target mRNAs in COPD [62]. Finally, to assess the potential interconnection of miRNAs discriminating each pathological entity, we performed in silico prediction of hsa-miR target genes. Bioinformatic prediction results revealed that the miRNAs analyzed may potentially be involved in the regulation of several lung cancer driver genes, such as BCL2, CYCS, E2F3, MCL1, MFN2, MYC, RNF115, SENP1, SLC1A5, SLC6A4, SLC7A11, and TMOD3, which are involved in cancer key regulatory pathways [63,64,65,66,67,68,69,70,71,72,73,74,75]. Since several authors have experimentally confirmed all miRNA-regulated genes, miRNAs may modulate these targets combined or individually, affecting different hallmarks of lung carcinogenesis. For instance, Lu et al. assessed the regulation of hsa-miR-17-5p on MFN2 expression using a luciferase reporter assay system. The authors reported that hsa-miR-17-5p regulates proliferation and apoptosis in human pulmonary artery smooth muscle cells (hPASMCs) treated with hypoxia through MFN2 modulation and binding to its 3'-UTR [76]. Christoffersen et al. showed that the 3’-UTR sequence of MYC revealed the presence of a perfectly complementary 6-nucleotide seed match to hsa-miR-34a -5p. Western blotting revealed that overexpression of hsa-miR-34a-5 p resulted in marked downregulation of endogenous MYC protein, emphasizing its significance as a tumor suppressor [77]. In summary, the present study identified distinct miRNAs in AMs obtained from BAL samples that could discriminate between NSCLC and COPD. One potential limitation was the small sample size used for miRNA analysis which does not allow to apply our findings to a broader population or perform a subanalysis according to COPD severity. Moreover, it might be interesting in the future to analyze the direct effect of smoke in the correlation between miRNAs and their targets linked to carcinogenesis. However, considering the nature of the lung samples used, our results, even if preliminary, could be of clinical relevance and lead to future studies involving larger populations. Our data showed common and specific deregulated miRNAs in NSCLC and COPD, allowing us to understand the underlying regulatory mechanisms involved in the pathogenesis of these smoking-related conditions and potentially providing new tools for diagnosis or pharmacological treatment.
Author Contributions
Conceptualization, Davida Mirra, Giuseppe Spaziano, Erika Cione and Bruno D’Agostino; Data curation, Giuseppe Spaziano, Ida Cerqua and Erika Cione; Formal analysis, Davida Mirra, Renata Esposito, Francesca Panico and Maddalena Falciani; Funding acquisition, Liberata Sportiello; Investigation, Davida Mirra, Renata Esposito, Francesca Panico, Antonio Squillante and Ida Cerqua; Methodology, Davida Mirra; Project administration, Giuseppe Spaziano and Bruno D’Agostino; Resources, Luca Gallelli; Supervision, Luca Gallelli and Erika Cione; Validation, Giuseppe Spaziano, Liberata Sportiello and Luca Gallelli; Visualization, Renata Esposito; Writing – original draft, Davida Mirra; Writing – review & editing, Davida Mirra, Giuseppe Spaziano, Erika Cione and Bruno D’Agostino.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Institutional Review Board Statement
Institutional Review Board Statement: This study was part of a clinical trial recorded at clini-caltrials.gov (NCT04654104) and was approved by the local ethics committee “Calabria Centro” (n. 361 del 22 Ottobre 2020). This work was conducted in compliance with the Institutional Re-view Board/Human Subjects Research Committee requirements, the Declaration of Helsinki, and the Guidelines for Good Clinical Practice criteria.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Funding
This research received no funding.
References
- López-Campos, J.L.; Tan, W.; Soriano, J.B. Global burden of COPD. Respirology. 2016, 21(1):14-23. [CrossRef]
- Safiri, S.; Carson-Chahhoud, K.; Noori, M.; Nejadghaderi, S. A.; Sullman, M. J. M.; Ahmadian Heris, J.; Ansarin, K.; Mansournia, M. A.; Collins, G. S.; Kolahi, A. A.; Kaufman, J. S. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990-2019: results from the Global Burden of Disease Study 2019. BMJ, 2022, 378:e069679. [CrossRef]
- Pauwels, R. A.; Rabe, K. F. Burden and Clinical Features of Chronic Obstructive Pulmonary Disease (COPD). The Lancet 2004, 364 (9434), 613–620. [CrossRef]
- Kotlyarov, S. The Role of Smoking in the Mechanisms of Development of Chronic Obstructive Pulmonary Disease and Atherosclerosis. IJMS 2023, 24 (10), 8725. [CrossRef]
- Urbanek, K.; De Angelis, A.; Spaziano, G.; Piegari, E.; Matteis, M.; Cappetta, D.; Esposito, G.; Russo, R.; Tartaglione, G.; De Palma, R.; Rossi, F.; D’Agostino, B. Intratracheal Administration of Mesenchymal Stem Cells Modulates Tachykinin System, Suppresses Airway Remodeling and Reduces Airway Hyperresponsiveness in an Animal Model. PLoS ONE 2016, 11 (7), e0158746. [CrossRef]
- D’Agostino, B.; Advenier, C.; De Palma, R.; Gallelli, L.; Marrocco, G.; Abbate, G. F.; Rossi, F. The Involvement of Sensory Neuropeptides in Airway Hyper-Responsiveness in Rabbits Sensitized and Challenged to Parietaria Judaica: Sensory Neuropeptides in Airway Hyper-Responsiveness. Clinical & Experimental Allergy 2002, 32 (3), 472–479. [CrossRef]
- D’Agostino, B.; Marrocco, G.; De Nardo, M.; Calò, G.; Guerrini, R.; Gallelli, L.; Advenier, C.; Rossi, F. Activation of the Nociceptin/Orphanin FQ Receptor Reduces Bronchoconstriction and Microvascular Leakage in a Rabbit Model of Gastroesophageal Reflux: N/OFQ Effects in the Airways in a GER Animal Model. British Journal of Pharmacology 2005, 144 (6), 813–820. [CrossRef]
- Rouget, C.; Cui, Y. Y.; D’Agostino, B.; Faisy, C.; Naline, E.; Bardou, M.; Advenier, C. Nociceptin Inhibits Airway Microvascular Leakage Induced by HCl Intra-Oesophageal Instillation: Nociceptin and Gastro-Oesophageal Reflux. British Journal of Pharmacology 2004, 141 (6), 1077–1083. [CrossRef]
- Gallelli, L.; D’Agostino, B.; Marrocco, G.; De Rosa, G.; Filippelli, W.; Rossi, F.; Advenier, C. Role of Tachykinins in the Bronchoconstriction Induced by HCl Intraesophageal Instillation in the Rabbit. Life Sciences 2003, 72 (10), 1135–1142. [CrossRef]
- Durham, A. L.; Adcock, I. M. The Relationship between COPD and Lung Cancer. Lung Cancer 2015, 90 (2), 121–127. [CrossRef]
- Papi, A. COPD Increases the Risk of Squamous Histological Subtype in Smokers Who Develop Non-Small Cell Lung Carcinoma. Thorax 2004, 59 (8), 679–681. [CrossRef]
- Young, R. P.; Hopkins, R. J. Link between COPD and Lung Cancer. Respiratory Medicine 2010, 104 (5), 758–759. [CrossRef]
- Schetter, A. J.; Heegaard, N. H. H.; Harris, C. C. Inflammation and Cancer: Interweaving MicroRNA, Free Radical, Cytokine and P53 Pathways. Carcinogenesis 2010, 31 (1), 37–49. [CrossRef]
- Caramori, G.; Adcock, I. M.; Casolari, P.; Ito, K.; Jazrawi, E.; Tsaprouni, L.; Villetti, G.; Civelli, M.; Carnini, C.; Chung, K. F.; Barnes, P. J.; Papi, A. Unbalanced Oxidant-Induced DNA Damage and Repair in COPD: A Link towards Lung Cancer. Thorax 2011, 66 (6), 521–527. [CrossRef]
- Schaible, A. M.; Filosa, R.; Temml, V.; Krauth, V.; Matteis, M.; Peduto, A.; Bruno, F.; Luderer, S.; Roviezzo, F.; Di Mola, A.; de Rosa, M.; D’Agostino, B.; Weinigel, C.; Barz, D.; Koeberle, A.; Pergola, C.; Schuster, D.; Werz, O. Elucidation of the Molecular Mechanism and the Efficacy in Vivo of a Novel 1,4-Benzoquinone That Inhibits 5-Lipoxygenase. Br J Pharmacol 2014, 171 (9), 2399–2412. [CrossRef]
- Schaible, A. M.; Filosa, R.; Krauth, V.; Temml, V.; Pace, S.; Garscha, U.; Liening, S.; Weinigel, C.; Rummler, S.; Schieferdecker, S.; Nett, M.; Peduto, A.; Collarile, S.; Scuotto, M.; Roviezzo, F.; Spaziano, G.; de Rosa, M.; Stuppner, H.; Schuster, D.; D’Agostino, B.; Werz, O. The 5-Lipoxygenase Inhibitor RF-22c Potently Suppresses Leukotriene Biosynthesis in Cellulo and Blocks Bronchoconstriction and Inflammation in Vivo. Biochemical Pharmacology 2016, 112, 60–71. [CrossRef]
- Mark, N. M.; Kargl, J.; Busch, S. E.; Yang, G. H. Y.; Metz, H. E.; Zhang, H.; Hubbard, J. J.; Pipavath, S. N. J.; Madtes, D. K.; Houghton, A. M. Chronic Obstructive Pulmonary Disease Alters Immune Cell Composition and Immune Checkpoint Inhibitor Efficacy in Non–Small Cell Lung Cancer. Am J Respir Crit Care Med 2018, 197 (3), 325–336. [CrossRef]
- Punturieri, A.; Szabo, E.; Croxton, T. L.; Shapiro, S. D.; Dubinett, S. M. Lung Cancer and Chronic Obstructive Pulmonary Disease: Needs and Opportunities for Integrated Research. JNCI Journal of the National Cancer Institute 2009, 101 (8), 554–559. [CrossRef]
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P. N. Smoking Alters Alveolar Macrophage Recognition and Phagocytic Ability: Implications in Chronic Obstructive Pulmonary Disease. Am J Respir Cell Mol Biol 2007, 37 (6), 748–755. [CrossRef]
- de Groot, L. E. S.; van der Veen, T. A.; Martinez, F. O.; Hamann, J.; Lutter, R.; Melgert, B. N. Oxidative Stress and Macrophages: Driving Forces behind Exacerbations of Asthma and Chronic Obstructive Pulmonary Disease? American Journal of Physiology-Lung Cellular and Molecular Physiology 2019, 316 (2), L369–L384. [CrossRef]
- Vlahos, R. Role of Alveolar Macrophages in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2014, 5. [CrossRef]
- Finicelli, M.; Digilio, F. A.; Galderisi, U.; Peluso, G. The Emerging Role of Macrophages in Chronic Obstructive Pulmonary Disease: The Potential Impact of Oxidative Stress and Extracellular Vesicle on Macrophage Polarization and Function. Antioxidants 2022, 11 (3), 464. [CrossRef]
- Polverino, F.; Mirra, D.; Yang, C. X.; Esposito, R.; Spaziano, G.; Rojas-Quintero, J.; Sgambato, M.; Piegari, E.; Cozzolino, A.; Cione, E.; Gallelli, L.; Capuozzo, A.; Santoriello, C.; Berrino, L.; de-Torres, J. P.; Hackett, T. L.; Polverino, M.; D’Agostino, B. Similar Programmed Death Ligand 1 (PD-L1) Expression Profile in Patients with Mild COPD and Lung Cancer. Sci Rep 2022, 12 (1), 22402. [CrossRef]
- Franco, R.; Schoneveld, O.; Georgakilas, A. G.; Panayiotidis, M. I. Oxidative Stress, DNA Methylation and Carcinogenesis. Cancer Letters 2008, 266 (1), 6–11. [CrossRef]
- Kabesch, M.; Adcock, I. M. Epigenetics in Asthma and COPD. Biochimie 2012, 94 (11), 2231–2241. [CrossRef]
- Molina-Pinelo, S.; Pastor, M. D.; Suarez, R.; Romero-Romero, B.; Gonzalez De la Pena, M.; Salinas, A.; Garcia-Carbonero, R.; De Miguel, M. J.; Rodriguez-Panadero, F.; Carnero, A.; Paz-Ares, L. MicroRNA Clusters: Dysregulation in Lung Adenocarcinoma and COPD. European Respiratory Journal 2014, 43 (6), 1740–1749. [CrossRef]
- Mirra, D.; Cione, E.; Spaziano, G.; Esposito, R.; Sorgenti, M.; Granato, E.; Cerqua, I.; Muraca, L.; Iovino, P.; Gallelli, L.; D’Agostino, B. Circulating MicroRNAs Expression Profile in Lung Inflammation: A Preliminary Study. JCM 2022, 11 (18), 5446. [CrossRef]
- Lu, L.-F.; Liston, A. MicroRNA in the Immune System, MicroRNA as an Immune System. Immunology 2009, 127 (3), 291–298. [CrossRef]
- Iannone, F.; Montesanto, A.; Cione, E.; Crocco, P.; Caroleo, M. C.; Dato, S.; Rose, G.; Passarino, G. Expression Patterns of Muscle-Specific MiR-133b and MiR-206 Correlate with Nutritional Status and Sarcopenia. Nutrients 2020, 12 (2), 297. [CrossRef]
- Cannataro, R.; Caroleo, M. C.; Fazio, A.; La Torre, C.; Plastina, P.; Gallelli, L.; Lauria, G.; Cione, E. Ketogenic Diet and MicroRNAs Linked to Antioxidant Biochemical Homeostasis. Antioxidants 2019, 8 (8), 269. [CrossRef]
- Leidinger, P.; Keller, A.; Borries, A.; Huwer, H.; Rohling, M.; Huebers, J.; Lenhof, H. P.; Meese, E. Specific peripheral miRNA profiles for distinguishing lung cancer from COPD. Lung cancer 2011, 74(1), 41–47. [CrossRef]
- Keller, A.; Fehlmann, T.; Ludwig, N.; Kahraman, M.; Laufer, T.; Backes, C.; Vogelmeier, C.; Diener, C.; Biertz, F.; Herr, C.; Jörres, R. A.; Lenhof, H. P.; Meese, E.; Bals, R., COSYCONET Study Group. Genome-wide MicroRNA Expression Profiles in COPD: Early Predictors for Cancer Development. Genomics, proteomics bioinformatics 2018, 16(3), 162–171. [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 Family: A Potential Tumor Suppressor and Therapeutic Candidate in Cancer. J Exp Clin Cancer Res 2019, 38 (1), 53. [CrossRef]
- Lu, Y. MiR-223-5p Suppresses OTX1 to Mediate Malignant Progression of Lung Squamous Cell Carcinoma Cells. Computational and Mathematical Methods in Medicine 2021, 2021, 1–11. [CrossRef]
- Chen, H.; Luo, Y.; Lin, M.; Peng, X.; Liu, M.; Wang, Y.; Li, S.; Yang, D.; Yang, Z. Serum Exosomal miR -16-5p Functions as a Tumor Inhibitor and a New Biomarker for PD-L1 Inhibitor-dependent Immunotherapy in Lung Adenocarcinoma by Regulating PD-L1 Expression. Cancer Medicine 2022, 11 (13), 2627–2643. [CrossRef]
- Ye, T.; Changyu, S.; Limeng, Z.; Yuan, P. Clinical Significance of MiRNA - 106a in Non-Small Cell Lung Cancer Patients Who Received Cisplatin Combined with Gemcitabine Chemotherapy. Cancer Biology & Medicine 2018, 15 (2), 157. [CrossRef]
- Han, J.; Hu, J.; Sun, F.; Bian, H.; Tang, B.; Fang, X. MicroRNA-20a-5p Suppresses Tumor Angiogenesis of Non-Small Cell Lung Cancer through RRM2-Mediated PI3K/Akt Signaling Pathway. Mol Cell Biochem 2021, 476 (2), 689–698. [CrossRef]
- Zhang, X.; Li, Y.; Qi, P.; Ma, Z. Biology of MiR-17-92 Cluster and Its Progress in Lung Cancer. Int. J. Med. Sci. 2018, 15 (13), 1443–1448. [CrossRef]
- Sweat, Y.; Ries, R. J.; Sweat, M.; Su, D.; Shao, F.; Eliason, S.; Amendt, B. A. MiR-17 Acts as a Tumor Suppressor by Negatively Regulating the MiR-17-92 Cluster. Molecular Therapy - Nucleic Acids 2021, 26, 1148–1158. [CrossRef]
- Fathinavid, A.; Ghobadi, M.Z.; Najafi, A.; Masoudi-Nejad, A. Identification of Common MicroRNA between COPD and Non-Small Cell Lung Cancer through Pathway Enrichment Analysis. BMC Genom. Data 2021, 22, 41.
- Sokolowski, J.W., Jr.; Burgher, L.W.; Jones, F.L., Jr.; Patterson, J.R.; Selecky, P.A. Guidelines for fiberoptic bronchoscopy in adults. American Thoracic Society guidelines. Medical Section of the American Lung Association. Am. Rev. Respir. Dis. 1987, 136, 1066.
- de Torres, J.P.; Marín, J.M.; Casanova, C.; Cote, C.; Carrizo, S.; Cordoba-Lanus, E.; Baz-Dávila, R.; Zulueta, J.J.; Aguirre-Jaime, A.; Saetta, M.; et al. Lung Cancer in Patients with Chronic Obstructive Pulmonary Disease: Incidence and Predicting Factors. Am. J. Respir. Crit. Care Med. 2011, 184, 913–919.
- Park, H.Y.; Kang, D.; Shin, S.H.; Yoo, K.-H.; Rhee, C.K.; Suh, G.Y.; Kim, H.; Shim, Y.M.; Guallar, E.; Cho, J.; et al. Chronic Obstructive Pulmonary Disease and Lung Cancer Incidence in Never Smokers: A Cohort Study. Thorax 2020, 75, 506–509.
- Lofdahl, J. M. Bronchoalveolar Lavage in COPD: Fluid Recovery Correlates with the Degree of Emphysema. European Respiratory Journal 2005, 25 (2), 275–281. [CrossRef]
- Pavel, A. B.; Campbell, J. D.; Liu, G.; Elashoff, D.; Dubinett, S.; Smith, K.; Whitney, D.; Lenburg, M. E.; Spira, A. Alterations in Bronchial Airway MiRNA Expression for Lung Cancer Detection. Cancer Prevention Research 2017, 10 (11), 651–659. [CrossRef]
- Dou, L.; Han, K.; Xiao, M.; Lv, F. MiR-223-5p Suppresses Tumor Growth and Metastasis in Non-Small Cell Lung Cancer by Targeting E2F8. oncol res 2019, 27 (2), 261–268. [CrossRef]
- Roffel, M. P.; Bracke, K. R.; Heijink, I. H.; Maes, T. MiR-223: A Key Regulator in the Innate Immune Response in Asthma and COPD. Front. Med. 2020, 7, 196. [CrossRef]
- Schembri, F.; Sridhar, S.; Perdomo, C.; Gustafson, A. M.; Zhang, X.; Ergun, A.; Lu, J.; Liu, G.; Zhang, X.; Bowers, J.; Vaziri, C.; Ott, K.; Sensinger, K.; Collins, J. J.; Brody, J. S.; Getts, R.; Lenburg, M. E.; Spira, A. MicroRNAs as Modulators of Smoking-Induced Gene Expression Changes in Human Airway Epithelium. Proc. Natl. Acad. Sci. U.S.A. 2009, 106 (7), 2319–2324. [CrossRef]
- Wei, K.; Pan, C.; Yao, G.; Liu, B.; Ma, T.; Xia, Y.; Jiang, W.; Chen, L.; Chen, Y. MiR-106b-5p Promotes Proliferation and Inhibits Apoptosis by Regulating BTG3 in Non-Small Cell Lung Cancer. Cell Physiol Biochem 2017, 44 (4), 1545–1558. [CrossRef]
- Sharma, A.; Kumar, M.; Ahmad, T.; Mabalirajan, U.; Aich, J.; Agrawal, A.; Ghosh, B. Antagonism of Mmu-Mir-106a Attenuates Asthma Features in Allergic Murine Model. Journal of Applied Physiology 2012, 113 (3), 459–464. [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; Li, Y.; Li, G.; Xiong, W.; Guo, C.; Zeng, Z. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol Cancer 2019, 18 (1), 10. [CrossRef]
- Wei, J.; Jia, A.; Ma, L.; Wang, Y.; Qiu, L.; Xiao, B. MicroRNA-16 Inhibits the Proliferation and Metastasis of Human Lung Cancer Cells by Modulating the Expression of YAP. J BUON 2020, 25(2):862-868.
- Zhang, K.; Yang, L.; Wang, J.; Sun, T.; Guo, Y.; Nelson, R.; Tong, T. R.; Pangeni, R.; Salgia, R.; Raz, D. J. Ubiquitin-Specific Protease 22 Is Critical to in Vivo Angiogenesis, Growth and Metastasis of Non-Small Cell Lung Cancer. Cell Commun Signal 2019, 17 (1), 167. [CrossRef]
- Wang, N.; Zhan, T.; Ke, T.; Huang, X.; Ke, D.; Wang, Q.; Li, H. Increased Expression of RRM2 by Human Papillomavirus E7 Oncoprotein Promotes Angiogenesis in Cervical Cancer. Br J Cancer 2014, 110 (4), 1034–1044. [CrossRef]
- Mirra, D.; Esposito, R.; Spaziano, G.; La Torre, C.; Vocca, C.; Tallarico, M.; Cione, E.; Gallelli, L.; D’Agostino, B. Lung MicroRNAs Expression in Lung Cancer and COPD: A Preliminary Study. Biomedicines 2023, 11 (3), 736. [CrossRef]
- Danov, O.; Wolff, M.; Bartel, S.; Böhlen, S.; Obernolte, H.; Wronski, S.; Jonigk, D.; Hammer, B.; Kovacevic, D.; Reuter, S.; Krauss-Etschmann, S.; Sewald, K. Cigarette Smoke Affects Dendritic Cell Populations, Epithelial Barrier Function, and the Immune Response to Viral Infection With H1N1. Front. Med. 2020, 7, 571003. [CrossRef]
- Barclay, A. N.; Brown, M. H. The SIRP Family of Receptors and Immune Regulation. Nat Rev Immunol 2006, 6 (6), 457–464. [CrossRef]
- Zhu, D.; Pan, C.; Li, L.; Bian, Z.; Lv, Z.; Shi, L.; Zhang, J.; Li, D.; Gu, H.; Zhang, C.-Y.; Liu, Y.; Zen, K. MicroRNA-17/20a/106a Modulate Macrophage Inflammatory Responses through Targeting Signal-Regulatory Protein α. Journal of Allergy and Clinical Immunology 2013, 132 (2), 426-436.e8. [CrossRef]
- Huang, C.; Yu, M.; Yao, X. MicroRNA-17 and the Prognosis of Human Carcinomas: A Systematic Review and Meta-Analysis. BMJ Open 2018, 8 (5), e018070. [CrossRef]
- Lee, S.-H.; Goswami, S.; Grudo, A.; Song, L.; Bandi, V.; Goodnight-White, S.; Green, L.; Hacken-Bitar, J.; Huh, J.; Bakaeen, F.; Coxson, H. O.; Cogswell, S.; Storness-Bliss, C.; Corry, D. B.; Kheradmand, F. Antielastin Autoimmunity in Tobacco Smoking–Induced Emphysema. Nat Med 2007, 13 (5), 567–569. [CrossRef]
- Polverino, F.; Laucho-Contreras, M.; Rojas Quintero, J.; Divo, M.; Pinto-Plata, V.; Sholl, L.; de-Torres, J. P.; Celli, B. R.; Owen, C. A. Increased Expression of A Proliferation-Inducing Ligand (APRIL) in Lung Leukocytes and Alveolar Epithelial Cells in COPD Patients with Non Small Cell Lung Cancer: A Possible Link between COPD and Lung Cancer? Multidiscip Respir Med 2016, 11 (1), 17. [CrossRef]
- Qiu, C.; Chen, G.; Cui, Q. Towards the Understanding of MicroRNA and Environmental Factor Interactions and Their Relationships to Human Diseases. Sci Rep 2012, 2 (1), 318. [CrossRef]
- Xie, M.; Park, D.; Sica, G. L.; Deng, X. Bcl2-Induced DNA Replication Stress Promotes Lung Carcinogenesis in Response to Space Radiation. Carcinogenesis 2020, 41 (11), 1565–1575. [CrossRef]
- Liu, C.; Wei, X. Unraveling the Potential of Senescence-Related Genes in Guiding Clinical Therapy of Lung Adenocarcinoma Patients. Funct Integr Genomics 2023, 23 (2), 188. [CrossRef]
- Wu, L.; Wan, S.; Li, J.; Xu, Y.; Lou, X.; Sun, M.; Wang, S. Expression and Prognostic Value of E2F3 Transcription Factor in Non-small Cell Lung Cancer. Oncol Lett 2021, 21 (5), 411. [CrossRef]
- Zhang, J.; He, J.; Xia, J.; Chen, Z.; Chen, X. Delayed Apoptosis by Neutrophils from COPD Patients Is Associated with Altered Bak, Bcl-Xl, and Mcl-1 MRNA Expression. Diagn Pathol 2012, 7 (1), 65. [CrossRef]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-Myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer Res 2020, 40 (2), 609–618. [CrossRef]
- Zhang, J.; Pan, L.; Zhang, Q.; Zhao, Y.; Wang, W.; Lin, N.; Zhang, S.; Wu, Q. MFN2 Deficiency Affects Calcium Homeostasis in Lung Adenocarcinoma Cells via Downregulation of UCP4. FEBS Open Bio 2023, 13 (6), 1107–1124. [CrossRef]
- Luo, Z.; Ye, X.; Shou, F.; Cheng, Y.; Li, F.; Wang, G. RNF115-Mediated Ubiquitination of P53 Regulates Lung Adenocarcinoma Proliferation. Biochemical and Biophysical Research Communications 2020, 530 (2), 425–431. [CrossRef]
- Gao, C.; Xiao, F.; Zhang, L.; Sun, Y.; Wang, L.; Liu, X.; Sun, H.; Xie, Z.; Liang, Y.; Xu, Q.; Wang, L. SENP1 Inhibition Suppresses the Growth of Lung Cancer Cells through Activation of A20-Mediated Ferroptosis. Ann Transl Med 2022, 10 (4), 224–224. [CrossRef]
- Tu, Y.; Yao, S.; Chen, Q.; Li, W.; Song, Y.; Zhang, P. 5-Hydroxytryptamine Activates a 5-HT/c-Myc/SLC6A4 Signaling Loop in Non–Small Cell Lung Cancer. Biochimica et Biophysica Acta (BBA) - General Subjects 2022, 1866 (4), 130093. [CrossRef]
- Xue, Y.; Jiang, K.; Ou, L.; Shen, M.; Yang, Y.; Lu, J.; Xu, W. Targeting Sphingosine Kinase 1/2 by a Novel Dual Inhibitor SKI-349 Suppresses Non-Small Cell Lung Cancer Cell Growth. Cell Death Dis 2022, 13 (7), 602. [CrossRef]
- Zhang, W.; Sun, Y.; Bai, L.; Zhi, L.; Yang, Y.; Zhao, Q.; Chen, C.; Qi, Y.; Gao, W.; He, W.; Wang, L.; Chen, D.; Fan, S.; Chen, H.; Piao, H.-L.; Qiao, Q.; Xu, Z.; Zhang, J.; Zhao, J.; Zhang, S.; Yin, Y.; Peng, C.; Li, X.; Liu, Q.; Liu, H.; Wang, Y. RBMS1 Regulates Lung Cancer Ferroptosis through Translational Control of SLC7A11. Journal of Clinical Investigation 2021, 131 (22), e152067. [CrossRef]
- Hassanein, M.; Hoeksema, M. D.; Shiota, M.; Qian, J.; Harris, B. K.; Chen, H.; Clark, J. E.; Alborn, W. E.; Eisenberg, R.; Massion, P. P. SLC1A5 Mediates Glutamine Transport Required for Lung Cancer Cell Growth and Survival. Clinical Cancer Research 2013, 19 (3), 560–570. [CrossRef]
- Li, H.; Zhao, S.; Chen, X.; Feng, G.; Chen, Z.; Fan, S. MiR-145 Modulates the Radiosensitivity of Non-Small Cell Lung Cancer Cells by Suppression of TMOD3. Carcinogenesis 2022, 43 (3), 288–296. [CrossRef]
- Lu, Z.; Li, S.; Zhao, S.; Fa, X. Upregulated miR-17 Regulates Hypoxia-Mediated Human Pulmonary Artery Smooth Muscle Cell Proliferation and Apoptosis by Targeting Mitofusin 2. Med Sci Monit 2016, 22, 3301–3308. [CrossRef]
- Christoffersen, N. R.; Shalgi, R.; Frankel, L. B.; Leucci, E.; Lees, M.; Klausen, M.; Pilpel, Y.; Nielsen, F. C.; Oren, M.; Lund, A. H. (2010). p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ 2010, 17(2), 236–245. [CrossRef]
Figure 1.
Analysis of hsa-miR-223-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. **** p < 0.0001.
Figure 1.
Analysis of hsa-miR-223-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. **** p < 0.0001.
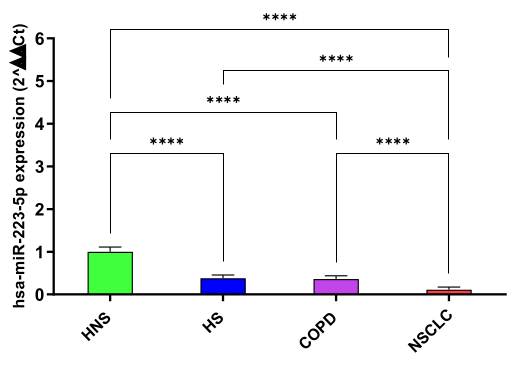
Figure 2.
Analysis of hsa-miR-16-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. **** p < 0.0001.
Figure 2.
Analysis of hsa-miR-16-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. **** p < 0.0001.
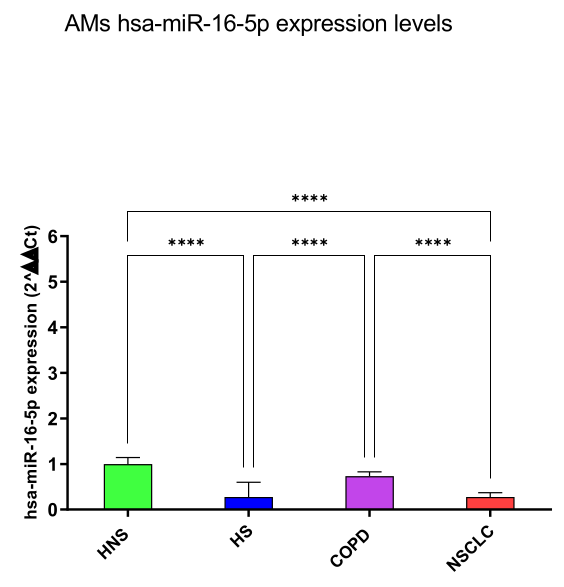
Figure 3.
Analysis of hsa-miR-20a-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. **** p < 0.0001.
Figure 3.
Analysis of hsa-miR-20a-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. **** p < 0.0001.
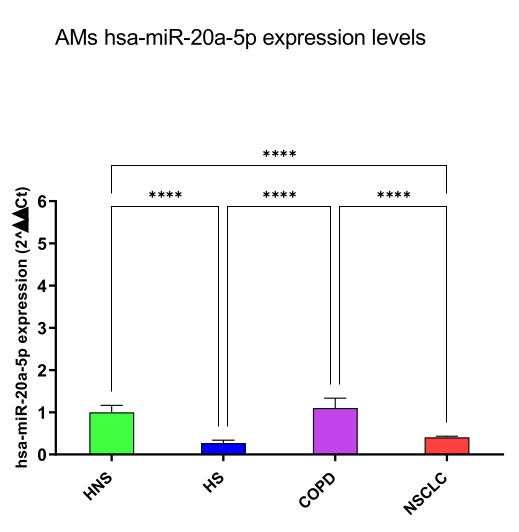
Figure 4.
Analysis of hsa-miR-17-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. * p < 0.05; **** p < 0.0001.
Figure 4.
Analysis of hsa-miR-17-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. * p < 0.05; **** p < 0.0001.
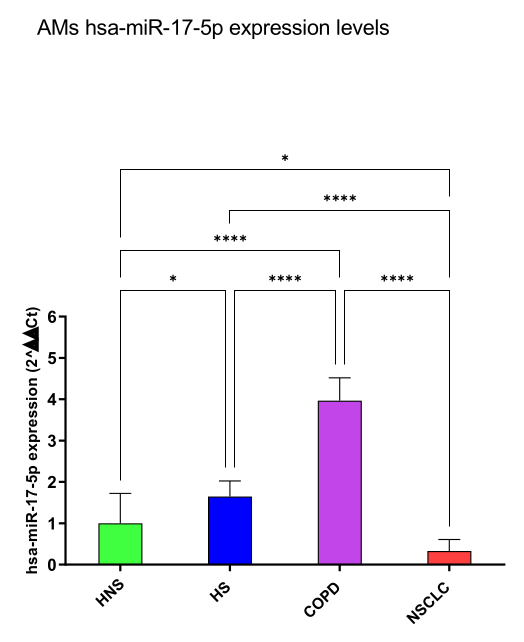
Figure 5.
Analysis of hsa-miR-34a-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. ** p < 0.01; **** p < 0.0001.
Figure 5.
Analysis of hsa-miR-34a-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. ** p < 0.01; **** p < 0.0001.
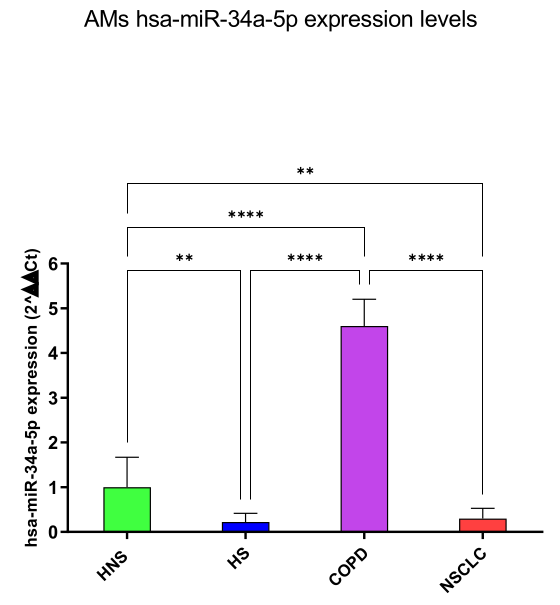
Figure 6.
Analysis of hsa-miR-106a-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. ** p < 0.01; **** p < 0.0001.
Figure 6.
Analysis of hsa-miR-106a-5p AMs expression levels in HNS (green column; biological replicates n = 9), HS (blue column; biological replicates n = 11), COPD (purple column; biological replicates n = 11) and NSCLC (red column; biological replicates n = 12). All samples were run in triplicate, and results are shown as means ± SD. The statistical tests used in these analyses were one-way analysis of variance followed by Tukey Multiple Comparison Test. ** p < 0.01; **** p < 0.0001.
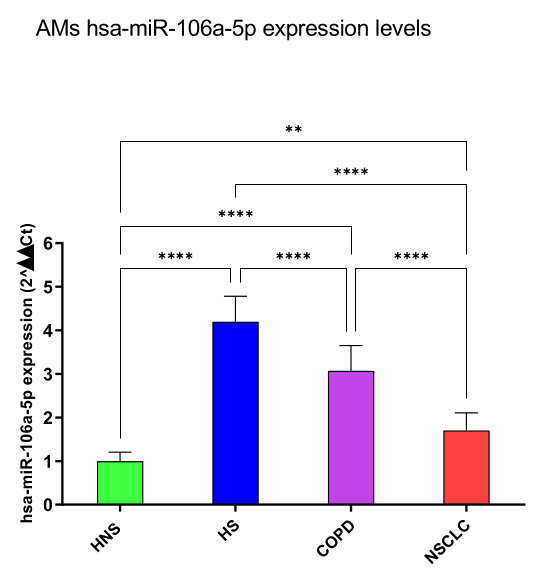

Table 1.
Demographic characteristics of the enrolled patients. Data are means ± SD unless specified. The statistical tests used in these analyses were one-way ANOVA followed by the Tukey Multiple Comparison Test. FEV1: forced expiratory volume in the first second; FVC, forced vital capacity; LABA: long-acting beta-agonists; SABA: short-acting beta-agonists; LAMA: long-acting muscarinic agents.
Table 1.
Demographic characteristics of the enrolled patients. Data are means ± SD unless specified. The statistical tests used in these analyses were one-way ANOVA followed by the Tukey Multiple Comparison Test. FEV1: forced expiratory volume in the first second; FVC, forced vital capacity; LABA: long-acting beta-agonists; SABA: short-acting beta-agonists; LAMA: long-acting muscarinic agents.
| Healthy Never Smokers |
Healthy Smokers |
COPD | NSCLC | P-value | |
|---|---|---|---|---|---|
| Total Partecipants (N) | 9 | 11 | 11 | 12 | |
| Age (SD) | 70.5 (15.2) | 52.5 (8.3) | 63.5 (6) | 75 (16.9) | NS |
| Gender (M/F) | 5/4 | 6/5 | 5/6 | 6/6 | NS |
| Smoking history (pack years) | NA | 35 | 28.3 | 31 | <0.001 |
|
Smoking habit (current/former smoker) |
NA | 8/3 | 11/0 | 6/6 | 0.0133 |
| FEV1 (% predicted) | 96% (3.2) | 91% (12) | 66% (14) | 88% (11) | 0.0438 |
| FEV/FVC | 76.1 (2.5) | 75 (1.4) | 59 (1.7) | 74 (3) | <0.0001 |
| Comorbidities | |||||
| Hypertension (%) | 5 (55.5%) | 5 (45.4%) | 3 (27.2%) | 5 (41.6%) | 0.0152 |
| Other cardiovascluar diseases (%) | 2 (22.2%) | 0 | 3 (27.2%) | 1 (12.5%) | NS |
| Diabetes Mellitus (%) | 1 (16.7%) | 0 | 0 | 1 (0.8%) | NS |
| Medications | |||||
| Inhaled corticosteroids (N, %) | 0 | 0 | 0 | 0 | NS |
| LABA/SABA/LAMA (N, %) | 0 | 0 | 11 (100%) | 3 (25%) | <0.0001 |
Table 2.
Bioinformatics tools for in silico analysis. Number of validated genes for each miRNA analyzed in miR Target Link 2.0 and Diana Tools databases.
Table 2.
Bioinformatics tools for in silico analysis. Number of validated genes for each miRNA analyzed in miR Target Link 2.0 and Diana Tools databases.
| Number of Target Genes | ||
|---|---|---|
| miRNA | miR Target Link 2.0 | DIANA Tools |
| hsa-miR-223-5p | 551 | 10 |
| hsa-miR-16-5p | 2,279 | 455 |
| hsa-miR-20a-5p | 1,659 | 611 |
| hsa-miR-17-5p | 1,817 | 136 |
| hsa-miR-34a-5p | 968 | 324 |
| hsa-miR-106a-5p | 1,166 | 435 |
Table 3.
Abbreviations, gene names, methods and tissue on which the miRNA selected were validated targets from miR Target Link Human database.
Table 3.
Abbreviations, gene names, methods and tissue on which the miRNA selected were validated targets from miR Target Link Human database.
| Abbreviation | Gene Name | Methods | Tissues | References(PMID) |
| BCL2 | BCL2 Apoptosis Regulator | Luciferase reporter assay, qRT-PCR, Western blot, Reporter assay, Proteomics analysis, Immunohistochemistry, Microarray, Sequencing, HITS-CLIP, Immunoblot, Immunoprecipitaion |
Cervix cells, gastric cells, Bone cells, Marrow cells, spleen, liver, kidney, lymph node, tracheal/bronchial epithelial cells, breast cells, ovary cells, embryonic kidney cells, gastric cancer cells, B cells, mesothelial cell, glioma cells |
17877811 18449891 18362358 17351108 17707831 20643754 20876285 19269153 16166262 19903841 20371350 23907579 22473208 24148817 25435430 26397135 26722459 |
| CYCS | Cytochrome C, Somatic |
Proteomics, PAR-CLIP, PCR array |
Breast cells, brain and liver |
18668040 23446348 28097098 |
| E2F3 | E2F Transcription Factor 3 |
CLASH. HITS-CLIP, luciferase reporter assay, Western blot |
Human embryonic kidney cells, B cells, stem cells | 23622248 22473208 17252019 |
| MCL1 |
MCL1 Apoptosis Regulator, BCL2 Family Member |
HITS-CLIP, microarray, Immunohistochemistry, Luciferase reporter assay, qRT-PCR, Western blot, PCR array | Human embryonic kidney cells, leukemic cells, liver | 22473208 18362358 23594563 28097098 |
| MFN2 |
Mitofusin 2 |
Proteomics, luciferase reporter assay, western blot, CLASH |
Breast cells, lungs, Human embryonic kidney cells | 18668040 27640178 23622248 |
| MYC | MYC Proto-Oncogene, BHLH Transcription Factor |
TRAP, Western blot, CLASH, Luciferase reporter assay, Western blot, Reporter assay; Western blot, qRT-PCR, Microarray; Sequencing. |
Bone cells, mouse embryonic fibroblasts, breast cells, kidney cells, cervical cells, human fibroblasts, oral ephitilium, stem cells, lymphoblastoid cells, bladder cells |
24510096 18695042 23622248 19696787 21294122 21297663 22159222 20371350 25572695 |
| RNF115 |
Ring Finger Protein 115 | HITS-CLIP | Kidney cells, cervical cells, Neuroblastoma cells, mouse embryonic fibroblasts, glial cells | 23313552 23824327 |
| SENP1 |
SUMO Specific Peptidase 1 |
HITS-CLIP, MIRT025428, PAR-CLIP |
Human embryonic kidney cells |
22473208 20371350 21572407 |
| SLC1A5 | Solute Carrier Family 12 Member 6 | HITS-CLIP | Neuronal mouse cells, mouse Primary Embryonic Fibroblasts | 23313552 |
| SLC6A4 |
Solute Carrier Family 6 Member 4 |
qRT-PCR, western blot, HITS-CLIP |
Lungs, brain |
22940131 23313552 23313552 |
| SLC7A11 |
Solute Carrier Family 7 Member 11 | PAR-CLIP HITS-CLIP |
Cervix cells, mouse neural progenitor cells, human retinal epithelial cells, primary mouse embryo fibroblasts, Human Embryonic Stem Cells, kidney cells | 23313552 22012620 20371350 21572407 |
| TMOD3 | Tropomodulin 3 | HITS-CLIP Proteomics |
Cervical cells colorectal cells |
23313552 21566225 |
Table 4.
miRNA gene interaction and possible biochemical pathways involved.
| Biochemical Pathways |
miRNA | Validated target genes |
|---|---|---|
| DNA replication- apoptosis | hsa-miR-16-5p hsa-miR-17-5p hsa-miR-20a-5p hsa-miR-34a-5p |
BCL2 |
| DNA damage telomere stress - senescence pathways |
hsa-miR-16-5p hsa-miR-17-5p hsa-miR-20a-5p hsa-miR-34a-5p |
CYCS |
|
Uncontrolled tumor growth and invasion |
hsa-miR-16-5p hsa-miR-17-5p hsa-miR-20a-5p hsa-miR-34a-5p |
E2F3 |
|
Apoptosis- Bcl2 pathway-drug resistance |
hsa-miR-16-5p hsa-miR-17-5p hsa-miR-20a-5p hsa-miR-34a-5p |
MCL1 |
| Promotion of tumor growth- drug resistance-poor survival |
hsa-miR-16-5p hsa-miR-17-5p hsa-miR-34a-5p hsa-miR-106a-5p |
MFN2 |
| Cancer cell growth and survival-drug resistance-poor survival |
hsa-miR-16-5p hsa-miR-17-5p hsa-miR-20a-5p hsa-miR-34a-5p |
MYC |
|
p53 pathway- proliferation and energy metabolism |
hsa-miR-20a-5p hsa-miR-16-5p hsa-miR-106a-5p hsa-miR-223-5p |
RNF115 |
| Cell cycle deregulation and cell proliferation-drug resistance | hsa-miR-20a-5p hsa-miR-16-5p hsa-miR-34a-5p hsa-miR-223-5p |
SENP1 |
| Cellular transformation and growth | hsa-miR-16-5p hsa-miR-20a-5p hsa-miR-16-5p hsa-miR-34a-5p hsa-miR-106a-5p |
SLC1A5 |
| C-MYC pathway-poor survival | hsa-miR-16-5p hsa-miR-20a-5p hsa-miR-16-5p hsa-miR-106a-5p |
SLC6A4 |
| Ferroptosis- tumor progression | hsa-miR-20a-5p hsa-miR-16-5p hsa-miR-106a-5p hsa-miR-223-5p |
SLC7A11 |
| Cancer progression- EGFR/PI3K/AKT or MAPK/ ERK signaling pathways | hsa-miR-20a-5p hsa-miR-16-5p hsa-miR-34a-5p hsa-miR-106a-5p |
TMOD3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



